 Nikhil Samarth
Nikhil Samarth Pooja Gulhane
Pooja Gulhane Shailza Singh
Shailza Singh- National Centre for Cell Science, NCCS Complex, Ganeshkhind, SP Pune University Campus, Pune, India
With a 5-year survival rate of only 15%, non-small cell lung cancer (NSCLC), the most common kind of lung carcinoma and the cause of millions of deaths annually, has drawn attention. Numerous variables, such as disrupted signaling caused by somatic mutations in the EGFR-mediated RAS/RAF/MAPK, PI3K/AKT, JAK/STAT signaling cascade, supports tumour survival in one way or another. Here, the tumour microenvironment significantly contributes to the development of cancer by thwarting the immune response. MicroRNAs (miRNAs) are critical regulators of gene expression that can function as oncogenes or oncosuppressors. They have a major influence on the occurrence and prognosis of NSCLC. Though, a myriad number of therapies are available and many are being clinically tested, still the drug resistance, its adverse effect and toxicity leading towards fatality cannot be ruled out. In this review, we tried to ascertain the missing links in between perturbed EGFR signaling, miRNAs favouring tumorigenesis and the autophagy mechanism. While connecting all the aforementioned points multiple associations were set, which can be targeted in order to combat NSCLC. Here, we tried illuminating designing synthetically engineered circuits with the toggle switches that might lay a prototype for better therapeutic paradigm.
1 Background
Cancer is anticipated to rank as the top cause of death and the single biggest obstacle to raising life expectancy in every country over the globe in the 21st century. Non-communicable diseases (NCDs) nowadays account for the majority of fatalities globally. According to World Health Organization (WHO) estimates from 2019, cancer is the first or second leading cause of deaths in 112 out of 183 countries (1). Worldwide, the incidence and death of cancer are rising quickly. Cancer transitions are especially noticeable in rising nations, where the disease is getting worse and the organization of frequent cancer types is changing at the same time. These cancers are frequently attributed to a so-called modernization of lifestyle, but the various cancer profiles in different nations and regions show that there is still significant geographic diversity, with local risk factors persisting in populations going through very different stages of social and economic development. A revision to the GLOBOCAN 2020 projections of cancer incidence and mortality provided by the International Agency for Research on Cancer, the number of new cancer cases worldwide is envisaged to be 19.3 million, and the number of cancer fatalities occurred to be close to 10.0 million. Lung cancer is the second most prevalent cancer diagnosed and the main cause of cancer mortality in 2020, accounting for roughly one in ten (11.4%) cancer diagnoses and one in five (18.0%) fatalities, with a predicted 2.2 million new cancer cases and 1.8 million deaths (1, 2). For both men and women, lung cancer is one of the most hazardous cancers. The fatality rate is higher than the sum of colon, breast, and pancreatic cancers. More than half of patients with lung cancer die within a year of being diagnosed. Lung cancer’s poor prognosis is a result of late diagnosis because there aren’t any obvious signs during the early stages of the disease’s progression. Lung cancers are divided into two subgroups according to their histological characteristics: non-small-cell lung cancer (NSCLC, which accounts for 85% of cases) and small-cell lung cancer (SCLC, which accounts for 15% of cases). Squamous-cell carcinoma, adenocarcinoma, and large-cell carcinoma are the three primary histological subtypes of NSCLC. Adenocarcinoma accounts for 40% to 70% and instances in the outer portion of the lung. Squamous-cell carcinoma accounts for 20%–30% of lung cancer cases and is typically seen in the centre of the lung and the remaining 10% -15% cases are of Large cell Carcinoma which can be found throughout the lung as shown in Figure 1 (3).
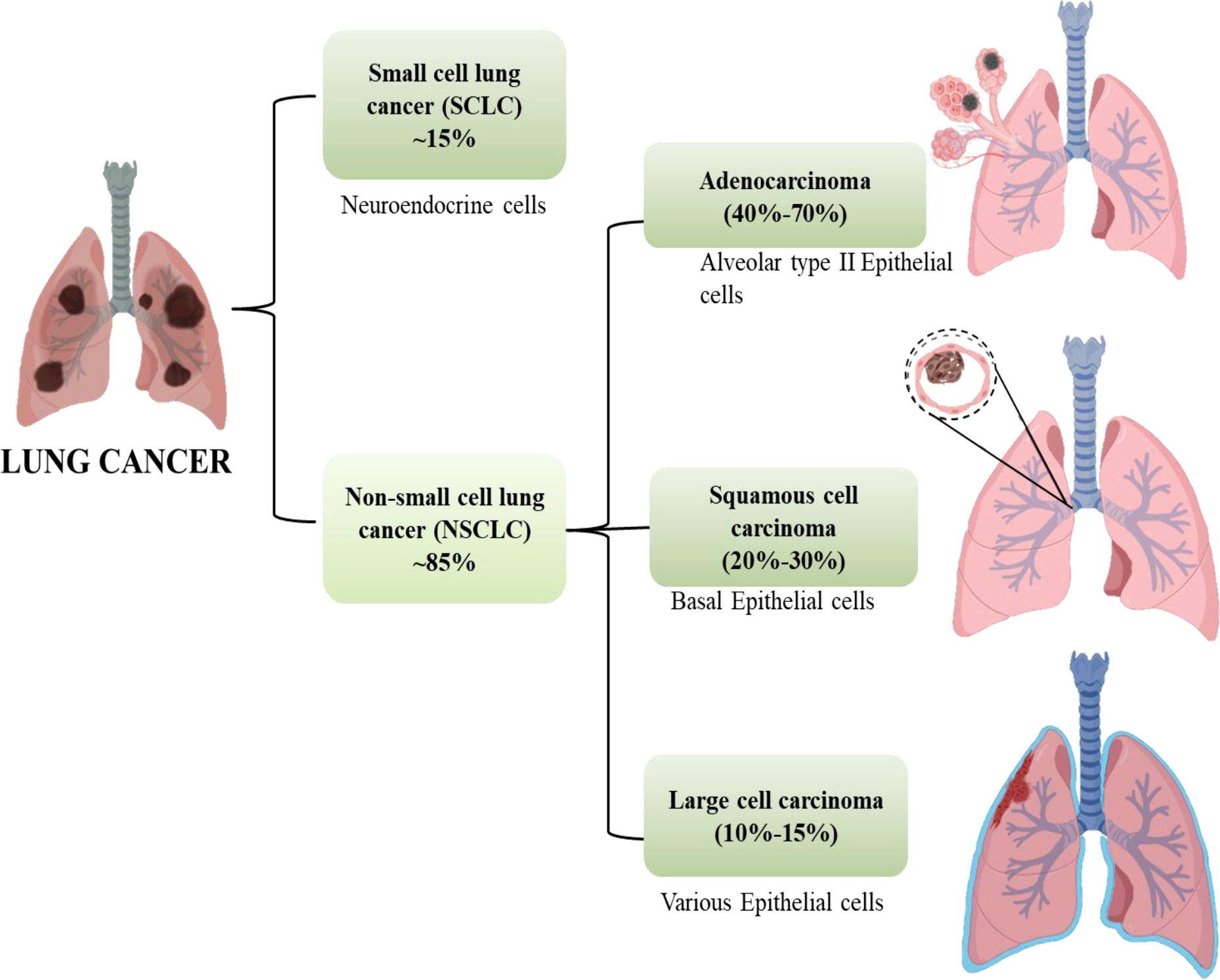
Figure 1 Subtypes of lung cancer.
2 Molecular events leading to pathogenesis in NSCLC
Tobacco has been a part of the cultural and economic structure since antiquity. Cigarette smoking was thought to be more important than occupational exposure in the general population in the cause of NSCLC. Cigarette smoking had a far greater impact than any other risk factor for lung cancer (4). The majority of NSCLCs are diagnosed in ex-smokers, implying that the accumulation of molecular damage caused by cigarette smoke sets in motion a chain of events that leads to cancer development even decades after smoking cessation (5). Human cells acquire a set of functional abilities as they transition from normal development states to neoplastic growth states. More specifically, these abilities are essential for human cells’ capacity to produce malignant tumors. The genetic and molecular changes that occur during carcinogenesis result in the “hallmarks of lung cancer” (Figure 2).
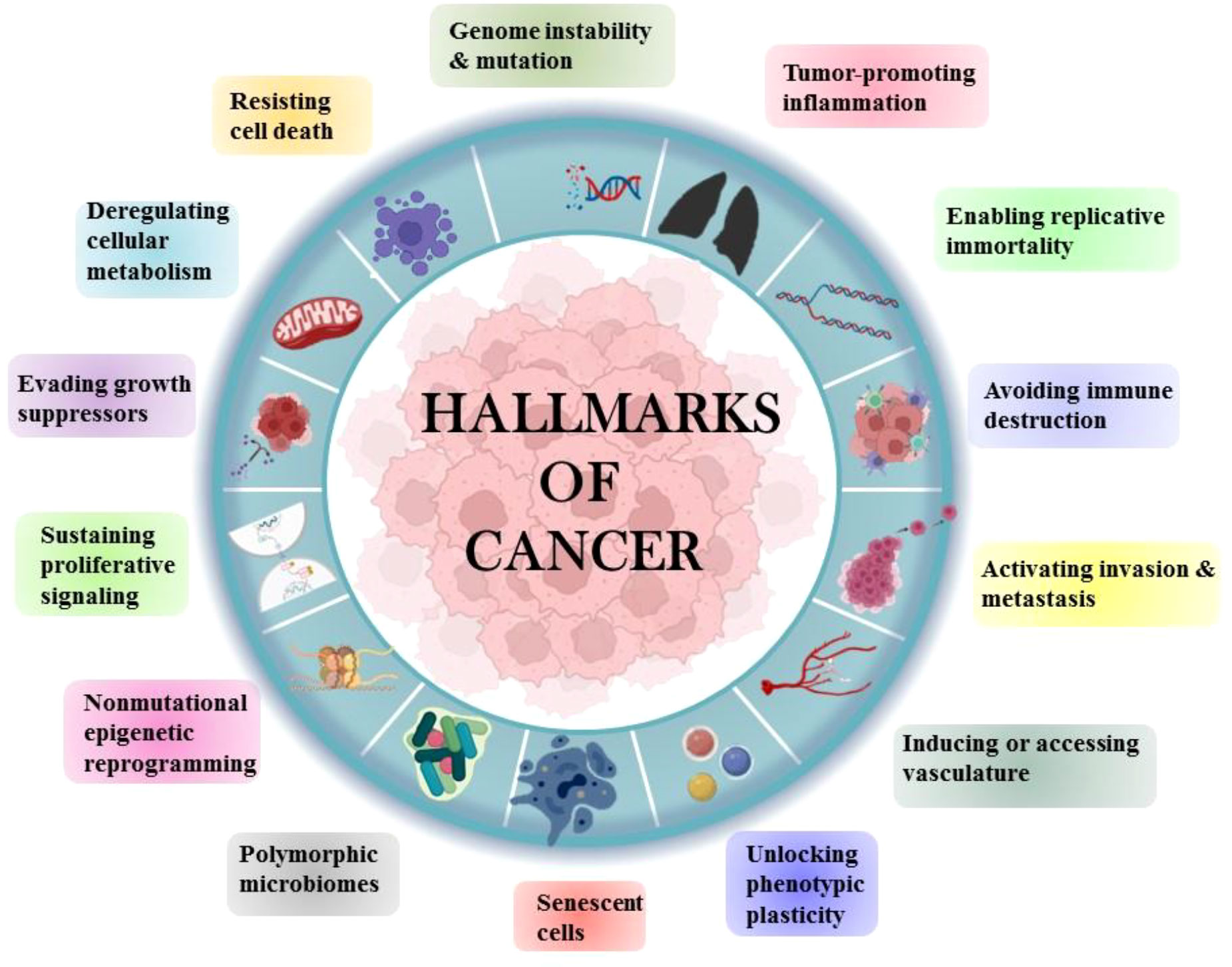
Figure 2 Hallmarks of Cancer.
2.1 Sustaining proliferative signaling
Numerous growth factors and their receptors are overexpressed by NSCLC cells and surrounding stromal cells, resulting in autocrine and paracrine growth stimulation loops. During tumor development proto-oncogenes encode several growth factors and receptors which are activated by various mechanisms. RAS, a member of an important signal transduction pathway, is one such proto-oncogene. The KRAS gene is mutated in 15% to 20% of all NSCLCs and approximately 30% of lung adenocarcinomas, with the most common mutation occurring at codon 12 (5). In NSCLC, two tyrosine kinase growth factor receptors, EGFR and Her2/Neu, are frequently overexpressed. EGFR and HER2/NEU somatic mutations have recently been reported in patients with LUAD (lung adenocarcinoma) among females and non-smokers (6, 7). Shigematsu et al. found that all mutations occurred within exons 18-21 of the EGFR tyrosine kinase domain (8).
2.2 Evading growth suppressors
For cancer cells to keep growing, it is imperative that they avoid antigrowth signals and must inhibit tumour suppressor genes (TSG), which are responsible for antigrowth signals. Cancer cells were found to have mutations in tumour suppressor genes, with p53 being the most frequently mutated one (9). More than 50% of patients with NSCLC have mutations that render the p53 tumour suppressor gene inactive (10). In addition, TSGs like RB1, CREBBP, KEAP1, STK11, CDKN2A, NOTCH1, and PTEN are particularly relevant to NSCLC. These TSG mutations typically exclude one another, showing that different genes can contribute to the development of cancer (11).
2.3 Avoiding immune destruction
The immune system’s capacity to restrain itself from attacking healthy body cells is a crucial component. This is accomplished by “checkpoint” proteins on immune cells, which function as switches that must be turned on (or off) in order to initiate an immune response. These checkpoints are sometimes used by cancer cells as a defence mechanism against immune system attacks. A group of receptors on the surface of activated T cells make up immune checkpoints. Immune checkpoint molecules prevent self-tolerance and immune homeostasis under “normal” physiological circumstances where it is important to prevent collateral tissue damage. However, by applying the “brakes” to host immune activation, they help to tolerate tumour cells (12, 13). More than 20 immune checkpoint modulators have been described for this mechanism, but programmed cell death receptor 1 (PD-1) and cytotoxic T lymphocyte associated protein-4 (CTLA-4) are particularly well-versed (14, 15). PD-L1, an immunosuppressive protein, is overexpressed in lung tumors, according to accumulating evidence, and inhibiting this pathway has provided long-lasting relief for some patients with advanced-stage NSCLC (16).
2.4 Enabling replicative immortality
A defining trait of tumour cells is replicative immortality or telomerase activation (17). The level of telomerase activity has been found noticeably higher in lung tumour cell lines in all histopathological subtypes of the tumor (18). According to the data, more than 80% of the NSCLCs studied expressed telomerase activity. Telomerase can stimulate tumour progression by ensuring the maintenance of telomeres above a critical short length, thereby preventing the induction of apoptosis or cellular senescence (19, 20). Chromosome instability brought on by telomere dysfunction may activate telomerase, activate oncogenes, and/or silence tumour suppressor genes, all of which work together to promote cancerous transformation, tumour growth, and drug resistance (21, 22).
2.5 Tumor promoting inflammation
When healthy cells are killed off by tumour cells, it results in inflammation that promotes the growth of the tumor. This causes the pro-inflammatory signals to be released from the beneficial cell contents into the interstitial tissue. This state will last as long as tumour cells grow (23). According to their cause, mechanism, severity, and outcome, the various types of inflammation contribute to the development of cancer, such as inflammation due to infection, environmental exposure, therapy induced inflammation and tumor associated inflammation. Tobacco smoke, silica-containing products, and asbestos can all increase the risk of developing NSCLC because the immune system is unable to get rid of these three substances, chronic inflammation will continue and as the inflammation progresses, tumors will form, migrate, grow, and differentiate from normal cells into different types of tumour cells (24).
2.6 Activating invasion and metastasis
When tumour cells develop the capacity to infiltrate the surrounding tissues, the invasion process is triggered as these motile cells pass through the extracellular matrix and basement membrane, developing into intravasation as they infiltrate the lymphatic or vascular circulation. The development of secondary tumors in distant organs results from a series of steps in the tumour metastasis process. These metastatic cells then travel through the circulatory system, invading the extracellular matrix and vascular basement membrane as part of the extravasation process, which is largely to blame for the mortality and morbidity associated with cancer (25). In NSCLC, the brain is one of the primary sites of lung carcinoma metastasis. Adenocarcinomas with epidermal growth factor receptor (EGFR) mutations and EML4ALK1 rearrangement are more frequently found to have brain metastases (26, 27). Metastatic NSCLC cells had elevated levels of the cell adhesion protein CD15 as TGF-α activates E-selectin (CD62E), it is also discovered to be overexpressed in hCMEC/D3 human brain endothelial cells. This raises the likelihood that metastatic NSCLC cells will attach to the brain endothelium, which will allow tumour cells to invade the tissue (28).
2.7 Inducing/accessing vasculature
The term “angiogenesis” refers to the process by which new blood and lymphatic vessels develop from an already-existing vasculature. This gives tumour cells the ability to take in nutrition in the form of nutrients and oxygen as well as the ability to expel metabolic waste. Solid tumors require a blood supply in order to develop beyond a few millimetres in size, and that is why angiogenesis is essential to the process of tumorigenesis (29). In relation to tumour vascular density, cancer metastasis, and prognosis, VEGF is the most potent and selective growth factor for endothelial cells. Non-small cell lung cancer patients have been shown to have high levels of circulating VEGF (30, 31). It is a growth factor that encourages vascular endothelial cells to proliferate and migrate while also inhibiting apoptosis and regulating their permeability. In NSCLC, it contributes to tumour growth by lymphangiogenesis, neoangiogenesis, lymph node spread, and high levels of VEGF have been linked to a poor prognosis (32, 33).
2.8 Genome instability and mutation
Several genes governing cell division and tumour suppression are damaged as a result of an increased propensity for genomic alterations and mutations brought on by the abnormal proliferation of cancer cells termed as Genomic instability. Since survival-enhancing mutations enhance the likelihood that those mutations will spread to subsequent cells, genomic instability has a tendency to accumulate in cancer cells (34). According to genomic research, Lung cancer has thousands of somatic mutations, copy number changes, and genome duplications. Due to exposure to tobacco carcinogens and germline genomic instability, lung cancer has a high somatic mutation rate, also the NSCLC initial tumors have a significant level of genetic variability (35, 36). Oncogenes, tumour suppressors, cyclins, cyclin-dependent kinases, and defects in cell cycle regulation are the main molecular mechanisms generating genomic instability in lung cancer. Genomic instability is also a result of defective spindle assembly checkpoint components including Bud and Mad family members, as well as aberrant DNA repair processes like BER, NER, TLS, HR, NHEJ, and MMR (37).
2.9 Resisting cell death
The systems used by cancer cells to detect damage or abnormalities may be changed, which would stop the appropriate signaling and activation of apoptosis. Cancer cells may potentially introduce flaws in downstream signaling or apoptosis-related proteins, which would also interfere with the normal apoptosis. Normally, the build-up of genetic abnormalities necessary to promote unchecked cell cycle progression and cancer would cause a cell to go through apoptosis. A crucial characteristic of cancer is its capacity to prevent cells from dying during apoptosis. Cancer cells harbour apoptotic defects, which are the root cause of treatment resistance in addition to carcinogenesis. It is possible that a deficiency in apoptotic signaling is the cause of NSCLC cells’ resistance to a variety of cytotoxic treatments (38). The “death domain,” a distinctive cytoplasmic region shared by members of the TNF receptor family, is essential for transferring the death signal from the cell’s surface to intracellular signaling pathways. Multiple degrees of death receptor signaling dysfunction can be present in human malignancies. These receptors’ surface expression can be down regulated or even completely missing at the receptor level, which prevents the death signal from reaching intracellular signaling cascades from the cell surface. Numerous tumours have been shown to have an increase in the ratio of anti- to pro-apoptotic Bcl-2 proteins, which has been linked to tumour cell survival and apoptosis resistance (39).
