 Carlos Murga-Zamalloa
Carlos Murga-Zamalloa Kedar Inamdar
Kedar Inamdar
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol., 20 December 2022
Sec. Hematologic Malignancies
Volume 12 - 2022 | https://doi.org/10.3389/fonc.2022.1099265
This article is part of the Research TopicChallenges in peripheral T-cell lymphomas: from biological advances to clinical applicabilityView all 16 articles
Mature T-cell lymphomas represent neoplastic expansions of T-cell lymphocytes with a post-thymic derivation. Most of these tumors feature aggressive clinical behavior and challenging histopathological diagnosis and classification. Novel findings in the genomic landscape of T-cell lymphomas are helping to improve the understanding of the biology and the molecular mechanisms that underly its clinical behavior. The most recent WHO-HAEM5 classification of hematolymphoid tumors introduced novel molecular and histopathological findings that will aid in the diagnostic classification of this group of neoplasms. The current review article summarizes the most relevant diagnostic features of peripheral T-cell lymphomas with an emphasis on the updates that are incorporated at the WHO-HAEM5.
Mature T and NK cell lymphomas represent approximately 5-10% of all non-Hodgkin lymphomas globally. With few exceptions, these groups of neoplasms are usually diagnosed at an advanced stage in adult patients. They feature an aggressive clinical course, with overall survival of 3 to 5 years after initial diagnosis (1–6). Currently, more than 20 different types of mature T-cell lymphomas (excluding primary cutaneous T-cell lymphomas) exist (Figure 1). The diagnosis is usually challenging due to the lack of specific molecular markers and overlapping morphological features.
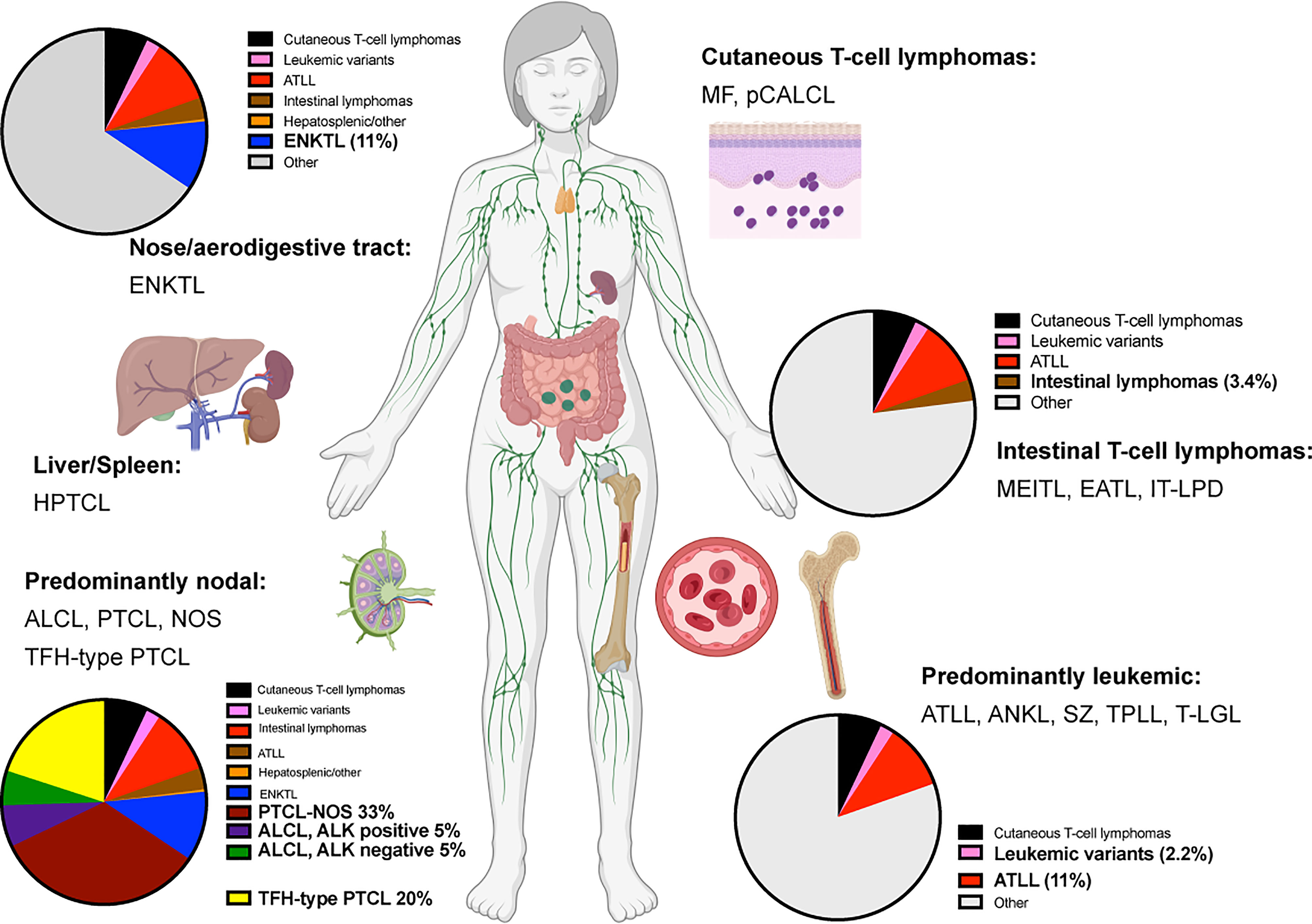
Figure 1 Predominant anatomical presentation and relative frequency of mature T-cell lymphomas. Abbreviations: Extranodal T/NK-cell lymphoma, nasal type (ENKTL); Hepatosplenic T-cell lymphoma (HPTCL); Anaplastic large cell lymphoma (ALCL); Peripheral T-cell lymphoma, non-otherwise specified (PTCL, NOS); T-follicular helper type peripheral T-cell lymphoma (TFH-PTCL); Mycosis Fungoides (MF); Primary cutaneous anaplastic large cell lymphoma (pCALCL); Monomorphic epitheliotropic intestinal T-cell lymphoma (MEITL), Enteropathy-associated T-cell lymphoma (EATL); Indolent T-cell lymphoproliferative disease of the gastrointestinal tract (IT-LPD); Adult T-cell leukemia/lymphoma (ATLL); Aggressive NK-cell leukemia (ANKL); Sèzary Syndrome (SZ); T-cell prolymphocytic lymphoma (T-PLL); T-cell large granular lymphoma (T-LGL).
The continuous discovery of the genomic landscape and mutational signatures of T-cell lymphomas is helping to identify novel molecular biomarkers that will improve the classification, patient risk stratification and introduce novel tailored therapies. The current review focuses on the relevant pathological and molecular findings that were incorporated in the upcoming World Health Organization classification of Haematolymphoid Tumours (WHO-HAEM5) (7). The updated WHO-HAEM5 has introduced organizational changes that aid in the differential diagnosis of mature T-cell lymphomas with a leukemic presentation. Modifications in the terminology of entities such as indolent T-cell lymphoproliferative disease of the GI tract and peripheral T-cell lymphoma of T-follicular helper origin are also included. Finally, the WHO-HAEM5 included novel genomic findings to help recognize aggressive forms in entities such as T-cell large granular leukemia (T-LGL) and Adult T-cell leukemia/lymphoma (ATLL).
This section encompasses the mature T-cell neoplasms characterized by predominant leukemic presentation (Table 1). This section also included Sèzary Syndrome to emphasize the primary site of presentation.
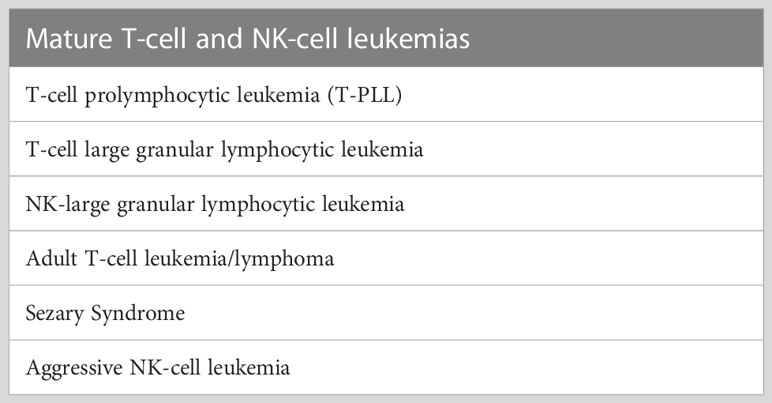
Table 1 List of disease entities with a predominant leukemic presentation.
Is characterized by the leukemic expansion of neoplastic post-thymic T-cell lymphocytes. More than 90% of the cases feature chromosomal abnormalities that involve the 14q32 or Xq28 loci associated with the increased cytoplasmic expression of TCL1A, TCL1B, or MTCP1. Immunophenotypically, the tumor cells are characterized by CD4 expression in 75% of the cases and positive expression of both CD4 and CD8 markers in 25% of the cases (8, 9). The WHO-HAEM5 incorporated diagnostic recommendations from the T-PLL international study group (TPLL-ISG) assembled in 2017 (10). The proposed guidelines indicate that at least 2 (out of 3) major criteria must be met to establish a diagnosis of T-PLL (Table 2). The major criteria include defining T-PLL characteristics, including peripheral blood/bone marrow involvement, specific genetic aberrancies (TCL1A/B and MTCP), and T-cell clonality (Table 2). Importantly, less than 10% of the cases will feature characteristic clinical features of T-PLL, yet genetic aberrancies of TCL1A, TCL1B, or MTCP are not identified. This group of patients is recognized as TCL1-family negative T-PLL. In these scenarios, the proposed diagnostic criteria indicate that at least one chromosomal abnormality specific to T-PLL or the involvement of T-PLL-specific sites (spleen, effusions, skin, and CNS) must be present to establish a definitive diagnosis of T-PLL (1 minor criteria, Table 2).
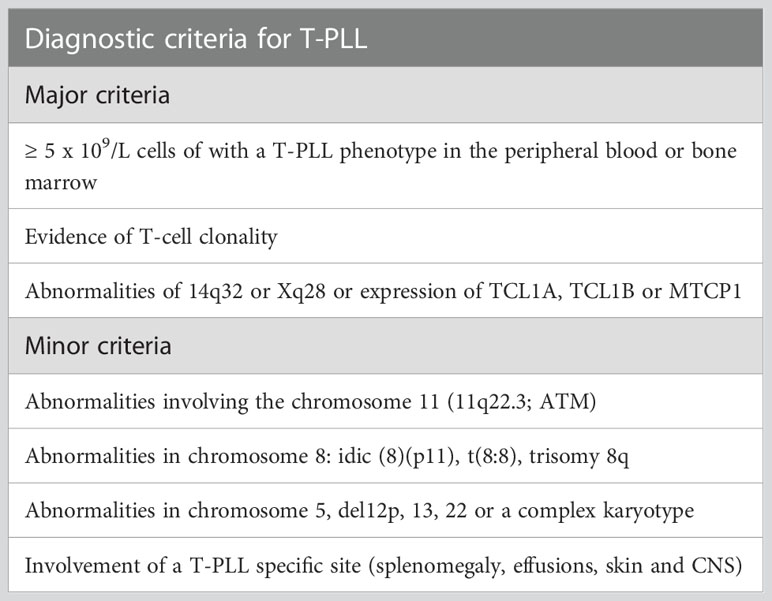
Table 2 Proposed diagnostic criteria for T-PLL by the T-PLL international study group (TPLL-ISG). Adapted from Staber et al. (10).
