 Yang Shi
Yang Shi Meiqi Wang3†
Meiqi Wang3† Dan Liu
Dan Liu Saif Ullah
Saif Ullah Xing Ma
Xing Ma Huiyu Yang
Huiyu Yang Bingrong Liu
Bingrong Liu- 1Department of Gastroenterology, The First Affiliated Hospital of Zhengzhou University, Zhengzhou, China
- 2State Key Laboratory of Esophageal Cancer Prevention and Treatment, Zhengzhou University, Zhengzhou, China
- 3Academy of Medical Sciences Zhengzhou University, Zhengzhou, China
- 4Department of Nuclear Medicine, The Affiliated Cancer Hospital of Zhengzhou University & Henan Cancer Hospital, Zhengzhou, China
The tumorigenesis of esophageal carcinoma arises from transcriptional dysregulation would become exceptionally dependent on specific regulators of gene expression, which could be preferentially attributed to the larger non-coding cis-regulatory elements, i.e. super-enhancers (SEs). SEs, large genomic regulatory entity in close genomic proximity, are underpinned by control cancer cell identity. As a consequence, the transcriptional addictions driven by SEs could offer an Achilles’ heel for molecular treatments on patients of esophageal carcinoma and other types of cancer as well. In this review, we summarize the recent findings about the oncogenic SEs upon which esophageal cancer cells depend, and discuss why SEs could be seen as the hallmark of cancer, how transcriptional dependencies driven by SEs, and what opportunities could be supplied based on this cancer-specific SEs.
Introduction
Esophageal carcinoma is the fourth most common gastrointestinal cancer worldwide. The incidence and mortality rates of esophageal carcinoma accounts for 11.75% and 14.99%, respectively (1). According to statistics from the International Researches Agency of Cancer (IRAC), the mortality rate of the cancer would increase 69% in China by 2040, while the main histological type is squamous cell carcinoma (ESCC) (2, 3). The standard-of-care regimens consisted of surgery, platinum-based chemotherapy, radiotherapy, and PD-1/PD-L1 blockade therapy (4, 5), which have improved the survival rate and patient’s quality of life. However, chemoradiotherapy resistance, postoperative recurrence and unresectable advanced lesions are still impeding the long-term survival of these patients. Given that the limited actionable drivers in ESCC, more effective avenues based on the clarification of the carcinogenesis are required. Recently, it is widely recognized that the disease-associated variations are bound up with enhancer regions, especially super-enhancers (SEs), which undoubtedly provides novel insights into therapeutic maneuvers.
Enhancers refer to non-coding part of genome that activate genes expression independent of orientation, distance, and location regarding its transcription start sites (TSSs). These regulatory elements always provide binding sites for multiple transcription factors (TFs) that can also be transcribed to produce non-coding enhancer RNAs (eRNAs). More recently, cluster of enhancers located genomic proximity were identified as the unique transcriptional single entity, known as SEs. SEs are underpinned by highly abundant and orchestrated interactions with transcription apparatus and active enhancer marks (Acetylation at lysine 27, H3K27ac & Monomethylation at lysine 4, H3K4me1). Notably, SEs have been demonstrated to dictate cell identity and disease and play a key role in carcinogenesis in a broad spectrum of tumors. Recently, it has been proposed that SEs have the potential to serve as valuable prognostic and therapeutic targets in cancer.
During the past decade, various types of cancer pathogenesis have been proved to be closely associated with SEs, such as oncogenes activation, dysregulated signaling pathways, and genetic mutations. Generally, cancer-specific SEs assembly are not presented in the corresponding non-cancerous tissues, such as C-Myc, INSM1 (6) and TAL1 (7). Broadly, these SEs activate the tumorigenic signaling pathways, promoting oncogenic transcriptions, and enriched in key TFs binding motifs. Fortunately, the sensitivity of SEs to perturbation has shown the promising therapeutic vulnerability in various types of cancer, including breast cancer (8), nasopharyngeal carcinoma (9), small cell lung cancer (10), medulloblastoma (11) and esophageal carcinoma (12). The critical oncogenes of ESCC, e.g. TP63, SOX2, KLF5 and ALDH3A1, have been shown to participate in core regulatory circuitry (CRC) driven by SEs (12). Besides, the pharmacological inhibition of cyclin dependent kinase 7 (CDK7), bromodomain-containing protein 4 (BRD4) and histone deacetylases (HDACs), has been applied in esophageal carcinoma treatment (13).
Therefore, not surprisingly, deregulation of SEs is fundamental mechanism of cancer, which offers an Achilles heel for diagnostic and therapeutic maneuvers (14). This review attempts to discuss SEs’ fundamental characteristics and roles in esophageal carcinoma, which would pave the way for SE-based diagnostic and therapeutic maneuvers.
Super-enhancers: identification, organization and functions
Identification of super-enhancers
Enhancers were firstly recognized as the cis-regulatory elements from simian virus 40 (SV40), which could prominently promote rabbit β-globulin transcription in HeLa cells (15, 16). In general, enhancers activate cell-type-specific gene expression regardless of their distance, position, and orientation with respective to the cognate promoter (17, 18). The elements of enhancer are bound up with critical TFs through their tissue-specific recognition motifs, thereby functioning as the platform to integrate signaling pathways and further dictate cell lineages (19). Mechanistically, the enrichment of master TFs on enhancers results in recruiting of the subunit of the Mediator complex (Med1), RNA polymerase II (Pol II), and the basal transcription apparatus, which is organized by looping between enhancers and their cognate promoters. Additionally, enhancer regions are mainly overlapped with DNase hypersensitive sites (DHS), and the active state of enhancers are dependent on the following combinations of histone modifications: enrichment with H3K27ac, H3K4me1, and deficiency of Trimethylation at lysine 4 (H3K4me3) (Table 1) (20).

Table 1 Histone modification markers and the related functional state of the regulatory elements.
SEs were firstly identified as the unique cluster of enhancers in close genomic proximity, which were densely occupied by master TFs Oct4/Pou5f1, Sox2, Nanog (OSN), Klf4, Esrrb, and Med1 in murine embryonic stem cells (mESCs) (21, 22). The Rank Ordering of Super-Enhancers (ROSE) algorithm had been proposed to separate typical-enhancers (TEs) from SEs, whose constituent enhancers were stitched together within 12.5 kb genomic regions enriched by input-normalized level of Med1 signal.
Structural features of super-enhancers
Liquid-liquid phase separation (LLPS) is a physicochemical process by which membraneless organelles are generated in eukaryotic cells, which could compartmentalize biochemical reactions within the dense phase (23–25). It has been demonstrated that SEs are phase-separated assemblies accumulated by exceptionally high densities of master TFs, co-activators, and RNA Pol II, dictating the roles in cell identity and disease, including cancers (26). The intrinsically disordered regions (IDRs) of the co-activators (BRD4 & Med1), driven by high-valency and weak-affinity interactions, are responsible for the formation of the phase-separated condensates at SEs, which could bring those cis-elements and the cognate promoter in close three-dimensional (3D) proximity and then facilitate SEs activation (Figure 1). This physical interaction between enhancer and promoter both involving in TEs and SEs are mediated by cohesin-associated CTCF (CCCTC-binding factor) loops. Additionally, the ubiquitously expressed TF Yin Yang 1 (YY1) has been also identified as the structural regulators contributing to this loop structure (27). The disruption of this structure is likely to result in activation of oncogenes outside the neighborhood by deletion of CTCF binding site, which are consistent with a tendency of liquid phase condensates to undergo fusion.
