 Waseem Nasr
Waseem Nasr Marie-Dominique Filippi
Marie-Dominique Filippi- 1Division of Experimental Hematology and Cancer Biology, Cincinnati Children’s Research Foundation, Cincinnati, OH, United States
- 2University of Cincinnati College of Medicine, Cincinnati, OH, United States
The disorders known as bone marrow failure syndromes (BMFS) are life-threatening disorders characterized by absence of one or more hematopoietic lineages in the peripheral blood. Myelodysplastic syndromes (MDS) are now considered BMF disorders with associated cellular dysplasia. BMFs and MDS are caused by decreased fitness of hematopoietic stem cells (HSC) and poor hematopoiesis. BMF and MDS can occur de novo or secondary to hematopoietic stress, including following bone marrow transplantation or myeloablative therapy. De novo BMF and MDS are usually associated with specific genetic mutations. Genes that are commonly mutated in BMF/MDS are in DNA repair pathways, epigenetic regulators, heme synthesis. Despite known and common gene mutations, BMF and MDS are very heterogenous in nature and non-genetic factors contribute to disease phenotype. Inflammation is commonly found in BMF and MDS, and contribute to ineffective hematopoiesis. Another common feature of BMF and MDS, albeit less known, is abnormal mitochondrial functions. Mitochondria are the power house of the cells. Beyond energy producing machinery, mitochondrial communicate with the rest of the cells via triggering stress signaling pathways and by releasing numerous metabolite intermediates. As a result, mitochondria play significant roles in chromatin regulation and innate immune signaling pathways. The main goal of this review is to investigate BMF processes, with a focus mitochondria-mediated signaling in acquired and inherited BMF.
Introduction
Ineffective hematopoiesis leading to the absence of one or more hematopoietic lineages in the peripheral blood represents broad and heterogeneous blood disorders comprised of bone marrow failure (BMF) and myelodysplastic (MDS) syndromes. Patients with BMF or MDS suffer from a severe reduction of one or more hematopoietic lineages in the peripheral blood, which is life-threatening (1–4) BMF can be inherited or acquired. The most common inherited BMFs include Fanconi anemia, Shwachman-Diamond syndrome, congenital amegakaryocytic thrombocytopenia, and reticular dysgenesis. Other inherited BMF are X-linked recessive dyskeratosis congenita and Blackfan-Diamond Anemia (5–7). MDS, which are now classified as acquired disorders that resemble BMF with a variety of cell dysplastic features, may occur de novo or secondary to BMF. MDS are classified in several groups based on established clinical and histopathological features, as defined by the World Health Organization: MDS with single lineage dysplasia, MDS with ring sideroblasts (MDS-RS), MDS with multilineage dysplasia, MDS with excess blasts (MDS-EB), MDS with isolated del(5q). BMF/MDS can also appear after allogenic or autologous hematopoietic stem cell transplantation (HSCT) (8, 9), as well as after myeloablative chemotherapy (10), and are called therapy-related BMF/MDS. Some patients develop secondary MDS/AML within 6 years of autologous HSCT (11). Although BMF/MDS are very heterogeneous, the same genomic mutations are frequently found in MDS patients such as mutations in genes related to RNA splicing (SF3B1, SRSF2, U2F1, ZRSR2), DNA methylation (TET2, DNMT3A, IDH1/IDH2), chromatin modification (ASXL1, EZH2), transcription regulation (RUNX1, BCOR), and DNA repair control (p53). These observations suggest that additional environmental factors largely contribute to disease development. Substantial clinical data have shown that hyperactivity of inflammatory cytokines, including TNFα, IL-6, and transforming growth factor–β (TGFβ), directly contribute to hematopoietic failure in BMF/MDS.1 (12–14), Chronic inflammation and enhanced innate immune signaling are also recognized as contributing factors of inefficient hematopoiesis (15–17). Interestingly, several evidence suggest that disruption of mitochondria is another preponderant factor in BMF/MDS development. Abnormal mitochondria have been linked to both acquired and hereditary BMF (18–24). Patients with MDS have transcriptional, morphological and functional dysregulation of their mitochondria, according to several studies (25–28). Some of these defects are the direct consequences of abnormal expression of nuclear-encoded mitochondrial genes. Others could arise in response to stress or the inflammatory milieu. The functions of mitochondria, which supply energy and metabolic activity in response to cellular demand (29), go well beyond energy production. Mitochondria communicate with the rest of the cell through activation of signaling pathways and control a broad range of cellular functions such as apoptosis, iron metabolism and heme production. In addition, mitochondria participate in the generation of metabolite intermediates, acetyl-CoA and S-Adenosyl-Methionine (SAM), used for epigenetic remodeling, as well as those for de novo biosynthetic processes, including nucleotides and fatty acids. Finally, mitochondria control the cellular response to stress, including inflammation stress. This review will discuss the emerging role of mitochondria as driver of de novo or secondary BMF/MDS. It will discuss the potential mechanism, direct or indirect, of how abnormal mitochondrial functions contribute to ineffective hematopoiesis.
MDS with sideroblasts are mitochondrial diseases
Mitochondria are home of heme synthesis
The first step in heme biosynthesis takes place into mitochondria and involves the condensation of succinyl-CoA and glycine to form δ-aminolevulinic acid (ALA) in the mitochondrial matrix. This reaction is catalyzed by ALA synthase (ALAS). There are two isoforms of ALAS, ALAS1 and ALAS2, which is found exclusively in erythroid cells. ALA is exported to the cytosol via SLC25A38 and ABCB10 where it is converted to coproporphyrinogen III (CPgenIII). CPgenIII is imported back into mitochondria, where it is converted to protoporphyrinogen IX by coproporphyrinogen oxidase (CPOX). Then, protoporphyrinogen IX is oxidized to protoporphyrin IX (PPIX) by protoporphyrinogen oxidase (PPOX). Finally, ferrous iron is incorporated into PPIX to form heme in the mitochondrial matrix, a reaction catalyzed by ferrochelatase (FECH) (30). T the expression of both Alas2 and FECH is controlled by iron, thus linking the regulation of heme biosynthesis in erythroid cells to the availability of iron. Iron is acquired by differentiating erythroid progenitors via transferrin receptor 1 (TfR1)-mediated endocytosis and transferred to mitochondria for heme synthesis via mitoferrin1 (MFRN1) and mitoferrin2 (MFRN2), expressed in erythroid and non-erythroid tissues, respectively. The generation of globin and heme levels in erythroid precursors is balanced by a cell membrane heme exporter known as feline leukemia virus subgroup C receptor 1 (FLVCR1). Flvcr1b, an isoform of Flvcr1 that is present in mitochondria, facilitates heme efflux into the cytoplasm (31).
MDS with RS: A mitochondrial disorder affecting the erythroid lineage
Sideroblastic anemia, congenital or acquired, are associated with MDS and are characterized by the presence of ring sideroblasts, which result from decreased heme production and excess iron deposit within mitochondria of erythroid cells (32). Mutations in genes related to heme synthesis are drivers of MDS-RS. Mutation in ALAS2 reduces protoporphyrin causing an accumulation of iron in mitochondria and subsequently cell death. Mutations in Ala carriers, ABCB7 and in Slc25a38, also reduces heme synthesis and causes MDS-RS. Germline mutation in the Glutaredoxin 5 [GLRX5] gene causes iron overload and is associated with sideroblastic-like microcytic anaemia. GLRX5 is a mitochondrial protein, which is involved in the biogenesis of iron-sulfur clusters.
The splicing factor SF3B1 is the most commonly mutated genes in MDS with the disease phenotype with ring sideroblasts (33, 34). SF3B1 Splicing factor 3b, together with splicing factor 3a and a 12S RNA unit, forms the U2 small nuclear ribonucleoproteins complex (U2 snRNP) and binds pre-mRNA upstream of the intron’s branch site in a sequence independent manner. SF3B1 mutations cause abnormal mitochondrial iron absorption and ineffective erythropoiesis (35). Initial studies found that SF3B1-mutant erythroblasts displayed larger quantities of mitochondria. When co-mutation with EZH2 occurs, mitochondrial membrane potential is abnormal and ROS are increased; likely driving cell death (1). The exact molecular mechanism behind the abnormal mitochondrial functions and iron deposition is being uncovered. Mutated-SF3B1 notably targets expression of genes involved in mitochondrial heme synthesis such as PPOX, TMEM14C and Abcb7, causing a block in protoporphyrin synthesis (36–39). Interestingly, SFB1 mutation confers proliferation advantage to the clone. In a remarkable study by Hsu (40), the clonal evolution of MDS was studied using iPSCs reprogrammed from patient samples and shows that the initial mutation is t (4, 12), followed by mutations in SF3B1, EZH2, and del(5q), in that sequence.
