 Farrah S. Bakr
Farrah S. Bakr Sean J. Whittaker
Sean J. Whittaker
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Oncol., 24 November 2022
Sec. Hematologic Malignancies
Volume 12 - 2022 | https://doi.org/10.3389/fonc.2022.1043254
This article is part of the Research TopicAdvances in the Understanding and Treatment of Cutaneous LymphomaView all 7 articles
Cutaneous T-cell lymphomas (CTCL) are a heterogeneous group of non-Hodgkin’s lymphomas (NHL) characterised by the clonal proliferation of malignant, skin homing T-cells. Recent advances have been made in understanding the molecular pathogenesis of CTCL. Multiple deep sequencing studies have revealed a complex genomic landscape with large numbers of novel single nucleotide variants (SNVs) and copy number variations (CNVs). Commonly perturbed genes include those involved in T-cell receptor signalling, T-cell proliferation, differentiation and survival, epigenetic regulators as well as genes involved in genome maintenance and DNA repair. In addition, studies in CTCL have identified a dominant UV mutational signature in contrast to systemic T-cell lymphomas and this likely contributes to the high tumour mutational burden. As current treatment options for advanced stages of CTCL are associated with short-lived responses, targeting these deregulated pathways could provide novel therapeutic approaches for patients. In this review article we summarise the key pathways disrupted in CTCL and discuss the potential therapeutic implications of these findings.
Cutaneous T-cell lymphomas are a heterogeneous group of Non-Hodgkin’s lymphomas of which Mycosis Fungoides (MF) is responsible for almost 50% of all primary cutaneous lymphomas (1). They display wide variation in relation to their clinical, histopathological, immunophenotypic and underlying biologic features (1, 2). CTCLs are amongst a wider group of mature T-cell malignancies of which the more common subtypes are Peripheral T-cell lymphoma, Not Otherwise Specified (PTCL nos) and systemic anaplastic large cell lymphoma (ALCL). A rare HTLV-1 driven subtype of T-cell lymphoma known as Adult T-Cell Leukaemia/Lymphoma (ATLL) shares several phenotypic and genetic features with Sézary syndrome (SS) (3). Though MF/SS are the focus of this review, we will compare and contrast genomic abnormalities of MF/SS with other CTCL variants and systemic T-cell lymphomas and highlight potential novel therapeutic strategies.
In recent years, multiple high-throughput next-generation sequencing (NGS) studies have identified a complex genomic landscape in MF/SS, including high rates of somatic non-synonymous variants (SNVs) and copy number variants (CNVs) (4–13). The majority of studies have utilised whole exome sequencing (WES) of peripheral blood samples enriched for CD4+ leukemic T-cells from SS patients (4–9, 12, 13). Fewer samples from advanced stage MF have been analysed (n=56) by WES or whole genome sequencing (WGS) platforms (4, 5, 7, 10–12). An independent analysis of genomic data published prior to 2017 encompassing 220 CTCL cases (186 SS; 25 MF; 9 CTCL NOS) has highlighted at least 55 putative driver genes affecting multiple signalling pathways (13). Interestingly, there is significant overlap between MF and SS in the pathways affected. Commonly perturbed genes include those involved in TCR signalling pathways (PLCG1; CARD11; CD28; RLTPR) and those that selectively up-regulate the NFkB pathway (13). Other disrupted pathways include the DNA damage response (TP53; POT1; ATM; BRAC1-2), chromatin modification (ARID1A; TRRAP; DNMT3A; TET2) and JAK STAT signalling (STAT5B; JAK3). Critically the aforementioned gene variants have been functionally validated confirming their driver gene status (4–8, 12–14). A summary of the pathways and their associated gene mutations is provided in Table 1.
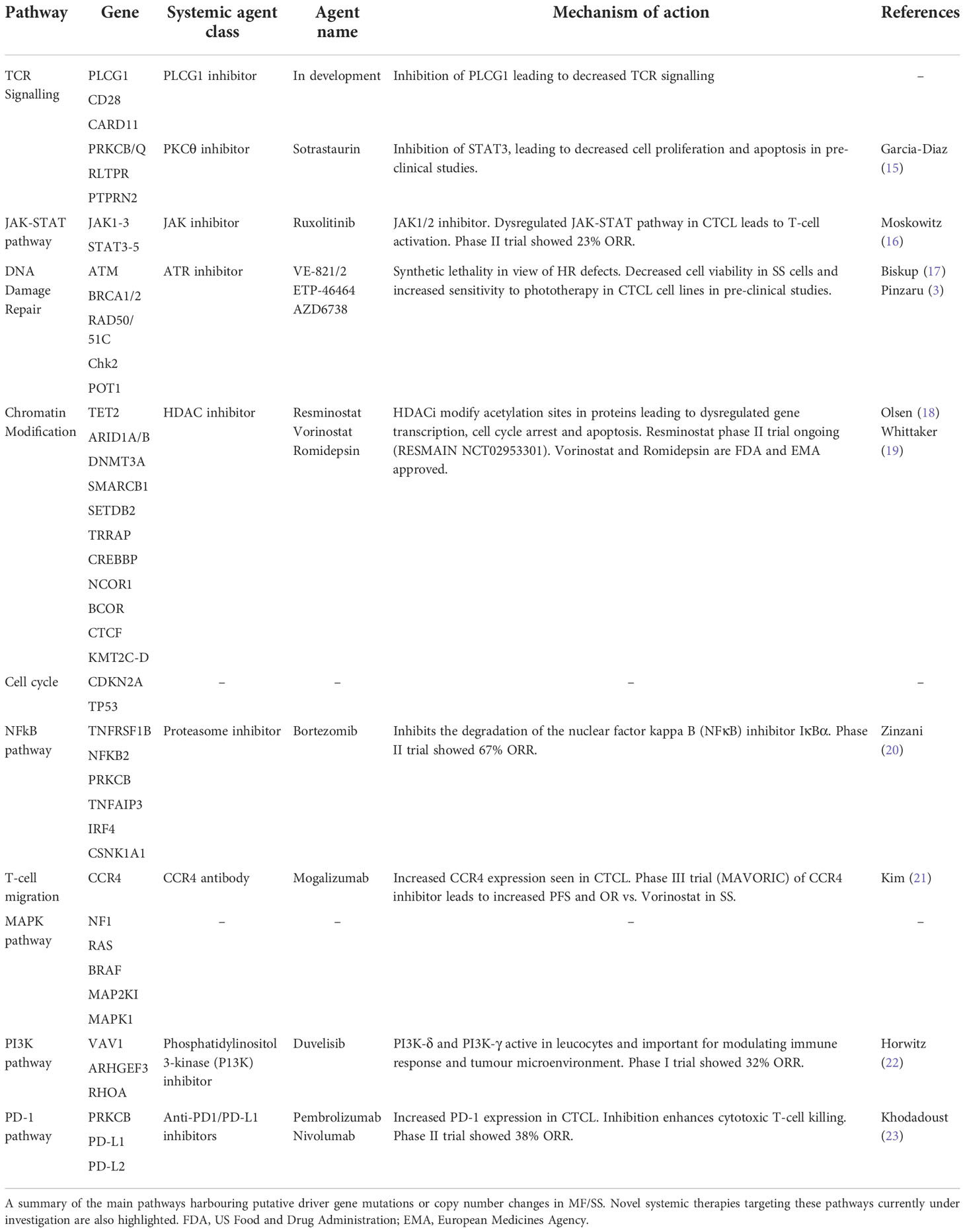
Table 1 Major aberrant pathways and associated emerging systemic therapies for advanced MF/SS.