Is defined by a persistent (at least 6 months) increase in the number of circulating large granular lymphocytes (2-20 x 109/L). About 40-50% of the patients will develop neutropenia, 30-40% anemias, and 10-25% splenomegaly. The updated WHO-HAEM5 incorporated novel molecular findings from two studies that established a correlation between the genomic landscape and clinical characteristics.
The first study (Sannikommu et al.) conducted a retrospective analysis of 224 patients with T-LGL (median follow-up of 36 months). It evaluated the clinical outcomes, therapy responses, and the mutational status of STAT3 (11). In this study, mutations in STAT3 were identified in 36% of the patients, and the most frequent change was Y640F-STAT3. The study demonstrated that patients with STAT3 mutations are more likely to feature neutropenia and anemias. However, no differences in overall survival were identified when patients were stratified according to the mutational status of STAT3.
The second (Barila et al.) is a retrospective study that evaluated 169 patients diagnosed with T-LGL between 1992 and 2018 (12). STAT3 mutations were identified in 60% of the patients with CD8+ T-LGL (n=65). However, mutations in STAT3 were not detected in patients with CD4+/CD8neg/dim T-LGL. STAT5b mutations were detected in 34% of CD4+/CD8neg/dim T-LGL patients and not in CD8+ T-LGL patients. Correlation with clinical characteristics showed that patients with STAT3 mutations were characterized by a higher frequency of neutropenia, anemia, and transfusion-dependent anemia. Multivariate analysis demonstrated that mutations in STAT3 were independently associated with reduced overall survival (267 months vs. not-reached).
The study by Barila et al. also included 36 patients with CLPD-NK (12). The clinical characteristics of CLPD-NK were like the T-LGL counterparts. Approximately 26% of the patients featured anemia, 48% neutropenia, and 13% splenomegaly. However, mutations in STAT3 were identified only in 6% of the patients, and STAT5b mutations were not detected. Expression of CD94 was identified in 94% of the CLPD-NK cases. In contrast, CD94 expression was detected in 14% of Tα/β T-LGL and 52% of Tγ/δ T-LGL cases. Due to the clinical similarities with T-LGL, the WHO-HAEM5 has renamed this entity NK-large granular lymphocytic leukemia (NK-LGL).
Is an aggressive T-cell neoplasm caused by infection of the human T-cell leukemia virus type 1 (HTLV-1). Therefore, it is predominantly diagnosed in HTLV-1 endemic areas including Japan, Central America, South America, and intertropical Africa (13). The neoplastic lymphocytes show variable morphology and range from small to large forms, with irregular nuclear contours, ‘flower-like cells,’ and occasionally vacuolated cytoplasm (14). CD3/CD4 positive, mature T-cells characterize the immunophenotype with co-expression of CD25, FoxP3, and lack of CD7 expression (14). The most common presentation (acute type, ~60%) and the chronic variant (~15% of cases) are characterized by leukemic involvement with hepatosplenomegaly. Predominantly nodal and cutaneous manifestations occur in the smoldering and lymphomatous variants (~25% of cases). Those two variants are more frequent in geographical areas where HTLV-1 is nonendemic, and the diagnosis is usually problematic because those subtypes can mimic cutaneous T-cell lymphomas or, more rarely, anaplastic large cell lymphoma (15). The prognosis is poor, regardless of the clinical presentation, and ranges from 1 year to 2 years after diagnosis. HTLV-1 serology testing is non-diagnostic of ATLL, and direct detection of viral transcripts in the neoplastic cells is critical for a definitive diagnosis in cases where the clinical and histopathological presentation is suggestive of different subtypes of T-cell lymphomas (15, 16).
The updated version of WHO-HAEM5 has incorporated novel genomic findings from two recent studies. The first study (Kogure et al.) identified recurrent loss-of-function mutations in the CIC-L/ATXN1 complex after performing whole-exome sequencing in 150 ATLL patients (17). The CIC-L gene encodes a transcriptional repressor that complexes with the ATXN1 protein. Mutations or structural variations in CIC-L and ATXN1 were mutually exclusive in ATLL patients, and in combination, alterations in the ATXN1 and CIC-L complex were identified in 53% of the cases (17). To support the oncogenic role of CIC-L/ATXN1 during the neoplastic expansion of ATLL, murine models demonstrated that conditional deletion of CIC-L in CD4+ T-cell lymphocytes was associated with the proliferation of CD4+CD25+CD127-FoxP3+ regulatory T-cells.
A second study evaluated the clinical outcomes and the genetic landscape of 463 ATLL patients (18). Patients with aggressive forms of ATLL displayed a higher number of mutations. The 3-year overall survival rate was 54% for patients with 0 to 1 mutation, 39% for those with 2-5 mutations, and 19% for those with more than 6 mutations (18). Multivariate analysis demonstrated that older age (more than 70 years), PRKCB mutations, and PD-L1 amplifications were independent prognostic factors for poor overall survival. Copy number amplifications of PD-L1 were more frequent in the aggressive group and were predictive of poor outcomes in patients with indolent and aggressive forms of the disease. Among the patients with indolent clinical behavior, IRF4 mutations, PD-L1 amplifications, and CDKN2A deletions predicted poor outcomes (18). Overall, these findings highlight the relevance of including genomic profiling for the classification and prognosis of ATLL.
Is a rare NK-cell neoplasm characterized by a fulminant clinical course with a median overall survival of 2 months or less after diagnosis. A leukemic presentation and occasional skin and CNS involvement characterize this entity. The WHO-HAEM5 included under this category the cases of NK/T cell lymphoma with an intravascular presentation that were previously considered a variant of extranodal NK/T-cell type lymphoma. In addition, novel clinical findings and mutational analysis are included under this category in the updated WHO-HAEM5.
A recent large retrospective evaluated the molecular profile and clinical features of 161 individuals diagnosed with ANKL in China (19). The peak incidence was 21 and 30 years old, with a median overall survival of 55 days. A group of the patients (16%) demonstrated a prolonged prodromal disease (subacute ANKL) that was characterized by fever, lymphocytosis, generalized lymphadenopathy, and hepatosplenomegaly (infectious mononucleosis-like symptoms). The prodromal phase has a median duration of 115 days, precedes the fulminant onset of ANKL, and the overall mean survival in this group of patients is 213 days. In contrast, patients without a prodromal phase (‘classic’ ANKL) have a median overall survival of 44 days. A targeted sequencing panel identified similar frequencies of mutations in the JAK-STAT pathway between the two groups. Importantly, mutations in TP53 were detected in 38% of the ‘classic’ ANKL patients, and no mutations in TP53 were identified in the subacute group (19).
A second study evaluated the genomic landscape of 10 ANKL patients utilizing whole-exome sequencing. The mutational spectrum of ANKL clustered separately from other related mature T-cell lymphomas with leukemic presentation (T-LGLL, T-PLL, and CPLD-NK) (20). The most recurrently mutated genes were DDX3X (29%) and STAT3 (21%), and mutations in TP53 were detected in a single patient. The most frequent mutations in ANKL were also seen in NK-T-cell lymphoma, nasal type (NKTCL); therefore, no specific mutations were identified in the ANKL that can help distinguish those cases from NKTCL (20).
Primary intestinal T-cell lymphomas constitute a diverse group of neoplasms predominantly derived from resident intestinal T-cell lymphocytes. Apart from NK-cell enteropathy, these neoplasms feature an aggressive clinical course and a poor prognosis. Within this group, the neoplastic proliferations that arise secondary to celiac disease are defined as enteropathy-associated T-cell lymphoma (EATL). EATL is an aggressive disease predominantly composed of CD8+ T-cells with the expression of TCR-α/β (21, 22). Monomorphic epitheliotropic intestinal T-cell lymphoma (MEITL) is more frequent in Asia and is composed of medium-sized lymphocytes with a homogeneous appearance, epitheliotropism, and frequent expression of CD56 (23–27) (Figure 2). The category of intestinal T-cell lymphomas non-otherwise specified (ITCL-NOS) remains as an entity that fails classification with the current schemes (Table 3; Figure 3), with very few case reports available (28–30). The WHO-HAEM5 has modified the designation of indolent-type T-cell lymphoproliferative disease of the GI tract and has incorporated novel molecular findings in NK-cell enteropathy.
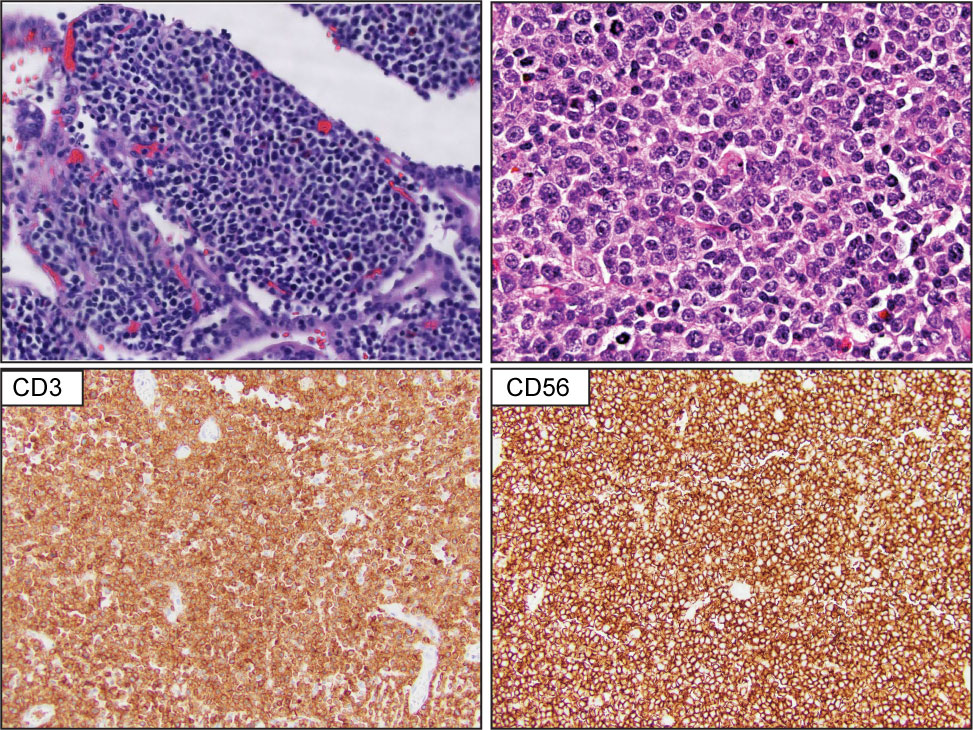
Figure 2 Monomorphic epitheliotropic intestinal T-cell lymphoma (MEITL). Upper panel show representative H&E images that highlight extensive epitheliotropism by medium size lymphocytes with a homogeneous appearance. The lower panel demonstrates that the atypical lymphocytes are composed of CD3-positive and CD56-positive T-cell lymphocytes.
