
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Oncol. , 01 December 2022
Sec. Cancer Genetics
Volume 12 - 2022 | https://doi.org/10.3389/fonc.2022.1064817
This article is part of the Research Topic Case Reports in Cancer Genetics : 2022 View all 16 articles
 Yanming Chen1†
Yanming Chen1† Qing Zhu1†Ye Wang2Xiaoxiao Dai3Ping Chen4Ailin Chen1Sujuan Zhou4,5Chungang Dai1Shengbin Zhao4
Qing Zhu1†Ye Wang2Xiaoxiao Dai3Ping Chen4Ailin Chen1Sujuan Zhou4,5Chungang Dai1Shengbin Zhao4 Sheng Xiao6
Sheng Xiao6 Qing Lan1*
Qing Lan1*Neurotrophic tyrosine receptor kinase (NTRK) rearrangements are oncogenic drivers of various types of adult and pediatric tumors, including gliomas. However, NTRK rearrangements are extremely rare in glioneuronal tumors. Here, we report a novel NTRK2 rearrangement in a 24-year-old female with dysembryoplastic neuroepithelial tumor (DNT), a circumscribed WHO grade I benign tumor associated with epilepsy. By utilizing targeted RNA next-generation sequencing (NGS), fluorescence in situ hybridization (FISH), reverse transcriptase PCR (RT-PCR), and Sanger sequencing, we verified an in-frame fusion between NTRK2 and the lipoma HMGIC fusion partner-like 3 (LHFPL3). This oncogenic gene rearrangement involves 5’ LHFPL3 and 3’ NTRK2, retaining the entire tyrosine kinase domain of NTRK2 genes. Moreover, the targeted DNA NGS analysis revealed an IDH1 (p.R132H) mutation, a surprising finding in this type of tumor. The pathogenic mechanism of the LHFPL3::NTRK2 in this case likely involves aberrant dimerization and constitutive activation of RTK signaling pathways.
The neurotrophic tyrosine receptor kinase (NTRK) family includes NTRK1, NTRK2, and NTRK3 which encode neurotrophin receptors TrkA, TrkB, and TrkC, respectively. Fusions involving the NTRK family are one of the most common mechanisms of oncogenic Trk activation (1) and occur sporadically in low-grade gliomas and glioblastoma (2). A variety of N-terminal gene fusion partners are described (3), which replace the ligand-binding site of the Trk, resulting in ligand-independent dimerization and phosphorylation of the Trk (4).
We describe here a novel NTRK2::LHFPL3 fusion in a Dysembryoplastic neuroepithelial tumor (DNT), a low-grade glioneuronal tumor. While the signaling pathways of TrkB are well-studied, not much is known about the LHFPL3. Lipoma HMGIC fusion partner-like 3 (LHFPL3) is a member of the superfamily of tetra-span transmembrane proteins. RNA-Seq expression data from GTEx showed a dominant LHFPL3 expression in the brain, including amygdala, anterior cingulate cortex, caudate, front cortex, hippocampus, hypothalamus, nucleus accumbens, putamen, and substantia nigra, while its expression is minimal in other non-brain tissues. Two human diseases have been linked to this group of genes. One LHFP-like gene is fused to the high mobility group AT-Hook 2 (HMGA2) in lipomas (5). Mutations in an LHFP-like gene result in deafness (6).
DNTs represent a type of glioneuronal and neuronal tumor (WHO, grade I) in the central nervous system (CNS) (7) and is the second most prevalent CNS neoplasm associated with epilepsy onset (8). DNT was first described by Daumas-Duport in 1988 (9), characterized by a multinodular growth of specific glioneuronal elements, with the columnar architecture of oligodendrocyte-like cells oriented perpendicular to the cortical surface and floating neurons in an abundant mucinous matrix (10, 11). DNT has shown genetic diversity and lacks molecular characteristics (12). Although studies have indicated FGFR1 mutations are more common in DNTs (10), other genetic alterations were also reported (13). While certain pediatric gliomas have been shown to harbor NTRK fusions with frequencies of 5–25% (14), only one case of mixed neuronal-glial tumors (MNGT) with SPECC1L::NTRK2 fusion was reported (12). Due to the exceeding rare NTRK rearrangement in this group of glioneuronal and neuronal tumors, we conducted a comprehensive integrating analysis of imaging, conventional histopathology, and molecular profiling.
