
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Oncol., 04 November 2022
Sec. Gastrointestinal Cancers: Gastric and Esophageal Cancers
Volume 12 - 2022 | https://doi.org/10.3389/fonc.2022.1061988
 Qingyi Zhang1†
Qingyi Zhang1† Xu Lin1†
Xu Lin1† Kan Jiang2†Jun Deng3
Kan Jiang2†Jun Deng3 Lei Ke1Ziheng Wu1Pinghui Xia1Qi Li3Li Yu1Pengzhi Ni1Wang Lv1
Lei Ke1Ziheng Wu1Pinghui Xia1Qi Li3Li Yu1Pengzhi Ni1Wang Lv1 Jian Hu1*
Jian Hu1*Background: Esophageal squamous cell carcinoma (ESCC) is an aggressive tumor with a 5-year survival rate of only 20%. More than 80% of ESCC patients possess TP53 mutation, which abolishes the G1/S checkpoint and accelerates the cell cycle. Thus, WEE1 and PKMYT1, regulators of G2/M phase in cell cycle, play essential roles in TP53-mutated cancer cells. PD0166285(PD) is a pyridopyrimidine compound that can inhibit WEE1 and PKMYT1 simultaneously, however, the effects of PD on ESCC, either as monotherapy or in combination therapy with radiotherapy, remain unclear.
Methods: To measure the anti-tumor efficacy of PD in ESCC cells, cell viability, cell cycle and cell apoptosis assays were examined in KYSE150 and TE1 cells with PD treatment. The combination therapy of PD and irradiation was also performed in ESCC cells to find whether PD can sensitize ESCC cells to irradiation. Vivo assays were also performed to investigate the efficacy of PD.
Results: We found that the IC50 values of PD among ESCC cells ranged from 234 to 694 nM, PD can regulate cell cycle and induce cell apoptosis in ESCC cells in a dose-dependent manner. When combined with irradiation, PD sensitized ESCC cells to irradiation by abolishing G2/M phase arrest, inducing a high ratio of mitosis catastrophe, eventually leading to cell death. We also demonstrated that PD can attenuate DNA damage repair by inhibiting Rad51, further research also found the interaction of WEE1 and Rad51. In vivo assays, PD inhibited the tumor growth in mice, combination therapy showed better therapeutic efficacy.
Conclusion: PD0166285 can exert antitumor effect by inhibiting the function of WEE1 and PKMYT1 in ESCC cells, and also sensitize ESCC cells to irradiation not only by abolishing G2/M arrest but also attenuating DNA repair directly. We believe PD0166285 can be a potent treatment option for ESCC in the future.
Esophageal carcinoma (EC), including esophageal cell carcinoma (ESCC) and esophageal adenocarcinoma (EAC), is the seventh most common cancer in the world, and the sixth in mortality overall (544,000 deaths) (1). In 2020, one in every 18 cancer deaths was due to EC. While the predominant type of EC worldwide is ESCC, EAC is dominant in the United States and western countries (2–4). The etiology of esophageal cancer is complex, smoking, alcohol abuse and nitrosamine intake are closely related to the incidence of ESCC, while gastroesophageal reflux, Barrett’s esophagus, and excess body weight cause EAC (5–8). China accounts for more than 50% of the global morbidity and mortality, and more than 90% of patients with EC in China are ESCC (9, 10). Owing to the nonspecific symptoms and biomarkers of EC, most patients are diagnosed at an advanced stage with poor diagnosis (11, 12). Although surgery is believed to be the best treatment for EC patients, multidisciplinary treatments are also considered to ensure optimal treatment. Unfortunately, the prognosis for esophageal cancer remains poor, with a 5-year survival rate of less than 20% (13, 14). Unlike EAC, which has more therapeutic targets and drugs, there are limited treatment options for ESCC. Thus, it is urgent to identify more potential therapeutic targets for ESCC patients.
WEE1 G2 checkpoint kinase (WEE1) and protein kinase membrane associated tyrosine/threonine 1 (PKMYT1) are members of the WEE1 kinase family, and act as dominant cell cycle regulators in G2/M checkpoint. WEE1 and PKMYT1 can arrest the cell cycle at G2/M phase by phosphorylating CDK1. WEE1 can only phosphorylate CDK1 at Tyr15, while PKMYT1 has dual activity for Thr14 and Tyr15 (15, 16). The overexpression of WEE1 and PKMYT1 has been reported in multiple cancers, such as lung cancer, glioblastoma, liver cancer, colorectal cancer, esophageal and so on (17–20). A high frequency of Tp53 mutation occurs in most tumors cell, which makes the G2/M checkpoint more important for tumor cells to repair DNA damage. Due to the properties of tumor cells and WEE1 protein kinase family, it’s possible to treat tumors by targeting WEE1 kinase family. There are several I/II phase clinical trials focusing on MK1775, a specific inhibitor of WEE1, showing promising efficacy and safety (21–24). However, there are very few studies investigate the anti-tumor efficacy of PKMYT1 and WEE1 co-inhibition. Large-scale genome sequencing shows that almost 90% of ESCC patients have TP53 mutation (25–28). A previous study has also demonstrated that inhibition of WEE1 can exert antitumor effect in ESCA (25). Hence, we hypothesize that inhibiting PKMYT1 and WEE1 simultaneously can be a promising therapeutic treatment method.
PD0166285 (PD) is a pyridopyrimidine compound that can simultaneously inhibit WEE1 and PKMYT1 with IC50 values of 24 and 72 nM, respectively (29). In TP53-deficient cells, PD can induce premature mitosis by inhibiting WEE1 and PKMYT1, which will further reduce DNA damage repair and induce mitotic catastrophe, eventually lead to cell death. A variety of studies demonstrated the anti-tumor and radiosensitizing effects of PD (29, 30). However, the effects of PD on ESCC, either as monotherapy or in combination therapy with radiotherapy, remain unclear.
In this study, We explored the anti-tumor effect of PD in ESCC cells in vitro and in vivo.
PD can also sensitize ESCC during radiotherapy by abrogating the G2/M checkpoint and decreasing DNA damage repair. Our study demonstrates that PD has potential therapeutic efficacy in ESCC, and may further improve the therapeutics outcomes of ESCC when combined with radiotherapy.
Human esophageal squamous carcinoma cells and normal esophageal epithelial cells (KYSE70, KYSE150, KYSE410, KYSE450, KYSE510, TE1, TE7, EC1, HEEC) were maintained in RPMI 1640 medium (HyClone, USA) with 10% fetal bovine serum (HyClone, USA), 1% penicillin/streptomycin (Life Technologies, USA). Cells were cultured at 37°C in a humidified atmosphere with 5% CO2. All cell lines were obtained from Shanghai Institute of Biochemistry (Shanghai, China) and were validated by mycoplasma testing. PD0166285 (PD) was purchased from MedChemExpress (MCE,USA), PD was dissolved in DMSO (MCE, USA) at 10mmol/mL and stored in -80°C。
Cell Counting Assay Kit-8 (CCK8) (MCE, USA) was used to measure cell viability. After treatment, cells were incubated with 10% CCK8 for 3 h, then measured by a microplate reader (Bio-Rad, USA) with an absorbance at 450 nm. Each experiment had six duplicate wells and was performed three times.
Cells were collected and lysed with RIPA Lysis Buffer (Beyotime, China), the concentration of proteins were measured by Bicinchoninic Acid (BCA) protein assay kit (Thermo, USA) and proteins were boiled in 1×SDS loading buffer. The proteins were separated by SDS-PAGE, and then transferred to NC membranes. After blocking, membranes were incubated overnight at 4°C with primary antibodies (antibodies were showed in Supplement Table1), after incubating with secondary antibodies, then the membranes were detected with a Bio-Rad Molecular Imager ChemiDoc XRS + System (Bio-Rad, USA).
Cells were collected and lysed with IP buffer. After centrifuging, the lysates were incubated with protein G magnetic beads for 1h at 4°C. The immunoprecipitation reaction were set up with equal quantities of the lysates (input, nonreactive antibody-Mouse/Rabbit IgG, and specific antibody), the input lysates were boiled with SDS immediately, the other lysates rotated overnight at 4°C. The beads were washed next day with wash buffer at least three times, and then boiled with SDS, followed by western blot.
Cells were plated in 6-well plates with different numbers (1000-5000 cells). 3 hours prior to irradiation, PD was added into the medium and the terminal concentration was 100 nM. Cells were irradiated with a Precision X-RAD 225 machine operating at 225 kV and 13.3 mA with a 2-mm Al filter (source-to-skin distance (SSD): 36 cm; dose rate: 1.3 Gy/min). Medium was changed 2 days after irradiation. 14 days after irradiation, the plates were fixed with 4% paraformaldehyde (PFA) for 15 min, and the stained with 0.2% crystal violet for 30 min, then washed with PBS. The linear quadratic (LQ) model (SF = exp(-αD-βD2)) was employed to fit the survival curves. Each experiment had three duplicate wells and was performed three times.
The cell cycle assay and cell apoptosis assay were performed with BD FACS Canto II (BD Biosciences, USA). Cell cycle was evaluated with Cell Cycle Kit (BD Biosciences, USA), 48 h after incubation with PD alone or 24 h after irradiation, the cells were collected for analysis, and followed the manufacturer’s instructions. Apoptosis assay was performed with a FITC Annexin V apoptosis detection kit I (BD Biosciences, USA), and 48h after treatment, cells were collected for assay. Cell cycle data was analyzed with Modfit (Verity Software House, USA), and apoptosis data was analyzed with Flowjo (BD Biosciences, USA). Flow cytometry results were obtained on the same machine at the same settings on the same day. All these assays were performed three times.