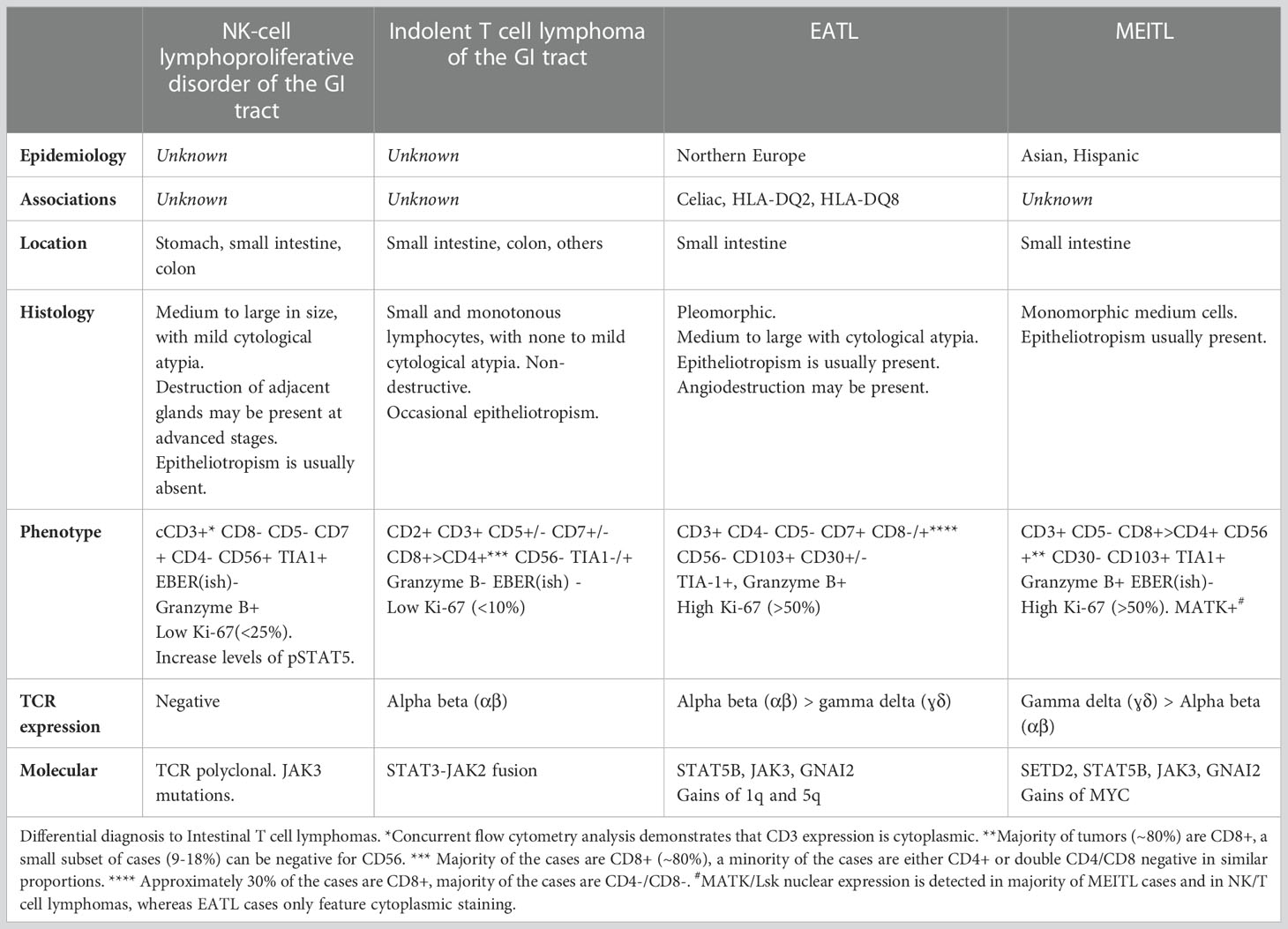
Table 3 Histological and molecular features of intestinal T-cell lymphomas. Adapted from Osmani et al. (27).
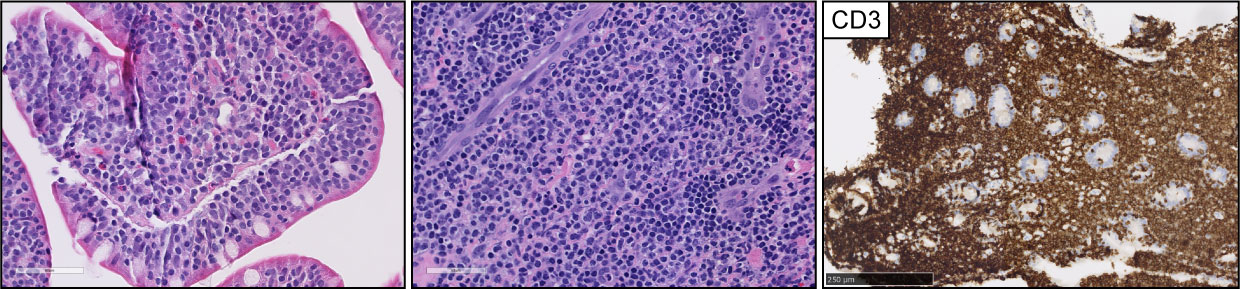
Figure 3 Intestinal T-cell lymphoma, non-otherwise specified (ITCL-NOS). H&E pictures demonstrate dense infiltrates of medium to large, atypical lymphocytes with epitheliotropism. The infiltrates were composed of CD3-positive T-cell lymphocytes with (not shown) aberrant CD7 and CD5 loss and negative expression of CD56.
Is morphologically characterized by superficial infiltrates composed of small lymphocytes with minimal cytological atypia that exhibit a Ki-67 proliferation index that is less than 10% (31). This entity is characterized by chronic and relapsing gastrointestinal symptoms, including diarrhea, dyspepsia, and vomiting (32, 33). In some cases, disease progression with an aggressive clinical course has been reported (34, 35). Therefore, the term ‘lymphoproliferative’ has been replaced by ‘lymphoma’ to highlight that this entity represents a clonal disease with a risk of transformation into more aggressive forms.
Has been renamed indolent NK-cell lymphoproliferative disorder of the GI tract (iNKLPD). This entity is characterized by non-specific gastrointestinal symptoms, including abdominal pain, constipation, and reflux, with a chronic/relapsing course (36). The tumor cells feature mild cytological atypia, and the Ki-67 proliferation index is usually in the 25%-50% range; in contrast to ENKTCL, these tumors are negative for EBV expression. Additional case series reports have confirmed the indolent clinical course, with no reported cases of progression to more advanced stages (37). A recent study in 7 patients identified somatic mutations in JAK3 in 30% of the patients; additional mutations in other genes, including IFG1R and AURKB, were also detected at lower frequencies (38). The same study demonstrated increased expression of phosphorylated STAT5 in 100% of cases (n = 7) (38), supporting the idea that this process constitutes a neoplastic lymphoproliferative disease.
Nodal involvement is the most frequent mode of presentation of T-cell lymphomas, and the three entities discussed below account for more than 60% of all mature T-cell neoplasms. Secondary nodal involvement by other T-cell lymphomas, such as ENKTL (discussed in 5.1) or cutaneous T-cell lymphomas, is not uncommon. However, their primary presentation site is different from nodal, and the involvement of lymph nodes can occur during the evolution of the disease.
ALCL is the third most common nodal T-cell lymphoma and accounts for approximately 11-13% of peripheral T-cell lymphomas (39, 40). Morphologically is characterized by large anaplastic cells, ‘hallmark cells,’ organized in a cohesive pattern, with uniform and strong expression of CD30 in more than 80% of the tumor cells (41) (Figure 4). Translocations involving the tyrosine kinase ALK are present in approximately 50% of ALCL cases. The uncontrolled activation of the ALK-kinase defines a phosphoproteomic and transcriptional signature that drives the oncogenic program of ALK+ ALCL cases (42–44), and ALK+ ALCL constitutes a specified entity. ALK-negative ALCL comprises a heterogenous group and is characterized by inferior clinical outcomes compared to ALK+ ALCL. Specific genomic rearrangements within this group, including DUSP22, and TP63 fusions, are associated with differential clinical outcomes (45–48). The WHO-HAEM5 has included novel genomic findings from two studies that will help understand the biology of ALCL.
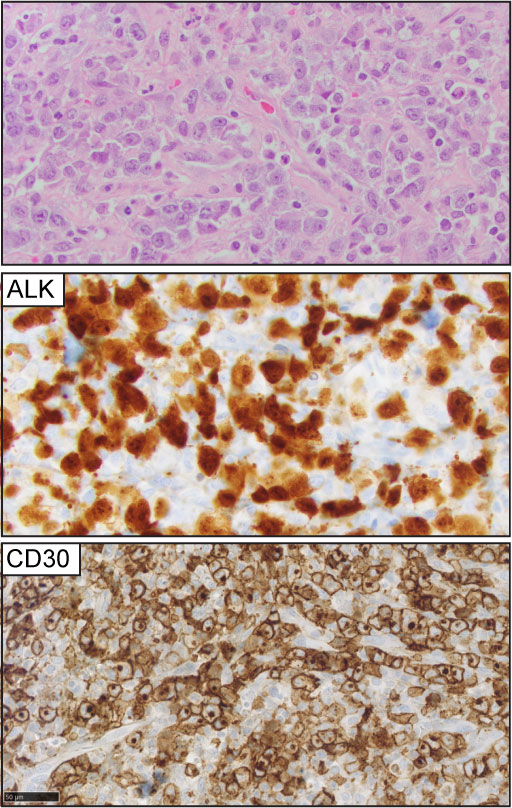
Figure 4 Anaplastic large cell lymphoma (ALCL). Representative (H&E) pictures of ALCL highlight the clustering of large, atypical lymphocytes with a variable amount of cytoplasm, open chromatin, and ‘kidney-shaped’ nuclei. ALK expression is detected in 50% of the cases (ALK+ ALCL). CD30 expression is homogeneously positive in at least 80% of the tumor cells.
Hapgood et al. (48) correlated the outcomes and molecular features of 62 ALK-negative ALCL patients. The findings demonstrated inferior clinical outcomes in patients with DUSP22 rearrangements (n = 12, 44% five-year OS) in comparison to ≥80% 5-year overall survival (OS) that was described in the previous case series (47). These findings indicate that additional molecular mechanisms can modify the outcomes of these patients. Consistent with this, a recent cohort study in 82 ALCL patients with systemic ALCL (ALK+ and ALK-) identified that mutations in TP53 and STAT3 are associated with a worse prognosis independent of ALK status (49).
The second study demonstrated the presence of specific super-enhancer (SE) regions enriched with BATF3 sites in ALCL cell lines and patient samples. The findings further revealed that the transcriptional activity of BATF3 can mediate the expression of IL2R. Importantly, increased expression of IL2R was predominantly detected in ALCL cases compared to different peripheral T-cell lymphomas, including AITL and PTCL-NOS. The findings indicate that chromatin remodeling may play a critical role during the growth of these groups of tumors and that IL2-dependent signaling plays a pivotal role in substituting T-cell receptor signaling in ALCL tumors (50).
Finally, the WHO-HAEM5 incorporated findings from a recent study by Fitzpatrick et al. that identified JAK2 fusions in 6% of systemic CD30+ ALK-negative T-cell lymphomas (n=97) (51). Among the cases with JAK2 translocations, three fulfilled the diagnostic criteria of ALK-negative ALCL, and the other three were diagnosed as CD30+ PTCL-NOS. All the cases with JAK2 translocations featured at least a proportion of ALCL-like large anaplastic tumor cells, in addition to Reed-Sternberg (RS)-like cells. Co-expression of CD30 and CD15 was present in the large anaplastic component in 80% of the cases. In all instances, the tumor cells were negative for PAX-5, and T-cell clonality was established. Due to the limited number of cases, a comparison of differential clinical outcomes was not possible (51). The presence of RS-like cells with co-expression of CD30 and CD15 can be a diagnostic pitfall, and recognition of this subset of cases is critical during diagnosis.
