 Xianwen Hu
Xianwen Hu Qi Huang1
Qi Huang1 Jiong Cai
Jiong Cai
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Oncol., 13 October 2022
Sec. Cancer Imaging and Image-directed Interventions
Volume 12 - 2022 | https://doi.org/10.3389/fonc.2022.1035800
This article is part of the Research TopicCase Reports in Cancer Imaging and Image-directed Interventions : 2022View all 10 articles
Introduction: Adult primary intracranial Ewing sarcomas (EWs)/primitive neuroectodermal tumors (PNETs) are extremely rare, with only 30 patients published before us. The imaging features and treatment strategies of primary intracranial EWs/PNETs are unclear due to its rarity. The aim of this study was to investigate the clinical features, imaging findings, treatment, survival analysis, and prognosis of adult EWs/PNETs, and a systematic review was conducted based on the patient we treated and published literature.
Case description: A 19-year-old male patient suffered from head pain due to an accidental fall on a motorcycle that occurred more than 10 days before going to the hospital, and underwent computed tomography (CT) examination; it was found that the left temporo-occipital fossa was occupied. Magnetic resonance imaging (MRI) was recommended to understand the nature of the lesion, and the result showed that it has a high probability of being a meningioma. He underwent surgical removal of the mass under general anesthesia, and surprisingly, postoperative pathology revealed EWs/PNET. The disease has a high degree of malignancy, and the patient developed multiple metastases throughout the body 5 years after surgery.
Conclusion: Primary intracranial EWs/PNETs in adult patients are rare, of which imaging findings should be considered as one of the differential diagnoses of meningioma, hemangiopericytoma, and malignant triton tumor. Larger solid-cystic masses with septum-like enhancement may be relatively specific imaging findings of intracranial EWs/PNETs. The prognosis of primary adult intracranial EWs/PNETs is poor. Radical tumor resection combined with radiotherapy and chemotherapy is currently the main and possibly the most effective treatment method.
Ewing sarcomas (EWs) and primitive neuroectodermal tumors (PNETs) share the same genetic and histological features; thus, they are collectively referred to as EWs/PNETs, which originate in the neuroectoderm and are mainly composed of primitive neuroectodermal cells (1). EWs/PNETs are highly malignant small round cell tumors with multi-directional differentiation potential, and can be divided into two types according to their origin in bone or soft tissue: intraosseous and extraosseous (2). About 85%–90% of the patients with EWs/PNET show the typical chromosomal translocation t(11;22)(q24;12), which leads to the fusion of the gene EWSR1 and the ETS family gene FLI1 (1, 3). The clinical manifestations of intracranial EWs/PNETs usually include headache, vomiting, dizziness, and other symptoms of intracranial hypertension, and some patients may manifest as eye movement disorders, decreased vision, and unsteady gait (4). Laboratory tests for intracranial EWs/PNETs are usually non-positive, of which imaging studies are often misdiagnosed as meningiomas due to their rarity and mostly dural origin, and the diagnosis of the disease is mainly based on pathology and immunohistochemistry. The prognosis of intracranial EWs/PNETs is poor, associated with age more than 14 years at diagnosis, initial tumor volume more than 200 ml, being male, development of metastases, etc. (2, 5, 6). The primary intracranial EWs/PNETs are extremely rare, the incidence of which is lower in adults than in children, and the published literature is only found in case or case series. Herein, we report a 19-year-old male patient with pathologically confirmed EWs/PNET originating in the left temporo-occipital fossa, who was discovered incidentally on imaging after an accidental fall. Moreover, we reviewed the published literature on adults (≥18 years of age) with intracranial EWs, summarized the radiological and clinical features of this rare tumor, and discussed its imaging differential diagnosis in detail.