The tumor tissue was fixed by formalin and embedded in paraffin. The embedded tumor tissue was cut into 5μm thick slices. For immunohistochemistry, the primary antibodies were utilized according to the manufacturer’s protocol. The immunohistochemical staining of pan-TRK (ab181560, Abcam, UK), Synaptophysin (36406, CST, USA), CD34 (Kit-0004, MXB, China), NSE (MAB-0791, MXB, China), Olig2 (ab109186, Abcam, UK), ATRX (sc-55584, Santa Cruz, USA), P53 (2527, CST, USA), IDH1 (R132H) (H09, Dianova, Germany), and Ki67 (9449, CST, USA) followed the protocol as the previous report (15).
Total RNA from fresh tumor tissue was extracted with TRIzol™ LS Reagent following the manufacturer’s instructions (10296010, ThermoFisher, Invitrogen, USA). 100ng total RNA was utilized for reverse transcription. End repairing and adaptor ligation were performed according to standard NGS protocols (E7771 and E6111, NEB, USA). PCR enrichment was performed using 390 gene-specific primers specific to a group of 63 genes commonly involved in solid tumors, and the enriched PCR products were sequenced in an Illumina NovaSeq 6000 platform (San Diego, USA). Sequencing results were analyzed with SeqNext software (JSI, Germany).
FISH was performed on 5µm paraffin slides of tumor tissue with dual-color break-apart probes for NTRK2 (Betrue, China). A total of 50 interphases were studied. FISH was applied as previously described (16).
Total RNA was extracted with TRIzol™ LS Reagent according to the manufacturer’s instructions (10296010, ThermoFisher, Invitrogen, USA). The RNA integrity was evaluated in agarose gel electrophoresis. cDNA was synthesized with random priming and SuperScript™ IV reverse transcriptase (18090050, ThermoFisher, USA). The first PCR was performed as the previous report (17). The primers were specific for LHFPL3 and NTRK2 (LHFPL3-F: 5’-CTTCAAAGCCGCCTCCTTCTT, NTRK2-R: 5’-TCCTGCTCAGGACAGAGGTTA). The first PCR condition was 95°C 3 min for 1 cycle followed by 35 cycles of 95°C 30s, 58°C 60 sec, and 72°C 60 sec, extend 72°C 5 min. 1μL of the first PCR product was utilized for nested PCR with the nested primers (LHFPL3-F: 5’-TCCGCTGCCTGCCTTGTGCTT, NTRK2-R: 5’-GCTGAACAAATGTGTCTGGC). Nested PCR condition was 95°C 3 min for 1 cycle followed by 40 cycles of 95°C 30s, 58°C 60 sec, and 72°C 60 sec, extend 72°C 5 min. Eventually, Sanger sequencing was performed to confirm the breakpoint on both LHFPL3 exon2 and NTRK2 exon16.
A 24-year-old female presented with a history of more than a decade of recurrent and unprovoked seizures. T2 weighted magnetic resonance imaging (MRI) of the brain showed an inhomogeneous hyperintense lesion located in the left frontal lobe (Figure 1A), measuring 2.0 × 1.5 × 1.7 cm in dimension, while T1 weighted MR image revealed hypo-intensity without peri-tumoral edema (Figure 1B). Contrast-enhanced MRI exhibited no significant enhancement (Figures 1C, D).
