 Kefeng Shen
Kefeng Shen Meilan Zhang
Meilan Zhang Jiachen Wang
Jiachen Wang Wei Mu
Wei Mu Jin Wang
Jin Wang Chunyan Wang
Chunyan Wang Shugang Xing
Shugang Xing Zhenya Hong
Zhenya Hong Min Xiao
Min Xiao
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Oncol. , 19 October 2022
Sec. Hematologic Malignancies
Volume 12 - 2022 | https://doi.org/10.3389/fonc.2022.1036511
Fanconi anemia (FA) genes play critical roles in the repair of DNA lesions. Non-FA (or underlying FA) patients harboring heterozygous germline FA gene mutations may also face an increased risk of developing bone marrow failure, primary immunodeficiency disease, and hereditary cancer predisposition syndromes. We report a female patient who suffered from ovarian cancer at 50 years of age. During the initial treatment, six cycles of docetaxel and carboplatin (DC) combination chemotherapy were administered followed by two cycles of docetaxel maintenance therapy. Then, she received a routine follow-up every 3 months for the next 3 years, and all the results of the examination and laboratory tests were normal. Unfortunately, at 54 years of age, she developed a secondary cancer of therapy-related (t-) chronic myelomonocytic leukemia (t-CMML). After two courses of a highly intensive induction chemotherapy regimen with DAC (decitabine) and HAA (homoharringtonine, cytarabine), the patient suffered from severe and persistent bone marrow failure (BMF). Targeted next-generation sequencing (NGS) of a panel of 80 genes was performed on her initial bone marrow aspirate sample and identified PTPN11, NRAS, and DNMT3A somatic mutations. In addition, RNA sequencing (RNA-seq) revealed a rare NUP98-HOXC11 fusion. Whole-exome sequencing (WES) verified RAD51C, BRIP1, PALB2, and FANCG heterozygous germline mutations of the FA pathway, which were further confirmed in buccal swab samples by Sanger sequencing. For this patient, we hypothesized that an altered FA pathway resulted in genomic instability, hypersensitivity to DNA-crosslinking agents or cytotoxic chemotherapeutics, and unsuccessful DNA damage repair. Consequently, she developed ovarian cancer and secondary t-CMML and then suffered from BMF and delayed post−chemotherapy bone marrow recovery after several chemotherapy courses. This case highlights the importance of genetic counseling in patients with hematopoietic neoplasms with high clinical suspicion for carrying cancer susceptibility gene mutations, which require timely diagnosis and personalized management.
The Fanconi anemia (FA) pathway (also known as the FA-BRCA pathway) is involved in the repair of DNA lesions by homologous recombination, which plays a vital role in the maintenance of genomic stability (1). To date, researchers have already identified germline mutations in 22 specific genes associated with the FA pathway, each accounting for an individual FA complementation group (2). Patients with FA gene mutation are hypersensitive to DNA damage and unable to successfully repair damaged DNA when exposed to DNA-crosslinking agents, cytotoxic chemotherapeutics, and ionizing radiation (3, 4). Throughout the lifetime of patients with an FA gene mutation, DNA damage increasing accumulates, which would lead to a complex clinically and genetically heterogeneous disorder characterized by developmental abnormalities, bone marrow failure (BMF), immune deficiency, and a high risk of developing various cancers (e.g., Fanconi anemia, breast/ovarian cancer, leukemia) (5–7).
Here, we present the case of a 54-year-old female patient with multiple FA gene mutations. She was healthy, and with a normal history in her early years. Unfortunately, she developed ovarian cancer, secondary t-CMML, and post−chemotherapy BMF sequentially in her fifties. Comprehensive genetic testing showed that many molecular variations (including FA gene germline mutations, RAS and epigenetic pathway somatic mutations, and NUP98-HOXC11 fusion) were highly linked to her serious and complex medical history (Figures 1A, B).