There is limited data available on chromosomal rearrangements in view of the small WGS datasets from advanced MF samples. However, complex patterns of chromosomal rearrangements and translocations with no recurrent balanced translocations have been frequently identified with specific gains (17q, 8q) and losses (10q, 17p) (24).
Mutations in PLCG1 and CARD11 are two of the most frequently observed in MF/SS. They appear to be mutually exclusive and occur in almost 30% of SS cases (13). These gain of function mutations increase downstream T-cell signalling specifically through enhanced NFkB, NFAT and AP1 transcriptional activity (12, 14). These transcription factors regulate the expression of genes involved in cell proliferation, survival and differentiation. Crucially, there is evidence that many of these variants induce constitutive activation of downstream T-cell signalling without T-cell stimulation (12, 14). Furthermore, CTLA4-CD28 and ICOS-CD28 gene fusions enhance CD28 dependent T-cell signalling, and RLTPR variants activate the NFkB pathway thereby increasing downstream TCR signalling (7, 13). The high prevalence of these of NFkB pathway gene variants in CTCL supported by functional data indicates that there is a critical selection pressure for activation of the NFkB pathway in the transformation of mature T-cells.
PLCG1 mutations have also been detected in other mature T-cell malignancies, notably HTLV-1 associated ATLL (25), PTCL(NOS) (26), hepato-splenic T-cell lymphomas (27) and AITL (28) with the PLCG1 p.S345F and R48W variants being two of the most frequently reported. In addition, mutations of the JAK-STAT, CD28, VAV1, DNMT3A and TET2 genes are also reported in other mature T-cell malignancies (4–9, 25–28).
Enhanced T-cell activation via the T-cell receptor and NFkB pathway, leads to downstream activation of multiple pathways including the Janus tyrosine kinase (JAK) Signal transducers and activators of transcription (STAT) pathway. These proteins have a multitude of functions including TĤ cell proliferation and differentiation, as well as gene regulation and epigenetic modification. Unlike the STAT proteins, gain of function mutations in the JAK proteins are infrequently observed. However, copy number gains of both STAT3 and STAT5B are common and associated with constitutive expression of these key transcription factors (29, 30).
Epigenetic changes include DNA and histone modifications which affect gene transcription and regulate cell differentiation. Furthermore, epigenetic modification is critical for sustaining the transcriptional memory for T-cells allowing rapid transcription of inducible genes upon activation (31, 32). Global hypomethylation is a consistent feature of malignancy and contributes to genomic instability but DNA methylation of gene promoters can lead to gene silencing. Crucially these changes are observed in MF/SS with evidence for hypomethylation of 7.8% of CpG sites in SS, and hypermethylation of 3.2% of CpG sites, specifically in the proximal region of promoters (33). There is extensive evidence that promoter hypermethylation leads to the silencing of specific tumour suppressor genes in MF/SS including those involved in cell cycle regulation (CDKN2A/2B) (34), DNA repair (MLH1 and MGMT) (35), apoptosis (FAS) (36) and JAK-STAT signalling (SHP-1) (37). Methylation of cytosine residues to 5-methylcytosine is mediated by DNA methyltransferases (DNMTs) and gain of function mutations of DNMT3A have been frequently identified in haematologic malignancies including MF/SS (4, 5, 8, 9). A second type of DNA methylation involves 5-hydroxymethylation of cytosine which is mediated by ten-eleven translocation 1-3 enzymes (TET1-3) but, in contrast to 5-methylcytosine, this is associated with enhanced gene expression (38). Loss of function TET2 mutations are well documented in SS (5, 7–9, 30). In SS, there is also mutational selection pressure for genes involved in other epigenetic modifications including IDH encoding isocitrate dehydrogenases which inhibit TET proteins, ARID1A/1B which affect chromatin modelling and MLL genes which mediate histone methyltransferases (4, 8, 9, 30). Large epigenomic studies in MF/SS have shown that the methylation pattern of leukemic T-cells in SS can be similar to that of regulatory T-cells and that there is almost universal activation of NFkB (39). Recent data suggest that hypermethylation of the hTERT promoter in MF/SS may be associated with telomerase activation (40).
MicroRNA (miRs), one of a group of non-coding RNA transcripts, are key post-transcriptional regulators of mRNA and are known to affect both the stability and translation of mRNA. In MF/SS, miR dysregulation has been observed with aberrant expression linked to abnormal DNA methylation of miR promoters as well as copy number changes (41).In addition, constitutive activation of STAT3/5 has been shown to enhance miR-155 and miR-21 expression leading to increased apoptosis resistance and Th2 proliferation (41).
The DNA damage response (DDR) consists of numerous complex and inter-dependent signalling pathways which either maintain cell viability by repair of DNA or direct the damaged cell to undergo senescence or programmed cell death. Inevitably this complex process is closely linked with pathways regulating the cell cycle, chromatin remodelling and apoptosis (42). Previous cytogenetic and array CGH studies in MF/SS identified complex structural and numerical chromosomal abnormalities (24). More recent WGS and WES studies have confirmed a high degree of genomic instability with over 60 gene aberrations reported across all 5 DDR pathways (5, 8–10). One study of 101 SS samples identified SNVs and/or CNVs affecting genes involved in DNA repair and telomere maintenance in over 50% of cases. Notably, SNVs and CNVs affecting genes involved in homologous recombination such as RAD51C, BRAC2 and POLD1, are also detected in MF/SS (9). TP53 is the most commonly mutated gene in CTCL with loss of function SNVs and deletions which lead to a significant detrimental effect on the DNA damage response and telomere stability.
Telomere dysregulation is a recognised feature of MF/SS where shortened telomeres have been observed (43, 44). Furthermore, mutations in POT1, encoding a telomere binding protein are also frequently detected in MF/SS and ATLL (4, 5, 8–11, 25, 30, 45) and studies suggest that these loss of function variants likely contribute to telomere dysfunction by abolishing telomere binding and inducing DNA damage at telomeres in the form of telomere induced foci (TIFs) (3, 46–51). Cell cycle dysregulation is a major contributor to tumorigenesis in MF/SS. Mutation or deletions have been reported in several cell cycle checkpoint and tumour suppressor genes including CDKN1, CDKN2A, CDKN2B, ATM, ATR, TP53, RB1 and PTEN (4, 5, 8–10). Loss of function ATM mutations have been reported in several T-cell lymphomas (25, 26, 52–60) including MF/SS (9–11) and ATR mutations are also observed in MF/SS and NK T-cell lymphoma (5, 8, 10, 11, 61). In view of the key role of these kinases in the DDR and in cell cycle regulation, pathway disruption either by gene mutation or as a result of telomere dysfunction is likely to contribute to the genomic instability seen in MF/SS.
As the vast majority of studies have been conducted in samples from advanced stages of MF or leukaemic SS samples, until recently there has been little insight into the driver events in early-stage disease. In solid malignancies, the use of mathematical modelling of WGS/WES data has deepened understanding of the evolution of genomic events and the impact of intra-tumour heterogeneity on therapeutic response (62). The use of paired plaques and tumours from MF patients has demonstrated that sub-clonal evolution is a feature of MF and is linked to disease progression, however, as yet these preliminary studies have failed to identify a common series of genomic events in early stages of disease (11).