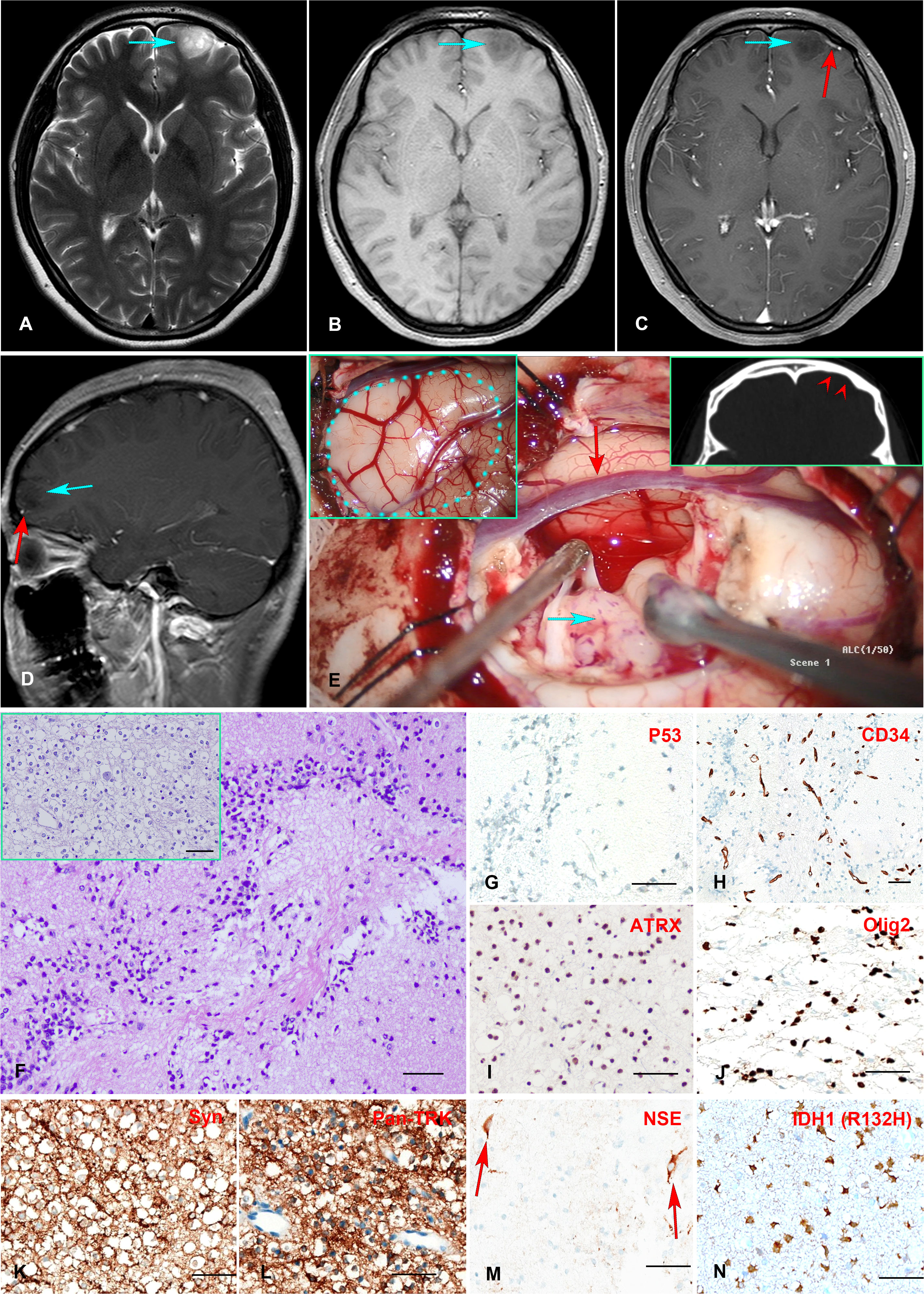
Figure 1 (A–E) Preoperative MR imaging and intraoperative stills. T2-weighted MRI revealed an inhomogeneous hyperintense lesion in the left frontal lobe (A). T1-weighted MRI showed a hypointensity of the lesion (B). Enhanced T1-weighted MR images indicated no enhancement of the mass, with a vessel (red arrow) underneath the tumor (C, D). The intraoperative stills revealed the tumor was regionally confined in a lobe. The dotted blue line in the panel delineated the region of the tumor. The red arrowheads indicate left frontal bone deformation and thinning caused by long-term tumor compression(E). (F–N) Histopathology and immunohistochemistry staining of the Formalin-fixed paraffin-embedded (FFPE) tissue tumor tissue sections. H&E staining revealed numerous round monotonous oligodendrocyte-like cells, with neurons floating in the matrix. The multinodular glial architecture was also observed (inserted) (F). Immunohistochemical staining indicates negative P53-negative expression (G), scattered positive CD34 expression in tumor cells in addition to vascular endothelial cells (H), positive ATRX expression (I), positive Olig2 expression (J), positive Syn and Pan-TRK expression (K, L), positive NSE expression in neurons (M), and positive mutant IDH1 (R132H) expression (N). Scale bar = 50μm.
