 Chao Li
Chao Li Daqin Feng1*
Daqin Feng1*- 1Department of Neurosurgery, The First Affiliated Hospital, Guangxi Medical University, Nanning, China
- 2Department of Neurosurgery, Beijing Tiantan Hospital, Capital Medical University, Beijing, China
Background: Mixed gangliocytoma-adenoma (MGA) is a rare tumor of pituitary gland. It’s difficult to distinguish it from pituitary adenoma by clinical manifestations, imaging features or serological testing. Thus, the histopathological examination is still the golden standard for diagnosis. Besides, studies on molecular level are still lacking.
Case information: In this case report, we described a 28-year-old male with MGA presenting as acromegaly, who suffered staging operation and post-operation gamma knife radiosurgery, but finally died of secondary hyperglycemic hyperosmolar collapse. A complete data including clinical, histopathological, ultrastructural and single-cell transcriptome level information were collected and analyzed.
Conclusion: This case report detailed the only clinical and molecular report of MGA following operation and radiotherapy. Complete clinical data enhanced the understanding of the diagnosis and treatment of this disease. Besides, the single-cell transcriptome sequencing analysis further disclosed the intra-tumoral heterogeneity and provided support for subsequent basic research.
Introduction
Mixed gangliocytoma-adenoma (MGA) is a kind of collision tumor of the pituitary gland, which was first written into World Health Organization (WHO) Classification of pituitary tumors in 2017 (1, 2). It is rarely seen among pituitary tumors, accounting for 0.29% approximately and often presenting the same kind of clinical symptoms as pituitary adenomas (3). Up to now, no more than 200 cases had been reported worldwide, and research on the diagnostic, therapeutic and molecular pathological aspects of the disease is still lacking (4). The aim of this study is to report a MGA, to be more precise, a growth hormone (GH) secreting type presenting as acromegaly. The clinical course, imaging features, blood hormone levels, histopathologic features, ultrastructural features and single-cell transcriptome characterization of the case were included in this study. This case report followed the CARE Guidelines (5).
Case description
Clinical course
The patient was a 20-year-old Chinese male who developed symptoms and signs of acromegaly within three years prior to hospitalization, including enlargement of the hands, feet, nose and jaw, thicker skin, deepening of the voice, and myocardial hypertrophy, etc. (Figure 1A). Over the one-month period before admition, he developed headache, hypertension, diabetes, reduced vision, visual field defects and polyuria. Blood hormone level screening showed the serum growth hormone level of him was over 40ng/mL (0.06-5.00ng/mL), and the Insulin-like growth factor-1(IGF-1) was 1567ng/mL(127~424ng/mL). The MRI revealed a solid-cystic lesion located in the enlarged and sunken sellar turcica, measuring about 44mm*27mm*37mm. The lesion presented with equal T1 and T2 signals, and showed significantly inhomogeneous enhancement. The MRI also showed an invasive behavior with mass effect on the optic chiasm, left cavernous sinus involvement, and internal carotid artery encased (Figure 1B).