Another study described the importance of the Retinoblastoma protein (pRb) gene, a crucial cell cycle regulator that controls the transition from the G1 to the S phase, in mitochondrial functions in erythroid cells. Deletion of Rb in erythroid cells caused poor erythropoiesis with dysplastic features due to abnormal mitochondrial biogenesis and cell cycle exit. Erythroid-specific deletion of pRb led to decreased expression of mitochondria-related genes, a reduction in mitochondrial membrane potential and a change in the ROS produced by the mitochondria. Expression of critical oxidative phosphorylation genes such Ndufa1 (complex 1, OXPHOS), Atp5s (ATP synthesis), and Cox7b (electron transfer), expression of the mitochondrial biogenesis gene PGC1b, of the mitochondrial antioxidant Prdx3, which is crucial for maintaining the balance of (ROS), as well as ALAS2, and ABCB were all decreased. In this Rb-deficiency mouse model, overexpressing PGC1b was sufficient to normalize the RBC counts, underscoring the critical role of mitochondria in the pathogenesis of the disease (27).
Finally, decreased FLVCR1 levels or increased expression of aberrant alternative splicing of FLVCR1 transcript are seen in DBA patients and a cellular model of DBA (41, 42). Downregulation of FLVCR1a and FLVCR1b results in an increase in oxidative stress, cell cycle arrest at G0/G1, and apoptosis due to heme accumulation. This is yet another illustration of how a flaw in mitochondrial homeostasis can result in ineffective erythropoiesis and BMF. Germline mutations in other genes, such as PUS1, YARS2, SLC19A2 and TRNT1, as well as mitochondrial DNA deletions, have been identified in distinct forms of inherited sideroblastic anemias (32) (Figure 1).
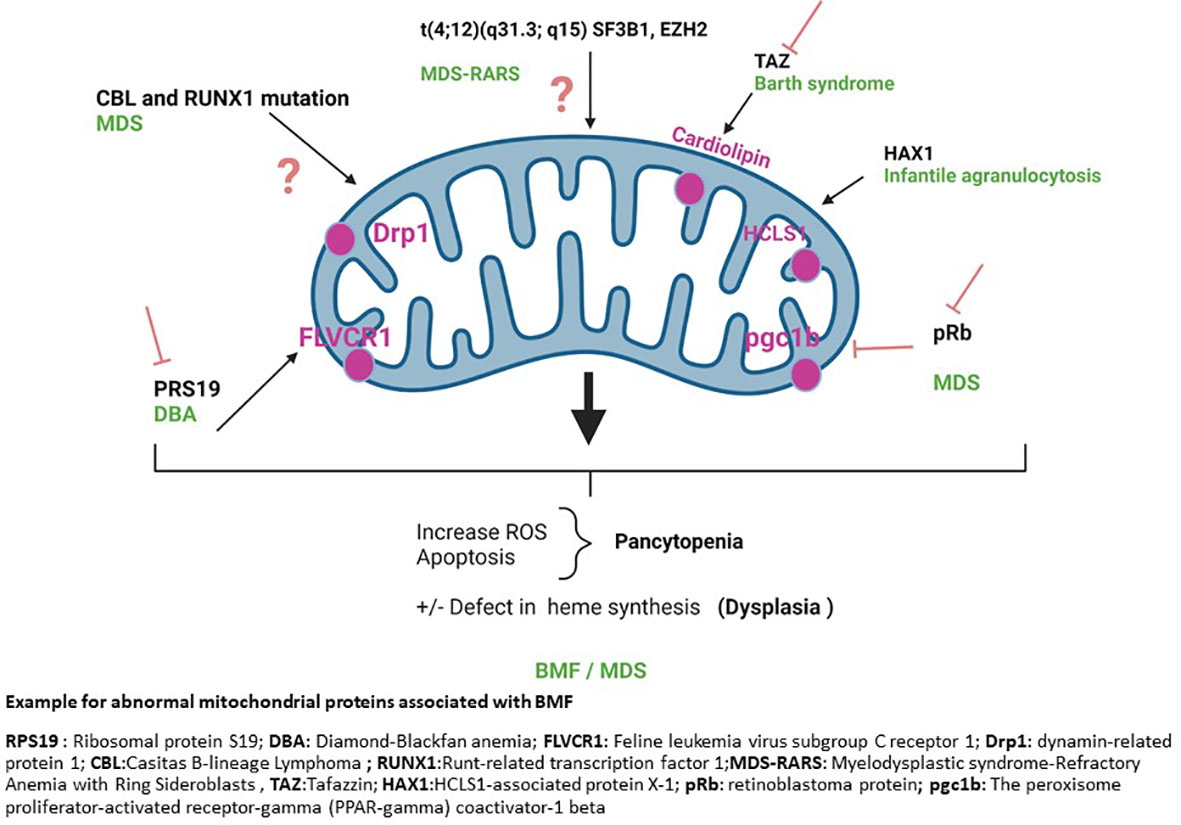
Figure 1 Example for abnormal mitochondrial proteins associated with BMF. RPS19, Ribosomal protein S19; DBA, Diamond-Blackfan anemia; FLVCR1, Feline leukemia virus subgroup C receptor 1; Drp1, dynamin-related protein 1; CBL, Casitas B-lineage Lymphoma; RUNX1, Runt-related transcription factor 1; MDS-RARS, Myelodysplastic syndrome-Refractory Anemia with Ring Sideroblasts, TAZ, Tafazzin; HAX1, HCLS1-associated protein X-1; pRb, retinoblastoma protein; pgc1b, The peroxisome proliferator-activated receptor-gamma (PPAR-gamma) coactivator-1 beta.
Mutations in genes that alter the generation of mitochondrially –produced metabolites cause MDS
Mitochondrial functions and epigenetic regulation are tightly linked in several ways. One way is through the tricarboxylic acid (TCA) cycle – a major mitochondrial metabolic pathway. The TCA cycle produces several intermediate metabolites, citrate, alpha-ketoglutarate (α-KG), itaconate, succinate, fumarate, malate and oxaloacetate, through a series of enzymatic reactions. When the TCA metabolites are coupled with the mitochondrial electron transport chain, TCA intermediates are used for subsequent metabolic reactions through oxidative phosphorylation (OXPHOS) to generate ATP. TCA metabolites also serve in non-metabolic signaling roles. For example, itaconate, succinate, fumarate have all been shown to alter the innate immune response. In addition, the TCA metabolites are directly involved in epigenetic regulation. Succinate and fumarate can directly inhibit the activity of histone or DNA demethylase. Alpha-KG is needed for the activity of DNA demethylase. Acetyl-CoA serve as donor group of histone acetylation; S-Adenosyl-Methionine (SAM) which serve as donor group for DNA or histone methylation is generated through a complex interaction between the mitochondrial one-carbon folate pathway and the methionine cycle (43). The tight connection between mitochondrial metabolism and chromatin regulation is one component of the preponderant, yet ill-understood, role of mitochondria in MDS pathogenesis. Mutations in the TCA enzyme IDHs (IDH1-IDH2) occur in about 7% of MDS cases, with IDH2 mutations being more frequent (about 4.5%) than IDH1 mutations (about 2.5%). (44) IDH2 mutations are particularly enriched in the RAEB subtype of MDS. IDH1/2 catalyzes the oxidation of isocitrate to oxalosuccinate within the TCA cycle, which is followed by decarboxylation of the carboxyl group beta to the ketone to form α-KG. This reaction also generates NADPH. 44 Mutations in IDH1/2 thus by impacting α-KG production alter the activity of metabolic enzymes that depend on α-KG availability, such as the DNA demethylase Tet2. Tet2 primarily catalyzes the oxidation of 5-methylcytosine (5mC) to 5-hydroxymethylcytosine (5hmC). Mutations in Tet2 are also associated with clonal hematopoiesis, increased risk of MDS progression, and poor prognosis in AML (45). Interestingly, IDH2 mutations are mutually exclusive with TET2 and SF3B1 mutations, but are frequently associated with SRSF2 mutations. Other known mutations in epigenetic regulators that are associated with clonal hematopoiesis and MDS are found in the DNA methylase DNMT3a (46). DNMT3A, catalyzes the methylation of CpG dinucleotides in genomic DNA, which is dependent on SAM availability. Hence, stressors that alter mitochondrial functions could easily contribute to disease development and participate in disease heterogeneity in a given genetic background. Any abnormality in mitochondrial functions that would cause abnormal production of SAM or aKG would also alter Tet2 or DNMT3a functions and could drive MDS pathogenesis without the need for somatic mutation in specific genes (see below).
