 Jens von den Driesch1†
Jens von den Driesch1† Jana Flöttmann1†
Jana Flöttmann1† Friedrich Prall2
Friedrich Prall2 Christina S. Mullins1
Christina S. Mullins1 Michael Linnebacher1*
Michael Linnebacher1* Florian Bürtin1
Florian Bürtin1- 1Clinic of General, Visceral, Vascular and Transplantation Surgery, University Medical Center Rostock, University of Rostock, Rostock, Germany
- 2Institute of Pathology, University Medical Center Rostock, University of Rostock, Rostock, Germany
Introduction: Medullary pancreatic carcinoma (MPC) is a rare subtype of pancreatic ductal adenocarcinoma. MPCs represent less than 1% of all pancreatic cancers, and, with only 26 cases in the literature, knowledge regarding drug response and treatment outcome is very limited.
Material and methods: We present the case of a 64-year-old male patient with MPC who was treated by left pancreatic resection and adjuvant chemotherapy. Due to local recurrence, the patient underwent intended curative reoperation. From both surgical specimens, patient-derived xenografts (PDXs) and, from the recurrence, a patient-derived cell line (PDCL) were established. We subsequently performed an in-depth characterization of this cell line including phenotypic characterization, surface protein expression, growth, and migratory performance as well as mutational analysis using whole-exome sequencing (WES). Additionally, in vitro drug sensitivity toward the standard-of-care chemotherapeutic regimen and selected targeted therapies was evaluated.
Results: The pathological and molecular properties of this rare MPC case observed in the patient’s tumors are preserved in the corresponding PDX and the PDCL of HROP68Tu2. Despite displaying an “immunogenic phenotype” with marked T-cell infiltration and a high-level expression of HLA II and Programmed death-ligand 1 (PD-L1), molecular analysis revealed microsatellite stability but a multitude of mutations affecting KRAS, TP53, KAT6B, FOXG1, RUNX1, and GRIK2 among others. Furthermore, HROP68Tu2 cells were susceptible toward 5-FU, irinotecan, oxaliplatin, gemcitabine, paclitaxel, and erlotinib as single agents, but only a moderate synergistic response was seen to the drugs of the FOLFIRINOX regimen. Even worse, the drugs of the two combinations gemcitabine plus paclitaxel and gemcitabine plus erlotinib showed antagonistic effects. Moreover, lapatinib, PRIMA-Met1, and olaparib selected as targeted therapeutics according to the mutational profiles and protein expression inhibited HROP68Tu2 cells’ growth.
Conclusion: This study illustrates the establishment of the first preclinical MPC models as well as the first in-depth characterization of an MPC PDCL. Since the scientific and clinical knowledge of this rare pancreatic cancer type is very limited, the presented models contribute to a better understanding of MPC and might be a valuable tool for the development of future treatment options.
1 Introduction
Pancreatic ductal adenocarcinoma (PDAC) is the fourth leading cause of cancer-related death in the United States and the seventh worldwide (1, 2). Despite an increasing incidence, especially in countries with a high human development index, the prognosis remains dismal with reported 5-year survival rates ranging from 4.2% to 10% (1–3). Since most patients present with advanced cancer stages, only 10%–16% of the patients are eligible for resection, currently the only curative treatment option. Yet, those treated by surgery also show a poor prognosis with a median survival of 18 months and a 5-year survival rate of 15%–25% due to early local recurrence or metastasis (1, 4, 5).
Recent survival improvements are mainly attributed to intensified adjuvant and palliative chemotherapy regimens. In the adjuvant setting, modified FOLFIRINOX, a drug regimen comprised of folinic acid, fluorouracil (5-FU), irinotecan, and oxaliplatin, as well as the combination of gemcitabine with capecitabine has shown improved survival compared to gemcitabine monotherapy (6, 7). In locally advanced or metastatic stages, FOLFIRINOX and gemcitabine plus nanoparticle-bound paclitaxel are first-line treatment options (8). Nevertheless, most PDACs are characterized by a markedly low susceptibility to chemotherapy mediated by a specific tumor microenvironment with a dense desmoplastic reaction and the infiltrates of immune-suppressive cell populations as well as by a low tumor mutational burden (TMB) (9–11). In contrast, pancreatic medullary carcinoma (MPC), a rare subtype of PDAC, appears pathologically cell rich and low in stroma and is often characterized by a high TMB and microsatellite instability (12, 13). To the best of our knowledge, our group established the first patient-derived xenograft (PDX) and patient-derived cell line (PDCL) from a recurrent medullary pancreatic carcinoma, named HROP68Tu2, with distinct clinical, pathological, and molecular characteristics. In addition to the morphological and molecular characterization of the PDCL, the response to therapeutic agents was evaluated in vitro. 5-FU, irinotecan, oxaliplatin, gemcitabine, paclitaxel, and erlotinib were tested as single agents and in the clinically established combinations FOLFIRINOX, gemcitabine combined with paclitaxel (GemPac), and gemcitabine plus erlotinib (GemErlo). Since whole exome sequencing (WES) revealed a distinct mutational profile, and the tumor cells expressed high levels of HER2 and EGFR, the targeted drugs lapatinib, olaparib, and PRIMA-Met1 were additionally tested.
2 Materials and methods
2.1 Patient-derived xenograft and cell line establishment
The collection and processing of tumor tissue have been approved by the institutional review board of the University Medical Center Rostock (A 2018-0054). All animal experiments have been approved by the Landesamt für Landwirtschaft, Lebensmittelsicherheit und Fischerei Mecklenburg-Vorpommern under the registration numbers LALLF M-V/TSD/7221.3-1-007/19.
The tumor processing and establishment of PDX and PDCL were conducted as described previously (14, 15). In brief, after surgical en bloc resection of the tumor, a small tumor piece irrelevant for the evaluation of the resection margin was removed, cleaned, cut into cubes of 3 × 3 × 3 mm under sterile conditions, and cryopreserved immediately. Afterward, tumor specimens were thawed, incubated in Matrigel® (Corning, New York, USA), and subcutaneously implanted into the flanks of NSG mice. After reaching the required tumor size, mice were euthanized and PDX were explanted and cut accordingly for re-engraftment, histological examination, and snap freezing.
For cell line establishment, tumor tissue was mechanically dissected and passed through a 100 µm cell strainer. After centrifugation and resuspension in PBS, cells were seeded in a collagen-coated 6-well plate in a culture medium including antibiotics and antimycotics and incubated at 37°C in a humidified atmosphere of 5% CO2. As stable growth was observed, cells were transferred into a 25 cm2 culture flask with a standard culture medium (DMEM/F12 (1:1), 5% fetal calf serum (FCS), and 2 mM L-glutamine; all cell culture reagents were from PAN-Biotech, Aidenbach, Germany. Cells were regularly tested for mycoplasma contamination using the PlasmoTest™—Mycoplasma Detection Kit (In vivoGen, San Diego, CA, USA) according to the manufacturer’s protocol.
2.2 Short tandem repeat analysis
The concordance of HROP68Tu2 PDCL cells and healthy donor tissue was confirmed by short tandem repeat (STR) analysis as previously described (14). In short, DNA from donor tissue and tumor cells was isolated and the fragments of D5S818, D7S820, D16S539, D13S317, vWA, TPOX, THO1, CSF1PO, and Amelogenin were PCR-amplified with fluorescence-labeled primers. Subsequently, samples were size-separated and analyzed by automated capillary electrophoresis (Thermo Fisher Scientific, Waltham, MA, USA) (14).
2.3 Growth and migration
For doubling time determination, 4 × 104 HROP68Tu2 cells were seeded in a coated 24-well plate and allowed to attach for 72 h. To determine the mass of the vital cells, a crystal violet assay was conducted as described previously (16). Cells were stained with 0.2% crystal violet every 24 h in quadruplicates for six consecutive days and absorption was measured at 540 nm using the plate reader Tecan Infinite (Tecan, Männedorf, Switzerland). Migration speed was determined by the scratch assay. Cells grown to 100% confluency in a 6-well plate were switched to a serum-free medium and scratched with a 10 µl pipette tip. Three distances between edges were measured every 24 h for four consecutive days at 10× magnification using an inverted microscope (Carl Zeiss AG, Jena, Germany). The experiment was performed in independent triplicates.