The patient received extensive tumor resection after careful preoperative planning. The tumor was soft and confined in the lobe (Figure 1E). A postoperative MRI confirmed complete resection of the tumor. The fresh tumor tissues were sent for histology and molecular profiling analysis. H&E staining revealed a multinodular glial element, and a honeycomb appearance of oligodendrocyte-like cells was observed, with neurons floating in the matrix (Figure 1F). Immunohistochemistry analysis showed positive ATRX nuclear stain and negative P53 expression (Figures 1G, I), consistent with the genomic analysis showed wild-type ATRX and TP53. Scattered CD34 expression was seen in tumor cells in addition to vascular endothelial cells (Figure 1H), which was consistent with a previous report showing frequent CD34 expression in mixed or diffuse DNT (18). Tumor cells also expressed synaptophysin (Syn) (Figure 1K), oligodendrocyte transcription factor 2 (Olig2) (Figure 1J), and Ki-67 positivity less than 5%. Neuron-specific enolase (NSE) expression was observed in the neurons as expected (Figure 1M). Taken together, the histopathological and molecular pathological profiles were consistent with a complex subtype DNT. Moreover, Anti-Pan TRK antibody staining revealed a strong signal in tumor cells, consistent with the TrkB rearrangement (Figure 1L).
An RNA NGS assay with 390 ‘bait’ probes targeting 63 genes commonly rearranged in solid tumors revealed an LHFPL3::NTRK2 transcript (Figure 2A). The LHFPL3::NTRK2 fusion gene contained the first two exons of LHFPL3 and the last 6 exons of NTRK2, starting at exon 16 (Figure 2B). FISH analysis with a break-apart NTRK2 probe confirmed the NTRK2 rearrangement in tumor cell touch preparation (Figures 2C, D).
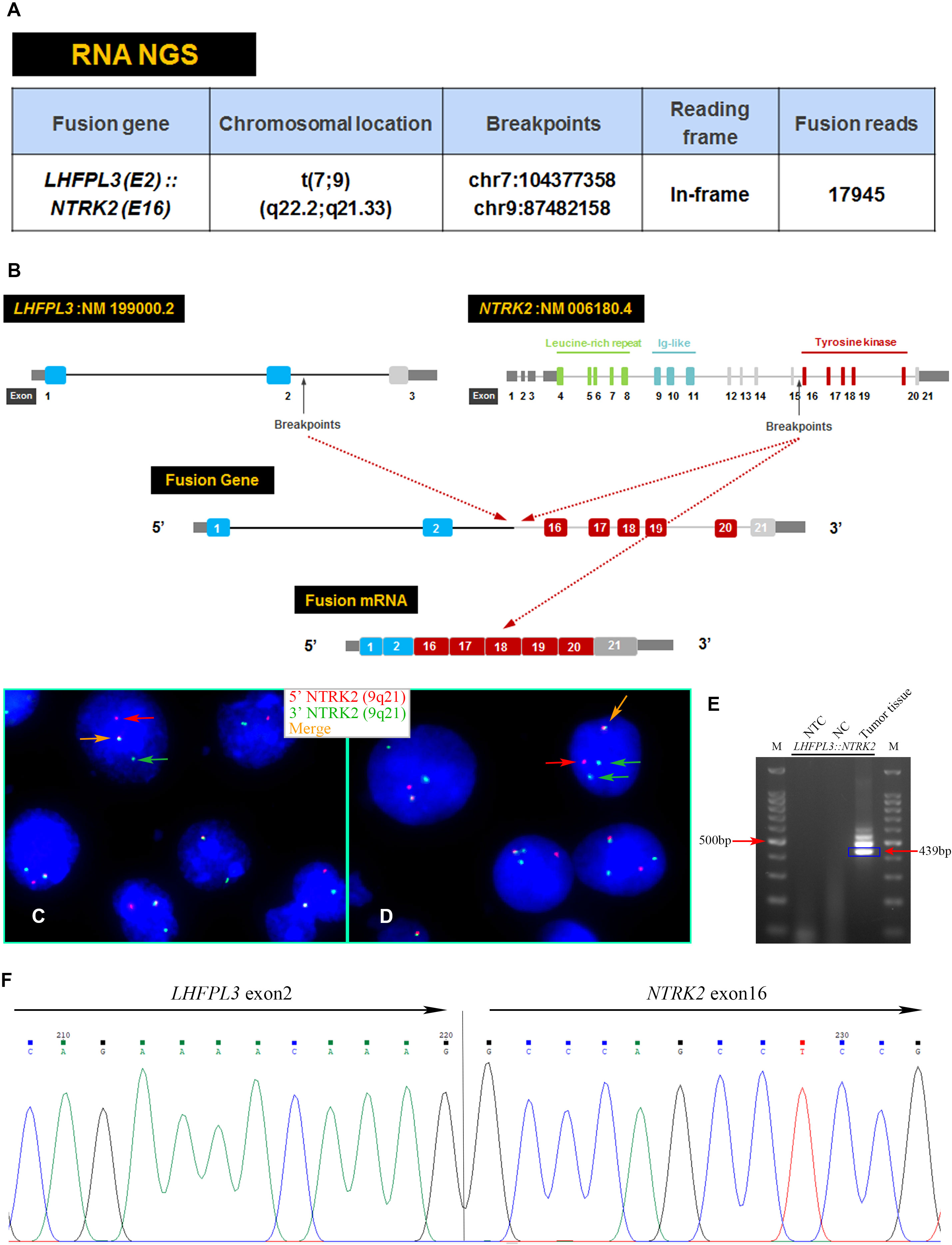