Cells were plated on glass slides that were put in 24-well plates. After irradiation, cells were washed 3 times with PBS and fixed with 4% PFA for 20 min. Then cells were blocked with 5% BSA for 30 min and incubated with γ-H2AX and Rad51 overnight at 4°C. Then cells were incubated with FITC-conjugated anti-rabbit antibody (Abcam, USA) for 1 h at room temperature. DAPI (Beyotime, China) was used for the final stain, and images were captured by a Leica TCS SP8 confocal microscope.
To further assess DNA damage, an alkaline comet assay was performed with a comet assay kit (Trevizen, USA) following the manufacturer’s instructions. We finally stained the slides with SYBR Green I, and images were captured with Olympus IX71 inverted microscope (Tokyo, Japan). The DNA damage was calculated by measuring the tail DNA% (percent DNA in the tail) and tail moment (TM = percentage of DNA in the tail × tail length). We measured at least 100 cells with CASP software.
The co-expressed genes of WEE1 were calculated by the R (version 3.6.3) package “stat” based on the TCGA project’s expression datasets of the ESCC transcriptome. Results were presented using the R package “ggplot2” (version 3.3.3). The statistical significance of correlations was assessed with Pearson’s rank correlation coefficient test. P < 0.05 were considered statistically significant.
Using the R package “Cluster Profiler”(version 3.14.3), gene enrichment analysis of Gene Ontology (GO) and Kyoto Encyclopedia of Genes Genome (KEGG) were performed on co-expressed genes of WEE1 with Pearson rho greater than 0.30. Pathways enriched were considered significant with p.adjust < 0.05 and q value < 0.2.
RNA-sequencing expression (level 3) profiles and corresponding clinical information for ESCC were downloaded from the TCGA dataset (https://portal.gdc.com).
R software GSVA package was used to analyze, choosing parameter as method=‘ssgsea’. The correlation between genes and pathway scores was analyzed by Spearman correlation. P values < 0.05 was considered statistically significant.
SiRNAs targeting Wee1 were synthesized by GenePharm (China), the sequences were list in Supplement Table2. JetPRIME (Polyplus-transfection, France) was used for transfection, KYSE150 cells were cultured in 6-well plates with 50% intensity. 48 h after transfection, cells were collected for subsequent assays.
All animal experiments were approved by the Committee of Animal Experimental Ethical Inspection of the First Affiliated Hospital, College of Medicine, Zhejiang University, and followed by the institutional Guidelines for animal care and use.
Six-week-old female nude mice were purchased and housed in an SPF environment. 1×106 KYSE150 cells were injected subcutaneously into the right flank of nude mice and when tumor volume reached 100-150mm3, mice were randomly divided into four groups (n=6), the irradiation group accepted a total dose of 12Gy radiation using an X-RAD225 small animal irradiator with a 0.95 Gy/min dose rate. PD was dissolved in the mixture (10% DMSO, 40% PEG300, 5% Tween-80 and 45% saline) with a final concentration of 2.5 mg/mL. Mice were injected intraperitoneally at the dose of 10mg/kg every two days. For combination therapy group, PD was injected 3 hours before irradiation. Tumor volumes were calculated every two days with a caliper and measured with the formula (L × W × W)/2. Mice were sacrificed after 2 weeks of treatment, and tumors and organs were collected for subsequent experiments.
Tumor tissues were formalin-fixed and paraffin-embedded, and 4-μm sections were cut. After dewaxing and hydration, slides were incubated with antigen retrieval solution. Then the slides were treated with 3% H2O2 and 5% goat serum. Slides were incubated with primary antibody (PKMYT1, 1:50, WEE1, 1:100) at 4°C overnight. After incubation with secondary biotinylated antibody and DAB stain, the slides were scanned by a Motic EasyScan (Motic, USA) with 200× magnification.
All statistical analyses were conducted using SPSS 23.0 for windows (SPSS Inc., Chicago, USA) and GraphPad Prism (GraphPad Software Inc, USA). Data are presented as the means ± standard deviation. P values < 0.05 indicate statistical significance. Each experiment was replicated thrice.
We measured the expressions of WEE1 and PKMTY1 in ESCC cell lines and normal esophageal epithelial cells, which showed high expressed Wee1 and PKMYT1 in ESCC cell lines (Figure 1B). To determine the anti-tumor efficacy of PD in ESCC cells, we evaluated the cell viability with different PD concentrations (Figure 1A). We found that the IC50 values of PD in ESCC cells ranged from 234 to 694 nM (Figures 1C, D, Supplementary Figure S1), which was lower than HEEC’s IC50 value (Figure 1E). This may be caused by the high expression of WEE1, PKMYT1, and TP53 mutation in esophageal cancer cells. We then measured the expression of p-CDK1 T14, p-CDK1 Y15 and CyclinB1 in KYSE150 and TE1 treated with PD [300 nM] at different time. Down-regulated p-CDK1 T14 and p-CDK1 Y15 can be observed 6 hours after PD was added. In the meantime, decreased protein levels of WEE1 and PKMYT1 were also observed (Figure 1F). Clonogenic assay with PD at dose escalating concentrations was also performed in KYSE150 and TE1 cells, increasing doses with PD caused decreased cell clones (Figures 1E-H).
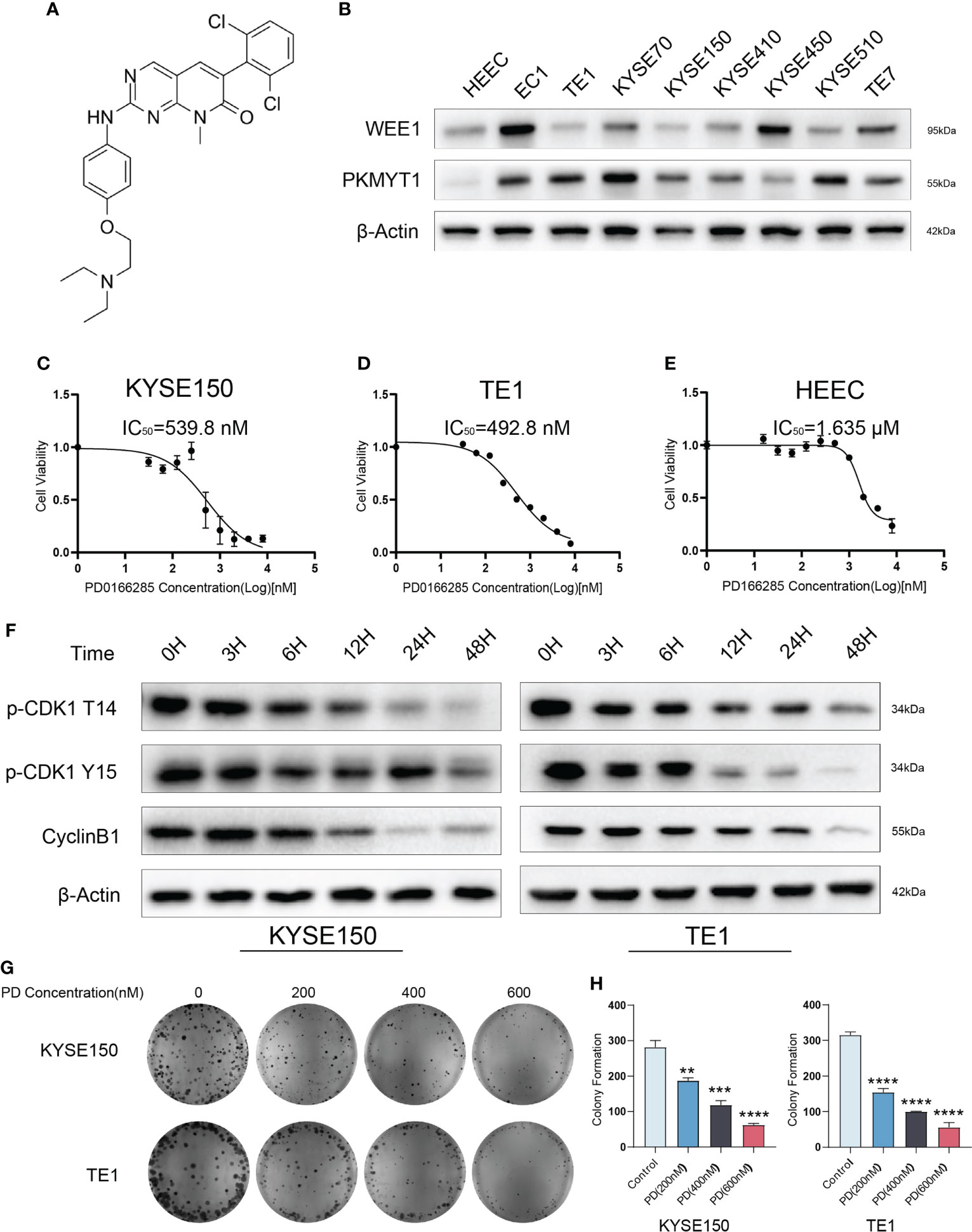
Figure 1 PD0166285 is effective in ESCC cells. (A) The Structural formula of PD0166285. (B) The expression of WEE1 and PKMYT1 are upregulated in ESCC cells. (C–E) The IC50 of PD in KYSE150, TE1 and HEEC cells. (F) PD inhibits WEE1 and PKMYT1 in a time-dependent manner. (G, H) The clonogenic assay shows PD inhibited cell proliferation. **P < 0.01, ***P < 0.001, ****P < 0.0001.