2.4 Flow cytometry
The cell surface proteins of HROP68Tu2 cells were analyzed by flow cytometry using FACS Calibur (BD Bioscience, Franklin Lakes, NJ, USA) and open-source FCSalyser software. A panel of FITC-, PE-, and APC-conjugated antibodies was used for direct staining, targeting CD13, CD15, CD29, CD40, CD54, CD58, CD66adecb, CD71, CD86, CD95, HLA class I, HLA class II (Immunotools, Friesoythe, Germany); CD26, CD80, CD178 (eBioscience, Thermo Fisher Scientific, MA, USA); CD152, CD227 (Biolegend, San Diego, CA, USA); CD90 (Dianova, Eching, Germany); and CD326 (Miltenyi Biotech, Bergisch-Gladbach, Germany). Pembrolizumab (anti-PD-1), atezolizumab (anti-PD-L1), cetuximab (anti-EGFR), trastuzumab (anti-HER2), mesothelin (Biolegend, San Diego, CA, USA), and RP215 (Santa Cruz Biotechnologies, Dallas, TX, USA) were used as primary antibodies and labeled with FITC-conjugated polyclonal anti-human and anti-mouse IgG antibodies (Immunotools) for indirect staining.
2.5 Whole exome sequencing and microsatellite instability (MSI) score
The WES of the donor tumor tissue of HROP68 (primary and local recurrence as well as PDX models) was conducted by Centogene (Rostock, Germany) according to the protocol described by Trujillano and colleagues (17). In brief, DNA extraction was done using the QIAcube instrument with the QIAamp DNA Blood Mini QIAcube Kit (Qiagen, Hilden, Germany). In the exome, the target regions were amplified, and raw sequence data were analyzed. The HiSeq4000 platform (Illumina, San Diego, CA, USA) was used for sequencing. The used exome version was Centogene’s “CentoXome,” which covers approximately 33 Mb CCDS and is based on the Twist Bio Human Core Exome. Bioinformatic analysis was conducted as described by Matschos et al. (14). Pathogenetic classification was verified by an automated query to ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/ accessed on 23.09.2022) followed by manual evaluation.
The MSI status of HROP68Tu2 was assessed using MSIsensor as described (18), a program that automatically detects somatic microsatellite changes, based on standard tumor-normal paired next- generation sequencing data. The number of somatic homopolymers and microsatellite sites is compared to the total number of sites. A percentage of 3.5%–20% somatic mutations is considered MSI-Low (MSI-L) and >20% MSI-High (MSI-H) (18).
2.6 In vitro drug response
The triplicates of HROP68Tu2 cells were seeded into a 96-well plate (1 × 104). After 24 h, chemotherapeutics were added with a serial dilution technique, sparing healthy controls. The medium and drugs, respectively, were changed after 96 h to avoid deterioration effects. After 168 h, the medium was discarded, and cells were rinsed with PBS and analyzed by the crystal violet assay as described above. All assays were performed in independent triplicates. Lapatinib, PRIMA-1Met and olaparib (Hölzel Diagnostika, Köln, Germany), 5-FU, irinotecan, oxaliplatin, gemcitabine, paclitaxel, and erlotinib as single agents plus the combinations FOLFIRINOX, GemPac, and GemErlo in molar ratios analogous to the clinical regimen were tested (FOLFIRINOX: 1.07 folinic acid, 1.00 irinotecan, 80.95 5-FU, 0.80 oxaliplatin; GemPac: 1.00 gemcitabine, 0.04 paclitaxel (19). GemErlo: 1.00 gemcitabine, 0.21 erlotinib (20)). Individual interactions between 5-FU, irinotecan, and oxaliplatin were not assessed as monotherapy or other combinations are not recommended for clinical use. To evaluate the drug interactions, combination indices (CIs) (CI > 1.3 indicates antagonism, and CI = 0.6–0.8 indicates moderate synergism (19)) were calculated using the following equation (19):
2.7 Statistics
IC50 and IC20 values were calculated and analyzed by an unpaired Student’s t-test using Graph Pad Prism 5 (Graph Pad Software, San Diego, CA, USA). P-values ≤ 0.05 were considered statistically significant.
3 Results
3.1 Clinical case
A 64-year-old man presented with an excessive weight loss of 13 kg and fatigue during the last 5 months. Contrast-enhanced computed tomography (CEPT) revealed a large tumor of the pancreatic tail (Figure 1), and the patient was referred to our clinic for primary resection. He had no history of smoking, alcohol abuse, or prior cancer and neither did his first-degree relatives. Pancreatic left resection with splenectomy removed a soft and fragile, encapsulated tumor mass. Adjuvant treatment comprised of two cycles modified FOLFIRINOX, subsequently switched to five cycles of gemcitabine due to 5-FU-induced coronary vasospasm. Follow-up CEPT exhibited local recurrence in the left upper abdomen 11 months after resection. Since no distant metastasis was diagnosed, radical en bloc resection including the left colonic flexure and adrenal gland was performed. Pathological examination stated the completeness of resection and confirmed the origin of this second tumor from the previously resected MPC. The patient presented with severe disorientation and motor aphasia 2 weeks after discharge. Brain magnetic resonance imaging revealed occipital and parietal metastases, which were then treated by stereotactic radiotherapy. Subsequently, palliative systemic therapy was commenced, adding up to four cycles of gemcitabine plus nab-paclitaxel and one cycle of gemcitabine monotherapy in total. Although additional treatment with pembrolizumab was scheduled, the patient succumbed early to rapidly progressive cerebral metastases 23 months after the primary resection.
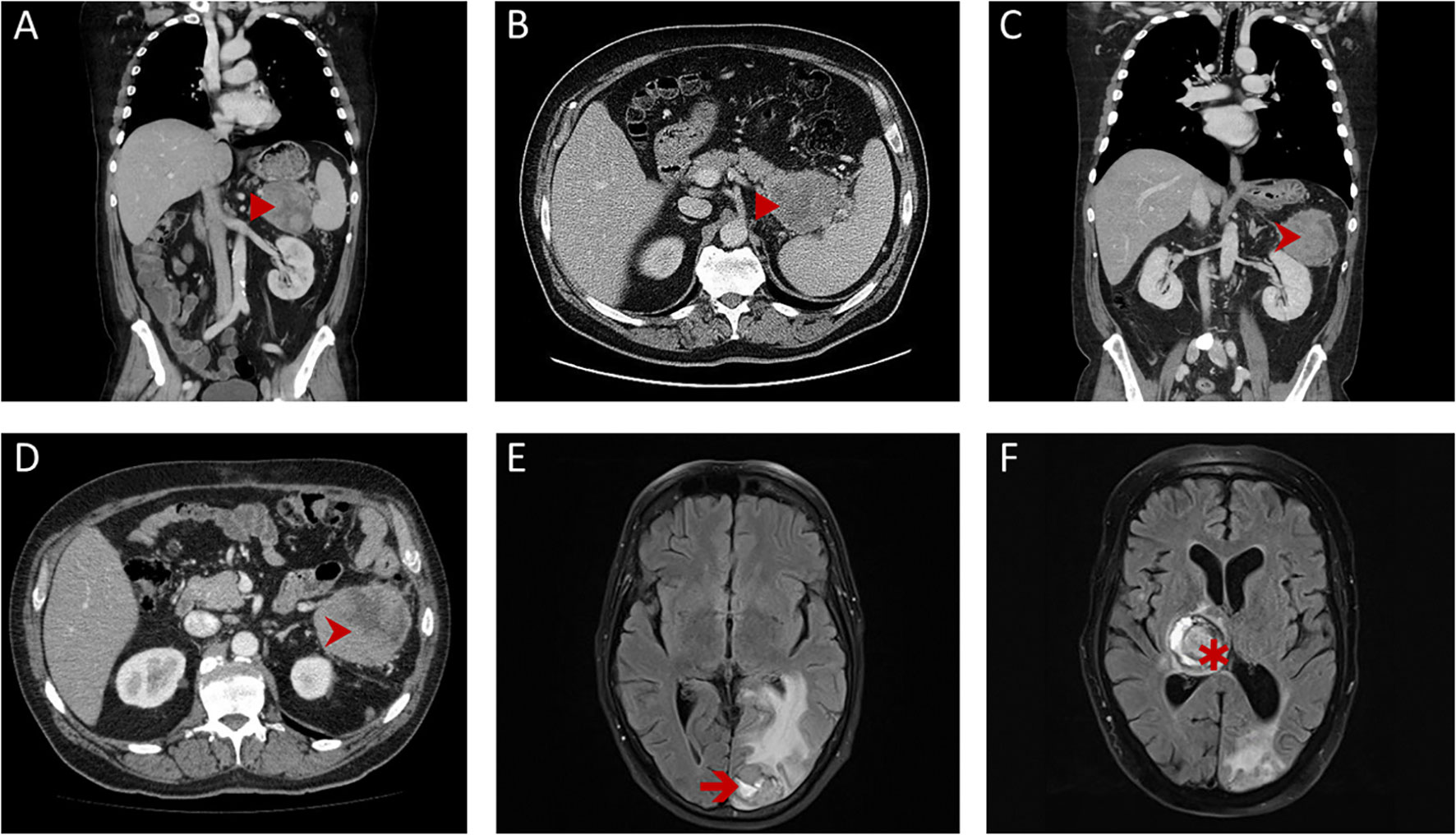
Figure 1 Radiological findings. First contrast-enhanced computed tomography (CEPT; A, B) of the thorax and abdomen revealed a large tumor (►) of the pancreatic tail, close to the splenic hilum with a pseudo-encapsulated appearance. Follow-up CEPT (C, D) showed isolated local recurrence (➤) with similar features in the left-upper abdomen and the retroperitoneum. Repeated magnetic resonance imaging scans of the brain showed a left occipital metastasis (➔) shortly after resection for local recurrence (E) and a rapidly progressing metastasis of the thalamus region (✱) over the course of palliative systemic treatment (F).
