 Jessica D. Daley
Jessica D. Daley Adam C. Olson
Adam C. Olson Kelly M. Bailey
Kelly M. Bailey
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Oncol. , 22 November 2022
Sec. Radiation Oncology
Volume 12 - 2022 | https://doi.org/10.3389/fonc.2022.1048705
This article is part of the Research Topic Targeting DNA Damage Response to Enhance Antitumor Innate Immunity in Radiotherapy View all 10 articles
Ewing sarcoma is a fusion-oncoprotein-driven primary bone tumor most commonly diagnosed in adolescents. Given the continued poor outcomes for patients with metastatic and relapsed Ewing sarcoma, testing innovative therapeutic approaches is essential. Ewing sarcoma has been categorized as a ‘BRCAness’ tumor with emerging data characterizing a spectrum of DNA damage repair defects within individual Ewing tumors, including the presence of EWSR1::FLI1 itself, recurrent somatic mutations, and rare germline-based defects. It is critical to understand the cumulative impact of various DNA damage repair defects on an individual Ewing tumor’s response to therapy. Further, in addition to DNA-damage-directed therapies, subsets of Ewing tumors may be more susceptible to DNA-damage/immunotherapy combinations given the significant cross-talk between DNA damage and inflammatory pathways in the tumor microenvironment. Here we review potential approaches utilizing DNA-damaging agents as modulators of the Ewing tumor immune microenvironment, with a focus on radiation and opportunities during disease metastasis and relapse.
Ewing sarcoma is the second most common bone tumor diagnosed in adolescents and young adults. Ewing sarcoma is driven by a fusion oncoprotein derived from the translocation of EWSR1 on chromosome 22 with an ETS family member, most commonly FLI1 on chromosome 11 (1). Patients with upfront metastatic or relapsed Ewing sarcoma continue to have very poor outcomes (2), and new therapeutic approaches continue to be in high demand. The exquisite sensitivity of Ewing tumors to DNA damage has been recognized for decades and DNA damaging agents such as chemotherapy and radiation continue to be the mainstays of Ewing sarcoma therapy, even for aggressive disease (3).
DNA damage can elicit significant alterations in tumor biology, including modulation of the tumor immune microenvironment (TIME). Historically, the impact of DNA damage on the Ewing TIME has been understudied given a paucity of tumor biopsies at the time of relapse and the lack of syngeneic or transgenic (immunocompetent) mouse models of Ewing sarcoma (4). DNA-damaging agents can promote immunogenicity through multiple mechanisms including increasing the neoantigen repertoire, increasing antigen presentation, and shifting the cytokine profile to promote an inflamed tumor microenvironment (5, 6). Understanding TIME alterations elicited by DNA damage specifically in Ewing sarcoma is a high priority, as TIME modulation during DNA damage may offer a new avenue for therapy for patients with aggressive disease. Therapeutically, it can be challenging to increase chemotherapy doses or add additional marrow-suppressive agents into existing chemotherapy backbones for the treatment of Ewing sarcoma, also highlighting why multi-modality approaches, such as TIME modulation, are in need.
Immunotherapy includes medications and cell-based therapies that broadly act by enhancing the anti-tumor immune response through various mechanisms (7) and have been utilized successfully in many adult carcinomas and soft tissue sarcomas (8, 9) (10). Clinical trials investigating single-agent immunotherapy, such as PD1 inhibition, have not demonstrated a significant clinical benefit in advanced Ewing sarcoma (11, 12). Given the importance of immunotherapy type and timing in disease response (13) such results are neither surprising nor discouraging when currently so little is known about the Ewing TIME. Primary Ewing sarcoma is known to have low overall immune infiltration compared to other tumors types. However, some studies have demonstrated a correlation between increased infiltration of CD8+ T cells and improved outcomes (14, 15). Our recent work demonstrated the Ewing TIME can evolve and demonstrate enhanced immune cell infiltration upon disease metastasis and relapse, possibly due to a combination of prior chemotherapy exposure and changes in tumor microenvironments (bone versus lung) (16). This work again highlights the need to better understand Ewing tumor immunobiology, especially in the setting of relapse.
In this mini-review we will discuss the layers of DNA damage repair defects in Ewing sarcoma, how DNA damaging agents can influence the TIME, and ways in which immunomodulation during DNA damage could provide new therapeutic opportunities for Ewing sarcoma in the future.
Ewing tumors demonstrate high sensitivity to DNA damage. DNA damaging agents, including doxorubicin and cyclophosphamide, have formed the chemotherapy backbone for the treatment of Ewing sarcoma since the first use of adjuvant therapy in the 1970s (17). Ewing sarcoma is also sensitive to radiation therapy (18). Decades later, a screen of hundreds of cancer cell lines seeking to identify biomarkers for targeted cancer agents discovered EWSR1::FLI1 was significantly associated with sensitivity to the PARP [Poly (ADP-ribose) polymerase] inhibitor (PARPi) olaparib (19). PARP1 is an enzyme involved in DNA damage repair and a drug target in BRCA-mutant cancers deficient in homologous recombination repair (20). PARP1 drives transcription and accelerates base excision repair (21, 22), and inhibition of PARP1 leads to cell death in cancers deficient in homologous repair by causing defects in the replication fork needed to repair DNA damage. Further studies elucidated that EWSR1::FLI1 interacts directly with PARP (20). Gorthi et al. demonstrated that expression of the EWSR1::FLI1 fusion oncoprotein correlated with increased chemosensitivity (23). Mechanistically, they found that EWSR1::FLI1 promotes R-loop accumulation, and ultimately deranges DNA damage repair machinery by impairing normal BRCA1 functionality. A study in 2002 by Spahn et al. also demonstrated that the N-terminal portion of EWSR1::FLI1 can interact with the C-terminal portion of BRCA1-Associated Ring Domain 1 (BARD1), thus providing another potential link between EWSR1::FLI1 and BRCA1 biology (24). Such studies provided rationale for phase II clinical trial of olaparib as single-agent therapy in patients with refractory Ewing sarcoma (25) and subsequent studies have demonstrated that sensitivity to PARP inhibition in Ewing sarcoma is increased in the setting of other DNA damaging agents (irinotecan, temozolomide) (26). Despite this, the overall clinical response of Ewing tumors to PARPi has been underwhelming. Lastly, elegant work has demonstrated the importance of the level of EWSR1::FLI1 fusion oncoprotein expression on Ewing cell behavior. EWSR1::FLI1 expression can vary between cells within a tumor. It is plausible that Ewing cells with low versus high EWS::FLI1 expression may demonstrate altered sensitivity to DNA damage (27–29), thus allowing for tumor cell subpopulation targeting.
In addition to DNA-damage-repair defects imparted by EWSR1::FLI1 in all Ewing tumors, there is the potential for Ewing tumors to harbor additional defects in DNA damage repair through the presence of somatic and germline variants or post-transcriptional modifications resulting in loss of protein expression. When comparing Ewing tumors to adult carcinomas, and even other pediatric primary bone tumors such as osteosarcoma, Ewing sarcoma demonstrates a very low tumor mutational burden (30–32). A handful of recurrent somatic variants, such as STAG2, CDKN2A, and TP53 have been reported in Ewing sarcoma (31, 32). Ewing tumors harboring one or more of these somatic mutations may demonstrate altered responses to DNA damage, as each of the corresponding proteins have been shown to participate in DNA damage repair through different mechanisms. For example, in vitro studies of STAG2-deficient glioblastomas demonstrated increased sensitivity to PARP inhibition (33). In Ewing sarcoma, loss of STAG2 expression can be secondary to STAG2 somatic mutations or loss of protein expression in the absence of a mutation (34).