2.10 Deregulating cellular metabolism
Cancer cells have altered cellular metabolism because they require a large amount of energy to grow rapidly. Aerobic glycolysis has an aberrant role in the energy production of cancer cells. They also have other abnormal metabolic traits, such as enhanced fatty acid production and elevated rates of glutamine metabolism, in addition to their dependence on glycolysis. Recent research has connected a number of characteristics of cancer cells, including dysregulated Warburg-like glucose metabolism, fatty acid production, and glutaminolysis, to therapeutic resistance in the treatment of cancer (40). In growing cells, glucose is a vital nutrient needed for numerous metabolic pathways. It’s possible that cancer gains from a lot of glucose. In true scenario, greater incidence and fatality rates in several forms of cancer, including NSCLC, have been connected to high blood sugar levels and a clinical diagnosis of diabetes (41). Given that the cancer cells can derive vital metabolites and energy primarily from glucose fermentation, even under aerobic conditions, Warburg hypothesized that hyperglycemia might hasten the development of cancer cells (42). Tyrosine kinase oncogenic activity has been found to be the primary driver of certain advanced NSCLC cancers. Growing evidence suggests that these kinases also cause metabolic alterations in tumour cells. Numerous studies have focused on the precise mechanisms by which oncogenic EGFR signaling rewires the metabolism of NSCLCs, and it is known that these pathways affect the control of glucose metabolism (43).
2.11 Emerging hallmarks and enabling characteristics
2.11.1 Unlocking phenotypic plasticity
Cells ability to differentiate is typically limited in comparison to the massive amount of development and differentiation that occurs during organogenesis. This constraint allows cells to remain organized and functional within their tissue. Cells in cancer, on the other hand, go through molecular and phenotypic changes that allow them to take on different identities along a phenotypic spectrum known as cellular plasticity. Changes in cellular phenotype are important in cancer progression because they can aid in tumor initiation, metastasis, immune invasion, chemoresistance, and tumour progression (44, 45). There are three types of phenotypic plasticity that can be activated during cancer. Dedifferentiation occurs when a normal cell reverts to a progenitor-like state. Blocked differentiation occurs when differentiated progenitor cells are prevented from entering a non-proliferative state. Trans differentiation occurs when cells that were previously committed to one differentiation pathway switch to another (46, 47).
2.11.2 Non mutational epigenetic reprogramming
Permanent genetic alterations, such as point mutations, deletions, translocations, amplifications, and epigenetic modifications that impact various chromatin-dependent processes, including histone modifications, DNA methylation patterns, and microRNA regulation, are the cause of the onset and progression of lung cancer. Over 50% of all human gene promoter sequences contain CpG dinucleotide islands, which make up just 1% of the human genome yet are sites of DNA methylation in eukaryotes. Increased mitotic recombination and consequent chromosomal instability are brought on by a substantial decrease in cytosine methylation at these repeated regions in cancer cells. In addition, abnormal cytosine methylation affects the transcriptional regulation of other genes involved in processes including DNA repair, apoptosis, the epithelial-mesenchymal transition (EMT), cellular motility, invasion, and metastasis (48, 49).
2.11.3 Polymorphic microbiomes
The commensal microbiota has come to light as an important biomarker and regulator of tumorigenesis and cancer therapy response. Tumor-associated dysregulation of the local microbiome in the lung has been discovered in lung cancer patients and mouse models, which influences cancer progression by shaping the tumor microenvironment and modulating the activity of tumor-infiltrating immune cells. Lung microbiota has the ability to regulate specific oncogenic pathways that are directly responsible for carcinogenesis. It is possible that changes in bacteria-derived molecules caused by dysbiosis in the tumor microenvironment can affect lung cancer cell metabolism and oncogenic signaling (50, 51). The expression of genes involved in the PI3K and ERK1/2 signaling pathway was shown to increase after the human lung adenocarcinoma cell line A549 was stimulated in vitro with bacterial products isolated from lung cancer patient enriched bacteria. This result was consistent with the transcriptomic changes seen in lung cancer patients compared to healthy people (52). Upregulation of the PI3K pathway has been identified as an early event in the process of lung tumorigenesis, implying a direct link between lung microbiota and oncogenesis (53).
2.11.4 Senescent cells
Cellular senescence is a long-lasting and irreversible cell cycle arrest with secretory features, macromolecular damage; altered metabolism that occurs in response to various stresses and more often activates a persistent DNA-damage response. It is usually viewed as an endogenous tumour suppressor mechanism due to the induction of a long-lasting and generally irreversible cell cycle arrest. Senescent malignant and non-malignant cells, on the other hand, remain metabolically active and can secrete a slew of mainly pro-inflammatory cytokines, chemokines, growth factors, and matrix-remodeling proteases known as the senescence-associated secretory phenotype (SASP). This SASP shields malignant cells from immune clearance and influences the tumour microenvironment and ultimately promotes cancer relapse and metastasis (54–57).
3 Tumor microenvironment
In the past 25 years, a breakthrough change has occurred in the approach to cognizance biology of solid tumors. Earlier the focus of cancer research was almost restricted to some aspects like individual tumor cells, the transformation processes and mechanisms that conveyed their malignancy, but recent research has centred towards tumour in its whole complexity (58) Cancers are not just masses of malignant cells but sophisticated reprobate organs, which attract the other cells and alter them (59). TME consists of cellular (cancer cell, stromal fibroblast, endothelial cell, immune cells) and non-cellular components (ECM components) (Figure 3) (60). The interaction between Tumour cells and microenvironment (TME) plays a crucial role in cancer progression (NSCLC) (61, 62). The cancer cell communicates with the microenvironment through ECM, also cell-cell contacts or via mediators which allow these contacts such as cytokines, enzymes, chemokines, and exosomes (63).
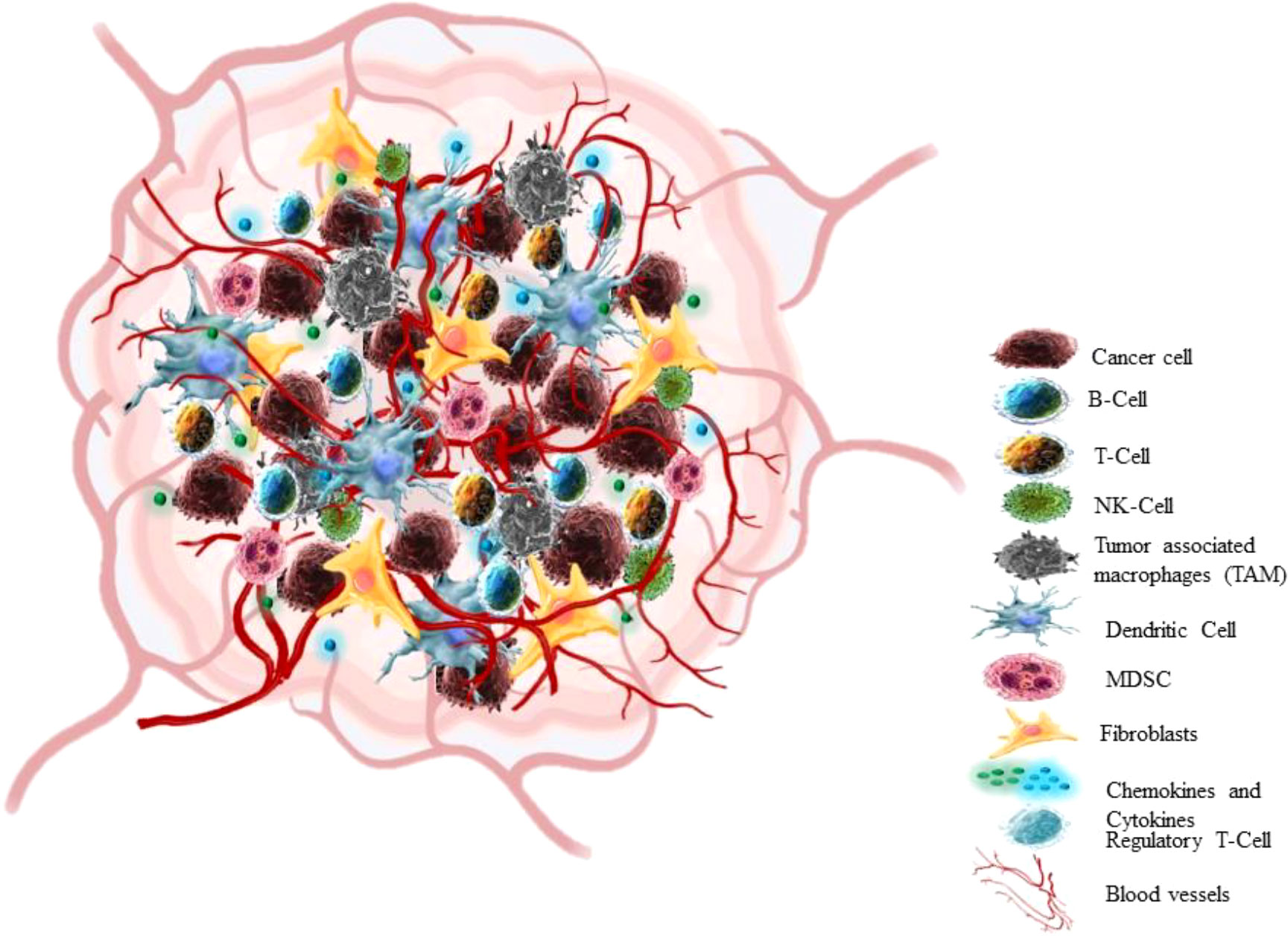
Figure 3 Tumor microenvironment.
A tumor microenvironment has four characteristics such poor nutrients, hypoxia, high acidity and the immunosuppressive microenvironment (64). Tumor cells also show deregulated metabolism due to both intrinsic (such as the genetic makeup of cancer cells) and extrinsic factors (such as oxygen tension, nutrition availability, and pH). The cancer cells scavenge nutrients in the microenvironment leading to suppression of antitumor immune response (65). Thus, the reprogrammed metabolic pathway in cancer cells satisfies particular requirements for energy, biomass, redox homeostasis, and cellular communication (66). The NSCLC is a heterogeneous tumor at genetic and cellular level (62). Numerous studies have characterized TME of lung tumors at a single cell resolution. Lambrechts et al., reported 52,698 cells in TME transcriptome using scRNA seq, out of these 75% cells originated from the lung tumor and also presented thorough classification of stromal cells with various pathways in NSCLC patients (67). Fengying Wu et al, also analysed tissue biopsy sample of 42 NSCLC patients particularly from stage III/IV using scRNA seq, in order to study tumor heterogeneity in advanced NSCLC (68). These studies have enlisted different immune cells in the NSCLC microenvironment.
3.1. Cancer-associated fibroblasts
Cancer-associated fibroblasts (CAFs) are a very diverse population of stromal cells that play a significant role in the microenvironment of solid tumors (69) such as development, progression, and metastasis of tumor. The Fibroblasts can be broadly categorized into two states: quiescent and activated, despite the fact that they show substantial context-dependent phenotypic and functional variety. The cancer cells lead to the activation of fibroblasts in the TME through the release of TGFβ, PDGF, EGF, CTGF, and FGF (70). The cancer cells and CAFs are referred as ‘Partners in crime’ and inflammations support their interactions (71). These CAFs can also secrete mediators like cytokines, growth factors, CAF-specific proteins and exosomes contributing to proliferation and chemoresistance to lung cancer. The VCAM1 derived from CAFs stimulates proliferation and metastasis in A549 and H358 lung cancer cells via AKT and MAPK signaling (72). CAFs isolated from lung cancer tissue promote metastasis by secreting IL-6, which activates JAK2 and STAT3 signaling and also induces EMT via increasing vimentin expression in lung cancer cells both in vitro and in vivo (73). This CAFs leads to an immunosuppressive and growth-promoting character by modulating different immune cells in TME (74). The CAFs isolated from lung tumor patients show expression of inhibitory ligands such as PDL-2 and FASL, inhibition of these ligands can restores T cell cytotoxicity (75).
3.2 Endothelial cells
Endothelial and vascular blood vessels provide nutrients to tumor tissues, also serve as cancer niche cells and build an environment that supports cancer (76) via encouraging cancer cell migration, invasion and metastasis. They show plasticity, and can alter the cell’s fate. As the tumor progress, endothelial cells go through a process known as “endothelial-mesenchymal transition” to transform into cancer-associated fibroblasts (77). Due to hypoxic environment, these cells express various angiogenic factors like hypoxia inducible factor (HIF), vascular endothelial growth factor A (VEGFA), platelet-derived growth factor (PDGF) or angiopoietin 2 (ANGPT2) along with proangiogenic chemokines and receptors to begin neo-angiogenesis (78). These Tumor Endothelial Cells (TEC) releases cytokines which reduces the cytotoxic responses of the immune cells. The VEGF and Notch signaling pathways may be more active in TECs as compared to normal tissue endothelial cells, which is responsible for the elevation of angiogenesis (79) SIRT1 increases vessel density and negatively regulates DLL4/Notch signaling and deacetylation of Notch1 intracellular domain (N1IC) which promotes growth of Lewis lung carcinoma (LLC) xenograft derived vascular endothelial cells (80).
3.3 Immune cells
The important components of the tumour microenvironment are immune cells. They can either inhibit tumour growth or enhance it, depending on the circumstances, in which they interact with the tumour microenvironment. Immune cells can be broadly divided into two groups viz., innate immune cells and adaptive immune cells (77). From the available literature evidence, it is now clear that the innate immune response affects the TME by regulating T cell fate and also significantly shapes the TME. Macrophages (Ms), dendritic cells (DCs), neutrophils, myeloid derived suppressor cells (MDSCs), natural killer cells (NKs), and innate lymphoid cells (ILCs) are the innate immune cells. Mechanistically, cytokines in the TME control immunological processes, which result in suppressed immune reactions that direct tumour growth. It is crucial to gain a thorough understanding of innate immune cells and apply that knowledge to emerging treatments that intend dysregulated cells in the TME (81). Stankovic et al., 2019 has reported the presence of different immune cells in NSCLC using flow cytometry from a patient sample which were B cell, T cell, natural killer cell, macrophages, dendritic cells (82). Further the immune cells in the NSCLC microenvironment are discussed in following sections.
3.3.1 Macrophages
The extraordinary plasticity of macrophages allows them to adapt their physiology in response to environmental signals, giving rise to numerous subpopulations of macrophages with diverse roles. Tumor-associated macrophages or TAMs, are found to be increased in the stromal compartment of a variety of solid tumors and orchestrate to the development, angiogenesis, and metastasis of cancer (83). In NSCLC microenvironment, TAM acts as an immunosuppressive cell and favors proliferation and metastasis of NSCLC (84). Macrophages can typically be polarized into M1 or M2 macrophages. Classically activated macrophages, often referred to as M1-polarized macrophages. TAMs are believed to more closely resemble M2-polarized macrophages, also referred to as alternatively activated macrophages, TAMs are crucial in establishing the link between inflammation and cancer (85). NOX4 is highly expressed in NSCLC cells, which recruits TAM (M2 Macrophage) through production of different cytokines that are dependent on ROS/PI3K signaling, thus promotes NSCLC growth (86). In NSCLC patients, high density of TAM is associated with poor survival and vice versa. Studies have reported that TGF‐β secreted by TAM increases SOX9 expression through C-jun/SMAD3 pathway leading to metastasis of NSCLC (87).
TAMs, which comprise the majority of immunoregulatory cells in tumors, help to suppress cytotoxic T lymphocyte (CTL) responses in tumour microenvironments. In murine tumor models, proliferation of CD8+ T cell is suppressed by TAMs, which rely on L-arginine metabolism via inducible nitric oxide synthase (iNOS) or arginase I, which leads to production of reactive oxygen species (ROS). TAMs also produce IL-10, which induces the expression of PD-L1 (costimulatory molecule) in monocytes, and can suppress CTL responses. Additionally, TAM derived IL-10, indoleamine 2,3-dioxygenase, prostaglandin E2 (PGE2) and CCL17, CCL18, and CCL22 results in significant roles in the activation of Tregs and inhibition of T cells respectively in the tumour microenvironment (85).
3.3.2 Dendritic cell
Dendritic cells (DCs) are important for the initiation of antigen specific immune response and tolerance, as a result exploiting DC manipulation has a great potential for generating effective antitumor immunity (88). DCs are classified into three types such as conventional DCs (cDCs),plasmacytoid dendritic cells (pDC) and monocyte derived DCs (moDCs) (89). Further, conventional DCs (cDCs), are of two types’ conventional type 1 DCs (cDC1s) and conventional type 2 DCs (cDC2s). cDC1s present antigen to CD8+ T cells while cDC2s activates the CD4+ T cell response and pDC produces large amounts of interferon-α (90). DCs derived from NSCLC patients are immunosuppressive as it up regulates the expression of co-inhibitory molecule B7-H3, thus stimulating the T cell suppressive effects of DCs (91). NSCLC cells modulate the CD1c+ DC subsets functioning through regulating CD205 and CD103 expression on CD1+c DCs, which could be one aspect of NSCLC induced immunosuppressive microenvironment (92, 93). The co-culture of DCs with lung carcinoma cells (LCC) or T cells showed increased production of TGF‐β which induces generation of CD4+CD25+ FOXP3+ regulatory T (Treg) cells to suppress T cell proliferation (94).
3.3.3 T regulatory cell
The key immunosuppressive cells such as regulatory T cells (Tregs) support tumour growth by impeding the effector immune response (95). In the tumour microenvironment (TME), traditional T cell can activate and differentiate the Tregs cells, which shows potent immunosuppressive role, impede antitumor immunity which in turn contributes to the growth of tumors. The T regulatory cells are divided into two types based on biological characteristics, natural Treg (nTreg) cells, which naturally develop in thymus and Inducible Treg (iTreg) cells, derived from the naive T cells present in the periphery (96). The self-antigens in thymus induces the natural Treg cells while the iTreg cells, induced by the antigens in the periphery, are generated as a result of particular cytokine in TME. Basically, T regulatory cells are the subgroup of CD4+ T cells, which are separated from other immune cells by the expression of FOXP3 (97). Tregs are prevalent in NSCLC and suggest an increased chance of recurrence of disease in the early stage and also acts as a blockade to Immune checkpoint inhibition (ICIs) in Lung cancer (98). The study has shown Foxp3+ regulatory T cells are critical for lung tumorigenesis in K-ras mutant mice model (99). Enlisted are the mechanisms through which Tregs play immunosuppressive roles. 1). Tregs releases inhibitory cytokines like TGF-β, IL-10 and IL-35 which block immune function through these and other associated pathways.Tregs can also block CD8+ T cell and DC function via membrane-bound TGF-β, which governs the body’s antitumor immunological function. 2). Treg cells produce granzymes and perforin which mediates cytotoxicity of effector cells. 3). They interfere with the cell metabolism, which ultimately affects function T cells by different modes like the deprivation of IL-2 (100), increasing production of adenosine via continuous expression CD39 and CD73 on Tregs (101). 4). Entire causes suppressive environment along MDSC (102).
3.3.4 Myeloid derived suppressor cell
About 30 years ago, monocytes and neutrophils with significant immunosuppressive properties were originally described, and later given the designation myeloid-derived suppressor cells (MDSCs) to emphasize their distinct position among myeloid cells (103). MDSCs are divided into subtypes as polymorphonuclear MDSCs (PMN-MDSCs) and monocytic MDSCs (M-MDSCs) and play inhibitory role in lung cancer (104). This immunosuppressive cell steadily accumulates in tumor tissue and regulates the immune response against tumor by interacting with innate and adaptive immune cell (105). NSCLC patient study has shown endoplasmic reticulum stress in neutrophils increases the expression of Lectin-type oxidized LDL receptor-1 (LOX-1) and convert these neutrophils to suppressive PMN-MDSCs (106). Patients with non-small cell lung cancer have B7-H3 predominantly expressed on intratumoral CD14+HLA-DR/low MDSC and is associated with NSCLC progression (107). In lung cancer, MDSCs impairs the B cell response via inhibiting IL-7 and STAT5 signalling which is critical for the development and differentiation of B cells, therefore can promote immune escape (108).
In TME, these MDSCs are manifest to inflammatory and hypoxic surrounding, As an outcome, there is increase in arginase 1 (Arg1) and iNOS due to hypoxia-inducible factor (HIF)-1, as well as a decrease in ROS production, also increase in the inhibitory protein PD-L1 on the surface of MDSCs (109).
3.3.5 Natural killer cell
NK cells were initially supposed to be big granular lymphocytes that naturally show cytotoxicity against the tumor cells. Later, it was discovered that NK cells have a distinct lymphocyte lineage that have both cytotoxic and cytokine-producing effector activities (110) but in lung cancer they are capable of relevant cytokine production rather than cytotoxicity (111). In NSCLC microenvironment, TGF-β mediates the pro-angiogenic NK phenotype via production of VEGF, PIGF and IFN-γ (112).