3.2 Pathology
The histologic examination of the primary tumor led to a diagnosis of MPC: the tumor was composed of solid sheets and nests of large polygonal neoplastic epithelial cells’ paucity of stromal components with polymorphous vesicular nuclei (Figure 2) in a stroma-poor background. Tumor borders were pushing and sharply delineated, and a brisk lymphocytic and histiocytic infiltrate was seen. A high mitotic index of 80%–90% and fairly large areas of tumor necrosis attested to a rapidly growing neoplasm. The tumor cells were immunohistochemically positive using a pan-cytokeratin cocktail (AE1/3) as well as Ber-EP4 and CK7 positive, whereas CK20, CDX2, HMB45, Hepar-1, LCA, and AFP immunostains were negative. Mismatch repair protein MLH1 and MSH2 expression was retained, molecular MSI testing (Bethesda markers plus the mononucleotide marker CAT25 (21) additionally confirmed a microsatellite-stable tumor. PD-L1 (clone 22C3) immunostaining was positive in approximately 20% of the tumor cells, albeit membranous immunostaining was weak, mostly; the tumor-infiltrating lymphocytes and histiocytes were PD-L1 positive throughout. This allowed the conclusion that this MPC was immunogenic.
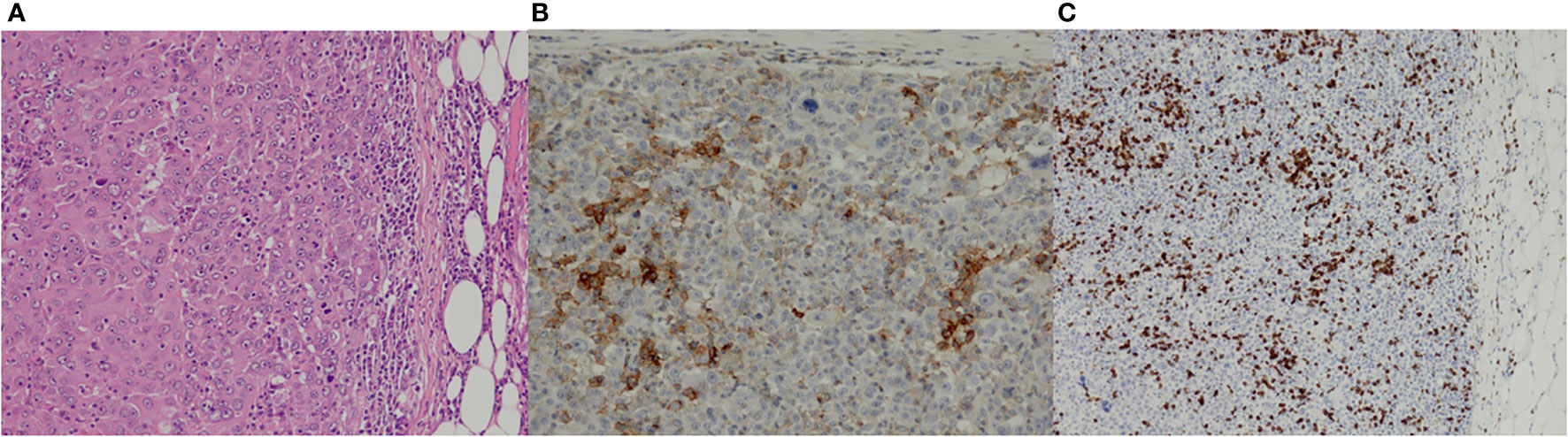
Figure 2 Pathological findings of the primary cancer. Hematoxylin–eosin stain (A; ×10) showed the solid sheets and nests of large polygonal neoplastic epithelial cells with a paucity of stromal components. Immunostaining for PD-L1 (B; ×20) and CD3 (C; ×10) reflected the immunogenic properties of the tumor.
3.3 Patient-derived xenograft establishment
Successful engraftment, defined as tumor outgrowth to a target volume of 1,500 mm3, was observed after the implantation of the primary resected tumor (HROP68Tu1) as well as the local recurrence (HROP68Tu2). In both cases, two out of two mice showed tumor outgrowth in at least one flank. HROP68Tu2 reached the target size 168 days after implantation. The growth kinetics of HROP86Tu2 compared well to those of other successfully engrafted pancreatic PDXs (n = 12) established from our group (Figure 3A). To confirm histopathological congruency with the original tumor and to exclude a potential murine lymphoma, a common pitfall in xenograft development, histologic sections (hematoxylin–eosin and PD-L1 immunostaining) were reviewed by an experienced pathologist (Figures 3B, C).
