
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 22 November 2022
Sec. Cancer Metabolism
Volume 12 - 2022 | https://doi.org/10.3389/fonc.2022.1042196
 Shadia Hamoud Alshahrani1
Shadia Hamoud Alshahrani1 Yousif Saleh Ibrahim2
Yousif Saleh Ibrahim2 Abduladheem Turki Jalil3
Abduladheem Turki Jalil3 Abdelgadir Alamin Altoum4
Abdelgadir Alamin Altoum4 Harun Achmad5Rahman S. Zabibah6
Harun Achmad5Rahman S. Zabibah6 Gamal A. Gabr7,8
Gamal A. Gabr7,8 Andrés Alexis Ramírez-Coronel9,10,11
Andrés Alexis Ramírez-Coronel9,10,11 Ameer A. Alameri12Qutaiba A. Qasim13
Ameer A. Alameri12Qutaiba A. Qasim13 Sajad Karampoor14*
Sajad Karampoor14* Rasoul Mirzaei15*
Rasoul Mirzaei15*MicroRNAs (miRNAs) are emerging as a significant modulator of immunity, and their abnormal expression/activity has been linked to numerous human disorders, such as cancer. It is now known that miRNAs potentially modulate the production of several metabolic processes in tumor-associated immune cells and indirectly via different metabolic enzymes that affect tumor-associated signaling cascades. For instance, Let-7 has been identified as a crucial modulator for the long-lasting survival of CD8+ T cells (naive phenotypes) in cancer by altering their metabolism. Furthermore, in T cells, it has been found that enhancer of zeste homolog 2 (EZH2) expression is controlled via glycolytic metabolism through miRNAs in patients with ovarian cancer. On the other hand, immunometabolism has shown us that cellular metabolic reactions and processes not only generate ATP and biosynthetic intermediates but also modulate the immune system and inflammatory processes. Based on recent studies, new and encouraging approaches to cancer involving the modification of miRNAs in immune cell metabolism are currently being investigated, providing insight into promising targets for therapeutic strategies based on the pivotal role of immunometabolism in cancer. Throughout this overview, we explore and describe the significance of miRNAs in cancer and immune cell metabolism.
Metabolism is an energy production mechanism that cells use to maintain cellular balance, proliferation, and differentiation (1). Aerobic glycolysis, known as the Warburg effect, is a distinctive metabolic feature of tumor metabolism (2). Cancer cells, unlike normal cells, generate most of their energy through increased rates of glycolysis in the cytosol rather than pyruvate oxidation in the mitochondria (3). Over the last decade, there has been a growing emphasis on immunometabolism, or the function of metabolism in stimulating and controlling immune cells (4, 5). Due to the apparent essential role of intracellular metabolism in a set of cellular and biochemical processes to obtain and use nutrients, including lipids, proteins, and carbohydrates, to generate energy via adenosine triphosphate (ATP), as well as other biomolecules, it is not surprising that energy generation via cellular metabolism would be a significant determinant of the destiny and activity of immune cells (4, 6). Furthermore, aberrant cellular metabolism leads to immune cell malfunction, which is frequently connected with immunological-related diseases, such as cancer (7–9).
miRNAs are endogenous (~22nt) RNAs that can control many biological functions (10). Compelling evidence suggests that miRNAs play an essential part in energy metabolism, notably lipid and glucose metabolism and also amino acid biosynthesis (11). Furthermore, miRNAs may identify and modify metabolic components at the transcriptional level, which is essential in both non-neoplastic and tumor cells (12). For instance, miR-143 controls glycolysis by affecting Hexokinase 2 (HK2), which phosphorylates glucose to generate Glucose 6-phosphate (G6P), therefore allocating glucose to the glycolytic process. Malignant cell metabolism might also be changed to avoid apoptosis and promote cell proliferation and survival. The Warburg effect, in which miRNA dysregulation leads to higher glycolysis, is the best-characterized metabolic phenotype found in cancer cells (13, 14). However, a growing body of evidence suggests that miRNAs are also heavily engaged in the metabolic regulation of immune cells in cancer. miRNAs seem to have an impact on the Phosphoinositide 3-kinase (PI3K)/protein kinase B (AKT) network, which is a signaling system that regulates glucose metabolism via reducing phosphatase and tensin homolog (PTEN) activities, which might increase the expression of interleukin (IL)-4, IL-10, transforming growth factor-beta (TGF-β), and arginase, and found in the inflammatory zone (FIZZ) in M2 macrophages (15–17). This review presents recent results on the action of miRNAs in cancer metabolism, focusing on immune cells.
Cellular metabolism is a complex metabolic process that turns metabolites into biological activity (18). Catabolic pathways at the center of this physiological network decompose substances to create energy, which is subsequently utilized to fuel biochemical mechanisms and perform mechanical activity. Metabolism generates energy for cellular operations by producing ATP (19). Metabolism decomposes nutrients like protein, sugar, and fat into smaller units like glucose, fatty acids, and amino acids. This procedure has the potential to produce energy (19). Metabolism transforms simpler components like lipids, proteins, and nucleotides into macromolecules (i.e., anabolism). Energy is required for this procedure. Furthermore, metabolism involves cellular tasks other than anabolism, catabolism, and energy, such as cellular signaling and gene transcription (19). Metabolites, for instance, act as precursors for posttranslational protein modification to induce changes in protein functionality or control epigenetics to induce alterations in gene expression.
Immunometabolism, or the change in metabolic processes and activity of immune cells, is a new field of study that has gained international interest in the last decade (20). Metabolic reprogramming leads to variations in biosynthetic processes and metabolite quantities that occur throughout the proliferation and differentiation (21). Metabolic rewiring occurs in all cell types, including cancer and immunological cells (21). Because they govern downstream transcriptional and posttranscriptional processes, these modifications are crucial for a proper immune reaction, and their deregulation may impair growth, effector functions, and propagation (22). In summary, immune cells will increase the expression of aerobic glycolysis in an inflammatory setting (as observed in B cells, effector T helper type 1 (Th1), T helper 17 (Th17) cells, natural killer (NK) cells, dendritic cells, and M1 macrophages), whereas metabolism based on oxidative phosphorylation (OXPHOS) typically facilitates an anti-inflammatory characteristic (as detected in regulatory T (Treg) cells and M2 macrophages) (22). The metabolic rate of resting or naive cells is normally low in the form of low glycolysis and OXPHOS (22). M0 macrophages differentiate into M1 and M2 phenotypes, while naïve CD4+ T cells become effector T cells or Tregs, and quiescent NK cells become activated. In fact, each cell subtype has distinct metabolic requirements and cytokine repertoires (22). There will be much to learn about metabolic and immune control in future research into disease states. In addition to systemic metabolic alterations, innate metabolic changes are related to building an immune reaction and functional programming inside a cell (23). One of the most prominent of these changes is the stimulation of anaerobic glycolysis, a common characteristic of T cells, dendritic cells (DCs), and macrophages involved in proinflammatory stimulation (24–26).
The Warburg effect is tumor cells’ most well-known metabolic switching mechanism (27). Compared to normal cells, cancer cells prefer glycolysis to mitochondrial OXPHOS, even when oxygen levels are high (28). Since glycolysis is inefficient for ATP synthesis, its output is considerably faster, supplying energy to cancer cells for growth and proliferation. Moreover, glycolysis provides tumor cells with various building components for biomass production (29). The oncogene c-MYC and the hypoxia-inducible factor-1 (HIF-1) facilitate the development of essential enzymes that boost aerobic glycolysis, the most significant of which are HK2, pyruvate kinase 2 (PKM2), glucose transporter 1 (GLUT1), and lactate dehydrogenase A (LDHA) (28–30). The upregulation of GLUT1 promotes glucose absorption by tumor cells. An increase in the expression of HK2 converts glucose into G6P, the first step in glycolysis, and elevates its flow to the pentose phosphate pathway (PPP), causing the formation of nicotinamide adenine dinucleotide phosphate oxidase (NADPH). NADPH is required for anaerobic activities, such as nucleotide synthesis, as well as body defense against reactive oxygen species (ROS) (28, 29).
Many studies have shown comparable increases in glutamine uptake by tumor cells as well as improvements in the tricarboxylic acid (TCA) cycle, PPP, lipid catabolism, and other metabolic activities observed in rapidly proliferating normal cells (31). In this context, the primary distinction between tumor and propagating normal cells is that the latter’s intake of fuel molecules is rigorously managed by growth factor signaling (32). On the other hand, tumor cells absorb glucose and glutamine from the extracellular media due to mutations in key metabolic regulatory genes, including c-MYC, AKT, and PI3K (31). Modifications in the function of these and other genes cause cell-autonomous increases in the abundance and the function of membrane transporters and metabolic enzymes, allowing for increased glycolysis and glutaminolysis in the absence of external stimuli (33). This allows efficient coupling of energy and biosynthesis requirements for uncontrolled cell growth.
However, in certain tumor cells, even in the presence of oxygen, glucose enters the cytoplasm and performs a chemical process catalyzed by lactate dehydrogenase and pyruvate kinase to create lactate and nicotinamide adenine dinucleotide + (NAD+) rather than entering the tricarboxylic acid (TCA) cycle, as is the case in normal cells (34, 35). Compared to glycolysis, the TCA cycle generates almost 20 times the amount of energy from a single glucose molecule (36). In contrast to the mitochondrial TCA cycle, glycolysis may simultaneously convert one glucose molecule into 10-100 times as much ATP (2). This efficient strategy may be selected by tumor cells because glycolysis improves tumor cell competitiveness in a nutrient-poor niche (2). Furthermore, the pentose phosphate pathway (PPP) (another branch of glycolysis) can be used to generate pentose. In this process, neither ATP nor any other form of cellular energy is produced or utilized. G-6-P dehydrogenase (G6PDH) and 6-phosphogluconate dehydrogenase (6PGDH) accelerate the conversion of glucose-6-phosphate (G-6-P), a byproduct of glycolysis into nicotinamide adenine dinucleotide phosphate (NADPH) and ribose-5-phosphate (R-5-P) (37). Glycolysis may restart using R-5-P when it is converted back into fructose-6-phosphate (F-6-P) and glyceraldehyde-3-phosphate (G-3-P). One of the universal electron carriers, NADPH, includes minimizing intracellular oxidation by controlling the transformation of oxidized glutathione to decreased glutathione and interacting with a broad range of enzymes. It accomplishes this by transporting electrons and hydrogen released by the energy of sunlight (38). Reactive oxygen species (ROS) and other oxides are byproducts of several cellular metabolic activities; they attach to proteins and are degraded in the presence of NADPH, which maintains the function of these proteins (38). Additionally, R-5-P generated by PPP may give a rapid rate of synthesis of nucleic acid for cancerous cells (38).
A metabolic fuel, glutamine enables rapidly replicating cells to fulfill their increasing demands for ATP, biochemical precursors, and reductants (39). Solute carrier family 1 member 5 (SLC1A5) is responsible for transporting glutamine into the cell; GLS then catalyzes the dehydrogenation process in the mitochondria to produce glutamate. GLUD, alanine, or aspartate transaminase (TAs) transform glutamate to -KG, a byproduct of the TCA cycle (40). Furthermore, signaling molecules such as RAS, MP-activated protein kinase (AMPK), and protein kinase B (PKB) A activate glycolytic enzymes and drive lactate generation, forcing tumor cells to consume glutamine to fulfill their enhanced energy demands (41). In addition to supplying energy, glutamine is essential for tumor cell biogenesis. When glutamine reaches the cell, the activity of GLS and GLUD produces ammonia, which may be employed immediately in the production of purines and pyrimidines (42). It has since been discovered that glutamine creates -KG, a type of raw material used in the production of purines and pyrimidines, via a redox interaction with aspartic acid (43, 44). Glutamate catabolism in tumor cells promotes the production of nucleotide precursors, which aids in tumorigenesis and replication. Glutamine also serves as a nitrogen source in protein production; isotope tracking indicated that glutamine supplies almost half of the non-essential amino acids needed by cancer cells for protein biosynthesis (40, 42). Finally, in hypoxic circumstances, cancer cells may utilize glutamine to create citric acid and fatty acids via reducing carboxylation processes while also constructing dihydro-orotate acid to alleviate the negative consequences of ammonia on cancer cells (45, 46).
The highly active metabolic processes of tumor cells can cause significant differences in the form of nutrients and other small molecules in the tumor microenvironment (TME) (47). This can have severe consequences for the immune system. Increased metabolic activities of cancer cells and impaired angiogenesis within the TME may lead to nutrient deficiency as well as a hypoxic environment, leading to metabolic competition between tumor cells and invading immune cells (48–50). Similarly, in animal models, glucose uptake and the effective function of anti-tumor CD4+ T cells are inversely related to the glycolytic activity of tumor cells, and access to glucose in the TME increases cytokine production from anti-tumor CD8+ T cells (48). The key effector component of the antitumor response is composed of CD4+ T cells (CD4+ Conv) and CD8+ effector T cells (Teff), while non-proliferative naive CD4+ and CD8+ T cells identify their relevant antigens in the setting of costimulatory signaling. In fact, they become proliferate and activate metabolic characteristics to facilitate rapid expansion (51–53). Even though many early studies identified increased aerobic glycolysis as a characteristic of T cell activation, it is now obvious that increased TCA cycle metabolism, as well as OXPHOS, are indeed essential aspects of CD4+ and CD8+ T cell stimulation. While TCA cycle metabolism seems to increase after 24 h of stimulation, aerobic glycolysis increases more rapidly within six hours (53–55).
Tumor necrosis factor alpha (TNF-α), IL-12, and interferon-gamma (IFN-γ) induce metabolic rewiring of effector T cells via glycolytic signaling pathways, such as PI3K–AKT–mammalian target of rapamycin (mTOR) (53, 56, 57). This glycolytic transition enables rapid ATP generation, NAD+ renewal, and nucleotide biosynthesis, all of which are essential for effector function, cytokine secretion, and cell growth. IL-2 autocrine secretion boosts GLUT1 expression and the activation of the PI3K pathway (56, 58). When the T-cell receptor (TCR) is activated, opposing moderators of PI3K signaling (PTEN) and PI3K Interacting Protein 1 (PIK3IP1)/transmembrane inhibitor of PI3K (TrIP) are downregulated, allowing metabolic rewiring (59, 60). PI3K phosphorylates AKT, stimulating the expression of GLUT1 to convey glucose and regulate glycolytic enzymes, including LDHA, HK2, and PKM2 (54, 56). Furthermore, mTOR and c-MYC activation boost HIF-1 expression to support glycolysis and reduce oxygen consumption rate (OCR) in CD8+ T cells (54). As a result, the deletion of HIF in CD8+T cells lowers their penetration into cancerous tissues and, as a result, enhances tumor development (61). A current finding explored more information about the process of glycolysis as it relates to the stimulation of T cells such as CD4 + and CD8 + T cells. Several studies underlined two separate phases; the first is rapid glycolysis, which begins a few minutes after TCR activation and is controlled by pyruvate dehydrogenase kinase 1 (PDHK1), which diminishes OXPHOS but stimulates lactate generation independently of glucose intake. CD28, the PI3K axis, and HIF-1 activation will sustain glycolysis at a high rate (52).
Myeloid cells also have a function in tumor immunity (62). Macrophages, which are produced from circulatory monocytes, help to phagocytose dead cells and secrete cytokines (62). Through the major histocompatibility complex (MHC), they also can deliver antigens (tumor peptides) to naïve T lymphocytes via MHC molecules. Activated macrophages, such as T cells, are divided into subgroups. Whether macrophages transform into classical anticancer M1-like morphologies or an alternative suppressive M2-like phenotype is influenced by the metabolic shift that happens during infections or malignancies. The metabolic pattern will determine macrophage polarization and impact people with cancer prognosis (63, 64). TAMs (tumor-associated macrophages) have been demonstrated to exhibit an M2-like phenotype in cancer. Therefore, the oxidative metabolism of protumoral M2-like macrophages increases their proliferation at the expense of M1. M2 cells can develop their immunosuppressive phenotype by undergoing a metabolic transition in response to anti-inflammatory markers such as IL-4 and IL-10 and glucocorticoid activation. They boost their oxygen consumption rate to facilitate their expansion (65). Their oxidative metabolism is supported by an increase in the amount and content of mitochondria, as evidenced by the increased expression of succinate dehydrogenase A (SDHA) (65–67). In the setting of M2 polarization, fatty acid oxidation (FAO) is the primary energy source for the TCA cycle. The interplay between the signal transducer and activator of transcription (STAT6) and peroxisome proliferator-activated receptors (PPARs) is induced by IL-4 stimulation, which enhances CD36 expression to bind and translocate fatty acids in cells (65, 68). Many investigations have demonstrated that lipid oxidation is required for M2-like/TAM development. FAO is necessary for tumor progression, propagation, and migration via IL-1 production (62). Unlike M1 macrophages, which use Nitric Oxide Synthase 2 (NOS2) to create NO and destroy cancer cells, M2-TAMs metabolize arginine to polyamines through increasing Arginase 1 expression (62). Arginase 1 activity promotes cancer growth, spread, and angiogenesis and has been associated with a poor prognosis in various types of cancer (69–72). Further details are depicted in Table 1.

Table 1 Metabolic reaction of anti-tumoral and pro-tumoral immune cells in cancer.
miRNAs are short non-coding RNAs that silence their target mRNAs after transcription through transcriptional silencing or mRNA degradation (80, 81). miRNA-mediated gene expression modulation can occur via mRNA breakdown or translational suppression. miRNAs attach to the target mRNA’s 3-untranslated region (3-UTR) via incorrect base-pairing and can control a wide range of genetic components simultaneously (82–84). As a result, miRNAs, such as transcriptional regulators, could function as master gene modulators and collaborate to establish gene expression profiles in cells (85). miRNAs play roles in various biochemical processes, including growth, differentiating, propagation, apoptosis, and induced pluripotent stem cells (86).