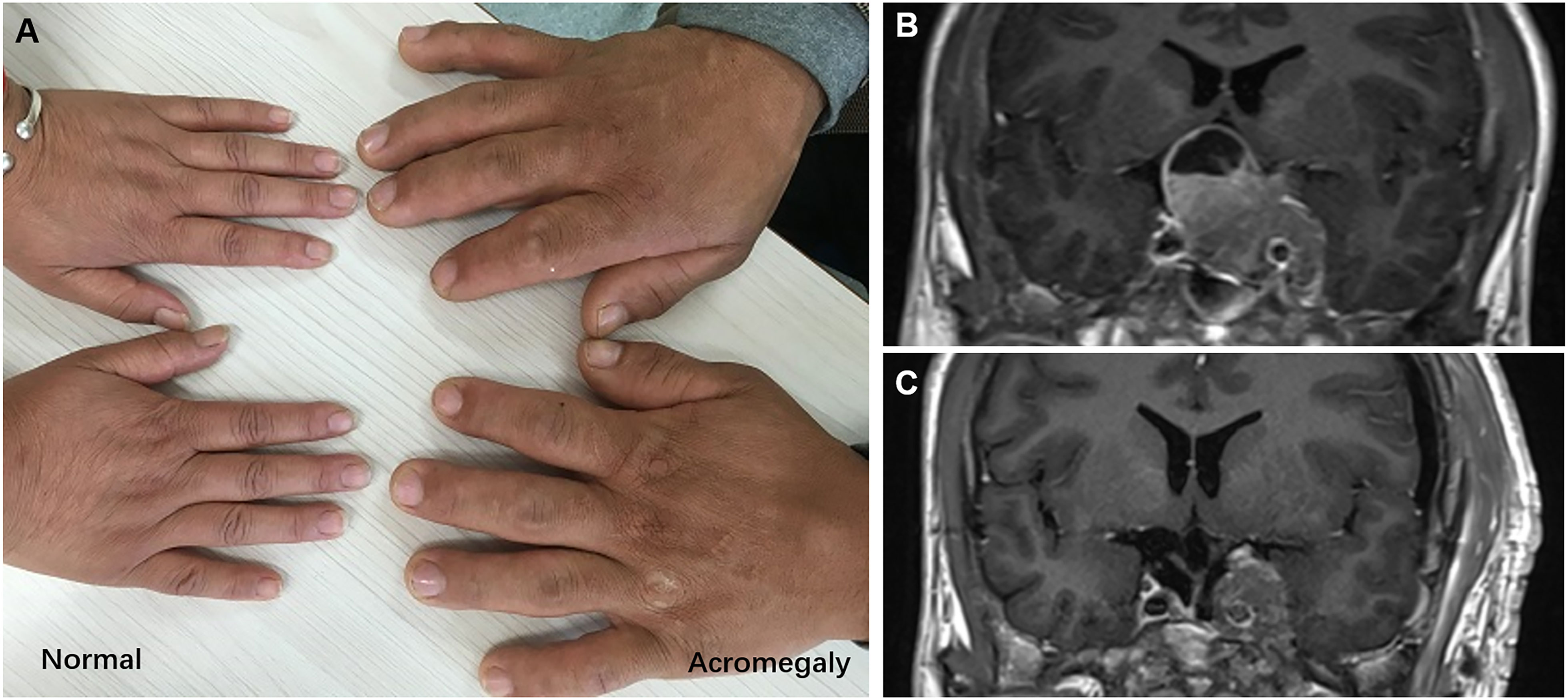
Figure 1 (A) The hands of normal and acromegaly people. (B) Preoperative MRI, 44mm× 27mm×37mm in size, with left cavernous sinus involvement, and internal carotid artery encased. (C) Postoperative MRI, show the residual tumor encircling the left internal carotid artery.
Due to the relatively large size of the tumor and the location both within the sellar and supra-sellar, it was difficult to remove it at once. So, the doctor suggested a staged tumor resection plan. For the first time, the tumor was removed from the sellar using a transnasal-sphenoidal approach. Intraoperatively, the tumor was seen to be grayish-red in color, with a heterogeneous texture and a relatively rich blood supply. The operation was successful with no postoperative complications like cerebrospinal fluid leakage. After the first surgery, the serum GH level decreased to some extent but was not well controlled and remained at about 18ng/mL (0.06-5.00ng/mL). Eleven months later, the patient underwent a second surgery with the frontolateral approach to remove the suprasellar and parasellar part of the tumor. However, due to the hard texture of the tumor encircling the left internal carotid artery, some of the tumor body remained (Figure 1C), and the postoperative serum GH level still did not drop to normal level which was about 7ng/mL (0.06-5.00ng/mL). Three months after surgery, the patient further underwent gamma knife radiation therapy. Unluckily, it did not show significant efficacy after treatment. Then, the doctor suggested a growth inhibitor analog injection therapy, but the patients denied due to the weak economic condition. Four months later, the patient developed a severe hyperosmolar hyperglycemic syndrome with blood glucose exceeding 35 mmol/L, resulting in electrolyte disturbances and secondary coma. Finally, the patient died of ineffective resuscitation in 33 months after the first surgery. Time line of the clinical course was shown in Figure S1.
Histopathological examination
The hematoxylin-eosin (HE) staining revealed cap-like gangliocytoma cells surrounding the adenoma cells (Figure 2A). Further immunohistochemistry profile showed strongly positive for GH and Synapsin, scattered positive for Prolactin(PRL) and Ck8/18, and about 5% monoclonal antibody (MIB)-1 proliferative index positivity (Figures 2B–D). Immunofluorescent staining show strong positive for Pituitary transcript factor 1(Pit-1) (Figure 2E), which further proved its Pit-1 lineage origin. These findings are characteristic of the diagnosis of MGA. Further, the ultrastructure observed by electron microscope showed that tumor cells were densely distributed, with nuclei of different sizes, intracytoplasmic fibrous vesicles, round secretory granules within the fibrous vesicles, and scattered capillaries in the interstitium (Figure 2F). The suspicious gangliocytoma cells were large and contain abundant cytoplasm and Nissl granules.