Because of the link between mitochondria and epigenetics, altered mitochondrial metabolism is a common characteristics of MDS/BMF that drives disease phenotype. Interestingly, in addition to exhibiting specific metabolic alterations that result from the genetic context, MDS have a common abnormal metabolic signature. Elegant studies from the Huang’s lab showed that hypoxia-inducible factor 1α (HIF1A) transcriptional signature is generally activated in MDS patient bone marrow stem/progenitors, in major MDS-associated mutations (Dnmt3a, Tet2, Asxl1, Runx1, and Mll1). 48 Remarkably, using inducible activation of HIF1A signaling mouse model, they show that HIF1A is sufficient to induce dysplastic and cytopenic MDS phenotypes. On the other hand, both genetic and chemical inhibition of HIF1A signaling rescues MDS phenotypes in a mouse model of MDS, indicating that elevated HIF1A is necessary for MDS phenotype. Therefore, metabolic changes associated with HIF1A are central pathobiologic mediators of MDS. Two other important observations were that HIF1A signature is also associated with enrichment in several inflammatory/immune response–related pathways. Plus, it renders a state of pseudohypoxia and mitochondrial dysfunction in which expressions of nuclear-encoded mitochondrial genes, notably the electron transport chain complex II that is normally important for OXPHOS, are downregulated. In this context, metabolites of the TCA cycle, aKG, succinate, fumarate and malate, accumulate – thus further altering cellular functions (47).
Abnormal mitochondrial dynamics contributes to MDS
Mitochondria are very dynamic organelles, whose numbers and organization can vary greatly. (48–50) The mitochondrial network can be organized into interconnected and fused filaments, or into fragmented and smaller unit (48). Mitochondrial fusion is controlled by mitofusins 1 and 2 (Mfn1 and Mfn2) and optic atrophy 1 and 3 (Opa1, Opa3). Mitochondrial fission is regulated by dynamin-related protein 1 (Drp1) and fission protein 1 (Fis1) (48–50). Mitochondrial dynamism is important to adapt cells to energy demand. When energy demand is high, mitochondria are fused and mitochondrial oxidative phosphorylation [OXPHOS] is favored. Mitochondrial fusion also enables ‘mixing’ mitochondrial membrane proteins for repair mechanisms. A recent study describes the role of aberrant mitochondrial fission as driver of MDS. They found a MDS patient harboring a mutation in both the E3 ubiquitin-protein ligase CBL gene and the transcription factor RUNX1 gene. In a mouse model of CBL exon deletion with RUNX1 mutants, that recapitulated clinically relevant MDS phenotypes, HSC and progenitor cells exhibited excessive mitochondrial fragmentation that was caused by enhanced activity of the mitochondrial fission regulator DRP1. The subsequent elevation in ROS production and inflammatory signals promoted the development of dysplasia and impaired granulopoiesis (18). Other studies have reported abnormal mitochondrial structure, or abnormal mitochondrial biogenesis and mitophagy in BMF, notably in FA. In this case, they found that FA genes are required for selective autophagy, which removes unwanted cytoplasmic contents including mitochondria such that FA gene deficiency results in impaired virophagy and antiviral host defense, decreased Parkin-mediated mitophagy, and increased mitochondrial ROS-dependent inflammasome activation (51). Loss of FA-gene-mediated selective autophagy may contribute to the pathophysiology of FA-gene associated diseases.
Role of reactive oxygen species in developing BMF/MDS
One common characteristic of BMF/MDS is the higher susceptibility of myeloid progenitors to apoptosis. Excessive myeloid cell apoptosis contributes to peripheral cytopenias even when the bone marrow is hypercellular. Numerous variables, including internal or external reactive oxygen species (ROS), can cause apoptosis. ROS comprise radical and non-radical molecules (52), and are often released as a byproduct of oxidative phosphorylation or during mitochondrial stress conditions. To counteract ROS, HSC express enzymatic and nonenzymatic defensive mechanisms, such as superoxide dismutase, glutathione peroxidase, myeloperoxidase, Vitamins C, E, and reduced glutathione (GSH) (53). When compared to controls, MDS patients have significantly higher levels of intracellular peroxides in lymphocytes, erythroid precursors, monocytes and granulocytes, as well as a considerably lower superoxide/peroxides ratio and GSH levels, resulting in oxidative stress and subsequent macromolecule and organelle damages (52, 54).
Chronic oxidative stress is also found in FA cells due to increased DNA damage. This is associated with mitochondrial damage and OXPHOS dysfunction. In fact, spontaneous mitochondrial fragmentation occurs in FA cells that leads to change in mitochondrial distribution, shape, and integrity (55, 56). HSPCs deficient in the FA protein Fancd2 rely on increased mitochondrial translation for survival and proliferation (57). The changes in mitochondrial structure are also accompanied by changes in metabolism. FA cells exhibit lower OXPHOS, increased glycolytic flux and decreased glutaminolysis (23). We know that a balance between glycolysis and OXPHOS is necessary for HSPC differentiation. Quiescent HSC rely mostly on anaerobic glycolysis and lysosomal functions for their energy needs. HSC activation and commitment to differentiation are associated with increased aerobic glycolysis, mitochondrial activation and increased OXPHOS. Hence, abnormal mitochondrial function in FA cells could substantially impact the ability of HSPC to differentiate, further contributing the FA pathogenesis (23). Similarly, a study focused on SBDS gene that affects ribosome biogenesis, mitotic spindle assembly, chemotaxis, and ROS generation, shows that lower expression of SBDS causes defective mitochondria and elevated ROS (58) (Figure 1).
Other mitochondrial abnormalities linked to inherited BMF
Mitochondrial abnormalities are found in a variety of hematologic phenotypes. The Pearson syndrome (24, 59), is a multisystem mitochondrial respiratory chain disorder caused by a single large scale mitochondrial DNA deletion. Patients present with pancytopenia sideroblastic anemia and exocrine pancreatic insufficiency. The Barth syndrome, which presents with neutropenia in addition to musculoskeletal defects and cardiomyopathy, is another mitochondrial disorder (22, 60).The Barth syndrome is caused by a mutation in the gene TAZ. TAZ encodes for the mitochondrial phospholipid transacylase tafazzin. TAZ controls the production of tetralinoleoyl cardiolipin, a mitochondrial membrane-specific lipid. When tafazzin is knocked down by shRNA in mice, tetralinoleoyl cardiolipin levels are drastically reduced, and monolysocardiolipins accumulate in mitochondria (61).The aberrant buildup of monolysocardiolipins causes mitochondrial dysfunction (62) leading to enhanced mitochondrial membrane potential breakdown, abnormal cytochrome c release, caspase-3 activation (63) and cellular death, including in myeloid precursors and neutrophils (64). Similarly, in the Kostmann disease also known as infantile agranulocytosis (severe congenital neutropenia) nonsense mutations in mitochondrial-associated antiapoptotic protein (HCLS1) lead to premature stop codons, loss of function, and frequently, total loss of protein expression, causing acute neutropenia that is often accompanied by neurologic and cognition impairments (65, 66). Finally, BMF and MDS have been linked to mutations in mitochondrial DNA (67) (Figure 1).
It has long been known that mitochondria can be taken up by cells and can transfer from cell to cell, in vivo or in vitro. Because of this, mitochondrial replacement or supplementation in cells is being proposed as novel therapeutic approach of mitochondrial diseases. A recent study shows that mitochondrial augmentation in human CD34+ cells from healthy donors or patients with mitochondrial DNA disorder can prove beneficial (68). The group described a method of ex vivo transfer of HSPC with normal exogenous mitochondria, that they termed mitochondrial augmentation therapy (MAT). They show that MAT can improve mitochondrial content and oxygen consumption of healthy and diseased HSPCs. Importantly, they used xenotransplant in immunodeficient NSGS mice to show that MAT confers HSPCs from a patient with an mtDNA disorder superior human engraftment (68).
Secondary BMF/MDS following hematopoietic stress from bone marrow transplantation: Importance of abnormal mitochondrial functions
Secondary MDS and AML are becoming more widely known as late complications of stem cell transplantation (69). The incidence of treatment-related MDS and AML is between 5 percent and 20 percent 5-10 years after an autologous stem cell transplantation (ASCT) (10). Among all cancers, MDS development occurred in 35% of non-Hodgkin’s lymphoma patients who receive ASCT (70). In a Japanese study, 1.38% lymphoma patients receiving ASCT developed secondary myeloid dysplasia 3 years after transplant; 0.37% lymphoma patients receiving allogeneic SCT developed secondary MDS 3 years later (71). The causes and mechanisms behind the development of secondary BMF/MDS are multiple and could be cell intrinsic or arising from damage in the bone marrow microenvironment.
One clear cell intrinsic mechanism is linked to the therapy of the original disorder. There is strong evidence that alkylating anti-leukemic drugs, or whole body irradiation (TBI) used pre-transplantation cause chromosomal damage that can result in MDS/AML (72).