Several lines of evidence suggest that miRNAs act as a key function in metabolic activities, notably lipid and glucose metabolism, as well as amino acid biosynthesis (11). Furthermore, miRNAs may identify and modify metabolic components at the transcriptional level, which is vital in both cancer and normal cells (12). Tumor cell metabolism may be altered to avoid apoptosis and promote cell proliferation and survival. The Warburg effect, in which miRNA dysregulation leads to higher glycolysis, is the most robust metabolic phenotype found in cancer cells (13, 14). miRNAs have a role in tumor cell metabolism regulation by modulating the expression of target genes whose compounds either explicitly govern metabolic machinery or indirectly modify the transcription of metabolic enzymes, so acting as master controllers (10). This section will summarize and discuss the involvement of miRNAs in several metabolic processes in cancer (Table 2).
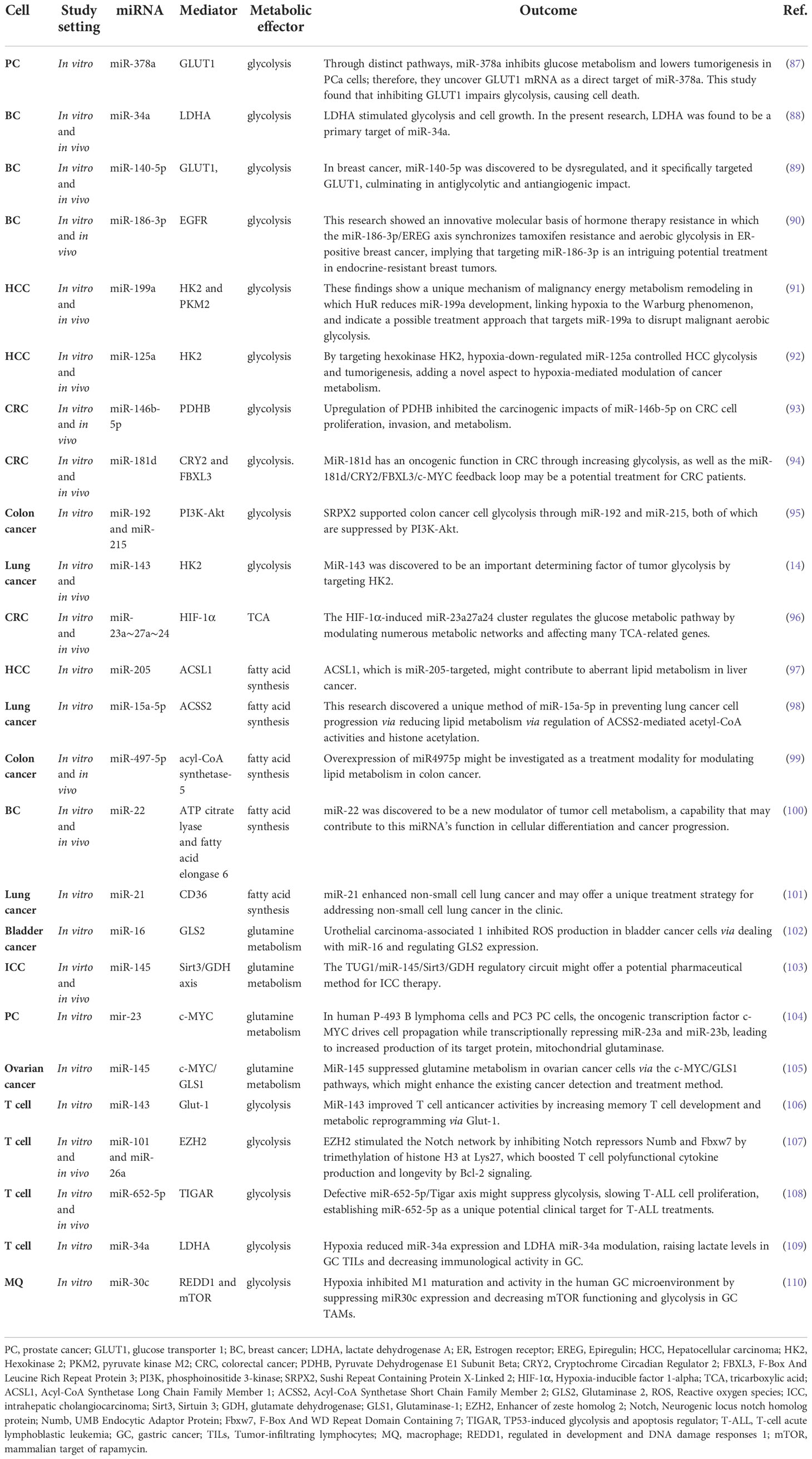
Table 2 miRNAs reprogrammed metabolic pathways in tumor and tumor-associated immune cells.
miRNAs can regulate metabolic pathways, several of which are altered in malignancies and other cellular activities (111). The downstream targets of several distinct miRNAs have been directly or indirectly linked to alterations in tumor metabolism. According to research, miRNAs govern the irreversible processes of glycolysis, particularly the critical enzymes (Figures 1, 2) (112). For example, miR-143 controls glycolysis by addressing Hexokinase 2 (HK2), which phosphorylates glucose to create G6P, so dedicating glucose to anaerobic glycolysis (14). HK2, coding for the first rate-limiting enzyme of glycolysis, is among the top list of genes predicted and potentially regulated by multiple miRNAs, including miR-143. Besides, the miR-200 family, which includes miR-200a, miR-200b, and miR-200c, has been shown to control phosphoglucose isomerase, which is also associated with tumorigenesis (113). Current results suggest a role of miR-200s in PGI/AMF-induced EMT, and thus approaches for up-regulation of miR-200s could be a novel therapeutic strategy for the treatment of highly invasive breast cancer. Furthermore, miR-17-92 regulates the amounts of enolase (ENO) 1, ENO2, phosphoglycerate kinase 1 (PGK1), and triosephosphate isomerase 1 (114). A quantitative mass spectrometry analysis indicated that oxysterol-binding-protein-related-protein 8 (ORP8) targets miR-143 (115). miR-155 by stimulating the STAT3 (a transcriptional activator for HK2) can block miR-143 (116). Additionally, miR-143 decreases the expression of HK2 in both primary keratinocytes and cell lines generated from head and neck squamous cell carcinoma (117). Furthermore, HK2 has been identified as a miR-143 target, suggesting that miR-143 may influence glucose metabolism in colon cancer cells (118). Similarly, miR-143 has also been found as a determinant factor in cancer glycolysis in human lung cancer by addressing HK2 (14).
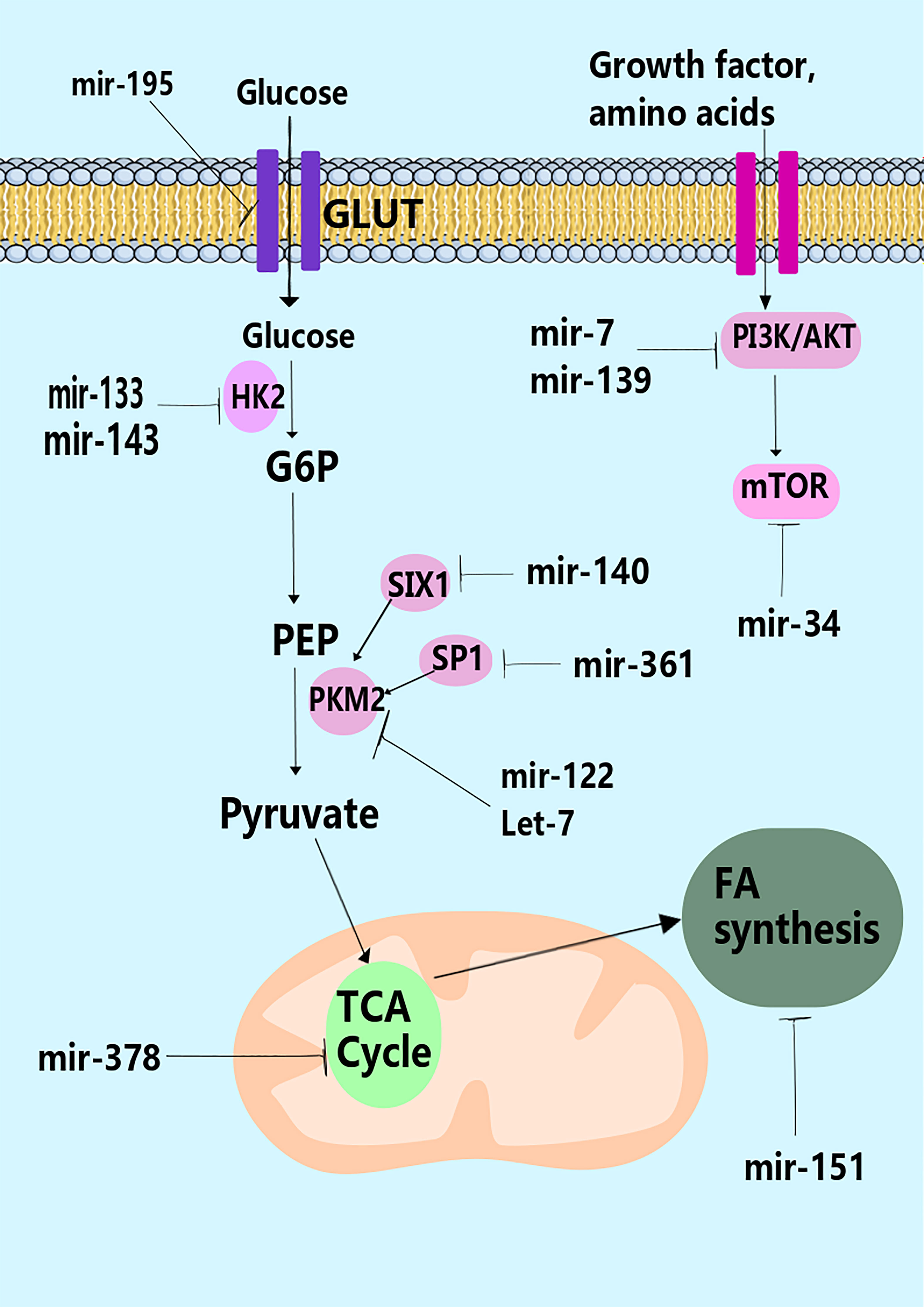
Figure 1 miRNAs and control of metabolism. miRNAs have been shown to regulate many stages in metabolism through modulation of the expression of key metabolic enzymes (e.g., hexokinase-2 by miR-133), transporters (e.g., GLUT by miR195-5p), or pathways that control metabolism such as the PI3K pathway (e.g., miR-19, miR-7, and miR-139).
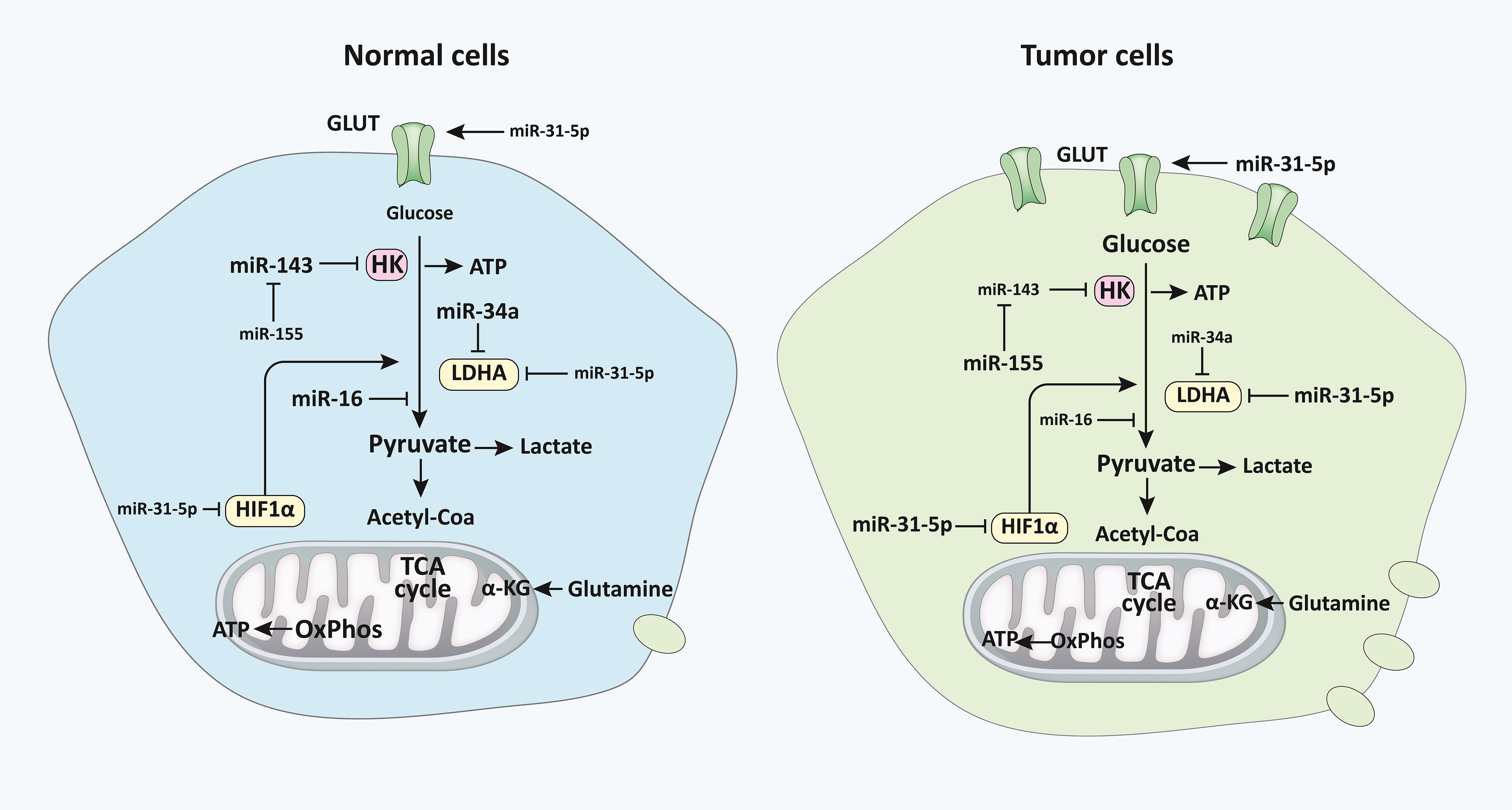
Figure 2 miRNAs regulate glucose metabolism in normal vs malignant cells. The diagram represents the primary alternative energy metabolism pathways in human cells and their proportionate use in normal quiescent cells and tumor and highly proliferative cells, which are depicted via varying degrees of clarity. The letter size distinguishes the frequency of recognized regulatory miRNAs, their primary mRNA targets (HK, LDHA, and HIF1), as well as particular indirect miRNA regulatory interplays with other miRNAs or pathway interactions. Modulatory connections are represented by referring arrowheads, whereas repressive interactions are represented by inhibiting arrows. The designations of pathways and metabolites are emphasized in bold and thicker lines to indicate their predominance in each condition (33). miRNA, microRNA; HK, Hexokinase; LDHA, lactate dehydrogenase A; HIF1, Hypoxia-inducible factor 1.
Through carefully fusing external stimuli with the availability of amino acids and intracellular energy, the mammalian target of rapamycin (TOR) regulates the cellular development and metabolic functions of all eukaryotic cells (119). The expression of various glycolytic enzymes, including GLUT1, LDHB, and PKM2, is increased when mTOR is activated (120, 121). In response to growth factors and PI3K/AKT signaling, the activation of mTOR signaling causes an increase in HIF-1 protein synthesis, which leads to the overexpression of glycolytic enzymes (122, 123). Some studies have found a vital function for miRNAs in controlling mTOR through PI3K/AKT-independent and dependent pathways. Nagaraja et al. (124) showed that miR-100 directly targets mTOR and actively suppresses its signaling in ovarian cancer. A negative relationship was discovered between miR-193a-3p and mTOR by Fornari et al. (125). They found that miR-193a-3p directly repressed the translation of mTOR1 and c-met in hepatocellular carcinoma, which altered the cell cycle, increased invasiveness, and made cancer cells more susceptible to doxorubicin therapy and hypoxia-induced apoptosis (125). Goa et al. (126) found that miR-126 prevents the growth of tumor cells by explicitly targeting the p85b subunit of PI3K in a different investigation. Since PI3K/AKT triggers downstream signals to mTOR for cellular proliferation, miR-126 loss increases tumor formation (126). Similarly, Pineau et al. (127) demonstrated a negative connection between miR-221 and the expression of DDIT4, a regulator of the PI3K/AKT/mTOR pathway in liver cancer cell lines. Several miRNAs might alter glucose homeostasis via mechanisms other than aerobic glycolysis. MiR-375 is one such miRNA that may regulate glucose homeostasis in the body via modulating insulin release (111). In β cells, the upregulation mentioned above limits insulin exocytosis through processes that are independent of transmembrane Ca2+ fluxes and intracellular Ca2+ signaling (128). Several earlier studies have shown that miR-375 regulates glucose homeostasis in pancreatic β cells via modulating the phosphatidylinositol 3-kinase (PI3K) signaling pathway (129). MiR-375 expression was significantly reduced in pancreatic cancer cells (130, 131). As a result, the precise impact of downregulation on glucose metabolism and possible targets in cancer cells has not been discovered.
Multiple pathways branch off from glycolysis, the first of which is the PPP (132). Because NADPH and nucleic acid precursors are required for cancer transformation in PPP, it is vital for glucose catabolic and anabolic pathways. Glycolysis can be directed to the PPP, which provides NADPH to cells while storing reduced glutathione (GSH) and ribose-5-phosphate, a substrate for nucleotide synthesis (133). Co-regulation of glycolysis and flux to the PPP is critical for tumorigenesis and/or malignancy. The conversion of G6P into PPP, in particular, enhances cancer aggressiveness by producing ribose 5-phosphate and reducing equivalents such as NADPH, which is used in lipogenesis and also mitigation of oxidative stress (134). In lung tumors, miR-1 and miR-206 mediate the increase in G6P and subsequent decrease in nuclear factor erythroid-related factor 2 (NRF2) function (135). The levels of G6PD are increased in some tumors, and the enzyme is regulated by miR-206 and miR-1 in cervical tumors associated with papillomavirus infections, while miR-1 downregulates G6PD in malignant tumors (136–138). Moreover, in HCC specimens, the amounts of G6PD are inversely correlated with miR-1 and the liver-specific miR-122, and the reduction of expression of these two miRNAs enhances tumorigenesis (52). A simultaneous decline in the expression levels of miR-122 and miR-1 is expected to contribute to the dysregulation of glucose metabolism in HCC cells, resulting in fast tumor development. It has been revealed that miR-122 depletion has a larger influence on HCC due to its prevalence in the benign liver and drastic reduction in HCC.