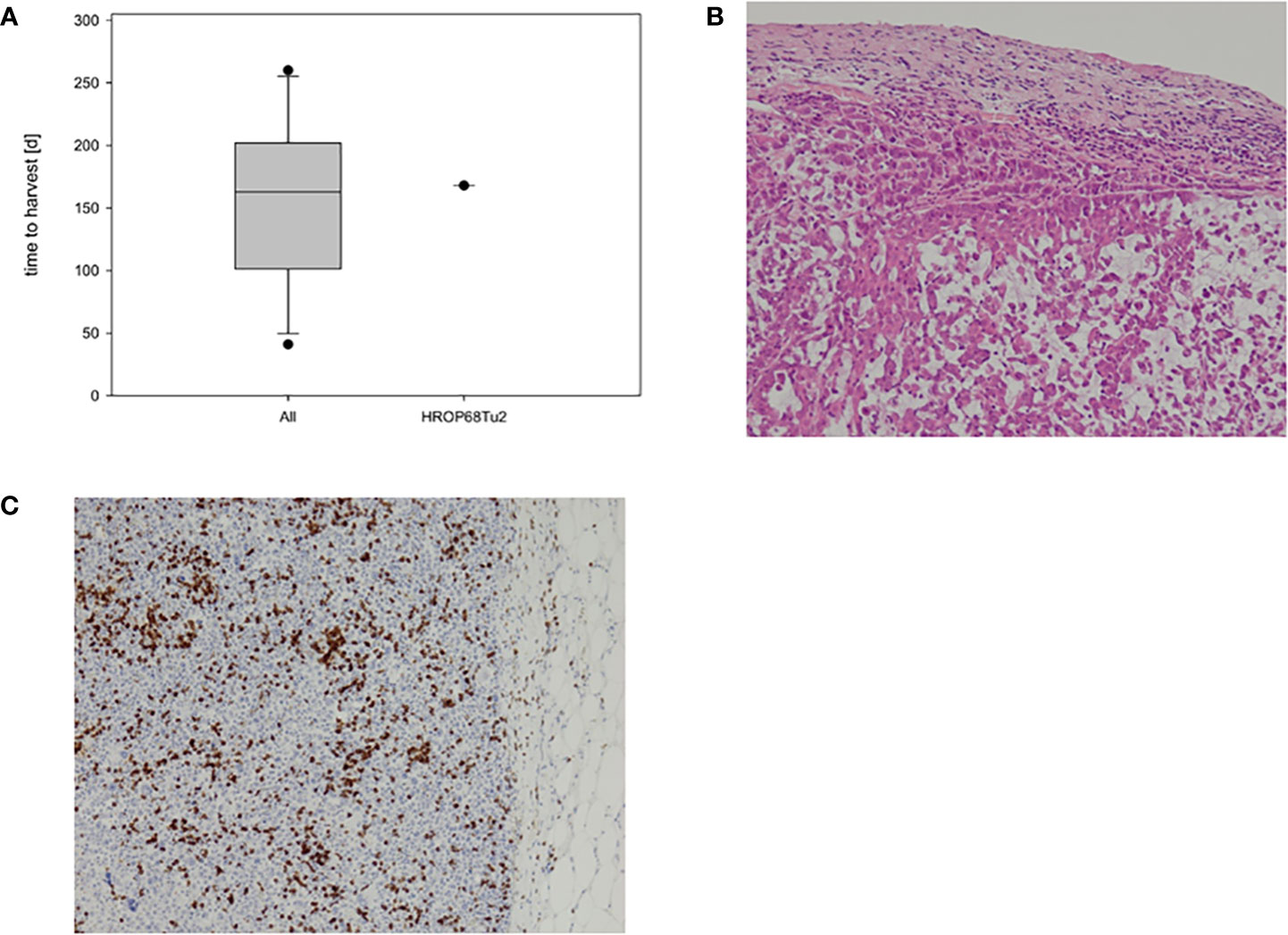
Figure 3 HROP68Tu2 PDX. Growth kinetics of HROP68Tu2 (A) compared to other pancreatic PDXs with successful outgrowth in passage fT0 (n = 12). The in vivo growth of HROP68Tu2 did not differ from the median growth rate of other pancreatic PDX. The hematoxylin–eosin stain of the PDX (B; 20x) revealed a close resemblance to the original donor tumor (B). Tumor cells showed strong immunostaining for PD-L1 (C).
3.4 Morphology, growth kinetics, and migration of HROP68Tu2 cell line
A permanent 2-dimensional (2-D) cell line was established by immediate incubation of freshly resected tissue obtained from the secondary surgery (HROP68Tu2) as described before (22). After adaption to standard culture conditions, HROP68Tu2 cells were passaged more than 40 times as a permanent cell line. Cells were regularly tested negative for mycoplasma contamination. Cross-contamination or mix-up was excluded by STR analysis, confirming the donor–patient origin. HROP68Tu2 cells grew evenly as an adherent monolayer with heterogeneous cell size and morphology (Figure 4). Doubling time in the exponential growth phase was 89.56 h ( ± 17.46). Morphology and growth did not change significantly during 40 consecutive passages.
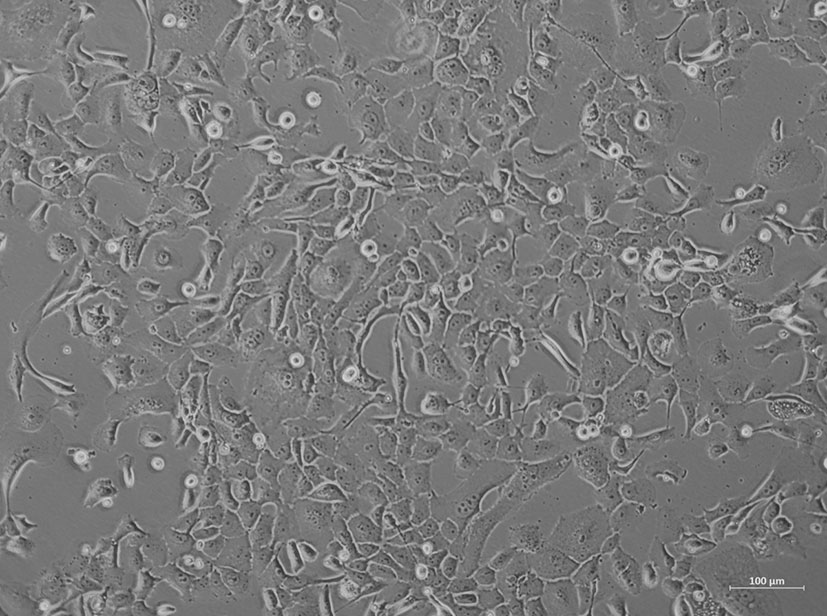
Figure 4 Cell morphology of the primary cell line HROP68Tu2 in passage 11. Picture was taken using an inverted microscope at ×40 magnification.
Cells migrated with 1.78 µm/h ( ± 0.84) in a classical scratch assay. The migratory behavior appeared to be invasive as cells detached from the scratch’s edges and migrated into the scratch as single cells (Figure 5).

Figure 5 Scratch assay. Cells were scratched using a 10 µl pipet tip. Pictures were taken using an inverted microscope at ×10 magnification.
3.5 Flow cytometric characterization
HROP68Tu2 cells´ expressions of surface proteins were evaluated by flow cytometry (Figure 6). Common epithelial and tumor markers were expressed to a variable extent: CD326 (EpCAM) (98.6% ± 1.7, MFI: 40.6 ± 3.1), CD227 (MUC-1) (58.4% ± 2.8, MFI: 7.8 ± 0.1), and CD66adecb (CEA) (38.2% ± 0.2, MFI: 5.0 ± 0.2). However, the epithelial markers CD15 (1.3% ± 0.6, MFI: 0.4 ± 0.3) and CD26 (7.3% ± 1.2, MFI: 0.4 ± 0.2) could not be detected. The adhesion marker CD29 (Integrin β - 1) was highly expressed (99.81% ± 0.01, MFI: 221.28 ± 19.23). A common pattern of the Fas receptor (CD95) (65.3% ± 11.6, MFI: 1.7 ± 0.3) and aberrant Fas ligand (CD178) (2.5% ± 0.9, MFI: 0.7 ± 0.3) expression was detected. HLA I expression was preserved homogeneously and with high intensity (99.7% ± 0.1, MFI: 165.6 ± 38.4). An average of 40.9% (± 12.0) of HROP68Tu2 cells expressed HLA II, albeit with low intensity (MFI: 1.8 ± 0.3). Furthermore, the cells expressed several proteins that are targets of antibody-based therapies. Matching the pathology report of the patient tumor, but even more so of the PDX, HROP68Tu2 cells showed homogeneous and high PD-L1 expression (95.9% ± 2.2, MFI: 26.5 ± 0.8). Furthermore, CD90 (84.0% ± 3.2, MFI: 5.1 ± 0.6) and CD40 (TNFRS5) (41.3 ± 1.83, MFI: 4.6 ± 0.3) expression was observed.