Increased inflammation following treatment could also be a contributing factor. Inflammatory factors are altered after BMT. Among those factors is the transforming growth factor beta (TGFβ). TGFβ is known to suppress cellular growth and to contribute to ineffective hematopoiesis (73–77). The allogeneic and autologous stem cell transplantation conditioning protocols decrease TGFβ production (78, 79). After roughly 7 weeks of bone marrow repopulation, the plasma level of TGFβ returns to normal (80, 81). Interestingly, this is seen in mouse model as well. Research from our lab has demonstrated that while TGFβ levels are lower in the bone marrow microenvironment, TGFβ protein and signaling are enhanced in HSPC after bone marrow transplantation. In this context, TGFβ acts through p38MAPK to impair HSC self-renewal and cause ineffective hematopoiesis after bone marrow transplantation (13). Increased inflammatory cytokines and associated inflammatory signals are a common characteristic of BMD/BMF. TGFβ plasma levels are elevated in hematopoietic cells of patients with myelodysplastic syndromes MDS (82–84). TGFβ signaling is also elevated in FA patients (85, 86). In a Fanca-deficient mouse model, challenge with polyinosinic:polycytidylic acid (pIC) leads to changing DNA repairing system via enhanced TGFβ signaling and causes BMF due to increased DNA mutations (75). TGFβ signaling inhibition restored hematopoiesis in this mouse model. TGFβ is also elevated in Shwachman-Bodian-Diamond Condition (SBDS) and Diamond Blackfan anemia (DBA) 2. Blocking the TGFβ pathway using a small molecule inhibitor or a TGF-family ligand trap can ameliorate the inefficient erythropoiesis also found in SDS or Diamond Blackfan anemia patients (87–89).
Overall, the role of inflammation in BMF or MDS development is now established. Toll-like receptors (TLRs) or their signaling effectors are often overexpressed in MDS samples compared to healthy controls, enhancing a type I interferon response through NFkB, MAPK, and IRF3 (12, 90, 91). The inflammasome is elevated in BMF/MDS patients and contributes to ineffective hematopoiesis (92–94). The inflammasome is a multiprotein complex composed of the sensor of damage associated molecular patterns (DAMPs), ie NLRP3 Nod-like receptor, an adaptor protein apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC), and caspase-1 causing the release of interleukin-1b (IL-1b) and IL-18 and cell death by pyroptosis (95). Inflammatory milieu and cell death it creates contribute to pancytopenia and ineffective hematopoiesis. In addition, inflammation could provide a selective advantage to mutated HSC clones, as seen in a model of Dnmt3a-loss of functions in which chronic infection drives clonal expansion of the Dnmt3a-mutant clones via INFy (96). Therefore, increased inflammation and/or inflammatory cytokines following transplantation or hematopoietic stress could be a factor contributing to secondary BMF/MDS.
Viral infections activate DNA and RNA sensing pathways to trigger innate immune signaling pathways that converge on an interferon response. The DNA-sensing pathway, cGAS/STING, activates NFkB, IRF3 to clear the viral infection. In response to viral RNA, the innate immune response starts with cytosolic viral RNA sensor retinoic acid inducible gene-I (RIG-I). Then, RIG-I engages the adaptor protein MAVS (Mitochondrial AntiViral Signaling). In turn, MAVS, which is anchored onto mitochondria, triggers a sequence of signaling that converge onto NFkB, IRF3 or the inflammasome (97–99). These pathways have been involved in MDS, directly or indirectly. DDX41 can activate cGAS/STING; mutation in DDX41 are associated with MDS (100). It is important to note that numerous viruses have been connected to the formation of MDS, including CMV (101), HTLV-1 (102), parvovirus B19 (103), and HHV-6 (104). In a very interesting retrospective study on lymphoma patients who developed secondary AML following HSCT indicates that at the time of stem cell transplantation, 1% of patients who received auto-SCT and 5% of patients who received allo-SCT had infections (71). The hypothesis that MDS could start as a viral infection was suggested more than 20 years ago (105). The infection could trigger dysregulated cytokine production in the BM microenvironment, providing optimal growth support to stem cells harboring a mutation (8, 9, 106, 107)., as recent studies are now demonstrating (96).
Stress-induced abnormal mitochondria. Interestingly, abnormal expression of nuclear-encoded mitochondrial genes is a predicting factor of therapy-related MDS (108). Consistent with this, we have shown that HSC keep abnormal mitochondria after BMT, indicating that the stress of BMT permanently alters mitochondria and HSC functions (109). The mechanisms causing alteration in mitochondrial functions in HSC following transplantation involves deregulation in mitochondrial dynamism and decreased expression of mitochondrial genes (109). Inability to remove abnormal mitochondria could contribute to secondary BMF/MDS in multiple ways. Abnormal mitochondria likely cause abnormal metabolism, including abnormal TCA cycle and OXPHOS that could mimic IDH or Tet2 mutation phenotypes. Abnormal mitochondria could contribute to secondary BMF/MDS via activation of innate immune signaling. Indeed, mitochondria serve as platform of innate immune signaling. Activation of numerous innate immune pathways occurs at the plasma membrane of mitochondria and depends on mitochondrial regulation (110, 111). For instance, activation of the inflammasome can depend on ROS production from stressed mitochondria (112). Viral infections, as seen above, lead to MAVS activation (97–99). MAVS activation requires mitochondrial polarization (i.e., established mitochondria membrane potential [MMP]) and is enhanced by mitochondrial fusion (111). Conversely, termination of the innate immune response is mediated by removal of mitochondria via mitophagy (113–115). Finally, mitochondrial stress is often accompanied by an abnormal release of mitochondrial DNA, which could activate DNA sensing pathways and subsequently innate immune signaling. Hence, abnormal mitochondria could be a mediator of inflammation following transplantation directly or in the context of added infection, and thus create an inflammatory a context for a mutated clone to expand.
The case for combinatorial effects: possible interactions between dysregulated TGFB, defective mitochondria and innate immune pathways as causal factors of secondary BMF/MDS. TGFβ upregulation and mitochondria abnormality occur in tandem in many BMFs. The source or consequence of this relationship is not completely clear. Our group recently reported the possible link between overexpression of TGFβ and mitochondria in the development of BMF/MDS (106). Using an TGFβ overexpressing mouse model, we demonstrated that elevated TGFβ signaling alone is not sufficient to cause BMF or MDS. However, elevated TGFβ signaling plus challenge with the double-stranded RNA pIC cause chronic pancytopenia, bone marrow dysplasia, increased hematopoietic stem and progenitor cell pools, which are phenotypes to human BMF. We further showed that elevated TGFβ plus pIC challenge alters mitochondrial functions with an elevated mitochondrial membrane potential and mitochondrial content. The gene expression profile of HSC shows persistent changes in the transcription profile in HSC from overexpressed TGFβ mice challenged with pIC that includes nuclear-encoded mitochondrial genes. Only overexpressed TGFβ HSC had higher expression of mrpl46 and other genes essential for the regulation of mitochondrial translation following pIC stress. This phenotype was associated with elevated levels of reactive oxygen species, and caspase-1 activation (106). Our findings imply that bone marrow failure with dysplastic features can occur without a prior genetic damage when chronic enhanced TGFβ signaling changes the acute immune response to pIC. Because pIC triggers an innate immune response mimicking a viral response, TGFβ may alter the innate immune pathways by modifying mitochondrial response, thus leading to development of an environment favored for BMF/MDS initiation and progression. These findings suggest a combinatorial effect between TGFβ and mitochondrial-mediated innate immune pathways could contribute to secondary BMF/MDS (Figure 2).
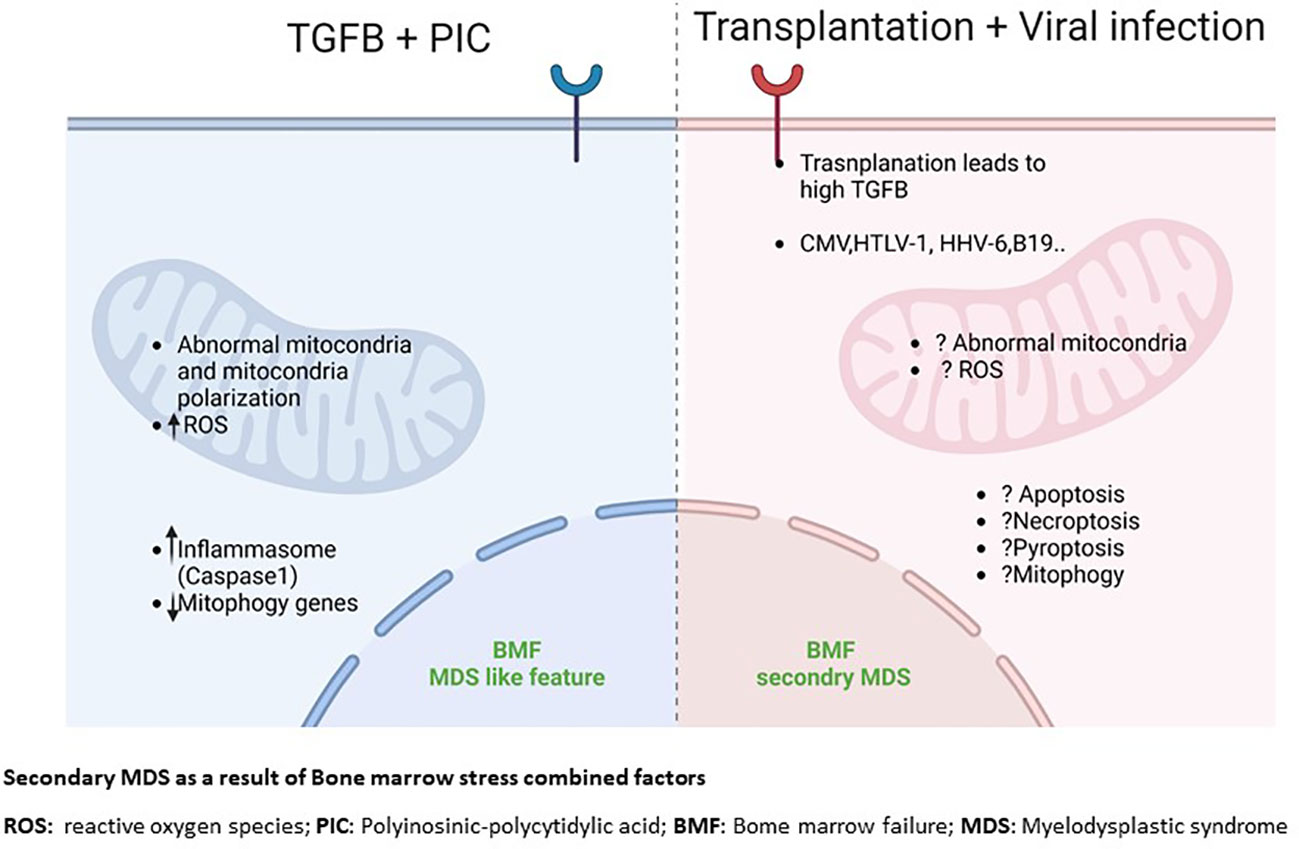
Figure 2 Secondary MDS as a result of Bone marrow stress combined factors. ROS, reactive oxygen species; PIC, Polyinosinic-polycytidylic acid; BMF, Bome marrow failure; MDS, Myelodysplactic syndrome.