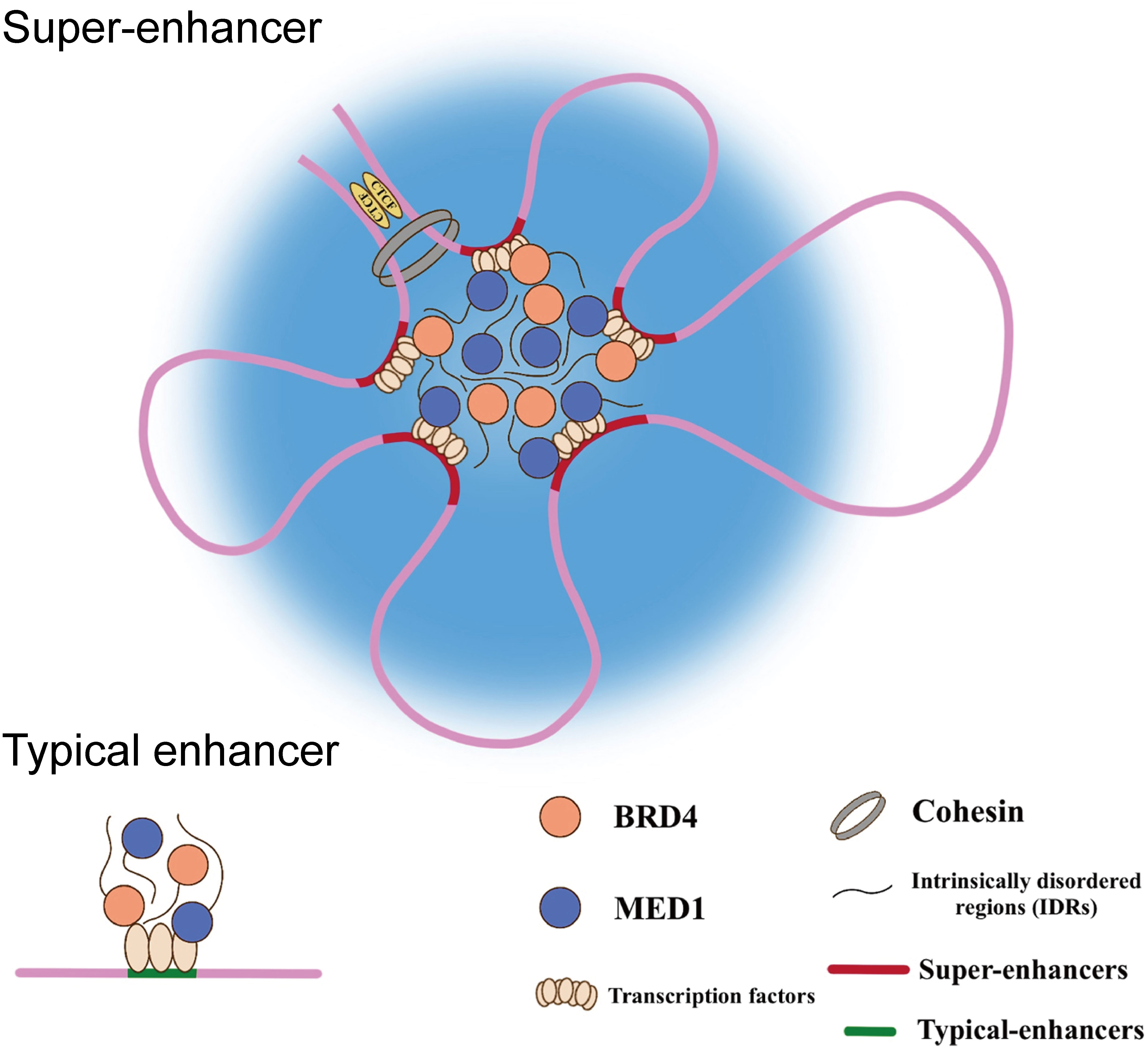
Figure 1 Schematic diagram of the phase-separated condensates at super-enhancers.
Functional properties of super-enhancers
The main features of SEs had been summarized as following Richard A. Young et al. and colleagues: (i) high-density occupancy of master TFs, co-activators and chromatin remodelers (Table 1), (ii) large genomic spanning, (iii) ability to exceptionally activate transcription, (iv) sensitivity to perturbation and (v) dictate cell identity and disease (28). Based on these observations, the oncogenic role of SEs usually caused by genetic variations are proposed to drive the transcriptional addiction in cancer.
The transcriptional activities of SEs exhibit an order-of-magnitude higher than TEs and the individual constituent enhancers within SEs, which also showing highly context-dependent manner under rigorous genetic regulation (29). And more interestingly, the cooperativity of SEs constituents is neither additively nor synergistically, suggesting that the “modus operandi” of each component under highly precise and complex regulation by other component (30).
Establishment of oncogenic super-enhancers in esophageal carcinoma
Generally, inappropriate SEs are acquired de novo during tumorigenesis compared with the counterpart normal tissues, which driving expression of the critical oncogenes. The malignant transformation and maintenance underpinned by tumorigenic SEs could be seen as one of the core tenets of cancer biology (28, 31, 32). Mechanically, the formation of oncogenic SEs may stem from (i) focal amplification, (ii) genomic rearrangements, (iii) single nucleotide polymorphisms (SNPs), (iv) disruption of topological associating domain (TADs).
Genetic variations harboring super-enhancers constituents
Somatic copy number alterations (SCNAs) are common mechanism of oncogenesis driven by SEs. For example, focal amplification peak harboring SEs identified by profiling of H3K27ac in esophageal carcinoma (chr13:73880413-74042621, about ~162 kb), which subsequently proven to activate the oncogene KLF5 expression. And these cancer-specific focal amplification peaks enriched in SEs have also been identified in several types of cancers, including head and neck squamous cell carcinoma, colorectal carcinoma, and liver hepatocellular carcinoma as well (33). Additionally, genomic rearrangements could change the natural genomic context resulting in close proximity between oncogene promoter and their SEs, which ectopically activates gene expression (34). This phenomenon is also described as “enhancer hijacking”, by which oncogenic SEs formed in colorectal carcinoma, medulloblastoma, and acute myeloid leukemia as well (11, 35, 36). Besides, SNPs have been reported to promote tumorigenesis by disrupting the activities of SEs, including acquired oncogenic or abrogating protective allele within master TFs binding sites (37, 38).
These genomic variants provide novel insights into the carcinogenesis of esophageal carcinoma, which deserves further investigation.
Hijacking of super-enhancers by topological associating domain disruption
TADs have been recognized as the self-interacting genomic region, which demonstrates higher interconnection frequency than the outer regions (39). It has become clear that the main function of TADs is to insulate promoters from distal enhancers or SEs, which conducted by binding of insulator CCCTC-binding factor (CTCF) in cooperation with cohesin complex at their TAD boundaries (40). Although TADs structures are conserved in mammalian, disruption of TADs boundaries caused by genetic or epigenetic could be convenient for abnormal interactions between enhancer/SEs and promoters, which is undoubtedly could be laid the foundation for tumorigenesis.
It has been reported that TADs boundaries disruption could be caused by recurrent mutations of CTCF and cohesin binding sites, which were identified in esophageal carcinoma as well as other types of cancers, including liver hepatocellular carcinoma, colorectal carcinoma, and gastric carcinoma (41, 42). Furthermore, the boundaries disruption is also exemplified by epigenetic dysregulation in glioma by increasing hypermethylation in CTCF site followed by its reduced binding activity (43). Modification of 3D genome by TADs disruption could activate oncogene expression driven by inadvertent SEs-promoter looping, which might be utilized as the novel candidates for targeting the oncogenic SEs in esophageal carcinoma.
Principles of SEs-driven oncogenic transcription dependencies
Transcriptional core regulatory circuitries
The ESC master TFs OSN have previously been demonstrated to bind to their own genes or those of the others in mESCs, which forms an autoregulatory feed-froward loop, i.e., CRC (44, 45). The constituents of this interconnected loop were subsequently extended with the other core TFs, Klf4 and Esrrb, both of which prominent for the maintenance of ESC state (Figure 2A) (21). Thus, CRC plays a critical role in the reprogram of somatic cell into induced pluripotent stem cells (iPSCs), and its dysregulation is undoubtedly involved in cancer (12, 46).
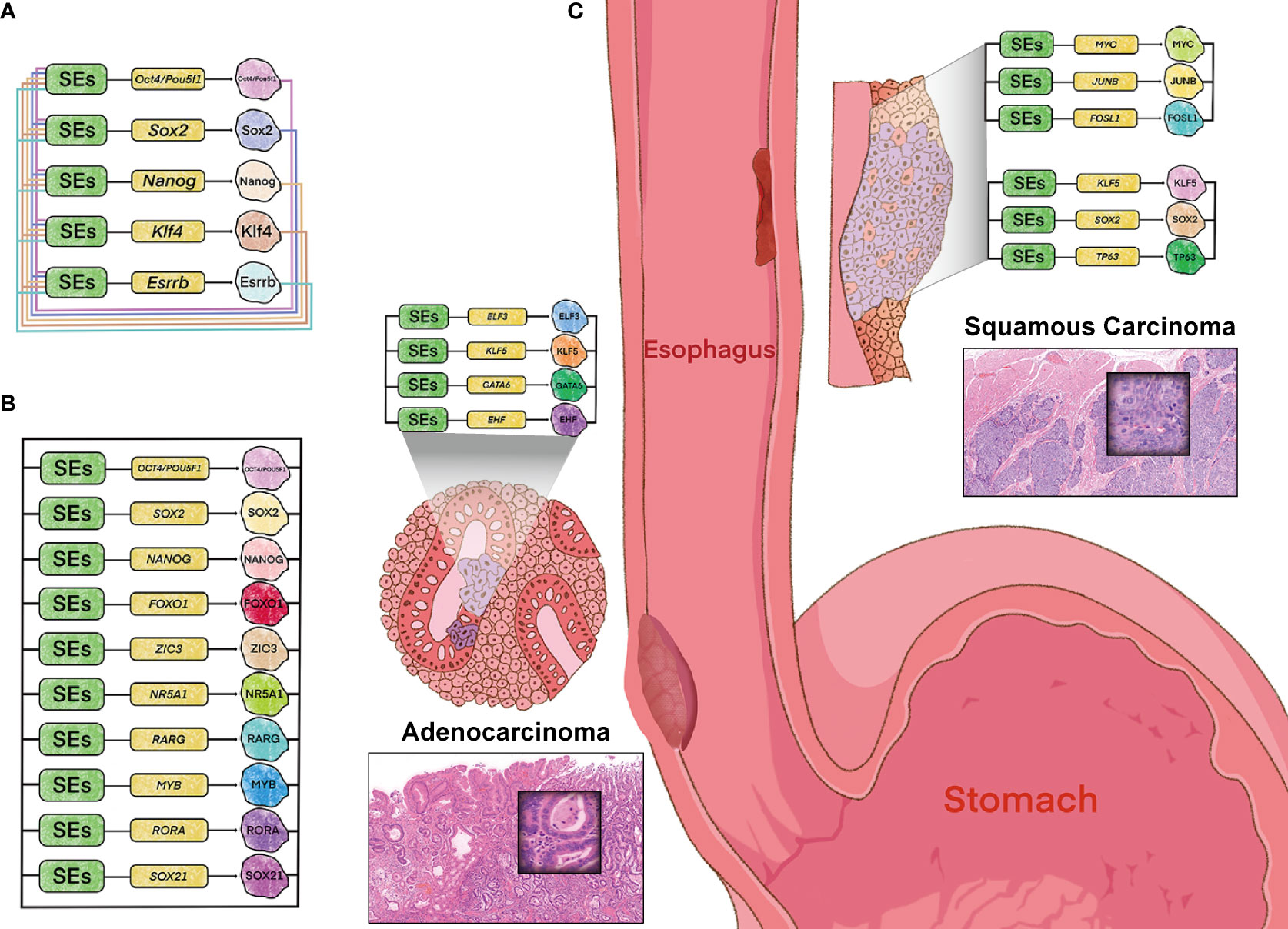
Figure 2 Model of core regulatory circuitry and its implications in esophageal tumorigenesis. (A) CRC model in murine embryonic stem cells. (B) CRC model in human embryonic stem cells identified by “CRC Mapper” algorithm. (C) Histopathology- and cell-type-specific CRC in esophageal carcinoma, including adenocarcinoma (left) and squamous cell carcinoma (right). Hematoxylin-eosin stain (magnification x 100).