A series of mutational signatures have been identified which are linked to a combination of intrinsic and extrinsic mutagens, and have been associated with specific malignancies (63, 64). The presence of these mutational signatures in clonally expanded cell populations is determined by assessing the six substitution types and their 5’ and 3’ nucleotide context giving 96 different trinucleotide mutation types. A recent meta-analysis of whole exome sequencing (WES) data from 403 patients across several T-cell NHL subtypes, including 6 MF/SS studies has revealed that the UV signature (signature 7) was exclusively present in MF/SS. It accounted for the mutational burden in 52% of MF and 23% of SS cases (65). In addition, these C>T/CC>TT mutations at dipyrimidine sites had a significant bias towards the untranscribed strand which is a feature of transcription-coupled nucleotide excision repair associated with UV-induced mutations (66). Crucially, the presence of this signature (7.5-88% of the overall SNVs) in CD4+ cells isolated from the blood of SS patients suggests that these malignant T-cells either circulate freely from the skin to the blood compartment or develop from skin resident memory T-cells. A significant proportion of patients analysed (41%) were treatment naïve and the detection of a mutational signature is dependent on the presence of a clonal population suggesting that the malignant T-cell in MF/SS accumulates UV associated mutations before transformation and clonal expansion.
Similar to other malignancies linked to exogenous mutagens (e.g. non-melanoma and melanoma skin cancers and smoking-associated lung cancers), MF/SS exhibit a very high mutational load, and are unique amongst other types of T-cell lymphoma which carry a much lower mutational burden. These data confirm the significant contribution of environmental UV exposure to the mutational burden in MF and SS and UV is a likely causal factor in the transformation of T-cells that are either circulating through or resident in skin.
Primary cutaneous CD30+ anaplastic large cell lymphoma (pcALCL) has similar genomic alterations to its systemic counterpart albeit at much lower frequency. Most fail to express anaplastic lymphoma kinase (ALK), but have an excellent prognosis (67). In a small proportion of patients mutations of JAK1 and/or STAT3 and NPM1-TYK2 gene fusions been reported in pcCD30+ALCL (68).
Chromosomal rearrangements involving the DUSP22-IRF4 (MUM1) locus on 6p25.3 have also been identified in both pcCD30+ ALCL (25%) and less commonly (5%) in lymphomatoid papulosis (LYP) (69), but MUM1 expression is not specific for this rearrangement.
The majority of cases harbour one of two homozygous loss of function germline HAVCR2 variants (pTyr82Cys and p.Ile97Met) observed in Polynesian/East Asian and European origin respectively (70). HAVCR2 encodes T-cell immunoglobulin mucin 3 (TIM-3) is expressed by CD8+ T-cells and NK cells and regulates peripheral tolerance, innate immunity and inflammatory responses. Somatic variants have also been detected in genes involved in epigenetic modification (TET2, ARID1B), the PI3K/AKT/mTOR and JAK-STAT pathways (71).
Similar to MF/SS, mutations in the JAK/STAT, MAPK, MYC and chromatin modification pathways have been detected, but interestingly, TCR-CD28 signalling pathway mutations have also been reported (72). In addition, panniculitic Vδ2 T-cell lymphomas do not show germline mutations of HAVCR2 as seen in a majority of αβ SPTCL patients (72).
The prognosis for patients with MF/SS is variable even amongst patients with the same stage of disease. This has been partly addressed by the proposal of clinical prognostic models such as the CLIPi (73, 74) which is currently the subject of a multi-centre prospective study. In view of the genomic heterogeneity observed in MF/SS, it is likely that a more accurate prognostic model will require analysis of genetic clusters which may include a combination of gene mutations including SNVs and CNVs. There is some evidence that specific genomic clusters can be defined in SS (75), however, there has been no clinical correlation with patient outcomes to date. Furthermore, the paucity of genomic data in MF highlights the need for further NGS studies, particularly in early stage disease.
In view of the genomic heterogeneity in MF/SS reflecting the underlying high rate of UV signature mutations, a single targeted treatment option is unlikely to be effective. However, a deeper understanding of the genomic landscape could provide insight into potential therapeutic approaches especially in early stage disease (76). Stratification of individual patients according to mutational profile/deregulated pathway could allow the use of existing treatments such as Ipilimumab (CD28-CTLA4 fusion), Ruxolitinib and Tofacitinib (JAK mutations or JAK2 fusions as detected in rare aggressive cytotoxic CTCL variants) (77, 78) and Bortezomib (NFkB pathway) (79). Patients with abnormalities of epigenetic regulation such as DNMT3A and TET2 could be selected for treatment with demethylating agents such as 5-Azactidine and/or HDACi, whilst those with RHOA mutations could be eligible for PI3K inhibitors (Duvelisib). However, an alternative is to consider a tumour agnostic approach and the genomic landscape of MF/SS including marked genomic instability suggest that targeting the DDR pathway might be a productive strategy. A summary of emerging treatments targeting the various dysregulated pathways in MF/SS is provided in Table 1.
In view of the immune dysregulation profile seen in advanced MF/SS with a diminished Th1 immune response (IFN-gamma, IL-12) and skewing towards a Th2 immune profile (increased IL-4, IL-5 and/or IL-13) (80), interferons (IFN-alpha and IFN-gamma) were amongst the first immunotherapies to be used. IFN-alpha has been shown to stimulate antitumour cytotoxicity by activating CD8+ T-cells and NK cells and restores the Th1/Th2 balance by reducing IL-4 and IL-5 production by malignant T-cells (81, 82). IFN-gamma acts similarly to IFN-alpha by activating CD8+ and NK cells and increasing Th1 cytokine profile (80). IFN-alpha is EMA approved and effective in early stage patients who are refractory to skin-directed therapies (83), in which case it can be combined with phototherapy (84). Several clinical trials have highlighted the potential use of recombinant IL-12 as a novel immunotherapy with encouraging results (85–87).
There are currently several trials underway targeting checkpoint molecules (88, 89). Mogamulizumab, an anti-CCR4 monoclonal antibody has recently been approved by FDA and EMA following a phase III clinical trial which showed increased progression free survival (PFS) compared with the HDAC inhibitor, Vorinostat (21). Recently, it has emerged that Mogamulizumab also contributes to efficient immune restoration involving CD8+ as well as stem and memory CD4+ cells (90).
Pembrolizumab and Nivolumab inhibit the PD-1 receptor which enhances cytotoxic T-cell killing and have shown clinical responses in phase I and II trials (23, 91, 92). Whilst the high mutational burden of MF/SS would suggest that immunotherapies should be successful, the modest responses from these phase II trials (93) highlight the challenges of using a PD-1 inhibitor on a T-cell lymphoma which can express PD-1. Specifically there are two scenarios: PD-1 expression might reflect underlying gain of function mutations which would benefit from inhibition and there is emerging data suggesting that PD-1 mutations can be detected in some MF/SS patients (94), or PD-1 inhibition might reverse the tumour cell exhaustion leading to activation and proliferation of malignant T-cells. Whilst this second scenario has not yet been seen in MF/SS patients receiving PD-1 inhibitors, a phase II trial of Nivomumab in ATLL was discontinued because of rapid disease progression (95).
Targeting of CD47 with intralesional or intravenous TTI-621 has been used in patients with relapsed or refractory MF/SS in recent phase I trials with encouraging results (96, 97). Recent reports have shown that the CD39-CD73-adenosine pathway generates an immunosuppressive tumour microenvironment in SS and this provides a potential option to use emerging novel therapeutic approaches to target this pathway possibly in combination with checkpoint inhibitors or Mogamulizumab (98, 99).