The interplay among natural killer cells and cancer cells exist, this interaction varies consistently with the development of NK cells, cancer progression and metastasis. The NK cells identify tumour cells by using a variety of surface molecule and activate them. In response to immune control, NK cells show their activity through releasing cytokines (IFN-γ, TNF-α, GM-CSF, IL-5), ADCC and also producing memory NK cells. But, the persistent exposure of NK cells with the cancer cells, alter the NK cells to immunosuppressive state through tumour derived molecules (PGE-2, PDL-1 and adenosine) and stromal cell (fibroblast, macrophage and monocyte) educated by the tumour which in turn leads to immune escape phenomena (113).
3.3.6 B cell
B cells have the ability to directly present antigens to CD4+ and CD8+ T cells, shaping their ability to mount antigen-specific immune responses in the context of tumor microenvironment. In addition, depending on the constitution of the tumor microenvironment, the types of B cells present, and the antibodies they produce, tumor-infiltrating B cells may have both protumor and antitumor effects (114). In NSCLC patients, tumor-infiltrating B cells and CD4+ T cells inhabit in Tertiary lymphoid structures (TLS) and these are associated with better prognosis (115). The study has revealed tumor-infiltrated B cells subtypes and their various roles in the development of NSCLC using single-cell RNA sequencing of NSCLC patient sample (116).
3.3.7. T cell
In TME, Tumor infiltrating T lymphocytes, mainly CD4+ and CD8+ T cells and their immunoregulatory cytokines define adaptive immunity. Naive CD8+ T cells engaged with antigen-presenting cells (APC) through peptide-major histocompatibility complex I (MHC I) and T-cell receptors (TCR) on T cell further co-stimulatory signals causes conversion of CD8+ T cells into cytotoxic T cell which fight against tumor and intracellular pathogens (117). The presence of PD1 and CTLA4 on T cells interact with PDL1 and CD80/CD86 on APC respectively, ultimately inhibiting T cell activation (118). Expression inhibitory receptor on T cell leads to dysfunctional state of T cells, thus abolishing immune response against cancer cells (119). In NSCLC patients, tumor infiltrating CD8+ T cells expresses inhibitory receptors such as PD-1, Tim-3, CTLA-4, LAG-3, and BTLA which are correlated to progression of disease (120). The PD-1 targeted therapies in NSCLC patients showed an increased number of Ki-67+PD-1+CD8 T cells (15). The immune cells have a multifaceted role in the nsclc microenvironment and miRNAs are crucial players in modulating biology of tumor microenvironment. The miRNAs and their role in NSCLC is extensively discussed in the below section.
4 MicroRNA as regulator in TME
MicroRNA (miRNAs) are endogenous small non-coding RNA (22 nucleotides),which regulates the gene expression (121). The first miRNA was discovered in Caenorhabditis elegans in 1993 which represses lin-14 mRNA (122) but it took over a decade to understand its crucial functions in gene regulation. The molecular aspects of miRNA function and biogenesis are tightly regulated at several levels and have been extensively studied (123). Canonical pathway is a dominant pathway for miRNA biogenesis. The pri-miRNA transcript is produced by RNA polymerase II/III. This pri-miRNA is cleaved by a microprocessor complex which consists of DGCR8 and Drosha to form pre-miRNA. Further pre-miRNA exported from nucleus to cytoplasm with the help of exportin 5 (XPO5)/RanGTP complex and again cleaved by Dicer to form mature miRNA duplex (124). The RNA-induced silencing complex (RISC) consist of mature miRNAs and Argonaute (Ago) proteins, which further regulates post-transcriptional gene silencing by inducing messenger RNA (mRNA) degradation or translational inhibition (125). The canonical pathway for biogenesis of miRNA is shown in Figure 4. The complementary sequence between the seed sequence of each unique miRNA and the target mRNAs determines the specification of target mRNA (126).
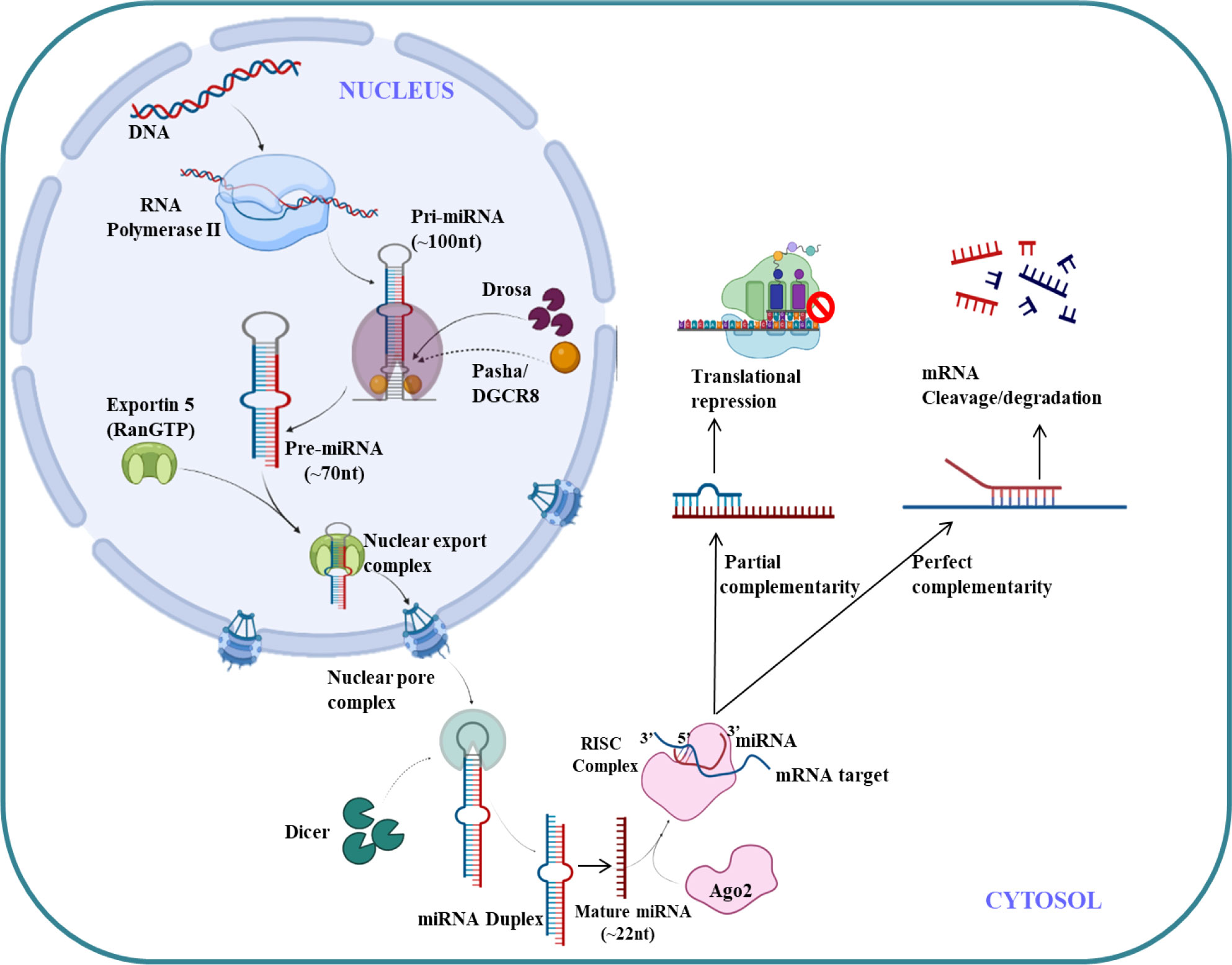
Figure 4 miRNA biogenesis and regulation.
In 2002, Calin et al. were the first to uncover a linkage between miRNA dysregulation and cancer (127). Alterations in miRNA are entailed in cancer initiation and progression. The differential expression of miRNA genes in cancer and normal cells are due to the presence of these genes in cancer associated genomic regions (128). In 2006, Volinia et al. reported miRNA signature in solid cancer and validated the predicted targets of differentially expressed miRNAs as a classic oncogenes or tumor suppressors (129), as the miRNAs regulate gene expression at post transcriptional level, they have a significant impact on a broad range of pathways, however their influence is more on developmental and oncogenic pathways (128, 130). The different mechanism for dysregulation of miRNA in cancer includes various aspects such as amplification or deletion of miRNA genes, transcription factors controls miRNA expression, the dysregulated epigenetic modulation of miRNA and lastly, aberrant expression of any enzymes of miRNA biogenesis pathway could cause abnormal expression of miRNA (131).
Based on the important role that miRNAs play in lung cancer pathogenesis, they are considered to be prospective prognostic markers. There are mounting evidences which suggests that miRNAs are dysregulated in human cancer including non-small cell lung cancer (NSCLC), and may act as oncogene or tumor suppressor miRNA (132). Takamizawa et al. were the first to report the reduced expression of let-7 in lung cancer and also showed in vitro overexpression of let-7 in A549 cell line inhibited growth of lung cancer cells (133).
The abnormal expression of miRNAs affects the different hallmarks of cancer (131). In the TME, miRNA are supposed to connect the tumor cells and immune cells in the surrounding, thus, the dysregulated expression of miRNAs in cancer cell can directly have an impact on the immune microenvironment. For instance, miRNA can direct the production of chemokines or cytokines by tumor cells, which in turn influences immune cells. However, exosomes are crucial for miRNA transport, and can also have an impact on the immunological microenvironment since miRNA of tumor cells can be transferred via exosomes to immune cells (134).
5 miRNAs involved in EGFR signaling cascade
There are extensive reports which suggest the role of miRNA in angiogenesis, proliferation, cell metabolism and modulating immune cell response in non-small cell lung cancer. Aside from various environmental parameters, it is important to understand the molecular abnormalities in lung cancer development as the cancer cannot grow without involvement of any molecular abnormalities. Although, a number of signaling axes have been implicated in the pathogenesis of lung cancer, it seems that the fatality is due to the overlap of oncogenic pathways (135). Thus the studies have been done to investigate the various signaling axes which led to the pathogenesis in lung cancer. It has been reported in earlier studies that overexpression of Epidermal growth factor receptor (EGFR) is found in 50% of NSCLC and it is associated with poor prognosis (136). The majority of EGFR kinase domain mutations are found at exons 18 to 21, which increases EGFR’s kinase activity and triggers over activation of downstream signaling pathways, the three major downstream signaling pathways such as phosphatidylinositol 3-kinase (PI3K)/Akt/mTOR, interleukin 6(IL-6)/Janus kinase (JAK)/signal transducer and activator of transcription 3 (STAT3), mitogen-activated protein kinases (MAPK)/extracellular signal-regulated kinases (ERK) which supports tumorigenesis of NSCLC (3, 136). Approximately 90% of all EGFR mutations are of the two types, L858R (point mutations in exon 21 that result in a leucine to arginine substitution at codon 858) and exon 19 deletion (in-frame deletions in exon 19) (137). miRNA expression patterns that are abnormal can be employed as prognostic and diagnostic indicators in NSCLC. The development of NSCLC is strongly influenced by EGFR signaling pathways and miRNAs. Numerous studies have demonstrated the dual function of miRNAs as either oncogenes or oncosuppressors, and that it primarily depends on their direct targets and the activated downstream signaling pathways (138, 139). It was discovered that EGFR, MET, and the miRNA cluster 23a27a24-2 were biologically connected, with miR-27a controlling MET, EGFR, and Sprouty2 in a variety of NSCLC cell lines. These findings suggest that miR-27a can inhibit MET and EGFR by directly or indirectly targeting their 3’ UTRs via Sprouty2 (140). Luciferase assays revealed that EGFR was a direct target of miR-134, and its overexpression in NSCLC cells inhibited EGFR expression (141, 142). Another miR-218-5p also inhibits EGFR in A549 and H1975 cell lines (143). To determine the miRNA-dependent EGFR signaling cascade, database-dependent miRNA target prediction was carried out. This model demonstrated that the circulating miR-145, miR-199a-5p, and miR-495 have favorable correlations with the EGFR ligand EGF. It follows that miRNAs can control several EGFR signaling pathway elements, including the ligand, receptor, or downstream signaling molecules (144, 145). According to findings, mutant K-Ras negatively affects miR-199b expression in NSCLC cells by overexpressing it and silencing it. Overexpressing mutant K-Ras inhibits miR-199b expression, while silencing it increases it. MiR-199b functions as an anticancer agent by concurrently blocking the Akt and ERK pathways by specifically targeting numerous Akt and ERK pathway components in NSCLC (146). Our previous system biology study reported miR-520c-3p could target AKT signaling in NSCLC (147). Downregulated miR-16 involved in lung cancer proliferation and invasion through ERK/MAPK signaling which inhibits MEK1 (148). miR-1258 inhibits NSCLC proliferation via downregulating GRB2 expression both in vitro and in vivo (149). Similarly miR-181a-5p downregulates Kras in A549 cells, thus inhibiting NSCLC proliferation (150). miRNAs are also known for their regulatory role in NSCLC metabolism.miR-33b also affects the glucose metabolism in NSCLC by downregulating LDHA enzyme (151) and miR-144 targets GLUT1 thus involved in Warburg effect in lung cancer (152). Perturbed EGFR signaling leading towards autophagy and key regulatory miRNA in EGFR signaling in NSCLC is represented in Figure 5.
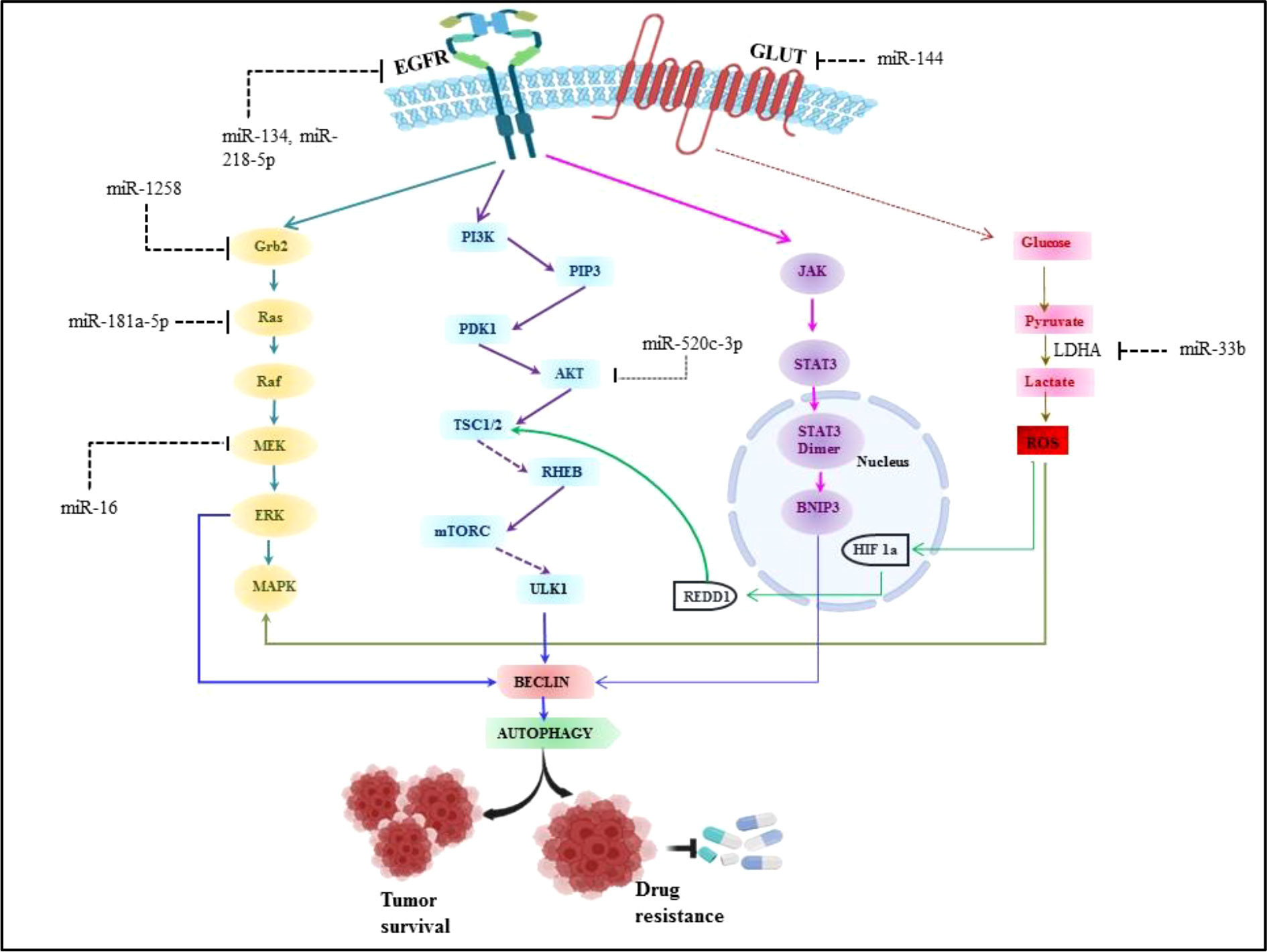
Figure 5 Perturbed EGFR signaling leading towards autophagy and key regulatory miRNA in EGFR signalling in NSCLC.
Furthermore, miR-224 promotes tumor growth in NSCLC both in vitro and in vivo by directly targeting the tumor suppressors TNFAIP1 and SMAD4 (153). Specifically miR-218 downregulate the IL-6 receptor and JAK3, thus, by overexpression miR-218 in NSCLC cells prevents cell proliferation, invasion, and colony formation (154). miR-196b-5p downregulates the tumor suppressors, GATA6 and TSPAN12 expression. Report suggested that TSPAN12 knockdown H1299/shTSPAN12 cells were injected in nude mice, resulting in significantly increased tumor size (155). Additional research revealed that miR-7-5p inhibits NSCLC tumor metastasis by targeting NOVA2. miR-7-5p mimic and negative control was injected in the A549 xenograft model, leading to decrease in tumor growth and metastasis. while NOVA2 overexpression reduces the inhibitory effect of miR-7-5p on lung cancer cells (156). Similarly, miR-187-5p downregulates CYP1B1 which is involved in the development and metastasis of NSCLC cells (157).
6 miRNA as immunomodulator in NSCLC microenvironment
Several miRNAs have been reported to regulate the cells in the NSCLC microenvironment (Figure 6). CAFs, being one of them, contributing towards proliferation and drug resistance in NSCLC via miR-1. miR-1 mediates SDF-1 expression, which upregulates CXCR4 expression, leading to increased expression of NF-κB and Bcl-xL in A549 and 95D lung cancer cells (158). The study has shown miRNAs plays role in conversion of normal fibroblast in CAFs in NSCLC patient, with this aspect miR-1, miR-206 were down regulated and miR-31 was upregulated in CAFs of Lung regulating FOXO3a/VEGF/CCL2 signaling (159). Moreover miR-101 is downregulated in CAFs of lung cancer which targets the CXCL12 expression thus overexpression of this miRNA could suppress the CAFs to promote cancer cell proliferation (160). miR-21 and miR-103a involved in stimulating the polarization of M1 to M2 phenotype in lung cancer, also activates AKT-STAT3 signaling axis while tumor suppressor miR-1207-5p and miR-155 stimulates M1 polarization (161). The previous study also suggests that extracellular vesicles derived from hypoxia lung cancer causes increased M2 polarization via transfer of miR-103a. This miR-103a decreases PTEN level, ultimately increasing activation of AKT and STAT3, also expressing many immunosuppressive and pro angiogenic factors (162). miR-20a down regulates the expression PTEN and up regulates PDL1 expression thus leading to proliferation of NSCLC (163). The Natural killer cells isolated from patients with NSCLC showed reduced expression of miR-130a and elevated expression of STAT3. miR-130a can target STAT3, thus overexpression of miR-130a enhances NK-92 cells’ killing activity against A549 cells (164). TGF-β is a potent immunosuppressive found in the NSCLC microenvironment and study reported that TGF-β induced miRNA-183 expression represses DAP12 in NK cells affecting cytotoxic response of NK cell against tumor cell (165). Nannan Song et al. have reported the role of miR-138-5p in NSCLC. This miRNA downregulates CCND3, Ki67, CCD20, MCM in A549/3LL cells and is also found to regulate DCs maturation in nude and C57BL/6 mouse models bearing A549 and 3LL cells respectively. Furthermore miR-138-5p downregulates PDL1 in A549 and PD1 on DCs (166).