Trio-occupancy of OSN has also been well-studied in hESCs (human ESCs), which dominates the pluripotency contributing to human fibroblasts differentiated into the induced pluripotent identity. And this model has been complemented for additional seven key TFs by “CRC Mapper” algorithm, including FOXO1, ZIC3, NR5A1, RARG, MYB, RORA, and SOX21 (Figure 2B) (47). The principle of CRC identification by “CRC Mapper” has been characterized as the following three properties: TFs (i) encoded by SEs-assigned genes, (ii) binding to SEs of their own genes, (iii) forming fully interconnected feed-froward loops with other TFs by binding to their SEs.
In accordance with this strategy, cancer-type and -subtype specificity of CRC models have been identified in esophageal cancer and other malignancies. For example, KLF5 has been proven to be collaborated with ELF3, GATA6 and EHF in EAC (46), while cooperated with TP63 and SOX2 in ESCC to form CRC (12), respectively (Figure 2C). And SOX2 has also been identified co-regulated with KLF4, EGR1 and NOTCH1 in Glioblastoma (48). These cancer-type and -subtype specific CRCs driven by SEs orchestrate the oncogenic transcriptional addiction, while offers therapeutic vulnerabilities due to perturbation sensitivity of their own SEs. These tissue-specific and cancer-specific CRCs driven by SEs orchestrate the oncogenic transcriptional addiction, which offers therapeutic vulnerabilities consistent with the perturbation sensitivity of their own SEs.
Furthermore, cell-type-specific CRC observed in types of cancers is in line with tumor heterogeneity, one of the hallmarks of malignancy. For example, the other CRC (MYC/JUNB/FOSL1) has been identified in ESCC, different with the KLF5/SOX2/TP63 circuitry (Figure 2C). This heterogeneity of CRC coincides with the two major genomic molecular subtypes in ESCC, which are presumably dominated by the two CRCs respectively. And different CRCs have also been identified in other types of cancers, including medulloblastomas and acute myeloid leukemia (49, 50). The heterogeneity of CRC highlights cell-type-specific property of SEs, which sheds light on the novel therapeutic possibilities.
Concentration of oncogenic signaling pathways on super-enhancers
The constituents within SEs have been demonstrated to be heavily loaded with terminal TFs of the Wnt, Tgfb-1, and Lif signaling pathways in mESCs, showing the preferential affections upon SE-assigned genes compared with genes regulated by TEs (51). Therefore, SEs could serve as a platform to converging multiple signaling pathways, dictating the development and disease state of cells, especially oncogenesis (52). For example, EAC-specific SEs has been proposed to be loaded with tumor-promoting TFs LIF, which contributes its malignant features by activating STAT3 and PI3K/AKT signaling pathways (46). Similarly, the cancer-subtype-specific SEs are densely bound up with its specific master TFs TP63 in ESCC, which participates in the formation of CRC and promotes cancer cell proliferation via PI3K/AKT signaling pathways (12, 53, 54). These findings have also been clarified in other types of cancer, e.g., colorectal carcinoma (51), pancreatic carcinoma (55) and osteosarcoma (56). Taken together, these lines of evidence have been proposed that these oncogenic SEs occupied by key TFs pertaining to the critical signaling pathways upon which cancer cell depend, which offers the therapeutic vulnerabilities for esophageal carcinoma.
The occupancy of CDK7 at super-enhancers in esophageal carcinoma
The cyclin-dependent kinases (CDKs) of mammals comprise two main subfamilies with unique properties associated with cell cycle (CDKs 1-6 & 14-18) and transcriptional regulation (CDKs 7-13 & 19-20) (57). The unique functional repertoire of CDK7, the critical component of CDKs, is based on the regulation of transcription and cell cycle progression. Generally, CDK7 could activate transcriptional initiation and elongation by combining with transcription factor II H (TFIIH), while it could also control cell cycle progression by virtue of forming CDK-activating kinase (CAK) (58, 59).
Studies have demonstrated that the dysregulation of CDK7 was involved in various types of cancers and considered to be positively correlated with the aggressive clinicopathological features of these cancers, including esophageal cancer, hepatocellular carcinoma, gastric cancer, and colorectal cancer (13, 60–62). In ESCC, an immunohistochemical (IHC) analysis demonstrated that elevated expression of CDK7 was observed in over 80% samples which was associated with high tumor grade and poor prognosis. Notably, inhibited proliferation and decreased chemotherapeutic resistance have been observed when CDK7 gene was silenced (63). Besides, the level of CDK7 was higher in ESCC tissues with lymph node metastases compared to control group and positively correlated with tumor metastasis and patients’ overall survival (64).
The overabundance of CDK7 within SEs regions offer the opportunities of blockade therapies in a lineage-specific cancer cell manner. THZ1, a covalent inhibitor of CDK7, was proposed to have a preferential impact on a plethora of oncogenes driven by SEs and could cause the disruption of specific transcriptional programs. For example, Chipumuro et al. found that this inhibitor could suppress the transcription of amplified Mycn to suppress the neuroblastoma cells proliferation and the sensitivity to THZ1 was related to preferentially decreased expression of SE-driven oncogenes (65). Subsequent studied showed that esophageal cancer, lung cancer and prostate cancer were sensitive to THZ1 treatment at low nanomolar range (10, 13, 66). Additionally, THZ1 could also cause downregulation of SEs-associated functional long noncoding RNAs acting as competing endogenous (ce-lncRNAs), such as HOTAIR, XIST, SNHG5, and LINC00094 (67), which are associated with expression of cancer hallmark genes. Notably, LINC00094, as a novel oncogenic lncRNA could be activated by master TFs, e.g., TCF3 and KLF5, and positively correlate with poor prognosis. Upon inhibiting these TFs by THZ1, the level of LINC00094 is downregulated, which could cause tumor regression in ESCC. Thus, these studies have demonstrated THZ1 could be utilized as the crosshair of cancer drug discovery (57).
Similar to CDK7, CDK9 is mainly responsible for oncogenic transcription, e.g., as MCL-1 and C-Myc, by binding to elongation complex (p-TEFb) (68) and knocking down CDK9 has shown an excellent antineoplastic activity in hematologic and solid tumors. SNS-032, a selective inhibitor against CDK family as well can inhibit transcription initiation and elongation by targeting both CDK7 and CKD9, with the IC50 values of 62 and 4 nM, respectively (69–71). It could suppress the lung and lymph node metastasis as well as inhibit ESCC proliferation (72). These findings indicate that SNS-032 play an antineoplastic role in ESCC and is expected to enter clinical trials to validate its efficacy, especially in those with metastasis.
The occupancy of BRD4 at super-enhancers in esophageal carcinoma
The bromo domain and extra-terminal domain (BET) family contain BRD2, BRD3, BRD4, and testis-specific BRDT, which are characterized by acetylation recognition and transcription regulation (73). The well-known member of the BET family, BRD4 was initially recognized as the component of Mediator complex, and subsequently the general regulator for RNA Pol II by virtue of recruitment with transcriptional elongation factor P-TFEb. Consistently, the genomic profiling of BRD4 demonstrated that it is mainly enriched at active enhancers and promoters, which is not surprisingly complicated in critical cellular processes, such as embryogenesis (74) and cancer development (75–82). More importantly, the tumorigenic transcriptional activation of BRD4 are preferentially to regulate SE associated oncogenes, such as C-Myc and BCL2 (83, 84)
Emerging evidence have demonstrated that inhibition of BRD4 occupancy result in SEs disruption and subsequently suppress its related oncogenes expression (73). Up till now, about 20 BRD4 inhibitors have been entered into clinical trials, some of which showed valuable therapeutics for several cancers, including hematological malignancies and non-small cell lung cancer (85). The critical oncogenes within these cancers showed highly sensitivity to JQ1, one of the promising anti-tumor BET inhibitors (22, 86). For instance, JQ1 has been demonstrated to inhibit ESCC proliferation by inhibiting C-Myc amplification (86). For another, it could also block recruitment of BRD4 on the promoter of aurora kinases A and B (AURKA/B) to trigger cellular senescence, which provided a novel action manner of BRD4 in esophageal carcinoma (87). In esophageal adenocarcinoma (EAC), high expression of YAP1 has been reported to be observed and positively associated with poor prognosis. JQ1 is capable of blocking BRD4 binding to the YAP1 promoter and suppress Hippo/YAP1 signaling (88), which could be synergistically enhanced when traditional chemotherapeutic agents, e.g., docetaxel, are added both in vitro and in vivo (88). Notably, cell cycle arrest, especially in G1 phase, has been observed in all the studies above. These studies also indicated that JQ1 might be a promising drug in esophageal carcinoma treatment.