Conclusions and future directions
The formation of BMF involves numerous crucial interrelated factors, including genetics, proteomics, nutrition, cellular signaling, metabolism, and interaction between the HSC and other stromal cells. Mitochondria are emerging as important factors in the pathogenesis of BMF/MDS. How exactly mitochondria contribute to BMF/MDS remain to be analyzed in detail. We need to further understand the potential effects of damaged mitochondria on BMF/MDS development, including the potential consequences on HSC metabolism in disease context and how metabolic changes contribute to disease development. Examining this will need to be done both in the context of de novo BMF/MDS and secondary BMF/MDS. How the stress of bone marrow transplantation, with or without viral infection, alters mitochondrial functions is another area of interest. The fact that mitochondria serve as platform of innate immune signaling is very intriguing and will need to be examined in detail, as abnormal mitochondria could represent an important mechanism of hyperinflammation associated with BMF/MDS. Finally, the development of mitochondrial transfer or metabolic reprogramming through metabolite addition could be complimentary to current therapeutics, and need to be carefully evaluated. Therefore, a fuller understanding of interplay between mitochondrial functions and inflammation is essential for both our fundamental understanding of HSC biology and BMF/MDS pathogenesis as well as for the developmental of novel therapies. It will be important to systematically investigate the role of mitochondrial functions and associated metabolism in BMF/MDS.
Author contributions
WN and M-DF equally contributed to writing the manuscript. M-DF edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Department of Defense award BM 190093 to M-DF.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Bagby GC, Fleischman AG. The stem cell fitness landscape and pathways of molecular leukemogenesis. Front Biosci (Schol Ed). (2011) 3:487–500. doi: 10.2741/s167
2. Leguit RJ, van den Tweel JG. The pathology of bone marrow failure. Histopathology. (2010) 57(5):655–70. doi: 10.1111/j.1365-2559.2010.03612.x
3. Shimamura A, Alter BP. Pathophysiology and management of inherited bone marrow failure syndromes. Blood Rev (2010) 24(3):101–22. doi: 10.1016/j.blre.2010.03.002
4. Young NS, Calado RT, Scheinberg P. Current concepts in the pathophysiology and treatment of aplastic anemia. Blood. (2006) 108(8):2509–19. doi: 10.1182/blood-2006-03-010777
5. Nepal M, Che R, Ma C, Zhang J, Fei P. FANCD2 and DNA damage. Int J Mol Sci (2017) 18(8):1804. doi: 10.3390/ijms18081804
6. Da Costa L, Leblanc T, Mohandas N. Diamond-blackfan anemia. Blood. (2020) 136(11):1262–73. doi: 10.1182/blood.2019000947
7. Tsai FD, Lindsley RC. Clonal hematopoiesis in the inherited bone marrow? Failure syndromes. Blood. (2020) 136(14):1615–22. doi: 10.1182/blood.2019000990
8. Friedberg JW, Neuberg D, Stone RM, Alyea E, Jallow H, LaCasce A, et al. Outcome in patients with myelodysplastic syndrome after autologous bone marrow transplantation for non-hodgkin’s lymphoma. J Clin Oncol (1999) 17(10):3128–35. doi: 10.1200/JCO.1999.17.10.3128
9. Stone RM, Neuberg D, Soiffer R, Takvorian T, Whelan M, Rabinowe SN, et al. Myelodysplastic syndrome as a late complication following autologous bone marrow transplantation for non-hodgkin’s lymphoma. J Clin Oncol (1994) 12(12):2535–42. doi: 10.1200/JCO.1994.12.12.2535
10. Brown JR, Yeckes H, Friedberg JW, Neuberg D, Kim H, Nadler LM, et al. Increasing incidence of late second malignancies after conditioning with cyclophosphamide and total-body irradiation and autologous bone marrow transplantation for non-hodgkin’s lymphoma. J Clin Oncol (2005) 23(10):2208–14. doi: 10.1200/JCO.2005.05.158
11. Krishnan A, Bhatia S, Slovak ML, Arber DA, Niland JC, Nademanee A, et al. Predictors of therapy-related leukemia and myelodysplasia following autologous transplantation for lymphoma: an assessment of risk factors. Blood (2000) 95(5):1588–93. doi: 10.1182/blood.V95.5.1588.005k38_1588_1593
12. Barreyro L, Chlon TM, Starczynowski DT. Chronic immune response dysregulation in MDS pathogenesis. Blood (2018) 132(15):1553–60. doi: 10.1182/blood-2018-03-784116
13. Hinge A, Xu J, Javier J, Mose E, Kumar S, Kapur R, et al. p190-b RhoGAP and intracellular cytokine signals balance hematopoietic stem and progenitor cell self-renewal and differentiation. Nat Commun (2017) 8:14382. doi: 10.1038/ncomms14382
14. Pietras EM, Mirantes-Barbeito C, Fong S, Loeffler D, Kovtonyuk LV, Zhang S, et al. Chronic interleukin-1 exposure drives haematopoietic stem cells towards precocious myeloid differentiation at the expense of self-renewal. Nat Cell Biol (2016) 18(6):607–18. doi: 10.1038/ncb3346
15. Leimkühler NB, Schneider RK. Inflammatory bone marrow microenvironment. Hematology/the Educ Program Am Soc Hematol Am Soc Hematol Educ Program (2019) 2019(1):294–302. doi: 10.1182/hematology.2019000045
16. Mekinian A, Grignano E, Braun T, Decaux O, Liozon E, Costedoat-Chalumeau Nc , et al. Systemic inflammatory and autoimmune manifestations associated with myelodysplastic syndromes and chronic myelomonocytic leukaemia: a French multicentre retrospective study. Rheumatol (Oxford). (2016) 55(2):291–300. doi: 10.1093/rheumatology/kev294
17. Trowbridge JJ, Starczynowski DT. Innate immune pathways and inflammation in hematopoietic aging, clonal hematopoiesis, and MDS. J Exp Med (2021) 218(7):e20201544. doi: 10.1084/jem.20201544
18. Aoyagi Y, Hayashi Y, Harada Y, Choi K, Matsunuma N, Sadato D, et al. Mitochondrial fragmentation triggers ineffective hematopoiesis in myelodysplastic syndromes. Cancer Discovery (2022) 12(1):250–69. doi: 10.1158/2159-8290.CD-21-0032
19. Cappelli E, Ravera S, Vaccaro D, Cuccarolo P, Bartolucci M, Panfoli I, et al. Mitochondrial respiratory complex I defects in fanconi anemia. Trends Mol Med (2013) 19(9):513–4. doi: 10.1016/j.molmed.2013.07.008
20. Cui X, Wang J, Cai Z, Wang J, Liu K, Cui S, et al. Complete sequence analysis of mitochondrial DNA and telomere length in aplastic anemia. Int J Mol Med (2014) 34(5):1309–14. doi: 10.3892/ijmm.2014.1898
21. Kim HR, Shin MG, Kim MJ, Kim HJ, Shin JH, Suh SP, et al. Mitochondrial DNA aberrations of bone marrow cells from patients with aplastic anemia. J Korean Med Sci (2008) 23(6):1062–7. doi: 10.3346/jkms.2008.23.6.1062
22. Beecher G, Fleming MD, Liewluck T. Hereditary myopathies associated with hematological abnormalities. Muscle Nerve. (2022) 65(4):374–90. doi: 10.1002/mus.27474
23. Cappelli E, Cuccarolo P, Stroppiana G, Miano M, Bottega R, Cossu V, et al. Defects in mitochondrial energetic function compels fanconi anaemia cells to glycolytic metabolism. Biochim Biophys Acta Mol Basis Dis (2017) 1863(6):1214–21. doi: 10.1016/j.bbadis.2017.03.008