To further investigate the underlying mechanism of PD in ESCC cells. We treated KYSE150 and TE1 with escalating PD concentrations (200 nM, 400 nM, 600 nM). after drug exposure (48 h), cells were collected for flow cytometry to detect cell cycle and cell apoptosis. PD interrupted the regular cell cycle, G2/M ratio of the cell cycle decreased gradually with increased PD concentrations (Figures 2A-C). We also collected protein for Western blot assay at the same time, the biomarkers of the cell cycle were detected and the results were consistent with the results obtained with flow cytometry (Figures 2B-D). Cell apoptosis assay indicated that PD induced cell apoptosis in a dose-dependent manner. With PD concentration reaching 600 nM, almost half of the cells died, which is consistent with our cell viability experiments above (Figures 2E-G). Western blot showed apoptosis-related proteins changed gradually with the increased drug concentration (Figures 2F-H).
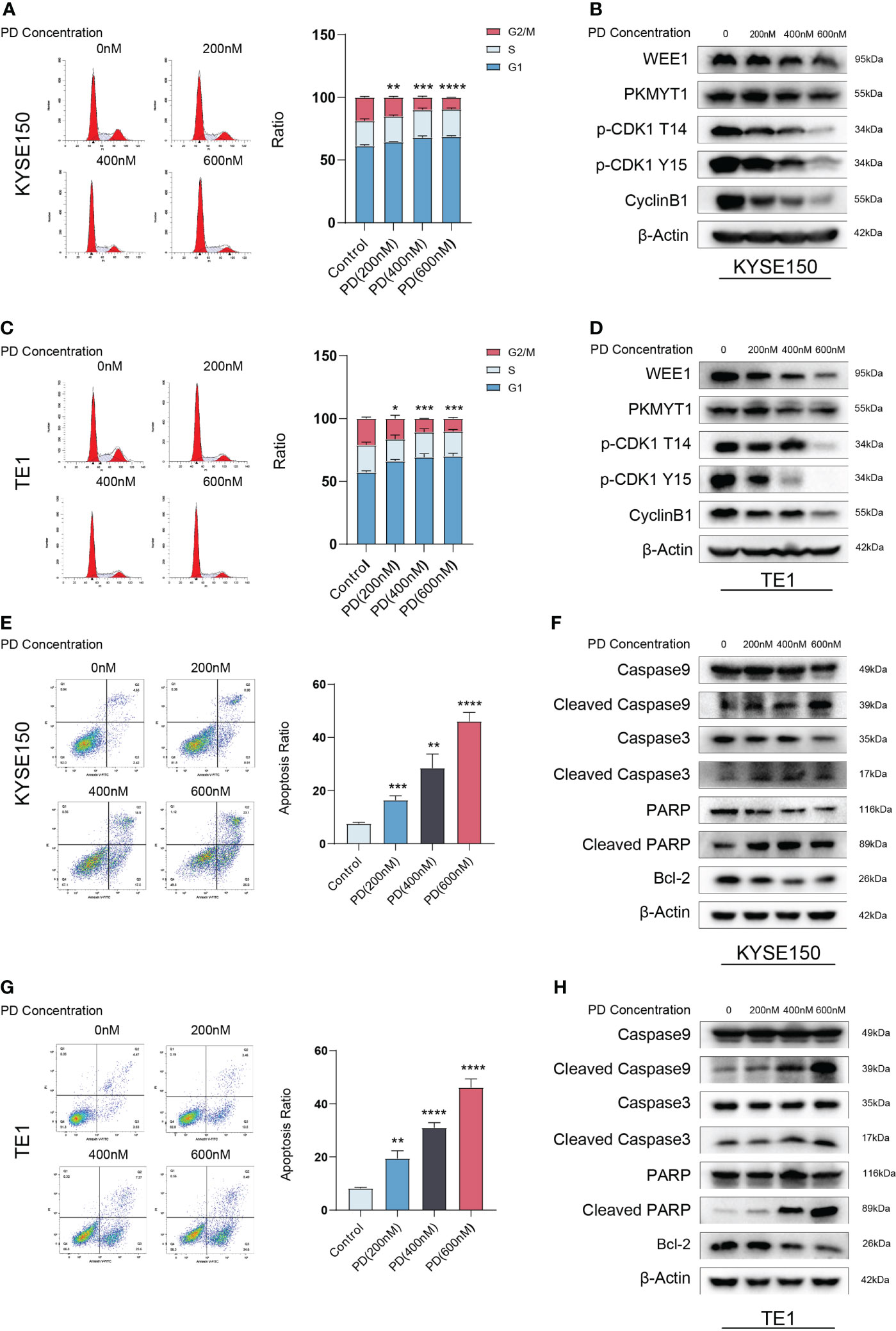
Figure 2 PD exerts antitumor effects in a dose-dependent manner. (A, C) PD decreased the G2/M ratio in KYSE150 and TE1 cells. (B, D) PD inhibits the protein levels of WEE1, PKMYT1, p-CDK1-T14, p-CDK1-Y15 and CyclinB1 in a dose-dependent manner. (E, G) PD induces cell apoptosis in ESCC cells with escalating PD concentrations. (F, H) PD induces changes in the expression of apoptosis-related proteins. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001..
Previous research showed cell cycle arrest in cells treated with irradiation, cell cycle arrests in all phases to repair DNA damages, including single-strand break repair (SSBR), double-strand break repair (NHEJ), Homologous recombination (HR), and so on. Since G1 arrest depends on the full function of P53, G2/M arrest means much more important in P53-deficient cells. We observed significant G2/M blockage in ESCC. ESCC cells had a high rate of TP53 mutation. Therefore, we hypothesized that PD could sensitize ESCC cells to radiotherapy by abrogating G2/M blockage. We performed a clonogenic assay with escalating radiation doses, PD (100 nM) was added into the medium 3 hours before irradiation, and we exchanged the medium 48 hours after irradiation. Cells were cultured for 14 days, then fixed and stained with a crystal violet solution. There were few clones in plates treated with radiotherapy and PD, Sensitization enhancement ratio (SER) value was calculated and both greater than 1. (Figures 3A, B). We then determined the effect of PD on the cell cycle after irradiation. Cells were grown in six-well plates and added PD 3 hours before irradiation, cells were collected 24 hours after irradiation for flow cytometry and western blot. Flow cytometry showed that PD abrogated the G2/M arrest in ESCC cells, and the ratio of the G2/M phase decreased significantly (Figures 3C, D). In the meantime, PD also inhibited the protein levels of the G2/M phase checkpoint (Figures 3E-F). Disruption of the cell cycle also leads to cell apoptosis, where PD showed an accumulative effect in combination with irradiation in ESCC cells (Figures 4A, B), and the results of western blot were consistent with the results from the flow cytometry assays. The expression of caspase9, caspase3, bcl2, and parp were downregulated, while cleaved-caspase9, cleaved-caspase3, cleaved-parp were upregulated (Figures 4C, D).
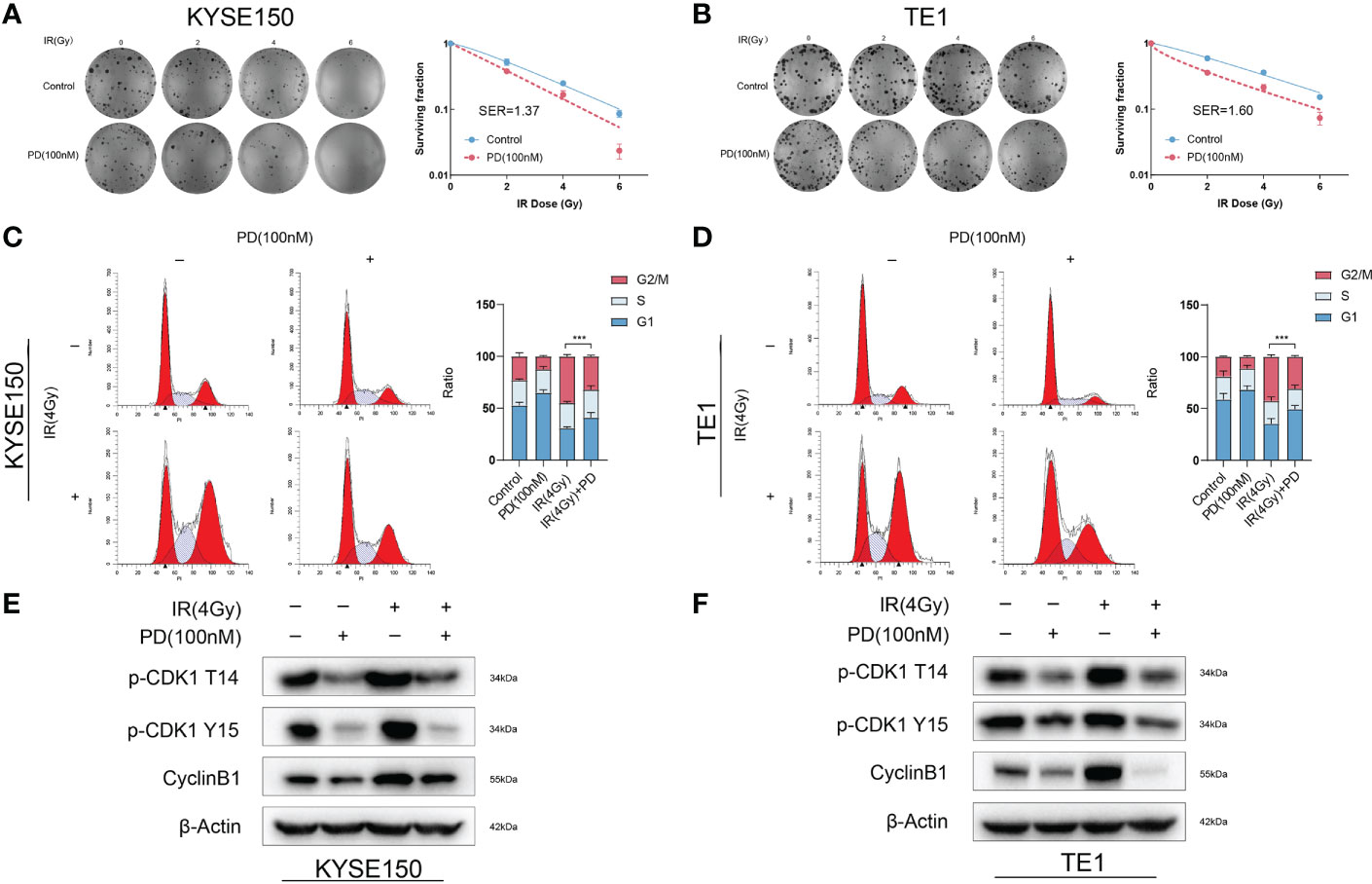
Figure 3 PD sensitizes ESCC cells to irradiation and disrupts the G2/M arrest due to irradiation. (A, B) Clonogenic survival assays of KYSE150 and TE1 cells with different doses of irradiation. (C, D) PD disrupts the G2/M arrest induced by irradiation, reducing the G2/M ratio of cell cycle in ESCC cells. (E, F) PD induces altered expression of G2/M phase-related cyclins. ***P < 0.001..