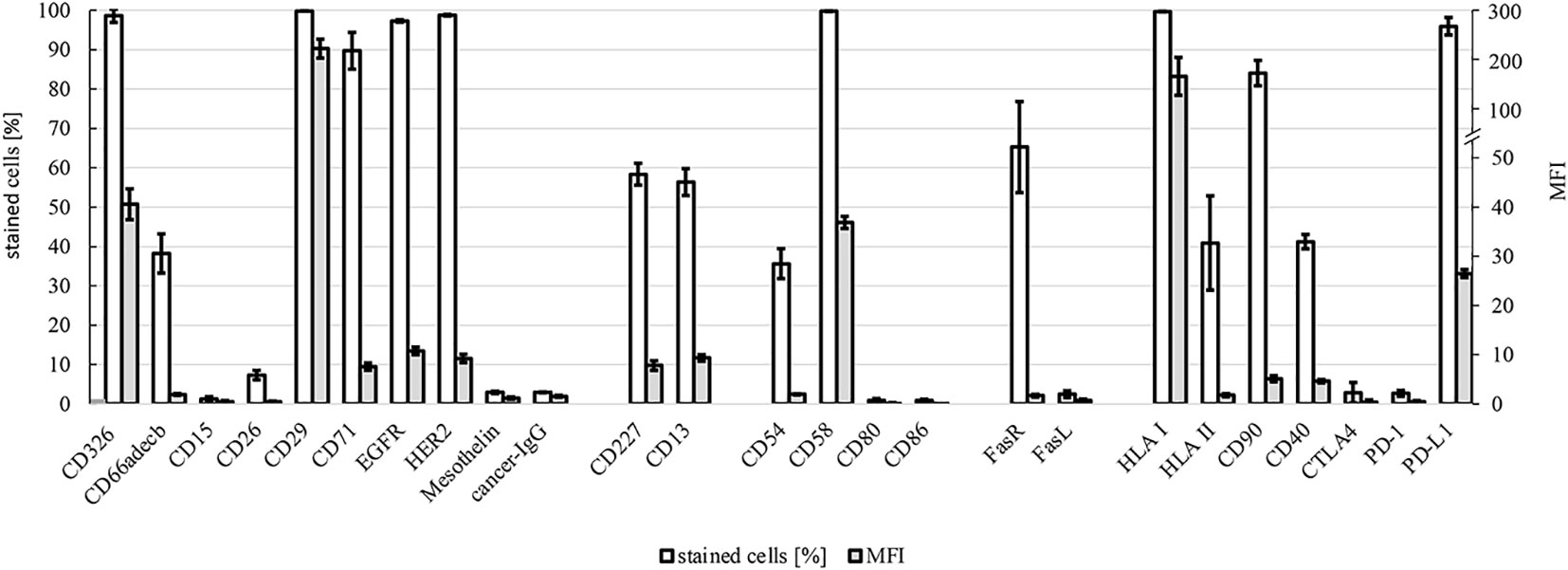
Figure 6 Flow cytometry. Expression of surface markers in HROP68Tu2 cells: tumor markers/signaling/proliferation (CD326, CD66adecb, CD15, CD26, CD29, CD71, EGFR, HER2, mesothelin, and cancer IgG), drug resistance (CD227 and CD13), adhesion/cell–cell interaction (CD54, CD58, CD80, and CD86), death ligand and receptor (CD95 and CD178), and immunogenicity/immunosuppression (HLA I, HLA II, CD90, CD40, CTLA4, PD-1, and PD-L1) were assessed by flow cytometry using FACS Calibur. Positively stained cells are given in % ± SEM and MFI (mean fluorescence intensity) ± SEM.
3.6 Whole exome sequencing: Somatic mutations and MSI scoring
The overall number of somatic mutations was determined by the WES analysis of HROP68 tumor tissues (primary cancer and local recurrence), subtracting alterations from the reference genome observed in the normal DNA of the patient. In sum, there was a relatively high TMB with 486 somatic mutations for HROP68Tu1 and 729 for HROPTu2, as well as an even higher number of 1,498 for HROP68Tu1 T1 M2 and 1,964 for HROP68Tu2 T0 M2. The higher numbers on the PDX models likely reflect the high tumor cell purity and possibly the very short ischemic time between collection and snap freezing. No mutations were observed in the following MMR genes: MLH1, MSH2, MSH6, PMS2, and EPCAM (CD326). In addition, we discovered a non-coding POLD1 mutation in the PDX tissue of HROP68Tu2. Furthermore, the WES revealed a high amount of frameshift mutations, a total number of 92 in the primary tumor and 113 in the local recurrence, HROP68Tu1 T1 M2: 192 and HROP68Tu2 T0 M2: 154.
As can be depicted from Table 1, several mutations classified as pathogenic affecting tumor-relevant genes including KRAS and TP53 could be identified. The mutations in BRCA1 and TP53 were subsequently included as targets in a precision oncology fashion sensitivity testing.

Table 1 Mutational profiling of the HROP68 primary tumor (HROP68Tu1), HROP68 recurrent tumor (HROP68Tu2), and their PDX models (HROP68Tu1 T1 M2 and HROP68Tu2 T0 M2).
Moreover, WES data were also used to calculate an MSI score, which was determined by MSIsensor to be 16.28% for the primary tumor and 13.77% for HROP68Tu2. In comparison: 23 other pancreatic cancer cases in our biobank had a mean MSI score of 3.09 (range 0.52 to 6.38). Taking the limit of MSIsensor for MSI-H of at least 20% into account, this analysis confirmed the absence of MSI-H, thereby matching the pathological findings. Still, when considering the relatively high TMB, the overall number of mutations reflecting frameshifts in coding genomic regions as identified by MSIsensor analysis, we consider HROP68 as an MPC with a low level of MSI (MSI-L).
The WES data obtained from Centogene as. vcf files are available upon reasonable request.
3.7 In vitro drug response
The treatment response of HROP68Tu2 was tested toward selected clinically approved therapeutics and several novel targeted drugs selected according to the observed somatic mutations and membrane receptors expressed by HROP68Tu2 cells. These were lapatinib due to the EGFR and HER2 expression, PRIMA-1Met because of the TP53 mutation, and, similarly, olaparib due to the BRCA1 mutation. HROP68Tu2 cells were sensitive toward all tested drugs within the therapeutic range as defined by achievable human plasma levels (Table 2).
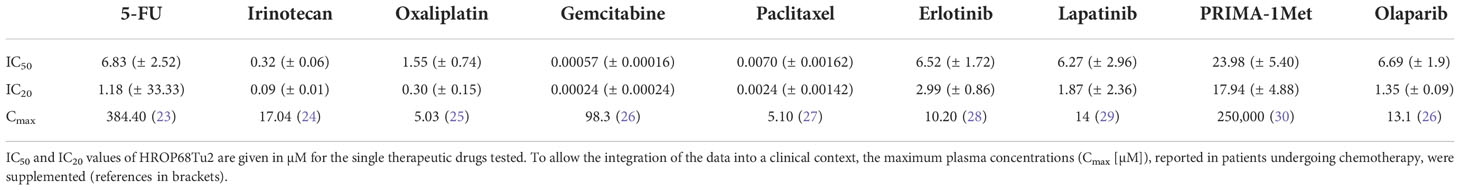
Table 2 Chemosensitivity of HROP68Tu2 toward single drugs: IC50 and IC20 values.
Next, the response to the clinically used combination therapy FOLFIRINOX was analyzed. Although a moderate synergistic effect of the drugs 5-FU, irinotecan, and oxaliplatin was observed, chemosensitivity to FOLFIRINOX compared to 5-FU alone was not significantly increased (p = 0.118). The interaction of the drugs involved can be judged by calculating a combination index (CI), which was in this case 0.71, thereby indicating a moderate synergistic effect for FOLFIRINOX (Table 3).
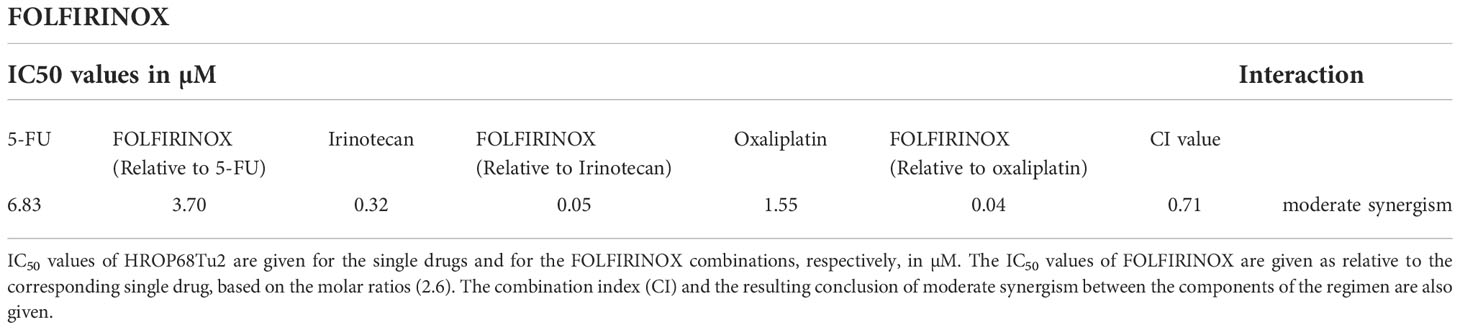
Table 3 Drug interaction of FOLFIRINOX.