Perhaps the most interesting option would be tumour agnostic therapies targeting the DNA damage response (DDR) pathways which show considerable promise in various solid malignancies often as maintenance therapies after platinum-based chemo regimens (42). The DDR pathways are a vast network of over 450 proteins. These therapeutic approaches build on the success of PARP inhibitors inducing synthetic lethality in homologous recombination (HR) deficient malignancies (due to BRCA1 or BRCA2 loss). In view of the somatic mutations or deletions of HR genes (including ATM, BRCA1, BRCA2, Chk2, RAD50, RAD51C), these could be repositioned for use in MF/SS patients (Figure 1). Several ATR inhibitors are in early clinical development for use in both solid and haematological malignancies (100, 101). There is increasing evidence that ATR inhibition may be a potential therapeutic target in MF/SS. Small molecule inhibitors of ATR and Chk1 (VE-821/2 and Chir-124) have been shown to sensitise MF/SS cell lines to phototherapy by inducing apoptosis (17). In addition, cells overexpressing POT1 mutants (p.F62V and p.K90E) treated with an ATR inhibitor (ETP-46464) resulted in significant abrogation of TIFs (3). ATM inhibition has also been shown to overcome HDAC inhibitor resistance in both B and T-cell derived lymphomas including MF/SS, providing a rationale for combination therapy (102). Acting immediately downstream of ATR, targeting of Chk1 is effective in MYC-driven tumours including B-cell lymphomas owing to MYC-induced replication stress (103). Amplification of the MYC oncogene is one of the most commonly observed aberrations in MF/SS (4) which could increase replication stress in these cells and hence increase their sensitivity to ATR and/or Chk1 inhibitors. Furthermore, Chk1 inhibitors synergise with a number of therapeutic agents to induce cell death in MF/SS including the proteasome inhibitor, Ixazomib (104) and phototherapy (17).
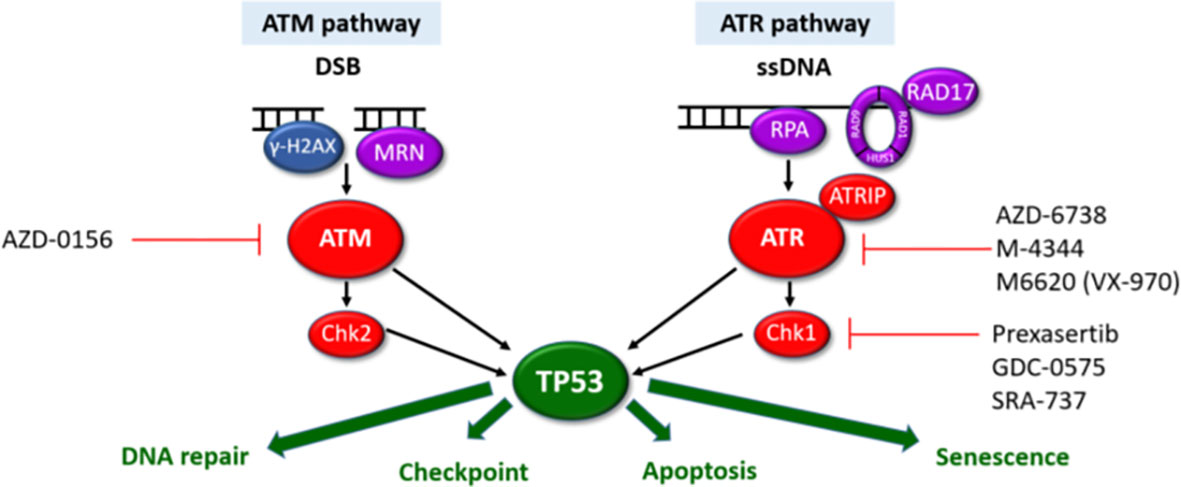
Figure 1 ATM and ATR DDR pathways. The ATM and ATR pathways respond to different types of DNA damage with separate sensors (purple), mediators (blue) and transducers (red). Synthetic lethality can be exploited for cells with aberrant DDR pathways. Cells harbouring LOF mutations in one pathway have increased reliance on the other pathway for DNA damage repair. Inhibition of the intact pathway prevents p53-mediated DNA repair, cell cycle arrest and apoptosis. This results in accumulation of unrepaired DNA damage, mitotic catastrophe triggering p53-independent cell death. Pharmacological agents targeting components of the ATM/ATR pathways currently in clinical development are highlighted.
Despite the wide range of treatment options currently available for MF/SS, therapeutic responses are invariably in the region of 30% and usually short lived (105), highlighting the need for a better understanding of the pathogenic mechanisms which would enable the development of more targeted therapies. A deeper understanding of dysregulated pathways and immunology in recent years has facilitated the development of several novel drugs currently in clinical trials. In addition to modulating pathways such as the JAK-STAT and NFĸB pathways and immune checkpoints, targeting genomic instability and the DDR present an exciting novel treatment approach for T-cell malignancies such as MF/SS.
FB and SW planned and wrote the manuscript. Figure and table was generated by FB. All authors contributed to the article and approved the submitted version.
Open access fees are provided by King’s College London via the grant code: ST461.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer VG declared a past collaboration with the author SW to the handling editor.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Bradford PT, Devesa SS, Anderson WF, Toro JR. Cutaneous lymphoma incidence patterns in the united states: A population-based study of 3884 cases. Blood (2009) 113:5064–73. doi: 10.1182/blood-2008-10-184168
2. Kim EJ, Hess S, Richardson SK, Newton S, Showe LC, Benoit BM, et al. Immunopathogenesis and therapy of cutaneous T cell lymphoma. J Clin Invest (2005) 115(4):798–812. doi: 10.1172/JCI200524826
3. Pinzaru AM, Hom RA, Beal A, Phillips AF, Ni E, Cardozo T, et al. Telomere replication stress induced by POT1 inactivation accelerates tumorigenesis. Cell Rep (2016) 15:2170–84. doi: 10.1016/j.celrep.2016.05.008
4. Choi J, Goh G, Walradt T, Hong BS, Bunick CG, Chen K, et al. Genomic landscape of cutaneous T cell lymphoma. Nat Genet (2015) 47:1011–9. doi: 10.1038/ng.3356
5. Da Silva Almeida AC, Abate F, Khiabanian H, Martinez-Escala E, Guitart J, Tensen CP, et al. The mutational landscape of cutaneous T cell lymphoma and sézary syndrome. Nat Genet (2015) 47:1465–70. doi: 10.1038/ng.3442
6. Prasad A, Rabionet R, Espinet B, Zapata L, Puiggros A, Melero C, et al. Identification of gene mutations and fusion genes in patients with sézary syndrome. J Invest Dermatol (2016) 136:1490–9. doi: 10.1016/j.jid.2016.03.024
7. Ungewickell A, Bhaduri A, Rios E, Reuter J, Lee CS, Mah A, et al. Genomic analysis of mycosis fungoides and sézary syndrome identifies recurrent alterations in TNFR2. Nat Genet (2015) 47:1056–60. doi: 10.1038/ng.3370
8. Wang L, Ni X, Covington KR, Yang BY, Shiu J, Zhang X, et al. Genomic profiling of sézary syndrome identifies alterations of key T cell signaling and differentiation genes. Nat Genet (2015) 47:1426–34. doi: 10.1038/ng.3444
9. Woollard WJ, Pullabhatla V, Lorenc A, Patel VM, Butler RM, Bayega A, et al. Candidate driver genes involved in genome maintenance and DNA repair in sézary syndrome. Blood (2016) 127:3387–97. doi: 10.1182/blood-2016-02-699843
10. McGirt LY, Jia P, Baerenwald DA, Duszynski RJ, Dahlman KB, Zic JA, et al. Whole-genome sequencing reveals oncogenic mutations in mycosis fungoides. Blood (2015) 126:508–19. doi: 10.1182/blood-2014-11-611194