Although a variety of small-molecule BET inhibitors have been entered in clinical researches, however, the off-target and side-effects cannot be neglected (89). Proteolysis targeting chimeric (PROTAC) technology have been utilized as an effective degradation tool over the years, which can ubiquitinate the disease-causing proteins by hijacking E3 ligases to achieve the anti-tumor effects (90). Compared to the general small-molecule inhibitors, the hetero-bifunctional molecule based on PROTAC technology demonstrated degradation of BET protein at low nanomolar dose with negligible side-effects (91). The first oral PROTACs (ARV-110, NCT03888612 & ARV-471, NCT04072952) have achieved encouraging benefit for prostate and breast cancer treatment in clinical trials (92). The newly developed GNE987, and PROTAC pan-BET degrader, have been showing good potency against several cancers, including hematological malignancies (93, 94), neuroblastoma (95), and prostate carcinoma (96). Additionally, MZ1, a PROTAC BET degrader, was previously identified unexpectedly degradation of BRD4 over other members of BET family, such as BRD2 and BRD3 (97). However, MZ1 have been proposed to suppress ESCC migration by degrading BRDT, initially recognized as testis-specific protein, rather than BRD2, BRD3 and BRD4. The migratory inhibition of ESCC by MZ1 resulted from down-regulation of ΔNp63 target genes enriched within SEs (98).
The enrichment of histone acetylation at super-enhancers in esophageal carcinoma
Histone H3K27ac has been proposed to separate the active enhancers from poised enhancers occupied by H3K4me1 alone (99). The occupancy of H3K27ac facilitates the chromatin accessibility, which is responsible for exceptionally higher enrichment of master TFs, co-activators, and transcriptional machinery on SEs. Therefore, the dysregulated H3K27ac within SEs region could drive the oncogenesis in several types of cancer.
Histone acetylation is a relative steady-state controlled by two families of enzymes: histone acetyltransferases (HATs) and histone deacetylases (HDACs) (100, 101). Numerous studies have demonstrated that the disturbed equilibrium of histones acetylation is closely related with the tumorigenicity of esophageal carcinoma (Table 2). Furthermore, the tumor-promoting genes activated by hyperacetylation have also been identified in esophageal carcinoma (Table 3). The dysregulated histone acetylation of esophageal carcinoma could be accounted for the oncogenesis driven by SEs, which densely bound up with the active enhancer marker H3K27ac.
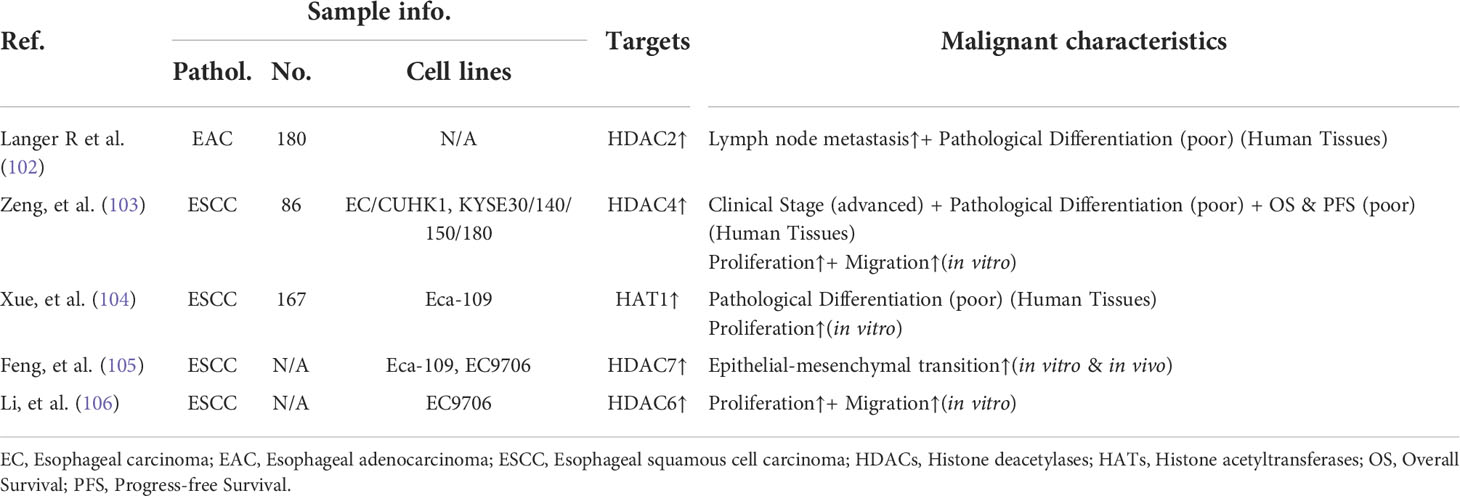
Table 2 The correlations between dysregulated HDACs (and/or HATs) and the advanced oncogenic features of esophageal carcinoma.
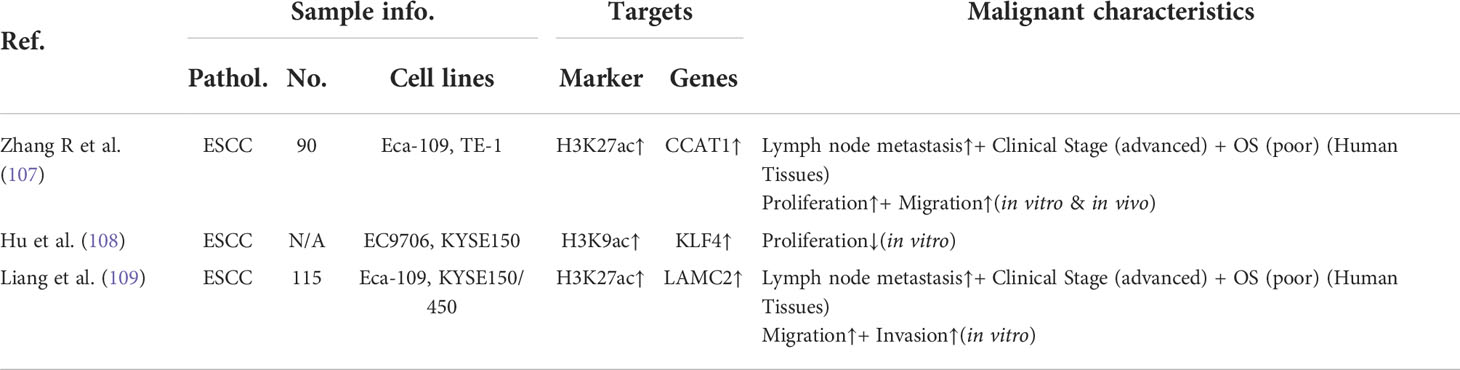
Table 3 The associations between dysregulated histone acetylation modifications and advanced oncogenic features.
Based on this knowledge, the aberrant histone acetylation has been addressed as the alternative avenues for cancer treatment, especially those driven by oncogenic SEs. For example, the aberrantly expressed gene SIRT7, NAD-dependent deacetylase, was activated by SEs in non-alcoholic fatty liver disease (NAFLD)-associated hepatocellular carcinoma (HCCs). Depletion of SIRT7 associated SE could suppress the tumorigenicity both in vitro and in vivo (110). And several types of HDAC inhibitors (HDACis) have been shown as the promising therapeutic strategies against esophageal carcinoma (Table 4). Therefore, the recruitment of exceptionally high histone acetylation modification, especially H3K27ac, within the oncogenic SEs could contribute their vulnerabilities to HDACis for patients of esophageal carcinoma.