A third layer of DNA-damage-repair deficiency to consider in Ewing sarcoma derives from germline pathogenic variants. Multiple sequencing studies of pediatric cancers have noted a small fraction of germline pathogenic variants in patients with Ewing sarcoma (35, 36). In a germline variant analysis of sequencing data from 175 patients with Ewing sarcoma, likely pathogenic variants were identified in 13.1% of patients (37). In the variants found, involving 22 different genes, a strong enrichment for DNA repair pathways and DNA double-strand break repair was noted on pathway analysis. Our work and others continue to add to the growing number of germline variants in DNA damage repair genes noted in patients with Ewing sarcoma (38, 39). Our group’s prior work demonstrated that loss of additional DNA damage repair machinery, such as BARD1 expression, can indeed confer Ewing cells more susceptible to DNA damage as compared wuth the sensitivity imparted by the presence of EWSR1::FLI1 alone (40). Figure 1 depicts a brief summary of the spectrum of DNA damage repair deficiencies in Ewing sarcoma.
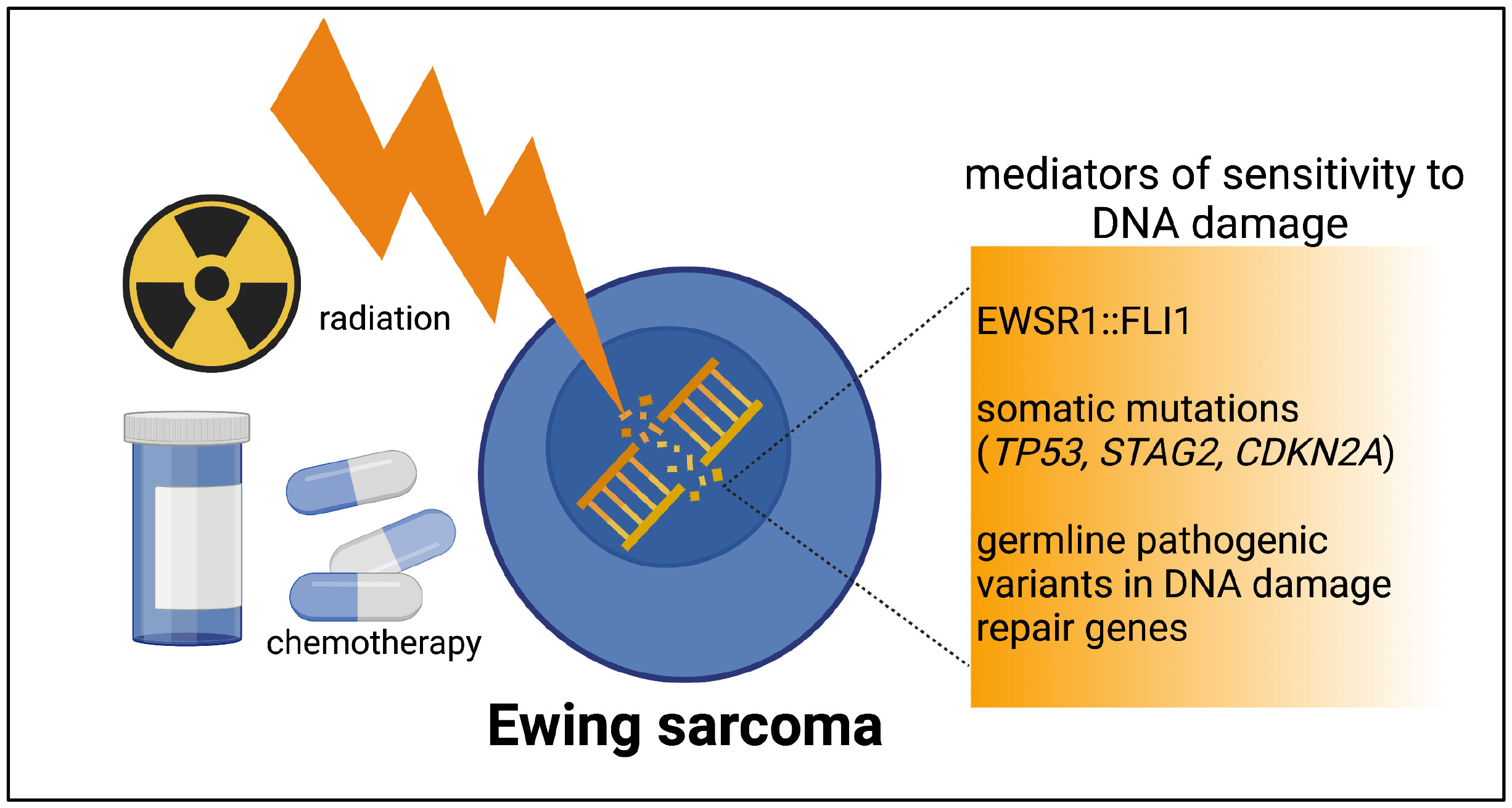
Figure 1 The spectrum of DNA damage repair deficiencies in Ewing tumors. Ewing tumors all have a level of DNA damage repair deficiency imparted by EWSR1::FLI1. The presence of one or more recurrent somatic mutations or rare germline pathogenic variants have the potential to contribute an additional level of DNA damage repair deficiency in a subset of Ewing tumors. Figure created by biorender.com.
Given the spectrum of DNA damage repair defects in Ewing sarcoma, DNA damaging agents will continue to be a mainstay of therapy. Following the original treatment schema with doxorubicin and cyclophosphamide discussed above, in the 1980s it was noted that ifosfamide and etoposide, which also exert their anti-neoplastic effect through induction of DNA damage, were effective in treating patients with relapsed Ewing sarcoma (41). This led to the development of the National Cancer Institute protocol INT-0091 (CCG-7881 and POG-8850)
in which ifosfamide and etoposide were added to the standard therapy backbone. Improved overall and event free survival was seen in patients with newly diagnosed, localized Ewing sarcoma using this five-drug approach (42). Alternating cycles of VDC (vincristine, doxorubicin, cyclophosphamide) and IE (ifosfamide and etoposide) thus remain the standard of care for patients with upfront localized or metastatic Ewing sarcoma. AEWS0031 later demonstrated that shortening the time between cycles (interval compression) provides additional benefit (43).
In addition to chemotherapy, radiation is also an important component of Ewing sarcoma treatment. Radiation is a curative-intent treatment modality option for local control, either as definitive treatment or as adjuvant treatment following surgical resection. The commonly prescribed radiation dose for definitive treatment of primary tumors is 55-60 Gy in 1.8-2 Gy fractions (44). The most recent Children’s Oncology Group protocols for Ewing sarcoma recommend 45 Gy be delivered to the original tumor volume with an additional 10.8 Gy boost delivered to the post-induction chemotherapy volume (3). Gross residual disease post-surgical resection is treated with 55.8 Gy, and microscopic disease treated with 50.4 Gy. There has recently been data suggesting that dose escalation up to a total dose of 70.2 Gy may be of benefit in improved local control (45), although this strategy has not been widely adopted to date. More recent studies show that hypofractionation (5-10 Gy doses over 5-10 fractions) may be as or more effective at treating sarcomas, including Ewing sarcoma (46).