A 19-year-old male patient suffered from head pain due to an accidental fall on a motorcycle that occurred more than 10 days before going to the hospital, and underwent computed tomography (CT) examination; a mass was found in the left temporo-occipital fossa. He was admitted to our hospital for further diagnosis and treatment. His parents were healthy and no family members had a history of similar diseases. Magnetic resonance imaging (MRI) was recommended to understand the nature of the lesion, and the result showed that it has a high probability of being a meningioma (as shown in Figure 1). No obvious positive signs were found in physical examination, and all laboratory indexes were within the normal reference value range. Based on the established diagnosis of a left temporo-occipital fossa space-occupying lesion, the patient underwent surgical resection of the tumor under general anesthesia. During the operation, the left temporoparietal horseshoe-shaped skin flap was taken, and the scalp was incised in full thickness to expose the skull and then a bone flap with a size of about 6 cm × 6 cm was removed. After the bone flap was taken out, it was found that the dura of about 2 cm × 2 cm was invaded by the tumor and broke through, and the inner plate of the covering skull was not invaded by the tumor. The dura was cut along the edge of the tumor invading the dura, and the tumor was removed together. The tumor tissue was gray, with clear demarcation from the surrounding brain tissue, abundant blood supply, soft texture, and a size of about 4.0 cm × 5.0 cm × 4.0 cm. Postoperatively, the resected tumor tissue was sent for pathological examination, and the results showed that there were diffusely distributed round cells in the tumor (as shown in Figure 2). Immunohistochemical