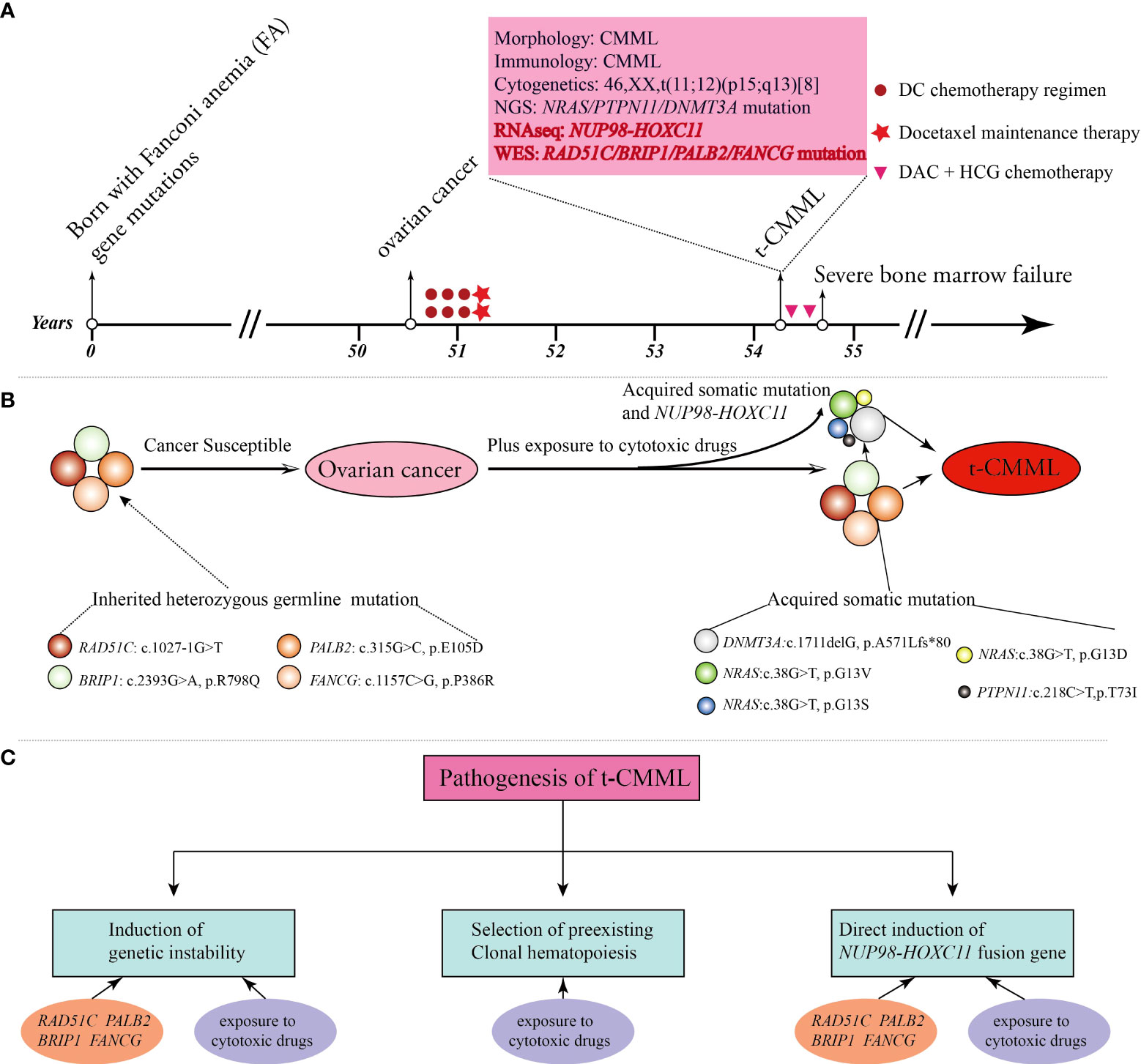
Figure 1 A brief summary of the medical history and comprehensive molecular-genetic analysis. (A) Patient timeline of key clinical events and laboratory test results. (B) The patient was born with four Fanconi anemia (FA) gene mutations and germline predisposition to cancers. She developed ovarian cancer, and then the altered FA pathway plus exposure to cytotoxic drugs promoted the emergence of acquired somatic mutations and the NUP98-HOXC11 fusion, all of which contributed to the development of secondary t-CMML. (C) Potential biological mechanisms contributing to the pathogenesis of t-CMML.