Although morphologically and immunophenotypically indistinguishable from ALK-negative ALCL, is a distinct clinical entity primarily associated with textured breast implants, generally associated with an indolent clinical course. It is a rare disease with an estimated risk of 1 case in 4000-30,000 women undergoing breast implant surgeries. The median interval from surgery to the development of BIA-ALCL is 8-11 years (52). Diagnosis is often made in the peri-implant effusion fluid as the initial specimen. Cytospin preparations from the fluid show large, pleomorphic tumor cells with prominent nucleoli, abundant vacuolated cytoplasm, and irregular cytoplasmic membranes. Hallmark cells seen in other ALCL types are frequently present. Capsulectomy specimens show varying degrees of infiltration by the tumor cells. In the early stage, they often line the capsule, whereas, in the most advanced stage, they infiltrate through the fibrous capsule to form a mass. Axillary lymph nodes can be involved in approximately 20-30% of cases (53). Capsular invasion, mass formation, and lymph node involvement are adverse prognostic factors (53, 54).
In the updated WHO-HAEM5, BIA-ALCL was upgraded to a definite entity based on its unique clinical, genomic, and molecular features. A comprehensive study of the genetic subtype of BIA-ALCL cases by Feldman et al. (55) shows that there is oncogenic activation of the JAK-STAT3 signaling pathway caused by mutations of somatic mutations of STAT3, JAK1, and JAK2. Laurent et al. (56) also demonstrated that the genomic alterations in BIA-ALCL include loss-of-function modifications of the epigenetic modifiers KMT2C, KMT2D, CHD2, and CREBBP, in up to 74% of cases.
The WHO-HAEM5 highlighted the role of allergic inflammation mediated by the secretion of IL-13 in the pathogenesis of BIA-ALCL (57). The frequent presence of eosinophils and mast cells in the tumor microenvironment of BIA-ALCL further supports the role of allergic inflammation during the pathogenesis of the disease (58, 59).
Finally, WHO-HAEM5 incorporated the role of immune evasion during the disease progression of BIA-ALCL. Tabanelli et al. (60) evaluated PD-L1 and pSTAT expression and PD-L1 copy number alterations (CNAs) in a cohort of 9 BIA-ALCL cases. The findings indicated that 56% of BIA-ALCL overexpressed PD-L1, and 33% of cases harbored CNAs at 9p24.1. Consistent with this, the results also demonstrated variable proportions of PD1+ T-cells and PD-L1+ tumor-associated macrophages (TAMs) in both PD-L1+ and PD-L1-negative BIA-ALCLs, suggesting the presence of an active PD-1/PD-L1 axis (60).
Peripheral T-cell lymphomas derived from T-follicular helper (T-FH) lymphocytes encompass a spectrum of aggressive neoplasms with poor clinical outcomes (61). Two entities in this group are characterized by specific morphological features: angioimmunoblastic T-cell lymphoma (AITL) and Follicular T-cell lymphoma (F-TH). A third group comprises neoplastic expansions of T-follicular helper T-cell lymphocytes that do not fit the characteristic morphological patterns of AITL and T-FH lymphoma. This group was defined in the latest WHO-HAEMR4 as Nodal Peripheral T-cell lymphoma with TFH phenotype (PTCL-TFH).
AITL is the second most common type of nodal T-cell lymphoma and accounts for 15-20% of peripheral T-cell lymphomas. Morphologically is characterized by effacement of the nodal architecture, with an expansion of arborizing post-endothelial venules with clusters of neoplastic lymphocytes with clear cytoplasm (Figure 5). These areas characteristically feature an expansion of follicular dendritic cell meshworks. The diagnosis can be difficult because the tumor cells represent a minority of the infiltrates, and the tumor microenvironment features positive RS-like cells for EBV with eosinophilic infiltrates (62).
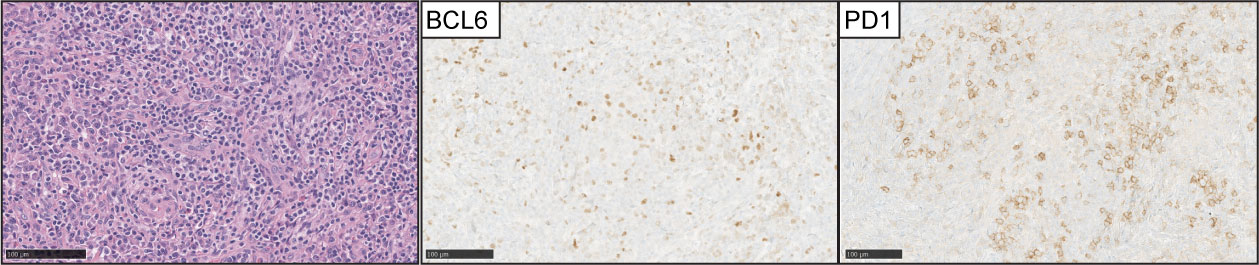
Figure 5 Angioimmunoblastic T-cell lymphoma (AITL). (Left panel) H&E images show characteristic branching of post-endothelial venules and adjacent lymphocytes with clear cell cytoplasm. Expression of BCL-6 and PD1 is detected in the neoplastic T-cell lymphocytes in the majority of cases.
Is morphologically distinct from AITL as it doesn’t feature expanded post-endothelial venules surrounded by T-cell lymphocytes with clear cytoplasm. In contrast, two main architectural patterns are described; the most common pattern resembles progressive transformation of the germinal centers (PTGC-L), where the atypical lymphocytes are expanded in poorly defined nodules that are surrounded by numerous B-cell lymphocytes. The second pattern is characterized by well-demarcated nodular expansion of atypical lymphocytes, resembling B-cell follicular lymphoma. Consistent with a T-FH immunophenotype, the atypical cells are CD4 positive and feature co-expression of ICOS, PD1, CD10, BCL-6, and CXCL13; importantly, the expression of one or two of the T-FH markers may be absent. CD21 and CD23 highlight distorted follicular dendritic cell (FDC) meshwork’s overlapping the atypical cells; in contrast to AITL, the FDCs are not expanded or associated with proliferating post-endothelial cells (63–66).
Was a new disease entity introduced in WHO-HAEM4R. This group of cases does not feature the characteristic morphological features of AITL or F-TH lymphoma (Figure 6). However, the expression of at least two (preferentially 3) T-FH markers in addition to CD4 is required for classification. ICOS and PD-1 are expressed in most cases. However, ICOS is expressed in 43-52% of the PTCL-NOS cases, and PD-1 is expressed in approximately 60% of PTCL-NOS cases (67, 68). CD10, BCL6, and CXCL13 are the most specific markers and rarely are expressed in PTCL-NOS cases (68).
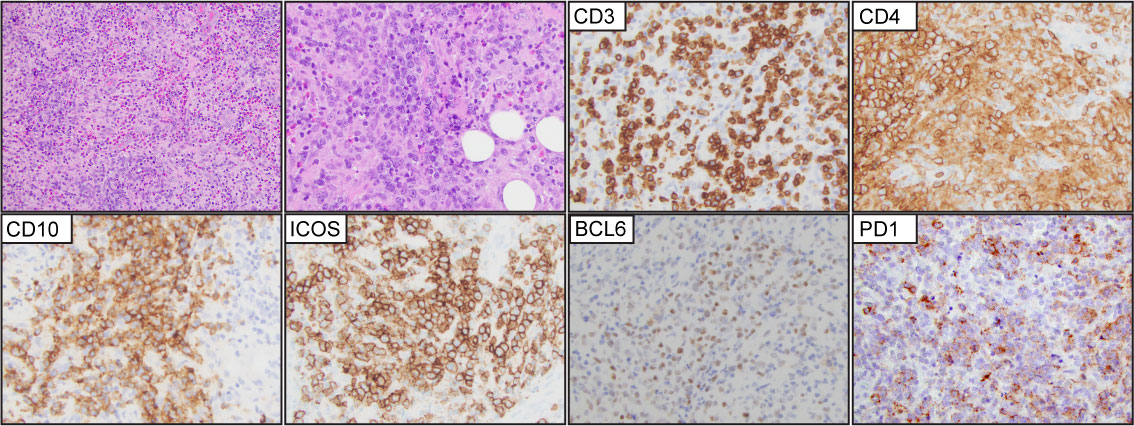
Figure 6 T-follicular helper type peripheral T-cell lymphoma (PTCL-TFH). Representative (H&E) images demonstrate effacement of the nodal architecture by medium to large, atypical lymphocytes that are organized in small clusters with numerous background histiocytes and eosinophils. The tumor cells are positive for CD3, CD4, CD10, ICOS, BCL6, and PD1.
The family of peripheral T-cell lymphomas with a T-follicular helper (T-FH) origin share similar clinical features and are characterized by disseminated disease at diagnosis with a 5-year overall survival of 30-35% (61). The morphological features can transition between T-FH subtypes over subsequent biopsies or transform into PTCL-NOS. The genetic profile includes similar frequencies of mutations in DNMT3A, RHOA, and TET2 (61, 69, 70), and IDH2 mutations are predominantly detected in AITL. The gene expression profiles are similar, and these groups of neoplasms cluster in proximity and apart from other peripheral T-cell lymphomas, including ALCL and PTCL-NOS (69). However, recent studies show that a subset of PTCL-TFH cases features gene expression profiles closer to the PTCL-NOS group (69). To emphasize that this group of neoplasms constitutes a spectrum with morphological plasticity, this family of neoplasms has been designated nodal T-follicular helper lymphomas (nTFH) in the updated WHO-HAEM5. Therefore, the previous designations of ‘angioimmunoblastic T-cell lymphoma,’ ‘follicular T-cell lymphoma,’ and ‘nodal peripheral T-cell lymphoma with TFH phenotype’ will be renamed nTFHL angioimmunoblastic type (nTFHL-AI), nTFHL follicular-type (nTFHL-F) and nTFHL non-otherwise specified (nTFHL-NOS).
This group remains the most prevalent in the United States and constitutes a ‘waste-basket’ category when other disease entities are excluded from the current classification schemes. Recent study series that correlated clinicopathological features with genomic/molecular data has helped to identify distinct groups that are no longer included within this classification, such as Nodal EBV+ T/NK-cell lymphomas and Nodal T-follicular helper lymphomas, NOS.
Previous studies demonstrated that the expression of the transcription factor GATA-3 is enriched in a subset of PTCL-NOS cases with characteristic transcriptional profiles, worse overall survival, and resistance to chemotherapy (71–74). A second, albeit more heterogeneous subtype, highly expresses the transcription factor TBX21 and is similarly enriched for its gene targets. An immunohistochemistry algorithm that includes CD183, CCR4, GATA-3, and TBX21 has been proposed to identify these cases (75) (Figure 7). The upregulated expression of GATA-3 is secondary to the engagement of T-cell receptors in the neoplastic cells, and this is likely secondary to specific interactions with the tumor microenvironment (76–78). Due to insufficient clinicopathological and prognostic findings, the current WHO-HAEM5 did not recognize this group of tumors as a specific subtype of PTCL.