11. Iyer A, Hennessey D, O’Keefe S, Patterson J, Wang W, Wong GKS, et al. Branched evolution and genomic intratumor heterogeneity in the pathogenesis of cutaneous T-cell lymphoma. Blood Adv (2020) 4(11):2489–500. doi: 10.1182/bloodadvances.2020001441
12. Vaqué JP, Gómez-López G, Monsálvez V, Varela I, Martínez N, Pérez C, et al. PLCG1 mutations in cutaneous T-cell lymphomas. Blood (2014) 123:2034–43. doi: 10.1182/blood-2013-05-504308
13. Park J, Yang J, Wenzel AT, Ramachandran A, Lee WJ, Daniels JC, et al. Genomic analysis of 220 CTCLs identifies a novel recurrent gain-of-function alteration in RLTPR (p.Q575E). Blood (2017) 130(12):1430–40. doi: 10.1182/blood-2017-02-768234
14. Patel VM, Flanagan CE, Martins M, Jones CL, Butler RM, Woollard WJ, et al. Frequent and persistent PLCG1 mutations in sézary cells directly enhance PLCγ1 activity and stimulate NFκB, AP-1, and NFAT signaling. J Invest Dermatol (2020) 140(2):380–9.e4. doi: 10.1016/j.jid.2019.07.693
15. García-Díaz N., Casar B., Alonso-Alonso R., Quevedo L., Rodríguez M., Ruso-Julve F., et al. (2022). PLCγ1/PKCθ downstream signaling controls cutaneous T-cell lymphoma development and progression. J. Invest. Dermatol. 142 (5), 1391–1400.e15. doi: 10.1016/j.jid.2021.09.024
16. Moskowitz AJ, Ghione P, Jacobsen E, Ruan J, Schatz JH, Noor S, et al. (2021). A phase 2 biomarker-driven study of ruxolitinib demonstrates effectiveness of JAK/STAT targeting in T-cell lymphomas. Blood 138(26):2828–37. doi: 10.1182/blood.2021013379
17. Biskup E, Naym DG, Gniadecki R. Small-molecule inhibitors of ataxia telangiectasia and Rad3 related kinase (ATR) sensitize lymphoma cells to UVA radiation. J Dermatol Sci (2016) 84(3):239–47. doi: 10.1016/j.jdermsci.2016.09.010
18. Olsen EA, Kim YH, Kuzel TM, Pacheco TR, Foss FM, Parker S, et al. Phase IIb multicenter trial of vorinostat in patients with persistent, progressive, or treatment refractory cutaneous T-cell lymphoma. J Clin Oncol (2007) 25(21): 3109–15. doi: 10.1200/JCO.2006.10.2434
19. Whittaker SJ, Demierre MF, Kim EJ, Rook AH, Lerner A, Duvic M, et al. Final results from a multicenter, international, pivotal study of romidepsin in refractory cutaneous T-cell lymphoma. J Clin Oncol (2010) 28(29):4485–91. doi: 10.1200/JCO.2010.28.9066
20. Zinzani PL, Musuraca G, Tani M, Stefoni V, Marchi E, Fina M, et al. Phase II trial of proteasome inhibitor bortezomib in patients with relapsed or refractory cutaneous T-cell lymphoma. J Clin Oncol (2007) 25(27):4293–97. doi: 10.1200/JCO.2007.11.4207
21. Kim YH, Bagot M, Pinter-Brown L, Rook AH, Porcu P, Horwitz SM, et al. Mogamulizumab versus vorinostat in previously treated cutaneous T-cell lymphoma (MAVORIC): An international, open-label, randomised, controlled phase 3 trial. Lancet Oncol (2018) 19(9):1192–204. doi: 10.1016/S1470-2045(18)30379-6
22. Horwitz SM, Koch R, Porcu P, Oki Y, Moskowitz A, Perez M, et al. Activity of the PI3K-δ,γ inhibitor duvelisib in a phase 1 trial and preclinical models of T-cell lymphoma. Blood (2018) 131(8):888–98. doi: 10.1182/blood-2017-08-802470
23. Khodadoust MS, Rook AH, Porcu P, Foss F, Moskowitz AJ, Shustov A, et al. Pembrolizumab in relapsed and refractory mycosis fungoides and sézary syndrome: A multicenter phase II study. J Clin Oncol (2020) 38(1):20–8. doi: 10.1200/JCO.19.01056
24. Mao X, Lillington DM, Czepulkowski B, Russell-Jones R, Young BD, Whittaker S. Molecular cytogenetic characterization of sézary syndrome. Genes Chromosom Cancer (2003) 36(3):250–60. doi: 10.1002/gcc.10152
25. Kataoka K, Nagata Y, Kitanaka A, Shiraishi Y, Shimamura T, Yasunaga JI, et al. Integrated molecular analysis of adult T cell leukemia/lymphoma. Nat Genet (2015) 47:1304–15. doi: 10.1038/ng.3415
26. Palomero T, Couronné L, Khiabanian H, Kim MY, Ambesi-Impiombato A, Perez-Garcia A, et al. Recurrent mutations in epigenetic regulators, RHOA and FYN kinase in peripheral T cell lymphomas. Nat Genet (2014) 46(2):166–70. doi: 10.1038/ng.2873
27. McKinney M, Moffitt AB, Gaulard P, Travert M, De LL, ANM R, et al. The genetic basis of hepatosplenic T-cell lymphoma. Cancer Discov (2017) 7(4):369–79. doi: 10.1158/2159-8290.CD-16-0330
28. Sakata-Yanagimoto M, Enami T, Yoshida K, Shiraishi Y, Ishii R, Miyake Y, et al. Somatic RHOA mutation in angioimmunoblastic T cell lymphoma. Nat Genet (2014) 46(2):171–5. doi: 10.1038/ng.2872
29. Crescenzo R, Abate F, Lasorsa E, Tabbo’ F, Gaudiano M, Chiesa N, et al. Convergent mutations and kinase fusions lead to oncogenic STAT3 activation in anaplastic large cell lymphoma. Cancer Cell (2015) 27(4):516–32. doi: 10.1016/j.ccell.2015.03.006
30. Kiel MJ, Sahasrabuddhe AA, Rolland DCM, Velusamy T, Chung F, Schaller M, et al. Genomic analyses reveal recurrent mutations in epigenetic modifiers and the JAK-STAT pathway in sézary syndrome. Nat Commun (2015) 6:8470. doi: 10.1038/ncomms9470
31. Antignano F, Zaph C. Regulation of CD4 T-cell differentiation and inflammation by repressive histone methylation. Immunol Cell Biol (2015) 93(3):245–52. doi: 10.1038/icb.2014.115
32. Dunn J, McCuaig R, Tu WJ, Hardy K, Rao S. Multi-layered epigenetic mechanisms contribute to transcriptional memory in T lymphocytes. BMC Immunol (2015) 16:27. doi: 10.1186/s12865-015-0089-9
33. van Doorn R, Slieker RC, Boonk SE, Zoutman WH, Goeman JJ, Bagot M, et al. Epigenomic analysis of sézary syndrome defines patterns of aberrant DNA methylation and identifies diagnostic markers. J Invest Dermatol (2016) 136(9):1876–84. doi: 10.1016/j.jid.2016.03.042
34. Scarisbrick JJ, Woolford AJ, Calonje E, Photiou A, Ferreira S, Orchard G, et al. Frequent abnormalities of the P15 and P16 genes in mycosis fungoides and sezary syndrome. J Invest Dermatol (2002) 118(3):493–9. doi: 10.1046/j.0022-202x.2001.01682.x