Different glucose metabolite patterns have been discovered in colorectal cancer (CRC) cases. A growing body of research indicates that pro-inflammatory mediators and critical rate-limiting enzymes of glycometabolic pathways may control impaired glucose metabolism in CRC, leading to distinct metabolic characteristics of cancer cells (139). However, the influence of miRNAs on glucose metabolism in CRC remained unclear. Qiu et al. (139) explored possible glycometabolism-regulating miRNAs in CRC by analyzing current miRNA expression datasets. They found miR-124 to be a potent regulator of PPP and nucleotide synthesis in CRC cells by targeting phosphoribosyl pyrophosphate synthetase 1 (PRPS1) and ribose 5-phosphate isomerase A (RPIA). Significantly, PRPS1 and RPIA, which are simultaneously elevated in CRC samples, might serve as potential markers for the poorly unknown CRC prognosis. Numerous investigations have shown that 6-Phosphogluconate dehydrogenase (6PGD) is upregulated in many malignancies (140). 6PGD has been identified as a functioning target of miR-206 and miR-613 in lung tumor cells, and miR-206 and miR-613 are thought to influence 6PGD expression and metabolic switching in cisplatin-resistant ovarian and lung tumor cells (141, 142). As a result, the uneven control of PPP and how cancer cells avoid PPP modulation constitute a new target for cancer diagnostics and therapy.
The TCA cycle is a common metabolic pathway in aerobic organisms. The TCA cycle is the last metabolic pathway for major nutrients (carbohydrate, lipid, and amino acid) as well as the primary hub of lipid, amino acid, and carbohydrate metabolism (143). The TCA cycle is the principal ATP generation process. It regulates energy production during mitochondrial respiration and is essential for glucose metabolism. A growing body of research has connected the primary enzyme activities of one-carbon metabolism and the TCA cycle to a wide range of malignancies (143).
Metabolic mediators are generated through glycolytic flux fuel biosynthetic pathways that create amino acids and nucleotides (144). The primary carbon source for replenishing the byproducts of the TCA cycle is glutamine. Glutaminase (GLS) converts glutamine into glutamate, which then enters the TCA cycle as α-ketoglutarate to provide energy for the cell. GLS was discovered to be a target of miR-23 (145). ATP citrate lyase (ACL) can metabolize glutamine-derived citrate to acetyl-CoA (AcCoA) and oxaloacetate (OAA). AcCoA contributes to de novo fatty acid (FA) production, while OAA can be converted into nucleotides and amino acids (146). These processes are responsible for producing macromolecules and organelles essential for cell growth. In cancer, several metabolic mediators have been identified; a glucose-dependent community that produces lactate and a lactate-dependent community that consumes lactate generated by neighboring cells (147). Tumor cells utilize glucose for fuel under hypoxic circumstances inside the tumor stroma and release lactate as a byproduct utilized by other cells. The hypoxia response system works by increasing the activity of glucose transporters and glycolytic enzymes (148). Let-7, miR-107, and miR-34 control glycolysis by addressing LDHA by p53 (149). Likewise, miR-181a and miR-183 could target isocitrate dehydrogenase 1/2 (IDH1/2) and TCA cycle enzymes (Figure 1) (150, 151). In addition, miR-378 affects the TCA cycle in breast cancer by modulating the expression of peroxisome proliferator-activated receptor-alpha, GA binding protein transcription factor, subunit (GABPA), co-activator 1-alpha (PGC-1α), and estrogen-related receptor gamma (ERRγ) (152). The regulation of PGC-1 by miR-23a may potentially assist the metabolic shift from OXPHOS to anaerobic glycolysis to produce anabolic precursors and support tumor cell growth (153). Until recently, the importance of the TCA cycle and the control of its activity was underestimated (140). Pyruvate dehydrogenase protein X component (PDHX) was shown to be a primary target of miR-26a in CRC cells, reported Chen and collaborators (154), and it is known that miR-26a may modify PDHX expression through specifically targeting the 3′UTR of PDHX. PDHX, which is found in the mitochondrial matrix, is a non-catalytic member of the PDH complex. It plays an essential role in the energy homeostasis that occurs in the mitochondria (155). Under some circumstances of aerobic metabolism, the PDH complex acts as a catalyst for the oxidative elimination of glucose and pyruvate. Of note, it has been discovered that miR-26a is responsible for regulating glucose metabolism in CRC cells. This occurs because it prevents pyruvate from converting into acetyl CoA and initiates the tricarboxylic acid cycle (154).
It has recently been proven that lactate may be taken up in the TCA cycle, serve as a source of energy, and even operate as an oncometabolite with signaling capabilities (156). Lactate was previously known as a “metabolic waste product” produced by aerobic glycolysis (156). Although lactate was first recognized exclusively as a waste product of anaerobic cellular metabolism, it is now known that many cells constantly use lactate as fuel under strictly aerobic conditions (157). For instance, Wei et al. (158) demonstrated that the lactate generation produced by miR-181a leads to increased cellular proliferation. Similarly, high lactate levels have been shown to induce an aggressive phenotype in breast cancer cells (159). During aerobic glycolysis in cancer cells, most pyruvate is converted into lactate instead of AcCoA, which enters the TCA cycle in mitochondria (112). The lactate produced in the cytosol is subsequently released outside of the cells by monocarboxylate transporters (MCTs). Most of the released lactate can eventually be taken up in the liver and converted to glucose before being recycled back into tumors (112). However, lactate has also been hypothesized as an energy source for tumors or stromal cells under aerobic conditions. No known significant cancer-associated miRNAs affect LDHA, which converts pyruvate into lactate. However, miR-124, miR-29a, and miR-29b can target both human and mouse MCT1 3’-UTRs, possibly by controlling the propensity of tumor cells to release lactate (160). Among these, miR-29b is associated with a region that is frequently amplified in breast cancer, while miR-124 is widely downregulated in medulloblastoma (161). CD147, a widely expressed plasma membrane glycoprotein, regulates MCT1 and MCT4 in the plasma membrane (162). Let-7b, a tumor suppressor miRNA that inhibits cancer cell migration and invasion, as well as tumorigenesis, regulates the expression levels of CD147 (163).
Tumor cells have different lipid metabolism patterns, in which fatty acids are mainly synthesized from scratch (164). Mitochondrial function inside the pyruvate-citrate shuttle is required for fatty acid, cholesterol production, and protein acetylation. Recent advancements in current knowledge of the control of lipid metabolism reveal that miRNAs play key roles in regulating the metabolism of both cholesterol and fatty acids (165). Several miRNAs have been described to regulate lipid metabolism, including miR-122, miR-370, miR-378, miR-335, miR-125a-5p, and miR-33 (165). MiR-122 is essential in regulating cholesterol and fatty acid metabolism (166). MiR-122 has been shown to cause the formation of cholesterol-rich membrane domains and ER-associated lipid droplets (LDs). Silencing miR-122 may significantly change the balance of lipid accumulation and metabolic activity (167, 168).
Glutamine is an amino acid preferred by tumor cells, and its oxidative metabolism fuels the TCA cycle (169). It is a vital nitrogen and carbon supplier for the de novo generation of nucleotides, lipids, non-essential amino acids, anapleurotic intermediates in the TCA cycle, lipids, and nucleotides GSH components (40, 170). Proliferating tumor cells require high levels of glutamine (171). Many cancer cells and TME-related compounds, including miRNAs, modulate glutamine absorption and metabolism. The oncosuppressor miR-137 targets alanine, serine, cysteine, and glutamate transporter (ASCT2), negatively correlated with ASCT2 in pancreatic ductal adenocarcinoma, pancreatic cancer, glioblastoma, and CRC (134). Epigenetic downregulation of miR-137 results in increased glutamine uptake, allowing tumor cells to grow in an unfavorable microenvironment due to abundant glutamine availability (172). Upon entry, GLS converts glutamine to glutamate, an enzyme directly targeted by miR-203 and miR-153, both of which are dysregulated in melanoma and glioblastoma (173, 174). Lately, it has been shown that miR-145 inhibits glutaminolysis in ovarian cancer cells by decreasing the expression of c-MYC, which lowers GLS transcription (105). Low c-MYC levels and lower glutamine consumption might explain why miR-145 upregulation in ovarian tumors has a less proliferative capacity and is less aggressive. Also, c-Myc may direct the reprogramming of glutamine metabolism through miR-18a activation. The rate-limiting enzyme of glutathione synthesis in liver cancer is the glutamate-cysteine ligase catalytic subunit (GCLC), which is downregulated by c-Myc, lowering GSH bioavailability (175). Because active c-MYC can increase glutamine bioavailability and favor tumor cells, c-MYC/miR-18a-regulated cells are more susceptible to oxidative damage due to the lack of GSH synthesis from glutamine.
Glutamate is converted to alpha-ketoglutarate (α-KG) by glutamate dehydrogenase to fuel the TCA cycle (GDH). Transaminase enzymes, such as glutamate-oxaloacetate transaminase, can also metabolize glutamine to alpha-KG (GOT1). GOT1 activation is critical for prolonged cell growth in pancreatic ductal adenocarcinoma (176). MiR-9-5p could function as a tumor suppressor by directly targeting GOT1, reducing pancreatic cancer cell growth and metastasis while influencing glutamine-dependent NADPH generation and redox balance (177). A particular miRNA has many targets in glutamine metabolism, as it does in glucose metabolism. The depletion of miR-122, for example, causes ASCT2 and GLS to be upregulated in HCC. It has been found that ASCT2, GLS, and miR-122 can be regarded as prognostic markers (178). miR-122 levels in individuals are negatively associated with ASCT2/GLS, while upregulation of ASCT2 and GLS correlates with poor prognosis (178).
miRNAs have the potential to directly modulate several metabolic processes through various metabolic enzymes in tumor-associated immune cells and tumor-associated signaling processes, thereby influencing cancer growth (Table 2) (179–182). For example, hypoxia in malignant cell exosomes increases the production of some biomolecules (such as miR-let-7a), which suppresses the AKT/mTOR axis, resulting in the transition from glucose metabolism to OXPHOS metabolism (181). In TAMs, the suppression of AKT/mTOR reduces glucose catabolism and increases tumor blood vessel development (181, 183). Similarly, hypoxia impedes mTOR function in TAMs by decreasing the expression of miR-30c expression, which enhances the development, activity, and metabolic activity of M1 macrophages through targeting controlled advancement and DNA damage responses 1 (REDD1), ultimately leading to gastric cancer pathogenesis and progression (110). Various studies have shown that miRNAs mainly act on vital physiological enzymes or energetic nutrient transporters and affect the growth and development of T cells (184, 185). In this line, Zhang and colleagues (106) discovered miR-143 as a T-cell metabolic modulator that lowers glucose absorption by inhibiting the glucose receptor GLUT1. They discovered that miR-143 enhances memory T cell development and metabolic rewiring by particularly lowering glycolysis by TCR-dependent stimulation (106). This section summarizes new research on miRNAs involved in the immune regulation of cell metabolic processes in tumors.
Metabolically, naïve T cells or T cells that have not encountered their antigen rely more on OXPHOS, whereas activated cytotoxic T cells (CD8+) switch to the glycolytic pathway (186). Wells and colleagues (172) found let-7 miRs as essential modulators of naïve CD8+ T cell phenotypic maintenance via T metabolic activity in this scenario. Let-7 controls glycolysis and protein synthesis simultaneously with cell proliferation and differentiation by reducing the transcriptional activity of critical metabolic enzymes (Ldha, Pkm, Tpi, Hk2, Pfk1, and Gpd2), GLUT1, GLUT2, and the synthesis of the Yars enzyme (187). The amount of Let-7 decreases with T-cell activation, c-MYC is suppressed, and energy metabolism switches from OXPHOS to glycolysis, leading to an appropriate CTL response to virus-infected cells or cells presenting cancer antigens (188). MiR-155 is a well-studied miR involved in immunological reactions, particularly T cell responses, where it affects cytokine as well as interferon signaling through suppressor of cytokine signaling 1 (SOCS1) and STAT1 (106, 189). Recently, miR-155 has also been linked to T-cell metabolism. Using suppressing the inositol 5-phosphatase Ship1, a regulator of the mTOR pathway, Monnot and colleagues demonstrated that miR-155 increases mTOR activity and, consequently, glycolysis of CD8+ T cells and enhances T cell proliferation and effector functions (190). Exogenous induction of miR-155 in antigen-specific CD8+ T cells enhances antitumor efficacy toward low-affinity antigens in OVA-expressing melanoma cells (190). Collectively, these data support the results of Dudda and colleagues, showing that miR-155 upregulation may enhance T cell adoptive-transfer treatment in malignancy (189). Memory T cells, including CD8+ T cells and CD4+ Tregs, rely on enhanced OXPHOS and lipid oxidation rather than glycolysis, whereas cytotoxic CD8+ T cells demand glycolysis (186). Zhang and colleagues (69) demonstrated that miR-143 promotes central T memory development by blocking glycolysis by GLUT1. Notably, an immunomodulatory metabolite IDO and its derivative kynurenine, which are typically formed inside the TME, reduce miR-143 expression, showing a reciprocal link between miR-143 and metabolism (48). Tumor cells interact with activated T cells in the TME to consume resources such as glucose, which is required to sustain anaerobic glycolysis. Finally, tumor-induced glucose deprivation reduces anti-tumoral T-cell function, resulting in tumor growth (50, 107). Similarly, tumor-induced glucose deprivation increased miR-101 and miR-26a expression levels in CD8+ T cells, which leads to the downregulation of their common target, the methyltransferase EZH2, a crucial enzyme of the epigenetic polycomb repressive complex 2 (PRC2). All in all, decreased EZH2 function diminishes anti-cancer CD8+ T cell responses, promoting immunological subverting (107).
EZH2 is the catalytic component of the polycomb-group family that trimethylates histone H3 at Lys27 (H3K27me3) (191, 192). The restricted epigenetic signature H3K27me3 controls the transcription process in tumor cells (192). According to scientific literature, EZH2 is dedicated to developing Th1 and T helper 2 (Th2) cells in mice (193, 194). Zhao and colleagues (107) showed that EZH2 modulates effector T cell functionality and longevity in T lymphocytes. In the TME, EZH2 is an important target and detector of anaerobic glycolysis breakdown. Moreover, in T cells, researchers discovered that EZH2 transcription is regulated via anaerobic glycolysis metabolism through miRNAs, rendering it functional and therapeutically crucial in ovarian cancer patients (107). Considering the physiological and molecular relevance of EZH2 in polyfunctional T cells in tumors, it is critical to assess how T cell EZH2 is regulated in the TME and becomes hypoglycemic due to the Warburg effect. Significantly, Zhao and colleagues (107) discovered that primary ovarian tumor cells downregulate EZH2 expression and limit multifunctional cytokine production and lifespan of effector T cells, which may be restored by glucose feeding and 2-deoxyglucose mimetic agents. This finding shows that glycolytic metabolism regulates T cell EZH2 in the context of cancer. According to a study performed by Zhao and colleagues (107), it has been shown that the glycolytic shift affects the activation of memory T cells, multiplication, and expression of IFN-γ in animal models (24, 57, 195). Another mechanism of action research shows that cancer cells maintain high levels of miR-26a and miR-101 expression in effector T cells.
Consequently, miR-26a and miR-101 repress EZH2 expression and impair the function of effector T cells. In conclusion, the current findings point to a metabolic target and tumor immune evasion scheme. Malignant cells inhibit T cell EZH2 activity by limiting aerobic glycolysis, reducing T cell-mediated anti-cancer immunity. miRNA expression is dynamic in naïve, memory, and effector CD8+ T cells throughout T cell development (196). A particular miRNA has been linked to the modulation of CD8+ T cell development. miR-17-92, for example, promotes CD8+ T memory cell growth and the formation of final effectors via the PI3K/AKT/mTOR axis (197). mTOR also induces the formation of HIF-1a and MYC, leading to the glucose metabolism of T cells. Nonetheless, the significance of miRNAs in the complex regulation of T metabolism in anti-cancer properties is not entirely understood. In this context, Zhang and colleagues (106) verified Glut-1 as a prominent target of miR-143 in this function to investigate the molecular mechanisms involved in enhancing the anti-proliferative activity of miR-143 in T cells. Glut-1 is the major glucose-transporter isoform in T cells, acting as a key metabolic monitoring station in glycolysis (198). Studies discovered that Glut-1 knockdown and miR-143 upregulation reduced glycolysis. A previous study has discovered that miR-143 controls tumor cell glycolysis by influencing HK2, a key glycolytic pathway enzyme (14, 199). MiR-143 was shown to suppress HK2 expression in T cells (106). T cells have a greater baseline OCR than differentiated effector cells (200). Zhang and colleagues (106) found that miR-143 upregulation resulted in greater baseline OCR and maximum respiration in T cells, which may be crucial for T cell memorization. Moreover, the dynamical expression pattern of miR-143 throughout T cell growth corresponded to the different types of energy used in naive, effector, and T memory cells (Figure 3). Naïve T cells create energy by breaking down amino acids, glucose, and fatty acids to fuel OxPhos. In addition, Zhang and colleagues (106) discovered that the upregulation of miR-143 increased the expression of Carnitine palmitoyltransferase I A (CPT1A). These findings suggest that miR-143 promotes Tm cell development by altering metabolic activity.