A 54-year-old female patient was admitted to the department of hematology in our hospital because of scattered ecchymosis on limb skin in July 2020. A complete blood count (CBC) test showed hyperleukocytosis and monocytosis with a white blood cell count (WBC) of 100.1×109/L, monocyte count of 47.1×109/L, hemoglobin level (HGB) of 72.0 g/L, and platelet count (PLT) of 16.0×109/L. An abdominal ultrasound scan showed that the thickness of the spleen was approximately 3.2 cm. Contrast-enhanced CT scans of the whole abdomen indicated no signs of ovarian cancer recurrence. Peripheral blood smear examination revealed monocytosis with an increased monocyte percentage of 56.0% and naïve monocyte percentage of 15.0% in total nucleated cells. Morphological examination of a bone marrow (BM) aspirate revealed dysplasia in granulocytic and megakaryocytic cell lineages and an increased naïve monocyte percentage (approximately 10%) (Figure 2A). BM biopsy analysis revealed marked hypercellularity (approximately 90%) with prominent naïve monocytosis (Figure S1; Supplemental Material). Multiparametric flow cytometry analysis of BM aspirates showed that abnormal myeloid blasts with a percentage of 4.9% in total nucleated cells expressed the markers CD34, CD117, CD38, CD9dim, CD13dim, and CD33 and that naïve monocytes with a percentage of 8.4% in total nucleated cells expressed the markers CD64bri, CD11cbri, CD15dim, and CD14neg (Figure S2; Supplemental Material). Cytogenetic analysis revealed an abnormal karyotype of “46, XX, t (11; 12)(p15; q13) [8]” (Figure 2B).
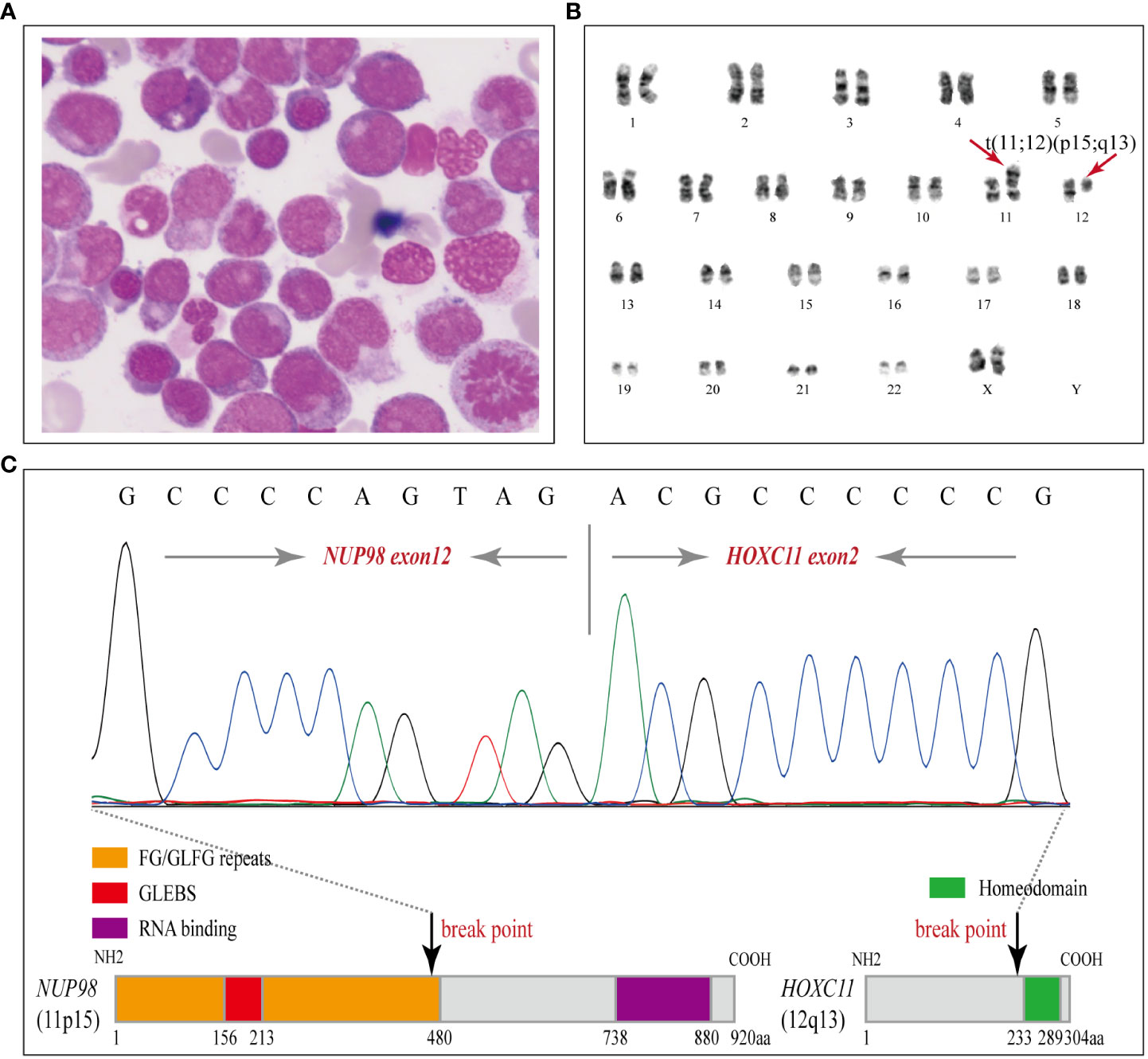
Figure 2 Morphology, karyotyping and molecular characterization of the NUP98-HOXC11 fusion. (A) A diagnostic bone marrow (BM) aspirate smear revealed dysplasia in granulocytic and megakaryocytic cell lineages and enriched naïve monocytes (Wright stained, × 1000). (B) Karyotyping performed on the diagnostic BM revealed 46, XX, t(11;12)(p15;q13) [8]. (C) Upper panel, Sanger sequencing was performed to verify the NUP98-HOXC11 fusion transcript containing exon 12 of NUP98 fused to exon 2 of HOXC11. Lower panel, a schematic diagram showing the NUP98-HOXC11 in-frame fusion and phenylalanine-glycine/glycine-leucine-phenylalanine-glycine (FG/GLFG) repeats and Gle2-binding sequence (GLEBS) domain in the amino-terminal portion of NUP98 retained in the fusion protein.