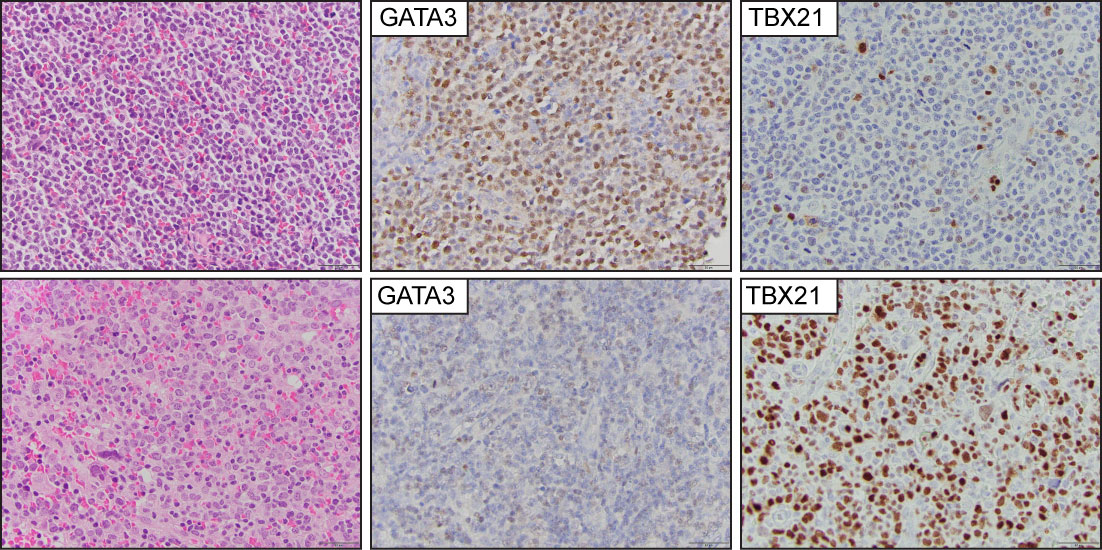
Figure 7 Peripheral T-cell lymphoma, non-otherwise specified (PTCL-NOS). Representative images from two PTCL-NOS cases. The upper panel shows a case classified in the GATA3 positive group, and (the lower panel) highlights a case within the TBX21 group.
The contribution of EBV during the development of lymphoproliferative diseases is well-recognized (79). Importantly, EBV-infected B-cell or T-cells can be identified in the tumor microenvironment of several T-cell lymphomas, including AITL, ATLL, or PTCL-NOS. However, those are likely the result of dysregulated immunosurveillance derived from the primary lymphoma rather than a primary EBV-driven mechanism.
In the setting of immunosuppression, EBV+ T-cell lymphomas are not as common as their B-cell counterparts. However, T-cell lymphoproliferative disorders that occur secondary to EBV infection in immunocompetent hosts are characterized by markedly aggressive behavior and many of those feature dismal outcomes. This category includes T and NK-cell-derived lymphoproliferative disorders in the setting of chronic active EBV infection (T/NK type CAEBV). T/NK type CAEBV will feature EBV+ T or NK-cell expansions that do not feature definitive morphological or immunophenotypic features compatible with a T or NK-cell type lymphoma diagnosis. However, the clinical course is very similar to T-cell lymphomas and is characterized by hepatitis, vasculitis, hemophagocytic syndrome, and end-organ damage (80–84). Aggressive NK-cell leukemia (ANKL) also features EBV+ lymphoma cells and is discussed in a different section (2.5). The WHO-HAEM5 has incorporated novel molecular findings in extranodal T/NK type lymphoma (ENKTL) and included nodal EBV+ T/NK-cell lymphoma as a new disease entity.
ENKTL is an aggressive type of lymphoproliferative disorder predominantly derived from NK cells, with a minority of cases from cytotoxic T-cell origin. Approximately 80% of the cases are localized in the nasal cavity or nasopharynx (85). The remaining 20% of the cases will involve other sites that include skin, testicles, and salivary glands. Morphologically the neoplastic infiltrate can show a broad spectrum of appearances ranging from small cells with minimal atypia to medium to large pleomorphic cells with marked cellular atypia (86, 87) (Figure 8). Necrosis is observed in approximately 60%-80% of the cases, and angiocentricity is present in nearly 50% of the cases (85, 86, 88). Virtually all the cases are EBV+ in more than 80% of the tumor cells (85, 87, 88). An NK-cell immunophenotype is the most frequently observed and is characterized by negative expression of surface CD3, with positive expression of cytoplasmic CD3ε, CD56, and the cytotoxic markers TIA-1 and granzyme B. Approximately 20% of true NK-cell origin tumors (germline TCRγ) will feature negative expression of CD56, however, expression of cytotoxic markers is always present (87–89). Cases with a T-cell origin defined by monoclonal TCRγ rearrangement can account for 20%-40%, and approximately 40% of those will lack CD56 expression (85, 89). However, EBV negative cases with negative expression of CD56 should be designated as PTCL-NOS (87). In recognition that a subset of these cases involves extra-nodal sites other than the nasal cavity, the WHO-HAEM5 has removed the term ‘nasal-type’ from this designation. In addition, the WHO-HAEM5 mentioned novel genomic findings that highlight the relevance of STAT3 mutations in ENKTL and the therapeutic role of PD-L1.
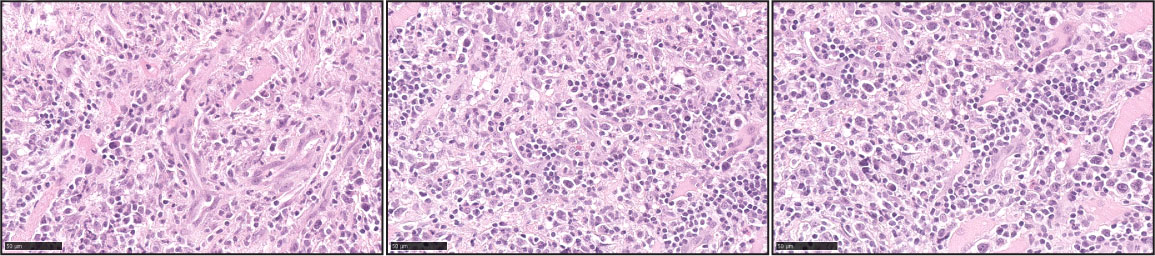
Figure 8 Extranodal T/NK-cell lymphoma, nasal type (ENKTL). Three representative pictures of ENTKL (H&E) highlight the pleomorphism of the atypical infiltrates. The atypical infiltrates can be predominantly composed of small lymphocytes or medium to large atypical forms. Angiocentricity is commonly identified.
A recent study by Song et al. conducted targeted gene capture sequencing in 171 PTCLs, and JAK/STAT mutations were identified in 78% of the ENKTL cases (n=109) (90). Mutations in STAT3 were predominantly detected in ENKTL cases (21%), in contrast to ALCL (3.7%) and PTCL-NOS cases (3.8%). The mutations identified in STAT3 were associated with increased transcriptional activity and higher expression of PD-L1 in-vitro. However, the evaluation of primary ENKTL samples showed that 93% of the cases (n=30) featured increased expression of PD-L1, suggesting that additional molecular mechanisms are involved in PD-L1 expression (90). Consistent with this, Bi et al. demonstrated that the expression of the EBV latent membrane protein 1 (LMP1) promotes PD-L1 expression in ENKTL cell lines in-vitro (91).
Finally, the therapeutic potential of PD-L1 in ENKTL has been evaluated in a recent phase 2 clinical trial that tested the efficacy of the IgG1-PD-L1 antibody avelumab as a single agent in refractory/relapsed (R/R) ENKTL (92). This study by Jin et al. demonstrated a 24% complete response rate in R/R patients that received avelumab (92). The patient responses to avelumab were associated with higher expression of PD-L1 in the tumor cells. However, response rates were not related to the levels of serum soluble PD-L1 (92).
This novel designation encompasses EBV+ NK/T cell lymphomas with a predominant nodal presentation. These tumors are characteristically diagnosed at an advanced stage and feature dismal clinical outcomes with overall survival of 1.5-3.5 months after diagnosis (93–95). This new entity differs from ENKTCL with secondary nodal involvement, as the former will primarily be present at extranodal sites. Cases of chronic active EBV infection of T-cell or NK-cell type (T/NK- type CAEBV) are also excluded from this category, as those do not feature morphological features consistent with a lymphoproliferative neoplasm. Finally, the presence of leukemic involvement by neoplastic cells with an NK-cell immunophenotype will be best classified as ANKL.
Other nodal T-cell lymphomas can feature a subset of tumor cells positive for EBV. Therefore, the minimal threshold for the number of EBV+ neoplastic cells varies among the published case series and ranges between 40%-50% (93–95). Immunophenotypically, approximately 60% of the cases will show tumor cells with a T-cell immunophenotype that are positive for CD8 and TβF1. The tumor cells will be less frequently positive for CD4 (93–95). Tumor cells with a characteristic NK-cell immunophenotype have been described. However, those account for less than 10% of the cases described (93, 94, 96).
This group of neoplasms features gene-expression profiles distinct from ENKTL with frequent loss of 14q11.2 and upregulation of PD-L1 (CD174) (93). Functional analysis of the gene-expression profiles demonstrates enrichment for NF-κB signaling (97). Consistent with this and in contrast to PTCL-NOS cases, predominant expression of BIRC3 and p50 is detected (97). Frequent mutations in TET2 (64%, n=14), followed by PIK3CD (33%, n=14) and STAT3 (19%, n=14) (97) characterize the genomic landscape of EBV+ NTNKCL.
Hepatosplenic T-cell lymphoma (HSTCL) is a rare but aggressive type of extranodal T-cell lymphoma that accounts for 1-2% of all peripheral T-cell lymphomas. The tumor cells involve the liver, spleen, and bone marrow in an exclusive intrasinusoidal distribution, often engorging and distending the cords and sinuses in the involved organs (39, 98). The morphology of the atypical infiltrates is characterized by monotonous, medium-sized lymphocytes with irregular nuclear contours, a moderate amount of agranular pale cytoplasm, and inconspicuous nucleoli. In a subset of the cases, the neoplastic cells feature blastoid chromatin, especially at advanced stages (99–101). Immunophenotypically, the neoplastic cells express pan T-cell markers CD2, CD3, and CD7, with frequent co-expression of CD56 and aberrant loss of CD5. A subset of cases is CD8+. However, the majority are negative for both CD4 and CD8. Expression of the cytotoxic granule-associated proteins, TIA1, and granzyme M is commonly observed. Expression of TCR-γ/δ is frequently observed (~75%), while ~25% of cases are TCR-α/β, and in a small number of instances (~5%), TCR-silent (102).
Several cytogenetic abnormalities are reported in HSTCL. The most common are isochromosome 7q [i(7q)] and trisomy 8 and are found in approximately 63% and 50% of cases, respectively (103). A unique molecular signature is identified in HSTCL cases. This is characterized by overexpression of genes encoding NK-cell-associated molecules (FOS and VAV3), the sphingosine-1-phosphatase receptor 5 (S1PR5) involved in cell trafficking, the tyrosine kinase SYK, with down-regulation of tumor suppressor gene AIM1 (104).
While HSTCL predominantly occurs in immunocompetent individuals, 20-30% of cases occur in chronic immune suppression or immune dysregulation associated with autoimmune disorders (99, 100, 105). The WHO-HAEM4 reported that this disease is mainly diagnosed in adolescents and young adults. However, a recent report from the prospective T-cell lymphoma project identified that more than 50% of the patients in their cohort were older than 60 years old. Thus, the WHO-HAEM5 acknowledged that the disease is diagnosed in adolescents, young adults, and older individuals (98).