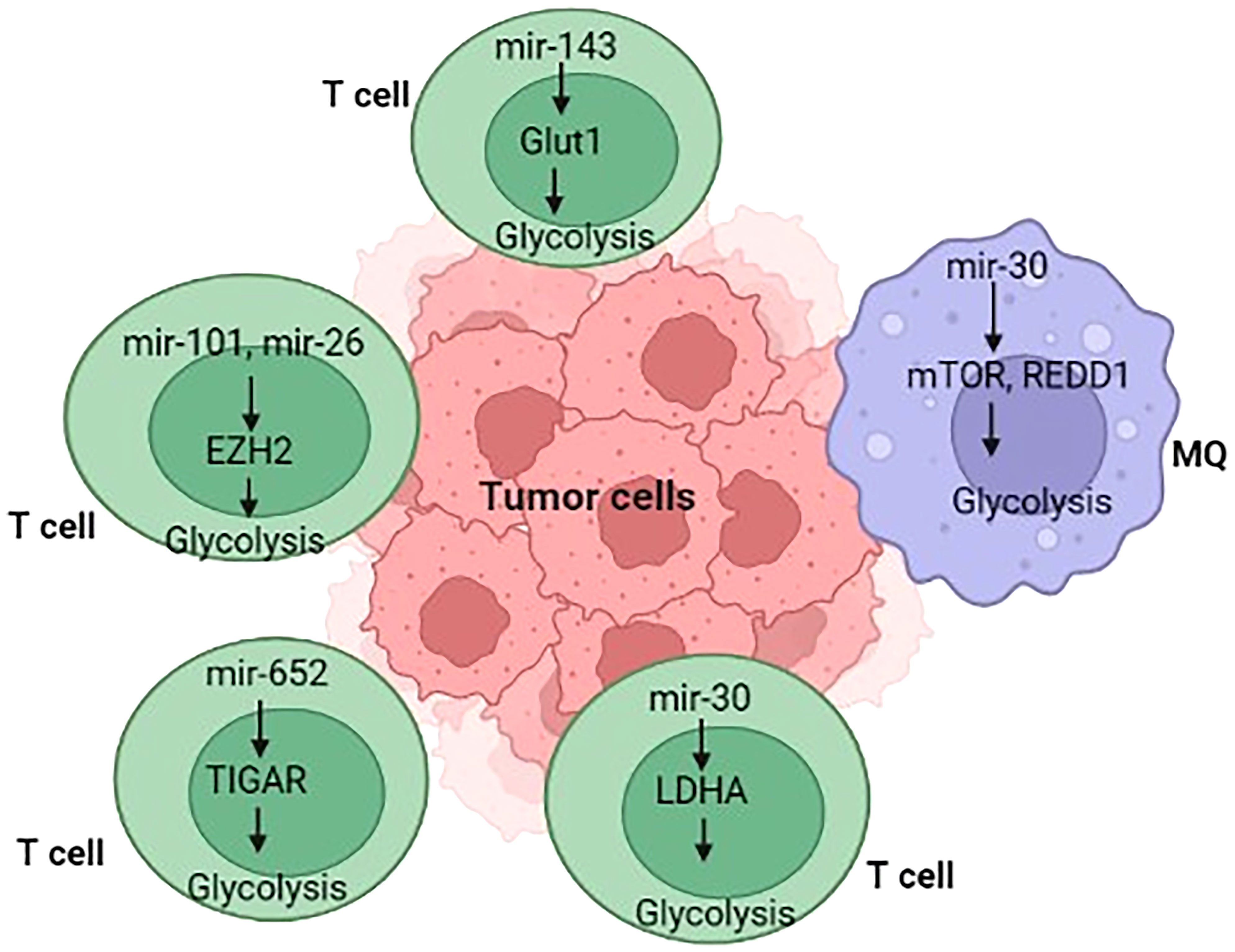
Figure 3 Overview of immunometabolism in tumor microenvironment.
Additionally, enhanced glycolysis is observed in T-cell acute lymphoblastic leukemia (T-ALL) and is induced by oncogenes, including runt-related transcription factor 2 (RUNX2) neurogenic and locus notch homolog protein (NOTCH) (201–203). The effectiveness of glycolysis in T-ALL is reversed or reduced, suggesting that the mechanisms of glycolysis in T-ALL need to be investigated (201–203). Given that miRs have been shown to alter glycolysis levels by targeting genes encoding glycolysis rate-limiting enzymes, including phosphofructo-2-kinase/fructose-2,6-bisphosphatase 3 (PFKFB3), GLUT-1, and Phosphofructokinase-1 (PFK-1). Liu and colleagues (108) discovered that defective miR-652-5p might restrict glycolysis via targeting TP53-induced glycolysis and apoptosis regulator (TIGAR), explaining the decrease of PFKFB3 expression and the molecular basis of T-ALL.
Macrophages play a significant role in normal homeostasis and contribute to self-limited inflammatory response throughout infection; however, these favorable activities might be overshadowed by sustained activation signals, resulting in dysfunctional macrophage behavior, which can have pathological results (204). Like M2 macrophages, TAMs increase revascularization, reduce antigen presentation, attenuate inflammatory factor secretion, and block T-cell infiltration into the tumor (205). Before polarization, macrophages have a quiescent metabolic activity that is fuelled by the TCA cycle and depends on OXPHOS for energy (206). Master transcription regulators, including nuclear factor-κB (NF-κB) and HIF-1α, are activated by macrophage polarizing signals to modify proinflammatory gene expression and metabolic reprogramming (207, 208). Inflammatory macrophages change their metabolism programming to prefer aerobic glycolysis over OXPHOS throughout polarization (26, 209, 210). Even though aerobic glycolysis, commonly known as the Warburg effect, is less effective than OXPHOS in producing ATP, the increased glucose flow into cells results in faster energy synthesis and the creation of additional biosynthetic intermediates (211).
miRNAs have been found to control several aspects of macrophage responses, such as those that increase or decrease inflammatory responses in response to polarizing stimuli (206). Recently, a large body of evidence has evolved that miRNAs are involved in modulating metabolic activities that affect macrophage activities (212). Tumor hypoxia is induced by increased metabolic rates and oxygen demand, which may contribute to the formation of persistent hypoxic conditions and the activation of HIF (213). TAMs display phenotypic characteristics similar to M2 macrophages under hypoxic circumstances (214). Hypoxia promotes the production of certain macromolecules in cancer cell exosomes, including miR-let-7a, which reduces glucose metabolism (through the associated AKT/mTOR pathway), culminating in a switch from glucose to OxPhos metabolism (Figures 3, 4) (181). miRNAs also have an impact on the PI3K/AKT axis. PI3K/AKT regulates glucose metabolism by reducing PTEN function, which might increase gene expression levels in M2 macrophages, including YM1, Fizz, ARG, TGF-β, IL-4, and IL-10 (15–17). miRNAs, including miR-32, miR-101, miR-301a-3p, and miR-103a, regulate the PI3K/AKT pathway, triggering apoptosis in cancer cells and hastening tumor formation and spread (216–219). TAMs can re-metabolize waste produced by cancer cells during metabolism, resulting in alterations in activity and phenotype. TAMs can attach to cancer cells and help them spread. Increased miR-543 expression stabilizes HIF-1 and enhances the glucose uptake ability of cancer cells (220). In cancer cells, miR-181b, miR-143, miR-4458, and miR-199a-5p enhance metastasis by boosting the function of HK2 (14, 221, 222). The downregulation of miRNAs in cancer cells, including miR-124, miR-152, miR-let-7a, and miR-139-5p, increases the expression of PKM2 expression, increasing tumor malignancy (223–226). In these situations, the glycolytic metabolite of cancer cells can induce TAMs to switch their metabolism into M2 macrophages and cooperate to accelerate tumorigenesis. Zhihua et al. (110) discovered that miR-30c promotes glycolysis and M1 polarization in human gastric cancer cells by regulating the mTOR pathway in TAMs. MiR-30c activates the PI3K/AKT/mTOR pathway by targeting the REDD1 gene, which is a negative regulator of mTOR. In cancer, hypoxia reduces miR-30c expression and decreases the proportion of anti-tumor M1 macrophages. Apart from the importance of macrophage polarization, Wenes and colleagues (227) found that TAMs lacking significant glycolytic REDD1 compete with endothelial cells for glucose consumption. Immune and tumor cells impact each other not just physiologically but by fighting for the same resources (like glucose), and they often produce miRNAs that affect metabolic pathways. Binenbaum and colleagues (228) have shown that in pancreatic adenocarcinoma, miRNA-enriched exosomes are produced by TAMs and absorbed by tumor cells, where they cause resistance to gemcitabine owing to a change in disease metabolism. While the precise mechanism is uncertain, miR-365, among other miRNAs, plays a critical role in this operation by raising the expression of pyrimidine metabolism, thus increasing the triphosphate-nucleotide pool that interferes with gemcitabine for DNA integration in tumor cells. Furthermore, miR-365 results in the activation of cytidine deaminase, an enzyme responsible for the catabolization of gemcitabine.
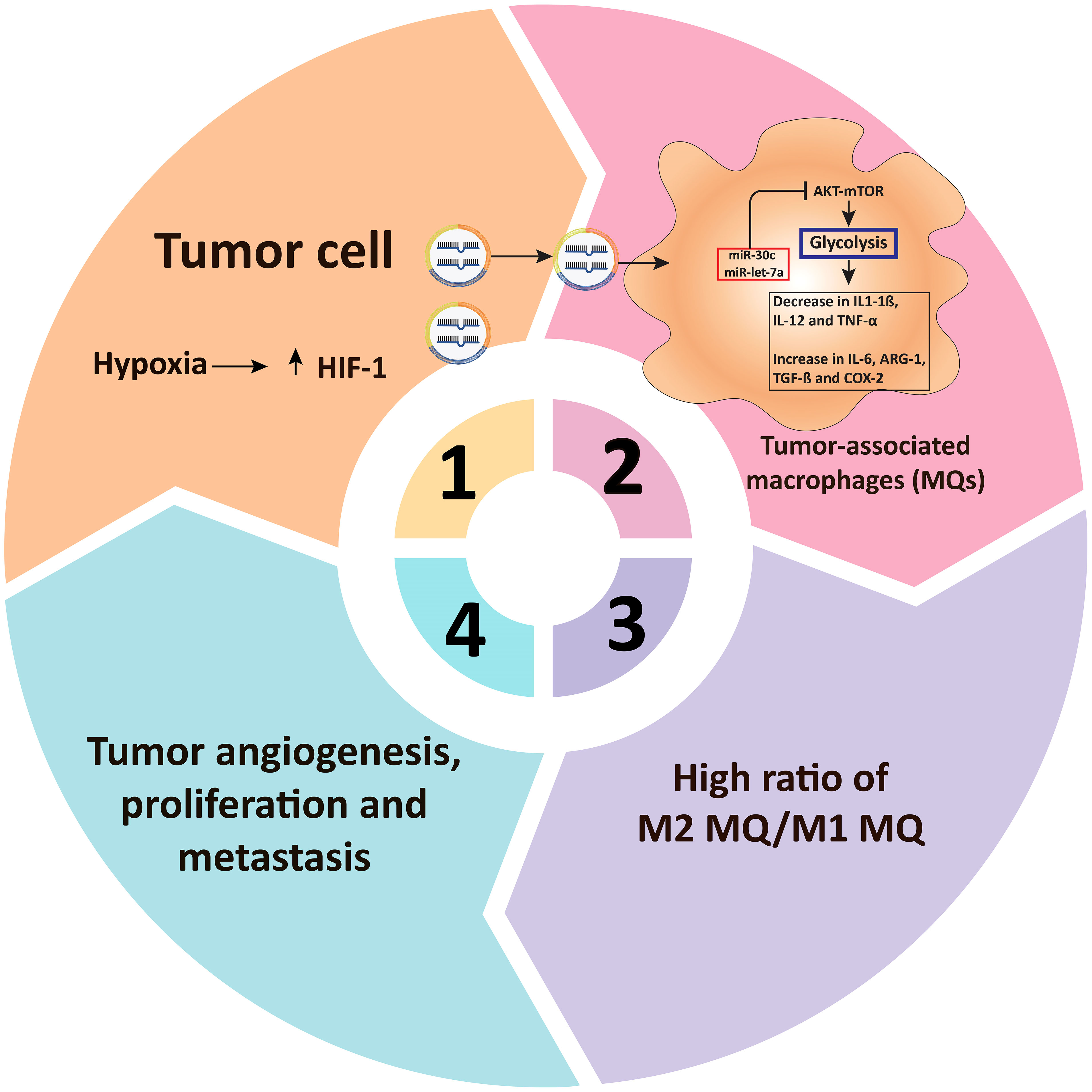
Figure 4 miRNAs regulate glucose metabolism in tumor-associated macrophages. In TAMs, microRNAs influence glucose metabolism. Tumor cells’ respiration is less than their oxygen demand, culminating in hypoxia as well as HIF-1 overexpression. Extracellular vesicles (including miR-30c and miR-let-7a, among several others) are secreted into TAMs through hypoxia-induced cancer cells, suppressing the glycolysis-related metabolic pathway AKT/mTOR in TAMs. Decreased glucose metabolism then raises M2-type associated factors including TGF-β, COX-2, IL-6, and ARG-1, reduces M1-type related factors including TNF-α, IL-12, and IL-1β, resulting in M2-type macrophage rewiring and tumor growth and angiogenesis (215). TAM, Tumor-associated macrophages: HIF-1α, Hypoxia-inducible factor 1-alpha; mTOR, mammalian target of rapamycin; ARG-1, Arginase 1; IL, Interleukin; COX-2, cyclooxygenase 2; TGF-β, Transforming growth factor beta; TNF-α, Tumor necrosis factor alpha.
Cellular metabolism influences the function of immune cells, activation, cytokine release, and anti-cancer or antiviral activity (47). A complete understanding of metabolic processes includes functional distinctions between quiescent and active TME immune cells. In this regard, metabolites (self-produced or immunometabolites), cancer cells or oncometabolite, and TME circumstances can all affect these cells (229). These substances have bioenergetic, immunological, and carcinogenic properties and can control the expression of inflammatory genes (230, 231). Succinate and citrate have pro-inflammatory effects, whereas α-KG, itaconate, and fumarate have immunosuppressive abilities (230). Most of these metabolites increase during the immune response and alter the balance between immune activation and suppression. Although high amounts of 2-Hydroxyglutarate have also been reported in cytogenetically typical malignancies, somatic mutations in cytosolic IDH1 can contribute to the development of oncometabolite 2-Hydroxyglutarate (229). IDH1 mutations were found in less than half of the individuals with elevated 2-hydroxyglutarate; the rest had mutations in IDH2, a mitochondrial homolog of IDH1. Succinate, 2-Hydroxyglutarate, and fumarate increase tumorigenesis by modulating cell signaling and influening chemotherapy and radiotherapy responsiveness via epigenetic processes (232, 233).
Besides, several cell metabolites, including tryptophan, are required to survive tumor-infiltrating lymphocytes (TILs) (234). Indoleamine 2,3-dioxygenase 1 (IDO1) is a tryptophan metabolism rate-limiting enzyme that converts tryptophan into kynurenine and 3-hydroxyanthranilic acid (235). IDO1 overexpression and tryptophan deficiency might lead to the dysfunction of effector T cells and tumor immune evasion (236). Upregulated miR-218 protects cervical cancer cells from immunological attack by increasing IDO1 levels (237). Lou and colleagues (238) discovered that miR-448 acts as a tumor suppressor in colon cancer cells by regulating downstream IDO1. An in vitro study showed that aberrant production of miR-448 is beneficial in allowing TILs to perform their full spectrum of activities (238). Huang and colleagues (239) showed that miR-153 expression is a key determinant in T cell chimeric antigen receptor (CAR) efficacy in colon cancer models. In colon cancer, miR-153 specifically targeted IDO1, enhanced CAR T cells’ cytotoxicity, and reduced tumor growth (239).
Recently, there has additionally been a rise in attention to deciphering the function of miRNAs in the modulation of anti-cancer immunity and how this may affect the effectiveness of various cancer therapies. This interest has been sparked by recent developments (240). Furthermore, defense mechanisms may have both pro- and anti-oncogenic effects, and the physiological communication between immune and cancerous cells in the TME is crucial in deciding how cancer will proceed. miRNAs are not only significant components of immune reaction mechanisms but also play a role in regulating and modulating a variety of interplay between immune cells and tumor cells (241).
In this context, immune checkpoint compounds are critical components in the process of modulating immunological responses (242). miRNAs, including miR-424, miR-200, miR-138–5p, miR-34a-5p, and miR-513, regulate the expression of PD-L1 and PD-1, respectively (242). Hence, the degrees of expression of these miRNAs might influence the interaction between the PD-1 receptor and the PD-L1 ligand, which in turn can modify the activities of T cells (243). MiR-138–5p is also thought to play a role in controlling some other immune checkpoint molecules known as CTLA-462. CTLA-462 is a molecule that, when it interacts with its receptors (either CD80 or CD86) on antigen-presenting cells (APC) like dendritic cells and macrophages, suppresses the function of effector T cells while simultaneously facilitating the development of regulatory T cells (Treg cells) (244). It has been shown that some miRNAs and immune checkpoint inhibitors may influence the effectiveness of other immunotherapies, including CAR T cells. miR-153 suppressed the expression of IDO in a human colon cancer xenograft cancer, which improved the impact of CAR T cells that targeted the epidermal growth factor receptor (239). Hence, to design anti-cancer therapeutic options that are both successful and tolerable and are founded on miRNAs, it is vital to properly evaluate the precise functions that miRNAs play in the TME.
Two of the most significant molecules controlling gene expression are transcription factors, or TFs, and miRNAs. At the level of transcription, TFs control the expression of genes by attaching to promoter regions, while miRNAs regulate gene expression at the post-transcriptional level by attaching to 3’ untranslated regions (245). Modulating TFs and miRNAs may either promote or inhibit tumorigenesis (246). It is significant to mention that TFs and miRNAs can control each other. They establish a feed-forward loop if they coordinate their regulation of a shared target gene (FFL). By participating in this process, major regulatory units called FFLs might continue to establish gene regulatory circuits. Aberrantly controlled TF-miRNA-mediated FFLs have been identified in various complex diseases such as CRC, glioblastoma, schizophrenia, and other diseases (245). dChip-GemiNI is the technique suggested by Yan et al. (247) to find common and particular TF-miRNA FFLs among the five different kinds of cancer. Using pan-cancer data, a more in-depth examination of TF-miRNA modulation showed 26 dysregulated FFLs across 13 different types of cancer, as well as anticipated potential genes and therapeutic targets (248).