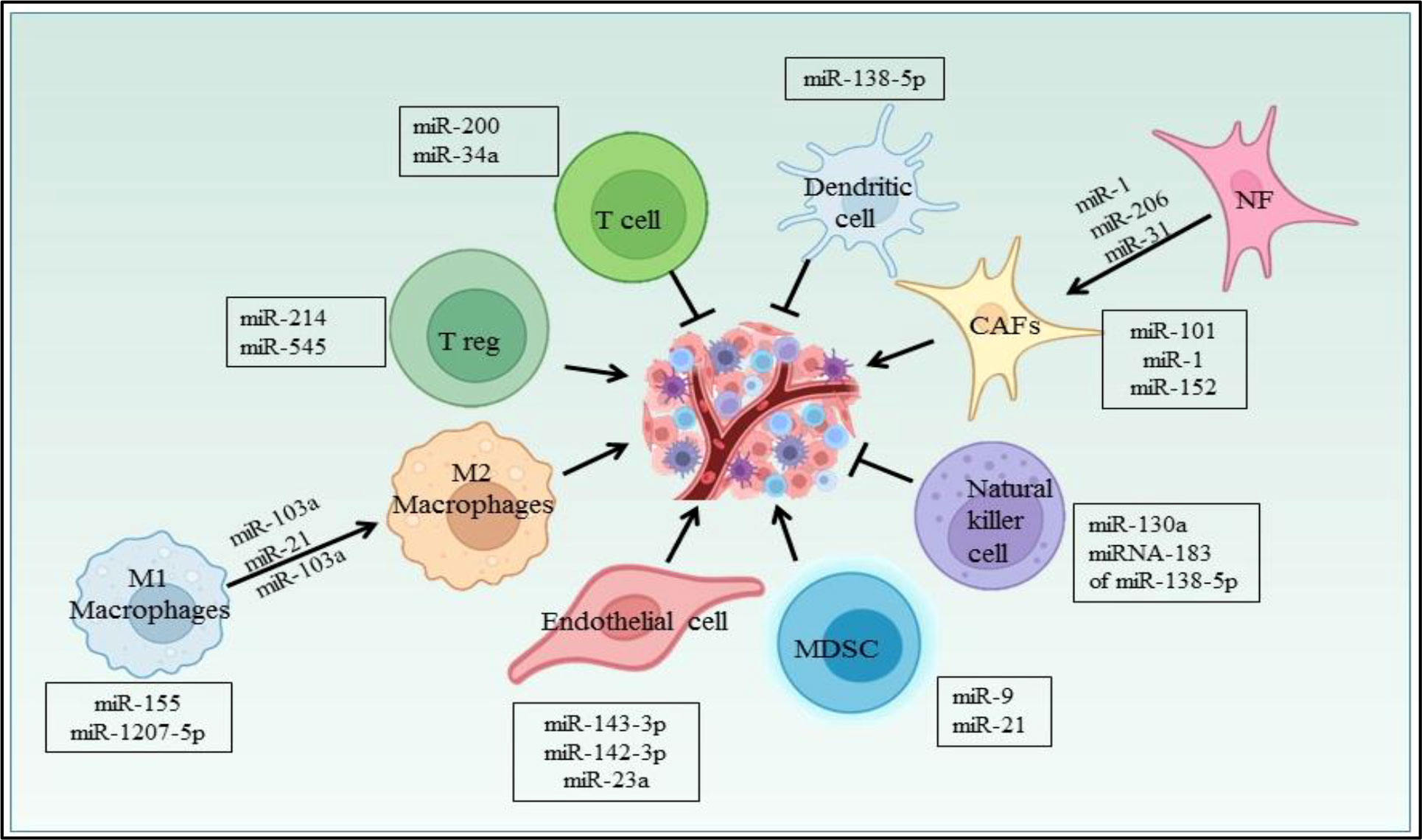
Figure 6 miRNA regulating different cells in the NSCLC microenvironment.
In lung cancer, miR-21 regulates MDSCs accumulation and activity. miR-21 downregulates RUNX1 and upregulates YAP in lewis lung cancer mice model promoting immunosuppressive role of MDSC (167). Also miR-9 regulates differentiation and function of MDSC via targeting RUNX1. miR-9 downregulation in MDSCs inhibits in vivo tumor growth in lewis lung cancer mouse model (168). Yuan Yin et al. showed LLC cells secretes miR-214 which targets PTEN in recipient CD4+ T cells results in promotion of Treg expansion (169). The radiotherapy treatment on lewis lung cancer tumor inhibits Treg recruitment via increasing miR-545 expression (170). miR-34a mimic MRX34, the first miRNA based therapeutic which entered in first phase clinical trial as an anticancer drug (171). The studies also suggest the miR-34a mimic can modulate the tumor infiltrating immune cells. In NSCLC mouse model, specifically it was shown that in vivo injection of MRX34 generates the lower expression of PD-L1, but MRX34 can also boost tumor infiltration of CD8+ T cells and reduce CD8+PD1+ T cells (172). miR-152 downregulates fibroblast growth factor 2 (FGF2) which in turn suppresses NSCLC proliferation (173). The study showed PD-L1 expression is regulated by the miR-200/ZEB1 axis in NSCLC (174). Previously reported that miR-34a, tumor suppressor downregulates CDK6 expression and high expression of miR-34a is associated with a prolonged progression-free survival in NSCLC patients (175). miRNAs also play a role in NSCLC angiogenesis. miR-519c targets HIF-1α which is an angiogenesis regulator (176). In hypoxia, lung cancer cells secretes exosomes, in which miR-23a is upregulated and targets PHD1 and 2 contributing to accumulation of HIF-1α,in turn to angiogenesis (177).
7 Autophagy
Autophagy is an intracellular catabolic degradation process in which cytoplasmic macromolecules, aggregated proteins, damaged organelles, or pathogens are delivered to lysosomes and digested by lysosomal hydrolases to generate nucleotides, amino acids, fatty acids, sugars, and ATP before being recycled into the cytosol. Eliminating aggregated proteins and damaged organelles, autophagy is a highly conserved catabolic cellular event. The dynamic process entails induction, autophagosome nucleation, development of the double membrane, sealing and fusion with the lysosome, and breakdown of absorbed components. In order to degrade and recycle non-functional cellular components as an internal source of nutrition, cells typically have basal levels of autophagy. The amount of autophagy may significantly increase in response to a variety of stressors and stresses, including deprivation, hypoxia, and medication, in order to supply intracellular nutrition and remove toxic components (178, 179). Initiation, vesicle nucleation, vesicle maturation, vesicle fusion, and cargo degradation are the five steps of the autophagy pathway. It starts with the activation of the Unc-51-like kinase 1 (ULK1) complex, which includes ULK1, ULK2, autophagy-related gene 13 (ATG13), focal adhesion kinase interacting protein 200 kDa (FIP200), and ATG101. Activated ULK1 kinase, mediates the phosphorylation and activation of the Beclin1-VPS34 (a class III phosphatidylinositol 3-kinase (PI3K)) complex and eventually instigate vesicle formation. Various protein conjugation events are required during the maturation process to help cleave pro-LC3 to soluble LC3-I, which is then conjugated to lipid phosphatidylethanolamine (PE) on the surface of the emerging autophagosome. After being conjugated to lipid, LC3-I is inserted onto the surface of emerging autophagic vesicles. The autophagosome is what happens when the isolation membrane is enclosed, and is then transported to the perinuclear region where the lysosome subsists. Here certain membrane-tethering complexes, SNAREs and Syntaxins aid in autophagosome-lysosome fusion. Finally, after the fusion lysosomal hydrolases degrade autophagic cargo, and recycled contents are discharged via nutrient transporters, fueling cell growth (180).
7.1. Environmental based accountabilities of autophagy in NSCLC
It appears that there are two possible outcomes for the role of autophagy in tumorigenesis. It appears to play a role in tumour suppression in the early stages of cancer (181, 182), and in later stages, it allows cancer cells to survive metabolic stress and hypoxia in solid tumors (183). According to a research by Rao et al. in a mouse model of non-small cell lung carcinogenesis they concluded that autophagy plays a dual role in oncogenesis. Cancer cells need to be in the best possible shape to withstand a hostile environment caused by hypoxia, as well as the lack of nutrients and trophic stimuli, particularly at advanced stages of tumour progression. However, autophagy is frequently inhibited during early oncogenesis, raising the possibility that autophagy is an oncosuppressive mechanism (184). The environment does affect autophagy’s function in cancer, and its upregulation is required for cancer cells to survive in hypoxic tumour regions (185).
7.2 Autophagy modifies the NSCLC tumor microenvironment
Literature evidence suggests that the tumor microenvironment is affected by autophagy. TME consists of several factors like hypoxia, inflammation, and cytokines. Under stressful situations in cancer microenvironments, autophagy meets the need for cellular energy and avoids cytotoxicity. Cancer cells could be able to adapt and survive in low oxygen environments by using the stress response when hypoxic circumstances are present. TME uses autophagy to provide the metabolic requirements of cancer stem cells, sound immune cells, cancer associated fibroblasts, angiogenesis, neural connections, and extracellular matrix (186).
Autophagy is proposed as a cell death mechanism, programmed cell death type II, and has been shown to play a greater variety of pathophysiological roles in many disease processes, including cancer; it also aids cells to clear damaged proteins, organelles, pathogens, or aggregates. Autophagy’s potential to modulate cell death makes it a therapeutic target in cancer (179). By controlling their target genes, microRNAs (miRNAs) were frequently shown to be dysregulated in NSCLC and were strongly linked to the formation, progression, and metastasis of the disease.
8 miRNA and autophagy
In addition to genetic factors, epigenetic mechanisms like microRNA networks are crucial for controlling autophagy-related pathways. Numerous miRNAs can alter autophagy and the processes that depend on it in a number of disorders. More studies have shown a connection between autophagy-regulating miRNAs and chemoresistance/chemosensitivity in lung cancer cells (187, 188). As previously mentioned, mTORC1 is inhibited in response to a variety of stress-inducing signals, which activates ULK1/2 and initiates autophagy. It has been demonstrated that miR-520a-3p, miR-513b, and miR-145 alter the mTOR signaling cascade to reduce NSCLC cell proliferation and migration (189, 190). In NSCLC tissue,miR-21 is highly expressed and regulates autophagy in A549 cells through AMPK/ULK1 signaling pathway (191). By directly downregulating HMGB1 and subsequently activating the PI3K/Akt/mTOR pathway, miR-142-3p overexpression prevents starvation-induced autophagy of NSCLC cells. Additionally, miR-142-3p overexpression decreases autophagy triggered by anticancer drugs and raises the chemosensitivity of NSCLC in vitro and in vivo (192). Studies indicate that microRNAs and treatment resistance in NSCLC patients are closely related. In a study, it was discovered that patients with gefitinib-resistant NSCLC have decreased expression of miRNA-153-3p. The downregulation of ATG5 caused by the overexpression of miRNA-153-3p decreased autophagy and increased gefitinib sensitivity in NSCLC (193).
9 Therapeutic roadmap
The last ten years have seen a significant advancement in NSCLC therapy, allowing for a more tailored choice of alternatives. Treatment options are influenced by immunologic and molecular characteristics. Patients with EGFR mutations, ALK rearrangements, ROS1 rearrangements, BRAF mutations, NTRK mutations, and high levels of PD-L1 for instance, should receive FDA-approved targeted therapy (Table 1) or immunotherapy as their first line of treatment. RET, MET, and HER2 in NSCLC are three additional oncogenic driver mutations that are intriguing therapeutic targets. The immune system’s ability to block PD-1, PD-L1, and CTLA-4 is also expanding (194).
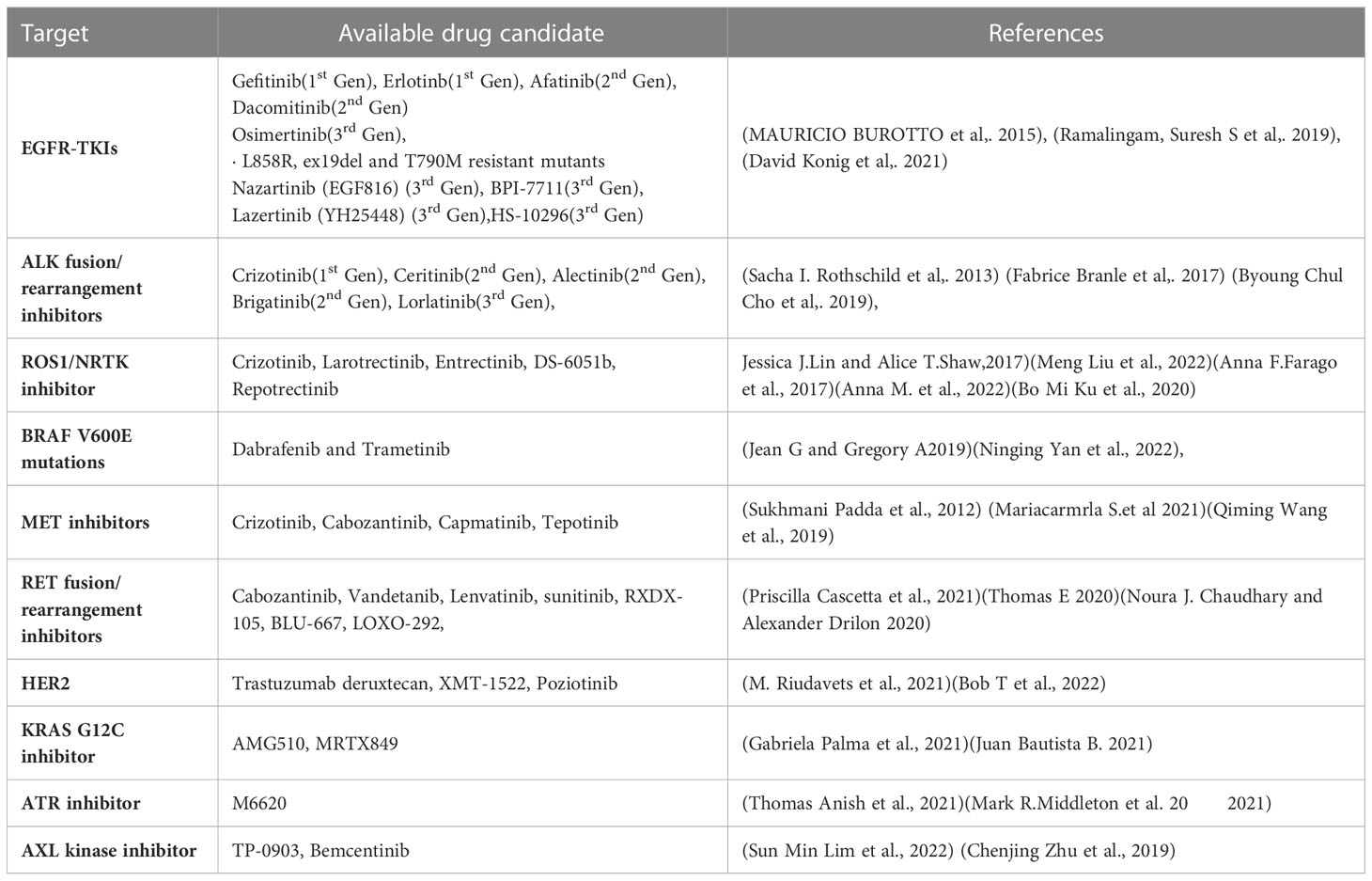
Table 1 Available targeted therapies.
9.1 Targeted therapies
9.1.1 Epidermal growth factor receptor tyrosine kinase inhibitors
EGFR is a member of the receptor tyrosine kinase family, which also includes HER2/ERBB2/NEU, HER3/ERBB3, and HER4/ERBB4. EGFR mutations are more prevalent in never smokers. These mutations affect EGFR exons 18–21, and 90% of them are exon 19 deletions or exon 21 L858R point mutations. Gefitinib is the first EGFR Tyrosine Kinase inhibitor investigated for use in advanced NSCLC. At present, FDA has approved the use of gefitinib, erlotinib, afatinib, dacomitinib, and osimertinib as first-line treatments for patients with metastatic NSCLC (195–197).
9.1.2 ALK fusion/rearrangement inhibitors
About 2-7% of patients with advanced NSCLC carry the fusion oncogene EML4-ALK; these patients are often younger and never smokers. The first recognised targeted treatment for ALK-positive advanced NSCLC is crizotinib. Alectinib is currently the first-line therapy of choice. Another next-generation ALK inhibitor, brittinib, outperformed crizotinib in terms of Progression free survival (198). A potent ALK TKI, Ensartinib (X-396) has also demonstrated inhibitory efficacy against MET, BAL, Axl, EPHA2, LTK, ROS1, and SLK (199).
9.1.3 ROS1/NRTK inhibitor
About 1-2% of NSCLC cases involve ROS1 rearrangement, which mainly affects younger people who have never smoked. The tyrosine kinase receptor is encoded for by the ROS1 gene, which is located on chromosome 6 (198). NTRK1 gene fusions can also involve NTRK2 or NTRK3 and is present in all forms of tumors. Such fusions represent roughly 1% of NSCLC (200). For metastatic ROS1 positive NSCLC, crizotinib received approval in March 2016. For ROS1, ALK, and NTRK gene fusions, entrectinib is an oral TKI. The use of entrectinib for the treatment of advanced or recurrent NSCLC with NTRK fusions in adults and children was first authorized in Japan in June 2019. Recently, the FDA approved entrectinib for use in both these indications and for advanced NSCLC patients who test positive for ROS1 (200).
9.1.4 BRAF V600E mutations
1-3% of NSCLC patients with a linked history of smoking had an activating BRAF mutation. Based on an open-label trial, the combination of dabrafenib and trametinib was approved for the treatment of metastatic NSCLC with BRAF V600 E mutations (201).
9.1.5 MET inhibitors
Hepatocyte growth factor binds to the tyrosine kinase receptor MET. Characteristic anomalies that result in enhanced MET signalling activity include MET gene amplification and exon 14-skipping mutations. Only 3% of instances of NSCLC had an isolated MET exon 14 mutation, however 15-20% of EGFR mutation-positive NSCLC patients have this acquired EGFR TKI resistance pathway instead. Highly selective MET inhibitors include capmatinib and tepotinib (202, 203).
9.1.6 RET fusion/rearrangement inhibitors
The RET rearrangement leads in ligand independent homodimerization and activation of the RET tyrosine kinase by autophosphorylation. It is composed of the RET intracellular kinase domain and the coiled coil domain of the partner gene. As a result, the phosphoinositide 3-kinases (PI3K)/AKT, mitogen-activated protein kinase (MAPK), and signal transducer and activator of transcription 3 (STAT3) pathways are activated, increasing cell proliferation, survival, migration, and differentiation (204). Experiments have been performed on multi-targeted TKIs (MKIs), including cabozantinib, vandetanib, lenvatinib, and sunitinib, which target RET fusion-driven NSCLCs. The most investigated RET inhibitors include cabozantinib and vandetanib, both of which exhibited a response rate in patients with RET-rearranged NSCLC between 20 and 50% (205).
9.1.7 HER2 inhibitors
2-4% of NSCLC patients have HER2 alterations, which primarily show up as protein overexpression, gene amplification, or gene mutation. Female, never-smoker, and lung adenocarcinoma patients with HER2 modifications had a higher risk of developing brain metastases than individuals without HER2 abnormalities or other genetic mutations (206). Pyrotinib is an oral pan-TKI that blocks HER1, HER2, and HER4 (108).
9.1.8 KRAS G12C inhibitor
One of the most frequent oncologic driver mutations in advanced NSCLC is the KRAS mutation. About 14% of lung adenocarcinomas have the KRAS G12C mutation. The median survival is reduced in NSCLC patients who also carry KRAS. AMG510 is a small, promising molecule that locks KRAS G12C in an inactive GDP-bound state, specifically inhibiting it. Another KRAS G12C inhibitor, MRTX844, is effective in treating advanced solid tumours with KRAS G12c mutations (207, 208).
9.1.9 ATR inhibitor
Inhibiting ATR signaling may make tumors more sensitive to chemotherapy that damages DNA because ATR is a crucial part of the DNA damage response. ATR is specifically inhibited by M6620. When combined with gemcitabine, berzosertib is well tolerated by patients with advanced solid tumors (209).