Contrary, combining gemcitabine with paclitaxel or erlotinib did not improve inhibitory effects compared to gemcitabine alone but rather showed antagonistic effects between gemcitabine and paclitaxel (GemPac; CI = 1.44, p>0.05) as well as gemcitabine and erlotinib (GemErlo; CI = 1.53, p>0.05) (Table 4).

Table 4 Drug interaction of GemPac and GemErlo.
4 Discussion
Patient-derived tumor models are the cornerstone of preclinical and translational research (14, 15, 22) and are valuable tools for the evaluation of novel drugs or treatment options for rare diseases (16, 31). The consequent biobanking of all resected pancreatic cancers at our department provided the opportunity to seize this rare case of primary and recurrent MPC, as well as to establish PDX from both the surgical specimen and a PDCL. The establishment of stable cell lines derived directly from the fresh surgical PDAC specimen is rarely successful, which can be mostly attributed to a low tumor–stroma ratio frequently causing fibroblastic overgrowth (32, 33). The positive model establishments of HROP68 might thus be mostly attributable to the high tumor–stroma ratio diagnosed in this case.
Generally, the evaluation of treatment efficacy for the individual PDAC patient is challenging since long-term survival remains an exception. This is explained by the intrinsic chemoresistance of PDAC, attributable to the large and dense tumor stroma, decreasing microvascularity and thereby drug delivery, to the epithelial cancer cell nests embedded into this stroma (34, 35). Due to the paucity of stromal components in HROP68, this relationship is unlikely in this case and might explain the observed drug responses of the present study. Compared to published data from many long-term established PDAC cell lines, HROP68Tu2 cells were highly responsive to gemcitabine but only moderately to 5-FU. Moreover, a moderate synergism was observed between the components of the FOLFIRINOX regimen. Contrary to that, an antagonistic interaction was observed for the combination of gemcitabine with both paclitaxel and erlotinib. However, due to the rareness of MPC, the comparison of these results with clinical data or even the retrospective translation of the presented case into the clinical context is hardly possible.
PDAC are bona fide non-immunogenic due to their low TMB; only 1%–2% of PDAC exhibit high levels of MSI, which is typically associated with a hypermutated phenotype (36). However, the associations of MSI to a medullary and mucinous histology have been described (37). Despite the high level of T-cell infiltration, the hallmark of ongoing immune recognition, observed in HROP68, the case was diagnosed as MSS both by immunostaining with a retained expression of the mismatch repair proteins MLH1 and MSH2, as well as by molecular microsatellite stability testing. Somehow challenging this diagnosis, WES analysis, however, identified a proportionally large number of somatic mutations, including a relevant fraction of insertions and deletions leading to frameshift mutations in coding regions. The latter translates to an elevated score in the MSIsensor analysis, albeit not reaching the 20% threshold to allow formally judging the case as MSI-H. Mutations of other candidate genes associated with a high TMB, like POLE (38), POLD1 (39), and MUTYH (40), could not be detected in the clinical samples, although the PDX HROP68Tu2 T0 M2 harbored a POLD1 mutation of uncertain significance. By applying a less rigorous tertiary data interpretation of the WES analysis, likely pathogenic mutations of DNA repair genes, known to be associated with an increased TMB and immunogenicity, like PALB2, ATR, and CHECK2 (41–43), were identified. However, when summing up all pieces of information, the uncommonly high TMB (44), combined with the MSI score, led us to the conclusion that HROP68 is an MSI-L MPC.
Although mutations of BRCA1/2 in PDAC are associated with increased survival due to a higher susceptibility to platinum-based chemotherapy (45, 46), this observation could not be confirmed for HROP68, harboring a BRCA1 mutation. While BRCA-mutated ovarian cancers respond well to the inhibitors of the poly-ADP ribose (PARPi), the PARPi olaparib improved progression-free survival in metastatic PDAC patients with a germline BRCA mutation but failed to improve overall survival (47, 48). In our study, olaparib had an inhibitory effect on HROP68Tu2 cells in vitro.
Similarly successful, HROP68Tu2 cells showed marked susceptibility to the tyrosine kinase inhibitors erlotinib and lapatinib, which were selected as targeted agents due to the homogeneous and high expression of EGFR and HER2. In the clinical setting, erlotinib led to small survival benefits, but lapatinib recently failed to improve the survival of patients with metastatic PDAC (49, 50).
The TP53 mutation displayed by HROP68 is classified as uncertain significance in the ClinVar database. However, it might be pivotal regarding the proliferation of HROP68 cells since it replaces a cysteine on the protein level and this, together with the loss of the second allele, likely results in a loss of proper protein function. Our data of the PRIMA-Met1 treatment response support this hypothesis since this drugs’ ability to restore wild-type p53 conformation has been shown for several mutant p53 proteins (51). The clear impact of PRIMA-Met1 on HROP68Tu2 cells would thus deliver evidence for the pathogenic character of the Cys277Phe p53 mutation with the dbSNP-ID rs763098116.
The immune landscape and interplay between various cell types contributing to immune evasion and a pro-inflammatory microenvironment are complex and not fully understood (52). On one hand, HROP68Tu2 showed a robust and homogeneous expression of HLA I as well as the expression of HLA II, CD58 (LFA3), and CD54 (ICAM1), allowing recognition by antigen-specific immune cells. On the other hand, the homogenous expression of PD-L1, CD90, and CD40 probably facilitated T-cell inactivation and likely immune evasion. PD-L1 expression is uncommon in PDACs and can only be found in 3% of cases (53). Although Le et al. (54) reported promising results for the efficiency of the PD-L1-inhibitor pembrolizumab for the treatment of different solid, MSI-positive tumors, the tumor response to pembrolizumab in MSI-positive PDCAs was rather modest with 18% in the KEYNOTE-158 Study (54, 55). Due to the uncommon combination of proteins expressed by HROP68 tumor cells, we consider it likely that an early application of checkpoint inhibitors in the clinical course of this patient might have improved the outcome, at least the overall survival time. It is one of the weak points of the current study that autologous immune analyses have not (yet) been performed in vitro using the HROP68Tu2 cell line and the peripheral as well as tumor-infiltrating T cells of the patient. Such analyses are planned, but, due to the restricted number of T cells available in our biobank, they shall be combined with an analysis of tumor-specific antigenic epitopes presented by HROP68Tu2 cells.
In summary and to the best of our knowledge, the PDX and PDCL of HROP68Tu2 are the first preclinical models established from an MPC. Although their preclinical implications are limited by the rarity of MPC, they are perfect tools for the establishment of novel drugs in the context of this rare subset of PDAC. Moreover, the observed good in vitro responses toward several drugs tested also emphasize the necessity to evaluate therapeutic options on a patient–individual level, i.e., precision oncology, in order to optimize the clinical outcome, especially for rare cancer cases.
Data availability statement
The original contributions presented in the study are publicly available. The data has been uploaded to the NCBI BioProject repository and can be found using accession number PRJNA915478.
Ethics statement
The studies involving human participants were reviewed and approved by Institutional review board of the University Medical Center Rostock (A 2018-0054). The patients/participants provided their written informed consent to participate in this study. The animal study was reviewed and approved by Landesamt für Landwirtschaft, Lebensmittelsicherheit und Fischerei Mecklenburg-Vorpommern.
Author contributions
Design and conception of the study: ML. Patients’ consent and clinical material acquisition: FB, ML, CM, and FP. Experiments: JvdD, JF, FB, and FP. Manuscript draft: JvdD, JF, FB, FP, CM, and ML. All authors contributed to the article and approved the submitted version.
Funding
This research was, in part, funded by grant number TBI-V-1-241-VBW-084 and TBI-V-240-VBW-084 from the state of Mecklenburg-Vorpommern.