To clarify the molecular mechanism, comprehensive genetic testing was conducted (Supplemental Material). Targeted next-generation sequencing (NGS) of a panel of 80 genes was performed on her initial BM aspirate sample (Illumina NextSeq550 platform, amplicon library prep, amplicon mean coverage: 1813.4×, percent Q30 bases: 97.7%, uniformity of coverage: 97.8%) and identified PTPN11 (T73I: 2.5%), NRAS (G12D: 3.2%, G12S: 7.7%, G13V: 22.4%), and DNMT3A (A571Lfs*80: 49.1%) somatic mutations. In addition, RNA sequencing (RNA-seq) identified a rare NUP98-HOXC11 fusion. The breakpoints of NUP98 and HOXC11 were located in intron 12 or intron 1, respectively, resulting in a NUP98-HOXC11 in-frame fusion transcript containing exon 12 of NUP98 fused to exon 2 of HOXC11. To confirm the NUP98-HOXC11 fusion, reverse transcription polymerase chain reaction (RT-PCR) was performed with a pair of primers, forward (at NUP98 exon 12): 5’-TCTTGGTACAGGAGCCTTTGG-3’, and reverse (at HOXC11 exon 2): 5’-GTTCCCGGATCTGGAATTTCG-3’, and then the amplified PCR products were purified to perform Sanger sequencing to verify the above finding (Figure 2C). Whole-exome sequencing (WES) of diagnostic BM samples identified four cancer susceptibility gene mutations associated with the FA-BRCA pathway, RAD51C (NM_058216.2: c.1027-1G>T), BRIP1 (NM_032043.2: c.2393G>A, p. R798Q), PALB2 (NM_024675.3: c.315G>C, p. E105D), and FANCG (NM_004629.1: c.1157C>G, p. P386R), which were further verified as heterozygous germline mutations by Sanger sequencing with buccal swab samples (Figure S3, Supplemental Material). RAD51C splicing mutation was predicted to be deleterious by the Human Splicing Finder and absent from the current Exome Aggregation Consortium (ExAC), 1000 Genomes Project (1000G), and Genome Aggregation Database (gnomAD), indicating that it may be a pathogenic/likely pathogenic variant. BRIP1, PALB2, and FANCG missense mutations were all rare in the current ExAC, 1000G, and gnomAD databases (Minor allele frequencies < 0.0001), and reported in ClinVar under “uncertain significance.” Furthermore, no other exonic variants were detected in genes associated with predisposition to cancer.
The patient’s past medical history provided the medical context that she had developed ovarian cancer with no history of cancer in her family in September 2016 (four years prior). Then she received six cycles of docetaxel and carboplatin (DC) combination chemotherapy [docetaxel, intravenous (I.V.) infusion by 60 minutes, 70 mg/m2, d1; carboplatin, I.V. infusion over 30 minutes, AUC (area under the plasma-concentration-versus-time curve) of 5 mg/mL/min, d1], followed by two cycles of docetaxel maintenance therapy (docetaxel, I.V. infusion by 60 minutes, 70 mg/m2, d1) after a definitive diagnosis from October 2016 to May 2017 in the medical oncology department of our hospital. After the primary treatment, she received a routine follow-up nearly every 3 months for the next 3 years. All the obtained results of the examination and laboratory tests were normal, and she had no signs or symptoms of ovarian cancer recurrence.