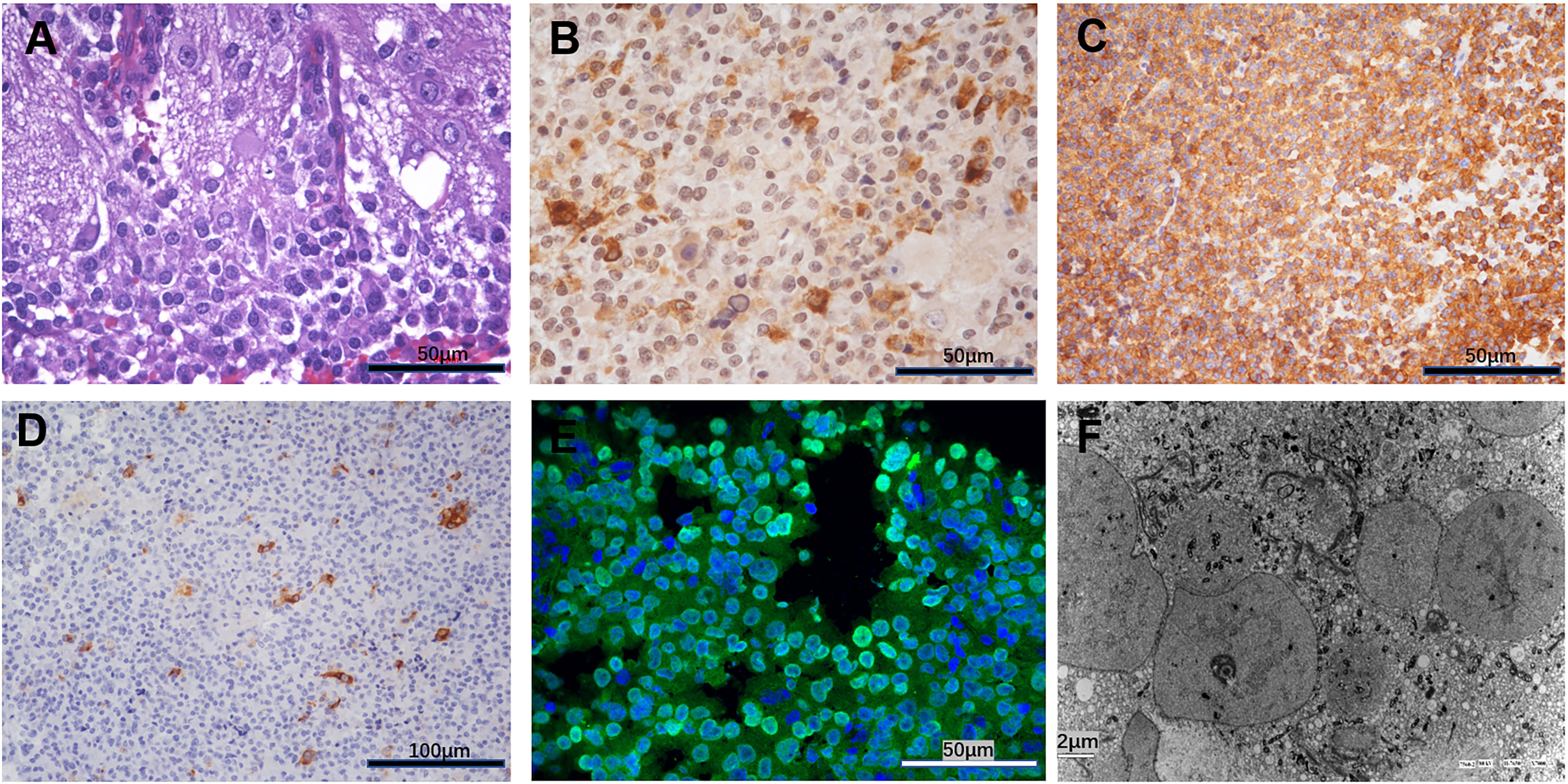
Figure 2 (A) Hematoxylin-eosin HE revealed cap-like gangliocytoma cells surrounding the adenoma cells. (B) Immunohistochemistry showed strongly positive for growth hormone. (C) Immunohistochemistry showed strongly positive for synapsin. (D) Immunohistochemistry showed scattered positive for Prolactin (PRL). (E) Immunofluorescent staining show strong positive for Pituitary Transcript Factor 1(Pit-1). (F) The ultrastructure observed by Electron microscope.