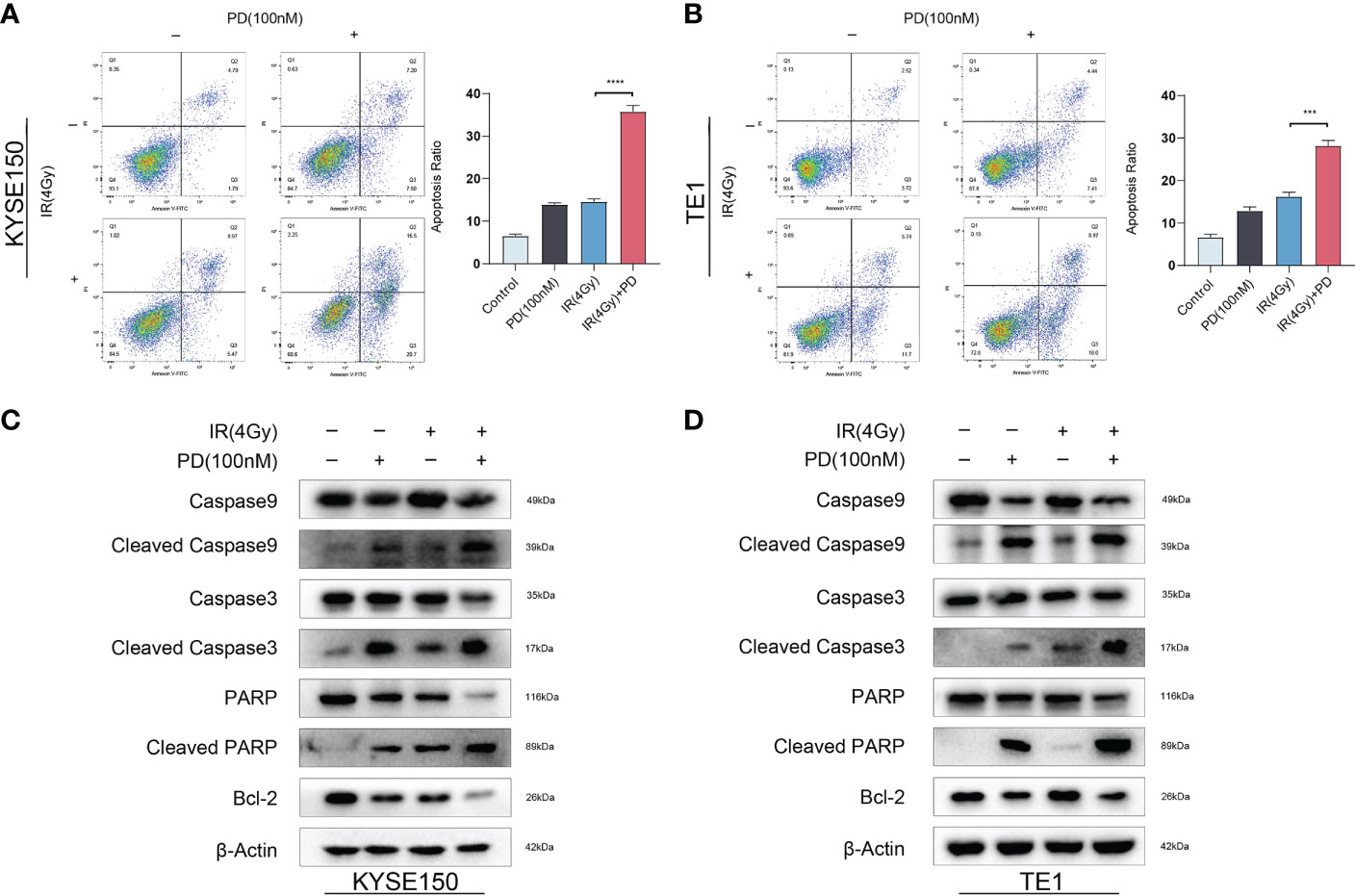
Figure 4 Combination therapy of PD and irradiation induces cell apoptosis. (A, B) The combination therapy of PD and irradiation increases the apoptosis ratio in ESCC cells. (C, D) The protein level of apoptosis-related altered with combination therapy. ***P < 0.001, ****P < 0.0001..
It’s known that sublethal DNA damage can induce mitotic catastrophe, especially in cells with irradiation. Mitotic catastrophe can be seen as a typical anti-cancer way of irradiation. PD inhibited the function of WEE1 and PKMYT1, then induced premature cell division, which disrupted the normal cell cycle and will ultimately induce mitotic catastrophe. We cultured cells in 14 mm slides, and treated them with irradiation alone or in combination with PD (100 nm). Cells were fixed and stained with Phalloidine and DAPI 24 hours after irradiation. Slides were scanned by confocal microscopy and analyzed to determine the percentage of cells with mitotic catastrophe (at least 100 cells were counted). The combination group had a higher percentage of mitotic catastrophe than irradiation alone (Figure 5A). We also detected the effect of PD on DNA damage repair, cells were fixed and stained with γ-H2AX and DAPI after irradiation (3 hours, 6hours,12 hours). Greater than 15 foci per nucleus with γ-H2AX stained was considered positive cells (cells with DNA damage). We observed that in 3h, the γ-H2AX peaked and then gradually decreased, but the combination group had more positive cells in 24h (Figures 5B, C). We also performed an alkaline comet assay to directly measure the effect of PD on DNA damage repair, as shown in figure (Figures 5D, E), the results showed that after irradiation for 24 h, the combination group significantly increased the comet tail DNA% and tail moment. All these observations indicated that PD attenuates DNA damage repair in ESCC cells.
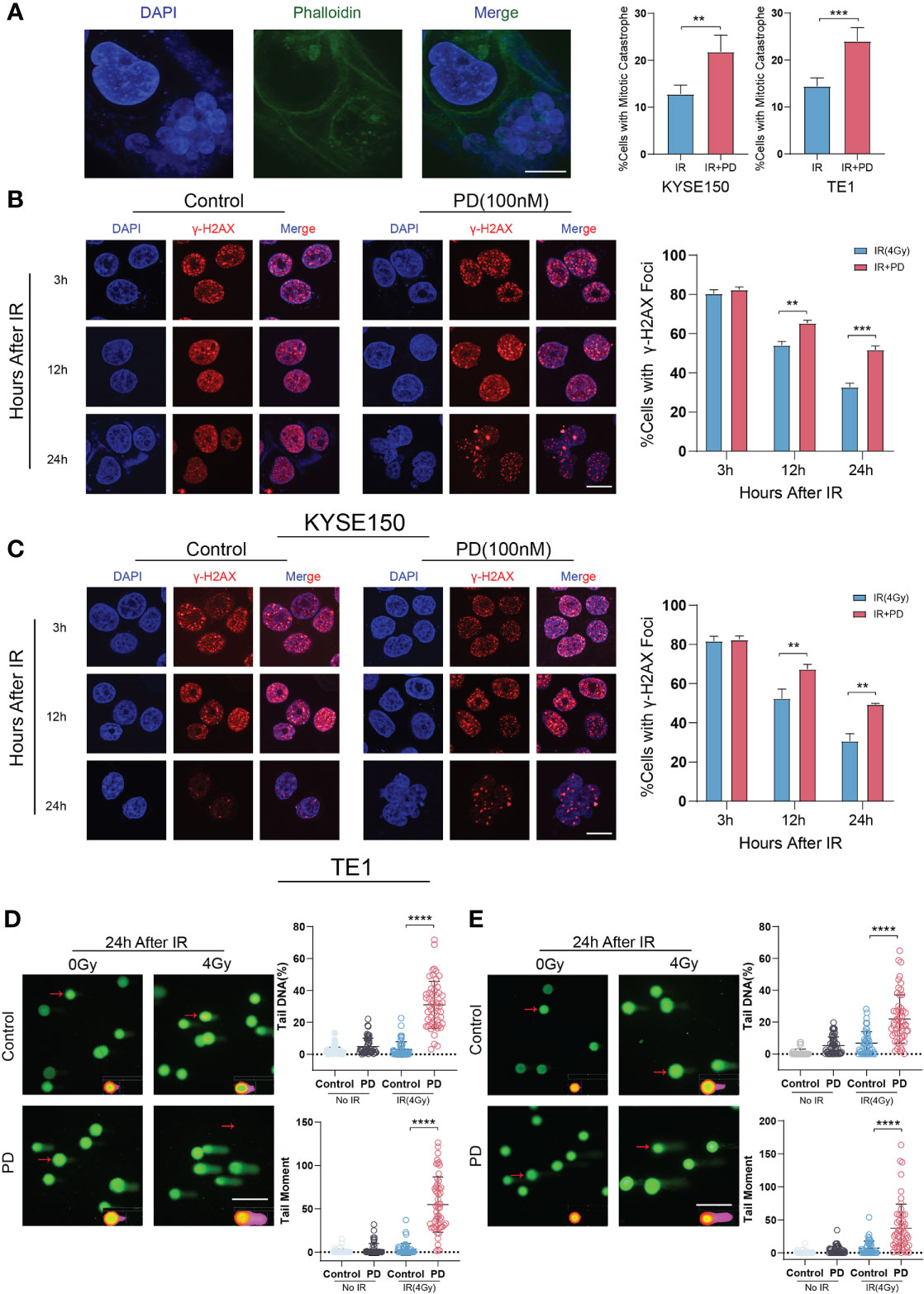
Figure 5 PD increases irradiation-induced DNA damage. (A) Irradiation induces mitotic catastrophe in ESCC cells, PD increases the proportion of mitotic catastrophe cells. (B, C) γ-H2AX foci is measured in ESCC cells at different time points with irradiation, combination therapy has more foci per nucleus. Scale bar = 10 μm. (D, E) Alkaline comet assay of KYSE150 and TE1 cells is performed at 0 h, 6 h and 24 h after irradiation. Comet tails and tail moments are also quantified at the indicated time points. Scale bars: 2 μm (A), 10 μm (B, C), 20 μm.(D, E) **P < 0.01, ***P < 0.001, ****P < 0.0001.