Based on the above results, the patient’s final diagnosis was t-CMML with the NUP98-HOXC11 fusion and secondary to primary ovarian cancer with inherited cancer predisposition gene mutations of FA genes. Considering the extremely poor prognosis, two courses of a highly intensive induction chemotherapy regimen with DAC (decitabine, 20 mg/m2, d1-5) and HAA (homoharringtonine, 2 mg/m2, d1-7; cytarabine, 10 mg/m2, q12h, d1-10) were administered from 2 July 2020 to 20 August 2020. Subsequently, the patient presented with obvious pancytopenia and suffered from severe and persistent bone marrow suppression and delayed post−chemotherapy bone marrow recovery with a poor response to G-CSF treatment. On 2 September 2020, a CBC test indicated remaining pancytopenia with a WBC of 0.9×109/L, HGB of 73.0 g/L, and PLT of 36.0×109/L. Then, the patient asked to be discharged and transferred to her local hospital for personal reasons.
Fanconi anemia (FA) is a rare human genetic disease that occurs following germline mutations associated with the FA pathway and is clinically characterized by malformations, BMF, immune deficiency, and an extremely high predisposition to various cancers. The “FA pathway” is also known as the “FA-BRCA pathway” because many of the FA genes are related to breast cancer (BRCA) and/or ovarian cancer (7, 8). FA patients display a wide spectrum of diverse clinical manifestations; even so, only a small number of them present a typical phenotype, and one−third of individuals with FA have a normal appearance (6). Thus, it is a major challenge for clinicians to make an accurate diagnosis for FA patients. Chromosome breakage analysis may be a gold standard test, which is, nonetheless, not widely applied due to its false-negative traits and the inaccessibility in some poorly equipped hospitals (9, 10). According to previous literature, non-FA (or underlying FA) patients carrying germline FA gene mutations also present with an increased risk of developing BMF, immune deficiency, and cancer susceptibility under some circumstances (5, 11, 12).
Our patient had no signs or symptoms of developmental abnormalities or organ defects, and unfortunately, she did not receive a chromosome breakage test. Genetic testing indicated that she was born with four FA-BRCA pathway gene mutations in RAD51C, BRIP1, PALB2, and FANCG. Moreover, RAD51C (RAD51 paralog C, alias FANCO), BRIP1 (BRCA1−interacting protein carboxy−terminal helicase 1, alias FANCJ), and PALB2 (partner and localizer of BRCA2, alias FANCN) have been fully implicated in an increased risk for breast and ovarian cancer (Table 1) (13–15). We hypothesize that the patient was possibly born with an increased predisposition to develop BMF, hematopoietic neoplasms, and breast and ovarian cancer due to a mild or moderate impaired FA-BRCA pathway.

Table 1 Association of defects in FA genes with predisposition to non-FA cancers.
At the cellular level, her cells are hypersensitive to DNA-crosslinking agents, cytotoxic chemotherapeutics, and ionizing radiation due to corrupted DNA damage repair (3, 5). Over time, cumulative genomic instability renders cells malignant by clonal evolution and neoplastic transformation through uncontrolled proliferation, escape from programmed cell death, and evasion of immune surveillance. Consequently, she developed ovarian cancer at 50 years of age and then received six cycles of docetaxel and carboplatin (DC) combination chemotherapy followed by two cycles of docetaxel maintenance therapy. Cytotoxic chemotherapeutic exposure aggravated the tendency of hematopoietic stem progenitor cells to undergo malignant transformation and BMF. Three years later, she suffered from a secondary cancer of t-CMML and severe and persistent BMF after two courses of an induction chemotherapy regimen with DAC-HAA. The potential biological mechanisms behind the pathogenesis of t-CMML could be generally summarized as follows (1). Genomic instability due to the RAD51C, BRIP1, PALB2, and FANCG germline mutations of the FA pathway and long-term exposure to cytotoxic drugs aggravated the disease. (2). The selective pressure of cytotoxic drugs promoted the emergence of somatic variants (DNMT3A, NRAS, and PTPN11 mutations) in subclone cells and led to the development of clonal hematopoiesis and evolution to hematopoietic malignancies (16, 17) (3). NUP98-HOXC11 fusion also impaired hematopoietic stem cell self-renewal and caused malignant transformation directly (18) (Figure 1C).