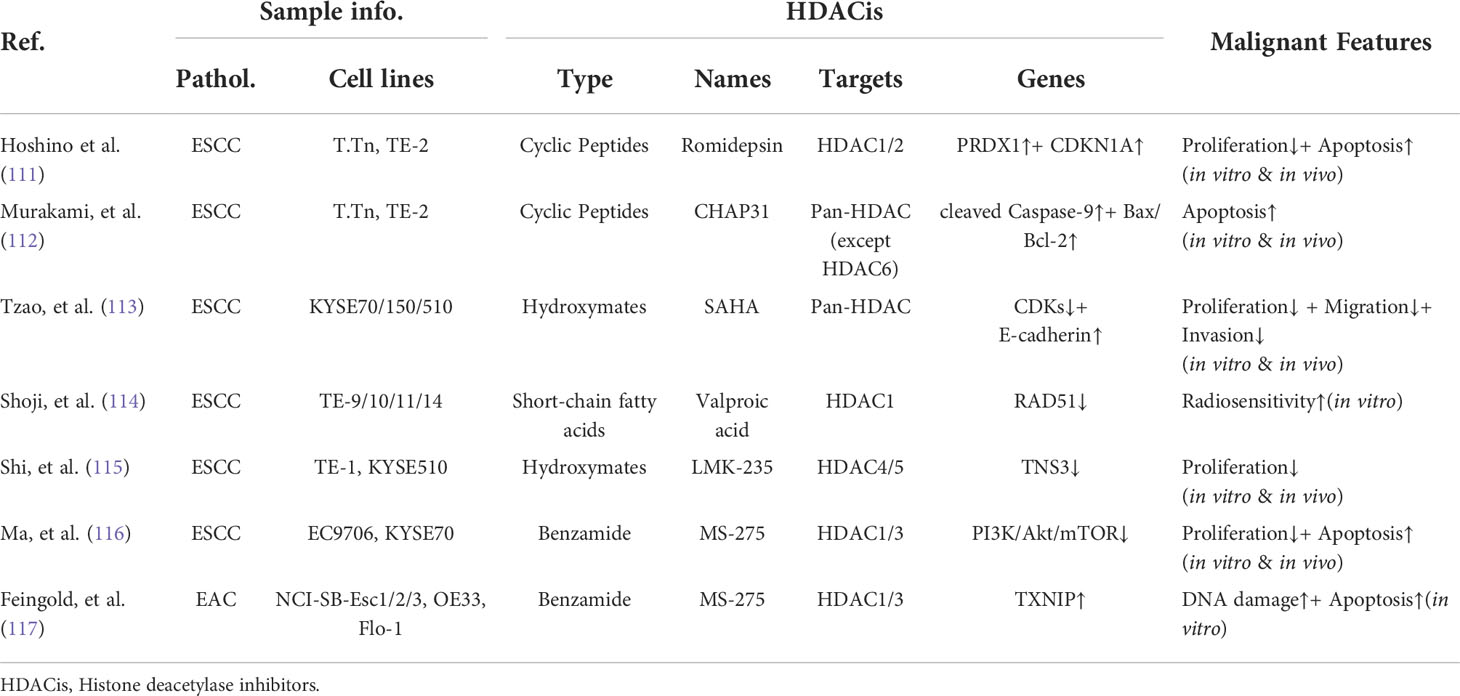
Table 4 The molecular mechanism of histone deacetylase inhibitors against esophageal carcinoma.
Conclusive remark
The concept of SEs and their function in physiology and disease has been established in the last decade. To date, there is no doubt that SEs acquired de novo are one of the hallmarks of esophageal carcinoma, and the available studies conclude the characteristics of the oncogenic SEs in esophageal carcinoma: (i) they are acquired based on genetic variants or TADs boundaries disruption, (ii) they drive the transcriptional additions depend on CRCs and convergence of tumorigenic signaling pathways, (iii) they could be targeted by inhibition of BRD4, CDK7, and histone acetylation, which exceptionally enriched within these cis-elements. However, there are still several unresolved points regarding SEs in esophageal carcinoma: (i) how the individual bona fide constituents selectively respond to targeted inhibition pertaining to the disproportionately enriched factors, (ii) how 3D genome structure of the SEs are influenced by the tumor-promoting signaling pathways, (iii) what are the critical transitions of SEs landscape during the process of precancerous lesions to cancer, (iv) what are the main differences of SEs profiling between squamous cell carcinoma and adenocarcinoma, (v) what are the clinical benefits based on the combinations of traditional treatments and novel therapeutic avenues targeting the subtype-specific or cell-type-specific CRCs. These insights about SEs-driven transcriptional dependencies in esophageal carcinoma, may shed light on the potential clinical application of the cancer-specific SEs.
Author contributions
BL, HY, and XM formulated the study concept and design; YS wrote the original manuscript draft; MW, DL, provided the technical and material support; SU, critically revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by the Zhongyuan talent program (ZYYCYU202012113).
Acknowledgments
We apologize to the authors whose study could not be cited for the limited spaces. We would like to thank Jiyu Zhang for his delicate and exquisite schematic graphs. And we also thank Zhixin Li for his comments on this manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Ferlay J, Colombet M, Soerjomataram I, Parkin DM, Piñeros M, Znaor A, et al. Cancer statistics for the year 2020: An overview. Int J Cancer (2021) 149:778–89. doi: 10.1002/ijc.33588
2. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: Globocan estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin (2021) 71(3):209–49. doi: 10.3322/caac.21660
3. Wei W, Zeng H, Zheng R, Zhang S, An L, Chen R, et al. Cancer registration in China and its role in cancer prevention and control. Lancet Oncol (2020) 21(7):e342–e9. doi: 10.1016/s1470-2045(20)30073-5
4. Qiao Y, Zhao C, Li X, Zhao J, Huang Q, Ding Z, et al. Efficacy and safety of camrelizumab in combination with neoadjuvant chemotherapy for escc and its impact on esophagectomy. Front Immunol (2022) 13:953229. doi: 10.3389/fimmu.2022.953229
5. Li ZC, Sun YT, Lai MY, Zhou YX, Qiu MZ. Efficacy and safety of pd-1 inhibitors combined with chemotherapy as first-line therapy for advanced esophageal cancer: A systematic review and network meta-analysis. Int Immunopharmacol (2022) 109:108790. doi: 10.1016/j.intimp.2022.108790
6. Perk J, Iavarone A, Benezra R. Id family of helix-Loop-Helix proteins in cancer. Nat Rev Cancer (2005) 5(8):603–14. doi: 10.1038/nrc1673
7. Mansour MR, Abraham BJ, Anders L, Berezovskaya A, Gutierrez A, Durbin AD, et al. Oncogene Regulation. An oncogenic super-enhancer formed through somatic mutation of a noncoding intergenic element. Sci (New York NY) (2014) 346(6215):1373–7. doi: 10.1126/science.1259037
8. Betancur PA, Abraham BJ, Yiu YY, Willingham SB, Khameneh F, Zarnegar M, et al. A Cd47-associated super-enhancer links pro-inflammatory signalling to Cd47 upregulation in breast cancer. Nat Commun (2017) 8:14802. doi: 10.1038/ncomms14802
9. Ke L, Zhou H, Wang C, Xiong G, Xiang Y, Ling Y, et al. Nasopharyngeal carcinoma super-Enhancer-Driven Etv6 correlates with prognosis. Proc Natl Acad Sci United States America (2017) 114(36):9683–8. doi: 10.1073/pnas.1705236114
10. Christensen CL, Kwiatkowski N, Abraham BJ, Carretero J, Al-Shahrour F, Zhang T, et al. Targeting transcriptional addictions in small cell lung cancer with a covalent Cdk7 inhibitor. Cancer Cell (2014) 26(6):909–22. doi: 10.1016/j.ccell.2014.10.019
11. Northcott PA, Lee C, Zichner T, Stutz AM, Erkek S, Kawauchi D, et al. Enhancer hijacking activates Gfi1 family oncogenes in medulloblastoma. Nature (2014) 511(7510):428–34. doi: 10.1038/nature13379
12. Jiang Y-Y, Jiang Y, Li C-Q, Zhang Y, Dakle P, Kaur H, et al. Tp63, Sox2, and Klf5 Establish a Core Regulatory Circuitry That Controls Epigenetic and Transcription Patterns in Esophageal Squamous Cell Carcinoma Cell Lines. Gastroenterology (2020) 159(4). doi: 10.1053/j.gastro.2020.06.050
13. Jiang Y-Y, Lin D-C, Mayakonda A, Hazawa M, Ding L-W, Chien W-W, et al. Targeting super-Enhancer-Associated oncogenes in oesophageal squamous cell carcinoma. Gut (2017) 66(8):1358–68. doi: 10.1136/gutjnl-2016-311818
14. Sengupta S, George RE. Super-Enhancer-Driven transcriptional dependencies in cancer. Trends In Cancer (2017) 3(4):269–81. doi: 10.1016/j.trecan.2017.03.006
15. Banerji J, Rusconi S, Schaffner W. Expression of a beta-globin gene is enhanced by remote Sv40 DNA sequences. Cell (1981) 27(2 Pt 1):299–308.