Radiation therapy is also a key component in the treatment of metastatic and relapsed Ewing sarcoma. For patients presenting with pulmonary metastases at diagnosis, there have been multiple studies demonstrating the survival benefit of whole lung irradiation after completion of chemotherapy (47). For patients presenting with bony metastases, outcomes are worse overall; however, radiation delivery to sites of metastatic disease is beneficial (48). Patients with solitary bone metastases benefited most from radiotherapy, with doses of up to 50 Gy to sites of bony metastases being utilized. As patients with Ewing sarcoma receiving radiation are often a higher-risk patient population (incomplete resections, metastatic disease, relapse, etc.), this is a group of high interest when considering immunotherapy interventions following post-DNA damage modulation of the Ewing TIME.
DNA damaging chemotherapeutic agents have been shown to induce immunogenicity through a variety of mechanisms (5). Given the low mutational burden of Ewing sarcoma, the mutagenic potential of DNA damaging agents is an appealing mechanism of enhancing immunogenicity by production of tumor neoantigens (49). Tumor neoantigens can induce increased anti-tumor T cell response which is again beneficial when combined with immunotherapy agents. However, increased neoantigens in the TIME are not always sufficient to induce immune response (50). DNA damaging agents additionally lead to release of damage associated molecular patterns (DAMPs) after cell death. DAMPs stimulate the recruitment of antigen-presenting cells to the site of cell death and further prime the TIME for an adaptive immune response. Doxorubicin and cyclophosphamide are utilized in the treatment of Ewing sarcoma and are known to induce immunogenic cell death (51). Cyclophosphamide additionally remains of particular interest as it has been shown to increase antigen presentation on tumor cells and expand dendritic cell populations that can promote T cell priming (52, 53).
An additional mechanism by which DNA damaging chemotherapeutics can increase anti-tumor immune response is through changes in the cytokine profile of the tumor environment. Cellular response to DNA damage includes activation of signaling pathways that lead to release of proinflammatory cytokines including IFN-α and cytokines triggered by activation of the NF-κB signaling pathway. Specifically, cyclophosphamide has been shown to induce IFN-γ and IL-2, pro-inflammatory cytokines that promote immunogenicity (53). Parkes et al. demonstrated that in a breast cancer model DNA-damage-repair defects lead to increased expression of the chemokines CXCL10 and CCL5 from tumor cells (16, 54).
The precise impact of chemotherapy commonly used in relapsed Ewing sarcoma [irinotecan and temozolomide (IT), topotecan and cyclophosphamide (TC), high dose ifosfamide (IFOS), and gemcitabine and docetaxel (GD) (55)] on Ewing tumor immunobiology is still largely unknown. PARP inhibitors have been shown to induce infiltration of CD8+ T cells in breast cancer, and the efficacy of PARP inhibition is due to recruitment of these cytotoxic T cells through the cGAS/STING pathway (56). In this model, depletion of CD8+ T cells decreased the efficacy of PARP inhibition. In addition to recruiting cytotoxic T cells, PARP inhibition has also been shown to increase the expression of immune checkpoint ligand PD-L1 on cancer cells (57). Our work has previously shown that PD-L1 and PD-L2 expression can be manipulated in Ewing cell subpopulations in response to inflammatory signaling (58).
In summary, the effect of DNA damaging chemotherapeutic agents used in the treatment of Ewing sarcoma can, in theory, manipulate the TIME; however, this is an understudied area. While chemotherapy has the temporary ability to alter the TIME, ultimately due to systemic effects, patients are largely overall immunosuppressed during therapy. Thus, focal delivery of DNA damage, such as radiation therapy, may be beneficial when considering immunotherapy combinations.
The interest in the immune-mediated effects of radiation date back to the 1980s when it was first noted that local radiation can lead to anti-tumor effect at distant sites of disease (59, 60). Subsequently, many studies have demonstrated that local radiation can produce systemic immune-mediated anti-tumor effects, though this is not a consistent finding in all studies (6, 61, 62). Studies examining the radiation anti-tumor effect in immunocompetent vs immunodeficient mouse models of melanoma have demonstrated that the presence of CD8+ cytotoxic T cells are necessary for this response (63). Radiation enhances the immune response to tumors through release of cytokines and chemokines in the tumor microenvironment following cell death (64). These cytokines and chemokines result in infiltration of effector immune cells (dendritic cells, macrophages, cytotoxic T cells) as well as immunosuppressive populations (Tregs, myeloid-derived suppressor cells) (65). Similar to the effect of chemotherapy described above, radiation induces immunogenic cell death leading to release of DAMPs. This leads to increased production and recruitment of proinflammatory cytokines and chemokines, including CXCL9, CXCL10, and CXCL11 (66). The generation of this proinflammatory environment is thought to lead to recruitment of effector T cells and may enhance the priming of T cells in the TIME. Recently, the essential role of natural killer (NK) cells in controlling the radiation-induced anti-tumor has been demonstrated (67).
In addition to promoting a proinflammatory TIME, radiation can also exert immunosuppressive effects. Tregs are a well described subset of CD4+ T cells that exert immunosuppressive effects on the TIME. In some adult carcinomas, radiation has been shown to increase Tregs and the subsequent production of immunosuppressive cytokines including TGF-β and IL-10 (68) TGF-β is known to be increased following radiation and is converted from its latent to active form by reactive species generated during radiation (69). TGF-β exerts immunosuppressive effects on the TIME and it has been shown that increase in TGF-β in the TIME can lead to decreased efficacy of immunotherapy through the exclusion of CD8+ T cells (70). Several studies have demonstrated that inhibition of the immunosuppressive pathways activated by localized radiation can improve radiation-induced tumor kill and anti-tumor immunity (71, 72).
An additional immunosuppressive cell population that can be induced/increased following radiation are myeloid-deprived suppressor cells (MDSCs). MDSCs are well described to promote tumor growth and survival and are known to be recruited into the TIME of pancreatic and prostate cancer immediately following radiation (73), with a decrease in this population seen at 1-2 weeks post radiation. TGF-β is known to induce differentiation of macrophages to an M2 phenotype which is protumor and immunosuppressive. These mechanisms of immunosuppression induced by radiation represent potential targets to improve the anti-tumor immune response induced by radiation.
Currently, relatively little is known about the specific impact of radiation on the Ewing sarcoma TIME. New therapeutic approaches for patients with metastatic and relapsed Ewing sarcoma are long overdue. While agents that induce tumor DNA damage clearly provide some benefit for the treatment of relapsed disease, they are rarely curative. Understanding which multi-modality therapeutic approaches may circumvent Ewing tumor cell resistance to single- modality therapies is a priority. It is possible that subsets of Ewing tumors in the DNA-damage- repair deficiency spectrum (Figure 1) could demonstrate differential responses to multi-modality therapy. Radiation therapy is often utilized in patients with high-risk (metastatic and relapsed) Ewing sarcoma, and given its potential to modulate the TIME, it is a logical treatment modality to consider in combination with immunomodulation (Figure 2). There has been growing interest in oncology to combine radiation with immunomodulatory agents to improve the anti-tumor immune response (74–76). Given the concurrent immune-stimulatory and immunosuppressive effects that radiation therapy can trigger in the TIME, there has been interest in combination therapies targeting both of these sequalae (77). Broadly speaking, logical categories of immunomodulators to preclinically study in combination with radiation for the treatment of Ewing sarcoma include: 1) immune checkpoint inhibitors (ICI), 2) cytokine modulators, and 3) cell-based therapies. Here we will briefly address each of these approaches.