We then examined several protein markers in DNA damage repair pathways by western blot. We observed that the expression of Rad51 was significantly downregulated while the expression of other proteins was unchanged or upregulated (Figure 6A). Thus, we assessed Rad51 foci formation by immunofluorescence. Cells were fixed and stained at several time points (after irradiation for 3 h, 12 h, 24 h), slides were scanned by confocal microscopy and analyzed. Less Rad51 foci were observed in the combination therapy group at 12 h and 24 h (Figures 6B, C).
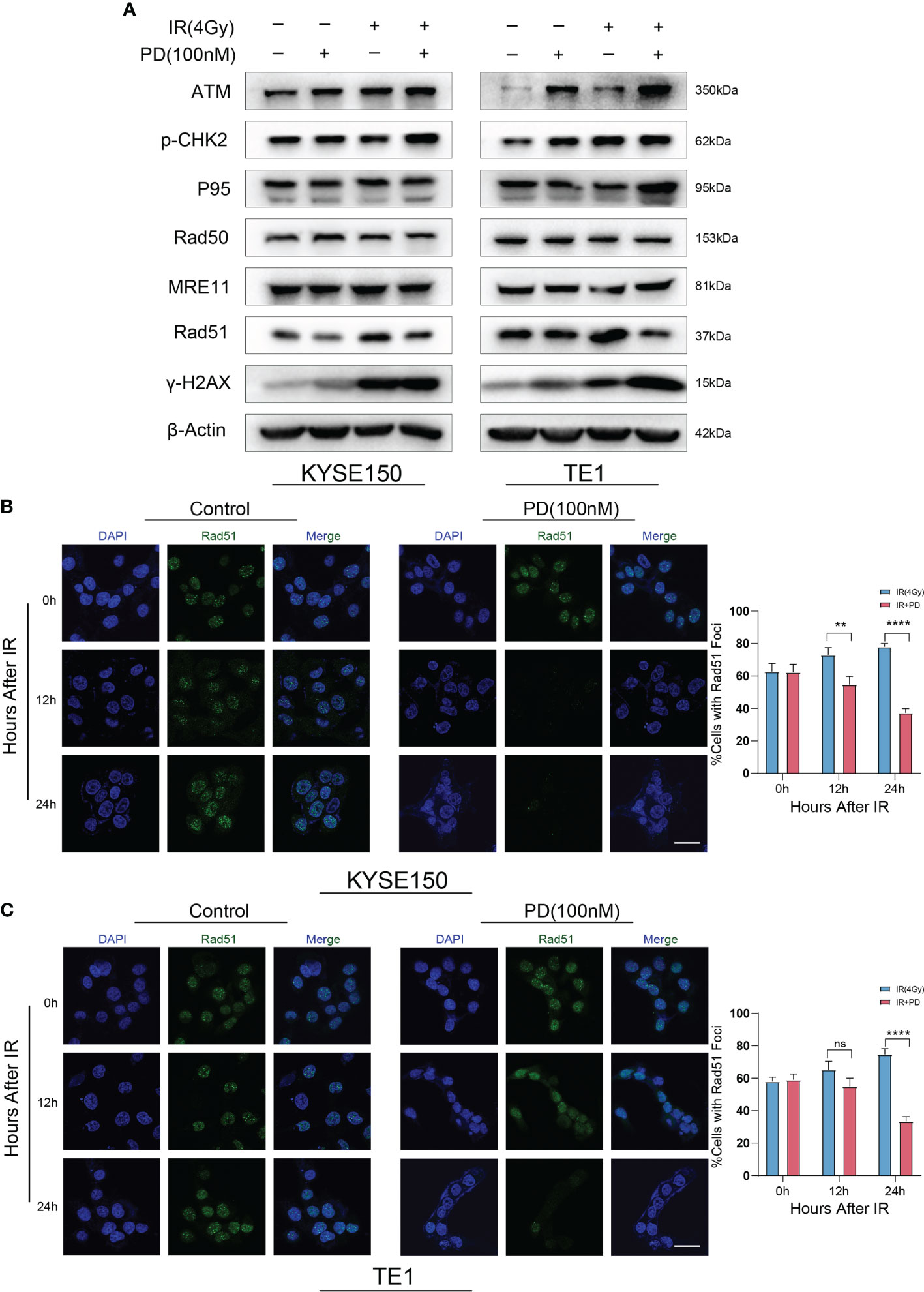
Figure 6 PD regulates DNA damage repair by Rad51. (A) DNA damage repair-related proteins are measured after irradiation, PD inhibits the expression of Rad51 in ESCC cells. (B) Rad51 foci is measured in ESCC cells at different time points with irradiation, combination therapy has few foci per nucleus. Scale bars: 20 μm (B, C). **P < 0.01, ****P < 0.0001, ns, no significance.
Based on TCGA database with ESCC, KEGG/GO enrichment analysis showed that WEE1 and DNA damage repair were related (Figure 7A). Gene Set Enrichment Analysis (GSEA) showed enrichment for DNA replication and cell cycle (Figures 7B, C). We then examined the expression correlations between WEE1 and Rad51 using the TCGA database and revealed that WEE1 expression was statistically positively associated with Rad51 (R = 0.293, p =0.008) (Figure 7D). We then transfected KYSE150 with siRNA to inhibit the expression of WEE1, and we also observed the downregulation of Rad51 (Figure 7E). We performed immunoprecipitation (IP) assay in KYSE150 cells, and obtained the interaction between WEE1 and Rad51 (Figure 7F). Decreased level of Rad51 was also obtained by IF in KYSE150 cells (Figure 7G). We proved that PD inhibited the function of Rad51 because of the inhibition of WEE1.
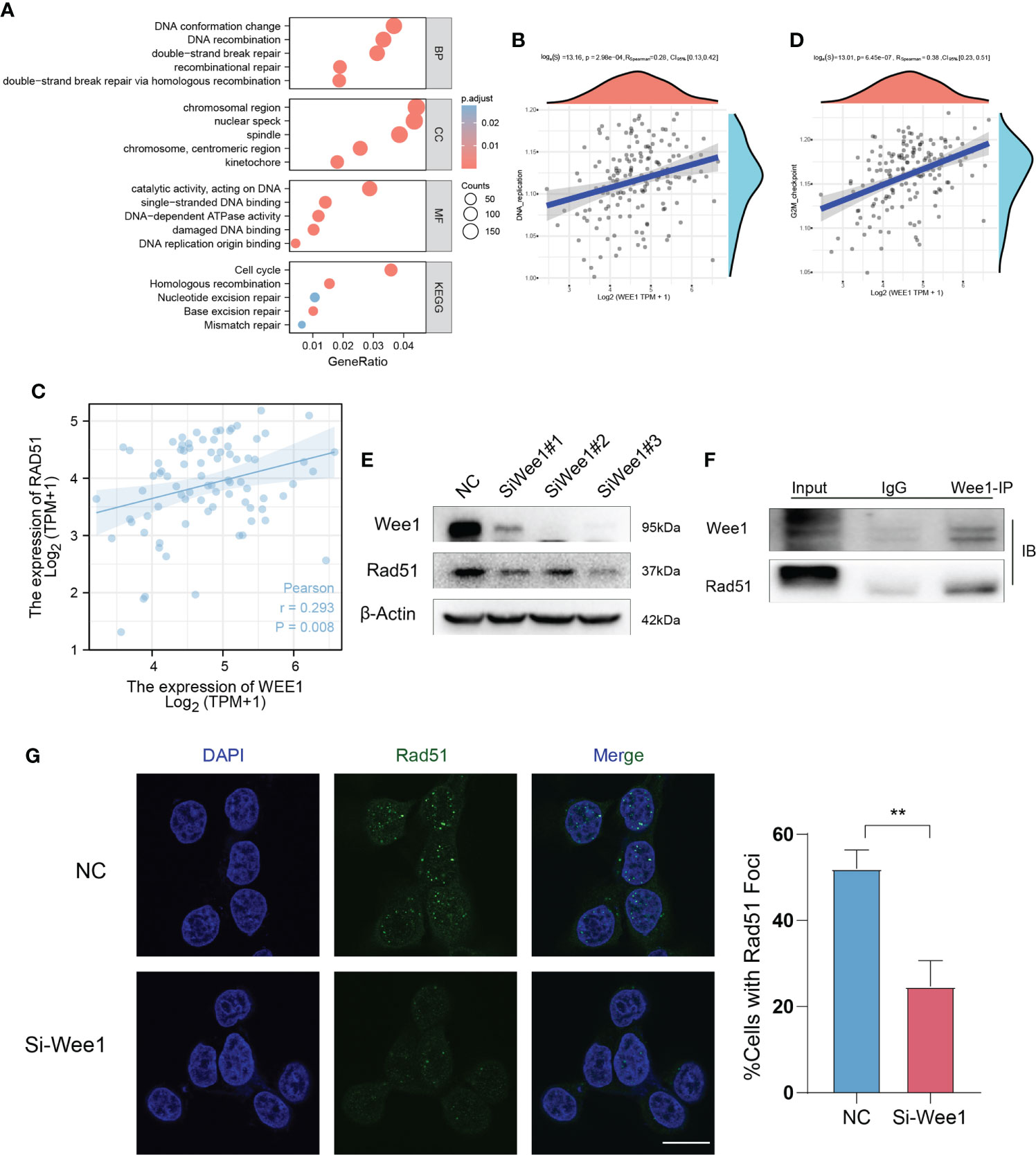
Figure 7 WEE1 interacts with Rad51 in ESCC cells. (A) KEGG/GO enrichment analysis shows that WEE1 and DNA damage repair is related. (B, C) Gene Set Enrichment Analysis (GSEA) shows enrichment for DNA replication and cell cycle. (D) WEE1 expression is statistically positively associated with Rad51. (E) Rad51 is downregulated with WEE1 inhibition in KYSE150 cells. (F) IP assay is performed to measure the interaction with WEE1 and Rad51 in KYSE150 cells. (G) Rad51 foci is measured in KYSE150 cells, Rad51 decreases in WEE1-inhibition cells. Scale bar = 10 μm (G).**P < 0.01.