It is well-established knowledge that non-coding RNAs, also known as ncRNAs, are responsible for a significant proportion of the human transcriptome. These non-coding RNAs mainly consist of miRNAs, long non-coding RNAs (lncRNAs), and circular RNAs (circRNAs) (249). Long non-coding RNA, also known as lncRNA, is a category of non-coding RNA with more than 200 nucleotides. According to several research findings, lncRNAs contribute significantly to the process of cancer formation in a number of different ways (250). Newly found non-coding RNA (ncRNA) with a structure consisting of a closed loop is called circular RNA (circRNA). Compared to the conventional form of linear RNA, circRNA is much more persistent because it is less susceptible to degradation by RNA exonuclease since it lacks terminal 5’ caps and 3’ polyadenylated tails (251). In cancer, it has been shown that CircRNAs can affect cellular metabolism regulation (252). Over the last several years, an ever-increasing number of scientists have committed their efforts to investigate ncRNAs’ biological activities. In addition, various computational algorithms have been created to forecast possible connections between ncRNAs and illness. This is something that is of paramount significance for the process of identifying biomarkers (253, 254).
In 2011, Salmena et al. (255) were the pioneers who proposed the competitive endogenous RNA (ceRNA) concept for the first time. A family of non-coding RNAs known as ceRNA can bind common miRNAs in a manner that competes and cross-regulates one another at the post-transcriptional stage (256, 257). This regulatory system, which is dependent on ceRNA, was found in several different malignancies. For instance, HER2 expression was controlled by the lncRNA HOTAIR thanks to its ability to compete for the miR-331-3p. This ultimately led to the formation of cancer (258). LncRNA H19 was shown to contribute to the development of oncogenic activities in gallbladder cancer through the alteration of miR-342-3p and FOXM1 (259). Additionally, circRNA ciRS-7 has the potential to behave as a “super sponge” for miR-7 and suppress the action of miR-7 (260). An increasing body of data suggests that the ceRNA regulatory network plays a role in the biological processes that contribute to the growth of HCC. These processes include propagation, dissemination, EMT, and chemotherapy resistance (261–264).
Throughout this overview, we emphasized that miRNAs play important roles in various fundamental processes such as immunity, gene regulation, and metabolism, especially in immune cells responding to cancer. We described the primary metabolic pathways used to fuel eukaryotic cells and the signaling networks and molecules that feed them, emphasizing the points at which they are known to be regulated by miRNAs. It has been shown how these processes are changed during the metabolic activity of tumors and how this affects immune cells invading the TME. The control of CD8+ T cell development has been ascribed to single miRNAs. For example, MiR-17-92 stimulates CD8+ T memory proliferation, differentiation, and terminal effector development through the PI3K/AKT/mTOR axis. mTOR additionally promotes the production of HIF-1a and MYC, leading to T-cell glucose metabolism. Notably, in human gastric cancer, miR-30c increased glycolysis and, consequently, M1 polarization through modulating the mTOR pathway in TAMs. MiR-30c activates the PI3K/AKT/mTOR axis in these cells by targeting the REDD1 gene, which is a potent inhibitor of mTOR. However, little data on the importance of miRNA-mediated metabolic wiring in immune cells are currently available in the cancer context. Unquestionably, a better knowledge of miRNA-mediated metabolic wiring of immune cells will provide new insights into the causes of immunologically related cancer and open new avenues for developing novel anticancer therapies. For example, by altering the profiles of miRNAs that target genes for critical metabolic enzymes or necessary nutrient transporters, the intracellular metabolism of immune cells can be manipulated, potentially altering metabolic rewiring and modifying immune functions. Overall, considering the importance of the regulatory role of miRNA in changing the metabolic profile of immune cells during cancer (influence on cancer fate and immune responses), future research will elucidate the precise role of these emerging regulatory elements in cancer for the discovery and potential development of novel therapeutic approaches for cancer treatment.
SA, YI, AJ, AAA, HA and RZ have the idea for and planned the study and contributed to the writing of the paper. GG, AR-C, AbAA, and QQ contributed to the writing of the manuscript. SK and RM contributed to the critical revision of the report. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Jang M, Kim SS, Lee J. Cancer cell metabolism: implications for therapeutic targets. Exp Mol Med (2013) 45:e45–5. doi: 10.1038/emm.2013.85
2. Liberti MV, Locasale JW. The warburg effect: How does it benefit cancer cells? Trends Biochem Sci (2016) 41:211–8. doi: 10.1016/j.tibs.2015.12.001
3. Smith RL, Soeters MR, Wüst RCI, Houtkooper RH. Metabolic flexibility as an adaptation to energy resources and requirements in health and disease. Endocr Rev (2018) 39:489–517. doi: 10.1210/er.2017-00211
4. Lee YS, Wollam J, Olefsky JM. An integrated view of immunometabolism. Cell (2018) 172:22–40. doi: 10.1016/j.cell.2017.12.025
5. Yao Q, Song Z, Wang B, Zhang JA. Emerging roles of microRNAs in the metabolic control of immune cells. . Cancer Lett (2018) 433:10–7. doi: 10.1016/j.canlet.2018.06.024
6. Hotamisligil GS. Foundations of immunometabolism and implications for metabolic health and disease. Immunity (2017) 47:406–20. doi: 10.1016/j.immuni.2017.08.009
7. McKinney E, Smith K. Metabolic exhaustion in infection, cancer and autoimmunity. Nat Immunol (2018) 19:213–21. doi: 10.1038/s41590-018-0045-y
8. Bantug GR, Galluzzi L, Kroemer G, Hess C. The spectrum of T cell metabolism in health and disease. Nat Rev Immunol (2018) 18:19–34. doi: 10.1038/nri.2017.99
9. Netea-Maier RT, Smit JW, Netea MG. Metabolic changes in tumor cells and tumor-associated macrophages: a mutual relationship. Cancer Lett (2018) 413:102–9. doi: 10.1016/j.canlet.2017.10.037
10. Chen B, Li H, Zeng X, Yang P, Liu X, Zhao X, et al. Roles of microRNA on cancer cell metabolism. J Trans Med (2012) 10:228. doi: 10.1186/1479-5876-10-228
11. Rottiers V, Näär AM. MicroRNAs in metabolism and metabolic disorders. Nat Rev Mol Cell Biol (2012) 13:239–50. doi: 10.1038/nrm3313
12. Pucci S, Mazzarelli P. MicroRNA dysregulation in colon cancer microenvironment interactions: the importance of small things in metastases. Cancer Microenviron (2011) 4:155–62. doi: 10.1007/s12307-011-0062-y
13. Cairns RA, Harris IS, Mak TW. Regulation of cancer cell metabolism. Nat Rev Cancer (2011) 11:85–95. doi: 10.1038/nrc2981
14. Fang R, Xiao T, Fang Z, Sun Y, Li F, Gao Y, et al. MicroRNA-143 (miR-143) regulates cancer glycolysis via targeting hexokinase 2 gene. J Biol Chem (2012) 287:23227–35. doi: 10.1074/jbc.M112.373084
15. Wang D, Wang X, Si M, Yang J, Sun S, Wu H, et al. Exosome-encapsulated miRNAs contribute to CXCL12/CXCR4-induced liver metastasis of colorectal cancer by enhancing M2 polarization of macrophages. Cancer Lett (2020) 474:36–52. doi: 10.1016/j.canlet.2020.01.005
16. Lu J, Xie L, Liu C, Zhang Q, Sun S. PTEN/PI3k/AKT regulates macrophage polarization in emphysematous mice. Scandinavian J Immunol (2017) 85:395–405. doi: 10.1111/sji.12545
17. Vergadi E, Ieronymaki E, Lyroni K, Vaporidi K, Tsatsanis C. Akt signaling pathway in macrophage activation and M1/M2 polarization. J Immunol (2017) 198:1006–14. doi: 10.4049/jimmunol.1601515
18. Fernandez-de-Cossio-Diaz J, Vazquez A. A physical model of cell metabolism. Sci Rep (2018) 8:8349. doi: 10.1038/s41598-018-26724-7
19. Chandel NS. Basics of metabolic reactions. Cold Spring Harbor Perspect Biol (2021) 13:a040527. doi: 10.1101/cshperspect.a040527
20. Pearce EJ, Pearce EL. Driving immunity: all roads lead to metabolism. Nat Rev Immunol (2018) 18:81–2. doi: 10.1038/nri.2017.139
21. O'Neill LA, Kishton RJ, Rathmell J. A guide to immunometabolism for immunologists. Nat Rev Immunol (2016) 16:553–65. doi: 10.1038/nri.2016.70
22. Traba J, Sack MN, Waldmann TA, Anton OM. Immunometabolism at the nexus of cancer therapeutic efficacy and resistance. Front Immunol (2021) 12:657293. doi: 10.3389/fimmu.2021.657293
23. O'Neill LA, Hardie DG. Metabolism of inflammation limited by AMPK and pseudo-starvation. Nature (2013) 493:346–55. doi: 10.1038/nature11862
24. Chang CH, Curtis JD, Maggi LB Jr., Faubert B, Villarino AV, O'Sullivan D, et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell (2013) 153:1239–51. doi: 10.1016/j.cell.2013.05.016
25. Everts B, Amiel E, Huang SC, Smith AM, Chang CH. TLR-driven early glycolytic reprogramming via the kinases TBK1-IKKε supports the anabolic demands of dendritic cell activation. Nat Immunol (2014) 15:323–32. doi: 10.1038/ni.2833
26. Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGettrick AF, Goel G, et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature (2013) 496:238–42. doi: 10.1038/nature11986
27. Warburg O. On respiratory impairment in cancer cells. Science (1956) 124:269–70. doi: 10.1126/science.124.3215.269
28. Phan LM, Yeung SC, Lee MH. Cancer metabolic reprogramming: importance, main features, and potentials for precise targeted anti-cancer therapies. Cancer Biol Med (2014) 11:1–19. doi: 10.7497/j.issn.2095-3941.2014.01.001
29. Li Z, Zhang H. Reprogramming of glucose, fatty acid and amino acid metabolism for cancer progression. Cell Mol Life Sci CMLS (2016) 73:377–92. doi: 10.1007/s00018-015-2070-4
30. Biswas SK. Metabolic reprogramming of immune cells in cancer progression. Immunity (2015) 43:435–49. doi: 10.1016/j.immuni.2015.09.001
31. Pavlova NN, Thompson CB. The emerging hallmarks of cancer metabolism. Cell Metab (2016) 23:27–47. doi: 10.1016/j.cmet.2015.12.006
32. Lum JJ, Bauer DE, Kong M, Harris MH, Li C, Lindsten T, et al. Growth factor regulation of autophagy and cell survival in the absence of apoptosis. Cell (2005) 120:237–48. doi: 10.1016/j.cell.2004.11.046
33. Gibson MS, Noronha-Estima C, Gama-Carvalho M. Therapeutic metabolic reprograming using microRNAs: From cancer to HIV infection. Genes (Basel) (2022) 13:273. doi: 10.3390/genes13020273
34. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the warburg effect: the metabolic requirements of cell proliferation. science (2009) 324:1029–33. doi: 10.1126/science.1160809
35. Rogatzki MJ, Ferguson BS, Goodwin ML, Gladden LB. Lactate is always the end product of glycolysis. Front Neurosci (2015) 9:22. doi: 10.3389/fnins.2015.00022
36. Locasale JW, Cantley LC. Altered metabolism in cancer. BMC Biol (2010) 8:1–3. doi: 10.1186/1741-7007-8-88
37. Jiang P, Du W, Wu M. Regulation of the pentose phosphate pathway in cancer. Protein Cell (2014) 5:592–602. doi: 10.1007/s13238-014-0082-8
38. Patra KC, Hay N. The pentose phosphate pathway and cancer. Trends Biochem Sci (2014) 39:347–54. doi: 10.1016/j.tibs.2014.06.005
39. Zhu L, Zhu X, Wu Y. Effects of glucose metabolism, lipid metabolism, and glutamine metabolism on tumor microenvironment and clinical implications. Biomolecules (2022) 12(4)580. doi: 10.3390/biom12040580
40. Altman BJ, Stine ZE, Dang CV. From Krebs to clinic: glutamine metabolism to cancer therapy. Nat Rev Cancer (2016) 16:619–34. doi: 10.1038/nrc.2016.71
41. Hensley CT, Wasti AT, DeBerardinis RJ. Glutamine and cancer: cell biology, physiology, and clinical opportunities. J Clin Invest (2013) 123:3678–84. doi: 10.1172/JCI69600
42. Yang L, Venneti S, Nagrath D. Glutaminolysis: a hallmark of cancer metabolism. Annu Rev Biomed Eng (2017) 19:163–94. doi: 10.1146/annurev-bioeng-071516-044546
43. Sullivan LB, Gui DY, Hosios AM, Bush LN, Freinkman E, Vander Heiden MG. Supporting aspartate biosynthesis is an essential function of respiration in proliferating cells. Cell (2015) 162:552–63. doi: 10.1016/j.cell.2015.07.017
44. Lane AN, Fan TW. Regulation of mammalian nucleotide metabolism and biosynthesis. Nucleic Acids Res (2015) 43:2466–85. doi: 10.1093/nar/gkv047
45. Jiang L, Shestov AA, Swain P, Yang C, Parker SJ, Wang QA, et al. Reductive carboxylation supports redox homeostasis during anchorage-independent growth. Nature (2016) 532:255–8. doi: 10.1038/nature17393
46. Wang Y, Bai C, Ruan Y, Liu M, Chu Q, Qiu L, et al. Coordinative metabolism of glutamine carbon and nitrogen in proliferating cancer cells under hypoxia. Nat Commun (2019) 10:1–14. doi: 10.1038/s41467-018-08033-9
47. Leone RD, Powell JD. Metabolism of immune cells in cancer. Nat Rev Cancer (2020) 20:516–31. doi: 10.1038/s41568-020-0273-y
48. Chang C-H, Qiu J, O'Sullivan D, Buck MD, Noguchi T, Curtis JD, et al. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell (2015) 162:1229–41. doi: 10.1016/j.cell.2015.08.016
49. Lukey MJ, Katt WP, Cerione RA. Targeting amino acid metabolism for cancer therapy. Drug Discov Today (2017) 22:796–804. doi: 10.1016/j.drudis.2016.12.003
50. Ho P-C, Bihuniak JD, Macintyre AN, Staron M, Liu X, Amezquita R, et al. Phosphoenolpyruvate is a metabolic checkpoint of anti-tumor T cell responses. Cell (2015) 162:1217–28. doi: 10.1016/j.cell.2015.08.012
51. Patel CH, Leone RD, Horton MR, Powell JD. Targeting metabolism to regulate immune responses in autoimmunity and cancer. Nat Rev Drug Discov (2019) 18:669–88. doi: 10.1038/s41573-019-0032-5
52. Menk AV, Scharping NE, Moreci RS, Zeng X, Guy C, Salvatore S, et al. Early TCR signaling induces rapid aerobic glycolysis enabling distinct acute T cell effector functions. Cell Rep (2018) 22:1509–21. doi: 10.1016/j.celrep.2018.01.040
53. Frauwirth KA, Riley JL, Harris MH, Parry RV, Rathmell JC, Plas DR, et al. The CD28 signaling pathway regulates glucose metabolism. Immunity (2002) 16:769–77. doi: 10.1016/S1074-7613(02)00323-0
54. Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D, et al. The transcription factor myc controls metabolic reprogramming upon T lymphocyte activation. Immunity (2011) 35:871–82. doi: 10.1016/j.immuni.2011.09.021
55. Gatza E, Wahl DR, Opipari AW, Sundberg TB, Reddy P, Liu C, et al. Manipulating the bioenergetics of alloreactive T cells causes their selective apoptosis and arrests graft-versus-host disease. Sci Trans Med (2011) 3:67ra8–8. doi: 10.1126/scitranslmed.3001975
56. Chapman NM, Boothby MR, Chi H. Metabolic coordination of T cell quiescence and activation. Nat Rev Immunol (2020) 20:55–70. doi: 10.1038/s41577-019-0203-y
57. Yang K, Shrestha S, Zeng H, Karmaus PW, Neale G, Vogel P, et al. T Cell exit from quiescence and differentiation into Th2 cells depend on raptor-mTORC1-mediated metabolic reprogramming. Immunity (2013) 39:1043–56. doi: 10.1016/j.immuni.2013.09.015