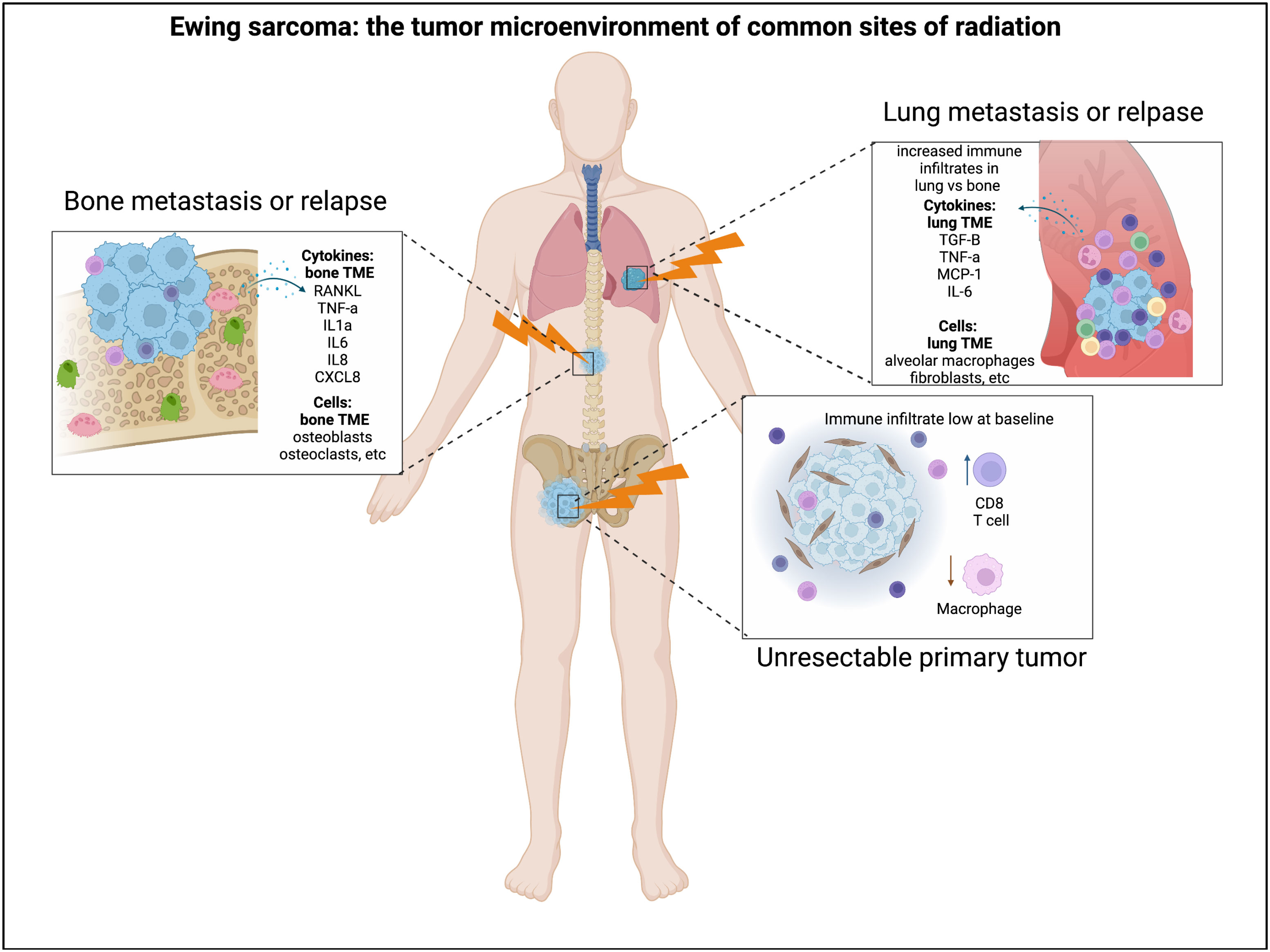
Figure 2 Radiation and the Ewing sarcoma tumor immune microenvironment. Radiation is often utilized for the treatment of Ewing sarcoma in the setting of unresectable primary tumors, lung metastases, relapse to the bone, etc. The Ewing tumor immune microenvironment can demonstrate differences in immune infiltration and cytokine abundance in these distinct microenvironments where radiation is utilized. Figure created by biorender.com.
The combination of radiation therapy and ICI in patients with advanced solid tumors has demonstrated promising early clinical results (74, 75). It has also been reported that the presence of DNA damage repair defects, such as germline BRCA 1/2 pathogenic variants, is correlated with increased expression of immunosuppressive ligands such as PD-1/PD-L1 (78), considered one marker of response to immune checkpoint inhibition. This association provides a rationale for preclinically testing the response of Ewing tumors with additional DNA damage repair defects to the combination of radiation and immune checkpoint inhibition.
In addition to examining ICIs, modulation of cytokines in the tumor microenvironment during radiation therapy is of great interest. While not every cytokine can be addressed in this mini-review, we will highlight two. TGF-β is an immunosuppressive cytokine that is increased in tumor microenvironments following radiation and has been shown to confer resistance to radiation (72). Inhibition of TGF-β during radiation has the potential to enhance the anti-tumor immune response (79). A second cytokine, IL-6, is known to be secreted by Ewing tumors (80, 81), and can be upregulated following radiation-induced DNA damage. Further, it is thought that the presence of IL-6 in the TME confers radiation resistance (82). IL-6 inhibitors are active in clinical trials as monotherapy for cancer (83), however, combination therapy with these inhibitors during radiation offers another therapeutic avenue worthy of preclinical testing.
Lastly, there is promise for delivering cell-based therapies in the setting of radiation. Chimeric antigen receptor T-cells (CAR-T) therapies have shown great success in the treatment of hematologic malignancies but have not seen the same success in solid tumors (84). Challenges have included identification of an ideal target antigen, cell trafficking to the tumor, and the overall immunosuppressive environment of solid tumors. Therapies targeting the DNA damage repair pathway have shown some success in solid tumors in improving response to CAR-T therapy through induction of a more pro-inflammatory TIME (85). Additionally, radiation therapy delivered prior to administration of CAR-T in a mouse model of glioblastoma demonstrated improved trafficking and efficacy of the CAR-T cells post-radiation (86). There is ongoing research in the field to identify a targetable antigen for cell based therapies for the treatment of Ewing sarcoma (87). In addition to CAR-T cell therapy, dendritic cell-based immunotherapy is a cell-based therapy that could logically be combined with DNA-damaging agents. Studies have demonstrated improved efficacy of dendritic cell vaccination when given in combination with radiation (88, 89). Lastly, as noted above, recent work has demonstrated the key role of NK cells in the radiation anti-tumor response. Understanding the role of NK cells in the TIME of Ewing tumors specifically during radiation is a priority (67, 90).