9.1.10 AXL kinase inhibitor
AXL is a receptor tyrosine kinase from the TAM family, together with MER and TYRO3. Despite the fact that AXL itself is not regarded to be a very strong oncogenic driver, overexpression of AXL has been shown to promote tumour cell growth, survival, invasion, metastasis, angiogenesis, epithelial to mesenchymal transition, and immune suppression. Bemcentinib is a very specific inhibitor of the AXL kinase (210).
9.2 Antibody-drug conjugates
Despite the fact that both targeted therapy and immunotherapy-based approaches have become the frontline gold standard of care for patients with advanced lung cancer, acquired resistance and disease progression are nevertheless frequently unavoidable. In this situation, chemotherapy is frequently used as a last resort, but it has a somewhat limited therapeutic index. The development of antibody-drug conjugates (ADCs) presents an enticing substitute. ADCs enable the targeted delivery of cytotoxic payloads to cancer cells by fusing the specificity of a monoclonal antibody with the cytotoxic effects of chemotherapy. Three essential parts make up an ADC construct: the payload, the linker, and the antibody. Each component of an ADC must be carefully created and engineered in order to produce a stable molecule that strikes a balance between efficacy and toxicity (211, 212). Ado-Trastuzumab Emtansine (T-DM1) is an ADC construct made of trastuzumab, an anti-HER2 monoclonal antibody, and emtansine (DM1), a microtubule inhibitor (213). A new ADC called trastuzumab Deruxtecan (T-DXd) combines trastuzumab, a humanized anti-HER2 monoclonal antibody, with deruxtecan, a topoisomerase inhibitor, through a protease-cleavable peptide linker (214). Trastuzumab, which is HER2-targeted, is coupled to the monomethyl Aurstatin F derivative duostatin-5 by a protease-cleavable valine citrulline linker in the drug A166 (139). A glycoprotein trans-membrane calcium signal transducer known as Trophoblast Cell-Surface Antigen (Trop-2) is thought to be a mediator of cell migration and anchorage-independent growth. A number of epithelial malignancies, including NSCLC and small cell lung cancer (SCLC), have been found to express it (215). The anti-Trop-2 monoclonal antibody sacituzumab is connected to the topoisomerase I inhibitor SN-38 via a hydrolysable, cleavable linker to form the anti-drug conjugate sacituzumab govitecan (216). A humanized anti-Trop2 mAb, a tetrapeptide-based cleavable linker, and the exatecan derivative DXd make up the drug datopotamab-deruxtecan (217). To fully utilize the capabilities of the upcoming ADC generation, numerous additional tactics are being investigated. ADCs with immunotherapy are the most alluring combo to encourage synergy.
9.3 Immunotherapy
Cancer immunotherapy functions by enabling our own immune systems to combat the disease. The immune system can be stimulated to target cancer cells by vaccination therapy (active immunotherapy) or immune blockade removal (passive immunotherapy) using checkpoint inhibitors. Nivolumab, an immune checkpoint inhibitor, was the first immunotherapy for metastatic NSCLC that was approved and is made entirely of human IgG4. It binds to the PD-1 receptor and prevents the tumour PD-L1 from attaching to the T cell PD-1 receptor, restoring anti-tumor immunity. Inhibitors of the PD-1 immune checkpoint include nivolumab and pembrolizumab. A fully humanized IgG1 anti-PD-L1 antibody is atezolizumab (218). Atezolizumab has just received approval for the first-line therapy of adult patients with metastatic non-squamous NSCLC who do not have EGFR or ALK genetic tumour abnormalities. It is used in conjunction with bevacizumab, paclitaxel, and carboplatin (219). Regardless of PD-L1 expression, first-line treatment with nivolumab with ipilimumab in advanced NSCLC patients exhibited better overall survival compared to individuals treated with chemotherapy. In patients with solid malignancies, including NSCLC, tislelizumab (anti-PD-1) exhibited evidence of anti-tumor effectiveness and was well tolerated. In addition to anti-PD-1/PD-L1 monotherapy, combination immunotherapy has also demonstrated effectiveness in patients with high PD-L1 expression (220). An immunoglobulin-like protein called Siglec-15 may be overexpressed in a variety of human malignancies. PD-L1 and siglec-15 expression are mutually exclusive and function as important immune suppressors. Monoclonal antibody NC 318 targets Siglec-15 to restore immune system normalcy (35). It has been proposed that interleukin 8 (IL-8) promotes immunological evasion. A human monoclonal antibody called BMS-986253 binds to and prevents the action of IL-8. A study revealed that BMS-986253 monotherapy was well tolerated in solid tumors with metastatic or irresectable disease (221). TNF receptor CD40 is a pro-inflammatory molecule. An antigen-presenting cell is activated by the humanized monoclonal antibody APX005M binding to CD40, which prompts the activation of T cells that are specifically tuned to respond to malignancy (222). Although recent developments in immunotherapy have been encouraging, additional pre-clinical and clinical research with an immunotherapy focus is needed to enhance patient clinical prognosis. Immunotherapy is already being used to treat NSCLC in certain circumstances.
Growing interest in modified-T-cell treatment, particularly that utilizing chimeric antigen receptor (CAR)-T cells, has been observed recently in a variety of solid malignancies. Clinical trials have shown promising outcomes, and the US Food and Drug Administration has authorized the use of anti-CD19 CAR-T cells for the treatment of hematological B-cell malignancies. Recent developments suggest that CAR-T cell therapy is also an effective method for treating NSCLC (223). But, T cell exhaustion is a condition that affects T cells that infiltrate lung tumors. According to a recent study, the NR4A transcription factor family was proven to have a significant impact on T cell exhaustion and to restrict CAR-T cell activity in solid tumors. They showed that CAR-T cells work better in mouse models without NR4A transcription factors, resulting in smaller tumors and higher survival rates (224).
Engineered immune-cell-based cancer treatments have shown strong success in B cell malignancies, but obstacles such as the scarcity of optimal targetable tumour antigens, tumor-mediated immunosuppression, and significant toxicity continue to limit their therapeutic efficacy and widespread use. By using synthetic biology, these difficulties can be solved and more powerful, effective adaptive medicines can be developed, allowing for the precise targeting of cancer cells while preserving healthy cells (225).
10 Synthetic biology as therapeutic approach for NSCLC
Synthetic biology pertains to engineering fundamentals which intend to amend living cells, permitting them to execute functions defined by the user and to obtain desired results (225). This field reconciles engineers and biologists to rewire and reprogram in organisms, through designing constructs for biomolecular components, pathways and networks (226). In 1980, Barbara Hobom introduced synthetic biology to define genetically engineered bacteria by recombinant DNA technology. Meanwhile in 2000, Eric Kool and other spokesperson reintroduced the term ‘synthetic biology’ at American Chemical Society’s yearly meeting which was held in San Francisco. Here, the name was used to define the synthesis of synthetic organic molecules that show role in living systems. Broadly, the word also has been used as ‘redesign life’ (227). The different scientific committee in world define the synthetic biology but there is no approved worldwide definition, the main characteristics of synthetic biology comprises “the de novo synthesis of genetic material and engineering-based method to develop components, organism and products” advocates by the report of the Secretariat of the Convention on Biological Diversity (2015) (228).
Synthetic biology’s only aim is to contribute towards the development of biomedical breakthroughs including escalating bacterial resistance to antibiotics, the rapid rise of novel infectious diseases and the emerging resistance to cancer drugs. Thus to solve these issues, synthetic biology anticipates the construction of specifically designed, easily controlled, and secure devices that would support human immune systems and correct metabolic anomalies (229). The first reports of genetic circuits that had been built to perform certain functions were reported in January 2000. Here, Collins and colleagues constructed a genetic toggle switch which is a synthetic, bistable gene-regulatory network in Escherichia coli and offered a straight forward theory that forecasts the conditions obligatory for bistability. The designed toggle switch comprises two constitutive promoters and two repressors, where each promoter regulates the expression of repressors that are antagonistic to one another (230). Researchers have created fundamental components and elements that allowed transcriptional, translational, and post-translational control of biological processes during the “first wave” of synthetic biology (231). It has exploded in the last ten years and is poised to revolutionize biotechnology and medicine (232). Synthetic biologists endeavor for useful qualities to optimize gene circuits like modularity, orthogonality, tunability, composability. The Design-build-test-learn optimization may be needed before the desired function is fulfilled, as the circuit behaviour rely on the specific cellular context (233). A research group has reported computational and experimental methods to characterize the circuits depending on growth factor exchange in two cell types such as macrophage and fibroblast, and discovered that the circuit is steady and resistant to perturbation (234). Many approaches with the use of synthetic biology have been investigated for treating cancer.
The most prevalent kind of synthetic circuit is the one based on transcriptional control, which comprises a DNA-binding component that can recognize a promoter DNA sequence and an actuator component that allows positive or negative transcription regulation. Cys2-His2 zinc finger domains are especially suitable for the creation of synthetic transcription factors as they are often arranged as covalent tandem repeats, enabling the identification of long asymmetrical sequences (235). Researcher have described the apparent modular recognition of DNA by transcription activator-like (TAL) effectors, which is more amenable to DNA targeting and this proteins contain tandem polymorphic amino acid repeats independently which specify a single, contiguous nucleotide in the DNA target (236). The types of synthetic circuit are shown in Figure 7. The engineered zinc-finger protein transcription factors (ZFP TFs) are designed to fuse the human GM-CSF gene regulatory region to upregulate the expression of GM-CSF inserted in ONYX-411 for cancer treatment (237). CRISPR/Cas9 technology is used in gene circuits as they regulate gene expression. In CRISPR/Cas9,via Watson-Crick base pairing between the guide RNA and the target DNA, Cas9 is able to identify its target site and this Cas9 can target virtually any DNA sequence by changing the guide RNA’s 20 nucleotide targeting sequence (238) which makes CRISPR/Cas9 easier to use. But soon a deactivated Cas9 (dCas9) introduced with D10A, H841A two point mutations which nullify the protein’s endonuclease activity while retaining its potency to associate with gRNA and to be recruited on a particular DNA sequence. dCas9 can transcriptionally inhibit or activate target genes which are accordingly known as CRISPRi or CRISPRa (239). The study has reported the use of CRISPR-Cas9 based synthetic circuit for bladder cancer therapeutics. The engineered modular AND gate circuit consists of a cancer specific promoter of hTERT gene with hUPII bladder specific promoter to specifically recognize the bladder cancer cell. The hUPII promoter drives gRNA targeting lacI while the hTERT promoter drives dCas9.The output transgene was released from LacI-mediated repression as a result of the dCas9-gRNA complex’s suppression of lacI. Because of this, the output transgene was only observed in bladder cancer cells, which had active hTERT and hUPII (240). The dual-promoter integrator is one example of a gene circuit that targets cancer. In DPI circuit inputs are two cancer-specific promoters and use a transcriptional AND gate inspired by the mammalian two-hybrid system for computing output (241).
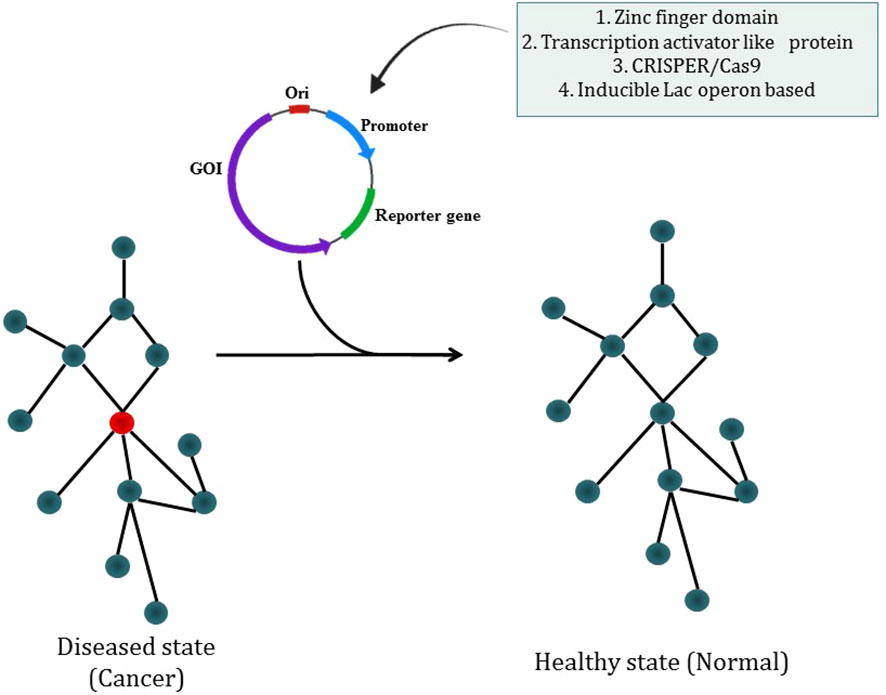
Figure 7 Synthetic biology modulating diseased state into a healthy state through different types of synthetic circuit.
RNA plays crucial regulatory roles in a variety of biological systems, thus scientists are designing the RNA based regulatory circuit in cells. An example for RNA based synthetic circuits for translation control is miRNA based circuits. MiRNAs are likely fine-tuners rather than two-state on/off controllers of gene expression in nature. Thus, synthetic biology could benefit from it. Franscisco et al. reported construction of a synthetic circuit with two module miRNA system in Chlamydomonas which was a negative switch for silencing the genes. This constructed circuit can work under both constitutive promoter and inducible promoter (242). Another study has reported the synthetic circuit with miR-146a in leishmaniasis using lac operon based system. The on and off state depends on the presence of an inducer such as IPTG which leads to miR-146 expression (243). Liliana Wroblewska designed a circuit which identifies miRNAs expressions in HeLa cells. miRNA such as miR-21(High), miR-141(low), miR-142-3p and miR-146a are mentioned as specific for HeLa cell and this identification helps to differentiate within different cells. Further driving the pro-apoptotic gene (BAX) expression as circuit output could specifically target HeLa cells (Liliana wroblewska 2015). However, it was also found that the designed synthetic sensor can sense the expression profile of particular protein such as p53 in cancer cell. They have shown the decline of p53 correlates with the increase in HSV-TK expression (244). Genetic circuits with post translational control has also been reported in study (245). The synthetic biology can modulate treatment landscape as one advantage of synthetic biology is the speed with which one can design a circuit with therapeutic candidates for cancer. Further preclinical studies are needed to authenticate particular genetic circuits.
11 Conclusion
Many believe that immunotherapy has great promise in the battle against a variety of malignancies. The immune system may be trained to spot cancer if the proper stimuli are applied. Checkpoint inhibitors, which effectively act as stop signs that prevent T cells from destroying cancer cells, are some of the greatest instances of immunotherapy. Antibodies have been created recently that essentially block such inhibitory signals, enabling the immune system to combat cancer. Despite the efficacy and potency of the available immunotherapies, the dearth of the tumor specific antigens or the substances which can trigger a specific immune response to a kind of cancer remains as a great hurdle. On a concluding note, instead of trying to treat the entire body systemically, considerably more specialized, targeted immunotherapies that operate locally at the tumour site are required. Additionally, a variety of immunotherapies can be included in an individual package, allowing for the stimulation of an array of immune responses. By customizing synthetic circuits that are capable to trigger the desired immune response more accurately as compared to the available immunotherapy patterns might prove as a better therapeutic regimen.
Author contributions
SS, NS and PG conceptualised the idea. NS and PG participated in writing of the manuscript along with figure preparation. SS, NS and PG edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
Department of Biotechnology, Ministry of Science and Technology, Government of India.