35. Scarisbrick JJ, Woolford AJ, Russell-Jones R, Whittaker SJ. Loss of heterozygosity on 10q and microsatellite instability in advanced stages of primary cutaneous T-cell lymphoma and possible association with homozygous deletion of PTEN. Blood (2000) 95(9):2937–42. doi: 10.1182/blood.v95.9.2937.009k15_2937_2942
36. Jones CL, Wain EM, Chu CC, Tosi I, Foster R, McKenzie RCT, et al. Downregulation of fas gene expression in sézary syndrome is associated with promoter hypermethylation. J Invest Dermatol (2010) 130(4):1116–25. doi: 10.1038/jid.2009.301
37. Van Doorn R, Zoutman WH, Dijkman R, De Menezes RX, Commandeur S, Mulder AA, et al. Epigenetic profiling of cutaneous T-cell lymphoma: Promoter hypermethylation of multiple tumor suppressor genes including BCL7a, PTPRG, and p73. J Clin Oncol (2005) 23(17):3886–96. doi: 10.1200/JCO.2005.11.353
38. Rasmussen KD, Helin K. Role of TET enzymes in DNA methylation, development, and cancer. Genes Dev (2016) 30(7):733–50. doi: 10.1101/gad.276568.115
39. Qu K, Zaba LC, Satpathy AT, Giresi PG, Li R, Jin Y, et al. Chromatin accessibility landscape of cutaneous T cell lymphoma and dynamic response to HDAC inhibitors. Cancer Cell (2017) 32(1):27–41.e4. doi: 10.1016/j.ccell.2017.05.008
40. Chebly A, Ropio J, Peloponese J, Poglio S, Prochazkova-Carlotti M, Cherrier F, et al. Exploring hTERT promoter methylation in cutaneous T-cell lymphomas. Mol Oncol (2021) 16(9):1931–46. doi: 10.1002/1878-0261.12946
41. Gluud M, Willerslev-Olsen A, Gjerdrum LMR, Lindahl LM, Buus TB, Andersen MH, et al. MicroRNAs in the pathogenesis, diagnosis, prognosis and targeted treatment of cutaneous T-cell lymphomas. Cancers (Basel) (2020) 12(5):1229. doi: 10.3390/cancers12051229
42. Pilié PG, Tang C, Mills GB, Yap TA. State-of-the-art strategies for targeting the DNA damage response in cancer. Nat Rev Clin Oncol (2019) 16(2):81–104. doi: 10.1038/s41571-018-0114-z
43. Wu K, Lund M, Bang K, Thestrup-Pedersen K. Telomerase activity and telomere length in lymphocytes from patients with cutaneous T-cell lymphoma. Cancer (1999) 86:1056–63. doi: 10.1002/(SICI)1097-0142(19990915)86:6<1056::AID-CNCR23>3.0.CO;2-#
44. Chevret E, Andrique L, Prochazkova-Carlotti M, Ferrer J, Cappellen D, Laharanne E, et al. Telomerase functions beyond telomere maintenance in primary cutaneous T-cell lymphoma. Blood (2014) 123:1850–9. doi: 10.1182/blood-2013-05-500686
45. Nagata Y, Kontani K, Enami T, Kataoka K, Ishii R, Totoki Y, et al. Variegated RHOA mutations in adult T-cell leukemia/lymphoma. Blood (2016) 127:596–604. doi: 10.1182/blood-2015-06-644948
46. Ramsay AJ, Quesada V, Foronda M, Conde L, Martinez-Trillos A, Villamor N, et al. POT1 mutations cause telomere dysfunction in chronic lymphocytic leukemia. Nat Genet (2013) 45:526–30. doi: 10.1038/ng.2584
47. Shi J, Yang XR, Ballew B, Rotunno M, Calista D, Fargnoli MC, et al. Rare missense variants in POT1 predispose to familial cutaneous malignant melanoma. Nat Genet (2014) 46:482–6. doi: 10.1038/ng.2941
48. Robles-Espinoza CD, Harland M, Ramsay AJ, Aoude LG, Quesada V, Ding Z, et al. POT1 loss-of-function variants predispose to familial melanoma. Nat Genet (2014) 46:478–81. doi: 10.1038/ng.2947
49. Calvete O, Martinez P, Garcia-Pavia P, Benitez-Buelga C, Paumard-Hernández B, Fernandez V, et al. A mutation in the POT1 gene is responsible for cardiac angiosarcoma in TP53-negative Li-fraumeni-like families. Nat Commun (2015) 6:8383. doi: 10.1038/ncomms9383
50. McMaster M, Sun C, Landi M, Savage S, Rotunno M, Yang X, et al. Germline mutations in protection of telomeres 1 in two families with Hodgkin lymphoma. Br J Haematol (2018) 181:372–7. doi: 10.1111/bjh.15203
51. Srivastava A, Miao B, Skopelitou D, Kumar V, Kumar A, Paramasivam N, et al. A germline mutation in the POT1 gene is a candidate for familial non-medullary thyroid cancer. Cancers (Basel) (2020) 12:1441. doi: 10.3390/cancers12061441
52. Koo GC, Tan SY, Tang T, Poon SL, Allen GE, Tan L, et al. Janus kinase 3-activating mutations identified in natural killer/T-cell lymphoma. Cancer Discov (2012) 2:591–7. doi: 10.1158/2159-8290.CD-12-0028
53. Stoppa-Lyonnet D, Soulier J, Laugé A, Dastot H, Garand R, Sigaux F, et al. Inactivation of the ATM gene in T-cell prolymphocytic leukemias. Blood (1998) 91(10):3920–6. doi: 10.1182/blood.v91.10.3920
54. Stilgenbauer S, Schaffner C, Litterst A, Liebisch P, Gilad S, Bar-Shira A, et al. Biallelic mutations in the ATM gene in T-prolymphocytic leukemia. Nat Med (1997) 3(10):1155–9. doi: 10.1038/nm1097-1155
55. Vořechovsky I, Luo L, Dyer MJS, Catovsky D, Amlot PL, Yaxley JC, et al. Clustering of missense mutations in the ataxia-telangiectasia gene in a sporadic T cell leukaemia. Nat Genet (1997) 17(1):96–9. doi: 10.1038/ng0997-96
56. Schatz JH, Horwitz SM, Teruya-Feldstein J, Lunning MA, Viale A, Huberman K, et al. Targeted mutational profiling of peripheral T-cell lymphoma not otherwise specified highlights new mechanisms in a heterogeneous pathogenesis. Leukemia (2015) 29(1):237–41. doi: 10.1038/leu.2014.261
57. Fang NY, Greiner TC, Weisenburger DD, Chan WC, Vose JM, Smith LM, et al. Oligonucleotide microarrays demonstrate the highest frequency of ATM mutations in the mantle cell subtype of lymphoma. Proc Natl Acad Sci USA (2003) 100(9):5372–7. doi: 10.1073/pnas.0831102100
58. Zehir A, Benayed R, Shah RH, Syed A, Middha S, Kim HR, et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med (2017) 23(6):703–13. doi: 10.1038/nm.4333
59. Wong TN, Miller CA, Jotte MRM, Bagegni N, Baty JD, Schmidt AP, et al. Cellular stressors contribute to the expansion of hematopoietic clones of varying leukemic potential. Nat Commun (2018) 9(1):455. doi: 10.1038/s41467-018-02858-0
60. Margolskee E, Jobanputra V, Jain P, Chen J, Ganapathi K, Nahum O, et al. Genetic landscape of T- and NK-cell post-transplant lymphoproliferative disorders. Oncotarget (2016) 7(25):37636–48. doi: 10.18632/oncotarget.9400