Single cell transcriptome sequencing analysis
Single-cell transcriptome sequencing analysis was performed on fresh tumor tissue obtained during two surgeries, 89 cells from the first surgery and 120 cells from the second surgery. First, a modified Single-cell Tagged Reverse Transcription sequencing (STRT-seq) technique (6) was applied and the single-cell RNA sequencing (scRNA-seq) data was obtained through Illumina sequencing. Then, information on chromosome copy number variation (CNV) was obtained by the InferCNV method based on scRNA-seq data, and a comparison of the CNV profiles of the two specimens revealed that two specimens had gain in chromosome 4, while chromosome 11 deletion was only detected in the specimen obtained from the first surgery, thus suggesting the possible existence of temporal and spatial heterogeneity within the tumor (Figure 3A). Then, all the 209 cells were analyzed by principal component analysis (PCA), and the results showed that the cells were almost clustered together, and only little part of cells (7/209) had cell cycle heterogeneity, which indicated that the heterogeneity within the tumor was small and about 3% of cells was in the process of mitosis (Figure 3B). At last, the differentially expressed genes of this case were analyzed and the biological processes (BP) corresponding to them were clarified by Gene Ontology (GO) enrichment analysis, in which the biological processes that were more involved were “Cytoskeleton Organization”, “anatomical structure development”, and “cellular component assembly involved in morphogenesis”(Figure 3C).
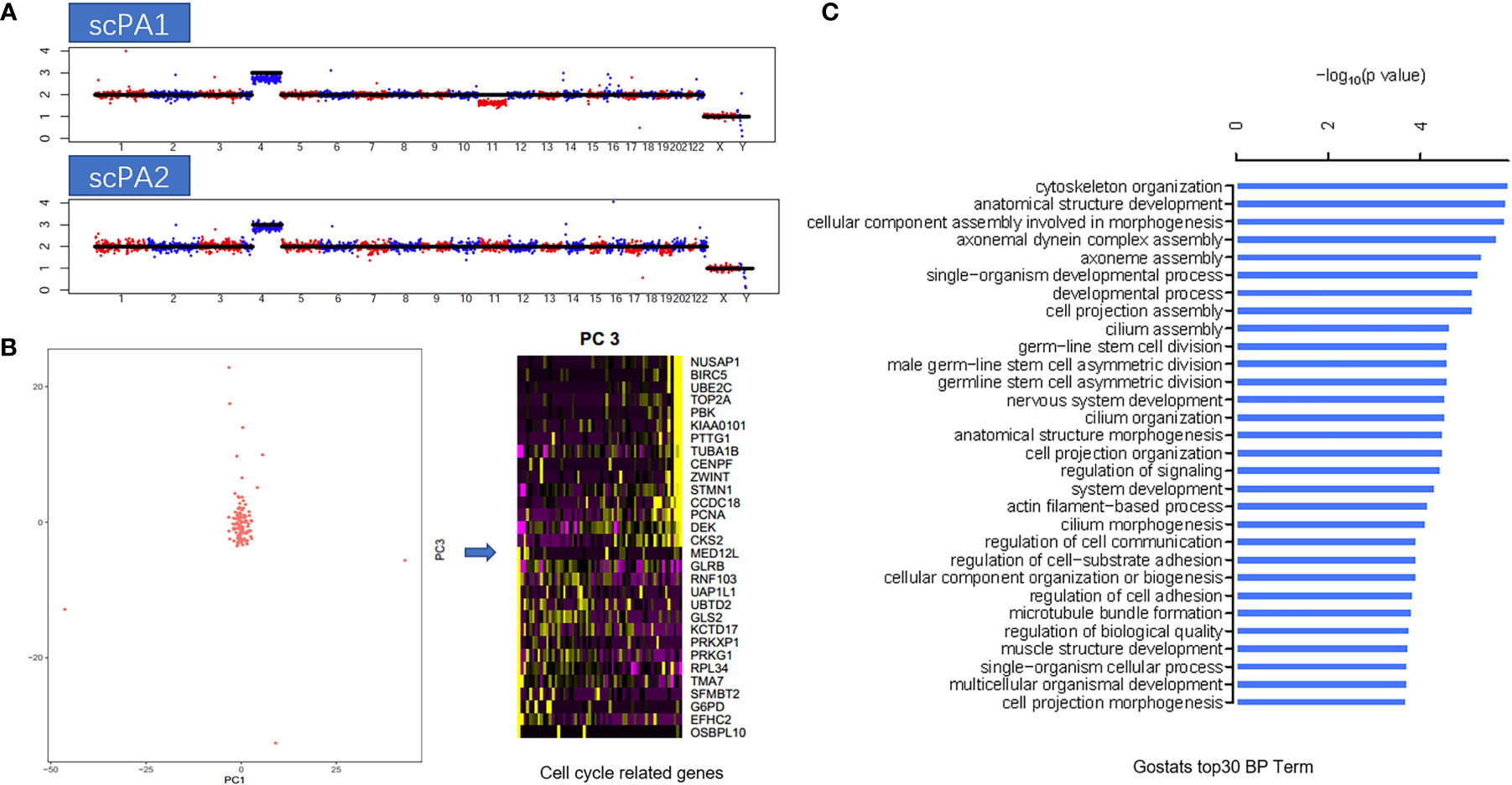
Figure 3 (A) Copy number variation (CNV) profiles of the two specimens: first surgery(scPA1) and second surgery(scPA2), show gain in chromosome 4 (both), and chromosome 11 deletion in scPA1 only, indicated the possible existence of temporal and spatial heterogeneity within the tumor. (B) Principal component analysis (PCA) showed that the cells were almost clustered together, and only little part of cells (7/209) had cell cycle heterogeneity (PC2 represented cell cycle related genes). (C) GO enrichment analysis of the differentially expressed genes of this case, the top 30 biological processes (BP) term were listed.