Acknowledgments
NS would like to thank University Grants Commission (UGC) for his Senior Research Fellowship. PG would like to thank her Junior Research Fellowship (JRF) from Department of Biotechnology, Ministry of Science and Technology, Government of India. The authors would also like to thank the Director, National Centre for Cell Science (NCCS) for supporting our research and Bioinformatics Facility.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Glossary
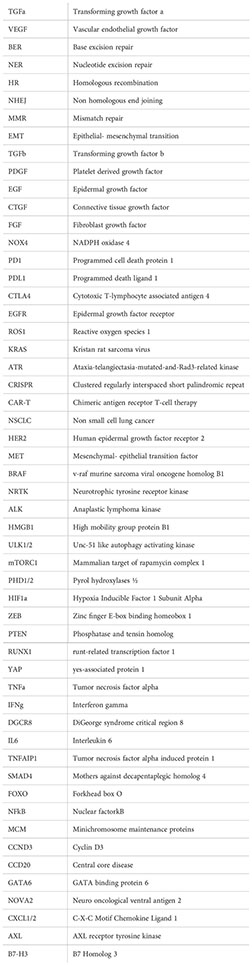
Abbreviations
- TGFα,Transforming growth factor α; VEGF, Vascular endothelial growth factor; BER, Base excision repair; NER, Nucleotide excision repair; HR, Homologous recombination; NHEJ, Non homologous end joining; MMR, Mismatch repair; EMT, Epithelial- mesenchymal transition; TGFβ, Transforming growth factor β; PDGF, Platelet derived growth factor; EGF, Epidermal growth factor; CTGF, Connective tissue growth factor; FGF, Fibroblast growth factor; NOX4, NADPH oxidase 4; PD1, Programmed cell death protein 1; PDL1, Programmed death ligand 1; CTLA4, Cytotoxic T-lymphocyte associated antigen 4; EGFR, Epidermal growth factor receptor; ROS1, Reactive oxygen species 1; KRAS, Kristan rat sarcoma virus; ATR, Ataxia-telangiectasia-mutated-and-Rad3-related kinase; CRISPR, Clustered regularly interspaced short palindromic repeat; CAR-T, Chimeric antigen receptor T-cell therapy; NSCLC, Non small cell lung cancer; HER2, Human epidermal growth factor receptor 2; MET, Mesenchymal- epithelial transition factor; BRAF, v-raf murine sarcoma viral oncogene homolog B1; NRTK, Neurotrophic tyrosine receptor kinase; ALK, Anaplastic lymphoma kinase; HMGB1, High mobility group protein B1; ULK1/2, Unc-51 like autophagy activating kinase; mTORC1, Mammalian target of rapamycin complex 1; PHD1/2, Pyrol hydroxylases ½; HIF1α, Hypoxia Inducible Factor 1 Subunit Alpha; ZEB, Zinc finger E-box binding homeobox 1; PTEN, Phosphatase and tensin homolog; RUNX1, runt-related transcription factor 1; YAP, yes-associated protein 1; TNFα, Tumor necrosis factor alpha; IFNγ, Interferon gamma; DGCR8, DiGeorge syndrome critical region 8; IL6, Interleukin 6; TNFAIP1, Tumor necrosis factor alpha induced protein 1; SMAD4, Mothers against decapentaplegic homolog 4; FOXO, Forkhead box O; NFκB, Nuclear factor-κB; MCM, Minichromosome maintenance proteins; CCND3, Cyclin D3; CCD20, Central core disease; GATA6, GATA binding protein 6; NOVA2, Neuro, oncological ventral antigen 2; CXCL1/2, C-X-C Motif Chemokine Ligand 1; AXL, AXL receptor tyrosine kinase; B7-H3, B7 Homolog 3
References
1. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries, CA. Cancer J Clin (2021) 71:209–49. doi: 10.3322/caac.21660
2. Thai AA, Solomon BJ, Sequist LV, Gainor JF, Heist RS. Lung cancer. The Lancet (2021) 398:535–54. doi: 10.1016/S0140-6736(21)00312-3
3. Liu G, Pei F, Yang F, Li L, Amin AD, Liu S, et al. Role of autophagy and apoptosis in non-small-cell lung cancer. Int J Mol Sci (2017) 18:1–24. doi: 10.3390/ijms18020367
4. Wynder EL, Graham EA. Tobacco smoking as a possible etiologic factor in bronchiogenic carcinoma. Bull World Health Organ (2005) 83:146–53. doi: 10.1001/jama.1985.03350440064033
5. Minna JD, Roth JA, Gazdar AF. Focus on lung cancer. Cancer Cell (2002) 1:49–52. doi: 10.1016/S1535-6108(02)00027-2
6. Herbst RS, Sandler AB. Overview of the current status of human epidermal growth factor receptor inhibitors in lung cancer, clin. Lung Cancer (2004) 6:S7. doi: 10.3816/clc.2004.s.009
7. Prabhakar CN. Epidermal growth factor receptor in non-small cell lung cancer, transl. Lung Cancer Res (2015) 4:110–8. doi: 10.3978/j.issn.2218-6751.2015.01.01
8. Shigematsu H, Lin L, Takahashi T, Nomura M, Suzuki M, Wistuba II, et al. Clinical and biological features associated with epidermal growth factor receptor gene mutations in lung cancers, J. Natl Cancer Inst (2005) 97:339–46. doi: 10.1093/jnci/dji055
10. Ahrendt SA, Hu Y, Buta M, McDermott MP, Benoit N, Yang SC, et al. p53 mutations and survival in stage I non-small-cell lung cancer: Results of a prospective study, J. Natl Cancer Inst (2003) 95:961–70. doi: 10.1093/jnci/95.13.961
11. Camargo Barros-Filho M, Guisier F, Rock LD, Becker-Santos DD, Sage AP, Marshall EA, et al. Tumour suppressor genes with oncogenic roles in lung cancer. Genes and Cancer (2019) (3):1–17. doi: 10.5772/intechopen.85017
12. Shi LZ, Goswami S, Fu T, Guan B, Chen J, Xiong L, et al. Blockade of CTLA-4 and PD-1 enhances adoptive t-cell therapy efficacy in an ICOS-mediated manner, cancer immunol. Res (2019) 7:1803–12. doi: 10.1158/2326-6066.CIR-18-0873
13. Wei SC, Duffy CR, Allison JP. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov (2018) 8:1069–86. doi: 10.1158/2159-8290.CD-18-0367
14. Anderson AC, Joller N, Kuchroo VK. Lag-3, Tim-3, and TIGIT: Co-inhibitory receptors with specialized functions in immune regulation. Immunity (2016) 44:989–1004. doi: 10.1016/j.immuni.2016.05.001
15. Kamphorst AO, Pillai RN, Yang S, Nasti TH, Akondy RS, Wieland A, et al. Proliferation of PD-1+ CD8 T cells in peripheral blood after PD-1-targeted therapy in lung cancer patients, proc. Natl Acad Sci USA (2017) 114:4993–8. doi: 10.1073/pnas.1705327114
16. Meyers DE, Bryan PM, Banerji S, Morris DG. Targeting the PD-1/PD-L1 axis for the treatment of non-small-cell lung cancer, curr. Oncol (2018) 25:e324–34. doi: 10.3747/co.25.3976
17. Blackburn EH. Switching and signaling at the telomere. Cell (2001) 106:661–73. doi: 10.1016/S0092-8674(01)00492-5
18. Cheol Lee J, Jong HS, Yoo CG, Koo Han S, Shim YS, Whan Kim Y. Telomerase activity in lung cancer cell lines and tissues. Lung Cancer (1998) 21:99–103. doi: 10.1016/S0169-5002(98)00034-8
19. Bernardes de Jesus B, Blasco MA. Telomerase at the intersection of cancer and aging. Trends in Genetics (2013) 29:513–20. doi: 10.1016/j.tig.2013.06.007
20. Dobija-Kubica K, Zalewska-Ziob M, Bruliński K, Rogoziński P, Wiczkowski A, Gawrychowska A, et al. Telomerase activity in non-small cell lung cancer. J Thorac Card Surg (2016) 13:15–20. doi: 10.5114/kitp.2016.58959
21. Heijne V, Gier D, The JL. Telomere dysfunction promotes non-reciprocal translocations. Nature (2000) 20070:641–5. doi: 10.1038/35020592
22. Rudolph KL, Millard M, Bosenberg MW, DePinho RA. Telomere dysfunction and evolution of intestinal carcinoma in mice and humans, nat. Genet (2001) 28:155–9. doi: 10.1038/88871
23. Zhang Q, Zhu B, Li Y. Resolution of cancer-promoting inflammation: A new approach for anticancer therapy, front. Immunol (2017) 8:71. doi: 10.3389/fimmu.2017.00071
24. Khusnurrokhman G, Wati FF. Tumor-promoting inflammation in lung cancer: A literature review, Ann. Med Surg (2022) 79:104022. doi: 10.1016/j.amsu.2022.104022
25. Fares J, Fares MY, Khachfe HH, Salhab HA, Fares Y. Molecular principles of metastasis: a hallmark of cancer revisited, signal transduct. Targeting Ther (2020) 5:1–17. doi: 10.1038/s41392-020-0134-x
26. Hendriks LEL, Smit EF, Vosse BAH, Mellema WW, Heideman DAM, Bootsma GP, et al. EGFR mutated non-small cell lung cancer patients: More prone to development of bone and brain metastases? Lung Cancer (2014) 84:86–91. doi: 10.1016/j.lungcan.2014.01.006
27. Popper HH. Progression and metastasis of lung cancer. Cancer and Metastasis Reviews (2016) 35:75–91. doi: 10.1007/s10555-016-9618-0
28. Zhu T, Bao X, Chen M, Lin R, Zhuyan J, Zhen T, et al. Mechanisms and future of non-small cell lung cancer metastasis, front. Oncol (2020) 10:585284. doi: 10.3389/fonc.2020.585284
29. Saman H, Raza SS, Uddin S, Rasul K. Inducing angiogenesis, a key step in cancer vascularization, and treatment approaches. Cancers (2020) 12:1–18. doi: 10.3390/cancers12051172
30. Imoto H, Osaki T, Taga S, Ohgami A, Ichiyoshi Y, Yasumoto K. Vascular endothelial growth factor expression in non-small-cell lung cancer: Prognostic significance in squamous cell carcinoma. J Thorac Cardiovasc Surg 115(5):1007–14. doi: 10.1016/S0022-5223(98)70398-8
31. Tamura M. Plasma VEGF concentration can predict the tumor angiogenic capacity in non-small cell lung cancer, oncol. Rep (2001) 8:1097–102. doi: 10.3892/or.8.5.1097
32. Shi W, Siemann DW. Angiogenesis and lung cancer. Adv Rad Oncol Lung Cancer (2011) 17–41. doi: 10.1007/174_2011_260
33. Hall RD, Le TM, Haggstrom DE, Gentzler RD. Angiogenesis inhibition as a therapeutic strategy in non-small cell lung cancer (NSCLC), transl. Lung Cancer Res (2015) 4:515–23. doi: 10.3978/j.issn.2218-6751.2015.06.09
34. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell (2000) 100:57–70. doi: 10.1016/s0092-8674(00)81683-9
35. Liang H, Chen Q, Hu Z, Zhou L, Meng Q, Zhang T, et al. Siglec15 facilitates the progression of non-small cell lung cancer and is correlated with spinal metastasis, Ann. Transl Med (2022) 10:281–1. doi: 10.21037/atm-22-764
36. De Bruin EC, McGranahan N, Mitter R, Salm M, Wedge DC, Yates L, et al. Spatial and temporal diversity in genomic instability processes defines lung cancer evolution. Science (2014) 80-.). 346:251–6. doi: 10.1126/science.1253462
37. Soca-Chafre G, Montiel-Dávalos A, de la Rosa-Velázquez IA, Caro-Sánchez CHS, Peña-Nieves A, Arrieta O. Multiple molecular targets associated with genomic instability in lung cancer. Int J Genomics (2019) 2019:9584504. doi: 10.1155/2019/9584504
38. Johnstone RW, Ruefli AA, Lowe SW. Apoptosis: A link between cancer genetics and chemotherapy. Int J Genom (2002) 108:153–64. doi: 10.1016/S0092-8674(02)00625-6
40. Zhao Y, Butler EB, Tan M. Targeting cellular metabolism to improve cancer therapeutics. Cell Death & Disease (2013) 4:e532–10. doi: 10.1038/cddis.2013.60
41. Rao Kondapally Seshasai S, Kaptoge S, Thompson A, Di Angelantonio E, Gao P, Sarwar N, et al. Fasting glucose, and risk of cause-specific death centers for disease control and prevention, Atlanta. N Engl J Med March (2011) 3:829–41. doi: 10.1056/NEJMoa1008862.Diabetes
42. Heiden MGV, Cantley LC, Thompson CB. Understanding the warburg effect: The metabolic requirements of cell proliferation. Science (2009) 80-.). 324:1029–33. doi: 10.1126/science.1160809
43. Makinoshima H, Takita M, Saruwatari K, Umemura S, Obata Y, Ishii G, et al. Signaling through the phosphatidylinositol 3-kinase (PI3K)/mammalian target of rapamycin (mTOR) axis is responsible for aerobic glycolysis mediated by glucose transporter in epidermal growth factor receptor (EGFR)-mutated lung adenocarcinoma. J Biol Chem (2015) 290:17495–504. doi: 10.1074/jbc.M115.660498
44. Yuan S, Norgard RJ, Stanger BZ. Cellular plasticity in cancer. Cancer Discov (2019) 9:837–51. doi: 10.1158/2159-8290.CD-19-0015
45. Hanahan D. Hallmarks of cancer: New dimensions. Cancer Discov (2022) 12:31–46. doi: 10.1158/2159-8290.CD-21-1059
46. Lee LJ, Papadopoli D, Jewer M, del Rincon S, Topisirovic I, Lawrence MG, et al. Cancer plasticity: The role of mRNA translation. Trends Cancer (2021) 7:134–45. doi: 10.1016/j.trecan.2020.09.005
47. Gupta PB, Pastushenko I, Skibinski A, Blanpain C, Kuperwasser C. Phenotypic plasticity: Driver of cancer initiation, progression, and therapy resistance. Cell Stem Cell (2019) 24:65–78. doi: 10.1016/j.stem.2018.11.011
48. Ansari J, Shackelford RE, El-Osta1 H. Epigenetics in non-small cell lung cancer: From basics to therapeutics. Transl Lung Cancer Res (2016) 5:155–71. doi: 10.21037/tlcr.2016.02.02
49. Deaton AM, Bird A. CpG islands and the regulation of transcription. Genes and Development (2011) 25:1010–22. doi: 10.1101/gad.2037511
50. Nicholson JK, Holmes E, Kinross J, Burcelin R, Gibson G, Jia W, et al. Host-gut microbiota metabolic interactions. Science (2012) 80-.). 336:1262–7. doi: 10.1126/science.1223813
51. Dong Q, Chen ES, Zhao C, Jin C. Host-microbiome interaction in lung cancer. Front Immunol (2021) 12:679829. doi: 10.3389/fimmu.2021.679829
52. Tsay JCJ, Wu BG, Badri MH, Clemente JC, Shen N, Meyn P, et al. Airway microbiota is associated with upregulation of the PI3K pathway in lung cancer. Am J Respir Crit Care Med (2018) 198:1188–98. doi: 10.1164/rccm.201710-2118OC
53. Gustafson AM, Soldi R, Anderlind C, Scholand MB, Qian J, Zhang X, et al. Airway PI3K pathway activation is an early and reversible event in lung cancer development. Sci Transl Med (2010) 2:26ra25. doi: 10.1126/scitranslmed.3000251
54. Gorgoulis V, Adams PD, Alimonti A, Bennett DC, Bischof O, Bishop C, et al. Cellular senescence: Defining a path forward. Cell (2019) 179:813–27. doi: 10.1016/j.cell.2019.10.005
55. Hernandez-Segura A, Nehme J, Demaria M. Hallmarks of cellular senescence. Exp Cell Res (2018) 28:436–53. doi: 10.1016/j.tcb.2018.02.001
56. Faget DV, Ren Q, Stewart SA. Unmasking senescence: context-dependent effects of SASP in cancer, nat. Rev Cancer (2019) 19:439–53. doi: 10.1038/s41568-019-0156-2
57. Domen A, Deben C, De Pauw I, Hermans C, Lambrechts H, Verswyvel J, et al. Prognostic implications of cellular senescence in resected non-small cell lung cancer. Transl Lung Cancer Res (2022) 11:1526–39. doi: 10.21037/tlcr-22-192
58. Henke E, Nandigama R, Ergün S. Extracellular matrix in the tumor microenvironment and its impact on cancer therapy. Front Mol Biosci (2020) 6:160. doi: 10.3389/fmolb.2019.00160
59. Balkwill FR, Capasso M, Hagemann T. The tumor microenvironment at a glance. J Cell Sci (2012) 125:5591–6. doi: 10.1242/jcs.116392
60. Baghban R, Roshangar L, Jahanban-Esfahlan R, Seidi K, Ebrahimi-Kalan A, Jaymand M, et al. Tumor microenvironment complexity and therapeutic implications at a glance. Cell Commun Signal (2020) 18:1–19. doi: 10.1186/s12964-020-0530-4
61. Tan Z, Xue H, Sun Y, Zhang C, Song Y, Qi Y. The role of tumor inflammatory microenvironment in lung cancer. Front Pharmacol (2021) 12:688625. doi: 10.3389/fphar.2021.688625
62. Chen Z, Fillmore CM, Hammerman PS, Kim CF, Wong KK. Non-small-cell lung cancers: A heterogeneous set of diseases. Nat Rev Cancer (2014) 14:535–46. doi: 10.1038/nrc3775
63. Yuan Y, Li H, Pu W, Chen L, Guo D, Jiang H, et al. Cancer metabolism and tumor microenvironment: fostering each other? Sci China Life Sci (2022) 65:236–79. doi: 10.1007/s11427-021-1999-2
64. Shi R, Tang YQ, Miao H. Metabolism in tumor microenvironment: Implications for cancer immunotherapy. MedComm (2020) 1:47–68. doi: 10.1002/mco2.6
65. Lyssiotis CA, Kimmelman AC. Metabolic interactions in the tumor microenvironment. Bmc Genomics (2017) 27:863–75. doi: 10.1016/j.tcb.2017.06.003
66. De Berardinis RJ, Chandel NS. Fundamentals of cancer metabolism. Sci Adv (2016) 2:1–18. doi: 10.1126/sciadv.1600200
67. Lambrechts D, Wauters E, Boeckx B, Aibar S, Nittner D, Burton O, et al. Phenotype molding of stromal cells in the lung tumor microenvironment. Nat Med (2018) 24:1277–89. doi: 10.1038/s41591-018-0096-5
68. Wu F, Fan J, He Y, Xiong A, Yu J, Li Y, et al. Single-cell profiling of tumor heterogeneity and the microenvironment in advanced non-small cell lung cancer. Nat Commun (2021) 12:1–11. doi: 10.1038/s41467-021-22801-0
69. Monteran L, Erez N. The dark side of fibroblasts: Cancer-associated fibroblasts as mediators of immunosuppression in the tumor microenvironment. Front Immunol (2019) 10:1835. doi: 10.3389/fimmu.2019.01835
70. Barrett R, Puré E. Cancer-associated fibroblasts: key determinants of tumor immunity and immunotherapy. Curr Opin Immunol (2020) 64:80–7. doi: 10.1016/j.coi.2020.03.004
71. Kuzet SE, Gaggioli C. Fibroblast activation in cancer: when seed fertilizes soil. Cell Tissue Res (2016) 365:607–19. doi: 10.1007/s00441-016-2467-x
72. Zhou Z, Zhou Q, Wu X, Xu S, Hu X, Tao X, et al. VCAM-1 secreted from cancer-associated fibroblasts enhances the growth and invasion of lung cancer cells through AKT and MAPK signaling. Cancer Lett (2020) 473:62–73. doi: 10.1016/j.canlet.2019.12.039
73. Wang L, Cao L, Wang H, Liu B, Zhang Q, Meng Z, et al. Cancer-associated fibroblasts enhance metastatic potential of lung cancer cells through IL-6/STAT3 signaling pathway. Oncotarget (2017) 8:76116–28. doi: 10.18632/oncotarget.18814
74. An Y, Liu F, Chen Y, Yang Q. Crosstalk between cancer-associated fibroblasts and immune cells in cancer. J Cell Mol Med (2020) 24:13–24. doi: 10.1111/jcmm.14745
75. Lakins MA, Ghorani E, Munir H, Martins CP, Shields JD. Cancer-associated fibroblasts induce antigen-specific deletion of CD8 + T cells to protect tumour cells. Nat Commun (2018) 9:1–9. doi: 10.1038/s41467-018-03347-0
76. Oya Y, Hayakawa Y, Koike K. Tumor microenvironment in gastric cancers. Cancer Sci (2020) 111:2696–707. doi: 10.1111/cas.14521
77. Anderson NM, Simon MC. The tumor microenvironment. Curr Biol (2020) 30:R921–5. doi: 10.1016/j.cub.2020.06.081
78. Nagl L, Horvath L, Pircher A, Wolf D. Tumor endothelial cells (TECs) as potential immune directors of the tumor microenvironment – new findings and future perspectives. Front Cell Dev Biol (2020) 8:766. doi: 10.3389/fcell.2020.00766
79. Yang D, Guo P, He T, Powell CA. Role of endothelial cells in tumor microenvironment. Clin Transl Med (2021) 11:19–22. doi: 10.1002/ctm2.450
80. Xie M, Liu M, He CS. SIRT1 regulates endothelial notch signaling in lung cancer. PloS One (2012) 7:1–11. doi: 10.1371/journal.pone.0045331
81. Hinshaw DC, Shevde LA. The tumor microenvironment innately modulates cancer progression. Cancer Res (2019) 79:4557–67. doi: 10.1158/0008-5472.CAN-18-3962
82. Stankovic B, Bjørhovde HAK, Skarshaug R, Aamodt H, Frafjord A, Müller E, et al. Immune cell composition in human non-small cell lung cancer. Front Immunol (2018) 9:3101. doi: 10.3389/fimmu.2018.03101
83. Goswami KK, Ghosh T, Ghosh S, Sarkar M, Bose A, Baral R. Tumor promoting role of anti-tumor macrophages in tumor microenvironment. Cell Immunol (2017) 316:1–10. doi: 10.1016/j.cellimm.2017.04.005
84. Sedighzadeh SS, Khoshbin AP, Razi S, Keshavarz-Fathi M, Rezaei N. A narrative review of tumor-associated macrophages in lung cancer: Regulation of macrophage polarization and therapeutic implications, transl. Lung Cancer Res (2021) 10:1889–916. doi: 10.21037/tlcr-20-1241
85. Yang L, Zhang Y. Tumor-associated macrophages: from basic research to clinical application. J Hematol Oncol (2017) 10:58. doi: 10.1186/s13045-017-0430-2
86. Zhang J, Li H, Wu Q, Chen Y, Deng Y, Yang Z, et al. Tumoral NOX4 recruits M2 tumor-associated macrophages via ROS/PI3K signaling-dependent various cytokine production to promote NSCLC growth. Redox Biol (2019) 22:101116. doi: 10.1016/j.redox.2019.101116