61. Dobashi A, Tsuyama N, Asaka R, Togashi Y, Ueda K, Sakata S, et al. Frequent BCOR aberrations in extranodal NK/T-cell lymphoma, nasal type. Genes Chromosom Cancer (2016) 55(5):460–71. doi: 10.1002/gcc.22348
62. McGranahan N, Swanton C. Clonal heterogeneity and tumor evolution: Past, present, and the future. Cell (2017) 168(4):613–28. doi: 10.1016/j.cell.2017.01.018
63. Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SAJR, Behjati S, Biankin AV, et al. Signatures of mutational processes in human cancer. Nature (2013) 500(7463):415–21. doi: 10.1038/nature12477
64. Kucab JE, Zou X, Morganella S, Joel M, Nanda AS, Nagy E, et al. A compendium of mutational signatures of environmental agents. Cell (2019) 177(4):821–36.e16. doi: 10.1016/j.cell.2019.03.001
65. Jones CL, Degasperi A, Grandi V, Amarante TD, Ambrose JC, Arumugam P, et al. Spectrum of mutational signatures in T-cell lymphoma reveals a key role for UV radiation in cutaneous T-cell lymphoma. Sci Rep (2021) 11(1):3962. doi: 10.1038/s41598-021-83352-4
66. Haradhvala NJ, Polak P, Stojanov P, Covington KR, Shinbrot E, Hess JM, et al. Mutational strand asymmetries in cancer genomes reveal mechanisms of DNA damage and repair. Cell (2016) 164(3):538–49. doi: 10.1016/j.cell.2015.12.050
67. Prieto-Torres L, Rodriguez-Pinilla SM, Onaindia A, Ara M, Requena L, Piris M. CD30-positive primary cutaneous lymphoproliferative disorders: Molecular alterations and targeted therapies. Haematologica (2019) 104(2):226–35. doi: 10.3324/haematol.2018.197152
68. Velusamy T, Kiel MJ, Sahasrabuddhe AA, Rolland D, Dixon CA, Bailey NG, et al. A novel recurrent NPM1-TYK2 gene fusion in cutaneous CD30-positive lymphoproliferative disorders. Blood (2014) 124(25):3768–71. doi: 10.1182/blood-2014-07-588434
69. Wada DA, Law ME, Hsi ED, Dicaudo DJ, Ma L, Lim MS, et al. Specificity of IRF4 translocations for primary cutaneous anaplastic large cell lymphoma: A multicenter study of 204 skin biopsies. Mod Pathol (2011) 24(4):596–605. doi: 10.1038/modpathol.2010.225
70. Gayden T, Sepulveda FE, Khuong-Quang DA, Pratt J, Valera ET, Garrigue A, et al. Germline HAVCR2 mutations altering TIM-3 characterize subcutaneous panniculitis-like T cell lymphomas with hemophagocytic lymphohistiocytic syndrome. Nat Genet (2018) 50(12):1650–7. doi: 10.1038/s41588-018-0251-4
71. Polprasert C, Takeuchi Y, Kakiuchi N, Yoshida K, Assanasen T, Sitthi W, et al. Frequent germline mutations of HAVCR2 in sporadic subcutaneous panniculitis-like T-cell lymphoma. Blood Adv (2019) 3(4):588–95. doi: 10.1182/bloodadvances.2018028340
72. Daniels J, Doukas PG, Escala MEM, Ringbloom KG, Shih DJH, Yang J, et al. Cellular origins and genetic landscape of cutaneous gamma delta T cell lymphomas. Nat Commun (2020) 11(1):1806. doi: 10.1038/s41467-020-15572-7
73. Agar NS, Wedgeworth E, Crichton S, Mitchell TJ, Cox M, Ferreira S, et al. Survival outcomes and prognostic factors in mycosis Fungoides/Sézary syndrome: Validation of the revised international society for cutaneous Lymphomas/European organisation for research and treatment of cancer staging proposal. J Clin Oncol (2010) 28:4730–9. doi: 10.1200/JCO.2009.27.7665
74. Benton EC, Crichton S, Talpur R, Agar NS, Fields PA, Wedgeworth E, et al. A cutaneous lymphoma international prognostic index (CLIPi) for mycosis fungoides and Sézary syndrome. Eur J Cancer (2013) 49(13):2859–68. doi: 10.1016/j.ejca.2013.04.018
75. Chang LW, Patrone CC, Yang W, Rabionet R, Gallardo F, Espinet B, et al. An integrated data resource for genomic analysis of cutaneous T-cell lymphoma. J Invest Dermatol (2018) 138(12):2681–3. doi: 10.1016/j.jid.2018.06.176
76. Hain C, Stadler R, Kalinowski J. Unraveling the structural variations of early-stage mycosis fungoides–CD3 based purification and third generation sequencing as novel tools for the genomic landscape in CTCL. Cancers (Basel) (2022) 14:4466. doi: 10.3390/cancers14184466
77. Lee K, Evans MG, Yang L, Ng S, Snowden C, Khodadoust M, et al. Primary cytotoxic T-cell lymphomas harbor recurrent targetable alterations in the JAK-STAT pathway. Blood (2021) 138(23):2435–40. doi: 10.1182/blood.2021012536
78. Bastidas Torres AN, Cats D, Out-Luiting JJ, Fanoni D, Mei H, Venegoni L, et al. Deregulation of JAK2 signaling underlies primary cutaneous CD8+ aggressive epidermotropic cytotoxic T-cell lymphoma. Haematologica (2022) 107(3):702–14. doi: 10.3324/haematol.2020.274506
79. Zhang Y, Lee D, Brimer T, Hussaini M, Sokol L. Genomics of peripheral T-cell lymphoma and its implications for personalized medicine. Front Oncol (2020) 10:898. doi: 10.3389/fonc.2020.00898
80. Guenova E, Watanabe R, Teague JE, Desimone JA, Jiang Y, Dowlatshahi M, et al. TH2 cytokines from malignant cells suppress TH1 responses and enforce a global TH2 bias in leukemic cutaneous T-cell lymphoma. Clin Cancer Res (2013) 19:3755–63. doi: 10.1158/1078-0432.CCR-12-3488
81. Yoo EK, Cassin M, Lessin SR, Rook AH. Complete molecular remission during biologic response modifier therapy for sézary syndrome is associated with enhanced helper T type 1 cytokine production and natural killer cell activity. J Am Acad Dermatol (2001) 45(2):208–16. doi: 10.1067/mjd.2001.116345
82. Suchin KR, Cassin M, Gottleib SL, Sood S, Cucchiara AJ, Vonderheid EC, et al. Increased interleukin 5 production in eosinophilic sézary syndrome: Regulation by interferon alfa and interleukin 12. J Am Acad Dermatol (2001) 44(1):28–32. doi: 10.1067/mjd.2001.109853
83. Gilson D, Whittaker SJ, Child FJ, Scarisbrick JJ, Illidge TM, Parry EJ, et al. British Association of dermatologists and U.K. cutaneous lymphoma group guidelines for the management of primary cutaneous lymphomas 2018. Br J Dermatol (2019) 180(3):496–526. doi: 10.1111/bjd.17240