t-CMML is a rare hematologic malignancy characterized by exposure to prior chemotherapy and/or radiation therapy used to treat a primary cancer (19). Takahashi K et al. (2013) performed the first systematic study to show the clinical characteristics and outcomes of t-CMML among a reasonably large number of 358 CMML patients (20). A total of 39 (11%) patients were defined as having t-CMML, and the median latency to develop t-CMML was 6 years (range: 1-32 years). The most common primary cancers in the t-CMML group were lymphoid malignancies (37%), followed by prostate cancer (31%) and breast or ovarian cancer (18%). They concluded that t-CMML was associated with higher-risk cytogenetics and manifests a poor prognosis. Subsequently, Subari S et al. (2015) and Patnaik MM et al. (2017) conducted two other large-scale studies of t-CMML in 265 and 497 CMML patients (21, 22), respectively, and they drew many conclusions similar to those of Takahashi K et al. To the best of our knowledge, no analysis of germline mutations in cancer susceptibility genes in t-CMML has been conducted, although systematic studies of the clinical characteristics and prognosis of the disease have been conducted. Thus, our case report may be the first relevant study.
NUP98 fusion was first described by Nakamura T et al. and Borrow J et al. (1996) in AML patients with recurrent t (7;11) (p15; p15) translocation that resulted in a NUP98-HOXA9 fusion gene (23, 24). Since then, it has been identified in a spectrum of hematologic malignancies, including AML, MDS, CML, T-ALL, and mixed-phenotype acute leukemia (MPAL), with a low incidence (<5%) (25). NUP98 fusion can induce abnormal transcriptional or epigenetic regulation and drive leukemogenesis through complex molecular mechanisms (18). In particular, NUP98 fusion is rarely reported in CMML patients, and we found only four relevant cases to date (Table 2). Currently, over 30 NUP98 fusion partners have been identified, but the HOXC11 partner is fairly rare. There are only four NUP98-HOXC11 fusion cases reported by Taketani T et al. (2002) and Gu BW et al. (2003) (30, 31), which identified one case in a pediatric patient and three in adult patients with de novo AML. To the best of our knowledge, this is the first report identifying a rare NUP98-HOXC11 fusion in a t-CMML patient.

Table 2 Summary of CMML cases with NUP98 fusions reported in the literature.
In summary, we report the first case of a non-FA (or underlying FA) patient born with multiple FA gene mutations who developed ovarian cancer and secondary t-CMML with a rare NUP98-HOXC11 fusion. Conventional chemotherapy and radiation therapy appear to be “double-edged swords”, as they kill cancer cells and induce secondary BMF, immune deficiency, and cancer (32). Particularly, it is difficult for clinicians to balance the clinical efficacy and adverse outcomes in cancer patients carrying cancer susceptibility gene mutations. From this case, we learn that cancer patients with high clinical suspicion for germline predisposition, such as secondary or early-onset hematologic malignancy patients, are desirable for appropriate genetic counseling and testing. Actually, those patients may benefit from timely diagnosis and personalized management, such as a better donor selection strategy, reduced-intensity conditioning for allogeneic hematopoietic stem cell transplantation, altered cytotoxic chemotherapeutics, and lifelong cancer surveillance.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/bioproject/PRJNA881251/.
The studies involving human participants were reviewed and approved by The Medical Ethics Committee of Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology. The patients/participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
KS collected and analyzed data, wrote the manuscript, and drew the figures. MZ performed RNAseq and analyzed data. JCW performed NGS and analyzed data. WM performed WES and analyzed data. JW performed morphological examination. CW performed flow cytometry and analyzed data. SX performed cytogenetic analysis. ZH and MX directed the research and reviewed the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the National Natural Science Foundation of China (No.81770211 to MX).