Discussion
One category of tumors highlighted in 2017 WHO classification is neuronal and paraneuronal tumors, which includes gangliocytoma and mixed gangliocytoma-adenoma, neurocytoma, paraganglioma and neuroblastoma, all of which are rare tumors, but they are of particular importance in the differential diagnosis of pituitary tumors (1, 2, 7, 8). This study highlights a case of mixed gangliocytoma-adenoma, which has a very low incidence, found to be only 0.29% (14/4891) in a retrospective German study that included 4891 pituitary lesions (3). Currently, there are three hypotheses regarding the pathogenesis of the disease: 1) Neuronal differentiation hypothesis. It is believed that most ganglion cell tumors in the sellar are the result of neuronal differentiation and chemosis in pituitary adenomas. Evidence supporting this hypothesis includes that both pituitary adenomas and ganglion cells can be observed microscopically (9). In a pathological study, Mikami et al. found that mixed gangliocytoma-adenoma is an intermediate morphological cell that lies between gangliocytoma and GH or PRL cell adenoma (10). It is generally believed that ganglion cells have no hormone-secreting capacity, but Li et al. found positive pituitary hormone expression in some tumor cells by performing neuron-related immunohistochemical staining of ganglion cell tumors in the sellar area (11). In addition, in vitro culture of anterior pituitary cells revealed that they can be converted into ganglion cells with different degrees of differentiation, providing a theoretical basis that pituitary adenomas can undergo neuronal transformation (12). 2) Residual ganglion cell hypothesis. It is believed that the ganglion cells that originally disappeared from the posterior pituitary gland due to excessive differentiation, like hypothalamic neurons, had the ability to release prohormone-releasing hormone, which can stimulate the occurrence of pituitary adenoma (13). At the same time, the ganglion cells are stimulated by the excessive secretion of prohormone-releasing hormone from the adenohypophysis and hypothalamic neurons, and eventually become tumorigenic (9). 3) Pituitary multipotent stem cell hypothesis (14). In 2006, Kontogeorgos et al. found that typical pituitary adenoma cells could express NFP, suggesting that pituitary adenomas have neuronal properties (15).
The clinical and imaging manifestations of mixed gangliocytoma-adenoma are essentially indistinguishable from pituitary adenoma, making preoperative diagnosis more difficult. This case presented clinically with acromegaly. On cranial MRI, the tumor was predominantly solid with occasional cystic changes. Also, it showed aggressive growth on imaging, invading the left cavernous sinus and encircling the internal carotid artery. So, a staged resection strategy was adopted. However, due to the tough texture of the cavernous sinus segment, a total dissection could not be achieved, and the oculomotor nerve was disturbed during the operation which leaded to postoperative oculomotor nerve palsy. In recent years, with the development of endoscopic technique, some medical centers had attempted to resecting the intrasellar and parasellar lesions through a combined endoscopic transsphenoidal and craniotomy strategy (16, 17). Three months after the postoperative interval, the patient underwent Gamma Knife treatment, but the results were not satisfactory. Gamma knife radiosurgery has emerged as a relatively safe management option for patients (e.g., when cavernous sinus or dural invasion prevents total resection) with adjuvant treatment after sub-total resection of the lesion, or as primary treatment for selected patients when the risk of surgery is considered too high. The reported incidence of new endocrine disorders after radiosurgery was typically in the range of 25% to 40%, with a lower incidence of hypopituitarism approaching 20% (18). With recent multicenter study series showing that functional pituitary adenomas require higher therapeutic doses than nonfunctional pituitary adenomas and that patients with high doses of pituitary stalk irradiation in Gamma Knife treatment are at higher risk for post-Gamma Knife treatment endocrine disorders, the risk burden of achieving long-term endocrine remission remains a concern in current treatment approaches (18, 19). Eventually, the patient died of sudden syncope due to secondary hyperglycemic hyperosmolar syndrome. It has been reported that 15% to 38% of patients with acromegaly develop reduced glucose tolerance or diabetes, and high GH leads to abnormal glucose metabolism through several pathways, with insulin resistance being the predominant mechanism (20). High GH, advanced age, long duration of disease, family history of diabetes, and hypertension are risk factors for the development of abnormal glucose tolerance in patients with acromegaly (21). Through the diagnosis and treatment process of this case, we further understood the refractory nature of mixed gangliocytoma-adenoma, especially the growth hormone type, which is often accompanied by systemic damage, and can greatly affect the patient’s quality and expectancy of life. The authors’ opinion was: 1) Surgical resection remains the preferred first-line treatment; after all, surgery can immediately reduce GH and IGF-1 to lower levels as well as improve signs and symptoms and obtain a pathologic histologic confirmation. 2) In surgery, resection of pituitary adenomas extending into the cavernous sinus is aggressively pursued; however, residual tumor in the cavernous sinus is inevitable. 3) The efficacy of Gamma knife treatment of residual or recurrent MGA presenting as acromegaly still needs to be tested. 4) It has also been shown that appropriate use of growth-inhibiting drugs during Gamma Knife treatment, while waiting for IGF-1 normalization, may allow patients with acromegaly to achieve endocrine remission. However, the cost of treatment with growth inhibitors is very high and imposes a heavy financial burden on patients. Therefore, it is important to explore effective lifelong alternative treatment options for patients with persistent acromegaly after surgical resection.