24. Wild KT, Goldstein AC, Muraresku C, Ganetzky RD. Broadening the phenotypic spectrum of Pearson syndrome: Five new cases and a review of the literature. Am J Med Genet A. (2020) 182(2):365–73. doi: 10.1002/ajmg.a.61433
25. Filanovsky K, Haran M, Mirkin V, Braester A, Shevetz O, Stanevsky A, et al. Peripheral blood cell mitochondrial dysfunction in myelodysplastic syndrome can be improved by a combination of coenzyme Q10 and carnitine. Mediterr J Hematol Infect Dis (2020) 12(1):e2020072. doi: 10.4084/MJHID.2020.072
26. Schildgen V, Wulfert M, Gattermann N. Impaired mitochondrial gene transcription in myelodysplastic syndromes and acute myeloid leukemia with myelodysplasia-related changes. Exp Hematol (2011) 39(6):666–75.e661. doi: 10.1016/j.exphem.2011.03.007
27. Sen T, Jain M, Gram M, Mattebo A, Soneji S, Walkley CR, et al. Enhancing mitochondrial function in vivo rescues MDS-like anemia induced by pRb deficiency. Exp Hematol (2020) 88:28–41. doi: 10.1016/j.exphem.2020.06.006
28. Ward GA, McGraw KL, Abbas-Aghababazadeh F, Meyer BS, McLemore AF, Vincelette ND, et al. Oxidized mitochondrial DNA released after inflammasome activation is a disease biomarker for myelodysplastic syndromes. Blood Adv (2021) 5(8):2216–28. doi: 10.1182/bloodadvances.2020003475
29. Nunnari J, Suomalainen A. Mitochondria: in sickness and in health. Cell. (2012) 148(6):1145–59. doi: 10.1016/j.cell.2012.02.035
30. Chiabrando D, Vinchi F, Fiorito V, Mercurio S, Tolosano E. Heme in pathophysiology: a matter of scavenging, metabolism and trafficking across cell membranes. Front Pharmacol (2014) 5:61. doi: 10.3389/fphar.2014.00061
31. Chiabrando D, Marro S, Mercurio S, Giorgi C, Petrillo S, Vinchi F, et al. The mitochondrial heme exporter FLVCR1b mediates erythroid differentiation. J Clin Invest. (2012) 122(12):4569–79. doi: 10.1172/JCI62422
32. Ducamp S, Fleming MD. The molecular genetics of sideroblastic anemia. Blood. (2019) 133(1):59–69. doi: 10.1182/blood-2018-08-815951
33. Yoshida K, Sanada M, Shiraishi Y, Nowak D, Nagata Y, Yamamoto R, et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature. (2011) 478(7367):64–9. doi: 10.1038/nature10496
34. Malcovati L, Papaemmanuil E, Ambaglio I, Elena C, Galli A, Della Porta MG, et al. Driver somatic mutations identify distinct disease entities within myeloid neoplasms with myelodysplasia. Blood. (2014) 124(9):1513–21. doi: 10.1182/blood-2014-03-560227
35. Malcovati L, Cazzola M. Recent advances in the understanding of myelodysplastic syndromes with ring sideroblasts. Br J Haematol (2016) 174(6):847–58. doi: 10.1111/bjh.14215
36. Clough CA, Pangallo J, Sarchi M, Ilagan JO, North K, Bergantinos R, et al. Coordinated missplicing of TMEM14C and ABCB7 causes ring sideroblast formation in SF3B1-mutant myelodysplastic syndrome. Blood. (2022) 139(13):2038–49. doi: 10.1182/blood.2021012652
37. Dolatshad H, Pellagatti A, Fernandez-Mercado M, Yip BH, Malcovati L, Attwood M, et al. Disruption of SF3B1 results in deregulated expression and splicing of key genes and pathways in myelodysplastic syndrome hematopoietic stem and progenitor cells. Leukemia. (2015) 29(5):1092–103. doi: 10.1038/leu.2014.331
38. Dolatshad H, Pellagatti A, Liberante FG, Llorian M, Repapi E, Steeples V, et al. Cryptic splicing events in the iron transporter ABCB7 and other key target genes in SF3B1-mutant myelodysplastic syndromes. Leukemia. (2016) 30(12):2322–31. doi: 10.1038/leu.2016.149
39. Ochi T, Fujiwara T, Ono K, Suzuki C, Nikaido M, Inoue D, et al. Exploring the mechanistic link between SF3B1 mutation and ring sideroblast formation in myelodysplastic syndrome. Sci Rep (2022) 12(1):14562. doi: 10.1038/s41598-022-18921-2
40. Hsu J, Reilly A, Hayes BJ, Clough CA, Konnick EQ, Torok-Storb B, et al. Reprogramming identifies functionally distinct stages of clonal evolution in myelodysplastic syndromes. Blood. (2019) 134(2):186–98. doi: 10.1182/blood.2018884338
41. Rey MA, Duffy SP, Brown JK, Kennedy JA, Dick JE, Dror Y, et al. Enhanced alternative splicing of the FLVCR1 gene in diamond blackfan anemia disrupts FLVCR1 expression and function that are critical for erythropoiesis. Haematologica. (2008) 93(11):1617–26. doi: 10.3324/haematol.13359
42. Mercurio S, Aspesi A, Silengo L, Altruda F, Dianzani I, Chiabrando D. Alteration of heme metabolism in a cellular model of diamond-blackfan anemia. Eur J Haematol (2016) 96(4):367–74. doi: 10.1111/ejh.12599
43. Martinez-Reyes I, Chandel NS. Mitochondrial TCA cycle metabolites control physiology and disease. Nat Commun (2020) 11(1):102. doi: 10.1038/s41467-019-13668-3
44. DiNardo CD, Jabbour E, Ravandi F, Takahashi K, Daver N, Routbort M, et al. IDH1 and IDH2 mutations in myelodysplastic syndromes and role in disease progression. Leukemia. (2016) 30(4):980–4. doi: 10.1038/leu.2015.211
45. Bowman RL, Levine RL. TET2 in normal and malignant hematopoiesis. Cold Spring Harb Perspect Med (2017) 7(8):a026518. doi: 10.1101/cshperspect.a026518
46. Ewalt M, Galili NG, Mumtaz M, Churchill M, Rivera S, Borot F, et al. DNMT3a mutations in high-risk myelodysplastic syndrome parallel those found in acute myeloid leukemia. Blood Cancer J (2011) 1(3):e9. doi: 10.1038/bcj.2011.7
47. Hayashi Y, Zhang Y, Yokota A, Yan X, Liu J, Choi K, et al. Pathobiological pseudohypoxia as a putative mechanism underlying myelodysplastic syndromes. Cancer Discovery (2018) 8(11):1438–57. doi: 10.1158/2159-8290.CD-17-1203
48. Cerveny KL, Tamura Y, Zhang Z, Jensen RE, Sesaki H. Regulation of mitochondrial fusion and division. Trends Cell Biol (2007) 17(11):563–9. doi: 10.1016/j.tcb.2007.08.006
49. Picard M, Shirihai OS, Gentil BJ, Burelle Y. Mitochondrial morphology transitions and functions: implications for retrograde signaling? Am J Physiol Regul Integr Comp Physiol (2013) 304(6):R393–406. doi: 10.1152/ajpregu.00584.2012
50. Roy M, Reddy PH, Iijima M, Sesaki H. Mitochondrial division and fusion in metabolism. Curr Opin Cell Biol (2015) 33:111–8. doi: 10.1016/j.ceb.2015.02.001
51. Sumpter R Jr., Sirasanagandla S, Fernandez AF, Wei Y, Dong X, Franco L, et al. Fanconi anemia proteins function in mitophagy and immunity. Cell. (2016) 165(4):867–81. doi: 10.1016/j.cell.2016.04.006
52. Turrens JF. Mitochondrial formation of reactive oxygen species. J Physiol (2003) 552(Pt 2):335–44. doi: 10.1113/jphysiol.2003.049478
53. Gonçalves AC, Alves V, Silva T, Carvalho C, Oliveira CR, Sarmento-Ribeiro AB. Oxidative stress mediates apoptotic effects of ascorbate and dehydroascorbate in human myelodysplasia cells in vitro. Toxicol In Vitro. (2013) 27(5):1542–9. doi: 10.1016/j.tiv.2013.03.009
54. Yang Y, Karakhanova S, Hartwig W, D'Haese JG, Philippov PP, Werner J, et al. Mitochondria and mitochondrial ROS in cancer: Novel targets for anticancer therapy. J Cell Physiol (2016) 231(12):2570–81. doi: 10.1002/jcp.25349
55. Capanni C, Bruschi M, Columbaro M, Cuccarolo P, Ravera S, Dufour C, et al. Changes in vimentin, lamin A/C and mitofilin induce aberrant cell organization in fibroblasts from fanconi anemia complementation group a (FA-a) patients. Biochimie. (2013) 95(10):1838–47. doi: 10.1016/j.biochi.2013.06.024