The authors would like to thank the patient and her family for consenting to report this rare case, Perfectgen Diagnostics (Ezhou, Hubei Province) for providing DNA sequencing service, and all the faculty and staff in our Clinical and Laboratory Unit for clinical and technical support.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2022.1036511/full#supplementary-material
1. Ceccaldi R, Sarangi P, D'Andrea AD. The fanconi anaemia pathway: New players and new functions. Nat Rev Mol Cell Biol (2016) 17(6):337–49. doi: 10.1038/nrm.2016.48
2. Taylor AMR, Rothblum-Oviatt C, Ellis NA, Hickson ID, Meyer S, Crawford TO, et al. Chromosome instability syndromes. Nat Rev Dis Primers (2019) 5(1):64. doi: 10.1038/s41572-019-0113-0
3. Helbling-Leclerc A, Garcin C, Rosselli F. Beyond DNA repair and chromosome instability-fanconi anaemia as a cellular senescence-associated syndrome. Cell Death Differ (2021) 28(4):1159–73. doi: 10.1038/s41418-021-00764-5
4. Longerich S, Li J, Xiong Y, Sung P, Kupfer GM. Stress and DNA repair biology of the fanconi anemia pathway. Blood (2014) 124(18):2812–9. doi: 10.1182/blood-2014-04-526293
5. Nepal M, Che R, Zhang J, Ma C, Fei P. Fanconi anemia signaling and cancer. Trends Cancer (2017) 3(12):840–56. doi: 10.1016/j.trecan.2017.10.005
6. Nalepa G, Clapp DW. Fanconi anaemia and cancer: An intricate relationship. Nat Rev Cancer (2018) 18(3):168–85. doi: 10.1038/nrc.2017.116
7. D'Andrea AD. Susceptibility pathways in fanconi's anemia and breast cancer. N Engl J Med (2010) 362(20):1909–19. doi: 10.1056/NEJMra0809889
8. Levy-Lahad E. Fanconi anemia and breast cancer susceptibility meet again. Nat Genet (2010) 42(5):368–9. doi: 10.1038/ng0510-368
9. Li N, Ding L, Li B, Wang J, D'Andrea AD, Chen J. Functional analysis of fanconi anemia mutations in China. Exp Hematol (2018) 66:32–41.e8. doi: 10.1016/j.exphem.2018.07.003
10. Mahmood R, Mahmood A, Khan SA, Jaffar R. An experience with 124 cases of fanconi anemia: clinical spectrum, hematological parameters and chromosomal breakage analysis. Am J Blood Res (2021) 11(5):498–503.
11. Bluteau O, Sebert M, Leblanc T, Peffault de Latour R, Quentin S, Lainey E, et al. A landscape of germline mutations in a cohort of inherited bone marrow failure patients. Blood (2018) 131(7):717–32. doi: 10.1182/blood-2017-09-806489
12. Bousfiha A, Jeddane L, Picard C, Al-Herz W, Ailal F, Chatila T, et al. Human inborn errors of immunity: 2019 update of the IUIS phenotypical classification. J Clin Immunol (2020) 40(1):66–81. doi: 10.1007/s10875-020-00758-x
13. Meindl A, Hellebrand H, Wiek C, Erven V, Wappenschmidt B, Niederacher D, et al. Germline mutations in breast and ovarian cancer pedigrees establish RAD51C as a human cancer susceptibility gene. Nat Genet (2010) 42(5):410–4. doi: 10.1038/ng.569
14. Moyer CL, Ivanovich J, Gillespie JL, Doberstein R, Radke MR, Richardson ME, et al. Rare BRIP1 missense alleles confer risk for ovarian and breast cancer. Cancer Res (2020) 80(4):857–67. doi: 10.1158/0008-5472.CAN-19-1991
15. Antoniou AC, Casadei S, Heikkinen T, Barrowdale D, Pylkäs K, Roberts J, et al. Breast-cancer risk in families with mutations in PALB2. N Engl J Med (2014) 371(6):497–506. doi: 10.1056/NEJMoa1400382
16. Bolton KL, Ptashkin RN, Gao T, Braunstein L, Devlin SM, Kelly D, et al. Cancer therapy shapes the fitness landscape of clonal hematopoiesis. Nat Genet (2020) 52(11):1219–26. doi: 10.1038/s41588-020-00710-0
17. Bowman RL, Busque L, Levine RL. Clonal hematopoiesis and evolution to hematopoietic malignancies. Cell Stem Cell (2018) 22(2):157–70. doi: 10.1016/j.stem.2018.01.011
18. Xu H, Valerio DG, Eisold ME, Sinha A, Koche RP, Hu W, et al. NUP98 fusion proteins interact with the NSL and MLL1 complexes to drive leukemogenesis. Cancer Cell (2016) 30(6):863–78. doi: 10.1016/j.ccell.2016.10.019
19. Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, et al. The 2016 revision to the world health organization classification of myeloid neoplasms and acute leukemia. Blood (2016) 127(20):2391–405. doi: 10.1182/blood-2016-03-643544