Currently, the diagnosis of MGA relies on postoperative pathological examination, including histomorphological observation and immunohistochemical staining (4, 22). Ganglion cells are distributed in clusters or scattered in the rich nerve fiber network, with eosinophilic cytoplasm and binucleated or multinucleated; while pituitary adenoma cells are diffusely distributed, with round or ovoid nuclei, uniformly slender and loose chromatin, granular cytoplasm, partially vacuolated cytoplasm, and abundant interstitial blood sinuses. Often, these neuronal tumors are situated cap-like over the adenomas, with no obvious boundary between them. Immunohistochemical staining reveals positive expression of markers of ganglicytoma (e.g. SYN, Ck8/18, CgA, S-100, etc.) and positive expression of pituitary cell hormones (e.g. GH, PRL, TSH, ACTH, LH, FSH, etc.). It has also been reported that GHRH is expressed in ganglion cells in MGA with acromegaly (23), and CRH is expressed in ganglion cells in MGA with Cushing’s syndrome (24). As for adenoma component in MGA, half is GH adenoma type as reported by Saeger in a Germany cohort with 14 cases included, but there are also reports related to prolactin adenoma, gonadotropin, and ACTH adenoma (3, 25, 26). In this case, tissue observation, immunohistochemistry, and electron microscopic ultrastructural observation were performed and were consistent with previous reports.
In addition, to explore the intratumoral heterogeneity of MGA, we also performed a pioneering single-cell transcriptome sequencing analysis of specimens from both surgeries to explore the tumor cell composition structure, intratumoral heterogeneity of the tumor, and functional types of cells at the single-cell level. Single-cell transcriptome sequencing is a relatively new technology for high-throughput transcriptome sequencing at the individual cell level, which can effectively address the challenges of cell heterogeneity and transcriptome heterogeneity within cell populations that are masked by bulk RNA-seq, and help discover new rare cell types and gain insight into the mechanisms of expression regulation during cell growth (27, 28). Through single-cell level studies, we found some temporal and spatial heterogeneity in the intratumoral cells of MGA, as evidenced by differences in the CNV profiles of the two specimens. However, at the transcriptomic level, the transcriptomic gene expression patterns of the cells obtained from the two specimens were extremely similar, with only a small fraction of cells in mitosis, which in turn suggested that MGA is a tumor with less intratumoral heterogeneity and inactive proliferation, which is consistent with previous studies of pituitary adenomas at the bulk level (29–31). We then focused on the functional properties of the adenomatous component of growth hormone in MGA, and obtained the more active biological processes within the tumor by GO analysis. However, unfortunately, no ganglion cells were found in the 209 single cells we obtained by analyzing their gene signature. We speculated that this might be related to the insufficient number of cells taken, therefore, we also intended to further search for ganglicytoma cells by increasing the specimen volume in future studies.
Conclusion
Mixed gangliocytoma-adenoma presenting as acromegaly is a rare CNS neoplasm which is not that benign, because it’s growth hormone over-secretion behavior can cause severe damage to various systems of the body, and once it shows an invasive growth pattern, it is hard to achieve anatomic and functional resection, thus seriously affects the quality and span of patients’ life. MRI findings are useful to make a surgery planning, but the histopathological examination, including a full immunohistochemistry panel, is essential to correctly diagnose MGA. With a high probability, single-cell molecular testing can help deeper understanding the pathogenesis of the disease, but more cases will be needed in the future.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Ethics statement
Written informed consent was obtained from the minor(s)’ legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
Author contributions
CL wrote the manuscript. DZ and DF reviewed and edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Natural Science Foundation of China (Grant No. 31671109).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2022.1088803/full#supplementary-material
Supplementary Figure 1 | The timeline of the clinical course of the case.