16. Benoist C, Chambon P. In vivo sequence requirements of the Sv40 early promotor region. Nature (1981) 290(5804):304–10. doi: 10.1038/290304a0
17. Buecker C, Wysocka J. Enhancers as information integration hubs in development: Lessons from genomics. Trends genetics: TIG (2012) 28(6):276–84. doi: 10.1016/j.tig.2012.02.008
18. Schaffner W. Enhancers, enhancers - from their discovery to today’s universe of transcription enhancers. Biol Chem (2015) 396(4):311–27. doi: 10.1515/hsz-2014-0303
19. Calo E, Wysocka J. Modification of enhancer chromatin: What, how, and why? Mol Cell (2013) 49(5):825–37. doi: 10.1016/j.molcel.2013.01.038
20. Gross DS, Garrard WT. Nuclease hypersensitive sites in chromatin. Annu Rev Biochem (1988) 57:159–97. doi: 10.1146/annurev.bi.57.070188.001111
21. Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, et al. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell (2013) 153(2):307–19. doi: 10.1016/j.cell.2013.03.035
22. Loven J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR, et al. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell (2013) 153(2):320–34. doi: 10.1016/j.cell.2013.03.036
23. Janmey PA, Hvidt S, Käs J, Lerche D, Maggs A, Sackmann E, et al. The mechanical properties of actin Gels. Elastic modulus and filament motions. J Biol Chem (1994) 269(51):32503–13. doi: 10.1016/S0021-9258(18)31663-6
24. Banani SF, Lee HO, Hyman AA, Rosen MK. Biomolecular condensates: Organizers of cellular biochemistry. Nat Rev Mol Cell Biol (2017) 18(5):285–98. doi: 10.1038/nrm.2017.7
25. Shin Y, Brangwynne CP. Liquid phase condensation in cell physiology and disease. Science (New York NY) (2017) 357(6357):eaaf4382. doi: 10.1126/science.aaf4382
26. Sabari BR, Dall’Agnese A, Boija A, Klein IA, Coffey EL, Shrinivas K, et al. Coactivator condensation at super-enhancers links phase separation and gene control. Science (2018) 361(6400):eaar3958. doi: 10.1126/science.aar3958
27. Weintraub AS, Li CH, Zamudio AV, Sigova AA, Hannett NM, Day DS, et al. Yy1 is a structural regulator of enhancer-promoter loops. Cell (2017) 171(7):1573–88.e28. doi: 10.1016/j.cell.2017.11.008
28. Hnisz D, Abraham BJ, Lee TI, Lau A, Saint-Andre V, Sigova AA, et al. Super-enhancers in the control of cell identity and disease. Cell (2013) 155(4):934–47. doi: 10.1016/j.cell.2013.09.053
30. Proudhon C, Snetkova V, Raviram R, Lobry C, Badri S, Jiang T, et al. Active and inactive enhancers cooperate to exert localized and long-range control of gene regulation. Cell Rep (2016) 15(10):2159–69. doi: 10.1016/j.celrep.2016.04.087
31. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell (2000) 100(1):57–70. doi: 10.1016/S0092-8674(00)81683-9
32. Hanahan D, Weinberg RA. Hallmarks of cancer: The next generation. Cell (2011) 144(5):646–74. doi: 10.1016/j.cell.2011.02.013
33. Zhang X, Choi PS, Francis JM, Imielinski M, Watanabe H, Cherniack AD, et al. Identification of focally amplified lineage-specific super-enhancers in human epithelial cancers. Nat Genet (2016) 48(2):176–82. doi: 10.1038/ng.3470
34. Herz HM. Enhancer deregulation in cancer and other diseases. BioEssays: News Rev Molecular Cell Dev Biol (2016) 38(10):1003–15. doi: 10.1002/bies.201600106
35. Groschel S, Sanders MA, Hoogenboezem R, de Wit E, Bouwman BAM, Erpelinck C, et al. A single oncogenic enhancer rearrangement causes concomitant Evi1 and Gata2 deregulation in leukemia. Cell (2014) 157(2):369–81. doi: 10.1016/j.cell.2014.02.019
36. Weischenfeldt J, Dubash T, Drainas AP, Mardin BR, Chen Y, Stutz AM, et al. Pan-cancer analysis of somatic copy-number alterations implicates Irs4 and Igf2 in enhancer hijacking. Nat Genet (2017) 49(1):65–74. doi: 10.1038/ng.3722
37. Oldridge DA, Wood AC, Weichert-Leahey N, Crimmins I, Sussman R, Winter C, et al. Genetic predisposition to neuroblastoma mediated by a Lmo1 super-enhancer polymorphism. Nature (2015) 528(7582):418–21. doi: 10.1038/nature15540
38. Kandaswamy R, Sava GP, Speedy HE, Beà S, Martín-Subero JI, Studd JB, et al. Genetic predisposition to chronic lymphocytic leukemia is mediated by a bmf super-enhancer polymorphism. Cell Rep (2016) 16(8):2061–7. doi: 10.1016/j.celrep.2016.07.053
39. Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, et al. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature (2012) 485(7398):376–80. doi: 10.1038/nature11082
40. Zuin J, Dixon JR, van der Reijden MI, Ye Z, Kolovos P, Brouwer RW, et al. Cohesin and ctcf differentially affect chromatin architecture and gene expression in human cells. Proc Natl Acad Sci United States America (2014) 111(3):996–1001. doi: 10.1073/pnas.1317788111
41. Katainen R, Dave K, Pitkänen E, Palin K, Kivioja T, Välimäki N, et al. Ctcf/Cohesin-binding sites are frequently mutated in cancer. Nat Genet (2015) 47(7):818–21. doi: 10.1038/ng.3335
42. Hnisz D, Weintraub AS, Day DS, Valton AL, Bak RO, Li CH, et al. Activation of proto-oncogenes by disruption of chromosome neighborhoods. Sci (New York NY) (2016) 351(6280):1454–8. doi: 10.1126/science.aad9024
43. Flavahan WA, Drier Y, Liau BB, Gillespie SM, Venteicher AS, Stemmer-Rachamimov AO, et al. Insulator dysfunction and oncogene activation in idh mutant gliomas. Nature (2016) 529(7584):110–4. doi: 10.1038/nature16490
44. Boyer LA, Lee TI, Cole MF, Johnstone SE, Levine SS, Zucker JP, et al. Core transcriptional regulatory circuitry in human embryonic stem cells. Cell (2005) 122(6):947–56. doi: 10.1016/j.cell.2005.08.020
45. Loh YH, Wu Q, Chew JL, Vega VB, Zhang W, Chen X, et al. The Oct4 and nanog transcription network regulates pluripotency in mouse embryonic stem cells. Nat Genet (2006) 38(4):431–40. doi: 10.1038/ng1760
46. Chen L, Huang M, Plummer J, Pan J, Jiang YY, Yang Q, et al. Master transcription factors form interconnected circuitry and orchestrate transcriptional networks in oesophageal adenocarcinoma. Gut (2020) 69(4):630–40. doi: 10.1136/gutjnl-2019-318325
47. Saint-André V, Federation AJ, Lin CY, Abraham BJ, Reddy J, Lee TI, et al. Models of human core transcriptional regulatory circuitries. Genome Res (2016) 26(3):385–96. doi: 10.1101/gr.197590.115
48. Riddick G, Kotliarova S, Rodriguez V, Kim HS, Linkous A, Storaska AJ, et al. A core regulatory circuit in glioblastoma stem cells links mapk activation to a transcriptional program of neural stem cell identity. Sci Rep (2017) 7:43605. doi: 10.1038/srep43605
49. Lin CY, Erkek S, Tong Y, Yin L, Federation AJ, Zapatka M, et al. Active medulloblastoma enhancers reveal subgroup-specific cellular origins. Nature (2016) 530(7588):57–62. doi: 10.1038/nature16546
50. Assi SA, Imperato MR, Coleman DJL, Pickin A, Potluri S, Ptasinska A, et al. Subtype-specific regulatory network rewiring in acute myeloid leukemia. Nat Genet (2019) 51(1):151–62. doi: 10.1038/s41588-018-0270-1
51. Hnisz D, Schuijers J, Lin CY, Weintraub AS, Abraham BJ, Lee TI, et al. Convergence of developmental and oncogenic signaling pathways at transcriptional super-enhancers. Mol Cell (2015) 58(2):362–70. doi: 10.1016/j.molcel.2015.02.014
52. Jia Q, Chen S, Tan Y, Li Y, Tang F. Oncogenic super-enhancer formation in tumorigenesis and its molecular mechanisms. Exp Mol Med (2020) 52(5):713–23. doi: 10.1038/s12276-020-0428-7
53. Ye S, Lee KB, Park MH, Lee JS, Kim SM. P63 regulates growth of esophageal squamous carcinoma cells Via the akt signaling pathway. Int J Oncol (2014) 44(6):2153–9. doi: 10.3892/ijo.2014.2374
54. Hu H, Xia SH, Li AD, Xu X, Cai Y, Han YL, et al. Elevated expression of P63 protein in human esophageal squamous cell carcinomas. Int J Cancer (2002) 102(6):580–3. doi: 10.1002/ijc.10739
55. Zhu X, Zhang T, Zhang Y, Chen H, Shen J, Jin X, et al. A super-enhancer controls tgf- B signaling in pancreatic cancer through downregulation of Tgfbr2. Cell signalling (2020) 66:109470. doi: 10.1016/j.cellsig.2019.109470
56. Lu B, He Y, He J, Wang L, Liu Z, Yang J, et al. Epigenetic profiling identifies lif as a super-Enhancer-Controlled regulator of stem cell-like properties in osteosarcoma. Mol Cancer Research: MCR (2020) 18(1):57–67. doi: 10.1158/1541-7786.Mcr-19-0470
57. Fisher RP. Cdk7: A kinase at the core of transcription and in the crosshairs of cancer drug discovery. Transcription (2019) 10(2):47–56. doi: 10.1080/21541264.2018.1553483
58. Coin F, Egly JM. Revisiting the function of Cdk7 in transcription by virtue of a recently described tfiih kinase inhibitor. Mol Cell (2015) 59(4):513–4. doi: 10.1016/j.molcel.2015.08.006
59. Schachter MM, Merrick KA, Larochelle S, Hirschi A, Zhang C, Shokat KM, et al. A Cdk7-Cdk4 T-loop phosphorylation cascade promotes G1 progression. Mol Cell (2013) 50(2):250–60. doi: 10.1016/j.molcel.2013.04.003
60. Huang JR, Qin WM, Wang K, Fu DR, Zhang WJ, Jiang QW, et al. Cyclin-dependent kinase 7 inhibitor Thz2 inhibits the growth of human gastric cancer in vitro and in vivo. Am J Trans Res (2018) 10(11):3664–76.