Figure 2 Characterization of the LHFPL3::NTRK2 fusion. (A) Targeted RNA NGS revealed an LHFPL3::NTRK2 fusion transcript with an intact reading frame. (B) A schematic diagram showed the breakpoints of LHFPL3 and NTRK2, with the fused gene retaining the tyrosine kinase domain of the NTRK2. (C, D) FISH with an NTRK2 split-apart probe showed the separation of the 5’ NTRK2 (Red) from the 3’ NTRK2 (Green), consistent with the NTRK2 gene rearrangement. (E) RT-PCR confirmed the expression of LHFPL3::NTRK2 fusion transcript. (F) Sanger sequencing of the PCR product confirmed the breakpoints of LHFPL3 and NTRK2 genes. NTC, No Template Control; NC, Negative Control.
RT-PCR analysis of tumor RNA with primers specific for LHFPL3 and NTRK2 showed 3 bands. The dominant band (Figure 2E) was Sanger sequenced and was identical to the transcript obtained from the RNA NGS. The other 2 minor bands were not sequenced, likely from differential splicing. A targeted DNA NGS (Figure 2F) analysis revealed an isocitrate dehydrogenase 1 (IDH1) mutation (p.R132H) (Figure 3A) and copy number gains of the chromosomes 7p, 9q, and X (Figure 3B). Immunohistochemistry analysis also confirmed the expression of mutant IDH1 (R132H) (Figure 1N). IDH1 mutations are often seen in diffuse gliomas including astrocytoma or oligodendroglioma but are rarely reported in DNTs (19). Other mutations, including ATRX and TP53 mutations in astrocytoma and chromosome 1p/19q co-deletion in oligodendroglioma, were not observed in this tumor.
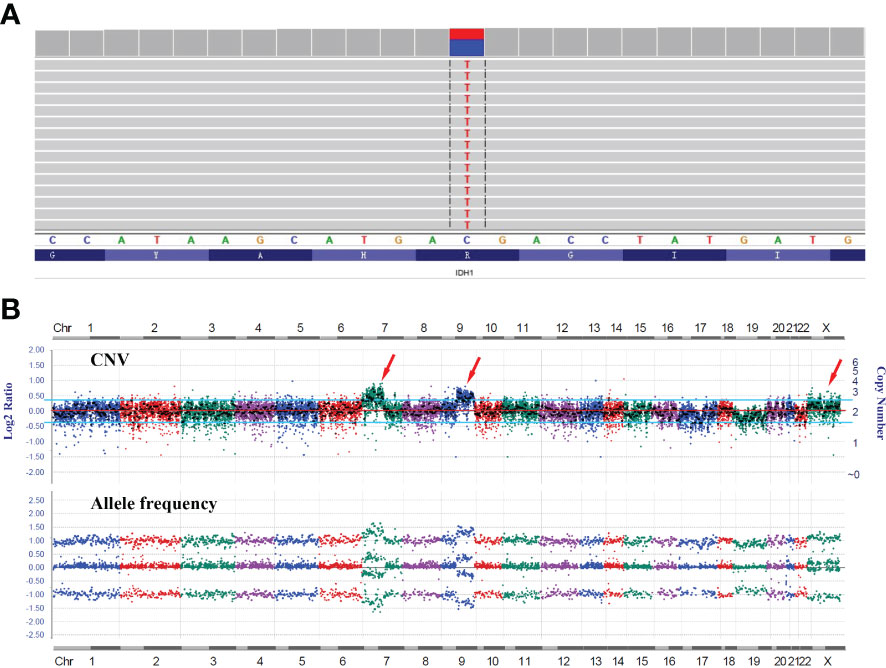
Figure 3 DNA NGS results of the tumor. (A) IGV image of the IDH1 mutation (p.R132H). (B) Copy number variation (CNV) analysis showed gains of the chromosomes 7p, 9q, and X with no evidence of 1p/19q co-deletion.