References
1. Mete O, Lopes MB. Overview of the 2017 WHO classification of pituitary tumors. Endocr Pathol (2017) 28(3):228–43. doi: 10.1007/s12022-017-9498-z
2. Lopes MBS. The 2017 world health organization classification of tumors of the pituitary gland: A summary. Acta Neuropathol (2017) 134(4):521–35. doi: 10.1007/s00401-017-1769-8
3. Saeger W, Lüdecke DK, Buchfelder M, Fahlbusch R, Quabbe HJ, Petersenn S. Pathohistological classification of pituitary tumors: 10 years of experience with the German pituitary tumor registry. Eur J endocrinol (2007) 156(2):203–16. doi: 10.1530/eje.1.02326
4. Cossu G, Brouland JP, La Rosa S, Camponovo C, Viaroli E, Daniel RT, et al. Comprehensive evaluation of rare pituitary lesions: A single tertiary care pituitary center experience and review of the literature. Endocr pathol (2019) 30(3):219–36. doi: 10.1007/s12022-019-09581-6
5. Riley DS, Barber MS, Kienle GS, Aronson JK, von Schoen-Angerer T, Tugwell P, et al. CARE guidelines for case reports: Explanation and elaboration document. J Clin Epidemiol (2017) 89:218–35. doi: 10.1016/j.jclinepi.2017.04.026
6. Natarajan KN. Single-cell tagged reverse transcription (STRT-seq). Methods Mol Biol (Clifton NJ) (2019) 1979:133–53. doi: 10.1007/978-1-4939-9240-9_9
7. Castillo M, Mukherji SK. Intrasellar mixed gangliocytoma-adenoma. AJR Am J roentgenol (1997) 169(4):1199–200. doi: 10.2214/ajr.169.4.9308499
8. Kamel OW, Horoupian DS, Silverberg GD. Mixed gangliocytoma-adenoma: A distinct neuroendocrine tumor of the pituitary fossa. Hum pathol (1989) 20(12):1198–203. doi: 10.1016/S0046-8177(89)80012-7
9. Geddes JF, Jansen GH, Robinson SF, Gmr E, Holton JL, Monson JP, et al. ‘Gangliocytomas’ of the pituitary: a heterogeneous group of lesions with differing histogenesis. Am J Surg pathol (2000) 24(4):607–13. doi: 10.1097/00000478-200004000-00017
10. Mikami S, Kameyama K, Takahashi S, Yoshida K, Kawase T, Sano T, et al. Combined gangliocytoma and prolactinoma of the pituitary gland. Endocr pathol (2008) 19(2):117–21. doi: 10.1007/s12022-008-9027-1
11. Li JY, Racadot O, Kujas M, Kouadri M, Peillon F, Racadot J. Immunocytochemistry of four mixed pituitary adenomas and intrasellar gangliocytomas associated with different clinical syndromes: acromegaly, amenorrhea-galactorrhea, cushing’s disease and isolated tumoral syndrome. Acta neuropathologica (1989) 77(3):320–8. doi: 10.1007/BF00687585
12. Vidal S, Horvath E, Bonert V, Shahinian K, Kovacs K. Neural transformation in a pituitary corticotroph adenoma. Acta neuropathologica (2002) 104(4):435–40. doi: 10.1007/s00401-002-0561-5
13. Koutourousiou M, Kontogeorgos G, Wesseling P, Grotenhuis AJ, Seretis A. Collision sellar lesions: Experience with eight cases and review of the literature. Pituitary. (2010) 13(1):8–17. doi: 10.1007/s11102-009-0190-2
14. Chen J, Hersmus N, Van Duppen V, Caesens P, Denef C, Vankelecom H. The adult pituitary contains a cell population displaying stem/progenitor cell and early embryonic characteristics. Endocrinol (2005) 146(9):3985–98. doi: 10.1210/en.2005-0185
15. Kontogeorgos G, Mourouti G, Kyrodimou E, Liapi-Avgeri G, Parasi E. Ganglion cell containing pituitary adenomas: Signs of neuronal differentiation in adenoma cells. Acta neuropathologica (2006) 112(1):21–8. doi: 10.1007/s00401-006-0055-y
16. Heng LJ, Jia D, Gong L, Zhang W, Ma J, Qu Y. Endoscopic endonasal resection of a mixed lesion of gangliocytoma and nonfunctioning pituitary adenoma. World neurosurg (2017) 106:1050.e1–.e6. doi: 10.1016/j.wneu.2017.05.064