87. Zhang S, Che D, Yang F, Chi C, Meng H, Shen J, et al. Tumor-associated macrophages promote tumor metastasis via the TGF-β/SOX9 axis in non-small cell lung cancer. Oncotarget (2017) 8:99801–15. doi: 10.18632/oncotarget.21068
88. Wculek SK, Cueto FJ, Mujal AM, Melero I, Krummel MF, Sancho D. Dendritic cells in cancer immunology and immunotherapy. Nat Rev Immunol (2020) 20:7–24. doi: 10.1038/s41577-019-0210-z
89. Peng X, He Y, Huang J, Tao Y, Liu S. Metabolism of dendritic cells in tumor microenvironment: For immunotherapy. Front Immunol (2021) 12:613492. doi: 10.3389/fimmu.2021.613492
90. Cell N, Cancer L, Rojiani MV, Kolhe R. Natural killer cells and dendritic Cells : Expanding clinical. Cancers (2021) 13(16):4037. doi: 10.3390/cancers13164037
91. Schneider T, Hoffmann H, Dienemann H, Schnabel PA, Enk AH, Ring S, et al. Non-small cell lung cancer induces an immunosuppressive phenotype of dendritic cells in tumor microenvironment by upregulating B7-H3. J Thorac Oncol (2011) 6:1162–8. doi: 10.1097/JTO.0b013e31821c421d
92. Chen H, Lin C, Lu C, Wang Y, Han R, Li L, et al. Metformin-sensitized NSCLC cells to osimertinib via AMPK-dependent autophagy inhibition. Clin Respir J (2019) 13:781–90. doi: 10.1111/crj.13091
93. Lu Y, Xu W, Gu Y, Chang X, Wei G, Rong Z, et al. Non-small cell lung cancer cells modulate the development of human CD1c+ conventional dendritic cell subsets mediated by CD103 and CD205. Front Immunol (2019) 10:2829. doi: 10.3389/fimmu.2019.02829
94. Dumitriu IE, Dunbar DR, Howie SE, Sethi T, Gregory CD. Human dendritic cells produce TGF-β1 under the influence of lung carcinoma cells and prime the differentiation of CD4 + CD25 + Foxp3 + regulatory T cells. J Immunol (2009) 182:2795–807. doi: 10.4049/jimmunol.0712671
95. Scott EN, Gocher AM, Workman CJ, Vignali DAA. Regulatory T cells: Barriers of immune infiltration into the tumor microenvironment. Front Immunol (2021) 12:702726. doi: 10.3389/fimmu.2021.702726
96. Li J, Eu JQ, Kong LR, Wang L, Lim YC, Goh BC, et al. Targeting metabolism in cancer cells and the tumour microenvironment for cancer therapy. Molecules (2020) 25:1–40. doi: 10.3390/molecules25204831
97. Paluskievicz CM, Cao X, Abdi R, Zheng P, Liu Y, Bromberg JS. T Regulatory cells and priming the suppressive tumor microenvironment. Front Immunol (2019) 10:2453. doi: 10.3389/fimmu.2019.02453
98. Principe DR, Chiec L, Mohindra NA, Munshi HG. Regulatory T-cells as an emerging barrier to immune checkpoint inhibition in lung cancer. Front Oncol (2021) 11:684098. doi: 10.3389/fonc.2021.684098
99. Granville CA, Memmott RM, Balogh A, Mariotti J, Kawabata S, Han W, et al. A central role for Foxp3+ regulatory T cells in K-ras-driven lung tumorigenesis. PloS One (2009) 4:1–8. doi: 10.1371/journal.pone.0005061
100. Spolski R, Li P, Leonard WJ. Biology and regulation of IL-2: from molecular mechanisms to human therapy. Nat Rev Immunol (2018) 18:648–59. doi: 10.1038/s41577-018-0046-y
101. Ohta A, Kini R, Ohta A, Subramanian M, Madasu M, Sitkovsky M. The development and immunosuppressive functions of CD4+ CD25+ FoxP3+ regulatory T cells are under influence of the adenosine-A2A adenosine receptor pathway. Front Immunol (2012) 3:190. doi: 10.3389/fimmu.2012.00190
102. Li C, Jiang P, Wei S, Xu X, Wang J. Regulatory T cells in tumor microenvironment: New mechanisms, potential therapeutic strategies and future prospects. Mol Cancer (2020) 19:1–23. doi: 10.1186/s12943-020-01234-1
103. Veglia F, Sanseviero E, Gabrilovich DI. Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nat Rev Immunol (2021) 21:485–98. doi: 10.1038/s41577-020-00490-y
104. Yang Z, Guo J, Weng L, Tang W, Jin S, Ma W. Myeloid-derived suppressor cells-new and exciting players in lung cancer. J Hematol Oncol (2020) 13:1–17. doi: 10.1186/s13045-020-0843-1
105. Zalfa C, Paust S. Natural killer cell interactions with myeloid derived suppressor cells in the tumor microenvironment and implications for cancer immunotherapy. Front Immunol (2021) 12:633205. doi: 10.3389/fimmu.2021.633205
106. Condamine T, Dominguez GA, Youn JI, Kossenkov AV, Mony S, Alicea-Torres K, et al. Gabrilovich, lectin-type oxidized LDL receptor-1 distinguishes population of human polymorphonuclear myeloid-derived suppressor cells in cancer patients. Sci Immunol (2016) 1:1–16. doi: 10.1126/sciimmunol.aaf8943
107. Zhang G, Huang H, Zhu Y, Yu G, Gao X, Xu Y, et al. A novel subset of B7-H3+CD14+HLA-DFT–/low myeloid-derived suppressor cells are associated with progression of human NSCLC. Oncoimmunology (2015) 4:1–12. doi: 10.4161/2162402X.2014.977164
108. Wang Y, Schafer CC, Hough KP, Tousif S, Duncan SR, Kearney JF, et al. Myeloid-derived suppressor cells impair b cell responses in lung cancer through IL-7 and STAT5. J Immunol (2018) 201:278–95. doi: 10.4049/jimmunol.1701069
109. Kumar V, Patel S, Tcyganov E, Gabrilovich DI. The nature of myeloid-derived suppressor cells in the tumor microenvironment. Trends Immunol (2016) 37:208–20. doi: 10.1016/j.it.2016.01.004
110. Vivier E, Tomasello E, Baratin M, Walzer T, Ugolini S. Functions of natural killer cells. Nat Immunol (2008) 9:503–10. doi: 10.1038/ni1582
111. Carrega P, Ferlazzo G. Natural killers are made not born: How to exploit NK cells in lung malignancies. Front Immunol (2017) 8:277. doi: 10.3389/fimmu.2017.00277
112. Bruno A, Focaccetti C, Pagani A, Imperatori AS, Spagnoletti M, Rotolo N, et al. The proangiogenic phenotype of natural killer cells in patients with non-small cell lung cancer, neoplasia (United states). Neoplasia (2013) 15:133–42. doi: 10.1593/neo.121758
113. Wu SY, Fu T, Jiang YZ, Shao ZM. Natural killer cells in cancer biology and therapy. Mol Cancer (2020) 19:1–26. doi: 10.1186/s12943-020-01238-x
114. Sharonov GV, Serebrovskaya EO, Yuzhakova DV, Britanova OV, Chudakov DM. B cells, plasma cells and antibody repertoires in the tumour microenvironment. Nat Rev Immunol (2020) 20:294–307. doi: 10.1038/s41577-019-0257-x
115. Germain C, Gnjatic S, Tamzalit F, Knockaert S, Remark R, Goc J, et al. Presence of b cells in tertiary lymphoid structures is associated with a protective immunity in patients with lung cancer. Am J Respir Crit Care Med (2014) 189:832–44. doi: 10.1164/rccm.201309-1611OC
116. Yuan M, Huang LL, Chen JH, Wu J, Xu Q. The emerging treatment landscape of targeted therapy in non-small-cell lung cancer, signal transduct. Jorjani Biomed (2019) 8(3):1–3. doi: 10.1038/s41392-019-0099-9
117. Guisier F, Barros-Filho MC, Rock LD, Strachan-Whaley M, Marshall EA, Dellaire G, et al. Janus or hydra: The many faces of T helper cells in the human tumour microenvironment. Tumor Microenviron (2020) 1224:33–51. doi: 10.1007/978-3-030-35723-8_3
118. Waldman AD, Fritz JM, Lenardo MJ. A guide to cancer immunotherapy: from T cell basic science to clinical practice. Nat Rev Immunol (2020) 20:651–68. doi: 10.1038/s41577-020-0306-5
119. Zhang Z, Liu S, Zhang B, Qiao L, Zhang Y, Zhang Y. T Cell dysfunction and exhaustion in cancer. Front Cell Dev Biol (2020) 8:1–13. doi: 10.3389/fcell.2020.00017
120. Thommen DS, Schreiner J, Müller P, Herzig P, Roller A, Belousov A, et al. Progression of lung cancer is associated with increased dysfunction of T cells defined by coexpression of multiple inhibitory receptors. Cancer Immunol Res (2015) 3:1344–54. doi: 10.1158/2326-6066.CIR-15-0097
121. Bartel DP. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell (2004) 116:281–97. doi: 10.1016/S0092-8674(04)00045-5
122. Lee RC, Feinbaum RL, Ambros V, The C. Elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell (1993) 75:843–54. doi: 10.1016/0092-8674(93)90529-y
123. Treiber T, Treiber N, Meister G. Regulation of microRNA biogenesis and its crosstalk with other cellular pathways. Nat Rev Mol Cell Biol (2019) 20:5–20. doi: 10.1038/s41580-018-0059-1
124. O’Brien J, Hayder H, Zayed Y, Peng C. Overview of microRNA biogenesis, mechanisms of actions, and circulation. Front Endocrinol (Lausanne) (2018) 9:402. doi: 10.3389/fendo.2018.00402
125. Liu J, Carmell MA, Rivas FV, Marsden CG, Thomson JM, Song JJ, et al. Argonaute2 is the catalytic engine of mammalian RNAi. Science (2004) 80-.). 305:1437–41. doi: 10.1126/science.1102513
126. Lin PY, Yu SL, Yang PC. MicroRNA in lung cancer. Br J Cancer (2010) 103:1144–8. doi: 10.1038/sj.bjc.6605901
127. Calin GA, Dumitru CD, Shimizu M, Bichi R, Zupo S, Noch E, et al. Frequent deletions and down-regulation of micro-RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc Natl Acad Sci USA (2002) 99:15524–9. doi: 10.1073/pnas.242606799
128. Calin GA, Croce CM. MicroRNA signatures in human cancers. Nat Rev Cancer (2006) 6:857–66. doi: 10.1038/nrc1997
129. Volinia S, Calin GA, Liu CG, Ambs S, Cimmino A, Petrocca F, et al. A microRNA expression signature of human solid tumors defines cancer gene targets. Proc Natl Acad Sci USA (2006) 103:2257–61. doi: 10.1073/pnas.0510565103
130. Johnson SM, Grosshans H, Shingara J, Byrom M, Jarvis R, Cheng A, et al. RAS is regulated by the let-7 microRNA family. Cell (2005) 120:635–47. doi: 10.1016/j.cell.2005.01.014
131. Peng Y, Croce CM. The role of microRNAs in human cancer. Signal Transduct Target Ther (2016) 1:15004. doi: 10.1038/sigtrans.2015.4
132. Croce CM. Causes and consequences of microRNA dysregulation in cancer. Nat Rev Genet (2009) 10:704–14. doi: 10.1038/nrg2634
133. Takamizawa J, Konishi H, Yanagisawa K, Tomida S, Osada H, Endoh H, et al. Reduced expression of the let-7 microRNAs in human lung cancers in association with shortened postoperative survival. Cancer Res (2004) 64:3753–6. doi: 10.1158/0008-5472.CAN-04-0637
134. Xing Y, Ruan G, Ni H, Qin H, Chen S, Gu X, et al. Tumor immune microenvironment and its related miRNAs in tumor progression, front. Immunol (2021) 12:624725. doi: 10.3389/fimmu.2021.624725
135. Sanaei MJ, Razi S, Pourbagheri-Sigaroodi A, Bashash D. The PI3K/Akt/mTOR pathway in lung cancer; oncogenic alterations, therapeutic opportunities, challenges, and a glance at the application of nanoparticles. Transl Oncol (2022) 18:101364. doi: 10.1016/j.tranon.2022.101364
136. Sharma SV, Bell DW, Settleman J, Haber DA. Epidermal growth factor receptor mutations in lung cancer. Nat Rev Cancer (2007) 7:169–81. doi: 10.1038/nrc2088
137. Paz-Ares L, Soulières D, Moecks J, Bara I, Mok T, Klughammer B. Pooled analysis of clinical outcome for EGFR TKI-treated patients with EGFR mutation-positive NSCLC. J Cell Mol Med (2014) 18:1519–39. doi: 10.1111/jcmm.12278
138. Ahmed SH, Espinoza-Sánchez NA, El-Damen A, Fahim SA, Badawy MA, Greve B, et al. Small extracellular vesicle-encapsulated miR-181b-5p, miR-222-3p and let-7a-5p: Next generation plasma biopsy-based diagnostic biomarkers for inflammatory breast cancer. PloS One (2021) 16:1–30. doi: 10.1371/journal.pone.0250642
139. Ding L, Lan Z, Xiong X, Ao H, Feng Y, Gu H, et al. The dual role of microRNAs in colorectal cancer progression. Int J Mol Sci (2018) 19:1–15. doi: 10.3390/ijms19092791
140. Acunzo M, Romano G, Palmieri D, Laganá A, Garofalo M, Balatti V, et al. Cross-talk between MET and EGFR in non-small cell lung cancer involves miR-27a and Sprouty2. Proc Natl Acad Sci USA (2013) 110:8573–8. doi: 10.1073/pnas.1302107110
141. Mirzadeh Azad F, Naeli P, Malakootian M, Baradaran A, Tavallaei M, Ghanei M, et al. Two lung development-related microRNAs, miR-134 and miR-187, are differentially expressed in lung tumors. Gene (2016) 577:221–6. doi: 10.1016/j.gene.2015.11.040
142. Qin Q, Wei F, Zhang J, Wang X, Li B. miR-134 inhibits non-small cell lung cancer growth by targeting the epidermal growth factor receptor. J Cell Mol Med (2016) 20:1974–83. doi: 10.1111/jcmm.12889
143. Zhu K, Ding H, Wang W, Liao Z, Fu Z, Hong Y, et al. Tumor-suppressive miR-218-5p inhibits cancer cell proliferation and migration via EGFR in non-small cell lung cancer. Oncotarget (2016) 7:28075–85. doi: 10.18632/oncotarget.8576
144. Wang F, Chan LWC, Law HKW, Cho WCS, Tang P, Yu J, et al. Exploring microRNA-mediated alteration of EGFR signaling pathway in non-small cell lung cancer using an mRNA: MiRNA regression model supported by target prediction databases. Genomics (2014) 104:504–11. doi: 10.1016/j.ygeno.2014.09.004
145. Wang F, Meng F, Wang L, Wong SCC, Cho WCS, Chan LWC. Associations of mRNA:microRNA for the shared downstream molecules of EGFR and alternative tyrosine kinase receptors in non-small cell lung cancer. Front Genet (2016) 7:173. doi: 10.3389/fgene.2016.00173
146. Jin H, Jang Y, Cheng N, Li Q, Cui PF, Zhou ZW, et al. Restoration of mutant K-ras repressed miR-199b inhibits K-ras mutant non-small cell lung cancer progression. J Exp Clin Cancer Res (2019) 38:1–11. doi: 10.1186/s13046-019-1170-7
147. Gulhane P, Nimsarkar P, Kharat K, Singh S. Deciphering miR-520c-3p as a probable target for immunometabolism in non-small cell lung cancer using systems biology approach. Oncotarget (2022) 13:725–46. doi: 10.18632/oncotarget.28233
148. Chen TM, Xiao Q, Wang XJ, Wang ZQ, Hu JW, Zhang Z, et al. miR-16 regulates proliferation and invasion of lung cancer cells via the ERK/MAPK signaling pathway by targeted inhibition of MAPK kinase 1 (MEK1). J Int Med Res (2019) 47:5194–204. doi: 10.1177/0300060519856505
149. Jiang W, Wei K, Pan C, Li H, Cao J, Han X, et al. MicroRNA-1258 suppresses tumour progression via GRB2/Ras/Erk pathway in non-small-cell lung cancer. Cell Prolif (2018) 51:1–14. doi: 10.1111/cpr.12502
150. Ma Z, Qiu X, Wang D, Li Y, Zhang B, Yuan T, et al. MiR-181a-5p inhibits cell proliferation and migration by targeting kras in non-small cell lung cancer A549 cells, acta biochim. Biophys Sin (Shanghai) (2015) 47:630–8. doi: 10.1093/abbs/gmv054
151. Zhai S, Zhao L, Lin T, Wang W. Downregulation of miR-33b promotes non-small cell lung cancer cell growth through reprogramming glucose metabolism miR-33b regulates non-small cell lung cancer cell growth, J. Cell Biochem (2019) 120:6651–60. doi: 10.1002/jcb.27961
152. Liu M, Gao J, Huang Q, Jin Y, Wei Z. Downregulating microRNA-144 mediates a metabolic shift in lung cancer cells by regulating GLUT1 expression. Oncol Lett (2016) 11:3772–6. doi: 10.3892/ol.2016.4468
153. Cui R, Meng W, Sun HL, Kim T, Ye Z, Fassan M, et al. MicroRNA-224 promotes tumor progression in nonsmall cell lung cancer. Proc Natl Acad Sci U. S. A (2015) 112:E4288–97. doi: 10.1073/pnas.1502068112
154. Yang Y, Ding L, Hu Q, Xia J, Sun J, Wang X, et al. MicroRNA-218 functions as a tumor suppressor in lung cancer by targeting IL-6/STAT3 and negatively correlates with poor prognosis. Mol Cancer (2017) 16:1–13. doi: 10.1186/s12943-017-0710-z
155. Liang G, Meng W, Huang X, Zhu W, Yin C, Wang C, et al. MiR-196b-5p-mediated downregulation of TSPAN12 and GATA6 promotes tumor progression in non-small cell lung cancer. Proc Natl Acad Sci USA (2020) 117:4347–57. doi: 10.1073/pnas.1917531117
156. Xiao H. MiR-7-5p suppresses tumor metastasis of non-small cell lung cancer by targeting NOVA2. Cell Mol Biol Lett (2019) 24:60. doi: 10.1186/s11658-019-0188-3
157. Mao M, Wu Z, Chen J. MicroRNA-187-5p suppresses cancer cell progression in non-small cell lung cancer (NSCLC) through down-regulation of CYP1B1. Biochem Biophys Res Commun (2016) 478:649–55. doi: 10.1016/j.bbrc.2016.08.001
158. Li J, Guan J, Long X, Wang Y, Xiang X. Mir-1-mediated paracrine effect of cancer- associated fibroblasts on lung cancer cell proliferation and chemoresistance. Oncol Rep (2016) 35:3523–31. doi: 10.3892/or.2016.4714
159. Shen H, Yu X, Yang F, Zhang Z, Shen J, Sun J, et al. Reprogramming of normal fibroblasts into cancer-associated fibroblasts by miRNAs-mediated CCL2/VEGFA signaling. PloS Genet (2016) 12:1–20. doi: 10.1371/journal.pgen.1006244
160. Zhang J, Liu J, Liu Y, Wu W, Li X, Wu Y, et al. MiR-101 represses lung cancer by inhibiting interaction of fibroblasts and cancer cells by down-regulating CXCL12. Biomed Pharmacother (2015) 74:215–21. doi: 10.1016/j.biopha.2015.08.013
161. Iurca I, Tirpe A, Zimta AA, Moldovan C, Gulei D, Slabý O, et al. Macrophages interaction and MicroRNA interplay in the modulation of cancer development and metastasis. Front Immunol (2020) 11:870. doi: 10.3389/fimmu.2020.00870
162. Hsu YL, Hung JY, Chang WA, Jian SF, Lin YS, Pan YC, et al. Hypoxic lung-Cancer-Derived extracellular vesicle MicroRNA-103a increases the oncogenic effects of macrophages by targeting PTEN. Mol Ther (2018) 26:568–81. doi: 10.1016/j.ymthe.2017.11.016
163. Gong J, Shen Y, Jiang F, Wang Y, Chu L, Sun J, et al. MicroRNA-20a promotes non-small cell lung cancer proliferation by upregulating PD-L1 by targeting PTEN, oncol. Lett (2022) 23:1–8. doi: 10.3892/ol.2022.13269
164. Zhou X, Liu S, Liu J, Zhang Z, Mao X, Zhou H. MicroRNA-130a enhances the killing ability of natural killer cells against non-small cell lung cancer cells by targeting signal transducers and activators of transcription 3, biochem. Biophys Res Commun (2020) 523:481–6. doi: 10.1016/j.bbrc.2019.11.099
165. Donatelli SS, Zhou JM, Gilvary DL, Eksioglu EA, Chen X, Cress WD, et al. TGF-β-inducible microRNA-183 silences tumor-associated natural killer cells. Proc Natl Acad Sci U. S. A (2014) 111:4203–8. doi: 10.1073/pnas.1319269111
166. Song N, Li P, Song P, Li Y, Zhou S, Su Q, et al. MicroRNA-138-5p suppresses non-small cell lung cancer cells by targeting PD-L1/PD-1 to regulate tumor microenvironment. Front Cell Dev Biol (2020) 8:540. doi: 10.3389/fcell.2020.00540
167. Meng G, Wei J, Wang Y, Qu D, Zhang J. MiR-21 regulates immunosuppression mediated by myeloid-derived suppressor cells by impairing RUNX1-YAP interaction in lung cancer. Cancer Cell Int (2020) 20:1–16. doi: 10.1186/s12935-020-01555-7
168. Tian J, Rui K, Tang X, Ma J, Wang Y, Tian X, et al. MicroRNA-9 regulates the differentiation and function of myeloid-derived suppressor cells via targeting Runx1. J Immunol (2015) 195:1301–11. doi: 10.4049/jimmunol.1500209
169. Yin Y, Cai X, Chen X, Liang H, Zhang Y, Li J, et al. Tumor-secreted miR-214 induces regulatory T cells: A major link between immune evasion and tumor growth. Cell Res (2014) 24:1164–80. doi: 10.1038/cr.2014.121
170. Liao C, Xiao W, Zhu N, Liu Z, Yang J, Wang Y, et al. Radiotherapy suppressed tumor-specific recruitment of regulator T cells via up-regulating microR-545 in Lewis lung carcinoma cells. Int J Clin Exp Pathol (2015) 8:2535–44.