61. Wang C, Jin H, Gao D, Wang L, Evers B, Xue Z, et al. A crispr screen identifies Cdk7 as a therapeutic target in hepatocellular carcinoma. Cell Res (2018) 28(6):690–2. doi: 10.1038/s41422-018-0020-z
62. Zhou Y, Lu L, Jiang G, Chen Z, Li J, An P, et al. Targeting Cdk7 increases the stability of snail to promote the dissemination of colorectal cancer. Cell Death Differ (2019) 26(8):1442–52. doi: 10.1038/s41418-018-0222-4
63. Zhang J, Yang X, Wang Y, Shi H, Guan C, Yao L, et al. Low expression of cyclinh and cyclin-dependent kinase 7 can decrease the proliferation of human esophageal squamous cell carcinoma. Digestive Dis Sci (2013) 58(7):2028–37. doi: 10.1007/s10620-013-2597-x
64. Zhang J, Zhu J, Yang L, Guan C, Ni R, Wang Y, et al. Interaction with Ccnh/Cdk7 facilitates Ctbp2 promoting esophageal squamous cell carcinoma (Escc) metastasis Via upregulating epithelial-mesenchymal transition (Emt) progression. Tumour biology: J Int Soc Oncodevelopmental Biol Med (2015) 36(9):6701–14. doi: 10.1007/s13277-015-3354-x
65. Chipumuro E, Marco E, Christensen CL, Kwiatkowski N, Zhang T, Hatheway CM, et al. Cdk7 inhibition suppresses super-Enhancer-Linked oncogenic transcription in mycn-driven cancer. Cell (2014) 159(5):1126–39. doi: 10.1016/j.cell.2014.10.024
66. Rasool RU, Natesan R, Deng Q, Aras S, Lal P, Sander Effron S, et al. Cdk7 inhibition suppresses castration-resistant prostate cancer through Med1 inactivation. Cancer Discov (2019) 9(11):1538–55. doi: 10.1158/2159-8290.Cd-19-0189
67. Wang QY, Peng L, Chen Y, Liao LD, Chen JX, Li M, et al. Characterization of super-enhancer-associated functional lncRNAs acting as ceRNAs in ESCC. Mol Oncol (2020) 14(9):2203–30. doi: 10.1002/1878-0261.12726
68. Tong Z, Mejia A, Veeranki O, Verma A, Correa AM, Dokey R, et al. Targeting CDK9 and MCL-1 by a new CDK9/p-TEFb inhibitor with and without 5-fluorouracil in esophageal adenocarcinoma. Ther Adv Med Oncol (2019) 11:1758835919864850. doi: 10.1177/1758835919864850
69. Zhang J, Liu S, Ye Q, Pan J. Transcriptional inhibition by Cdk7/9 inhibitor sns-032 abrogates oncogene addiction and reduces liver metastasis in uveal melanoma. Mol Cancer (2019) 18(1):140. doi: 10.1186/s12943-019-1070-7
70. Chen R, Wierda WG, Chubb S, Hawtin RE, Fox JA, Keating MJ, et al. Mechanism of action of sns-032, a novel cyclin-dependent kinase inhibitor, in chronic lymphocytic leukemia. Blood (2009) 113(19):4637–45. doi: 10.1182/blood-2008-12-190256
71. Wu Y, Chen C, Sun X, Shi X, Jin B, Ding K, et al. Cyclin-dependent kinase 7/9 inhibitor sns-032 abrogates Fip1-Like-1 platelet-derived growth factor receptor A and bcr-abl oncogene addiction in malignant hematologic cells. Clin Cancer research: an Off J Am Assoc Cancer Res (2012) 18(7):1966–78. doi: 10.1158/1078-0432.Ccr-11-1971
72. Zeng H, Yang H, Song Y, Fang D, Chen L, Zhao Z, et al. Transcriptional inhibition by Cdk7/9 inhibitor sns-032 suppresses tumor growth and metastasis in esophageal squamous cell carcinoma. Cell Death Dis (2021) 12(11):1048. doi: 10.1038/s41419-021-04344-w
73. French CA. Small-molecule targeting of bet proteins in cancer. Adv Cancer Res (2016) 131:21–58. doi: 10.1016/bs.acr.2016.04.001
74. Toyama R, Rebbert ML, Dey A, Ozato K, Dawid IB. Brd4 associates with mitotic chromosomes throughout early zebrafish embryogenesis. Developmental dynamics : an official publication of the American Association of Anatomists (2008) 237(6):1636–44. doi: 10.1002/dvdy.21576
75. Donati B, Lorenzini E, Ciarrocchi A. Brd4 and cancer: Going beyond transcriptional regulation. Mol Cancer (2018) 17(1):1–13. doi: 10.1186/s12943-018-0915-9
76. Zuber J, Shi J, Wang E, Rappaport AR, Herrmann H, Sison EA, et al. Rnai screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature (2011) 478(7370):524–8. doi: 10.1038/nature10334
77. Dawson MA, Prinjha RK, Dittmann A, Giotopoulos G, Bantscheff M, Chan WI, et al. Inhibition of bet recruitment to chromatin as an effective treatment for mll-fusion leukaemia. Nature (2011) 478(7370):529–33. doi: 10.1038/nature10509
78. Ceribelli M, Kelly PN, Shaffer AL, Wright GW, Xiao W, Yang Y, et al. Blockade of oncogenic iκb kinase activity in diffuse Large b-cell lymphoma by bromodomain and extraterminal domain protein inhibitors. Proc Natl Acad Sci United States America (2014) 111(31):11365–70. doi: 10.1073/pnas.1411701111
79. Wu Q, Liu F, Ge M, Laster KV, Wei L, Du R, et al. Brd4 drives esophageal squamous cell carcinoma growth by promoting Rcc2 expression. Oncogene (2022) 41(3):347–60. doi: 10.1038/s41388-021-02099-4
80. Chen X, Ma Q, Shang Z, Niu Y. Super-enhancer in prostate cancer: Transcriptional disorders and therapeutic targets. NPJ Precis Oncol (2020) 4(1):31. doi: 10.1038/s41698-020-00137-0
81. Crawford NP, Alsarraj J, Lukes L, Walker RC, Officewala JS, Yang HH, et al. Bromodomain 4 activation predicts breast cancer survival. Proc Natl Acad Sci United States America (2008) 105(17):6380–5. doi: 10.1073/pnas.0710331105
82. Shimamura T, Chen Z, Soucheray M, Carretero J, Kikuchi E, Tchaicha JH, et al. Efficacy of bet bromodomain inhibition in kras-mutant non-small cell lung cancer. Clin Cancer research: an Off J Am Assoc Cancer Res (2013) 19(22):6183–92. doi: 10.1158/1078-0432.Ccr-12-3904
83. Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, et al. Bet bromodomain inhibition as a therapeutic strategy to target c-myc. Cell (2011) 146(6):904–17. doi: 10.1016/j.cell.2011.08.017
84. Peirs S, Frismantas V, Matthijssens F, Van Loocke W, Pieters T, Vandamme N, et al. Targeting bet proteins improves the therapeutic efficacy of bcl-2 inhibition in T-cell acute lymphoblastic leukemia. Leukemia (2017) 31(10):2037–47. doi: 10.1038/leu.2017.10
85. Lu T, Lu W, Luo C. A patent review of Brd4 inhibitors (2013-2019). Expert Opin Ther patents (2020) 30(1):57–81. doi: 10.1080/13543776.2020.1702645
86. Wang J, Liu Z, Wang Z, Wang S, Chen Z, Li Z, et al. Targeting c-myc: Jq1 as a promising option for c-Myc-Amplified esophageal squamous cell carcinoma. Cancer Lett (2018) 419:64–74. doi: 10.1016/j.canlet.2018.01.051
87. Xu JL, Yuan YJ, Lv J, Qi D, Wu MD, Lan J, et al. Inhibition of Brd4 triggers cellular senescence through suppressing aurora kinases in oesophageal cancer cells. J Cell Mol Med (2020) 24(22):13036–45. doi: 10.1111/jcmm.15901
88. Song S, Li Y, Xu Y, Ma L, Pool Pizzi M, Jin J, et al. Targeting hippo coactivator Yap1 through bet bromodomain inhibition in esophageal adenocarcinoma. Mol Oncol (2020) 14(6):1410–26. doi: 10.1002/1878-0261.12667
89. Yang Y, Wu Z, Chen P, Zheng P, Zhang H, Zhou J. Proteolysis-targeting chimeras mediate the degradation of bromodomain and extra-terminal domain proteins. Future med Chem (2020) 12(18):1669–83. doi: 10.4155/fmc-2017-0264
90. Wan Y, Yan C, Gao H, Liu T. Small-molecule protacs: Novel agents for cancer therapy. Future Med Chem (2020) 12(10):915–38. doi: 10.4155/fmc-2019-0340
91. Raina K, Crews CM. Chemical inducers of targeted protein degradation. J Biol Chem (2010) 285(15):11057–60. doi: 10.1074/jbc.R109.078105