Significant historical impediments to studying the influence of DNA damage on the immune microenvironment in Ewing sarcoma include, but are not limited to, the lack of an immunocompetent animal model of Ewing sarcoma (4) and the sparsity of samples from disease relapse and pre-/post- intervention biopsies. Recently, a genetically engineered zebrafish model of Ewing sarcoma has been developed, which may offer a new immunocompetent model (91), although studies specifically investigating immune interactions in this model have not yet been performed. A potentially valuable model for studying the TIME of Ewing sarcoma is the development of humanized (presence of human immune cells), immunocompetent mouse models, a focus of ongoing work by our group. Developing and validating a robust preclinical model to study the impact of DNA-damaging agents used clinically for the treatment of Ewing sarcoma on the TIME is a crucial and necessary step toward determining promising immunomodulatory agents to partner with radiation or chemotherapy in an attempt to improve the outcomes for patients with advanced disease. While DNA damage, such as radiation therapy, is the focus of this mini-review, the impact of other novel agents, such as tyrosine kinase inhibitors, agents targeting EWSR1::FLI1, etc., on the Ewing sarcoma TIME are also worthy of exploration and represent a limitation of this mini-review.
JD, AO, and KB contributed to the mini-review concept, initial manuscript writing, and editing. JD and KB generated the figures and completed formatting. All authors contributed to the article and approved the submitted version.
KB is supported by the National Institutes of Health (K08CA252178) and Alex’s Lemonade Stand (Innovator Award). KB would like to thank the Morden Foundation, CHEMOWARRIOR, and the UPMC Children’s Hospital Foundation for their support. The University of Pittsburgh holds a Physician-Scientist Institutional Award from the Burroughs Welcome Fund (JD). Research reported here has been supported by the National Institutes of Health under their award number 5K12HD052892-15, PI: Terence S. Dermody, MD, JD and AO would like to thank Pittsburgh Cure Sarcoma for their support.
AO receives research funding from Varian Medical Systems, Reflexion Medical and serves as a consultant for RenovoRx.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Delattre O, Zucman J, Plougastel B, Desmaze C, Melot T, Peter M, et al. Gene fusion with an ETS DNA-binding domain caused by chromosome translocation in human tumours. Nature (1992) 359(6391):162–5.doi: 10.1038/359162a0
2. Stahl M, Ranft A, Paulussen M, Bolling T, Vieth V, Bielack S, et al. Risk of recurrence and survival after relapse in patients with Ewing sarcoma. Pediatr Blood Cancer. (2011) 57(4):549–53. doi: 10.1002/pbc.23040
3. Leavey PJ, Laack NN, Krailo MD, Buxton A, Randall RL, DuBois SG, et al. Phase III trial adding vincristine-Topotecan-Cyclophosphamide to the initial treatment of patients with nonmetastatic Ewing sarcoma: A children's oncology group report. J Clin Oncol (2021) 39(36):4029–38. doi: 10.1200/JCO.21.00358
4. Minas TZ, Surdez D, Javaheri T, Tanaka M, Howarth M, Kang HJ, et al. Combined experience of six independent laboratories attempting to create an Ewing sarcoma mouse model. Oncotarget (2017) 8(21):34141–63. doi: 10.18632/oncotarget.9388
5. Brown JS, Sundar R, Lopez J. Combining DNA damaging therapeutics with immunotherapy: more haste, less speed. Br J Cancer (2018) 118(3):312–24. doi: 10.1038/bjc.2017.376
6. Burnette B, Weichselbaum RR. Radiation as an immune modulator. Semin Radiat Oncol (2013) 23(4):273–80. doi: 10.1016/j.semradonc.2013.05.009
7. Waldman AD, Fritz JM, Lenardo MJ. A guide to cancer immunotherapy: from T cell basic science to clinical practice. Nat Rev Immunol (2020) 20(11):651–68. doi: 10.1038/s41577-020-0306-5
8. Ferris RL, Blumenschein G Jr., Fayette J, Guigay J, Colevas AD, Licitra L, et al. Nivolumab for recurrent squamous-cell carcinoma of the head and neck. N Engl J Med (2016) 375(19):1856–67. doi: 10.1056/NEJMoa1602252
9. Wolchok JD, Chiarion-Sileni V, Gonzalez R, Rutkowski P, Grob JJ, Cowey CL, et al. Overall survival with combined nivolumab and ipilimumab in advanced melanoma. N Engl J Med (2017) 377(14):1345–56. doi: 10.1056/NEJMoa1709684
10. Birdi HK, Jirovec A, Cortes-Kaplan S, Werier J, Nessim C, Diallo JS, et al. Immunotherapy for sarcomas: New frontiers and unveiled opportunities. J Immunother Cancer (2021) 9(2): 1–11. doi: 10.1136/jitc-2020-001580
11. Tawbi HA, Burgess M, Bolejack V, Van Tine BA, Schuetze SM, Hu J, et al. Pembrolizumab in advanced soft-tissue sarcoma and bone sarcoma (SARC028): A multicentre, two-cohort, single-arm, open-label, phase 2 trial. Lancet Oncol (2017) 18(11):1493–501. doi: 10.1016/S1470-2045(17)30624-1
12. Davis KL, Fox E, Merchant MS, Reid JM, Kudgus RA, Liu X, et al. Nivolumab in children and young adults with relapsed or refractory solid tumours or lymphoma (ADVL1412): a multicentre, open-label, single-arm, phase 1-2 trial. Lancet Oncol (2020) 21(4):541–50. doi: 10.1016/S1470-2045(20)30023-1
13. Messenheimer DJ, Jensen SM, Afentoulis ME, Wegmann KW, Feng Z, Friedman DJ, et al. Timing of PD-1 blockade is critical to effective combination immunotherapy with anti-OX40. Clin Cancer Res (2017) 23(20):6165–77. doi: 10.1158/1078-0432.CCR-16-2677
14. Berghuis D, Santos SJ, Baelde HJ, Taminiau AH, Egeler RM, Schilham MW, et al. Pro-inflammatory chemokine-chemokine receptor interactions within the Ewing sarcoma microenvironment determine CD8(+) T-lymphocyte infiltration and affect tumour progression. J Pathol (2011) 223(3):347–57. doi: 10.1002/path.2819
15. Stahl D, Gentles AJ, Thiele R, Gutgemann I. Prognostic profiling of the immune cell microenvironment in Ewing s sarcoma family of tumors. Oncoimmunology (2019) 8(12):e1674113. doi: 10.1080/2162402X.2019.1674113
16. Cillo AR, Mukherjee E, Bailey NG, Onkar S, Daley J, Salgado C, et al. Ewing Sarcoma and osteosarcoma have distinct immune signatures and intercellular communication networks. Clin Cancer Res (2022). doi: 10.1158/1078-0432.CCR-22-1471
17. Nesbit ME Jr., Gehan EA, Burgert EO Jr., Vietti TJ, Cangir A, Tefft M, et al. Multimodal therapy for the management of primary, nonmetastatic ewing's sarcoma of bone: A long-term follow-up of the first intergroup study. J Clin Oncol (1990) 8(10):1664–74. doi: 10.1200/JCO.1990.8.10.1664
18. Brown AP, Fixsen JA, Plowman PN. Local control of ewing's sarcoma: an analysis of 67 patients. Br J Radiol (1987) 60(711):261–8. doi: 10.1259/0007-1285-60-711-261
19. Garnett MJ, Edelman EJ, Heidorn SJ, Greenman CD, Dastur A, Lau KW, et al. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature (2012) 483(7391):570–5. doi: 10.1038/nature11005
20. Brenner JC, Feng FY, Han S, Patel S, Goyal SV, Bou-Maroun LM, et al. PARP-1 inhibition as a targeted strategy to treat ewing's sarcoma. Cancer Res (2012) 72(7):1608–13. doi: 10.1158/0008-5472.CAN-11-3648
21. Rouleau M, Patel A, Hendzel MJ, Kaufmann SH, Poirier GG. PARP inhibition: PARP1 and beyond. Nat Rev Cancer (2010) 10(4):293–301. doi: 10.1038/nrc2812
22. Fong PC, Boss DS, Yap TA, Tutt A, Wu P, Mergui-Roelvink M, et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med (2009) 361(2):123–34. doi: 10.1056/NEJMoa0900212
23. Gorthi A, Romero JC, Loranc E, Cao L, Lawrence LA, Goodale E, et al. EWS-FLI1 increases transcription to cause r-loops and block BRCA1 repair in Ewing sarcoma. Nature (2018) 555(7696):387–91. doi: 10.1038/nature25748
24. Spahn L, Petermann R, Siligan C, Schmid JA, Aryee DN, Kovar H. Interaction of the EWS NH2 terminus with BARD1 links the ewing's sarcoma gene to a common tumor suppressor pathway. Cancer Res (2002) 62(16):4583–7.