56. Ravera S, Vaccaro D, Cuccarolo P, Columbaro M, Capanni C, Bartolucci M, et al. Mitochondrial respiratory chain complex I defects in fanconi anemia complementation group a. Biochimie. (2013) 95(10):1828–37. doi: 10.1016/j.biochi.2013.06.006
57. Chatla S, Du W, Wilson AF, Meetei AR, Pang Q. Fancd2-deficient hematopoietic stem and progenitor cells depend on augmented mitochondrial translation for survival and proliferation. Stem Cell Res (2019) 40:101550. doi: 10.1016/j.scr.2019.101550
58. Henson AL, JBt M, Alard P, Wattenberg MM, Liu JM, Ellis SR. Mitochondrial function is impaired in yeast and human cellular models of shwachman diamond syndrome. Biochem Biophys Res Commun (2013) 437(1):29–34. doi: 10.1016/j.bbrc.2013.06.028
59. Pearson HA, Lobel JS, Kocoshis SA, Naiman JL, Windmiller J, Lammi AT, et al. A new syndrome of refractory sideroblastic anemia with vacuolization of marrow precursors and exocrine pancreatic dysfunction. J Pediatr (1979) 95(6):976–84. doi: 10.1016/S0022-3476(79)80286-3
60. Steward CG, Groves SJ, Taylor CT, Maisenbacher MK, Versluys B, Newbury-Ecob RA, et al. Neutropenia in barth syndrome: characteristics, risks, and management. Curr Opin hematology. (2019) 26(1):6–15. doi: 10.1097/MOH.0000000000000472
61. Acehan D, Vaz F, Houtkooper RH, James J, Moore V, Tokunaga C, et al. Cardiac and skeletal muscle defects in a mouse model of human barth syndrome. J Biol Chem (2011) 286(2):899–908. doi: 10.1074/jbc.M110.171439
62. Clarke SL, Bowron A, Gonzalez IL, Groves SJ, Newbury-Ecob R, Clayton N, et al. Barth syndrome. Orphanet J Rare Dis (2013) 8:23. doi: 10.1186/1750-1172-8-23
63. Makaryan V, Kulik W, Vaz FM, Allen C, Dror Y, Dale DC, et al. The cellular and molecular mechanisms for neutropenia in barth syndrome. Eur J Haematol (2012) 88(3):195–209. doi: 10.1111/j.1600-0609.2011.01725.x
64. Kuijpers TW, Maianski NA, Tool AT, Becker K, Plecko B, Valianpour F, et al. Neutrophils in barth syndrome (BTHS) avidly bind annexin-V in the absence of apoptosis. Blood. (2004) 103(10):3915–23. doi: 10.1182/blood-2003-11-3940
65. Klein C, Grudzien M, Appaswamy G, Germeshausen M, Sandrock I, Schäffer AA, et al. HAX1 deficiency causes autosomal recessive severe congenital neutropenia (Kostmann disease). Nat Genet (2007) 39(1):86–92. doi: 10.1038/ng1940
66. Schäffer AA, Klein C. Genetic heterogeneity in severe congenital neutropenia: how many aberrant pathways can kill a neutrophil? Curr Opin Allergy Clin Immunol (2007) 7(6):481–94. doi: 10.1097/ACI.0b013e3282f1d690
67. Katada S, Mito T, Ogasawara E, Hayashi J, Nakada K. Mitochondrial DNA with a large-scale deletion causes two distinct mitochondrial disease phenotypes in mice. G3 (Bethesda). (2013) 3(9):1545–52. doi: 10.1534/g3.113.007245
68. Jacoby E, Ben Yakir-Blumkin M, Blumenfeld-Kan S, Brody Y, Meir A, Melamed-Book N, et al. Mitochondrial augmentation of CD34(+) cells from healthy donors and patients with mitochondrial DNA disorders confers functional benefit. NPJ Regener Med (2021) 6(1):58. doi: 10.1038/s41536-021-00167-7
69. Amigo ML, del Cañizo MC, Rios A, Garcia MA, Caballero MD, Martin A, et al. Diagnosis of secondary myelodysplastic syndromes (MDS) following autologous transplantation should not be based only on morphological criteria used for diagnosis of de novo MDS. Bone Marrow Transplant. (1999) 23(10):997–1002. doi: 10.1038/sj.bmt.1701757
70. Metheny L, Callander NS, Hall AC, Zhang MJ, Bo-Subait K, Wang HL, et al. Allogeneic transplantation to treat therapy-related myelodysplastic syndrome and acute myelogenous leukemia in adults. Transplant Cell Ther (2021) 27(11):923.e921–923.e912. doi: 10.1016/j.jtct.2021.08.010
71. Yamasaki S, Suzuki R, Hatano K, Fukushima K, Iida H, Morishima S, et al. Therapy-related acute myeloid leukemia and myelodysplastic syndrome after hematopoietic cell transplantation for lymphoma. Bone Marrow Transplant. (2017) 52(7):969–76. doi: 10.1038/bmt.2017.52
72. Armitage JO, Carbone PP, Connors JM, Levine A, Bennett JM, Kroll S. Treatment-related myelodysplasia and acute leukemia in non-hodgkin’s lymphoma patients. J Clin Oncol (2003) 21(5):897–906. doi: 10.1200/JCO.2003.07.113
73. Hu HH, Chen DQ, Wang YN, Feng YL, Cao G, Vaziri ND, et al. New insights into TGF-β/Smad signaling in tissue fibrosis. Chem Biol Interact (2018) 292:76–83. doi: 10.1016/j.cbi.2018.07.008
74. Blank U, Karlsson S. The role of smad signaling in hematopoiesis and translational hematology. Leukemia. (2011) 25(9):1379–88. doi: 10.1038/leu.2011.95
75. Larsson J, Karlsson S. The role of smad signaling in hematopoiesis. Oncogene. (2005) 24(37):5676–92. doi: 10.1038/sj.onc.1208920
76. Vaidya A, Kale VP. TGF-β signaling and its role in the regulation of hematopoietic stem cells. Syst Synth Biol (2015) 9(1-2):1–10. doi: 10.1007/s11693-015-9161-2
77. Hinge A, Filippi MD. Deconstructing the complexity of TGFβ signaling in hematopoietic stem cells: Quiescence and beyond. Curr Stem Cell Rep (2016) 2(4):388–97. doi: 10.1007/s40778-016-0069-x
78. Coomes SM, Moore BB. Pleiotropic effects of transforming growth factor-β in hematopoietic stem-cell transplantation. Transplantation. (2010) 90(11):1139–44. doi: 10.1097/TP.0b013e3181efd018
79. Liem LM, Fibbe WE, van Houwelingen HC, Goulmy E. Serum transforming growth factor-beta1 levels in bone marrow transplant recipients correlate with blood cell counts and chronic graft-versus-host disease. Transplantation. (1999) 67(1):59–65. doi: 10.1097/00007890-199901150-00009
80. Coomes SM, Wilke CA, Moore TA, Moore BB. Induction of TGF-beta 1, not regulatory T cells, impairs antiviral immunity in the lung following bone marrow transplant. J Immunol (2010) 184(9):5130–40. doi: 10.4049/jimmunol.0901871
81. Panoskaltsis-Mortari A, Taylor PA, Yaeger TM, Wangensteen OD, Bitterman PB, Ingbar DH, et al. The critical early proinflammatory events associated with idiopathic pneumonia syndrome in irradiated murine allogeneic recipients are due to donor T cell infusion and potentiated by cyclophosphamide. J Clin Invest. (1997) 100(5):1015–27. doi: 10.1172/JCI119612
82. Zhou L, McMahon C, Bhagat T, Alencar C, Yu Y, Fazzari M, et al. Reduced SMAD7 leads to overactivation of TGF-beta signaling in MDS that can be reversed by a specific inhibitor of TGF-beta receptor I kinase. Cancer Res (2011) 71(3):955–63. doi: 10.1158/0008-5472.CAN-10-2933
83. Zhou L, Nguyen AN, Sohal D, Ying Ma J, Pahanish P, Gundabolu K, et al. Inhibition of the TGF-beta receptor I kinase promotes hematopoiesis in MDS. Blood. (2008) 112(8):3434–43. doi: 10.1182/blood-2008-02-139824
84. Bhagat TD, Zhou L, Sokol L, Kessel R, Caceres G, Gundabolu K, et al. miR-21 mediates hematopoietic suppression in MDS by activating TGF-β signaling. Blood. (2013) 121(15):2875–81. doi: 10.1182/blood-2011-12-397067
85. Río P, Bueren JA. TGF-β: a master regulator of the bone marrow failure puzzle in fanconi anemia. Stem Cell Investig (2016) 3:75. doi: 10.21037/sci.2016.09.17