58. DiToro D, Winstead CJ, Pham D. Differential IL-2 expression defines developmental fates of follicular versus nonfollicular helper T cells. Science (2018) 361(6407):eaao2933. doi: 10.1126/science.aao2933
59. Uche UU, Piccirillo AR, Kataoka S, Grebinoski SJ, D'Cruz LM. PIK3IP1/TrIP restricts activation of T cells through inhibition of PI3K/Akt. J Exp Med (2018) 215:3165–79. doi: 10.1084/jem.20172018
60. Hawse WF, Sheehan RP, Miskov-Zivanov N, Menk AV, Kane LP. Cutting edge: Differential regulation of PTEN by TCR, akt, and FoxO1 controls CD4+ T cell fate decisions. J Immunol (2015) 194:4615–9. doi: 10.4049/jimmunol.1402554
61. Palazon A, Tyrakis PA, Macias D, Veliça P, Rundqvist H, Fitzpatrick S, et al. An HIF-1α/VEGF-A axis in cytotoxic T cells regulates tumor progression. Cancer Cell (2017) 32:669–683.e5. doi: 10.1016/j.ccell.2017.10.003
62. Luby A, Alves-Guerra M-C. Targeting metabolism to control immune responses in cancer and improve checkpoint blockade immunotherapy. Cancers (Basel) (2021) 13:5912. doi: 10.3390/cancers13235912
63. Murray PJ. Macrophage polarization. Annu Rev Physiol (2017) 79:541–66. doi: 10.1146/annurev-physiol-022516-034339
64. Mills CD, Ley K. M1 and M2 macrophages: the chicken and the egg of immunity. J innate Immun (2014) 6:716–26. doi: 10.1159/000364945
65. Huang SC-C, Everts B, Ivanova Y, O'sullivan D, Nascimento M, Smith AM, et al. Cell-intrinsic lysosomal lipolysis is essential for alternative activation of macrophages. Nat Immunol (2014) 15:846–55. doi: 10.1038/ni.2956
66. Yu Q, Wang Y, Dong L, He Y, Liu R, Yang Q, et al. Regulations of glycolytic activities on macrophages functions in tumor and infectious inflammation. Front Cell Infect Microbiol (2020) 10:287. doi: 10.3389/fcimb.2020.00287
67. Tavakoli S, Zamora D, Ullevig S, Asmis R. Bioenergetic profiles diverge during macrophage polarization: implications for the interpretation of 18F-FDG PET imaging of atherosclerosis. J Nucl Med Off Publicat Soc Nucl Med (2013) 54:1661–7. doi: 10.2967/jnumed.112.119099
68. Szanto A, Balint BL, Nagy ZS, Barta E, Dezso B, Pap A, et al. STAT6 transcription factor is a facilitator of the nuclear receptor PPARγ-regulated gene expression in macrophages and dendritic cells. Immunity (2010) 33:699–712. doi: 10.1016/j.immuni.2010.11.009
69. Zhang QW, Liu L, Gong CY, Shi HS, Zeng YH, Wang XZ, et al. Prognostic significance of tumor-associated macrophages in solid tumor: a meta-analysis of the literature. PloS One (2012) 7:e50946. doi: 10.1371/journal.pone.0050946
70. Czystowska-Kuzmicz M, Sosnowska A, Nowis D. Small extracellular vesicles containing arginase-1 suppress T-cell responses and promote tumor growth in ovarian carcinoma. Nat Commun (2019) 10:3000. doi: 10.1038/s41467-019-10979-3
71. Ma Z, Lian J, Yang M, Wuyang J, Zhao C, Chen W, et al. Overexpression of arginase-1 is an indicator of poor prognosis in patients with colorectal cancer. Pathol Res Pract (2019) 215:152383. doi: 10.1016/j.prp.2019.03.012
72. Grzywa TM, Sosnowska A, Matryba P, Rydzynska Z, Jasinski M, Nowis D, et al. Myeloid cell-derived arginase in cancer immune response. Front Immunol (2020) 11:938. doi: 10.3389/fimmu.2020.00938
73. Newton RH, Shrestha S, Sullivan JM, Yates KB, Compeer EB, Ron-Harel N, et al. Maintenance of CD4 T cell fitness through regulation of Foxo1. Nat Immunol (2018) 19:838–48. doi: 10.1038/s41590-018-0157-4
74. Hamilton SE, Jameson SC. CD8 T cell quiescence revisited. Trends Immunol (2012) 33:224–30. doi: 10.1016/j.it.2012.01.007
75. Dong L, He Y, Zhou S, Cao Y, Li Y, Bi Y, et al. HIF1α-dependent metabolic signals control the differentiation of follicular helper T cells. Cells (2019) 8:1450. doi: 10.3390/cells8111450
76. Wang F, Zhang S, Jeon R, Vuckovic I, Jiang X, Lerman A, et al. Interferon gamma induces reversible metabolic reprogramming of M1 macrophages to sustain cell viability and pro-inflammatory activity. EBioMedicine (2018) 30:303–16. doi: 10.1016/j.ebiom.2018.02.009
77. Freemerman AJ, Johnson AR, Sacks GN, Milner JJ, Kirk EL, Troester MA, et al. Metabolic reprogramming of macrophages: glucose transporter 1 (GLUT1)-mediated glucose metabolism drives a proinflammatory phenotype. J Biol Chem (2014) 289:7884–96. doi: 10.1074/jbc.M113.522037
78. Keating SE, Zaiatz-Bittencourt V, Loftus RM, Keane C, Brennan K, Finlay DK, et al. Metabolic reprogramming supports IFN-γ production by CD56bright NK cells. J Immunol (2016) 196:2552–60. doi: 10.4049/jimmunol.1501783
79. Donnelly RP, Loftus RM, Keating SE, Liou KT, Biron CA, Gardiner CM, et al. mTORC1-dependent metabolic reprogramming is a prerequisite for NK cell effector function. J Immunol (2014) 193:4477–84. doi: 10.4049/jimmunol.1401558
80. Mirzaei R, Zamani F, Hajibaba M, Rasouli-Saravani A, Noroozbeygi M, Gorgani M, et al. The pathogenic, therapeutic and diagnostic role of exosomal microrna in the autoimmune diseases. J Neuroimmunol (2021) 358:577640. doi: 10.1016/j.jneuroim.2021.577640
81. Mirzaei R, Babakhani S, Ajorloo P, Ahmadi RH, Hosseini-Fard SR, Keyvani H, et al. The emerging role of exosomal miRNAs as a diagnostic and therapeutic biomarker in mycobacterium tuberculosis infection. Mol Med (2021) 27:1–31. doi: 10.1186/s10020-021-00296-1
82. Mirzaei R, Mohammadzadeh R, Mirzaei H, Sholeh M, Karampoor S, Abdi M, et al. Role of microRNAs in staphylococcus aureus infection: potential biomarkers and mechanism. IUBMB Life (2020) 72:1856–69. doi: 10.1002/iub.2325
83. Kazemi S, Mirzaei R, Sholeh M, Karampoor S, Keramat F, Saidijam M, et al. microRNAs in human brucellosis: a promising therapeutic approach and biomarker for diagnosis and treatment. Immunity Inflamm Dis (2021) 9:1209–18. doi: 10.1002/iid3.519
84. Bouzari B, Mohammadi S, Bokov DO, Krasnyuk II, Hosseini-Fard SR, Hajibaba M, et al. Angioregulatory role of miRNAs and exosomal miRNAs in glioblastoma pathogenesis. Biomed Pharmacot (2022) 148:112760. doi: 10.1016/j.biopha.2022.112760
85. Mirzaei R, Mahdavi F, Badrzadeh F, Hosseini-Fard SR, Heidary M, Jeda AS, et al. The emerging role of microRNAs in the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection. Int Immunopharmacol (2021) 90:107204. doi: 10.1016/j.intimp.2020.107204
86. Hatziapostolou M, Polytarchou C, Iliopoulos D. miRNAs link metabolic reprogramming to oncogenesis. Trends Endocrinol Metab (2013) 24:361–73. doi: 10.1016/j.tem.2013.03.002
87. Cannistraci A, Hascoet P, Ali A, Mundra P, Clarke N, Pavet V, et al. MiR-378a inhibits glucose metabolism by suppressing GLUT1 in prostate cancer. Oncogene (2022) 41:1445–55. doi: 10.1038/s41388-022-02178-0
88. Xiao X, Huang X, Ye F, Chen B, Song C, Wen J, et al. The miR-34a-LDHA axis regulates glucose metabolism and tumor growth in breast cancer. Sci Rep (2016) 6:1–9. doi: 10.1038/srep21735
89. He Y, Deng F, Zhao S, Zhong S, Zhao J, Wang D, et al. Analysis of miRNA–mRNA network reveals miR-140-5p as a suppressor of breast cancer glycolysis via targeting GLUT1. Epigenomics (2019) 11:1021–36. doi: 10.2217/epi-2019-0072
90. He M, Jin Q, Chen C, Liu Y, Ye X, Jiang Y, et al. The miR-186-3p/EREG axis orchestrates tamoxifen resistance and aerobic glycolysis in breast cancer cells. Oncogene (2019) 38:5551–65. doi: 10.1038/s41388-019-0817-3
91. Zhang LF, Lou JT, Lu MH, Gao C, Zhao S, Li B, et al. Suppression of miR-199a maturation by HuR is crucial for hypoxia-induced glycolytic switch in hepatocellular carcinoma. EMBO J (2015) 34:2671–85. doi: 10.15252/embj.201591803
92. Jin F, Wang Y, Zhu Y, Li S, Liu Y, Chen C, et al. The miR-125a/HK2 axis regulates cancer cell energy metabolism reprogramming in hepatocellular carcinoma. Sci Rep (2017) 7:1–12. doi: 10.1038/s41598-017-03407-3
93. Zhu Y, Wu G, Yan W, Zhan H, Sun P. miR-146b-5p regulates cell growth, invasion, and metabolism by targeting PDHB in colorectal cancer. Am J Cancer Res (2017) 7:1136. doi: 10.1152/ajpcell.00554.2007
94. Guo X, Zhu Y, Hong X, Zhang M, Qiu X, Wang Z, et al. miR-181d and c-myc-mediated inhibition of CRY2 and FBXL3 reprograms metabolism in colorectal cancer. Cell Death Dis (2017) 8:e2958–8. doi: 10.1038/cddis.2017.300
95. Rui G, Gang S, Xiao-fei Y, Xin L, Jian Z, Jian X, et al. SRPX2 regulates colon cancer cell metabolism by miR-192/215 via PI3K-akt. Материалы IV Петербургского международного онкологического форума" Белые ночи 2018" (2018) 478–8. doi: 10.1242/jcs.02648
96. Jin F, Yang R, Wei Y, Wang D, Zhu Y, Wang X, et al. HIF-1α-induced miR-23a∼ 27a∼ 24 cluster promotes colorectal cancer progression via reprogramming metabolism. Cancer Lett (2019) 440:211–22. doi: 10.1016/j.canlet.2018.10.025
97. Cui M, Wang Y, Sun B, Xiao Z, Ye L, Zhang X. MiR-205 modulates abnormal lipid metabolism of hepatoma cells via targeting acyl-CoA synthetase long-chain family member 1 (ACSL1) mRNA. Biochem Biophys Res Commun (2014) 444:270–5. doi: 10.1016/j.bbrc.2014.01.051
98. Ni Y, Yang Y, Ran J, Zhang L, Yao M, Liu Z, et al. miR-15a-5p inhibits metastasis and lipid metabolism by suppressing histone acetylation in lung cancer. Free Radical Biol Med (2020) 161:150–62. doi: 10.1016/j.freeradbiomed.2020.10.009
99. Gharib E, Nasri Nasrabadi P, Reza Zali M. MiR-497-5p mediates starvation-induced death in colon cancer cells by targeting acyl-CoA synthetase-5 and modulation of lipid metabolism. J Cell Physiol (2020) 235:5570–89. doi: 10.1002/jcp.29488
100. Koufaris C, Valbuena G, Pomyen Y, Tredwell G, Nevedomskaya E, Lau C-H, et al. Systematic integration of molecular profiles identifies miR-22 as a regulator of lipid and folate metabolism in breast cancer cells. Oncogene (2016) 35:2766–76. doi: 10.1038/onc.2015.333
101. Ni K, Wang D, Xu H, Mei F, Wu C, Liu Z, et al. miR-21 promotes non-small cell lung cancer cells growth by regulating fatty acid metabolism. Cancer Cell Int (2019) 19:1–10. doi: 10.1186/s12935-019-0941-8
102. Li H-J, Li X, Pang H, Pan J-J, Xie X-J, Chen W. Long non-coding RNA UCA1 promotes glutamine metabolism by targeting miR-16 in human bladder cancer. Japan J Clin Oncol (2015) 45:1055–63. doi: 10.1093/jjco/hyv132
103. Zeng B, Ye H, Chen J, Cheng D, Cai C, Chen G, et al. LncRNA TUG1 sponges miR-145 to promote cancer progression and regulate glutamine metabolism via Sirt3/GDH axis. Oncotarget (2017) 8:113650. doi: 10.18632/oncotarget.21922
104. Gao P, Tchernyshyov I, Chang T-C, Lee Y-S, Kita K, Ochi T, et al. C-myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature (2009) 458:762–5. doi: 10.1038/nature07823
105. Li J, Li X, Wu L, Pei M, Li H, Jiang Y. miR-145 inhibits glutamine metabolism through c-myc/GLS1 pathways in ovarian cancer cells. Cell Biol Int (2019) 43:921–30. doi: 10.1002/cbin.11182
106. Zhang T, Zhang Z, Li F, Ping Y, Qin G, Zhang C, et al. miR-143 regulates memory T cell differentiation by reprogramming T cell metabolism. J Immunol (2018) 201:2165–75. doi: 10.4049/jimmunol.1800230
107. Zhao E, Maj T, Kryczek I, Li W, Wu K, Zhao L, et al. Cancer mediates effector T cell dysfunction by targeting microRNAs and EZH2 via glycolysis restriction. Nat Immunol (2016) 17:95–103. doi: 10.1038/ni.3313
108. Liu S, Wang H, Guo W, Zhou X, Shu Y, Liu H, et al. MiR-652-5p elevated glycolysis level by targeting TIGAR in T-cell acute lymphoblastic leukemia. Cell Death Dis (2022) 13:1–8. doi: 10.1038/s41419-022-04600-7
109. Ping W, Senyan H, Li G, Yan C, Long L. Increased lactate in gastric cancer tumor-infiltrating lymphocytes is related to impaired T cell function due to miR-34a deregulated lactate dehydrogenase a. Cell Physiol Biochem (2018) 49:828–36. doi: 10.1159/000493110
110. Zhihua Y, Yulin T, Yibo W, Wei D, Yin C, Jiahao X, et al. Hypoxia decreases macrophage glycolysis and M1 percentage by targeting microRNA-30c and mTOR in human gastric cancer. Cancer Sci (2019) 110:2368–77. doi: 10.1111/cas.14110
111. Mirzaei H, Hamblin MR. Regulation of glycolysis by non-coding RNAs in cancer: Switching on the warburg effect. Mol Ther - Oncolyt (2020) 19:218–39. doi: 10.1016/j.omto.2020.10.003
112. Singh PK, Mehla K, Hollingsworth MA, Johnson KR. Regulation of aerobic glycolysis by microRNAs in cancer. Mol Cell Pharmacol (2011) 3:125–34. doi: 10.1053/j.gastro.2015.07.050
113. Ahmad A, Aboukameel A, Kong D, Wang Z, Sethi S, Chen W, et al. Phosphoglucose isomerase/autocrine motility factor mediates epithelial-mesenchymal transition regulated by miR-200 in breast cancer cells. Cancer Res (2011) 71:3400–9. doi: 10.1158/0008-5472.CAN-10-0965
114. Taguchi A, Yanagisawa K, Tanaka M, Cao K, Matsuyama Y, Goto H, et al. Identification of hypoxia-inducible factor-1 alpha as a novel target for miR-17-92 microRNA cluster. Cancer Res (2008) 68:5540–5. doi: 10.1158/0008-5472.CAN-07-6460
115. Krüger M, Moser M, Ussar S, Thievessen I, Luber CA, Forner F, et al. SILAC mouse for quantitative proteomics uncovers kindlin-3 as an essential factor for red blood cell function. Cell (2008) 134:353–64. doi: 10.1016/j.cell.2008.05.033
116. Jiang S, Zhang LF, Zhang HW, Hu S, Lu MH, Liang S, et al. A novel miR-155/miR-143 cascade controls glycolysis by regulating hexokinase 2 in breast cancer cells. EMBO J (2012) 31:1985–98. doi: 10.1038/emboj.2012.45
117. Peschiaroli A, Giacobbe A, Formosa A, Markert E, Bongiorno-Borbone L, Levine A, et al. miR-143 regulates hexokinase 2 expression in cancer cells. Oncogene (2013) 32:797–802. doi: 10.1038/onc.2012.100
118. Gregersen LH, Jacobsen A, Frankel LB, Wen J, Krogh A, Lund AH. MicroRNA-143 down-regulates hexokinase 2 in colon cancer cells. BMC Cancer (2012) 12:232. doi: 10.1186/1471-2407-12-232
119. Yecies JL, Manning BD. mTOR links oncogenic signaling to tumor cell metabolism. J Mol Med (Berl) (2011) 89:221–8. doi: 10.1007/s00109-011-0726-6
120. Buller CL, Loberg RD, Fan MH, Zhu Q, Park JL, Vesely E, et al. 3rd, a GSK-3/TSC2/mTOR pathway regulates glucose uptake and GLUT1 glucose transporter expression. Am J Physiol Cell Physiol (2008) 295:C836–43. doi: 10.1152/ajpcell.00554.2007
121. Sun Q, Chen X, Ma J, Peng H, Wang F, Zha X, et al. Mammalian target of rapamycin up-regulation of pyruvate kinase isoenzyme type M2 is critical for aerobic glycolysis and tumor growth. Proc Natl Acad Sci USA (2011) 108:4129–34. doi: 10.1073/pnas.1014769108
122. Dekanty A, Lavista-Llanos S, Irisarri M, Oldham S, Wappner P. The insulin-PI3K/TOR pathway induces a HIF-dependent transcriptional response in drosophila by promoting nuclear localization of HIF-alpha/Sima. J Cell Sci (2005) 118:5431–41. doi: 10.1242/jcs.02648
123. Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer (2003) 3:721–32. doi: 10.1038/nrc1187
124. Nagaraja AK, Creighton CJ, Yu Z, Zhu H, Gunaratne PH, Reid JG, et al. A link between mir-100 and FRAP1/mTOR in clear cell ovarian cancer. Mol Endocrinol (Baltimore Md) (2010) 24:447–63. doi: 10.1007/s11060-012-1027-9
125. Fornari F, Milazzo M, Chieco P, Negrini M, Calin GA, Grazi GL, et al. MiR-199a-3p regulates mTOR and c-met to influence the doxorubicin sensitivity of human hepatocarcinoma cells. Cancer Res (2010) 70:5184–93. doi: 10.1158/0008-5472.CAN-10-0145