A variety of genomic alterations have been reported in DNTs, although diagnostically specific changes are not established. Nevertheless, fibroblast growth factor 1 receptor (FGFR1) has been frequently activated in DNTs (10, 20). Among FGFR1 alterations, internal tandem duplication (ITD) of the tyrosine kinase domain (TKD) is the most common mutation, which was reported in 40~60% of DNTs. In addition, hotspot missense mutations of FGFR1 were also reported. Other genomic alterations include BRAF p.V600E and copy number chromosome gains (21). The frequency of BRAF alterations in DNTs remains inconclusive among different studies (22, 23), with several studies failing to identify BRAF alterations in their DNTs cohorts (20, 24).
NTRK rearrangements in glioneuronal tumors were extremely rare. STRN1::NTRK2 and ARHGEF2::NTRK1 were reported in a single case of malignant glioneuronal tumor, respectively (25, 26). Alvarez-Breckenridge (27) described a case of low-grade glioneuronal tumor with BCAN::NTRK1 fusion. Surrey (12) reported a case of mixed neuronal-glial tumors (MNGT) with SPECC1L::NTRK2 fusion. Torre et al. reported a cohort of gliomas harboring NTRK fusions, most infantile or adult cases were histologically high-grade (89.7%, 26/29), while pediatric cases demonstrated high-grade histology (15.4%, 2/13) were rare. The median follow-up period after diagnosis was 23 months. During the follow-up period, 57.0% of the cases suffered tumor recurrence or progression (28).
IDH1/2 mutations are rare in DNTs, different from low-grade diffuse gliomas. In a cohort of 100 DNTs, 3 IDH1 mutations were observed (19). Jayapalan (29) reported a rosette-forming glioneuronal tumor with IDH1 mutation, which recurred in situ and progressed to glioblastoma 6 years after partial resection. Authors speculated that IDH1 mutation may lead to the malignant transformation of this type of benign glioneuronal tumor. Nevertheless, the biological and prognostic implications of IDH1/2 mutations in DNTs remain to be clarified.
In conclusion, we report a rare DNT with a novel LHFPL3::NTRK2 fusion and an IDH1 mutation. These findings provide additional evidence for a heterogeneous genomic profile of DNTs. Further functional evaluation of the LHFPL3::NTRK2 fusion oncoprotein will not only detail the oncogenic signaling mechanism but also shed light on the cellular function of LHFPL3, a CNS-specifically expressed gene with no studies.
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
This study was reviewed and approved by the Ethics Committee of the Second Affiliated Hospital of Soochow University. Written informed consent to participate in this study was provided by the participant herself.
YC designed this study. XD, PC, SuZ, and ShZ performed histopathological and molecular pathological examinations. XD and SX were responsible for the pathological diagnosis. QZ, YW, AC, and CD supported clinical data. QL and SX guided this work. All authors contributed to the article and approved the submitted version.
This study was funded by the National Natural Science Foundation of China (81602183).