25. Choy E, Butrynski JE, Harmon DC, Morgan JA, George S, Wagner AJ, et al. Phase II study of olaparib in patients with refractory Ewing sarcoma following failure of standard chemotherapy. BMC Cancer. (2014) 14:813. doi: 10.1186/1471-2407-14-813
26. Stewart E, Goshorn R, Bradley C, Griffiths LM, Benavente C, Twarog NR, et al. Targeting the DNA repair pathway in Ewing sarcoma. Cell Rep (2014) 9(3):829–41. doi: 10.1016/j.celrep.2014.09.028
27. Franzetti GA, Laud-Duval K, van der Ent W, Brisac A, Irondelle M, Aubert S, et al. Cell-to-cell heterogeneity of EWSR1-FLI1 activity determines proliferation/migration choices in Ewing sarcoma cells. Oncogene (2017) 36(25):3505–14. doi: 10.1038/onc.2016.498
28. Pedersen EA, Menon R, Bailey KM, Thomas DG, Van Noord RA, Tran J, et al. Activation of wnt/beta-catenin in Ewing sarcoma cells antagonizes EWS/ETS function and promotes phenotypic transition to more metastatic cell states. Cancer Res (2016) 76(17):5040–53. doi: 10.1158/0008-5472.CAN-15-3422
29. Seong BKA, Dharia NV, Lin S, Donovan KA, Chong S, Robichaud A, et al. TRIM8 modulates the EWS/FLI oncoprotein to promote survival in Ewing sarcoma. Cancer Cell (2021) 39(9):1262–78.e7. doi: 10.1016/j.ccell.2021.07.003
30. Crompton BD, Stewart C, Taylor-Weiner A, Alexe G, Kurek KC, Calicchio ML, et al. The genomic landscape of pediatric Ewing sarcoma. Cancer Discov (2014) 4(11):1326–41. doi: 10.1158/2159-8290.CD-13-1037
31. Tirode F, Surdez D, Ma X, Parker M, Le Deley MC, Bahrami A, et al. Genomic landscape of Ewing sarcoma defines an aggressive subtype with co-association of STAG2 and TP53 mutations. Cancer Discovery (2014) 4(11):1342–53. doi: 10.1158/2159-8290.CD-14-0622
32. Brohl AS, Solomon DA, Chang W, Wang J, Song Y, Sindiri S, et al. The genomic landscape of the Ewing sarcoma family of tumors reveals recurrent STAG2 mutation. PloS Genet (2014) 10(7):e1004475. doi: 10.1371/journal.pgen.1004475
33. Bailey ML, O'Neil NJ, van Pel DM, Solomon DA, Waldman T, Hieter P. Glioblastoma cells containing mutations in the cohesin component STAG2 are sensitive to PARP inhibition. Mol Cancer Ther (2014) 13(3):724–32. doi: 10.1158/1535-7163.MCT-13-0749
34. Shulman DS, Chen S, Hall D, Nag A, Thorner AR, Lessnick SL, et al. Adverse prognostic impact of the loss of STAG2 protein expression in patients with newly diagnosed localised Ewing sarcoma: A report from the children's oncology group. Br J Cancer (2022). doi: 10.1038/s41416-022-01977-2
35. Zhang J, Walsh MF, Wu G, Edmonson MN, Gruber TA, Easton J, et al. Germline mutations in predisposition genes in pediatric cancer. N Engl J Med (2015) 373(24):2336–46. doi: 10.1056/NEJMoa1508054
36. Parsons DW, Roy A, Yang Y, Wang T, Scollon S, Bergstrom K, et al. Diagnostic yield of clinical tumor and germline whole-exome sequencing for children with solid tumors. JAMA Oncol (2016) 2(5):616–24. doi: 10.1001/jamaoncol.2015.5699
37. Brohl AS, Patidar R, Turner CE, Wen X, Song YK, Wei JS, et al. Frequent inactivating germline mutations in DNA repair genes in patients with Ewing sarcoma. Genet Med (2017) 19(8):955–8. doi: 10.1038/gim.2016.206
38. Venier RE, Maurer LM, Kessler EM, Ranganathan S, McGough RL, Weiss KR, et al. A germline BARD1 mutation in a patient with Ewing sarcoma: Implications for familial testing and counseling. Pediatr Blood Cancer (2019) 66(9):e27824. doi: 10.1002/pbc.27824
39. Gillani R, Camp SY, Han S, Jones JK, Chu H, O'Brien S, et al. Germline predisposition to pediatric Ewing sarcoma is characterized by inherited pathogenic variants in DNA damage repair genes. Am J Hum Genet (2022) 109(6):1026–37. doi: 10.1016/j.ajhg.2022.04.007
40. Maurer L, Daley J, Mukherjee E, Venier R, Julian C, Bailey N, et al. BRCA1-associated RING domain-1 (BARD1) loss and GBP1 expression enhance sensitivity to DNA damage in Ewing sarcoma. Cancer Res Commun (2022), 220–32. doi: 10.1158/2767-9764.crc-21-0047
41. Kung FH, Pratt CB, Vega RA, Jaffe N, Strother D, Schwenn M, et al. Ifosfamide/etoposide combination in the treatment of recurrent malignant solid tumors of childhood. A pediatric oncology group phase II study. Cancer (1993) 71(5):1898–903. doi: 10.1002/1097-0142(19930301)71:5<1898::aid-cncr2820710529>3.0.co;2-q
42. Grier HE, Krailo MD, Tarbell NJ, Link MP, Fryer CJ, Pritchard DJ, et al. Addition of ifosfamide and etoposide to standard chemotherapy for ewing's sarcoma and primitive neuroectodermal tumor of bone. N Engl J Med (2003) 348(8):694–701. doi: 10.1056/NEJMoa020890