The WHO-HAEM5 also incorporated findings from a recent study by Mckinney et al. (106) that analyzed the genomic landscape of a large cohort of HSTCL cases with whole-exome sequencing. The results demonstrated frequent mutations in genes involving the JAK/STAT signaling pathway, with STAT3 and STAT5B being the most frequently mutated. In addition, mutations in PI3KCD and the epigenetic regulators SETD2, INO80, TET3, and SMARCA2 were observed in up to 62% of the cases (52, 106, 107).
The diagnosis and classification of T-cell lymphomas require a correlation between the histomorphology, molecular studies, and the site and manner of presentation. To date, few entities such as ALK+ ALCL or T-PLL display specific genetic aberrancies that are the predominant driving force of lymphoma progression and therefore constitute a distinctive feature for classification. In contrast, most T-cell lymphomas share morphological similarities and feature different frequencies of genetic alterations that either amplify the T-cell receptor signaling (e.g., NF-kB, RhoA, GATA3) or bypass it entirely (e.g., JAK/STAT, Pi3K). Therefore, a more significant proportion of mature T-cell lymphomas remain diagnosed in the unclassifiable (NOS) group. Currently, there is a limited understanding of the biology of T-cell neoplasms, which translates into limited successful therapeutic approaches, and these tumors remain an area of unmet need.
KI and CMZ wrote the manuscript. All the authors contributed to the article and approved the submitted version.
CMZ is supported by the American Society of Hematology/Robert Wood Johnson Foundation (Harold Amos Medical Faculty Development Program).
The authors declare that the research was conducted without any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling editor RW declared a past collaboration with the authors.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Oluwasanjo A, Kartan S, Johnson W, Alpdogan O, Gru A, Mishra A, et al. Peripheral T-cell lymphoma, not otherwise specified (PTCL-NOS). Cancer Treat Res (2019) 176:83–98. doi: 10.1007/978-3-319-99716-2_4
2. Broccoli A, Zinzani PL. Peripheral T-cell lymphoma, not otherwise specified. Blood (2017) 129(9):1103–12. doi: 10.1182/blood-2016-08-692566
3. Vega F. Pathology and pathogenesis of T-cell lymphoma. Clin Lymphoma Myeloma Leuk (2020) 20 Suppl 1:S89–93. doi: 10.1016/S2152-2650(20)30474-2
4. Phan A, Veldman R, Lechowicz MJ. T-Cell lymphoma epidemiology: the known and unknown. Curr Hematol Malig Rep (2016) 11(6):492–503. doi: 10.1007/s11899-016-0353-y
5. Lansigan F, Horwitz SM, Pinter-Brown LC, Rosen ST, Pro B, Hsi ED, et al. Outcomes for relapsed and refractory peripheral T-cell lymphoma patients after front-line therapy from the COMPLETE registry. Acta Haematol (2020) 143(1):40–50. doi: 10.1159/000500666
6. Armitage JO. The aggressive peripheral T-cell lymphomas: 2017. Am J Hematol (2017) 92(7):706–15. doi: 10.1002/ajh.24791
7. Alaggio R, Amador C, Anagnostopoulos I, Attygalle AD, Araujo IBO, Berti E, et al. The 5th edition of the world health organization classification of haematolymphoid tumours: Lymphoid neoplasms. Leukemia (2022) 36(7):1720–48. doi: 10.1038/s41375-022-01620-2
8. Herling M, Khoury JD, Washington LT, Duvic M, Keating MJ, Jones D. A systematic approach to diagnosis of mature T-cell leukemias reveals heterogeneity among WHO categories. Blood (2004) 104(2):328–35. doi: 10.1182/blood-2004-01-0002
9. Dearden C. How I treat prolymphocytic leukemia. Blood (2012) 120(3):538–51. doi: 10.1182/blood-2012-01-380139
10. Staber PB, Herling M, Bellido M, Jacobsen ED, Davids MS, Kadia TM, et al. Consensus criteria for diagnosis, staging, and treatment response assessment of T-cell prolymphocytic leukemia. Blood (2019) 134(14):1132–43. doi: 10.1182/blood.2019000402
11. Sanikommu SR, Clemente MJ, Chomczynski P, Afable MG, 2nd, Jerez A, Thota S, et al. Clinical features and treatment outcomes in large granular lymphocytic leukemia (LGLL). Leuk Lymphoma (2018) 59(2):416–22. doi: 10.1080/10428194.2017.1339880
12. Barila G, Teramo A, Calabretto G, Vicenzetto C, Gasparini VR, Pavan L, et al. Stat3 mutations impact on overall survival in large granular lymphocyte leukemia: a single-center experience of 205 patients. Leukemia (2020) 34(4):1116–24. doi: 10.1038/s41375-019-0644-0
13. Malpica L, Enriquez DJ, Castro DA, Pena C, Idrobo H, Fiad L, et al. Real-world data on adult T-cell Leukemia/Lymphoma in Latin America: A study from the grupo de estudio latinoamericano de linfoproliferativos. JCO Glob Oncol (2021) 7:1151–66. doi: 10.1200/GO.21.00084
14. Dahmoush L, Hijazi Y, Barnes E, Stetler-Stevenson M, Abati A. Adult T-cell leukemia/lymphoma: a cytopathologic, immunocytochemical, and flow cytometric study. Cancer (2002) 96(2):110–6. doi: 10.1002/cncr.10480
15. Khanlari M, Ramos JC, Sanchez SP, Cho-Vega JH, Amador A, Campuzano-Zuluaga G, et al. Adult T-cell leukemia/lymphoma can be indistinguishable from other more common T-cell lymphomas. the university of Miami experience with a large cohort of cases. Mod Pathol (2018) 31(7):1046–63. 10.1038/s41379-018-0037-3
16. Takatori M, Sakihama S, Miyara M, Imaizumi N, Miyagi T, Ohshiro K, et al. A new diagnostic algorithm using biopsy specimens in adult T-cell leukemia/lymphoma: combination of RNA in situ hybridization and quantitative PCR for HTLV-1. Mod Pathol (2021) 34(1):51–8. doi: 10.1038/s41379-020-0635-8
17. Kogure Y, Kameda T, Koya J, Yoshimitsu M, Nosaka K, Yasunaga JI, et al. Whole-genome landscape of adult T-cell leukemia/lymphoma. Blood (2022) 139(7):967–82. doi: 10.1182/blood.2021013568
18. Kataoka K, Iwanaga M, Yasunaga JI, Nagata Y, Kitanaka A, Kameda T, et al. Prognostic relevance of integrated genetic profiling in adult T-cell leukemia/lymphoma. Blood (2018) 131(2):215–25. doi: 10.1182/blood-2017-01-761874
19. Tang YT, Wang D, Luo H, Xiao M, Zhou HS, Liu D, et al. Aggressive NK-cell leukemia: clinical subtypes, molecular features, and treatment outcomes. Blood Cancer J (2017) 7(12):660. doi: 10.1038/s41408-017-0021-z
20. Dufva O, Kankainen M, Kelkka T, Sekiguchi N, Awad SA, Eldfors S, et al. Aggressive natural killer-cell leukemia mutational landscape and drug profiling highlight JAK-STAT signaling as therapeutic target. Nat Commun (2018) 9(1):1567. doi: 10.1038/s41467-018-03987-2
21. Di Sabatino A, Biagi F, Gobbi PG, Corazza GR. How I treat enteropathy-associated T-cell lymphoma. Blood (2012) 119(11):2458–68. doi: 10.1182/blood-2011-10-385559
22. Sieniawski M, Angamuthu N, Boyd K, Chasty R, Davies J, Forsyth P, et al. Evaluation of enteropathy-associated T-cell lymphoma comparing standard therapies with a novel regimen including autologous stem cell transplantation. Blood (2010) 115(18):3664–70. doi: 10.1182/blood-2009-07-231324
23. Tan SY, Ooi AS, Ang MK, Koh M, Wong JC, Dykema K, et al. Nuclear expression of MATK is a novel marker of type II enteropathy-associated T-cell lymphoma. Leukemia (2011) 25(3):555–7. doi: 10.1038/leu.2010.295
24. Tian S, Xiao SY, Chen Q, Liu H, Ping J. Monomorphic epitheliotropic intestinal T-cell lymphoma may mimic intestinal inflammatory disorders. Int J Immunopathol Pharmacol (2019) 33:2058738419829387. doi: 10.1177/2058738419829387
25. Tan SY, Chuang SS, Tang T, Tan L, Ko YH, Chuah KL, et al. (epitheliotropic intestinal T-cell lymphoma): A neoplasm of intra-epithelial T-cells with predominant CD8alphaalpha phenotype. Leukemia (2013) 27(8):1688–96. doi: 10.1038/leu.2013.41
26. Yi JH, Lee GW, Do YR, Jung HR, Hong JY, Yoon DH, et al. Multicenter retrospective analysis of the clinicopathologic features of monomorphic epitheliotropic intestinal T-cell lymphoma. Ann Hematol (2019) 98(11):2541–50. doi: 10.1007/s00277-019-03791-y
27. Osmani K, Shah E, Drumheller B, Webb S, Singh M, Rubinstein P, et al. CD30 + primary intestinal T-cell lymphoma (unclassified) masquerading as chronic inflammation: a case report. Diagn Pathol (2022) 17(1):53. doi: 10.1186/s13000-022-01237-0
28. Watanabe-Okochi N, Imai Y, Kimura H, Yamashita D, Hara S, Takahashi T. Intestinal T-cell lymphoma, NOS, presenting with sole peritoneal and mucosal lymphomatosis throughout abdominal cavity. J Clin Exp Hematop (2020) 60(3):117–20. doi: 10.3960/jslrt.20020
29. Sun J, Lu Z, Yang D, Chen J. Primary intestinal T-cell and NK-cell lymphomas: A clinicopathological and molecular study from China focused on type II enteropathy-associated T-cell lymphoma and primary intestinal NK-cell lymphoma. Mod Pathol (2011) 24(7):983–92. doi: 10.1038/modpathol.2011.45
30. Tang XF, Yang L, Duan S, Guo H, Guo QN. Intestinal T-cell and NK/T-cell lymphomas: A clinicopathological study of 27 Chinese patients. Ann Diagn Pathol (2018) 37:107–17. doi: 10.1016/j.anndiagpath.2018.10.004
31. Perry AM, Warnke RA, Hu Q, Gaulard P, Copie-Bergman C, Alkan S, et al. Indolent T-cell lymphoproliferative disease of the gastrointestinal tract. Blood (2013) 122(22):3599–606. doi: 10.1182/blood-2013-07-512830
32. van Vliet C, Spagnolo DV. T- and NK-cell lymphoproliferative disorders of the gastrointestinal tract: Review and update. Pathology (2020) 52(1):128–41. doi: 10.1016/j.pathol.2019.10.001
33. Matnani R, Ganapathi KA, Lewis SK, Green PH, Alobeid B, Bhagat G. Indolent T- and NK-cell lymphoproliferative disorders of the gastrointestinal tract: A review and update. Hematol Oncol (2017) 35(1):3–16. doi: 10.1002/hon.2317