To determine whether PD can sensitize ESCC cells to irradiation in vivo, we explored the combination therapy of PD and irradiation in vivo using nude mice xenografts with KYSE150 cells. When tumors grow to a certain size (100–150 mm3), mice were randomized to groups that were treated with vehicle, PD (10 mg/kg) alone, irradiation (12 Gy), or the combination of PD and irradiation (Figure 8A). Combination therapy significantly inhibited tumor growth (Figure 8B). We then performed western blot of the tumor lysates, PD reduced the proteins levels of p-CDK1 T14, p-CDK1 Y15 CyclinB1, and Rad51 (Figure 8C), which was consistent with our previous findings. The immunohistochemical staining of tumors showed similar results (Figure 8D). During the treatment, PD showed good safety and tolerability, and no mice died because of treatment. We collected the liver and kidneys of mice for hematoxylin and eosin (HE) staining, there was no obvious tissue damage in organs (Supplementary Figure S2), indicating the potential application of PD.()
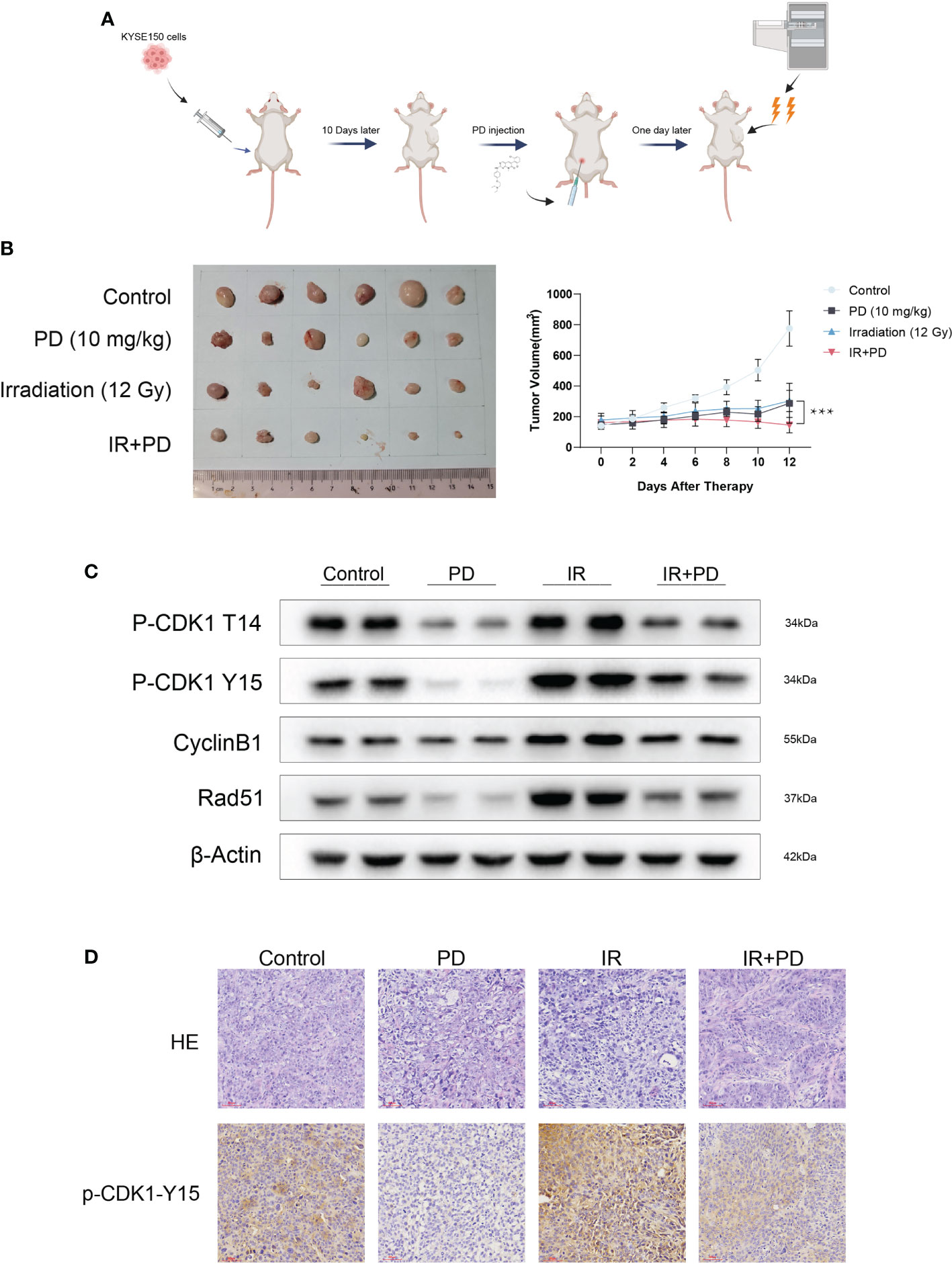
Figure 8 PD is effective with irradiation in vivo. (A) Schematic diagram of PD and irradiation combination therapy (created by BioRender). (B) Combination therapy inhibits tumor growth in vivo. (C) PD inhibits the function of WEE1 and PKMYT1 in vivo. (D) IHC shows PD inhibits the expression of p-CDK1-Y15 in vivo. Scale bar = 60 μm (D). ***P < 0.001.
Esophageal cancer (EC) is the seventh most common cancer worldwide, there are hundreds of thousands of new cases of EC each year, resulting in approximately half a million deaths (1). Radiotherapy is an accepted treatment in ESCC, especially for locally advanced ESCC or tumor recurrence; however, due to the heterogeneity of tumor, treatment leads to different outcomes (14, 31). Radiotherapy mainly treats tumors by DNA damage, which will arrest cell cycle to repair DNA damage. More than 90% ESCC patients have TP53 mutations, which abolish the G1/S checkpoint and make the G2/M checkpoint more important for DNA damage repair (26, 27).
WEE1 kinase family plays an indispensable role in mitosis and meiosis (15, 16, 24, 32). WEE1 inhibition has been proved to be a promising therapeutic target in TP3-mutated tumors. MK1775, as a specific inhibitor of WEE1, has been used in several preclinical studies and clinical trials (22–24, 33). Unfortunately, some patients didn’t respond to WEE1 inhibition as expected, and the mechanism of resistance is unclear (34, 35). Up-regulation of PKMYT1 was also observed in some patients with resistance to Wee1 inhibition, which may explain why some patients had no response to WEE1 inhibition. Evolving research reveals functional redundancy in WEE1 and PKMYT1, which ensures the activation of CDK1 and the coordination of cell cycle (36–39). PKMYT1, different from WEE1, can phosphorylate CDK1 at both Thr14 and Tyr15, and also act as an indispensable regulator of G2/M phase (15, 16). PKMYT1 implicates multiple tumors, overexpression of PKMYT1 frequently leads to poor prognosis (16, 19, 20). Thus, simultaneous inhibition of WEE1 and PKMYT1 is of great significance for the treatment of tumors.
PD0166285 is a compound of the pyridopyrimidine class that inhibits WEE1 and PKMYT1 activities at nanomolar concentrations (29). Our assays demonstrate that cells respond to PD 3-6 hours after dosing, the IC50 values of PD in ESCC cells range from 234 to 694 nM. PD can down-regulate the expression of p-CDK1 T14 and Y15 at the same time, which suggests that PD inhibits the normal activity of WEE1 and PKMYT1. Interestingly, we also reveal that PD inhibits the expression of WEE1 and PKMYT1 with escalating PD concentrations. We observe that PD regulates the cell cycle of ESCC cells and downregulates the G2/M ratio of cell cycle. Meanwhile, PD exhibits potent antitumor efficacy, where cell apoptosis can be induced by the compound in a dose-dependent manner.