86. Zhang H, Kozono DE, O’Connor KW, Vidal-Cardenas S, Rousseau A, Hamilton A, et al. TGF-β inhibition rescues hematopoietic stem cell defects and bone marrow failure in fanconi anemia. Cell Stem Cell (2016) 18(5):668–81. doi: 10.1016/j.stem.2016.03.002
87. Ashley RJ, Yan H, Wang N, Hale J, Dulmovits BM, Papoin J, et al. Steroid resistance in diamond blackfan anemia associates with p57Kip2 dysregulation in erythroid progenitors. J Clin Invest. (2020) 130(4):2097–110. doi: 10.1172/JCI132284
88. Suragani RN, Cadena SM, Cawley SM, Sako D, Mitchell D, Li R, et al. Transforming growth factor-β superfamily ligand trap ACE-536 corrects anemia by promoting late-stage erythropoiesis. Nat Med (2014) 20(4):408–14. doi: 10.1038/nm.3512
89. Joyce CE, Saadatpour A, Ruiz-Gutierrez M, Bolukbasi OV, Jiang L, Thomas DD, et al. TGFβ signaling underlies hematopoietic dysfunction and bone marrow failure in shwachman-diamond syndrome. J Clin Invest. (2019) 129(9):3821–6. doi: 10.1172/JCI125375
90. Starczynowski DT, Kuchenbauer F, Wegrzyn J, Rouhi A, Petriv O, Hansen CL, et al. MicroRNA-146a disrupts hematopoietic differentiation and survival. Exp Hematol (2011) 39(2):167–78.e164. doi: 10.1016/j.exphem.2010.09.011
91. Varney ME, Niederkorn M, Konno H, Matsumura T, Gohda J, Yoshida N, et al. Loss of tifab, a del(5q) MDS gene, alters hematopoiesis through derepression of toll-like receptor-TRAF6 signaling. J Exp Med (2015) 212(11):1967–85. doi: 10.1084/jem.20141898
92. Basiorka AA, McGraw KL, Eksioglu EA, Chen X, Johnson J, Zhang L, et al. The NLRP3 inflammasome functions as a driver of the myelodysplastic syndrome phenotype. Blood. (2016) 128(25):2960–75. doi: 10.1182/blood-2016-07-730556
93. Sallman DA, Cluzeau T, Basiorka AA, List A. Unraveling the pathogenesis of MDS: The NLRP3 inflammasome and pyroptosis drive the MDS phenotype. Front Oncol (2016) 6:151. doi: 10.3389/fonc.2016.00151
94. Wagner PN, Shi Q, Salisbury-Ruf CT, Zou J, Savona MR, Fedoriw Y, et al. Increased Ripk1-mediated bone marrow necroptosis leads to myelodysplasia and bone marrow failure in mice. Blood. (2019) 133(2):107–20. doi: 10.1182/blood-2018-05-847335
95. Swanson KV, Deng M, Ting JP. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol (2019) 19(8):477–89. doi: 10.1038/s41577-019-0165-0
96. Hormaechea-Agulla D, Matatall KA, Le DT, Kain B, Long X, Kus P, et al. Chronic infection drives Dnmt3a-loss-of-function clonal hematopoiesis via IFNgamma signaling. Cell Stem Cell (2021) 8(8):1428–42. doi: 10.1016/j.stem.2021.03.002
97. Hou F, Sun L, Zheng H, Skaug B, Jiang QX, Chen ZJ. MAVS forms functional prion-like aggregates to activate and propagate antiviral innate immune response. Cell. (2011) 146(3):448–61. doi: 10.1016/j.cell.2011.06.041
98. Subramanian N, Natarajan K, Clatworthy MR, Wang Z, Germain RN. The adaptor MAVS promotes NLRP3 mitochondrial localization and inflammasome activation. Cell. (2013) 153(2):348–61. doi: 10.1016/j.cell.2013.02.054
99. Zhou X, You F, Chen H, Jiang Z. Poly(C)-binding protein 1 (PCBP1) mediates housekeeping degradation of mitochondrial antiviral signaling (MAVS). Cell Res (2012) 22(4):717–27. doi: 10.1038/cr.2011.184
100. Jiang Y, Zhu Y, Liu ZJ, Ouyang S. The emerging roles of the DDX41 protein in immunity and diseases. Protein Cell (2017) 8(2):83–9. doi: 10.1007/s13238-016-0303-4
101. Raza A. Initial transforming event in myelodysplastic syndromes may be viral: Case for cytomegalovirus. Med Oncol (1998) 15(3):165–73. doi: 10.1007/BF02821935
102. Karlic H, Möstl M, Mucke H, Pavlova B, Pfeilstöcker M, Heinz R. Association of human T-cell leukemia virus and myelodysplastic syndrome in a central European population. Cancer Res (1997) 57(21):4718–21.
103. Urban C, Lackner H, Muller E, Benesch M, Strenger V, Sovinz P, et al. Stem cell transplantation in 6 children with parvovirus B19- induced severe aplastic anaemia or myelodysplastic syndrome. Klin Padiatr. (2011) 223(6):332–4. doi: 10.1055/s-0031-1287839
104. Kagialis-Girard S, Durand B, Mialou V, Pagès MP, Galambrun C, Bertrand Y, et al. Human herpesvirus 6 infection and transient acquired myelodysplasia in children. Pediatr Blood Cancer. (2006) 47(5):543–8. doi: 10.1002/pbc.20667
105. Raza A. Hypothesis: myelodysplastic syndromes may have a viral etiology. Int J hematology. (1998) 68(3):245–56. doi: 10.1016/S0925-5710(98)00051-6
106. Javier J, Hinge A, Bartram J, Xu J, Filippi MD. Transforming growth factor-β signaling modifies the hematopoietic acute inflammatory response to drive bone marrow failure. Haematologica. (2022) 107(6):1323–34. doi: 10.3324/haematol.2020.273292
107. Engels EA. Infectious agents as causes of non-Hodgkin lymphoma. Cancer Epidemiol Biomarkers Prev (2007) 16(3):401–4. doi: 10.1158/1055-9965.EPI-06-1056
108. Li L, Li M, Sun C, Francisco L, Chakraborty S, Sabado M, et al. Altered hematopoietic cell gene expression precedes development of therapy-related myelodysplasia/acute myeloid leukemia and identifies patients at risk. Cancer Cell (2011) 20(5):591–605. doi: 10.1016/j.ccr.2011.09.011
109. Hinge A, He J, Bartram J, Javier J, Xu J, Fjellman E, et al. Asymmetrically segregated mitochondria provide cellular memory of hematopoietic stem cell replicative history and drive HSC attrition. Cell Stem Cell (2020) 26(3):420–30.e426. doi: 10.1016/j.stem.2020.01.016
110. Breda CNS, Davanzo GG, Basso PJ, Saraiva Camara NO, Moraes-Vieira PMM. Mitochondria as central hub of the immune system. Redox Biol (2019) 26:101255. doi: 10.1016/j.redox.2019.101255
111. Kim SJ, Ahn DG, Syed GH, Siddiqui A. The essential role of mitochondrial dynamics in antiviral immunity. Mitochondrion. (2018) 41:21–7. doi: 10.1016/j.mito.2017.11.007
112. Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. (2011) 469(7329):221–5. doi: 10.1038/nature09663
113. Kim SH, Shin HJ, Yoon CM, Lee SW, Sharma L, Dela Cruz CS, et al. PINK1 inhibits multimeric aggregation and signaling of MAVS and MAVS-dependent lung pathology. Am J Respir Cell Mol Biol (2021) 64(5):592–603. doi: 10.1165/rcmb.2020-0490OC
114. Sun X, Sun L, Zhao Y, Li Y, Lin W, Chen D, et al. MAVS maintains mitochondrial homeostasis via autophagy. Cell Discovery (2016) 2:16024. doi: 10.1038/celldisc.2016.24
Keywords: bone marrow failure (BMF), mitochondria, TGF beta, innate immune signaling, myelodysplastic disorder (MDS)
Citation: Nasr W and Filippi M-D (2022) Acquired and hereditary bone marrow failure: A mitochondrial perspective. Front. Oncol. 12:1048746. doi: 10.3389/fonc.2022.1048746
Received: 19 September 2022; Accepted: 17 October 2022;
Published: 02 November 2022.
Edited by:
Makiko Mochizuki-Kashio, Tokyo Women’s Medical University, JapanReviewed by:
Yizhou Zheng, Chinese Academy of Medical Sciences and Peking Union Medical College, ChinaToshio Suda, National University of Singapore, Singapore
Copyright © 2022 Nasr and Filippi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marie-Dominique Filippi, bWFyaWUtZG9taW5pcXVlLmZpbGlwcGlAY2NobWMub3Jn