Acknowledgments
The authors thank Mathias Krohn for his excellent help with cell culture and performing the STR analysis. Furthermore, we would like to thank Centogene (Rostock, Germany) for performing whole exome sequencing.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2021. CA Cancer J Clin (2021) 71(1):7–33. doi: 10.3322/caac.21654
2. Arnold M, Abnet CC, Neale RE, Vignat J, Giovannucci EL, McGlynn KA, et al. Global burden of 5 major types of gastrointestinal cancer. Gastroenterology (2020) 159(2):335–349.e15. doi: 10.1053/j.gastro.2020.02.068
3. Bengtsson A, Andersson R, Ansari D. The actual 5-year survivors of pancreatic ductal adenocarcinoma based on real-world data. Sci Rep (2020) 10(1):16425. doi: 10.1038/s41598-020-73525-y
4. Latenstein AEJ, van Roessel S, van der Geest LGM, Bonsing BA, Dejong CHC, Groot Koerkamp B, et al. Conditional survival after resection for pancreatic cancer: A population-based study and prediction model. Ann Surg Oncol (2020) 27(7):2516–24. doi: 10.1245/s10434-020-08235-w
5. Huang L, Jansen L, Balavarca Y, Babaei M, van der Geest L, Lemmens V, et al. Stratified survival of resected and overall pancreatic cancer patients in Europe and the USA in the early twenty-first century: a large, international population-based study. BMC Med (2018) 16(1):125. doi: 10.1186/s12916-018-1120-9
6. Conroy T, Hammel P, Hebbar M, Ben Abdelghani M, Wei AC, Raoul J-L, et al. FOLFIRINOX or gemcitabine as adjuvant therapy for pancreatic cancer. N Engl J Med (2018) 379(25):2395–406. doi: 10.1056/NEJMoa1809775
7. Neoptolemos JP, Palmer DH, Ghaneh P, Psarelli EE, Valle JW, Halloran CM, et al. Comparison of adjuvant gemcitabine and capecitabine with gemcitabine monotherapy in patients with resected pancreatic cancer (ESPAC-4): a multicentre, open-label, randomised, phase 3 trial. Lancet (2017) 389(7):1011–24. doi: 10.1016/S0140-6736(16)32409-6
8. Tempero MA, Malafa MP, Al-Hawary M, Behrman SW, Benson AB, Cardin DB, et al. Pancreatic adenocarcinoma, version 2.2021, NCCN clinical practice guidelines in oncology. J Natl Compr Canc Netw (2021) 19(4):439–57. doi: 10.6004/jnccn.2021.0017
9. Martinez-Useros J, martin-galan m, Garcia-foncillas j. the match between molecular subtypes, histology and microenvironment of pancreatic cancer and its relevance for chemoresistance. Cancers (Basel) (2021) 13(2):322. doi: 10.3390/cancers13020322
10. Blando J, Sharma A, Higa MG, Zhao H, Vence L, Yadav SS, et al. Comparison of immune infiltrates in melanoma and pancreatic cancer highlights VISTA as a potential target in pancreatic cancer. Proc Natl Acad Sci U.S.A. (2019) 116(5):1692–7. doi: 10.1073/pnas.1811067116
11. Chalmers ZR, Connelly CF, Fabrizio D, Gay L, Ali SM, Ennis R, et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med (2017) 9(11):34. doi: 10.1186/s13073-017-0424-2
12. Goggins M, Offerhaus GJ, Hilgers W, Griffin CA, Shekher M, Tang D, et al. Pancreatic adenocarcinomas with DNA replication errors (RER+) are associated with wild-type K-ras and characteristic histopathology. poor differentiation, a syncytial growth pattern, and pushing borders suggest RER+. Am J Pathol (1998) 152(6):1501–7.
13. Wilentz RE, Goggins M, Redston M, Marcus VA, Adsay NV, Sohn TA, et al. Genetic, immunohistochemical, and clinical features of medullary carcinoma of the pancreas. Am J Pathol (2000) 156(5):1641–51. doi: 10.1016/S0002-9440(10)65035-3
14. Matschos S, Bürtin F, Kdimati S, Radefeldt M, Krake S, Prall F, et al. The HROC-Xenobank-A high quality assured PDX biobank of 100 individual colorectal cancer models. Cancers (Basel) (2021) 13(23):5882. doi: 10.3390/cancers13235882
15. Bürtin F, Matschos S, Prall F, Mullins CS, Krohn M, Linnebacher M. Creation and maintenance of a living biobank - how we do it. J Vis Exp (2021) 170:170. doi: 10.3791/62065
16. Wagner S, Beger NT, Matschos S, Szymanski A, Przybylla R, Bürtin F, et al. Tumour-derived cell lines and their potential for therapy prediction in patients with metastatic colorectal cancer. Cancers (Basel) (2021) 13(18):4717. doi: 10.3390/cancers13184717
17. Trujillano D, Bertoli-Avella AM, Kumar Kandaswamy K, Weiss ME, Köster J, Marais A, et al. Clinical exome sequencing: results from 2819 samples reflecting 1000 families. Eur J Hum Genet (2017) 25(2):176–82. doi: 10.1038/ejhg.2016.146
18. Niu B, Ye K, Zhang Q, Lu C, Xie M, McLellan MD, et al. MSIsensor: microsatellite instability detection using paired tumor-normal sequence data. Bioinformatics (2014) 30(7):1015–6. doi: 10.1093/bioinformatics/btt755
19. Begg SKS, Birnbaum DJ, Clark JW, Mino-Kenudson M, Wellner UF, Schilling O, et al. FOLFIRINOX versus gemcitabine-based therapy for pancreatic ductal adenocarcinoma: Lessons from patient-derived cell lines. Anticancer Res (2020) 40(7):3659–67. doi: 10.21873/anticanres.14355
20. Moore MJ, Goldstein D, Hamm J, Figer A, Hecht JR, Gallinger S, et al. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the national cancer institute of Canada clinical trials group. J Clin Oncol (2007) 25(15):1960–6. doi: 10.1200/JCO.2006.07.9525
21. Bianchi F, Galizia E, Catalani R, Belvederesi L, Ferretti C, Corradini F, et al. CAT25 is a mononucleotide marker to identify HNPCC patients. J Mol Diagn (2009) 11(3):248–52. doi: 10.2353/jmoldx.2009.080155
22. Mullins CS, Micheel B, Matschos S, Leuchter M, Bürtin F, Krohn M, et al. Integrated biobanking and tumor model establishment of human colorectal carcinoma provides excellent tools for preclinical research. Cancers (Basel) (2019) 11(10):1520. doi: 10.3390/cancers11101520
23. Bocci G, Danesi R, Di Paolo AD, Innocenti F, Allegrini G, Falcone A, et al. Comparative pharmacokinetic analysis of 5-fluorouracil and its major metabolite 5-fluoro-5,6-dihydrouracil after conventional and reduced test dose in cancer patients. Clin Cancer Res (2000) 6(8):3032–7.
24. Chabot GG. Clinical pharmacokinetics of irinotecan. Clin Pharmacokinet (1997) 33(4):245–59. doi: 10.2165/00003088-199733040-00001
25. Graham MA, Lockwood GF, Greenslade D, Brienza S, Bayssas M, Gamelin E. Clinical pharmacokinetics of oxaliplatin: a critical review. Clin Cancer Res (2000) 6(4):1205–18.