staining showed that tumor cells positively expressed CD99, Vimentin, Bcl-2, ki-67 (about 40%), weakly expressed CD56, SMA, and negatively expressed CD34, CgA, CK, Desmin, EMA, S100, etc. Ewing’s sarcoma breakpoint region 1 (EWSR1) gene rearrangement was identified by fluorescence in situ hybridization in a foreign hospital. Based on these findings, the patient was diagnosed with intracranial extraosseous EWs/PNET. He did not undergo further radiotherapy and chemotherapy after the operation. At the 5th year after the operation, the patient was re-admitted to the hospital because of multiple bone pains throughout the body. 18F-FDG PET/CT was recommended to evaluate the patient’s systemic condition, and the results revealed multiple high radioactive uptake foci throughout the body (as shown in Figure 3). Biopsy of the lesion at the iliac bone revealed the same disease as he had previously suffered. He then received a vincristine–cyclophosphamide–doxorubicin chemotherapy regimen and palliative radiation targeting the lesions of the humerus, ilium, and acetabulum. Unfortunately, he passed away 3 months after starting maintenance therapy with chemoradiotherapy.
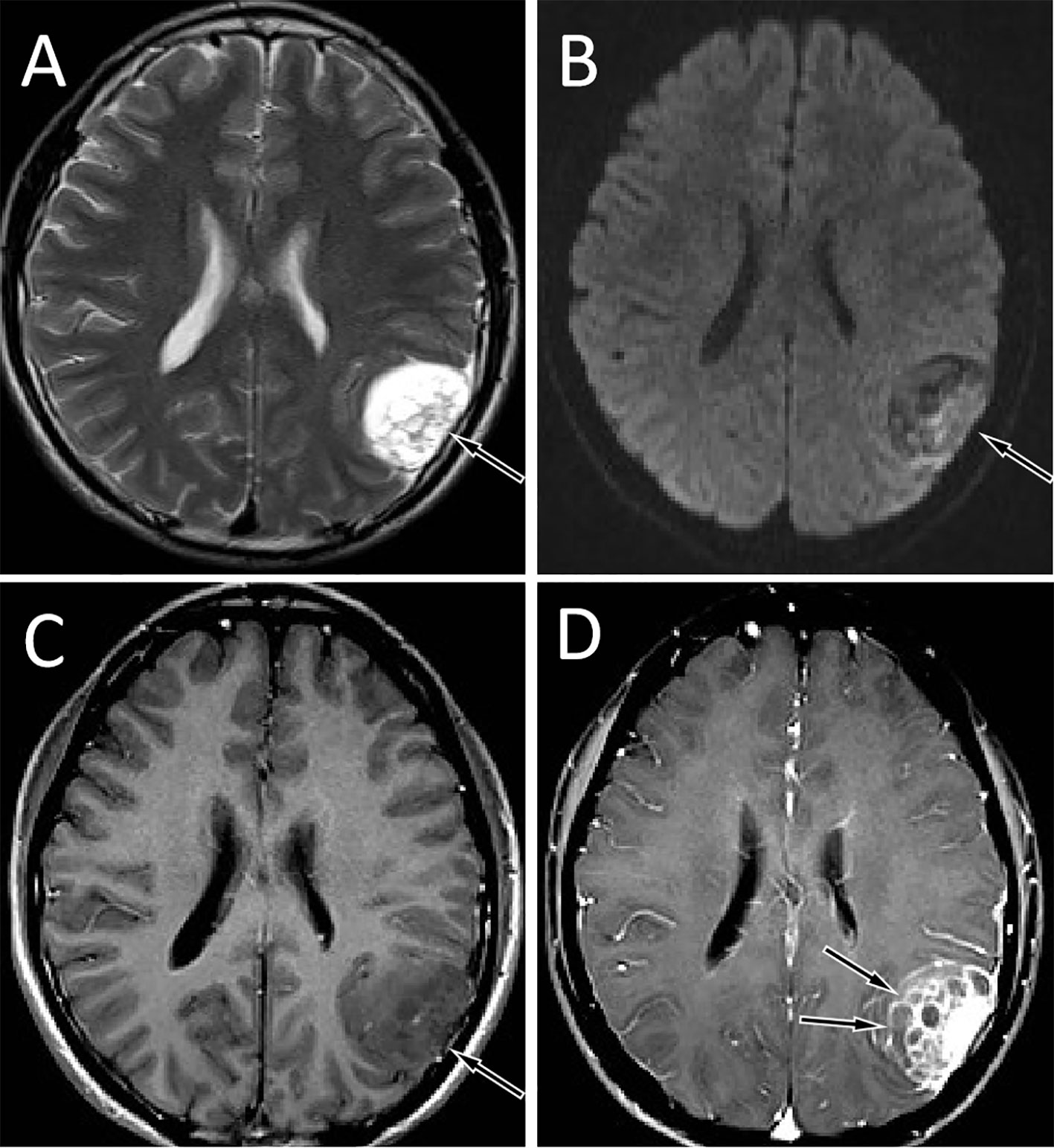
Figure 1 Brain MRI showed that the tumor was located in the left temporo-occipital fossa. T2WI sequence showed that the lesion was high signal (A, arrow). Diffusion-weighted imaging showed that the movement of water molecules was not restricted and showed heterogeneous low signal (B, arrow). The T1WI sequence showed that the lesion was slightly low signal (C, arrow), and the solid component of the tumor was significantly enhanced on contrast-enhanced T1WI, with multiple unenhanced cysts around it, and septum-like enhancement was seen in between (D, arrows).