Authors PC, SuZ, and ShZ were employed by Suzhou Sano Precision Medicine Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Vaishnavi A, Le AT, Doebele RC. TRKing down an old oncogene in a new era of targeted therapy. Cancer Discov (2015) 5:25–34. doi: 10.1158/2159-8290.CD-14-0765
2. Gambella A, Senetta R, Collemi G, Vallero SG, Monticelli M, Cofano F, et al. NTRK fusions in central nervous system tumors: A rare, but worthy target. Int J Mol Sci (2020) 21(3):1–24. doi: 10.3390/ijms21030753
3. Solomon JP, Benayed R, Hechtman JF, Ladanyi M. Identifying patients with NTRK fusion cancer. Ann Oncol (2019) 30:viii16–22. doi: 10.1093/annonc/mdz384
4. Cocco E, Scaltriti M, Drilon A. NTRK fusion-positive cancers and TRK inhibitor therapy. Nat Rev Clin Oncol (2018) 15(12):731–47. doi: 10.1038/s41571-018-0113-0
5. Petit MMR, Schoenmakers EFPM, Huysmans C, Geurts JMW, Mandahl N, Van De Ven WJM. LHFP, a novel translocation partner gene of HMGIC in a lipoma, is a member of a new family of LHFP-like genes. Genomics (1999) 57:438–41. doi: 10.1006/geno.1999.5778
6. Lerat J, Bonnet C, Cartault F, Loundon N, Jacquemont ML, Darcel F, et al. High prevalence of congenital deafness on reunion island is due to a founder variant of LHFPL5. Clin Genet (2019) 95:177–81. doi: 10.1111/cge.13460
7. Louis DN, Perry A, Wesseling P, Brat DJ, Cree IA, Figarella-Branger D, et al. The 2021 WHO classification of tumors of the central nervous system: a summary. Neuro Oncol (2021) 23(8):1231–51. doi: 10.1093/neuonc/noab106
8. Blumcke I, Aronica E, Urbach H, Alexopoulos A, Gonzalez-Martinez JA. A neuropathology-based approach to epilepsy surgery in brain tumors and proposal for a new terminology use for long-term epilepsy-associated brain tumors. Acta Neuropathologica (2014) 128:39–54. doi: 10.1007/s00401-014-1288-9
9. Daumas-Duport C, Scheithauer BW, Chodkiewicz J-P, Laws ER, Vedrenne C. Dysembryoplastic neuroepithelial tumor: A surgically curable tumor of young patients with intractable partial seizures. Neurosurgery (1988) 23:545–56. doi: 10.1227/00006123-198811000-00002
10. Sontowska I, Matyja E, Malejczyk J, Grajkowska W. Dysembryoplastic neuroepithelial tumour: insight into the pathology and pathogenesis. Folia Neuropathol (2017) 55(1):1–13. doi: 10.5114/fn.2017.66708
11. Caporalini C, Scagnet M, Moscardi S, Di Stefano G, Baroni G, Giordano F, et al. Dysembryoplastic neuroepithelial tumors: A single-institutional series with special reference to glutamine synthetase expression. Ann Diagn Pathol (2021) 54:151774. doi: 10.1016/j.anndiagpath.2021.151774
12. Surrey LF, Jain P, Zhang B, Straka J, Zhao X, Harding BN, et al. Genomic analysis of dysembryoplastic neuroepithelial tumor spectrum reveals a diversity of molecular alterations dysregulating the MAPK and PI3K/mTOR pathways. J Neuropathol Exp Neurol (2019) 78(12):1100–11. doi: 10.1093/jnen/nlz101
13. Lee D, Cho YH, Kang SY, Yoon N, Sung CO, Suh Y-L. BRAF V600E mutations are frequent in dysembryoplastic neuroepithelial tumors and subependymal giant cell astrocytomas. J Surg Oncol (2015) 111:359–64. doi: 10.1002/jso.23822
14. Amatu A, Sartore-Bianchi A, Siena S. NTRK gene fusions as novel targets of cancer therapy across multiple tumour types. ESMO Open (2016) 1:e000023. doi: 10.1136/esmoopen-2015-000023
15. Lan Q, Chen Y, Dai C, Li S, Fei X, Dong J, et al. Novel enhanced GFP-positive congenic inbred strain establishment and application of tumor-bearing nude mouse model. Cancer Sci (2020) 111(10):3626–38. doi: 10.1111/cas.14545
16. Panagopoulos I, Gorunova L, Andersen HK, Pedersen TD, Lomo J, Lund-Iversen M, et al. Genetic characterization of myoid hamartoma of the breast. Cancer Genomics Proteomics (2019) 16(6):563–8. doi: 10.21873/cgp.20158