43. Womer RB, West DC, Krailo MD, Dickman PS, Pawel BR, Grier HE, et al. Randomized controlled trial of interval-compressed chemotherapy for the treatment of localized Ewing sarcoma: A report from the children's oncology group. J Clin Oncol (2012) 30(33):4148–54. doi: 10.1200/JCO.2011.41.5703
44. Donaldson SS. Ewing Sarcoma: radiation dose and target volume. Pediatr Blood Cancer. (2004) 42(5):471–6. doi: 10.1002/pbc.10472
45. Laskar S, Sinha S, Chatterjee A, Khanna N, Manjali JJ, Puri A, et al. Radiation therapy dose escalation in unresectable Ewing sarcoma: Final results of a phase 3 randomized controlled trial. Int J Radiat Oncol Biol Phys (2022) 113(5):996–1002. doi: 10.1016/j.ijrobp.2022.04.024
46. Boyce-Fappiano D, Damron EP, Farooqi A, Mitra D, Conley AP, Somaiah N, et al. Hypofractionated radiation therapy for unresectable or metastatic sarcoma lesions. Adv Radiat Oncol (2022) 7(3):100913. doi: 10.1016/j.adro.2022.100913
47. Paulussen M, Braun-Munzinger G, Burdach S, Deneke S, Dunst J, Fellinger E, et al. [Results of treatment of primary exclusively pulmonary metastatic Ewing sarcoma. a retrospective analysis of 41 patients]. Klin Padiatr. (1993) 205(4):210–6. doi: 10.1055/s-2007-1025229
48. Khanna N, Pandey A, Bajpai J. Metastatic ewing's sarcoma: Revisiting the "Evidence on the fence". Indian J Med Paediatr Oncol (2017) 38(2):173–81. doi: 10.4103/ijmpo.ijmpo_24_17
49. Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature (2005) 434(7035):917–21. doi: 10.1038/nature03445
50. Galluzzi L, Buque A, Kepp O, Zitvogel L, Kroemer G. Immunogenic cell death in cancer and infectious disease. Nat Rev Immunol (2017) 17(2):97–111. doi: 10.1038/nri.2016.107
51. Bezu L, Gomes-de-Silva LC, Dewitte H, Breckpot K, Fucikova J, Spisek R, et al. Combinatorial strategies for the induction of immunogenic cell death. Front Immunol (2015) 6:187. doi: 10.3389/fimmu.2015.00187
52. Liu WM, Fowler DW, Smith P, Dalgleish AG. Pre-treatment with chemotherapy can enhance the antigenicity and immunogenicity of tumours by promoting adaptive immune responses. Br J Cancer (2010) 102(1):115–23. doi: 10.1038/sj.bjc.6605465
53. Sistigu A, Viaud S, Chaput N, Bracci L, Proietti E, Zitvogel L. Immunomodulatory effects of cyclophosphamide and implementations for vaccine design. Semin Immunopathol (2011) 33(4):369–83. doi: 10.1007/s00281-011-0245-0
54. Parkes EE, Walker SM, Taggart LE, McCabe N, Knight LA, Wilkinson R, et al. Activation of STING-dependent innate immune signaling by s-Phase-Specific DNA damage in breast cancer. J Natl Cancer Inst (2017) 109(1): 1–10. doi: 10.1093/jnci/djw199
55. McCabe M, Kirton L, Khan M, Fenwick N, Strauss SJ, Valverde C, et al. Phase III assessment of topotecan and cyclophosphamide and high-dose ifosfamide in rEECur: An international randomized controlled trial of chemotherapy for the treatment of recurrent and primary refractory Ewing sarcoma (RR-ES). J Clin Oncol (2022) 40(17_suppl):LBA2–LBA.
56. Pantelidou C, Sonzogni O, De Oliveria Taveira M, Mehta AK, Kothari A, Wang D, et al. PARP inhibitor efficacy depends on CD8(+) T-cell recruitment via intratumoral STING pathway activation in BRCA-deficient models of triple-negative breast cancer. Cancer Discov (2019) 9(6):722–37. doi: 10.1158/2159-8290.CD-18-1218
57. Jiao S, Xia W, Yamaguchi H, Wei Y, Chen MK, Hsu JM, et al. PARP inhibitor upregulates PD-L1 expression and enhances cancer-associated immunosuppression. Clin Cancer Res (2017) 23(14):3711–20.doi: 10.1158/1078-0432.CCR-16-3215
58. Bailey KM, Julian CM, Klinghoffer AN, Bernard H, Lucas PC, McAllister-Lucas LM. EWS-FLI1 low Ewing sarcoma cells demonstrate decreased susceptibility to T-cell-mediated tumor cell apoptosis. Oncotarget (2019) 10(36):3385–99.doi: 10.18632/oncotarget.26939
59. North RJ. Radiation-induced, immunologically mediated regression of an established tumor as an example of successful therapeutic immunomanipulation. preferential elimination of suppressor T cells allows sustained production of effector T cells. J Exp Med (1986) 164(5):1652–66.doi: 10.1084/jem.164.5.1652
60. Demaria S, Ng B, Devitt ML, Babb JS, Kawashima N, Liebes L, et al. Ionizing radiation inhibition of distant untreated tumors (abscopal effect) is immune mediated. Int J Radiat Oncol Biol Phys (2004) 58(3):862–70.doi: 10.1016/j.ijrobp.2003.09.012
61. Demaria S, Kawashima N, Yang AM, Devitt ML, Babb JS, Allison JP, et al. Immune-mediated inhibition of metastases after treatment with local radiation and CTLA-4 blockade in a mouse model of breast cancer. Clin Cancer Res (2005) 11(2 Pt 1):728–34.