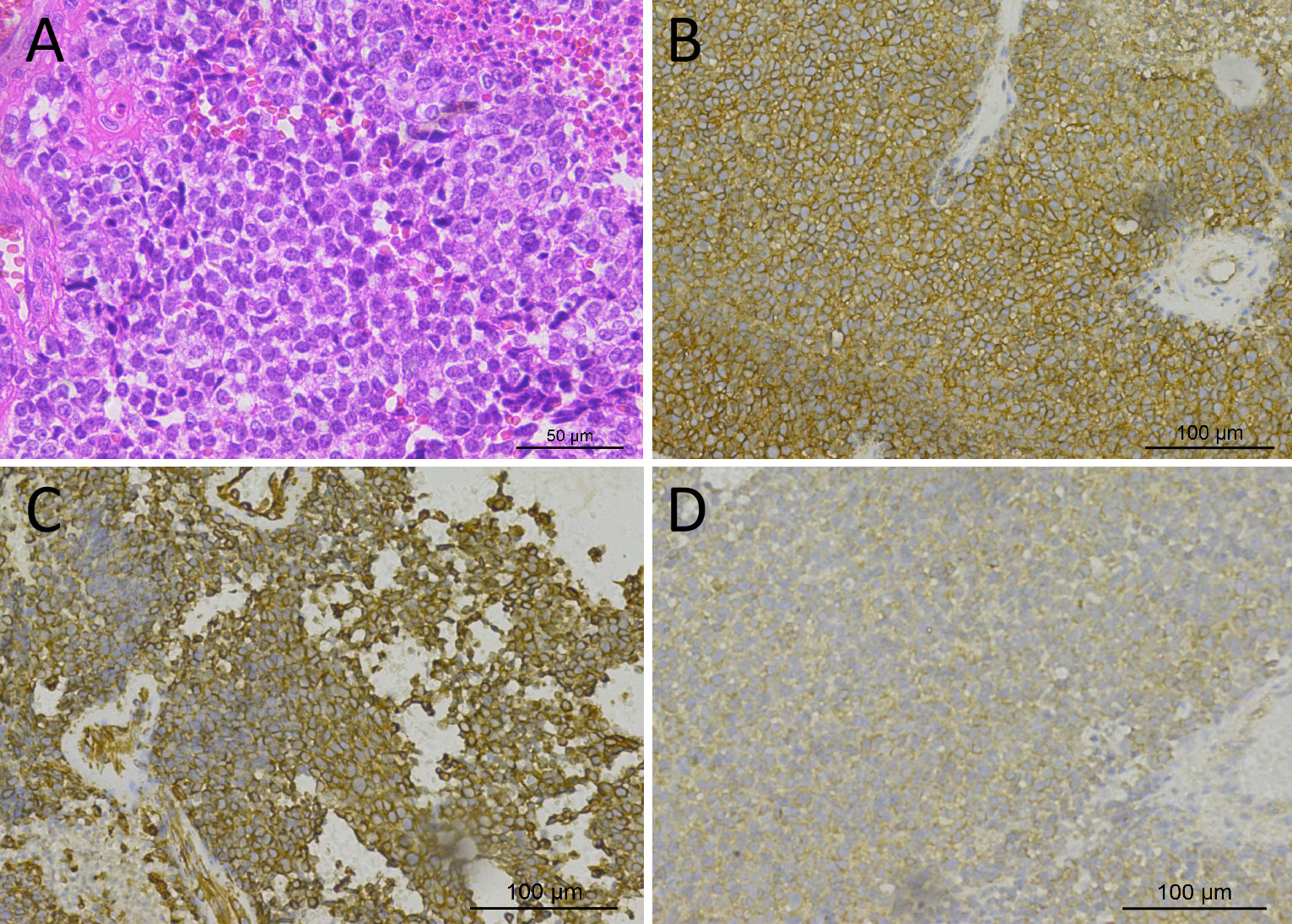
Figure 2 Hematoxylin–eosin staining showed that the tumor tissue was composed of small round cells with relatively uniform morphology, and the nuclei were deeply stained (A). Immunohistochemical staining showed that tumor cells positively expressed CD99 (B), Vimentin (C), and Bcl-2 (D).