92. Qi SM, Dong J, Xu ZY, Cheng XD, Qin JJ. Protac: An effective targeted protein degradation strategy for cancer therapy. Front Pharmacol (2021) 12:692574. doi: 10.3389/fphar.2021.692574
93. Sang X, Zhang Y, Fang F, Gao L, Tao Y, Li X, et al. Brd4 protac degradation agent gne-987 inhibits acute myeloid leukemia by targeting super enhancers. (2021). doi: 10.21203/rs.3.rs-1204848/v1
94. Sang X, Zhang Y, Fang F, Gao L, Tao Y, Li X, et al. BRD4 inhibitor GNE-987 exerts anticancer effects by targeting super-enhancer-related gene LYL1 in acute myeloid leukemia. J Immunol Res (2022) 2022:7912484. doi: 10.1155/2022/7912484
95. Chen YL, Li XL, Li G, Tao YF, Zhuo R, Cao HB, et al. BRD4 inhibitor GNE987 exerts anti-cancer effects by targeting super-enhancers in neuroblastoma. Cell biosci (2022) 12(1):33. doi: 10.1186/s13578-022-00769-8
96. Hu R, Wang WL, Yang YY, Hu XT, Wang QW, Zuo WQ, et al. Identification of a selective Brd4 protac with potent antiproliferative effects in ar-positive prostate cancer based on a dual Bet/Plk1 inhibitor. Eur J Med Chem (2022) 227:113922. doi: 10.1016/j.ejmech.2021.113922
97. Zengerle M, Chan KH, Ciulli A. Selective small molecule induced degradation of the bet bromodomain protein Brd4. ACS Chem Biol (2015) 10(8):1770–7. doi: 10.1021/acschembio.5b00216
98. Wang X, Kutschat AP, Yamada M, Prokakis E, Böttcher P, Tanaka K, et al. Bromodomain protein brdt directs Δnp63 function and super-enhancer activity in a subset of esophageal squamous cell carcinomas. Cell Death Differ (2021) 28(7):2207–20. doi: 10.1038/s41418-021-00751-w
99. Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, et al. Histone H3k27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci United States America (2010) 107(50):21931–6. doi: 10.1073/pnas.1016071107
100. Kurdistani SK, Grunstein M. Histone acetylation and deacetylation in yeast. Nat Rev Mol Cell Biol (2003) 4(4):276–84. doi: 10.1038/nrm1075
101. Seto E, Yoshida M. Erasers of histone acetylation: The histone deacetylase enzymes. Cold Spring Harbor Perspect Biol (2014) 6(4):a018713. doi: 10.1101/cshperspect.a018713
102. Langer R, Mutze K, Becker K, Feith M, Ott K, Höfler H, et al. Expression of class I histone deacetylases (Hdac1 and Hdac2) in oesophageal adenocarcinomas: An immunohistochemical study. J Clin Pathol (2010) 63(11):994–8. doi: 10.1136/jcp.2010.080952
103. Zeng L-S, Yang X-Z, Wen Y-F, Mail S-J, Wang M-H, Zhang M-Y, et al. Overexpressed Hdac4 is associated with poor survival and promotes tumor progression in esophageal carcinoma. Aging (Albany NY) (2016) 8(6):1236–49. doi: 10.18632/aging.100980
104. Xue L, Hou J, Wang Q, Yao L, Xu S, Ge D. Rnai screening identifies Hat1 as a potential drug target in esophageal squamous cell carcinoma. Int J Clin Exp Pathol (2014) 7(7):3898–907.
105. Feng Y, Ma Z, Pan M, Xu L, Feng J, Zhang Y, et al. Wnt5a promotes the metastasis of esophageal squamous cell carcinoma by activating the Hdac7/Snail signaling pathway. Cell Death Dis (2022) 13(5):480. doi: 10.1038/s41419-022-04901-x
106. Li N, Tie X-J, Liu P-J, Zhang Y, Ren H-Z, Gao X, et al. Effects of down-regulation of Hdac6 expression on proliferation, cell cycling and migration of esophageal squamous cell carcinoma cells and related molecular mechanisms. Asian Pacific J Cancer Prevention: APJCP (2013) 14(2):685–9. doi: 10.7314/APJCP.2013.14.2.685
107. Zhang E, Han L, Yin D, He X, Hong L, Si X, et al. H3k27 acetylation activated-long non-coding rna Ccat1 affects cell proliferation and migration by regulating Spry4 and Hoxb13 expression in esophageal squamous cell carcinoma. Nucleic Acids Res (2017) 45(6):3086–101. doi: 10.1093/nar/gkw1247
108. Hu C, Liu M, Zhang W, Xu Q, Ma K, Chen L, et al. Upregulation of Klf4 by methylseleninic acid in human esophageal squamous cell carcinoma cells: Modification of histone H3 acetylation through Hat/Hdac interplay. Mol Carcinog (2015) 54(10):1051–9. doi: 10.1002/mc.22174
109. Liang Y, Chen X, Wu Y, Li J, Zhang S, Wang K, et al. Lncrna Casc9 promotes esophageal squamous cell carcinoma metastasis through upregulating Lamc2 expression by interacting with the creb-binding protein. Cell Death Differ (2018) 25(11):1980–95. doi: 10.1038/s41418-018-0084-9
110. Wu F, Xu L, Tu Y, Cheung OKW, Szeto LLM, Mok MTS, et al. Sirtuin 7 super-enhancer drives epigenomic reprogramming in hepatocarcinogenesis. Cancer Lett (2022) 525:115–30. doi: 10.1016/j.canlet.2021.10.039
111. Hoshino I, Matsubara H, Hanari N, Mori M, Nishimori T, Yoneyama Y, et al. Histone deacetylase inhibitor Fk228 activates tumor suppressor Prdx1 with apoptosis induction in esophageal cancer cells. Clin Cancer Research: an Off J Am Assoc Cancer Res (2005) 11(21):7945–52. doi: 10.1158/1078-0432.Ccr-05-0840
112. Murakami K, Matsubara H, Hoshino I, Akutsu Y, Miyazawa Y, Matsushita K, et al. Chap31 induces apoptosis only Via the intrinsic pathway in human esophageal cancer cells. Oncology (2010) 78(1):62–74. doi: 10.1159/000292362
113. Tzao C, Jin JS, Chen BH, Chung HY, Chang CC, Hsu TY, et al. Anticancer effects of suberoylanilide hydroxamic acid in esophageal squamous cancer cells in vitro and in vivo. Dis esophagus: Off J Int Soc Dis Esophagus (2014) 27(7):693–702. doi: 10.1111/dote.12127
114. Shoji M, Ninomiya I, Makino I, Kinoshita J, Nakamura K, Oyama K, et al. Valproic acid, a histone deacetylase inhibitor, enhances radiosensitivity in esophageal squamous cell carcinoma. Int J Oncol (2012) 40(6):2140–6. doi: 10.3892/ijo.2012.1416
115. Shi Y, Xiang Z, Yang H, Khan S, Li R, Zhou S, et al. Pharmacological targeting of Tns3 with histone deacetylase inhibitor as a therapeutic strategy in esophageal squamous cell carcinoma. Aging (2021) 13(11):15336–52. doi: 10.18632/aging.203091
116. Ma S, Liu T, Xu L, Wang Y, Zhou J, Huang T, et al. Histone deacetylases inhibitor Ms-275 suppresses human esophageal squamous cell carcinoma cell growth and progression Via the Pi3k/Akt/Mtor pathway. J Cell Physiol (2019) 234(12):22400–10. doi: 10.1002/jcp.28805
117. Feingold PL, Surman DR, Brown K, Xu Y, McDuffie LA, Shukla V, et al. Induction of thioredoxin-interacting protein by a histone deacetylase inhibitor, entinostat, is associated with DNA damage and apoptosis in esophageal adenocarcinoma. Mol Cancer Ther (2018) 17(9):2013–23. doi: 10.1158/1535-7163.Mct-17-1240
Keywords: super-enhancers, esophageal carcinoma, core regulatory circuitry, transcriptional dependence, histone acetylation
Citation: Shi Y, Wang M, Liu D, Ullah S, Ma X, Yang H and Liu B (2022) Super-enhancers in esophageal carcinoma: Transcriptional addictions and therapeutic strategies. Front. Oncol. 12:1036648. doi: 10.3389/fonc.2022.1036648
Received: 04 September 2022; Accepted: 10 October 2022;
Published: 27 October 2022.
Edited by:
Zhendong Jin, Second Military Medical University, ChinaReviewed by:
Muhammad Azhar Nisar, Tulane University, United StatesXiaoqun Dong, Brown University, United States
Copyright © 2022 Shi, Wang, Liu, Ullah, Ma, Yang and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bingrong Liu, ZmNjbGl1YnJAenp1LmVkdS5jbg==; Huiyu Yang, aHVpeXV5YW5nQDEyNi5jb20=; Xing Ma, ZmNjbWF4QHp6dS5lZHUuY24=
†These authors have contributed equally to this work