62. Lugade AA, Moran JP, Gerber SA, Rose RC, Frelinger JG, Lord EM. Local radiation therapy of B16 melanoma tumors increases the generation of tumor antigen-specific effector cells that traffic to the tumor. J Immunol (2005) 174(12):7516–23.doi: 10.4049/jimmunol.174.12.7516
63. Lee Y, Auh SL, Wang Y, Burnette B, Wang Y, Meng Y, et al. Therapeutic effects of ablative radiation on local tumor require CD8+ T cells: changing strategies for cancer treatment. Blood (2009) 114(3):589–95.doi: 10.1182/blood-2009-02-206870
64. Luke JJ, Onderdonk BE, Bhave SR, Karrison T, Lemons JM, Chang P, et al. Improved survival associated with local tumor response following multisite radiotherapy and pembrolizumab: Secondary analysis of a phase I trial. Clin Cancer Res (2020) 26(24):6437–44.doi: 10.1158/1078-0432.CCR-20-1790
65. Barker HE, Paget JT, Khan AA, Harrington KJ. The tumour microenvironment after radiotherapy: mechanisms of resistance and recurrence. Nat Rev Cancer. (2015) 15(7):409–25.doi: 10.1038/nrc3958
66. Weichselbaum RR, Liang H, Deng L, Fu YX. Radiotherapy and immunotherapy: a beneficial liaison? Nat Rev Clin Oncol (2017) 14(6):365–79.doi: 10.1038/nrclinonc.2016.211
67. Walle T, Kraske JA, Liao B, Lenoir B, Timke C, von Bohlen Und Halbach E, et al. Radiotherapy orchestrates natural killer cell dependent antitumor immune responses through CXCL8. Sci Adv (2022) 8(12):eabh4050.doi: 10.1126/sciadv.abh4050
68. Dancea HC, Shareef MM, Ahmed MM. Role of radiation-induced TGF-beta signaling in cancer therapy. Mol Cell Pharmacol (2009) 1(1):44–56.doi: 10.4255/mcpharmacol.09.06
69. Barcellos-Hoff MH, Dix TA. Redox-mediated activation of latent transforming growth factor-beta 1. Mol Endocrinol (1996) 10(9):1077–83.doi: 10.1210/mend.10.9.8885242
70. Wang J, Xu Z, Wang Z, Du G, Lun L. TGF-beta signaling in cancer radiotherapy. Cytokine (2021) 148:155709.doi: 10.1016/j.cyto.2021.155709
71. Young KH, Gough MJ, Crittenden M. Tumor immune remodeling by TGFbeta inhibition improves the efficacy of radiation therapy. Oncoimmunology (2015) 4(3):e955696.doi: 10.4161/21624011.2014.955696
72. Vanpouille-Box C, Diamond JM, Pilones KA, Zavadil J, Babb JS, Formenti SC, et al. TGFbeta is a master regulator of radiation therapy-induced antitumor immunity. Cancer Res (2015) 75(11):2232–42.doi: 10.1158/0008-5472.CAN-14-3511
73. Vatner RE, Formenti SC. Myeloid-derived cells in tumors: effects of radiation. Semin Radiat Oncol (2015) 25(1):18–27. doi: 10.1016/j.semradonc.2014.07.008
74. Luke JJ, Lemons JM, Karrison TG, Pitroda SP, Melotek JM, Zha Y, et al. Safety and clinical activity of pembrolizumab and multisite stereotactic body radiotherapy in patients with advanced solid tumors. J Clin Oncol (2018) 36(16):1611–8.doi: 10.1200/JCO.2017.76.2229
75. Song HN, Jin H, Kim JH, Ha IB, Kang KM, Choi HS, et al. Abscopal effect of radiotherapy enhanced with immune checkpoint inhibitors of triple negative breast cancer in 4T1 mammary carcinoma model. Int J Mol Sci (2021) 22(19): 1–9.doi: 10.3390/ijms221910476
76. Demaria S, Guha C, Schoenfeld J, Morris Z, Monjazeb A, Sikora A, et al. Radiation dose and fraction in immunotherapy: one-size regimen does not fit all settings, so how does one choose? J Immunother Cancer (2021) 9(4): 1–15. doi: 10.1136/jitc-2020-002038
77. Bouquet F, Pal A, Pilones KA, Demaria S, Hann B, Akhurst RJ, et al. TGFbeta1 inhibition increases the radiosensitivity of breast cancer cells in vitro and promotes tumor control by radiation in vivo. Clin Cancer Res (2011) 17(21):6754–65. doi: 10.1158/1078-0432.CCR-11-0544
78. Strickland KC, Howitt BE, Shukla SA, Rodig S, Ritterhouse LL, Liu JF, et al. Association and prognostic significance of BRCA1/2-mutation status with neoantigen load, number of tumor-infiltrating lymphocytes and expression of PD-1/PD-L1 in high grade serous ovarian cancer. Oncotarget (2016) 7(12):13587–98.doi: 10.18632/oncotarget.7277
79. Ciardiello D, Elez E, Tabernero J, Seoane J. Clinical development of therapies targeting TGFbeta: current knowledge and future perspectives. Ann Oncol (2020) 31(10):1336–49.doi: 10.1016/j.annonc.2020.07.009
80. Lissat A, Joerschke M, Shinde DA, Braunschweig T, Meier A, Makowska A, et al. IL6 secreted by Ewing sarcoma tumor microenvironment confers anti-apoptotic and cell-disseminating paracrine responses in Ewing sarcoma cells. BMC Cancer. (2015) 15:552.doi: 10.1186/s12885-015-1564-7
81. Anderson JL, Titz B, Akiyama R, Komisopoulou E, Park A, Tap WD, et al. Phosphoproteomic profiling reveals IL6-mediated paracrine signaling within the Ewing sarcoma family of tumors. Mol Cancer Res (2014) 12(12):1740–54.doi: 10.1158/1541-7786.MCR-14-0159
82. Centurione L, Aiello FB. DNA Repair and cytokines: TGF-beta, IL-6, and thrombopoietin as different biomarkers of radioresistance. Front Oncol (2016) 6:175.doi: 10.3389/fonc.2016.00175
83. Kampan NC, Xiang SD, McNally OM, Stephens AN, Quinn MA, Plebanski M. Immunotherapeutic interleukin-6 or interleukin-6 receptor blockade in cancer: Challenges and opportunities. Curr Med Chem (2018) 25(36):4785–806.doi: 10.2174/0929867324666170712160621
84. Marofi F, Motavalli R, Safonov VA, Thangavelu L, Yumashev AV, Alexander M, et al. CAR T cells in solid tumors: Challenges and opportunities. Stem Cell Res Ther (2021) 12(1):81.doi: 10.1186/s13287-020-02128-1
85. Ji F, Zhang F, Zhang M, Long K, Xia M, Lu F, et al. Targeting the DNA damage response enhances CD70 CAR-T cell therapy for renal carcinoma by activating the cGAS-STING pathway. J Hematol Oncol (2021) 14(1):152.doi: 10.1186/s13045-021-01168-1
86. Weiss T, Weller M, Guckenberger M, Sentman CL, Roth P. NKG2D-based CAR T cells and radiotherapy exert synergistic efficacy in glioblastoma. Cancer Res (2018) 78(4):1031–43.doi: 10.1158/0008-5472.CAN-17-1788
87. Lin Z, Wu Z, Luo W. A novel treatment for ewing's sarcoma: Chimeric antigen receptor-T cell therapy. Front Immunol (2021) 12:707211.doi: 10.3389/fimmu.2021.707211
88. Teitz-Tennenbaum S, Li Q, Rynkiewicz S, Ito F, Davis MA, McGinn CJ, et al. Radiotherapy potentiates the therapeutic efficacy of intratumoral dendritic cell administration. Cancer Res (2003) 63(23):8466–75.
89. Kim KW, Kim SH, Shin JG, Kim GS, Son YO, Park SW, et al. Direct injection of immature dendritic cells into irradiated tumor induces efficient antitumor immunity. Int J Cancer (2004) 109(5):685–90.doi: 10.1002/ijc.20036
90. Omer N, Nicholls W, Ruegg B, Souza-Fonseca-Guimaraes F, Rossi GR. Enhancing natural killer cell targeting of pediatric sarcoma. Front Immunol (2021) 12:791206.doi: 10.3389/fimmu.2021.791206
Keywords: Ewing sarcoma, radiation, immunobiology, DNA damage, immunomodulation, relapse
Citation: Daley JD, Olson AC and Bailey KM (2022) Harnessing immunomodulation during DNA damage in Ewing sarcoma. Front. Oncol. 12:1048705. doi: 10.3389/fonc.2022.1048705
Received: 19 September 2022; Accepted: 26 October 2022;
Published: 22 November 2022.
Edited by:
Antonino De Paoli, Department of Radiation Oncology (IRCCS), ItalyReviewed by:
Alessandro De Vita, Scientific Institute of Romagna for the Study and Treatment of Tumors (IRCCS), ItalyCopyright © 2022 Daley, Olson and Bailey. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kelly M. Bailey, S2VsbHkuQmFpbGV5QGNocC5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.