Irradiation can directly cause DNA damage thus activate DNA damage checkpoint, which leads to cell cycle arrest. Due to the deficiency of TP53 and abolition of G1/S arrest, G2/M checkpoint is more dependent on DNA repair after irradiation. We have observed the upregulation of WEE1 and PKMYT1 in ESCC cells when treated with irradiation (24). We revealed that the administration of PD before irradiation can inhibit the phosphorylation of CDK1 and abolish G2/M phase arrest, which will decrease DNA repair and result in cell death both in vitro and in vivo. In addition to regulating the cell cycle, we also demonstrated that PD can directly regulate DNA repair pathways through inhibition of Rad51, an essential component of the homologous recombination repair pathway (40). Recent studies pointed out that the formation foci of Rad51 in cell is an important marker of HR repair (41, 42). In our research, PD significantly reduced the Rad51 foci formation and the protein expression simultaneously. In order to verify the mechanism, we performed genome sequencing to determine the relationship between WEE1 and Rad51 in ESCC. We demonstrate the expression of WEE1 correlates with Rad51, and after inhibiting WEE1 with siRNAs, the expression of Rad51 was reduced in the same time. The result of Immunoprecipitation further agrees with the previous results, we confirm the direct regulation between WEE1 and Rad51. Thus, we believe that PD can affect the sensitivity of ESCC to radiotherapy by regulating DNA repair in both direct and indirect manners (Figure 9).
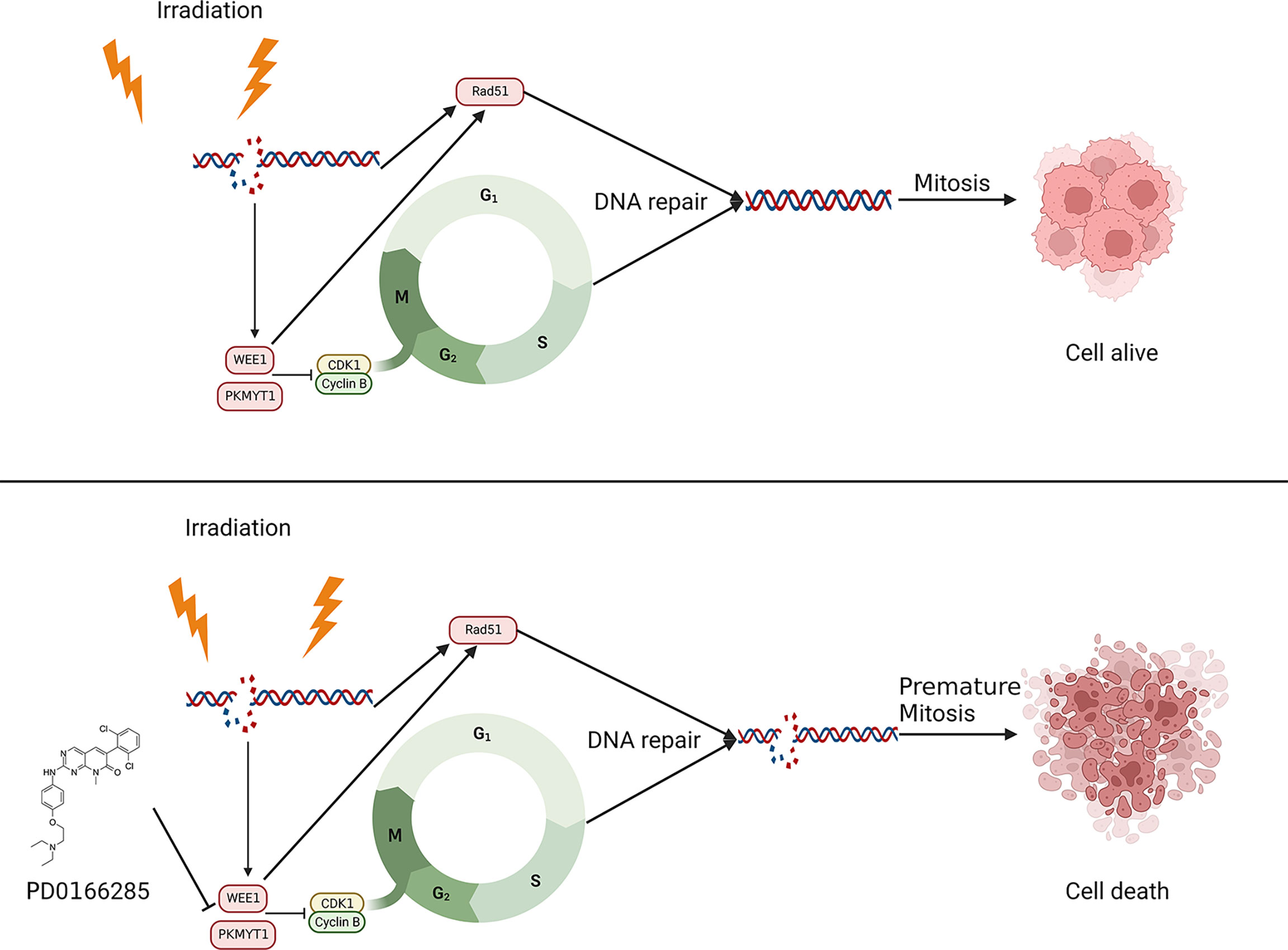
Figure 9 Schematic showing the potential therapeutic effects of PD toward ESCC.
In conclusion, our research demonstrate that PD0166285 can exert antitumor effect by inhibiting the function of WEE1 and PKMYT1 in ESCC cells, and also sensitize ESCC cells to irradiation not only by abolishing G2/M arrest but attenuating DNA repair directly. We believe PD0166285 can be a potent treatment option for ESCC in the future and further clinical trials are needed.
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
The animal study was reviewed and approved by Committee of Animal Experimental Ethical Inspection of the First Affiliated Hospital, College of Medicine, Zhejiang University.
QZ, XL and KJ contributions to project design and data acquisition. JD and QL analyzed the data. LK and ZW performed most assays, LY, PX, PN, WL revised the manuscript critically for content. JH proved this manuscript to publish. All authors contributed to the article and approved the submitted version.
The present study was supported by The Zhejiang Province Major Science and Technology Special Program Project (grant no. 2020C03058), The Zhejiang Province Lung Tumor Diagnosis and Treatment Technology Research Supported (grant no. JBZX-202007).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2022.1061988/full#supplementary-material
1. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin (2021) 71(3):209–49. doi: 10.3322/caac.21660
2. Ilson DH, van Hillegersberg R. Management of patients with adenocarcinoma or squamous cancer of the esophagus. Gastroenterology (2018) 154(2):437–51. doi: 10.1053/j.gastro.2017.09.048
3. Huang FL, Yu SJ. Esophageal cancer: Risk factors, genetic association, and treatment. Asian J Surg (2018) 41(3):210–5. doi: 10.1016/j.asjsur.2016.10.005
4. Smyth EC, Lagergren J, Fitzgerald RC, Lordick F, Shah MA, Lagergren P, et al. Oesophageal cancer. Nat Rev Dis Primers (2017) 3:17048. doi: 10.1038/nrdp.2017.48
5. Najafi F. Tobacco smoking and alcohol drinking: Two clinically significant risk factors for esophageal squamous cell carcinoma. Gastroenterology (2019) 157(3):897. doi: 10.1053/j.gastro.2019.04.054
6. Lee CH, Wu DC, Lee JM, Wu IC, Goan YG, Kao EL, et al. Carcinogenetic impact of alcohol intake on squamous cell carcinoma risk of the oesophagus in relation to tobacco smoking. Eur J Cancer (2007) 43(7):1188–99. doi: 10.1016/j.ejca.2007.01.039
7. Cook MB, Corley DA, Murray LJ, Liao LM, Kamangar F, Ye W, et al. Gastroesophageal reflux in relation to adenocarcinomas of the esophagus: a pooled analysis from the barrett's and esophageal adenocarcinoma consortium (BEACON). PLoS One (2014) 9(7):e103508. doi: 10.1371/journal.pone.0103508
8. Quante M, Graham TA, Jansen M. Insights into the pathophysiology of esophageal adenocarcinoma. Gastroenterology (2018) 154(2):406–20. doi: 10.1053/j.gastro.2017.09.046
9. Wei W, Zeng H, Zheng R, Zhang S, An L, Chen R, et al. Cancer registration in China and its role in cancer prevention and control. Lancet Oncol (2020) 21(7):e342–e9. doi: 10.1016/S1470-2045(20)30073-5
10. Chen W, Zheng R, Baade PD, Zhang S, Zeng H, Bray F, et al. Cancer statistics in China, 2015. CA Cancer J Clin (2016) 66(2):115–32. doi: 10.3322/caac.21338
11. Nguyen GH, Schetter AJ, Chou DB, Bowman ED, Zhao R, Hawkes JE, et al. Inflammatory and microRNA gene expression as prognostic classifier of barrett's-associated esophageal adenocarcinoma. Clin Cancer Res (2010) 16(23):5824–34. doi: 10.1158/1078-0432.CCR-10-1110
12. Mwachiro MM, Burgert SL, Lando J, Chepkwony R, Bett C, Bosire C, et al. Esophageal squamous dysplasia is common in asymptomatic kenyans: A prospective, community-based, cross-sectional study. Am J Gastroenterol (2016) 111(4):500–7. doi: 10.1038/ajg.2016.26
13. Herskovic A, Russell W, Liptay M, Fidler MJ, Al-Sarraf M. Esophageal carcinoma advances in treatment results for locally advanced disease: review. Ann Oncol (2012) 23(5):1095–103. doi: 10.1093/annonc/mdr433
14. Lagergren J, Smyth E, Cunningham D, Lagergren P. Oesophageal cancer. Lancet (2017) 390(10110):2383–96. doi: 10.1016/S0140-6736(17)31462-9
15. Mueller PR, Coleman TR, Kumagai A, Dunphy WG. Myt1: a membrane-associated inhibitory kinase that phosphorylates Cdc2 on both threonine-14 and tyrosine-15. Science (1995) 270(5233):86–90. doi: 10.1126/science.270.5233.86
16. Asquith CRM, Laitinen T, East MP. PKMYT1: a forgotten member of the WEE1 family. Nat Rev Drug Discovery (2020) 19(3):157. doi: 10.1038/d41573-019-00202-9
17. Mir SE, De Witt Hamer PC, Krawczyk PM, Balaj L, Claes A, Niers JM, et al. In silico analysis of kinase expression identifies WEE1 as a gatekeeper against mitotic catastrophe in glioblastoma. Cancer Cell (2010) 18(3):244–57. doi: 10.1016/j.ccr.2010.08.011