84. Stadler R, Otte HG, Luger T, Henz BM, Kühl P, Zwingers T, et al. Prospective randomized multicenter clinical trial on the use of interferon α-2a plus acitretin versus interferon α-2a plus PUVA in patients with cutaneous T-cell lymphoma stages I and II. Blood (1998) 92(10):3578–81. doi: 10.1182/blood.V92.10.3578
85. Rook AH, Wood GS, Yoo EK, Elenitsas R, Kao DMF, Sherman ML, et al. Interleukin-12 therapy of cutaneous T-cell lymphoma induces lesion regression and cytotoxic T-cell responses. Blood (1999) 94(3):902–8. doi: 10.1182/blood.v94.3.902.415k23_902_908
86. Kim YH, Hoppe RT, Rook AH, Maity A, Geskin LJ, Horowitz DP, et al. A single-arm PHASE 2A study of NM-IL-12 (rHu-IL12) in patients with mycosis fungoides-type CTCL (MF) undergoing low-dose TOTAL skin electron BEAM therapy (LD-TSEBT). Blood (2016) 128(22):4165. doi: 10.1182/blood.v128.22.4165.4165
87. Duvic M, Sherman ML, Wood GS, Kuzel TM, Olsen E, Foss F, et al. A phase II open-label study of recombinant human interleukin-12 in patients with stage IA, IB, or IIA mycosis fungoides. J Am Acad Dermatol (2006) 55(5):807–13. doi: 10.1016/j.jaad.2006.06.038
88. Ferenczi K, Fuhlbrigge RC, Pinkus JL, Pinkus GS, Kupper TS. Increased CCR4 expression in cutaneous T cell lymphoma. J Invest Dermatol (2002) 119(6):1405–10. doi: 10.1046/j.1523-1747.2002.19610.x
89. Samimi S, Benoit B, Evans K, Wherry EJ, Showe L, Wysocka M, et al. Increased programmed death-1 expression on CD4+ T cells in cutaneous T-cell lymphoma: Implications for immune suppression. Arch Dermatol (2010) 146(12):1382–8. doi: 10.1001/archdermatol.2010.200
90. Roelens M, de Masson A, Andrillon A, Ram-Wolff C, Biard L, Boisson M, et al. Mogamulizumab induces long term immune restoration and reshapes tumoral heterogeneity in sézary syndrome. Eur J Cancer (2021) 186(6):1010–25. doi: 10.1111/bjd.21018
91. Lesokhin AM, Ansell SM, Armand P, Scott EC, Halwani A, Gutierrez M, et al. Nivolumab in patients with relapsed or refractory hematologic malignancy: Preliminary results of a phase ib study. J Clin Oncol (2016) 34(23):2698–704. doi: 10.1200/JCO.2015.65.9789
92. Shen K, Liu Y, Cao X, Zhou D, Li J. Successful treatment of refractory sézary syndrome by anti-PD-1 antibody (nivolumab). Ann Hematol (2017) 96(4):687–8. doi: 10.1007/s00277-017-2929-6
93. Sivanand A, Surmanowicz P, Alhusayen R, Hull P, Litvinov IV, Zhou Y, et al. Immunotherapy for cutaneous T-cell lymphoma: Current landscape and future developments. J Cutan Med Surg (2019) 23(5):537–44. doi: 10.1177/1203475419867610
94. Park J, Daniels J, Wartewig T, Ringbloom KG, Martinez-Escala ME, Choi S, et al. Integrated genomic analyses of cutaneous T cell lymphomas reveal the molecular bases for disease heterogeneity. Blood (2021) 138(14):1225–36. doi: 10.1182/blood.2020009655
95. Ratner L, Waldmann TA, Janakiram M, Brammer JE. Rapid progression of adult T-cell leukemia–lymphoma after PD-1 inhibitor therapy. N Engl J Med (2018) 378(20):1947–8. doi: 10.1056/nejmc1803181
96. Querfeld C, Thompson JA, Taylor MH, DeSimone JA, Zain JM, Shustov AR, et al. Intralesional TTI-621, a novel biologic targeting the innate immune checkpoint CD47, in patients with relapsed or refractory mycosis fungoides or sézary syndrome: a multicentre, phase 1 study. Lancet Haematol (2021) 8(11):e808–17. doi: 10.1016/S2352-3026(21)00271-4
97. Johnson LDS, Banerjee S, Kruglov O, Viller NN, Horwitz SM, Lesokhin A, et al. Targeting CD47 in sézary syndrome with SIRPaFc. Blood Adv (2019). doi: 10.1182/bloodadvances.2018030577
98. Yakymiv Y, Marchisio S, Ortolan E, Bracci C, Senetta R, Rumore MR, et al. CD39/CD73 dysregulation and adenosine metabolism contribute to T cell immunosuppression in patients with sézary syndrome. Blood (2022) 30:blood.2022017259. doi: 10.1182/blood.2022017259
99. Sonigo G, Alizee B, Dumont M, Thonnart N, Ram-Wolff C, de Masson A, et al. Involvement of the CD39/CD73/adenosine pathway in T-cell proliferation and NK cell-mediated antibody-dependent cell cytotoxicity in sézary syndrome. Blood (2022) 139:2712–6. doi: 10.1182/blood.2021014782
100. Mei L, Zhang J, He K, Zhang J. Ataxia telangiectasia and Rad3-related inhibitors and cancer therapy: Where we stand. J Hematol Oncol (2019) 12(1):43. doi: 10.1186/s13045-019-0733-6
101. Gourley C, Balmaña J, Ledermann JA, Serra V, Dent R, Loibl S, et al. Moving from poly (ADP-ribose) polymerase inhibition to targeting DNA repair and DNA damage response in cancer therapy. J Clin Oncol (2019) 37(25):2257–69. doi: 10.1200/JCO.18.02050
102. Scotto L, Serrano XJ, Zullo K, Kinahan C, Deng C, Sawas A, et al. ATM Inhibition overcomes resistance to histone deacetylase inhibitor due to p21 induction and cell cycle arrest. Oncotarget (2020) 11(37):3432–42. doi: 10.18632/oncotarget.27723
103. Ferrao PT, Bukczynska EP, Johnstone RW, McArthur GA. Efficacy of CHK inhibitors as single agents in MYC-driven lymphoma cells. Oncogene (2012) 31(13):1661–72. doi: 10.1038/onc.2011.358
104. Ravi D, Beheshti A, Abermil N, Passero F, Sharma J, Coyle M, et al. Proteasomal inhibition by ixazomib induces CHK1 and MYC-dependent cell death in T-cell and Hodgkin lymphoma. Cancer Res (2016) 76(11):3319–31. doi: 10.1158/0008-5472.CAN-15-2477
Keywords: cutaneous T cell lymphoma (CTCL), dermatology, pathway targeted interventions, genomics, cutaneous lymphoma
Citation: Bakr FS and Whittaker SJ (2022) Advances in the understanding and treatment of Cutaneous T-cell Lymphoma. Front. Oncol. 12:1043254. doi: 10.3389/fonc.2022.1043254
Received: 13 September 2022; Accepted: 19 October 2022;
Published: 24 November 2022.
Edited by:
Jianjun Qiao, Zhejiang University, ChinaReviewed by:
Vieri Grandi, University of Florence, ItalyCopyright © 2022 Bakr and Whittaker. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sean J. Whittaker, c2Vhbi53aGl0dGFrZXJAa2NsLmFjLnVr
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.