26. Liston DR, Davis M. Clinically relevant concentrations of anticancer drugs: A guide for nonclinical studies. Clin Cancer Res (2017) 23(14):3489–98. doi: 10.1158/1078-0432.CCR-16-3083
27. Stage TB, Bergmann TK, Kroetz DL. Clinical pharmacokinetics of paclitaxel monotherapy: An updated literature review. Clin Pharmacokinet (2018) 57(1):7–19. doi: 10.1007/s40262-017-0563-z
28. Faivre L, Gomo C, Mir O, Taieb F, Schoemann-Thomas A, Ropert S, et al. A simple HPLC-UV method for the simultaneous quantification of gefitinib and erlotinib in human plasma. J Chromatogr B Analyt Technol BioMed Life Sci (2011) 879(23):2345–50. doi: 10.1016/j.jchromb.2011.06.026
29. Spector NL, Robertson FC, Bacus S, Blackwell K, Smith DA, Glenn K, et al. Lapatinib plasma and tumor concentrations and effects on HER receptor phosphorylation in tumor. PloS One (2015) 10(11):e0142845. doi: 10.1371/journal.pone.0142845
30. Bykov VJN, Zhang Q, Zhang M, Ceder S, Abrahmsen L, Wiman KG. Targeting of mutant p53 and the cellular redox balance by APR-246 as a strategy for efficient cancer therapy. Front Oncol (2016) 6:21. doi: 10.3389/fonc.2016.00021
31. Rivera M, Fichtner I, Wulf-Goldenberg A, Sers C, Merk J, Patone G, et al. Patient-derived xenograft (PDX) models of colorectal carcinoma (CRC) as a platform for chemosensitivity and biomarker analysis in personalized medicine. Neoplasia (2021) 23(1):21–35. doi: 10.1016/j.neo.2020.11.005
32. Xie D, Xie K. Pancreatic cancer stromal biology and therapy. Genes Dis (2015) 2(2):133–43. doi: 10.1016/j.gendis.2015.01.002
33. Rückert F, Aust D, Böhme I, Werner K, Brandt A, Diamandis EP, et al. Five primary human pancreatic adenocarcinoma cell lines established by the outgrowth method. J Surg Res (2012) 172(1):29–39. doi: 10.1016/j.jss.2011.04.021
34. Jacobetz MA, Chan DS, Neesse A, Bapiro TE, Cook N, Frese KK, et al. Hyaluronan impairs vascular function and drug delivery in a mouse model of pancreatic cancer. Gut (2013) 62(1):112–20. doi: 10.1136/gutjnl-2012-302529
35. Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D, Honess D, et al. Inhibition of hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science (2009) 324(5933):1457–61. doi: 10.1126/science.1171362
36. De' Angelis GL, Bottarelli L, Azzoni C, De' Angelis N, Leandro G, Di Mario F, et al. Microsatellite instability in colorectal cancer. Acta BioMed (2018) 89(9-S)::97–101. doi: 10.1186/s13045-020-00958-3
37. Luchini C, Brosens LAA, Wood LD, Chatterjee D, Shin JI, Sciammarella C, et al. Comprehensive characterisation of pancreatic ductal adenocarcinoma with microsatellite instability: histology, molecular pathology and clinical implications. Gut (2021) 70(1):148–56. doi: 10.1136/gutjnl-2020-320726
38. Kryklyva V, Ter Linden E, Kroeze LI, de Voer RM, van der Kolk BM, Stommel MWJ, et al. Medullary pancreatic carcinoma due to somatic POLE mutation: A distinctive pancreatic carcinoma with marked long-term survival. Pancreas (2020) 49(7):999–1003. doi: 10.1097/MPA.0000000000001588
39. He J, Ouyang W, Zhao W, Shao L, Li B, Liu B, et al. Distinctive genomic characteristics in POLE/POLD1-mutant cancers can potentially predict beneficial clinical outcomes in patients who receive immune checkpoint inhibitor. Ann Transl Med (2021) 9(2):129. doi: 10.21037/atm-20-7553
40. Robinson PS, Thomas LE, Abascal F, Jung H, Harvey LMR, West HD, et al. Inherited MUTYH mutations cause elevated somatic mutation rates and distinctive mutational signatures in normal human cells. Nat Commun (2022) 13(1):3949. doi: 10.1038/s41467-022-31341-0
41. Waddell N, Pajic M, Patch A-M, Chang DK, Kassahn KS, Bailey P, et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature (2015) 518(7540):495–501. doi: 10.1038/nature14169
42. Jiang M, Jia K, Wang L, Li W, Chen B, Liu Y, et al. Alterations of DNA damage response pathway: Biomarker and therapeutic strategy for cancer immunotherapy. Acta Pharm Sin B (2021) 11(10):2983–94. doi: 10.1016/j.apsb.2021.01.003
43. Parikh AR, He Y, Hong TS, Corcoran RB, Clark JW, Ryan DP, et al. Analysis of DNA damage response gene alterations and tumor mutational burden across 17,486 tubular gastrointestinal carcinomas: Implications for therapy. Oncologist (2019) 24(10):1340–7. doi: 10.1634/theoncologist.2019-0034
44. Qian Y, Gong Y, Fan Z, Luo G, Huang Q, Deng S, et al. Molecular alterations and targeted therapy in pancreatic ductal adenocarcinoma. J Hematol Oncol (2020) 13(1):130. doi: 10.1186/s13045-020-00958-3
45. Seeber A, Zimmer K, Kocher F, Puccini A, Xiu J, Nabhan C, et al. Molecular characteristics of BRCA1/2 and PALB2 mutations in pancreatic ductal adenocarcinoma. ESMO Open (2020) 5(6):e000942. doi: 10.1136/esmoopen-2020-000942
46. Pishvaian MJ, Blais EM, Brody JR, Lyons E, DeArbeloa P, Hendifar A, et al. Overall survival in patients with pancreatic cancer receiving matched therapies following molecular profiling: a retrospective analysis of the know your tumor registry trial. Lancet Oncol (2020) 21(4):508–18. doi: 10.1016/S1470-2045(20)30074-7
47. Miller RE, Leary A, Scott CL, Serra V, Lord CJ, Bowtell D, et al. ESMO recommendations on predictive biomarker testing for homologous recombination deficiency and PARP inhibitor benefit in ovarian cancer. Ann Oncol (2020) 31(12):1606–22. doi: 10.1016/j.annonc.2020.08.2102
48. Kindler HL, Hammel P, Reni M, van Cutsem E, Macarulla T, Hall MJ, et al. Overall survival results from the POLO trial: A phase III study of active maintenance olaparib versus placebo for germline BRCA-mutated metastatic pancreatic cancer. J Clin Oncol (2022) 6:JCO2101604. doi: 10.1200/JCO.21.01604
49. Safran H, Miner T, Bahary N, Whiting S, Lopez CD, Sun W, et al. Lapatinib and gemcitabine for metastatic pancreatic cancer. a phase II study. Am J Clin Oncol (2011) 34(1):50–2. doi: 10.1097/COC.0b013e3181d26b01
50. van Cutsem E, Vervenne WL, Bennouna J, Humblet Y, Gill S, van Laethem J-L, et al. Phase III trial of bevacizumab in combination with gemcitabine and erlotinib in patients with metastatic pancreatic cancer. J Clin Oncol (2009) 27(13):2231–7. doi: 10.1200/JCO.2008.20.0238
51. Bykov VJN, Issaeva N, Shilov A, Hultcrantz M, Pugacheva E, Chumakov P, et al. Restoration of the tumor suppressor function to mutant p53 by a low-molecular-weight compound. Nat Med (2002) 8(3):282–8. doi: 10.1038/nm0302-282
52. Goulart MR, Stasinos K, Fincham REA, Delvecchio FR, Kocher HM. T Cells in pancreatic cancer stroma. World J Gastroenterol (2021) 27(46):7956–68. doi: 10.3748/wjg.v27.i46.7956
53. Liang X, Sun J, Wu H, Luo Y, Wang L, Lu J, et al. PD-L1 in pancreatic ductal adenocarcinoma: a retrospective analysis of 373 Chinese patients using an in vitro diagnostic assay. Diagn Pathol (2018) 13(1):5. doi: 10.1186/s13000-017-0678-4
54. Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science (2017) 357(6349):409–13. doi: 10.1126/science.aan6733
55. Marabelle A, Le DT, Ascierto PA, Di Giacomo AM, de Jesus-Acosta A, Delord J-P, et al. Efficacy of pembrolizumab in patients with noncolorectal high microsatellite Instability/Mismatch repair-deficient cancer: Results from the phase II KEYNOTE-158 study. J Clin Oncol (2020) 38(1):1–10. doi: 10.1200/JCO.19.02105
Keywords: PDAC - pancreatic ductal adenocarcinoma, rare malignancy, medullary adenocarcinoma, precision medicine, patient-derived cell line
Citation: von den Driesch J, Flöttmann J, Prall F, Mullins CS, Linnebacher M and Bürtin F (2023) HROP68: A rare case of medullary pancreatic cancer—characterization and chemosensitivity of the first patient-derived cell line. Front. Oncol. 12:1082927. doi: 10.3389/fonc.2022.1082927
Received: 28 October 2022; Accepted: 23 November 2022;
Published: 20 January 2023.
Edited by:
Massimo Broggini, Mario Negri Institute for Pharmacological Research (IRCCS), ItalyReviewed by:
Michael Volkmar, Helmholtz Institute for Translational Oncology, German Cancer Research Center (DKFZ), GermanyAlfonso De Stefano, G. Pascale National Cancer Institute Foundation (IRCCS), Italy
Copyright © 2023 von den Driesch, Flöttmann, Prall, Mullins, Linnebacher and Bürtin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michael Linnebacher, TWljaGFlbC5saW5uZWJhY2hlckBtZWQudW5pLXJvc3RvY2suZGU=
†These authors have contributed equally to this work