18. Masaki T, Shiratori Y, Rengifo W, Igarashi K, Yamagata M, Kurokohchi K, et al. Cyclins and cyclin-dependent kinases: comparative study of hepatocellular carcinoma versus cirrhosis. Hepatology (2003) 37(3):534–43. doi: 10.1053/jhep.2003.50112
19. Zhang Q, Zhao X, Zhang C, Wang W, Li F, Liu D, et al. Overexpressed PKMYT1 promotes tumor progression and associates with poor survival in esophageal squamous cell carcinoma. Cancer Manag Res (2019) 11:7813–24. doi: 10.2147/CMAR.S214243
20. Jeong D, Kim H, Kim D, Ban S, Oh S, Ji S, et al. Protein kinase, membraneassociated tyrosine/threonine 1 is associated with the progression of colorectal cancer. Oncol Rep (2018) 39(6):2829–36. doi: 10.3892/or.2018.6371
21. Lheureux S, Cristea MC, Bruce JP, Garg S, Cabanero M, Mantia-Smaldone G, et al. Adavosertib plus gemcitabine for platinum-resistant or platinum-refractory recurrent ovarian cancer: a double-blind, randomised, placebo-controlled, phase 2 trial. Lancet (2021) 397(10271):281–92. doi: 10.1016/S0140-6736(20)32554-X
22. Liu JF, Xiong N, Campos SM, Wright AA, Krasner C, Schumer S, et al. Phase II study of the WEE1 inhibitor adavosertib in recurrent uterine serous carcinoma. J Clin Oncol (2021) 39(14):1531–9. doi: 10.1200/JCO.20.03167
23. Seligmann JF, Fisher DJ, Brown LC, Adams RA, Graham J, Quirke P, et al. Inhibition of WEE1 is effective in TP53- and RAS-mutant metastatic colorectal cancer: A randomized trial (FOCUS4-c) comparing adavosertib (AZD1775) with active monitoring. J Clin Oncol (2021) 39(33):3705–15. doi: 10.1200/JCO.21.01435
24. Moiseeva TN, Qian C, Sugitani N, Osmanbeyoglu HU, Bakkenist CJ. WEE1 kinase inhibitor AZD1775 induces CDK1 kinase-dependent origin firing in unperturbed G1- and s-phase cells. Proc Natl Acad Sci U S A (2019) 116(48):23891–3. doi: 10.1073/pnas.1915108116
25. Yang L, Shen C, Pettit CJ, Li T, Hu AJ, Miller ED, et al. Wee1 kinase inhibitor AZD1775 effectively sensitizes esophageal cancer to radiotherapy. Clin Cancer Res (2020) 26(14):3740–50. doi: 10.1158/1078-0432.CCR-19-3373
26. Cancer Genome Atlas Research Network, Analysis Working Group: Asan University, Brigham and Women's Hospital, Broad Institute, Brown University, et al. Integrated genomic characterization of oesophageal carcinoma. Nature (2017) 541(7636):169–75. doi: 10.1038/nature20805
27. Song Y, Li L, Ou Y, Gao Z, Li E, Li X, et al. Identification of genomic alterations in oesophageal squamous cell cancer. Nature (2014) 509(7498):91–5. doi: 10.1038/nature13176
28. Lin DC, Dinh HQ, Xie JJ, Mayakonda A, Silva TC, Jiang YY, et al. Identification of distinct mutational patterns and new driver genes in oesophageal squamous cell carcinomas and adenocarcinomas. Gut (2018) 67(10):1769–79. doi: 10.1136/gutjnl-2017-314607
29. Wang Y, Li J, Booher RN, Kraker A, Lawrence T, Leopold WR, et al. Radiosensitization of p53 mutant cells by PD0166285, a novel G(2) checkpoint abrogator. Cancer Res (2001) 61(22):8211–7.
30. Li J, Wang Y, Sun Y, Lawrence TS. Wild-type TP53 inhibits G(2)-phase checkpoint abrogation and radiosensitization induced by PD0166285, a WEE1 kinase inhibitor. Radiat Res (2002) 157(3):322–30. doi: 10.1667/0033-7587(2002)157[0322:wttigp]2.0.co;2
31. Cui Y, Chen H, Xi R, Cui H, Zhao Y, Xu E, et al. Whole-genome sequencing of 508 patients identifies key molecular features associated with poor prognosis in esophageal squamous cell carcinoma. Cell Res (2020) 30(10):902–13. doi: 10.1038/s41422-020-0333-6
32. Matheson CJ, Backos DS, Reigan P. Targeting WEE1 kinase in cancer. Trends Pharmacol Sci (2016) 37(10):872–81. doi: 10.1016/j.tips.2016.06.006
33. Guertin AD, Li J, Liu Y, Hurd MS, Schuller AG, Long B, et al. Preclinical evaluation of the WEE1 inhibitor MK-1775 as single-agent anticancer therapy. Mol Cancer Ther (2013) 12(8):1442–52. doi: 10.1158/1535-7163.MCT-13-0025
34. Do K, Wilsker D, Ji J, Zlott J, Freshwater T, Kinders RJ, et al. Phase I study of single-agent AZD1775 (MK-1775), a Wee1 kinase inhibitor, in patients with refractory solid tumors. J Clin Oncol (2015) 33(30):3409–15. doi: 10.1200/JCO.2014.60.4009
35. Takebe N, Naqash AR, O'Sullivan Coyne G, Kummar S, Do K, Bruns A, et al. Safety, antitumor activity, and biomarker analysis in a phase I trial of the once-daily Wee1 inhibitor adavosertib (AZD1775) in patients with advanced solid tumors. Clin Cancer Res (2021) 27(14):3834–44. doi: 10.1158/1078-0432.CCR-21-0329
36. Ayeni JO, Varadarajan R, Mukherjee O, Stuart DT, Sprenger F, Srayko M, et al. Dual phosphorylation of cdk1 coordinates cell proliferation with key developmental processes in drosophila. Genetics (2014) 196(1):197–210. doi: 10.1534/genetics.113.156281
37. Okumura E, Fukuhara T, Yoshida H, Hanada Si S, Kozutsumi R, Mori M, et al. Akt inhibits Myt1 in the signalling pathway that leads to meiotic G2/M-phase transition. Nat Cell Biol (2002) 4(2):111–6. doi: 10.1038/ncb741
38. Aarts M, Sharpe R, Garcia-Murillas I, Gevensleben H, Hurd MS, Shumway SD, et al. Forced mitotic entry of s-phase cells as a therapeutic strategy induced by inhibition of WEE1. Cancer Discovery (2012) 2(6):524–39. doi: 10.1158/2159-8290.CD-11-0320
39. Jin Z, Homola E, Tiong S, Campbell SD. Drosophila myt1 is the major cdk1 inhibitory kinase for wing imaginal disc development. Genetics (008) 180(4):2123–33. doi: 10.1534/genetics.108.093195
40. Godin S, Wier A, Kabbinavar F, Bratton-Palmer DS, Ghodke H, Van Houten B, et al. The shu complex interacts with Rad51 through the Rad51 paralogues Rad55-Rad57 to mediate error-free recombination. Nucleic Acids Res (2013) 41(8):4525–34. doi: 10.1093/nar/gkt138
41. Baumann P, West SC. Role of the human RAD51 protein in homologous recombination and double-stranded-break repair. Trends Biochem Sci (1998) 23(7):247–51. doi: 10.1016/s0968-0004(98)01232-8
Keywords: PD0166285, esophageal squamous cell carcinoma, WEE1, PKMYT1, radiotherapy
Citation: Zhang Q, Lin X, Jiang K, Deng J, Ke L, Wu Z, Xia P, Li Q, Yu L, Ni P, Lv W and Hu J (2022) PD0166285 sensitizes esophageal squamous cell carcinoma to radiotherapy by dual inhibition of WEE1 and PKMYT1. Front. Oncol. 12:1061988. doi: 10.3389/fonc.2022.1061988
Received: 05 October 2022; Accepted: 19 October 2022;
Published: 04 November 2022.
Edited by:
Mingzhou Guo, People’s Liberation Army General Hospital, ChinaReviewed by:
Zifu Li, Huazhong University of Science and Technology, ChinaCopyright © 2022 Zhang, Lin, Jiang, Deng, Ke, Wu, Xia, Li, Yu, Ni, Lv and Hu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jian Hu, ZHJfaHVqaWFuQHpqdS5lZHUuY24=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.