126. Guo C, Sah JF, Beard L, Willson JK, Markowitz SD, Guda K, et al. miR-126, suppresses the growth of neoplastic cells by targeting phosphatidylinositol 3-kinase signaling and is frequently lost in colon cancers. Genes Chromosomes Cancer (2008) 47:939–46. doi: 10.1002/gcc.20596
127. Pineau P, Volinia S, McJunkin K, Marchio A, Battiston C, Terris B, et al. miR-221 overexpression contributes to liver tumorigenesis. Proc Natl Acad Sci USA (2010) 107:264–9. doi: 10.1073/pnas.0907904107
128. Poy MN, Eliasson L, Krutzfeldt J, Kuwajima S, Ma X, Macdonald PE, et al. A pancreatic islet-specific microRNA regulates insulin secretion. Nature (2004) 432:226–30. doi: 10.1038/nature03076
129. El Ouaamari A, Baroukh N, Martens GA, Lebrun P, Pipeleers D, Van Obberghen E. miR-375 targets 3′-phosphoinositide–dependent protein kinase-1 and regulates glucose-induced biological responses in pancreatic β-cells. Diabetes (2008) 57:2708–17. doi: 10.2337/db07-1614
130. Bloomston M, Frankel WL, Petrocca F, Volinia S, Alder H, Hagan JP, et al. MicroRNA expression patterns to differentiate pancreatic adenocarcinoma from normal pancreas and chronic pancreatitis. Jama (2007) 297:1901–8. doi: 10.1001/jama.297.17.1901
131. Lee EJ, Gusev Y, Jiang J, Nuovo GJ, Lerner MR, Frankel WL, et al. Expression profiling identifies microRNA signature in pancreatic cancer. Int J Cancer (2007) 120:1046–54. doi: 10.1002/ijc.22394
132. Jin L, Zhou Y. Crucial role of the pentose phosphate pathway in malignant tumors. Oncol Lett (2019) 17:4213–21. doi: 10.3892/ol.2019.10112
133. Riganti C, Gazzano E, Polimeni M, Aldieri E, Ghigo D. The pentose phosphate pathway: an antioxidant defense and a crossroad in tumor cell fate. Free Radical Biol Med (2012) 53:421–36. doi: 10.1016/j.freeradbiomed.2012.05.006
134. Quirico L, Orso F, Cucinelli S, Paradzik M, Natalini D, Centonze G, et al. miRNA-guided reprogramming of glucose and glutamine metabolism and its impact on cell adhesion/migration during solid tumor progression. Cell Mol Life Sci CMLS (2022) 79:216–6. doi: 10.1007/s00018-022-04228-y
135. Singh A, Happel C, Manna SK, Acquaah-Mensah G, Carrerero J, Kumar S, et al. Transcription factor NRF2 regulates miR-1 and miR-206 to drive tumorigenesis. J Clin Invest (2013) 123:2921–34. doi: 10.1172/JCI66353
136. Cui J, Pan Y, Wang J, Liu Y, Wang H, Li H. MicroRNA-206 suppresses proliferation and predicts poor prognosis of HR-HPV-positive cervical cancer cells by targeting G6PD. Oncol Lett (2018) 16:5946–52. doi: 10.3892/ol.2018.9326
137. Hu T, Chang YF, Xiao Z, Mao R, Tong J, Chen B, et al. miR-1 inhibits progression of high-risk papillomavirus-associated human cervical cancer by targeting G6PD. Oncotarget (2016) 7:86103–16. doi: 10.18632/oncotarget.13344
138. He C, Yang J, Ding J, Li S, Wu H, Xiong Y, et al. Downregulation of glucose−6−phosphate dehydrogenase by microRNA−1 inhibits the growth of pituitary tumor cells. Oncol Rep (2018) 40:3533–42. doi: 10.1111/j.1742-4658.2012.08644.x
139. Qiu Z, Guo W, Wang Q, Chen Z, Huang S, Zhao F, et al. MicroRNA-124 reduces the pentose phosphate pathway and proliferation by targeting PRPS1 and RPIA mRNAs in human colorectal cancer cells. Gastroenterology (2015) 149:1587–1598.e11. doi: 10.1053/j.gastro.2015.07.050
140. Suriya Muthukumaran N, Velusamy P, Akino Mercy CS, Langford D. MicroRNAs as regulators of cancer cell energy metabolism. J Pers Med (2022) 12(8):1329. doi: 10.3390/jpm12081329
141. Chan B, VanderLaan PA, Sukhatme VP. 6-phosphogluconate dehydrogenase regulates tumor cell migration in vitro by regulating receptor tyrosine kinase c-met. Biochem Biophys Res Commun (2013) 439:247–51. doi: 10.1016/j.bbrc.2013.08.048
142. Zheng W, Feng Q, Liu J, Guo Y, Gao L, Li R, et al. Inhibition of 6-phosphogluconate dehydrogenase reverses cisplatin resistance in ovarian and lung cancer. Front Pharmacol (2017) 8:421. doi: 10.3389/fphar.2017.00421
143. Zhang Z, Zhu H, Li Q, Gao W, Zang D, Su W, et al. Gene expression profiling of tricarboxylic acid cycle and one carbon metabolism related genes for prognostic risk signature of colon carcinoma. Front Genet (2021) 12. doi: 10.3389/fgene.2021.647152
144. Tomasetti M, Amati M, Santarelli L, Neuzil J. MicroRNA in metabolic re-programming and their role in tumorigenesis. Int J Mol Sci (2016) 17:754. doi: 10.3390/ijms17050754
145. Rathore MG, Saumet A, Rossi JF, de Bettignies C, Tempé D, Lecellier CH, et al. The NF-κB member p65 controls glutamine metabolism through miR-23a. Int J Biochem Cell Biol (2012) 44:1448–56. doi: 10.1016/j.biocel.2012.05.011
146. DeBerardinis RJ, Cheng T. Q's next: the diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene (2010) 29:313–24. doi: 10.1038/onc.2009.358
147. Kennedy KM, Dewhirst MW. Tumor metabolism of lactate: the influence and therapeutic potential for MCT and CD147 regulation. Future Oncol (London England) (2010) 6:127–48. doi: 10.2217/fon.09.145
148. Semenza GL. HIF-1: upstream and downstream of cancer metabolism. Curr Opin Genet Dev (2010) 20:51–6. doi: 10.1016/j.gde.2009.10.009
149. Hermeking H. MicroRNAs in the p53 network: micromanagement of tumour suppression. Nat Rev Cancer (2012) 12:613–26. doi: 10.1038/nrc3318
150. Chu B, Wu T, Miao L, Mei Y, Wu M. MiR-181a regulates lipid metabolism via IDH1. Sci Rep (2015) 5:8801. doi: 10.1038/srep08801
151. Tanaka H, Sasayama T, Tanaka K, Nakamizo S, Nishihara M, Mizukawa K, et al. MicroRNA-183 upregulates HIF-1α by targeting isocitrate dehydrogenase 2 (IDH2) in glioma cells. J Meuro-oncol (2013) 111:273–83. doi: 10.15252/embr.201643068
152. Eichner LJ, Perry MC, Dufour CR, Bertos N, Park M, St-Pierre J, et al. miR-378(∗) mediates metabolic shift in breast cancer cells via the PGC-1β/ERRγ transcriptional pathway. Cell Metab (2010) 12:352–61. doi: 10.1016/j.cmet.2010.09.002
153. Safdar A, Abadi A, Akhtar M, Hettinga BP, Tarnopolsky MA. miRNA in the regulation of skeletal muscle adaptation to acute endurance exercise in C57Bl/6J male mice. PloS One (2009) 4:e5610. doi: 10.1371/journal.pone.0005610
154. Chen B, Liu Y, Jin X, Lu W, Liu J, Xia Z, et al. MicroRNA-26a regulates glucose metabolism by direct targeting PDHX in colorectal cancer cells. BMC Cancer (2014) 14:1–10. doi: 10.1186/1471-2407-14-443
155. Stacpoole PW. The pyruvate dehydrogenase complex as a therapeutic target for age-related diseases. Aging Cell (2012) 11:371–7. doi: 10.1111/j.1474-9726.2012.00805.x
156. de la Cruz-López KG, Castro-Muñoz LJ, Reyes-Hernández DO, García-Carrancá A, Manzo-Merino J. Lactate in the regulation of tumor microenvironment and therapeutic approaches. Front Oncol (2019) 9. doi: 10.3389/fonc.2019.01143
157. Brooks GA. The science and translation of lactate shuttle theory. Cell Metab (2018) 27:757–85. doi: 10.1016/j.cmet.2018.03.008
158. Wei Z, Cui L, Mei Z, Liu M, Zhang D. miR-181a mediates metabolic shift in colon cancer cells via the PTEN/AKT pathway. FEBS Lett (2014) 588:1773–9. doi: 10.1016/j.febslet.2014.03.037
159. Martinez-Outschoorn UE, Prisco M, Ertel A, Tsirigos A, Lin Z, Pavlides S, et al. Ketones and lactate increase cancer cell “stemness,” driving recurrence, metastasis and poor clinical outcome in breast cancer: achieving personalized medicine via metabolo-genomics. Cell Cycle (Georgetown Tex.) (2011) 10:1271–86. doi: 10.4161/cc.10.8.15330
160. Pullen TJ, da Silva Xavier G, Kelsey G, Rutter GA. miR-29a and miR-29b contribute to pancreatic beta-cell-specific silencing of monocarboxylate transporter 1 (Mct1). Mol Cell Biol (2011) 31:3182–94. doi: 10.1128/MCB.01433-10
161. Li KK, Pang JC, Ching AK, Wong CK, Kong X, Wang Y, et al. miR-124 is frequently down-regulated in medulloblastoma and is a negative regulator of SLC16A1. Hum Pathol (2009) 40:1234–43. doi: 10.1016/j.humpath.2009.02.003
162. Kirk P, Wilson MC, Heddle C, Brown MH, Barclay AN, Halestrap AP. CD147 is tightly associated with lactate transporters MCT1 and MCT4 and facilitates their cell surface expression. EMBO J (2000) 19:3896–904. doi: 10.1093/emboj/19.15.3896
163. Fu TY, Chang CC, Lin CT, Lai CH, Peng SY, Ko YJ, et al. Let-7b-mediated suppression of basigin expression and metastasis in mouse melanoma cells. Exp Cell Res (2011) 317:445–51. doi: 10.1016/j.yexcr.2010.11.004
164. Santos CR, Schulze A. Lipid metabolism in cancer. FEBS J (2012) 279:2610–23. doi: 10.1111/j.1742-4658.2012.08644.x
165. Fernández-Hernando C, Suárez Y, Rayner KJ, Moore KJ. MicroRNAs in lipid metabolism. Curr Opin Lipidol (2011) 22:86–92. doi: 10.1097/MOL.0b013e3283428d9d
166. Lynn FC. Meta-regulation: microRNA regulation of glucose and lipid metabolism. Trends Endocrinol Metab (2009) 20:452–9. doi: 10.1016/j.tem.2009.05.007
167. Esau C, Davis S, Murray SF, Yu XX, Pandey SK, Pear M, et al. miR-122 regulation of lipid metabolism revealed by in vivo antisense targeting. Cell Metab (2006) 3:87–98. doi: 10.1016/j.cmet.2006.01.005
168. Tsai WC, Hsu SD, Hsu CS, Lai TC, Chen SJ, Shen R, et al. MicroRNA-122 plays a critical role in liver homeostasis and hepatocarcinogenesis. J Clin Invest (2012) 122:2884–97. doi: 10.1172/JCI63455
169. Yoo HC, Yu YC, Sung Y, Han JM. Glutamine reliance in cell metabolism. Exp Mol Med (2020) 52:1496–516. doi: 10.1038/s12276-020-00504-8
170. Metallo CM, Gameiro PA, Bell EL, Mattaini KR, Yang J, Hiller K, et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature (2011) 481:380–4. doi: 10.1038/nature04431
171. Jin L, Alesi GN, Kang S. Glutaminolysis as a target for cancer therapy. Oncogene (2016) 35:3619–25. doi: 10.1038/onc.2015.447
172. Dong J, Xiao D, Zhao Z, Ren P, Li C, Hu Y, et al. Epigenetic silencing of microRNA-137 enhances ASCT2 expression and tumor glutamine metabolism. Oncogenesis (2017) 6:e356. doi: 10.1038/oncsis.2017.59
173. Liu Z, Wang J, Li Y, Fan J, Chen L, Xu R. MicroRNA-153 regulates glutamine metabolism in glioblastoma through targeting glutaminase. Tumour Biol J Int Soc Oncodevelopment Biol Med (2017) 39:1010428317691429. doi: 10.1177/1010428317691429
174. Chang X, Zhu W, Zhang H, Lian S. Sensitization of melanoma cells to temozolomide by overexpression of microRNA 203 through direct targeting of glutaminase-mediated glutamine metabolism. Clin Exp Dermatol (2017) 42:614–21. doi: 10.1111/ced.13119
175. Anderton B, Camarda R, Balakrishnan S, Balakrishnan A, Kohnz RA, Lim L, et al. MYC-driven inhibition of the glutamate-cysteine ligase promotes glutathione depletion in liver cancer EMBO Rep (2017) 18:569–85. doi: 10.15252/embr.201643068
176. Son J, Lyssiotis CA, Ying H, Wang X, Hua S, Ligorio M, et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature (2013) 496:101–5. doi: 10.1038/nature12040
177. Wang J, Wang B, Ren H, Chen W. miR-9-5p inhibits pancreatic cancer cell proliferation, invasion and glutamine metabolism by targeting GOT1. Biochem Biophys Res Commun (2019) 509:241–8. doi: 10.1016/j.bbrc.2018.12.114
178. Sengupta D, Cassel T, Teng KY, Aljuhani M, Chowdhary VK, Hu P, et al. Regulation of hepatic glutamine metabolism by miR-122. Mol Metab (2020) 34:174–86. doi: 10.1016/j.molmet.2020.01.003
179. Sartori T, Galvão dos Santos G, Nogueira-Pedro A, Makiyama E, Rogero MM, Borelli P, et al. Effects of glutamine, taurine and their association on inflammatory pathway markers in macrophages. Inflammopharmacology (2018) 26:829–38. doi: 10.1007/s10787-017-0406-4
180. Zhang P, Cao L, Zhou R, Yang X, Wu M. The lncRNA Neat1 promotes activation of inflammasomes in macrophages. Nat Commun (2019) 10:1–17. doi: 10.1038/s41467-019-09482-6
181. Park JE, Dutta B, Tse SW, Gupta N, Tan CF, Low JK, et al. Hypoxia-induced tumor exosomes promote M2-like macrophage polarization of infiltrating myeloid cells and microRNA-mediated metabolic shift. Oncogene (2019) 38:5158–73. doi: 10.1038/s41388-019-0782-x
182. Xu Z, Zhao L, Zhu L-Y, He M, Zheng L, Wu Y. MicroRNA-17, 20a regulates the proangiogenic function of tumor-associated macrophages via targeting hypoxia-inducible factor 2α. PloS One (2013) 8:e77890. doi: 10.1038/mtna.2016.72
183. Quinn JJ, Chang HY. Unique features of long non-coding RNA biogenesis and function. Nat Rev Genet (2016) 17:47–62. doi: 10.1038/nrg.2015.10
184. Henao-Mejia J, Williams A, Goff LA, Staron M, Licona-Limón P, Kaech SM, et al. The microRNA miR-181 is a critical cellular metabolic rheostat essential for NKT cell ontogenesis and lymphocyte development and homeostasis. Immunity (2013) 38:984–97. doi: 10.1016/j.immuni.2013.02.021
185. Warth SC, Hoefig KP, Hiekel A, Schallenberg S, Jovanovic K, Klein L, et al. Induced miR-99a expression represses mtor cooperatively with miR-150 to promote regulatory T-cell differentiation. EMBO J (2015) 34:1195–213. doi: 10.15252/embj.201489589
186. MacIver NJ, Michalek RD, Rathmell JC. Metabolic regulation of T lymphocytes. Annu Rev Immunol (2013) 31:259–83. doi: 10.1146/annurev-immunol-032712-095956
187. Wells AC, Daniels KA, Angelou CC, Fagerberg E, Burnside AS, Markstein M, et al. Modulation of let-7 miRNAs controls the differentiation of effector CD8 T cells. eLife (2017) 6:e26398. doi: 10.7554/eLife.26398
188. Kulyté A, Lorente-Cebrián S, Gao H, Mejhert N, Agustsson T, Arner P, et al. MicroRNA profiling links miR-378 to enhanced adipocyte lipolysis in human cancer cachexia. Am J Physiol Endocrinol Metab (2014) 306:E267–74. doi: 10.1152/ajpendo.00249.2013
189. Munn DH, Mellor AL. IDO in the tumor microenvironment: Inflammation, counter-regulation, and tolerance. Trends Immunol (2016) 37:193–207. doi: 10.1016/j.it.2016.01.002
190. Zhao X, Qu J, Sun Y, Wang J, Liu X, Wang F, et al. Prognostic significance of tumor-associated macrophages in breast cancer: a meta-analysis of the literature. Oncotarget (2017) 8:30576–86. doi: 10.18632/oncotarget.15736
191. Viré E, Brenner C, Deplus R, Blanchon L, Fraga M, Didelot C, et al. The polycomb group protein EZH2 directly controls DNA methylation. Nature (2006) 439:871–4. doi: 10.1038/nature04431
192. Cao R, Wang L, Wang H, Xia L, Erdjument-Bromage H, Tempst P, et al. Role of histone H3 lysine 27 methylation in polycomb-group silencing. Science (2002) 298:1039–43. doi: 10.1126/science.1076997
193. Tumes DJ, Onodera A, Suzuki A, Shinoda K, Endo Y, Iwamura C, et al. The polycomb protein Ezh2 regulates differentiation and plasticity of CD4(+) T helper type 1 and type 2 cells. Immunity (2013) 39:819–32. doi: 10.1016/j.immuni.2013.09.012
194. Tong Q, He S, Xie F, Mochizuki K, Liu Y, Mochizuki I, et al. Ezh2 regulates transcriptional and posttranslational expression of T-bet and promotes Th1 cell responses mediating aplastic anemia in mice. J Immunol (Baltimore Md. (2014) 1950) 192:5012–22. doi: 10.4049/jimmunol.1302943
195. Gubser PM, Bantug GR, Razik L, Fischer M, Dimeloe S, Hoenger G, et al. Rapid effector function of memory CD8+ T cells requires an immediate-early glycolytic switch. Nat Immunol (2013) 14:1064–72. doi: 10.1038/ni.2687