17. Shepard MJ, Elzoghby MA, Ghanim D, Lopes MBS, Jane JA Jr. Transsphenoidal surgery for mixed pituitary gangliocytoma-adenomas. World neurosurg (2017) 108:310–6. doi: 10.1016/j.wneu.2017.08.174
18. Pomeraniec IJ, Taylor DG, Cohen-Inbar O, Xu Z, Lee Vance M, Sheehan JP. Radiation dose to neuroanatomical structures of pituitary adenomas and the effect of gamma knife radiosurgery on pituitary function. J neurosurg (2019) 132(5):1499–506. doi: 10.3171/2019.1.JNS182296
19. Gupta T, Chatterjee A. Modern radiation therapy for pituitary adenoma: Review of techniques and outcomes. Neurol India (2020) 68(Supplement):S113–S22. doi: 10.4103/0028-3886.287678
20. Vilar L, Vilar CF, Lyra R, Lyra R, Naves LA. Acromegaly: Clinical features at diagnosis. Pituitary. (2017) 20(1):22–32. doi: 10.1007/s11102-016-0772-8
21. Hannon AM, Thompson CJ, Sherlock M. Diabetes in patients with acromegaly. Curr Diabetes Rep (2017) 17(2):8. doi: 10.1007/s11892-017-0838-7
22. Xiao P, Xue L, Peng JJ, Feng ST, Liao B, Wen JM. An intrasellar mixed gangliocytoma-adenoma including ependymal component, and review of the literature. BMJ Case Rep (2009) 2009:733–745. doi: 10.1136/bcr.11.2008.1200
23. Kurosaki M, Saeger W, Lüdecke DK. Intrasellar gangliocytomas associated with acromegaly. Brain tumor pathol (2002) 19(2):63–7. doi: 10.1007/BF02478929
24. Saeger W, Puchner MJ, Lüdecke DK. Combined sellar gangliocytoma and pituitary adenoma in acromegaly or cushing’s disease. a report of 3 cases. Virchows Archiv: an Int J pathol (1994) 425(1):93–9. doi: 10.1007/bf00193956
25. Sakata K, Fujimori K, Komaki S, Furuta T, Sugita Y, Ashida K, et al. Pituitary gangliocytoma producing TSH and TRH: A review of “Gangliocytomas of the sellar region”. J Clin Endocrinol Metab (2020) 105(10):3109–21. doi: 10.1210/clinem/dgaa474
26. Bódi I, Martin AJ, Connor SE, Thomas NW, Lantos PL. Mixed pituitary gangliocytoma/adenoma (prolactinoma) with histogenetic implications. Neuropathol Appl neurobiol (2002) 28(3):252–5. doi: 10.1046/j.1365-2990.2002.00392.x
27. Tang F, Barbacioru C, Wang Y, Nordman E, Lee C, Xu N, et al. mRNA-seq whole-transcriptome analysis of a single cell. Nat Methods (2009) 6(5):377–82. doi: 10.1038/nmeth.1315
28. Jovic D, Liang X, Zeng H, Lin L, Xu F, Luo Y. Single-cell RNA sequencing technologies and applications: A brief overview. Clin Trans Med (2022) 12(3):e694. doi: 10.1002/ctm2.694
29. Melmed S. Pituitary-tumor endocrinopathies. New Engl J Med (2020) 382(10):937–50. doi: 10.1056/NEJMra1810772
30. Barry S, Korbonits M. Update on the genetics of pituitary tumors. Endocrinol Metab Clinics North America (2020) 49(3):433–52. doi: 10.1016/j.ecl.2020.05.005
Keywords: mixed gangliocytoma-adenoma, pituitary tumors, acromegaly, single-cell transcriptome sequencing, case report
Citation: Li C, Feng D and Zhou D (2022) Case report: Clinical and single-cell transcriptome sequencing analysis of a mixed gangliocytoma-adenoma presenting as acromegaly. Front. Oncol. 12:1088803. doi: 10.3389/fonc.2022.1088803
Received: 03 November 2022; Accepted: 23 November 2022;
Published: 08 December 2022.
Edited by:
Zhaolun Cai, Sichuan University, ChinaReviewed by:
Wanchun Zhu, Shanghai Jiao Tong University, ChinaMaogui Li, Qilu Hospital, Shandong University, China
Zhaohui Zhu, Peking Union Medical College Hospital (CAMS), China
Copyright © 2022 Li, Feng and Zhou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dabiao Zhou, emhvdWRhYmlhb0BianR0aC5vcmc=; Daqin Feng, MTM4MDc3MTM5MThAMTYzLmNvbQ==