17. Chen Y, Wang Y, He Q, Wang W, Zhang T, Wang Z, et al. Integrative analysis of TP73 profile prognostic significance in WHO grade II/III glioma. Cancer Med (2021) 10(13):4644–57. doi: 10.1002/cam4.4016
18. Blümcke I, Wiestler OD. Gangliogliomas: An intriguing tumor entity associated with focal epilepsies. J Neuropathol Exp Neurol (2002) 61:575–84. doi: 10.1093/jnen/61.7.575
19. Thom M, Toma A, An S, Martinian L, Hadjivassiliou G, Ratilal B, et al. One hundred and one dysembryoplastic neuroepithelial tumors: An adult epilepsy series with immunohistochemical, molecular genetic, and clinical correlations and a review of the literature. J Neuropathol Exp Neurol (2011) 70:859–78. doi: 10.1097/NEN.0b013e3182302475
20. Rivera B, Gayden T, Carrot-Zhang J, Nadaf J, Boshari T, Faury D, et al. Germline and somatic FGFR1 abnormalities in dysembryoplastic neuroepithelial tumors. Acta Neuropathol (2016) 131(6):847–63. doi: 10.1007/s00401-016-1549-x
21. Kakkar A, Majumdar A, Kumar A, Tripathi M, Pathak P, Sharma MC, et al. Alterations in BRAF gene, and enhanced mTOR and MAPK signaling in dysembryoplastic neuroepithelial tumors (DNTs). Epilepsy Res (2016) 127:141–51. doi: 10.1016/j.eplepsyres.2016.08.028
22. Blumcke I, Coras R, Wefers AK, Capper D, Aronica E, Becker A, et al. Review: Challenges in the histopathological classification of ganglioglioma and DNT: microscopic agreement studies and a preliminary genotype-phenotype analysis. Neuropathol Appl Neurobiol (2019) 45(2):95–107. doi: 10.1111/nan.12522
23. Blumcke I, Aronica E, Becker A, Capper D, Coras R, Honavar M, et al. Low-grade epilepsy-associated neuroepithelial tumours - the 2016 WHO classification. Nat Rev Neurol (2016) 12(12):732–40. doi: 10.1038/nrneurol.2016.173
24. Matsumura N, Nobusawa S, Ito J, Kakita A, Suzuki H, Fujii Y, et al. Multiplex ligation-dependent probe amplification analysis is useful for detecting a copy number gain of the FGFR1 tyrosine kinase domain in dysembryoplastic neuroepithelial tumors. J Neurooncol (2019) 143(1):27–33. doi: 10.1007/s11060-019-03138-7
25. Boyer J, Birzu C, Bielle F, Goulas C, Savatovsky J, Karachi C, et al. Dramatic response of STRN-NTRK -fused malignant glioneuronal tumor to larotrectinib in adult. Neuro-Oncology (2021) 23:1200–2. doi: 10.1093/neuonc/noab080
26. Kurozumi K, Nakano Y, Ishida J, Tanaka T, Doi M, Hirato J, et al. High-grade glioneuronal tumor with an ARHGEF2-NTRK1 fusion gene. Brain Tumor Pathol (2019) 36(3):121–8. doi: 10.1007/s10014-019-00345-y
27. Alvarez-Breckenridge C, Miller JJ, Nayyar N, Gill CM, Kaneb A, D'Andrea M, et al. Clinical and radiographic response following targeting of BCAN-NTRK1 fusion in glioneuronal tumor. NPJ Precis Oncol (2017) 1(1):5. doi: 10.1038/s41698-017-0009-y
28. Torre M, Vasudevaraja V, Serrano J, DeLorenzo M, Malinowski S, Blandin AF, et al. Molecular and clinicopathologic features of gliomas harboring NTRK fusions. Acta Neuropathol Commun (2020) 8(1):107. doi: 10.1186/s40478-020-00980-z
Keywords: dysembryoplastic neuroepithelial tumors, NTRK2, LHFPL3, next-generation sequencing, IDH1
Citation: Chen Y, Zhu Q, Wang Y, Dai X, Chen P, Chen A, Zhou S, Dai C, Zhao S, Xiao S and Lan Q (2022) Case Report: A novel LHFPL3::NTRK2 fusion in dysembryoplastic neuroepithelial tumor. Front. Oncol. 12:1064817. doi: 10.3389/fonc.2022.1064817
Received: 08 October 2022; Accepted: 17 November 2022;
Published: 01 December 2022.
Edited by:
Emilio Cordova, Instituto Nacional de Medicina Genómica (INMEGEN), MexicoReviewed by:
Manabu Natsumeda, Niigata University, JapanCopyright © 2022 Chen, Zhu, Wang, Dai, Chen, Chen, Zhou, Dai, Zhao, Xiao and Lan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qing Lan, c3pscTAwNkAxNjMuY29t
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.