171. Bouchie A. First microRNA mimic enters clinic. Nat Biotechnol (2013) 31:577. doi: 10.1038/nbt0713-577
172. Cortez MA, Ivan C, Valdecanas D, Wang X, Peltier HJ, Ye Y, et al. PDL1 regulation by p53 via miR-34. J Natl Cancer Inst (2016) 108:1–9. doi: 10.1093/jnci/djv303
173. Cheng Z, Ma R, Tan W, Zhang L. MiR-152 suppresses the proliferation and invasion of NSCLC cells by inhibiting FGF2. Exp Mol Med (2014) 46:e112–9. doi: 10.1038/emm.2014.51
174. Chen L, Gibbons DL, Goswami S, Cortez MA, Ahn Y-H, Byers LA, et al. HHS public access. Nature Communications (2014) 5:5241. doi: 10.1038/ncomms6241
175. Hong JH, Roh KS, Suh SS, Lee S, Sung SW, Park JK, et al. The expression of microRNA-34a is inversely correlated with c-MET and CDK6 and has a prognostic significance in lung adenocarcinoma patients. Tumor Biol (2015) 36:9327–37. doi: 10.1007/s13277-015-3428-9
176. Cha ST, Chen PS, Johansson G, Chu CY, Wang MY, Jeng YM, et al. MicroRNA-519c suppresses hypoxia-inducible factor-1α expression and tumor angiogenesis. Cancer Res (2010) 70:2675–85. doi: 10.1158/0008-5472.CAN-09-2448
177. Hsu YL, Hung JY, Chang WA, Lin YS, Pan YC, Tsai PH, et al. Hypoxic lung cancer-secreted exosomal MIR-23a increased angiogenesis and vascular permeability by targeting prolyl hydroxylase and tight junction protein ZO-1. Oncogene (2017) 36:4929–42. doi: 10.1038/onc.2017.105
178. Liu Y, Wu L, Ao H, Zhao M, Leng X, Liu M, et al. Prognostic implications of autophagy-associated gene signatures in non-small cell lung cancer. Aging (2019) 11:11440–62. doi: 10.18632/aging.102544
179. Li X, He S, Ma B. Autophagy and autophagy-related proteins in cancer. Mol Cancer (2020) 19:1–16. doi: 10.1186/s12943-020-1138-4
180. Guo W, Du K, Luo S, Hu D. Recent advances of autophagy in non-small cell lung cancer: From basic mechanisms to clinical application. Front Oncol (2022) 12:861959. doi: 10.3389/fonc.2022.861959
181. Yue Z, Jin S, Yang C, Levine AJ, Heintz N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc Natl Acad Sci USA (2003) 100:15077–82. doi: 10.1073/pnas.2436255100
182. Takamura A, Komatsu M, Hara T, Sakamoto A, Kishi C, Waguri S, et al. Autophagy-deficient mice develop multiple liver tumors. Genes Dev (2011) 25:795–800. doi: 10.1101/gad.2016211
183. Degenhardt K, Mathew R, Beaudoin B, Bray K, Anderson D, Chen G, et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell (2006) 10:51–64. doi: 10.1016/j.ccr.2006.06.001
184. Rao S, Yang H, Penninger JM, Kroemer G. Autophagy in non-small cell lung carcinogenesis. Autophagy (2014) 10:529–31. doi: 10.4161/auto.27643
185. Guo JY, White E. Autophagy is required for mitochondrial function, lipid metabolism, growth, and fate of KRASG12D-driven lung tumors. Autophagy (2013) 9:1636–8. doi: 10.4161/auto.26123
186. Bustos SO, Antunes F, Rangel MC, Chammas R. Emerging autophagy functions shape the tumor microenvironment and play a role in cancer progression - implications for cancer therapy. Front Oncol (2020) 10:606436. doi: 10.3389/fonc.2020.606436
187. Rezaei S, Mahjoubin-Tehran M, Aghaee-Bakhtiari SH, Jalili A, Movahedpour A, Khan H, et al. Autophagy-related MicroRNAs in chronic lung diseases and lung cancer. Crit Rev Oncol Hematol (2020) 153:103063. doi: 10.1016/j.critrevonc.2020.103063
188. Petrek H, Yu AM. MicroRNAs in non-small cell lung cancer: Gene regulation, impact on cancer cellular processes, and therapeutic potential. Pharmacol Res Perspect (2019) 7:e00528. doi: 10.1002/prp2.528
189. Lv X, Li CY, Han P, Xu XY. MicroRNA-520a-3p inhibits cell growth and metastasis of non-small cell lung cancer through PI3K/AKT/mTOR signaling pathway. Eur Rev Med Pharmacol Sci (2018) 22:2321–7. doi: 10.26355/eurrev-201804-14822
190. Wang J, Sheng Z, Cai Y. Effects of microRNA-513b on cell proliferation, apoptosis, invasion, and migration by targeting HMGB3 through regulation of mTOR signaling pathway in non-small-cell lung cancer. J Cell Physiol (2019) 234:10934–41. doi: 10.1002/jcp.27921
191. Li S, Zeng X, Ma R, Wang L. MicroRNA-21 promotes the proliferation, migration and invasion of non-small cell lung cancer A549 cells by regulating autophagy activity via AMPK/ULK1 signaling pathway. Exp Ther Med (2018) 16:2038–45. doi: 10.3892/etm.2018.6370
192. Chen Y, Zhou X, Qiao J, Bao A. MiR-142-3p overexpression increases chemo-sensitivity of NSCLC by inhibiting HMGB1-mediated autophagy. Cell Physiol Biochem (2017) 41:1370–82. doi: 10.1159/000467896
193. Zhang W, Dong YZ, Du X, Peng XN, Shen QM. MiRNA-153-3p promotes gefitinib-sensitivity in non-small cell lung cancer by inhibiting ATG5 expression and autophagy. Eur Rev Med Pharmacol Sci (2019) 23:2444–52. doi: 10.26355/eurrev_201903_17391
194. Lin JJ, Cardarella S, Lydon CA, Dahlberg SE, Jackman DM, Jänne PA, et al. Five-year survival in EGFR-mutant metastatic lung adenocarcinoma treated with EGFR-TKIs. J Thorac Oncol (2016) 11:556–65. doi: 10.1016/j.jtho.2015.12.103
195. Burotto M, Manasanch EE, Wilkerson J, Fojo T. Gefitinib and erlotinib in metastatic non-small cell lung cancer: A meta-analysis of toxicity and efficacy of randomized clinical trials. Oncologist (2015) 20:400–10. doi: 10.1634/theoncologist.2014-0154
196. König D, Rothschild SI. Molecular diagnostic and precision medicine in non-small cell lung cancer. an update on the treatment of the most important actionable oncogenic driver alterations, heal. TIMES Oncol Hematol (2020) 5:16–27. doi: 10.36000/hbT.OH.2020.05.019
197. Ramalingam SS, Vansteenkiste J, Planchard D, Cho BC, Gray JE, Ohe Y, et al. Overall survival with osimertinib in untreated, EGFR -mutated advanced NSCLC. N Engl J Med (2020) 382:41–50. doi: 10.1056/nejmoa1913662
198. Shaw AT, Riely GJ, Bang YJ, Kim DW, Camidge DR, Solomon BJ, et al. Crizotinib in ROS1-rearranged advanced non-small-cell lung cancer (NSCLC): updated results, including overall survival, from PROFILE 1001. Ann Oncol (2019) 30:1121–6. doi: 10.1093/annonc/mdz131
199. Horn L, Mansfield AS, Szczęsna A, Havel L, Krzakowski M, Hochmair MJ, et al. First-line atezolizumab plus chemotherapy in extensive-stage small-cell lung cancer. N Engl J Med (2018) 379:2220–9. doi: 10.1056/nejmoa1809064
200. Drilon A, Siena S, Dziadziuszko R, Barlesi F, Krebs MG, Shaw AT, et al. Entrectinib in ROS1 fusion-positive non-small-cell lung cancer: integrated analysis of three phase 1–2 trials. Lancet Oncol (2020) 21:261–70. doi: 10.1016/S1470-2045(19)30690-4
201. Planchard D, Smit EF, Groen HJM, Mazieres J, Besse B, Helland Å., et al. Dabrafenib plus trametinib in patients with previously untreated BRAFV600E-mutant metastatic non-small-cell lung cancer: an open-label, phase 2 trial. Lancet Oncol (2017) 18:1307–16. doi: 10.1016/S1470-2045(17)30679-4
202. Awad MM, Oxnard GR, Jackman DM, Savukoski DO, Hall D, Shivdasani P, et al. MET exon 14 mutations in non-small-cell lung cancer are associated with advanced age and stage-dependent MET genomic amplification and c-met overexpression. J Clin Oncol (2016) 34:721–30. doi: 10.1200/JCO.2015.63.4600
203. Herrmann H, Bucksch H. Skipping, dict. Geotech. Eng. Geotech (2014) 5:1242–2. doi: 10.1007/978-3-642-41714-6_194053
204. Stinchcombe TE, Doebele RC, Wang X, Gerber DE, Horn L, Camidge DR. Preliminary clinical and molecular analysis results from a single-arm phase 2 trial of brigatinib in patients with disease progression after next-generation ALK tyrosine kinase inhibitors in advanced ALK+ NSCLC. J Thorac Oncol (2021) 16:156–61. doi: 10.1016/j.jtho.2020.09.018
205. Drilon A, Rekhtman N, Arcila M, Wang L, Ni A, Albano M, et al. Cabozantinib in patients with advanced RET-rearranged non-small-cell lung cancer: an open-label, single-centre, phase 2, single-arm trial. Lancet Oncol (2016) 17:1653–60. doi: 10.1016/S1470-2045(16)30562-9
206. Yang G, Xu H, Yang Y, Zhang S, Xu F, Hao X, et al. Pyrotinib combined with apatinib for targeting metastatic non-small cell lung cancer with HER2 alterations: a prospective, open-label, single-arm phase 2 study (PATHER2). BMC Med (2022) 20:1–9. doi: 10.1186/s12916-022-02470-6
207. Skoulidis F, Li BT, Dy GK, Price TJ, Falchook GS, Wolf J, et al. Sotorasib for lung cancers with KRAS p.G12C mutation. N Engl J Med (2021) 384:2371–81. doi: 10.1056/nejmoa2103695
208. Hallin J, Engstrom LD, Hargi L, Calinisan A, Aranda R, Briere DM, et al. The KRASG12C inhibitor MRTX849 provides insight toward therapeutic susceptibility of KRAS-mutant cancers in mouse models and patients. Cancer Discovery (2020) 10:54–71. doi: 10.1158/2159-8290.CD-19-1167
209. Middleton MR, Dean E, Evans TRJ, Shapiro GI, Pollard J, Hendriks BS, et al. Phase 1 study of the ATR inhibitor berzosertib (formerly M6620, VX-970) combined with gemcitabine ± cisplatin in patients with advanced solid tumours. Br J Cancer (2021) 125:510–9. doi: 10.1038/s41416-021-01405-x
210. Sang YB, Kim JH, Kim CG, Hong MH, Kim HR, Cho BC, et al. The development of AXL inhibitors in lung cancer: Recent progress and challenges, front. Oncol (2022) 12:811247. doi: 10.3389/fonc.2022.811247
211. Reuss JE, Gosa L, Liu SV. Antibody drug conjugates in lung cancer: State of the current therapeutic landscape and future developments, clin. Lung Cancer (2021) 22:483–99. doi: 10.1016/j.cllc.2021.07.011
212. Desai A, Abdayem P, Adjei AA, Planchard D. Antibody-drug conjugates: A promising novel therapeutic approach in lung cancer. Lung Cancer (2022) 163:96–106. doi: 10.1016/j.lungcan.2021.12.002
213. Verma S, Miles D, Gianni L, Krop IE, Welslau M, Baselga J, et al. Trastuzumab emtansine for HER2-positive advanced breast cancer. N Engl J Med (2012) 367:1783–91. doi: 10.1056/nejmoa1209124
214. Ogitani Y, Aida T, Hagihara K, Yamaguchi J, Ishii C, Harada N, et al. DS-8201a, a novel HER2-targeting ADC with a novel DNA topoisomerase I inhibitor, demonstrates a promising antitumor efficacy with differentiation from T-DM1, clin. Cancer Res (2016) 22:5097–108. doi: 10.1158/1078-0432.CCR-15-2822
215. Goldenberg DM, Cardillo TM, Govindan SV, Rossi EA, Sharkey RM. Erratum: Trop-2 is a novel target for solid cancer therapy with sacituzumab govitecan (IMMU-132), an antibody-drug conjugate (ADC). Oncotarget (2020) 11:942. doi: 10.18632/oncotarget.27512
216. Bardia A, Messersmith WA, Kio EA, Berlin JD, Vahdat L, Masters GA, et al. Sacituzumab govitecan, a trop-2-directed antibody-drug conjugate, for patients with epithelial cancer: final safety and efficacy results from the phase I/II IMMU-132-01 basket trial. Ann Oncol (2021) 32:746–56. doi: 10.1016/j.annonc.2021.03.005
217. Johnson M, Spira A, Yoh K, Heist R, Lisberg A, Greenberg J, et al. P47.05 a phase 2 study of datopotamab deruxtecan (Dato-DXd) in advanced NSCLC with actionable genomic alterations (TROPION-Lung05). J Thorac Oncol (2021) 16:S1098. doi: 10.1016/j.jtho.2021.08.498
218. Brahmer J, Reckamp KL, Baas P, Crinò L, Eberhardt WEE, Poddubskaya E, et al. Nivolumab versus docetaxel in advanced squamous-cell non–Small-Cell lung cancer. N Engl J Med (2015) 373:123–35. doi: 10.1056/nejmoa1504627
219. Socinski MA, Jotte RM, Cappuzzo F, Orlandi F, Stroyakovskiy D, Nogami N, et al. Atezolizumab for first-line treatment of metastatic nonsquamous NSCLC. N Engl J Med (2018) 378:2288–301. doi: 10.1056/nejmoa1716948
220. Insa A, Martín-Martorell P, Di Liello R, Fasano M, Martini G, Napolitano S, et al. Which treatment after first line therapy in NSCLC patients without genetic alterations in the era of immunotherapy? Crit Rev Oncol Hematol (2022) 169:103538. doi: 10.1016/j.critrevonc.2021.103538
221. Collins JM, Heery CR, Donahue RN, Palena C, Madan RA, Strauss J, et al. Phase I trial of BMS-986253, an anti-IL-8 monoclonal antibody, in patients with metastatic or unresectable solid tumors. J Clin Oncol (2018) 36:3091–1. doi: 10.1200/jco.2018.36.15_suppl.3091
222. Weiss SA, Djureinovic D, Jessel S, Krykbaeva I, Zhang L, Jilaveanu L, et al. A phase i study of APX005M and cabiralizumab with or without nivolumab in patients with melanoma, kidney cancer, or non-small cell lung cancer resistant to anti-PD-1/PD-L1. Clin Cancer Res (2021) 27:4757–67. doi: 10.1158/1078-0432.CCR-21-0903
223. Qu J, Mei Q, Chen L, Zhou J. Chimeric antigen receptor (CAR)-t-cell therapy in non-small-cell lung cancer (NSCLC): current status and future perspectives, cancer immunol. Immunother (2021) 70:619–31. doi: 10.1007/s00262-020-02735-0
224. Zhong S, Cui Y, Liu Q, Chen S. CAR-T cell therapy for lung cancer: A promising but challenging future. J Thorac Dis (2020) 12:4516–21. doi: 10.21037/jtd.2020.03.118
225. Wu MR, Jusiak B, Lu TK. Engineering advanced cancer therapies with synthetic biology. Nat Rev Cancer (2019) 19:187–95. doi: 10.1038/s41568-019-0121-0
226. Khalil AS, Collins JJ. Synthetic biology: Applications come of age, nat. Rev Genet (2010) 11:367–79. doi: 10.1038/nrg2775
228. Shapira P, Kwon S, Youtie J. Tracking the emergence of synthetic biology. Scientometrics (2017) 112:1439–69. doi: 10.1007/s11192-017-2452-5
229. Abil Z, Xiong X, Zhao H. Synthetic biology for therapeutic applications. Mol Pharm (2015) 12:322–31. doi: 10.1021/mp500392q
230. Gardner TS, Cantor CR, Collins JJ. Construction of a genetic toggle switch in escherichia coli. Nature (2000) 403:339–42. doi: 10.1038/35002131
231. Purnick PEM, Weiss R. The second wave of synthetic biology: From modules to systems. Nat Rev Mol Cell Biol (2009) 10:410–22. doi: 10.1038/nrm2698
232. Cameron DE, Bashor CJ, Collins JJ. A brief history of synthetic biology. Nat Rev Microbiol (2014) 12:381–90. doi: 10.1038/nrmicro3239
233. Kittleson JT, Wu GC, Anderson JC. Successes and failures in modular genetic engineering. Curr Opin Chem Biol (2012) 16:329–36. doi: 10.1016/j.cbpa.2012.06.009
234. Zhou X, Franklin RA, Adler M, Jacox JB, Bailis W, Shyer JA, et al. Circuit design features of a stable two-cell system. Cell (2018) 172:744–757.e17. doi: 10.1016/j.cell.2018.01.015
235. Beerli RR, Barbas CF. Engineering polydactyl zinc-finger transcription factors. Nat Biotechnol (2002) 20:135–41. doi: 10.1038/nbt0202-135
236. Bogdanove AJ, Voytas DF. TAL effectors: Customizable proteins for DNA targeting. Science (2011) 80-.). 333:1843–6. doi: 10.1126/science.1204094
237. Reik A, Gregory PD, Case CC. 628. an oncolytic adenovirus armed with a GM-CSF activating designed zinc-finger protein transcription factor: A potential cancer vaccine approach. Mol Ther (2004) 9:S238. doi: 10.1016/j.ymthe.2004.06.552
238. Schmidt F, Platt RJ. Applications of CRISPR-cas for synthetic biology and genetic recording. Curr Opin Syst Biol (2017) 5:9–15. doi: 10.1016/j.coisb.2017.05.008
239. Jusiak B, Cleto S, Perez-Piñera P, Lu TK. Engineering synthetic gene circuits in living cells with CRISPR technology. Trends in Biotechnology (2016) 34:535–47. doi: 10.1016/j.tibtech.2015.12.014
240. Liu Y, Zeng Y, Liu L, Zhuang C, Fu X, Huang W, et al. Synthesizing and gate genetic circuits based on CRISPR-Cas9 for identification of bladder cancer cells. Nat Commun (2014) 5:1–7. doi: 10.1038/ncomms6393
241. Nissim L, Bar-Ziv RH. A tunable dual-promoter integrator for targeting of cancer cells. Mol Syst Biol (2010) 6:1–9. doi: 10.1038/msb.2010.99
242. Navarro F, Baulcombe DC. miRNA-mediated regulation of synthetic gene circuits in the green AlgaChlamydomonas reinhardtii, ACS synth. Biol (2019) 8:358–70. doi: 10.1021/acssynbio.8b00393
243. Nimsarkar P, Ingale P, Singh S. Systems studies uncover miR-146a as a target in leishmania major infection model. Acs Omega (2020) 5:12516–26. doi: 10.1021/acsomega.0c01502
244. Mircetic J, Dietrich A, Paszkowski-Rogacz M, Krause M, Buchholz F. Development of a genetic sensor that eliminates p53 deficient cells. Nat. Commun (2017) 8:1–11. doi: 10.1038/s41467-017-01688-w
Keywords: NSCLC, EGFR, miRNA, autophagy, synthetic biology, EGFR-TKIs
Citation: Samarth N, Gulhane P and Singh S (2022) Immunoregulatory framework and the role of miRNA in the pathogenesis of NSCLC – A systematic review. Front. Oncol. 12:1089320. doi: 10.3389/fonc.2022.1089320
Received: 04 November 2022; Accepted: 02 December 2022;
Published: 21 December 2022.
Edited by:
Fabrizio Bianchi, IRCCS Casa Sollievo della Sofferenza Ospedale di San Pio da Pietrelcina, ItalyReviewed by:
Massimo Moro, National Cancer Institute Foundation (IRCCS), ItalyCopyright © 2022 Samarth, Gulhane and Singh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shailza Singh, c2luZ2hzQG5jY3MucmVzLmlu
†These authors have contributed equally to this work