196. Wu H, Neilson JR, Kumar P, Manocha M, Shankar P, Sharp PA, et al. miRNA profiling of naïve, effector and memory CD8 T cells. PloS One (2007) 2:e1020. doi: 10.1016/j.cmet.2006.05.009
197. Khan AA, Penny LA, Yuzefpolskiy Y, Sarkar S, Kalia V. MicroRNA-17∼ 92 regulates effector and memory CD8 T-cell fates by modulating proliferation in response to infections. Blood J Am Soc Hematol (2013) 121:4473–83. doi: 10.1038/s41467-020-17873-3
198. Chakrabarti R, Jung CY, Lee T-P, Liu H, Mookerjee BK. Changes in glucose transport and transporter isoforms during the activation of human peripheral blood lymphocytes by phytohemagglutinin. J Immunol (1994) 152:2660–8. doi: 10.1016/j.semcancer.2008.03.004
199. Miao Y, Zhang L-F, Guo R, Liang S, Zhang M, Shi S, et al. 18F-FDG PET/CT for monitoring the response of breast cancer to miR-143-based therapeutics by targeting tumor glycolysis. Mol Therapy-Nucleic Acids (2016) 5:e357. doi: 10.1038/mtna.2016.72
200. Kawalekar OU, O’Connor RS, Fraietta JA, Guo L, McGettigan SE, Posey AD Jr., et al. Distinct signaling of coreceptors regulates specific metabolism pathways and impacts memory development in CAR T cells. Immunity (2016) 44:380–90. doi: 10.1016/j.immuni.2016.01.021
201. Kishton RJ, Barnes CE, Nichols AG, Cohen S, Gerriets VA, Siska PJ, et al. AMPK is essential to balance glycolysis and mitochondrial metabolism to control T-ALL cell stress and survival. Cell Metab (2016) 23:649–62. doi: 10.1016/j.cmet.2016.03.008
202. Hu J, Wang T, Xu J, Wu S, Wang L, Su H, et al. WEE1 inhibition induces glutamine addiction in T-cell acute lymphoblastic leukemia. Haematologica (2021) 106:1816–27. doi: 10.1007/s11010-019-03571-2
203. Matthijssens F, Sharma ND, Nysus M, Nickl CK, Kang H, Perez DR, et al. RUNX2 regulates leukemic cell metabolism and chemotaxis in high-risk T cell acute lymphoblastic leukemia. J Clin Invest (2021) 131(6):e141566. doi: 10.1172/JCI141566
204. Wynn TA, Chawla A, Pollard JW. Macrophage biology in development, homeostasis and disease. Nature (2013) 496:445–55. doi: 10.1038/nature12034
205. Coffelt SB, Hughes R, Lewis CE. Tumor-associated macrophages: effectors of angiogenesis and tumor progression. Biochim Biophys Acta (2009) 1796:11–8. doi: 10.1016/j.bbcan.2009.02.004
206. Nelson MC, O'Connell RM. MicroRNAs: At the interface of metabolic pathways and inflammatory responses by macrophages. Front Immunol (2020) 11:1797–7. doi: 10.3389/fimmu.2020.01797
207. Kawai T, Akira S. TLR signaling. Cell Death Different (2006) 13:816–25. doi: 10.1038/sj.cdd.4401850
208. Newton K, Dixit VM. Signaling in innate immunity and inflammation. Cold Spring Harbor Perspect Biol (2012) 4(3):a006049. doi: 10.1101/cshperspect.a006049
209. Biswas SK, Mantovani A. Orchestration of metabolism by macrophages. Cell Metab (2012) 15:432–7. doi: 10.1016/j.cmet.2011.11.013
210. Rodríguez-Prados JC, Través PG, Cuenca J, Rico D, Aragonés J, Martín-Sanz P, et al. Substrate fate in activated macrophages: a comparison between innate, classic, and alternative activation. J Immunol (Baltimore Md. 1950) (2010) 185:605–14. doi: 10.1111/cpr.12510
211. Lunt SY, Vander Heiden MG. Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu Rev Cell Dev Biol (2011) 27:441–64. doi: 10.1146/annurev-cellbio-092910-154237
212. Krützfeldt J, Stoffel M. MicroRNAs: a new class of regulatory genes affecting metabolism. Cell Metab (2006) 4:9–12. doi: 10.1016/j.cmet.2006.05.009
213. Hoefflin R, Harlander S, Schäfer S, Metzger P, Kuo F, Schönenberger D, et al. HIF-1α and HIF-2α differently regulate tumour development and inflammation of clear cell renal cell carcinoma in mice. Nat Commun (2020) 11:1–21. doi: 10.1038/s41467-020-17873-3
214. Sica A, Larghi P, Mancino A, Rubino L, Porta C, Totaro MG, et al. Macrophage polarization in tumour progression. Semin Cancer Biol Elsevier (2008) 18:349–55. doi: 10.1016/j.semcancer.2008.03.004
215. Li J, Lu Z, Zhang Y, Xia L, Su Z. Emerging roles of non-coding RNAs in the metabolic reprogramming of tumor-associated macrophages. Immunol Lett (2021) 232:27–34. doi: 10.1016/j.imlet.2021.02.003
216. Hsu Y-L, Hung J-Y, Chang W-A, Jian S-F, Lin Y-S, Pan Y-C, et al. Hypoxic lung-cancer-derived extracellular vesicle microRNA-103a increases the oncogenic effects of macrophages by targeting PTEN. Mol Ther (2018) 26:568–81. doi: 10.1016/j.ymthe.2017.11.016
217. Wei X, Tang C, Lu X, Liu R, Zhou M, He D, et al. MiR-101 targets DUSP1 to regulate the TGF-β secretion in sorafenib inhibits macrophage-induced growth of hepatocarcinoma. Oncotarget (2015) 6:18389. doi: 10.18632/oncotarget.4089
218. Wang X, Luo G, Zhang K, Cao J, Huang C, Jiang T, et al. Hypoxic tumor-derived exosomal miR-301a mediates M2 macrophage polarization via PTEN/PI3Kγ to promote pancreatic cancer MetastasisTumor-promoting effects of hypoxic exosomal miR-301a. Cancer Res (2018) 78:4586–98. doi: 10.1158/0008-5472.CAN-17-3841
219. Bao L, Li X. MicroRNA-32 targeting PTEN enhances M2 macrophage polarization in the glioma microenvironment and further promotes the progression of glioma. Mol Cell Biochem (2019) 460:67–79. doi: 10.1007/s11010-019-03571-2
220. Zhang H, Guo X, Feng X, Wang T, Hu Z, Que X, et al. MiRNA-543 promotes osteosarcoma cell proliferation and glycolysis by partially suppressing PRMT9 and stabilizing HIF-1α protein. Oncotarget (2017) 8:2342. doi: 10.18632/oncotarget.13672
221. Guo W, Qiu Z, Wang Z, Wang Q, Tan N, Chen T, et al. MiR-199a-5p is negatively associated with malignancies and regulates glycolysis and lactate production by targeting hexokinase 2 in liver cancer. Hepatol (Baltimore Md.) (2015) 62:1132–44. doi: 10.1128/aem.45.3.748-754.1983
222. Tao T, Chen M, Jiang R, Guan H, Huang Y, Su H, et al. Involvement of EZH2 in aerobic glycolysis of prostate cancer through miR-181b/HK2 axis. Oncol Rep (2017) 37:1430–6. doi: 10.3892/or.2017.5430
223. Tang R, Yang C, Ma X, Wang Y, Luo D, Huang C, et al. MiR-let-7a inhibits cell proliferation, migration, and invasion by down-regulating PKM2 in gastric cancer. Oncotarget (2016) 7:5972–84. doi: 10.18632/oncotarget.6821
224. Taniguchi K, Sugito N, Kumazaki M, Shinohara H, Yamada N, Nakagawa Y, et al. MicroRNA-124 inhibits cancer cell growth through PTB1/PKM1/PKM2 feedback cascade in colorectal cancer. Cancer Lett (2015) 363:17–27. doi: 10.1016/j.canlet.2015.03.026
225. Wen Y-Y, Liu W-T, Sun H-R, Ge X, Shi Z-M, Wang M, et al. IGF-1-mediated PKM2/β-catenin/miR-152 regulatory circuit in breast cancer. Sci Rep (2017) 7:15897. doi: 10.1038/s41598-017-15607-y
226. Chen J, Yu Y, Chen X, He Y, Hu Q, Li H, et al. MiR-139-5p is associated with poor prognosis and regulates glycolysis by repressing PKM2 in gallbladder carcinoma. Cell Prolif (2018) 51:e12510. doi: 10.1111/cpr.12510
227. Wenes M, Shang M, Di Matteo M, Goveia J, Martín-Pérez R, Serneels J, et al. Macrophage metabolism controls tumor blood vessel morphogenesis and metastasis. Cell Metab (2016) 24:701–15. doi: 10.1016/j.cmet.2016.09.008
228. Binenbaum Y, Fridman E. Transfer of miRNA in macrophage-derived exosomes induces drug resistance in pancreatic adenocarcinoma. Cancer Res (2018) 78:5287–99. doi: 10.1158/0008-5472.CAN-18-0124
229. Ward PS, Patel J, Wise DR, Abdel-Wahab O, Bennett BD, Coller HA, et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell (2010) 17:225–34. doi: 10.1016/j.ccr.2010.01.020
230. Ryan DG, Murphy MP, Frezza C, Prag HA, Chouchani ET, O'Neill LA, et al. Coupling Krebs cycle metabolites to signalling in immunity and cancer. Nat Metab (2019) 1:16–33. doi: 10.1038/s42255-018-0014-7
231. Mills EL, Kelly B, O'Neill LAJ. Mitochondria are the powerhouses of immunity. Nat Immunol (2017) 18:488–98. doi: 10.1038/ni.3704
232. Xiang K, Jendrossek V, Matschke J. Oncometabolites and the response to radiotherapy. Radiat Oncol (2020) 15:197. doi: 10.1186/s13014-020-01638-9
233. Weng CY, Kao CX, Chang TS. Immuno-metabolism: The role of cancer niche in immune checkpoint inhibitor resistance. Int J Mol Sci (2021) 22(3)1258. doi: 10.3390/ijms22031258
234. Yi M, Xu L, Jiao Y, Luo S, Li A, Wu K. The role of cancer-derived microRNAs in cancer immune escape. J Hematol Oncol (2020) 13:25. doi: 10.1186/s13045-020-00848-8
235. Cheong JE, Sun L. Targeting the IDO1/TDO2-KYN-AhR pathway for cancer immunotherapy - challenges and opportunities. Trends Pharmacol Sci (2018) 39:307–25. doi: 10.1016/j.tips.2017.11.007
236. Michael-Marler S, Brown ML, Siebeling RJ. Eradication of Arizona hinshawii from artificially infected turtle eggs. Appl Environ Microbiol (1983) 45:748–54. doi: 10.1128/aem.45.3.748-754.1983
237. Fontcuberta J, Borrell M, Felez J, Elez I, López M, Remacha A, et al. [Changes in fibrinolysis in liver cirrhosis]. Sangre (1982) 27:727–36. doi: 10.1038/nbt.2890
238. Lou Q, Liu R, Yang X, Li W, Huang L, Wei L, et al. miR-448 targets IDO1 and regulates CD8(+) T cell response in human colon cancer. J Immunother Cancer (2019) 7:210. doi: 10.1186/s40425-019-0691-0
239. Huang Q, Xia J, Wang L, Wang X, Ma X, Deng Q, et al. miR-153 suppresses IDO1 expression and enhances CAR T cell immunotherapy. J Hematol Oncol (2018) 11:58. doi: 10.1186/s13045-018-0600-x
240. Dragomir M, Chen B, Fu X, Calin GA. Key questions about the checkpoint blockade-are microRNAs an answer? Cancer Biol Med (2018) 15:103. doi: 10.20892/j.issn.2095-3941.2018.0006
241. Mehta A, Baltimore D. MicroRNAs as regulatory elements in immune system logic. Nat Rev Immunol (2016) 16:279–94. doi: 10.1038/nri.2016.40
242. Cortez MA, Anfossi S, Ramapriyan R, Menon H, Atalar SC, Aliru M, et al. Role of miRNAs in immune responses and immunotherapy in cancer. Genes Chromosomes Cancer (2019) 58:244–53. doi: 10.1002/gcc.22725
243. Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol (2008) 26:677–704. doi: 10.1146/annurev.immunol.26.021607.090331
244. Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell (2015) 27:450–61. doi: 10.1016/j.ccell.2015.03.001
245. Qin G, Mallik S, Mitra R, Li A, Jia P, Eischen CM, et al. MicroRNA and transcription factor co-regulatory networks and subtype classification of seminoma and non-seminoma in testicular germ cell tumors. Sci Rep (2020) 10:852. doi: 10.1038/s41598-020-57834-w
246. Zhao M, Kim P, Mitra R, Zhao J, Zhao Z. TSGene 2.0: an updated literature-based knowledgebase for tumor suppressor genes. Nucleic Acids Res (2016) 44:D1023–31. doi: 10.3892/or.2015.3904
247. Yan Z, Shah PK, Amin SB, Samur MK, Huang N, Wang X, et al. Integrative analysis of gene and miRNA expression profiles with transcription factor–miRNA feed-forward loops identifies regulators in human cancers. Nucleic Acids Res (2012) 40:e135–5. doi: 10.1093/nar/gks395
248. Jiang W, Mitra R, Lin C-C, Wang Q, Cheng F, Zhao Z. Systematic dissection of dysregulated transcription factor–mirna feed-forward loops across tumor types. Briefings Bioinf (2016) 17:996–1008. doi: 10.1093/bib/bbv107
249. Chan JJ, Tay Y. Noncoding RNA: RNA regulatory networks in cancer. Int J Mol Sci (2018) 19:1310. doi: 10.3390/ijms19051310
250. Bhan A, Soleimani M, Mandal SS. Long noncoding RNA and cancer: a new paradigm. Cancer Res (2017) 77:3965–81. doi: 10.1158/0008-5472.CAN-16-2634
251. Jeck WR, Sharpless NE. Detecting and characterizing circular RNAs. Nat Biotechnol (2014) 32:453–61. doi: 10.1016/j.imlet.2021.02.003
252. Yu T, Wang Y, Fan Y, Fang N, Wang T, Xu T, et al. CircRNAs in cancer metabolism: a review. J Hematol Oncol (2019) 12:1–10. doi: 10.1038/s41590-018-0157-4
253. Lan W, Lai D, Chen Q, Wu X, Chen B, Liu J, et al. LDICDL: LncRNA-disease association identification based on collaborative deep learning. IEEE/ACM Trans Comput Biol Bioinf (2020) 5:224–30. doi: 10.1016/j.it.2012.01.007
254. Chen X, Sun L-G, Zhao Y. NCMCMDA: miRNA–disease association prediction through neighborhood constraint matrix completion. Briefings Bioinf (2021) 22:485–96. doi: 10.1038/s41577-019-0203-y
255. Salmena L, Poliseno L, Tay Y, Kats L, Pandolfi PP. A ceRNA hypothesis: the Rosetta stone of a hidden RNA language? Cell (2011) 146:353–8. doi: 10.3390/cells8111450
256. Taulli R, Loretelli C, Pandolfi PP. From pseudo-ceRNAs to circ-ceRNAs: a tale of cross-talk and competition. Nat Struct Mol Biol (2013) 20:541–3. doi: 10.1038/nsmb.2580
257. Yao R-W, Wang Y, Chen L-L. Cellular functions of long noncoding RNAs. Nat Cell Biol (2019) 21:542–51. doi: 10.1038/s41556-019-0311-8
258. Liu X-H, Sun M, Nie F-Q, Ge Y-B, Zhang E-B, Yin D-D, et al. Lnc RNA HOTAIR functions as a competing endogenous RNA to regulate HER2 expression by sponging miR-331-3p in gastric cancer. Mol Cancer (2014) 13:1–14. doi: 10.1186/1476-4598-13-92
259. Wang S-H, Ma F, Tang Z-H, Wu X-C, Cai Q, Zhang M-D, et al. Long non-coding RNA H19 regulates FOXM1 expression by competitively binding endogenous miR-342-3p in gallbladder cancer. J Exp Clin Cancer Res (2016) 35:1–12. doi: 10.1186/s13046-016-0436-6
260. Peng L, Yuan XQ, Li GC. The emerging landscape of circular RNA ciRS-7 in cancer. Oncol Rep (2015) 33:2669–74. doi: 10.3892/or.2015.3904
261. Wu L, Pan C, Wei X, Shi Y, Zheng J, Lin X, et al. lncRNA KRAL reverses 5-fluorouracil resistance in hepatocellular carcinoma cells by acting as a ceRNA against miR-141. Cell Communicat Signaling (2018) 16:1–15. doi: 10.1186/s12964-018-0260-z
262. Liu Z, Wang Y, Wang L, Yao B, Sun L, Liu R, et al. Long non-coding RNA AGAP2-AS1, functioning as a competitive endogenous RNA, upregulates ANXA11 expression by sponging miR-16-5p and promotes proliferation and metastasis in hepatocellular carcinoma. J Exp Clin Cancer Res (2019) 38:1–15. doi: 10.1186/s13046-019-1188-x
263. Huang G, Liang M, Liu H, Huang J, Li P, Wang C, et al. CircRNA hsa_circRNA_104348 promotes hepatocellular carcinoma progression through modulating miR-187-3p/RTKN2 axis and activating wnt/β-catenin pathway. Cell Death Dis (2020) 11:1–14. doi: 10.1038/s41419-020-03276-1
Keywords: MicroRNAs, cancer, metabolism, immunometabolism, immune cell
Citation: Alshahrani SH, Ibrahim YS, Jalil AT, Altoum AA, Achmad H, Zabibah RS, Gabr GA, Ramírez-Coronel AA, Alameri AA, Qasim QA, Karampoor S and Mirzaei R (2022) Metabolic reprogramming by miRNAs in the tumor microenvironment: Focused on immunometabolism. Front. Oncol. 12:1042196. doi: 10.3389/fonc.2022.1042196
Received: 12 September 2022; Accepted: 24 October 2022;
Published: 22 November 2022.
Edited by:
Juan Carlos Gallardo-Pérez, Instituto Nacional de Cardiologia Ignacio Chavez, MexicoReviewed by:
Wanchen Ning, Southern Medical University, ChinaCopyright © 2022 Alshahrani, Ibrahim, Jalil, Altoum, Achmad, Zabibah, Gabr, Ramírez-Coronel, Alameri, Qasim, Karampoor and Mirzaei. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sajad Karampoor, a2FyYW1wb3VyLnNAaXVtcy5hYy5pcg==; c2FqYWRrYXJhbXBvdXIxOTg3QGdtYWlsLmNvbQ==; Rasoul Mirzaei, cmFzdWwubWljcm85MkBnbWFpbC5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.