20. Takahashi K, Pemmaraju N, Strati P, Nogueras-Gonzalez G, Ning J, Bueso-Ramos C, et al. Clinical characteristics and outcomes of therapy-related chronic myelomonocytic leukemia. Blood (2013) 122(16):2807–920. doi: 10.1182/blood-2013-03-491399
21. Subari S, Patnaik M, Alfakara D, Gangat N, Elliott M, Hogan W, et al. Patients with therapy-related CMML have shorter median overall survival than those with De novo CMML: Mayo clinic long-term follow-up experience. Clin Lymphoma Myeloma Leuk (2015) 15(9):546–9. doi: 10.1016/j.clml.2015.06.002
22. Patnaik MM, Vallapureddy R, Yalniz FF, Hanson CA, Ketterling RP, Lasho TL, et al. Therapy related-chronic myelomonocytic leukemia (CMML): Molecular, cytogenetic, and clinical distinctions from de novo CMML. Am J Hematol (2018) 93(1):65–73. doi: 10.1002/ajh.24939
23. Nakamura T, Largaespada DA, Lee MP, Johnson LA, Ohyashiki K, Toyama K, et al. Fusion of the nucleoporin gene NUP98 to HOXA9 by the chromosome translocation t(7;11)(p15;p15) in human myeloid leukaemia. Nat Genet (1996) 12(2):154–8. doi: 10.1038/ng0296-154
24. Borrow J, Shearman AM, Stanton VP Jr, Becher R, Collins T, Williams AJ, et al. The t(7;11)(p15;p15) translocation in acute myeloid leukaemia fuses the genes for nucleoporin NUP98 and class I homeoprotein HOXA9. Nat Genet (1996) 12(2):159–67. doi: 10.1038/ng0296-159
25. Michmerhuizen NL, Klco JM, Mullighan CG. Mechanistic insights and potential therapeutic approaches for NUP98-rearranged hematologic malignancies. Blood (2020) 136(20):2275–89. doi: 10.1182/blood.2020007093
26. Hatano Y, Miura I, Nakamura T, Yamazaki Y, Takahashi N, Miura AB. Molecular heterogeneity of the NUP98/HOXA9 fusion transcript in myelodysplastic syndromes associated with t(7;11)(p15;p15). Br J Haematol (1999) 107(3):600–4. doi: 10.1046/j.1365-2141.1999.01754.x
27. Wong KF, So CC, Kwong YL. Chronic myelomonocytic leukemia with t(7;11)(p15;p15) and NUP98/HOXA9 fusion. Cancer Genet Cytogenet (1999) 115(1):70–2. doi: 10.1016/s0165-4608(99)00085-0
28. Soler G, Kaltenbach S, Dobbelstein S, Broccardo C, Radford I, Mozziconacci MJ, et al. Identification of GSX2 and AF10 as NUP98 partner genes in myeloid malignancies. Blood Cancer J (2013) 3(7):e124. doi: 10.1038/bcj.2013.20
29. Hayashi Y, Harada Y, Kagiyama Y, Nishikawa S, Ding Y, Imagawa J, et al. NUP98-HBO1-fusion generates phenotypically and genetically relevant chronic myelomonocytic leukemia pathogenesis. Blood Adv (2019) 3(7):1047–60. doi: 10.1182/bloodadvances.2018025007
30. Taketani T, Taki T, Shibuya N, Kikuchi A, Hanada R, Hayashi Y. Novel NUP98-HOXC11 fusion gene resulted from a chromosomal break within exon 1 of HOXC11 in acute myeloid leukemia with t(11;12)(p15;q13). Cancer Res (2002) 62(16):4571–4.
31. Gu BW, Wang Q, Wang JM, Xue YQ, Fang J, Wong KF, et al. Major form of NUP98/HOXC11 fusion in adult AML with t(11;12)(p15;q13) translocation exhibits aberrant trans-regulatory activity. Leukemia (2003) 17(9):1858–64. doi: 10.1038/sj.leu.2403036
Keywords: Fanconi anemia gene, hereditary cancer predisposition syndromes, ovarian cancer, therapy-related CMML (t-CMML), NUP98-HOXC11 fusion
Citation: Shen K, Zhang M, Wang J, Mu W, Wang J, Wang C, Xing S, Hong Z and Xiao M (2022) Inherited heterozygous Fanconi anemia gene mutations in a therapy-related CMML patient with a rare NUP98-HOXC11 fusion: A case report. Front. Oncol. 12:1036511. doi: 10.3389/fonc.2022.1036511
Received: 04 September 2022; Accepted: 28 September 2022;
Published: 19 October 2022.
Edited by:
Mohamed A. Yassin, Hamad Medical Corporation, QatarReviewed by:
Najim Ameziane, Cincinnati Children’s Hospital Medical Center, United StatesCopyright © 2022 Shen, Zhang, Wang, Mu, Wang, Wang, Xing, Hong and Xiao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Min Xiao, eGlhb21pbkB0amgudGptdS5lZHUuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.