34. Perry AM, Bailey NG, Bonnett M, Jaffe ES, Chan WC. Disease progression in a patient with indolent T-cell lymphoproliferative disease of the gastrointestinal tract. Int J Surg Pathol (2019) 27(1):102–7. doi: 10.1177/1066896918785985
35. Soderquist CR, Patel N, Murty VV, Betman S, Aggarwal N, Young KH, et al. Genetic and phenotypic characterization of indolent T-cell lymphoproliferative disorders of the gastrointestinal tract. Haematologica (2020) 105(7):1895–906. doi: 10.3324/haematol.2019.230961
36. Mansoor A, Pittaluga S, Beck PL, Wilson WH, Ferry JA, Jaffe ES. NK-cell enteropathy: a benign NK-cell lymphoproliferative disease mimicking intestinal lymphoma: clinicopathologic features and follow-up in a unique case series. Blood (2011) 117(5):1447–52. doi: 10.1182/blood-2010-08-302737
37. Xia D, Morgan EA, Berger D, Pinkus GS, Ferry JA, Zukerberg LR. NK-cell enteropathy and similar indolent lymphoproliferative disorders: A case series with literature review. Am J Clin Pathol (2019) 151(1):75–85. doi: 10.1093/ajcp/aqy108
38. Xiao W, Gupta GK, Yao J, Jang YJ, Xi L, Baik J, et al. Recurrent somatic JAK3 mutations in NK-cell enteropathy. Blood (2019) 134(12):986–91. doi: 10.1182/blood.2019001443
39. Vose J, Armitage J, Weisenburger D, International TCLP. International peripheral T-cell and natural killer/T-cell lymphoma study: pathology findings and clinical outcomes. J Clin Oncol (2008) 26(25):4124–30. doi: 10.1200/JCO.2008.16.4558
40. Yoon SE, Song Y, Kim SJ, Yoon DH, Chen TY, Koh Y, et al. Comprehensive analysis of peripheral T-cell and natural killer/T-cell lymphoma in Asian patients: A multinational, multicenter, prospective registry study in Asia. Lancet Reg Health West Pac (2021) 10:100126. doi: 10.1016/j.lanwpc.2021.100126
41. Vega F, Medeiros LJ. A suggested immunohistochemical algorithm for the classification of T-cell lymphomas involving lymph nodes. Hum Pathol (2020) 102:104–16. doi: 10.1016/j.humpath.2020.05.006
42. Murga-Zamalloa CA, Mendoza-Reinoso V, Sahasrabuddhe AA, Rolland D, Hwang SR, McDonnell SR, et al. NPM-ALK phosphorylates WASp Y102 and contributes to oncogenesis of anaplastic large cell lymphoma. Oncogene (2017) 36(15):2085–94. doi: 10.1038/onc.2016.366
43. McDonnell SR, Hwang SR, Rolland D, Murga-Zamalloa C, Basrur V, Conlon KP, et al. Integrated phosphoproteomic and metabolomic profiling reveals NPM-ALK-mediated phosphorylation of PKM2 and metabolic reprogramming in anaplastic large cell lymphoma. Blood (2013) 122(6):958–68. doi: 10.1182/blood-2013-01-482026
44. Murga-Zamalloa C, Lim MS. ALK-driven tumors and targeted therapy: focus on crizotinib. Pharmgenomics Pers Med (2014) 7:87–94. doi: 10.2147/PGPM.S37504
45. Agnelli L, Mereu E, Pellegrino E, Limongi T, Kwee I, Bergaggio E, et al. Identification of a 3-gene model as a powerful diagnostic tool for the recognition of ALK-negative anaplastic large-cell lymphoma. Blood (2012) 120(6):1274–81. doi: 10.1182/blood-2012-01-405555
46. Parrilla Castellar ER, Jaffe ES, Said JW, Swerdlow SH, Ketterling RP, Knudson RA, et al. ALK-negative anaplastic large cell lymphoma is a genetically heterogeneous disease with widely disparate clinical outcomes. Blood (2014) 124(9):1473–80. doi: 10.1182/blood-2014-04-571091
47. Pedersen MB, Hamilton-Dutoit SJ, Bendix K, Ketterling RP, Bedroske PP, Luoma IM, et al. DUSP22 and TP63 rearrangements predict outcome of ALK-negative anaplastic large cell lymphoma: A Danish cohort study. Blood (2017) 130(4):554–7. doi: 10.1182/blood-2016-12-755496
48. Hapgood G, Ben-Neriah S, Mottok A, Lee DG, Robert K, Villa D, et al. Identification of high-risk DUSP22-rearranged ALK-negative anaplastic large cell lymphoma. Br J Haematol (2019) 186(3):e28–31. doi: 10.1111/bjh.15860
49. Lobello C, Tichy B, Bystry V, Radova L, Filip D, Mraz M, et al. STAT3 and TP53 mutations associate with poor prognosis in anaplastic large cell lymphoma. Leukemia (2021) 35(5):1500–5. doi: 10.1038/s41375-020-01093-1
50. Liang HC, Costanza M, Prutsch N, Zimmerman MW, Gurnhofer E, Montes-Mojarro IA, et al. Super-enhancer-based identification of a BATF3/IL-2R-module reveals vulnerabilities in anaplastic large cell lymphoma. Nat Commun (2021) 12(1):5577. doi: 10.1038/s41467-021-25379-9
51. Fitzpatrick MJ, Massoth LR, Marcus C, Vergilio JA, Severson E, Duncan D, et al. JAK2 rearrangements are a recurrent alteration in CD30+ systemic T-cell lymphomas with anaplastic morphology. Am J Surg Pathol (2021) 45(7):895–904. doi: 10.1097/PAS.0000000000001708
52. Kucuk C, Jiang B, Hu X, Zhang W, Chan JK, Xiao W, et al. Activating mutations of STAT5B and STAT3 in lymphomas derived from gammadelta-T or NK cells. Nat Commun (2015) 6:6025. doi: 10.1038/ncomms7025
53. Ferrufino-Schmidt MC, Medeiros LJ, Liu H, Clemens MW, Hunt KK, Laurent C, et al. Clinicopathologic features and prognostic impact of lymph node involvement in patients with breast implant-associated anaplastic Large cell lymphoma. Am J Surg Pathol (2018) 42(3):293–305. doi: 10.1097/PAS.0000000000000985
54. Clemens MW, Medeiros LJ, Butler CE, Hunt KK, Fanale MA, Horwitz S, et al. Complete surgical excision is essential for the management of patients with breast implant-associated anaplastic Large-cell lymphoma. J Clin Oncol (2016) 34(2):160–8. doi: 10.1200/JCO.2015.63.3412
55. Oishi N, Brody GS, Ketterling RP, Viswanatha DS, He R, Dasari S, et al. Genetic subtyping of breast implant-associated anaplastic large cell lymphoma. Blood (2018) 132(5):544–7. doi: 10.1182/blood-2017-12-821868
56. Laurent C, Nicolae A, Laurent C, Le Bras F, Haioun C, Fataccioli V, et al. Gene alterations in epigenetic modifiers and JAK-STAT signaling are frequent in breast implant-associated ALCL. Blood (2020) 135(5):360–70. doi: 10.1182/blood.2019001904
57. Kadin ME, Morgan J, Xu H, Epstein AL, Sieber D, Hubbard BA, et al. IL-13 is produced by tumor cells in breast implant-associated anaplastic large cell lymphoma: implications for pathogenesis. Hum Pathol (2018) 78:54–62. doi: 10.1016/j.humpath.2018.04.007
58. Chen CY, Lee JB, Liu B, Ohta S, Wang PY, Kartashov AV, et al. Induction of interleukin-9-Producing mucosal mast cells promotes susceptibility to IgE-mediated experimental food allergy. Immunity (2015) 43(4):788–802. doi: 10.1016/j.immuni.2015.08.020
59. Burd PR, Thompson WC, Max EE, Mills FC. Activated mast cells produce interleukin 13. J Exp Med (1995) 181(4):1373–80. doi: 10.1084/jem.181.4.1373
60. Tabanelli V, Corsini C, Fiori S, Agostinelli C, Calleri A, Orecchioni S, et al. Recurrent PDL1 expression and PDL1 (CD274) copy number alterations in breast implant-associated anaplastic large cell lymphomas. Hum Pathol (2019) 90:60–9. doi: 10.1016/j.humpath.2019.05.007
61. Dobay MP, Lemonnier F, Missiaglia E, Bastard C, Vallois D, Jais JP, et al. Integrative clinicopathological and molecular analyses of angioimmunoblastic T-cell lymphoma and other nodal lymphomas of follicular helper T-cell origin. Haematologica (2017) 102(4):e148–e51. doi: 10.3324/haematol.2016.158428
62. Xie Y, Jaffe ES. How I diagnose angioimmunoblastic T-cell lymphoma. Am J Clin Pathol (2021) 156(1):1–14. doi: 10.1093/ajcp/aqab090
63. Ruiz SJ, Cotta CV. Follicular helper T-cell lymphoma: a b-cell-rich variant of T-cell lymphoma. Ann Diagn Pathol (2015) 19(4):187–92. doi: 10.1016/j.anndiagpath.2015.03.008
64. Huang Y, Moreau A, Dupuis J, Streubel B, Petit B, Le Gouill S, et al. Peripheral T-cell lymphomas with a follicular growth pattern are derived from follicular helper T cells (TFH) and may show overlapping features with angioimmunoblastic T-cell lymphomas. Am J Surg Pathol (2009) 33(5):682–90. doi: 10.1097/PAS.0b013e3181971591
65. Kiiskila J, Uotila P, Haapasaari KM, Kauppila S, Teppo HR, Kuusisto MEL, et al. Incidence and clinicopathological features of follicular T-cell lymphoma in Finland: a population-based immunohistochemical study. Hum Pathol (2021) 117:79–87. doi: 10.1016/j.humpath.2021.07.012
66. Lisa M, Verma PK, Nishi, Shuchismita, Hussain MA. Follicular T-cell lymphoma. J Cancer Res Ther (2021) 17(6):1568–71. doi: 10.4103/jcrt.JCRT_486_19
67. Marafioti T, Paterson JC, Ballabio E, Chott A, Natkunam Y, Rodriguez-Justo M, et al. The inducible T-cell co-stimulator molecule is expressed on subsets of T cells and is a new marker of lymphomas of T follicular helper cell-derivation. Haematologica (2010) 95(3):432–9. doi: 10.3324/haematol.2009.010991
68. Basha BM, Bryant SC, Rech KL, Feldman AL, Vrana JA, Shi M, et al. Application of a 5 marker panel to the routine diagnosis of peripheral T-cell lymphoma with T-follicular helper phenotype. Am J Surg Pathol (2019) 43(9):1282–90. doi: 10.1097/PAS.0000000000001315
69. Amador C, Bouska A, Wright G, Weisenburger DD, Feldman AL, Greiner TC, et al. Gene expression signatures for the accurate diagnosis of peripheral T-cell lymphoma entities in the routine clinical practice. J Clin Oncol (2022), JCO2102707. doi: 10.1200/JCO.21.02707
70. Miyoshi H, Sakata-Yanagimoto M, Shimono J, Yoshida N, Hattori K, Arakawa F, et al. RHOA mutation in follicular T-cell lymphoma: Clinicopathological analysis of 16 cases. Pathol Int (2020) 70(9):653–60. doi: 10.1111/pin.12981
71. Zhang W, Wang Z, Luo Y, Zhong D, Luo Y, Zhou D. GATA3 expression correlates with poor prognosis and tumor-associated macrophage infiltration in peripheral T cell lymphoma. Oncotarget (2016) 7(40):65284–94. doi: 10.18632/oncotarget.11673
72. Wang T, Lu Y, Polk A, Chowdhury P, Murga-Zamalloa C, Fujiwara H, et al. T-Cell receptor signaling activates an ITK/NF-kappaB/GATA-3 axis in T-cell lymphomas facilitating resistance to chemotherapy. Clin Cancer Res (2017) 23(10):2506–15. doi: 10.1158/1078-0432.CCR-16-1996
73. Wang T, Feldman AL, Wada DA, Lu Y, Polk A, Briski R, et al. GATA-3 expression identifies a high-risk subset of PTCL, NOS with distinct molecular and clinical features. Blood (2014) 123(19):3007–15. doi: 10.1182/blood-2013-12-544809
74. Geng X, Wang C, Gao X, Chowdhury P, Weiss J, Villegas JA, et al. GATA-3 is a proto-oncogene in T-cell lymphoproliferative neoplasms. Blood Cancer J (2022) 12(11):149. doi: 10.1038/s41408-022-00745-y
75. Amador C, Greiner TC, Heavican TB, Smith LM, Galvis KT, Lone W, et al. Reproducing the molecular subclassification of peripheral T-cell lymphoma-NOS by immunohistochemistry. Blood (2019) 134(24):2159–70. doi: 10.1182/blood.2019000779
76. Murga-Zamalloa C, Wilcox RA. GATA-3 in T-cell lymphoproliferative disorders. IUBMB Life (2020) 72(1):170–7. doi: 10.1002/iub.2130
77. Wilcox RA. A three-signal model of T-cell lymphoma pathogenesis. Am J Hematol (2016) 91(1):113–22. doi: 10.1002/ajh.24203
78. Wilcox RA, Wada DA, Ziesmer SC, Elsawa SF, Comfere NI, Dietz AB, et al. Monocytes promote tumor cell survival in T-cell lymphoproliferative disorders and are impaired in their ability to differentiate into mature dendritic cells. Blood (2009) 114(14):2936–44. doi: 10.1182/blood-2009-05-220111
79. Montes-Mojarro IA, Fend F, Quintanilla-Martinez L. EBV and the pathogenesis of NK/T cell lymphoma. Cancers (Basel) (2021) 13(6). doi: 10.3390/cancers13061414
80. Suzuki K, Ohshima K, Karube K, Suzumiya J, Ohga S, Ishihara S, et al. Clinicopathological states of Epstein-Barr virus-associated T/NK-cell lymphoproliferative disorders (severe chronic active EBV infection) of children and young adults. Int J Oncol (2004) 24(5):1165–74. doi: 10.3892/ijo.24.5.1165
81. Okano M, Kawa K, Kimura H, Yachie A, Wakiguchi H, Maeda A, et al. Proposed guidelines for diagnosing chronic active Epstein-Barr virus infection. Am J Hematol (2005) 80(1):64–9. doi: 10.1002/ajh.20398
82. Montes-Mojarro IA, Kim WY, Fend F, Quintanilla-Martinez L. Epstein - Barr virus positive T and NK-cell lymphoproliferations: Morphological features and differential diagnosis. Semin Diagn Pathol (2020) 37(1):32–46. doi: 10.1053/j.semdp.2019.12.004
83. Bollard CM, Cohen JI. How I treat T-cell chronic active Epstein-Barr virus disease. Blood (2018) 131(26):2899–905. doi: 10.1182/blood-2018-03-785931
84. Yonese I, Sakashita C, Imadome KI, Kobayashi T, Yamamoto M, Sawada A, et al. Nationwide survey of systemic chronic active EBV infection in Japan in accordance with the new WHO classification. Blood Adv (2020) 4(13):2918–26. doi: 10.1182/bloodadvances.2020001451
85. Li S, Feng X, Li T, Zhang S, Zuo Z, Lin P, et al. Extranodal NK/T-cell lymphoma, nasal type: a report of 73 cases at MD Anderson cancer center. Am J Surg Pathol (2013) 37(1):14–23. doi: 10.1097/PAS.0b013e31826731b5
86. Kuo TT, Shih LY, Tsang NM. Nasal NK/T cell lymphoma in Taiwan: a clinicopathologic study of 22 cases, with analysis of histologic subtypes, Epstein-Barr virus LMP-1 gene association, and treatment modalities. Int J Surg Pathol (2004) 12(4):375–87. doi: 10.1177/106689690401200410
87. Ng SB, Lai KW, Murugaya S, Lee KM, Loong SL, Fook-Chong S, et al. Nasal-type extranodal natural killer/T-cell lymphomas: a clinicopathologic and genotypic study of 42 cases in Singapore. Mod Pathol (2004) 17(9):1097–107. doi: 10.1038/modpathol.3800157
88. Ko YH, Ree HJ, Kim WS, Choi WH, Moon WS, Kim SW. Clinicopathologic and genotypic study of extranodal nasal-type natural killer/T-cell lymphoma and natural killer precursor lymphoma among koreans. Cancer (2000) 89(10):2106–16. doi: 10.1002/1097-0142(20001115)89:10<2106::AID-CNCR11>3.0.CO;2-G
89. Pongpruttipan T, Sukpanichnant S, Assanasen T, Wannakrairot P, Boonsakan P, Kanoksil W, et al. Extranodal NK/T-cell lymphoma, nasal type, includes cases of natural killer cell and alphabeta, gammadelta, and alphabeta/gammadelta T-cell origin: a comprehensive clinicopathologic and phenotypic study. Am J Surg Pathol (2012) 36(4):481–99. doi: 10.1097/PAS.0b013e31824433d8
90. Song TL, Nairismagi ML, Laurensia Y, Lim JQ, Tan J, Li ZM, et al. Oncogenic activation of the STAT3 pathway drives PD-L1 expression in natural killer/T-cell lymphoma. Blood (2018) 132(11):1146–58. doi: 10.1182/blood-2018-01-829424
91. Bi XW, Wang H, Zhang WW, Wang JH, Liu WJ, Xia ZJ, et al. PD-L1 is upregulated by EBV-driven LMP1 through NF-kappaB pathway and correlates with poor prognosis in natural killer/T-cell lymphoma. J Hematol Oncol (2016) 9(1):109. doi: 10.1186/s13045-016-0341-7
92. Kim SJ, Lim JQ, Laurensia Y, Cho J, Yoon SE, Lee JY, et al. Avelumab for the treatment of relapsed or refractory extranodal NK/T-cell lymphoma: an open-label phase 2 study. Blood (2020) 136(24):2754–63. doi: 10.1182/blood.2020007247
93. Ng SB, Chung TH, Kato S, Nakamura S, Takahashi E, Ko YH, et al. Epstein-Barr Virus-associated primary nodal T/NK-cell lymphoma shows a distinct molecular signature and copy number changes. Haematologica (2018) 103(2):278–87. doi: 10.3324/haematol.2017.180430
94. Jung KS, Cho SH, Kim SJ, Ko YH, Kim WS. Clinical features and treatment outcome of Epstein-Barr virus-positive nodal T-cell lymphoma. Int J Hematol (2016) 104(5):591–5. doi: 10.1007/s12185-016-2068-1
95. Jeon YK, Kim JH, Sung JY, Han JH, Ko YH. Hematopathology study group of the Korean society of p. Epstein-Barr virus-positive nodal T/NK-cell lymphoma: an analysis of 15 cases with distinct clinicopathological features. Hum Pathol (2015) 46(7):981–90. doi: 10.1016/j.humpath.2015.03.002
96. Takahashi E, Asano N, Li C, Tanaka T, Shimada K, Shimada S, et al. Nodal T/NK-cell lymphoma of nasal type: a clinicopathological study of six cases. Histopathology (2008) 52(5):585–96. doi: 10.1111/j.1365-2559.2008.02997.x
97. Wai CMM, Chen S, Phyu T, Fan S, Leong SM, Zheng W, et al. Immune pathway upregulation and lower genomic instability distinguish EBV-positive nodal T/NK-cell lymphoma from ENKTL and PTCL-NOS. Haematologica (2022) 107(8):1864–79. doi: 10.3324/haematol.2021.280003
98. Foss FM, Horwitz SM, Civallero M, Bellei M, Marcheselli L, Kim WS, et al. Incidence and outcomes of rare T cell lymphomas from the T cell project: hepatosplenic, enteropathy associated and peripheral gamma delta T cell lymphomas. Am J Hematol (2020) 95(2):151–5. doi: 10.1002/ajh.25674
99. Belhadj K, Reyes F, Farcet JP, Tilly H, Bastard C, Angonin R, et al. Hepatosplenic gammadelta T-cell lymphoma is a rare clinicopathologic entity with poor outcome: Report on a series of 21 patients. Blood (2003) 102(13):4261–9. doi: 10.1182/blood-2003-05-1675
100. Wu H, Wasik MA, Przybylski G, Finan J, Haynes B, Moore H, et al. Hepatosplenic gamma-delta T-cell lymphoma as a late-onset posttransplant lymphoproliferative disorder in renal transplant recipients. Am J Clin Pathol (2000) 113(4):487–96. doi: 10.1309/YTTC-F55W-K9CP-EPX5
101. Vega F, Medeiros LJ, Bueso-Ramos C, Jones D, Lai R, Luthra R, et al. Hepatosplenic gamma/delta T-cell lymphoma in bone marrow. A sinusoidal neoplasm with blastic cytologic features. Am J Clin Pathol (2001) 116(3):410–9. doi: 10.1309/BM40-YM6J-9T3X-MH8H
102. Yabe M, Medeiros LJ, Tang G, Wang SA, Ahmed S, Nieto Y, et al. Prognostic factors of hepatosplenic T-cell lymphoma: Clinicopathologic study of 28 cases. Am J Surg Pathol (2016) 40(5):676–88. doi: 10.1097/PAS.0000000000000614
103. Weidmann E. Hepatosplenic T cell lymphoma. a review on 45 cases since the first report describing the disease as a distinct lymphoma entity in 1990. Leukemia (2000) 14(6):991–7. doi: 10.1038/sj.leu.2401784
104. Travert M, Huang Y, de Leval L, Martin-Garcia N, Delfau-Larue MH, Berger F, et al. Molecular features of hepatosplenic T-cell lymphoma unravels potential novel therapeutic targets. Blood (2012) 119(24):5795–806. doi: 10.1182/blood-2011-12-396150
105. Vega F, Medeiros LJ, Gaulard P. Hepatosplenic and other gammadelta T-cell lymphomas. Am J Clin Pathol (2007) 127(6):869–80. doi: 10.1309/LRKX8CE7GVPCR1FT
106. McKinney M, Moffitt AB, Gaulard P, Travert M, De Leval L, Nicolae A, et al. The genetic basis of hepatosplenic T-cell lymphoma. Cancer Discovery (2017) 7(4):369–79. doi: 10.1158/2159-8290.CD-16-0330
Keywords: T-cell lymphoma, WHO, classification, diagnosis, molecular diagnosis, biology, pathology
Citation: Murga-Zamalloa C and Inamdar K (2022) Classification and challenges in the histopathological diagnosis of peripheral T-cell lymphomas, emphasis on the WHO-HAEM5 updates. Front. Oncol. 12:1099265. doi: 10.3389/fonc.2022.1099265
Received: 15 November 2022; Accepted: 05 December 2022;
Published: 20 December 2022.
Edited by:
Ryan Wilcox, University of Michigan, United StatesReviewed by:
Anamarija Perry, University of Michigan, United StatesCopyright © 2022 Murga-Zamalloa and Inamdar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Carlos Murga-Zamalloa, Y2F0dG9AdWljLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.