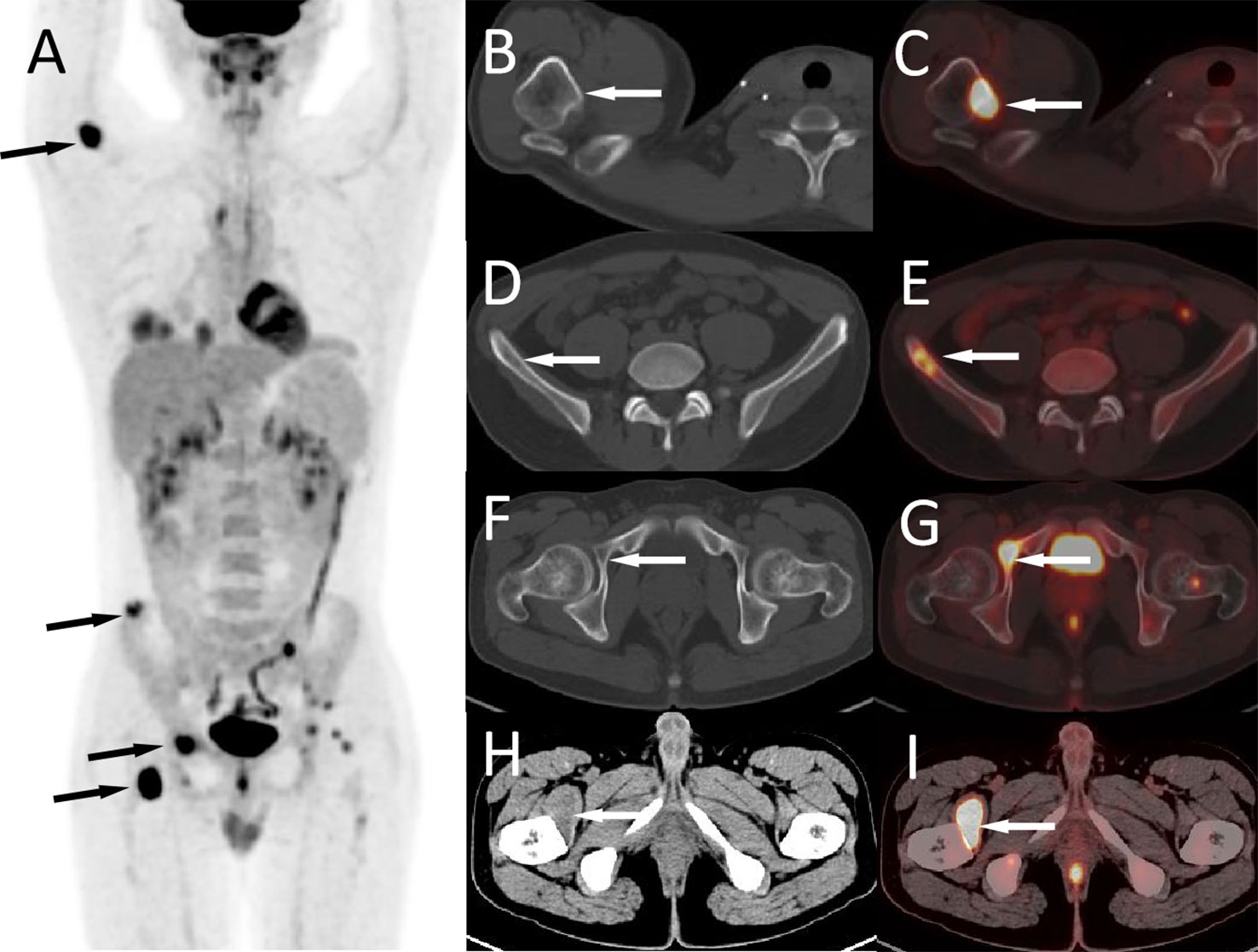
Figure 3 The patient’s 18F-FDG PET/CT at 5 years after operation; the maximum intensity projection figure showed multiple FDG concentration foci throughout the body (A, arrows). Axial figures showed that the right humerus (B, CT; C, PET/CT fusion), ilium (D, CT; E, PET/CT fusion), and acetabulum (F, CT; G, PET/CT fusion) can be seen with iso-density or slightly hypodense bone destruction areas with high radioactive uptake (arrows). Moreover, a hypodense mass with FDG accumulation was seen in the right iliopsoas muscle (H, CT; I, PET/CT fusion, arrows).
Primary intracranial EWs/PNET case reports and case series published in PubMed, Embase, and Web of Science databases as of 1 June 2022 were searched, with language restrictions limited to English. The following keywords were used: Ewing’s sarcoma, EWs, primitive neuroectodermal tumors, PNET, intracranial, and dura mater. The first author, publication year and country, as well as the patient’s age, gender, main clinical symptoms, CT and MRI imaging findings, treatment methods, and follow-up results were recorded for each case (as shown in Table 1).
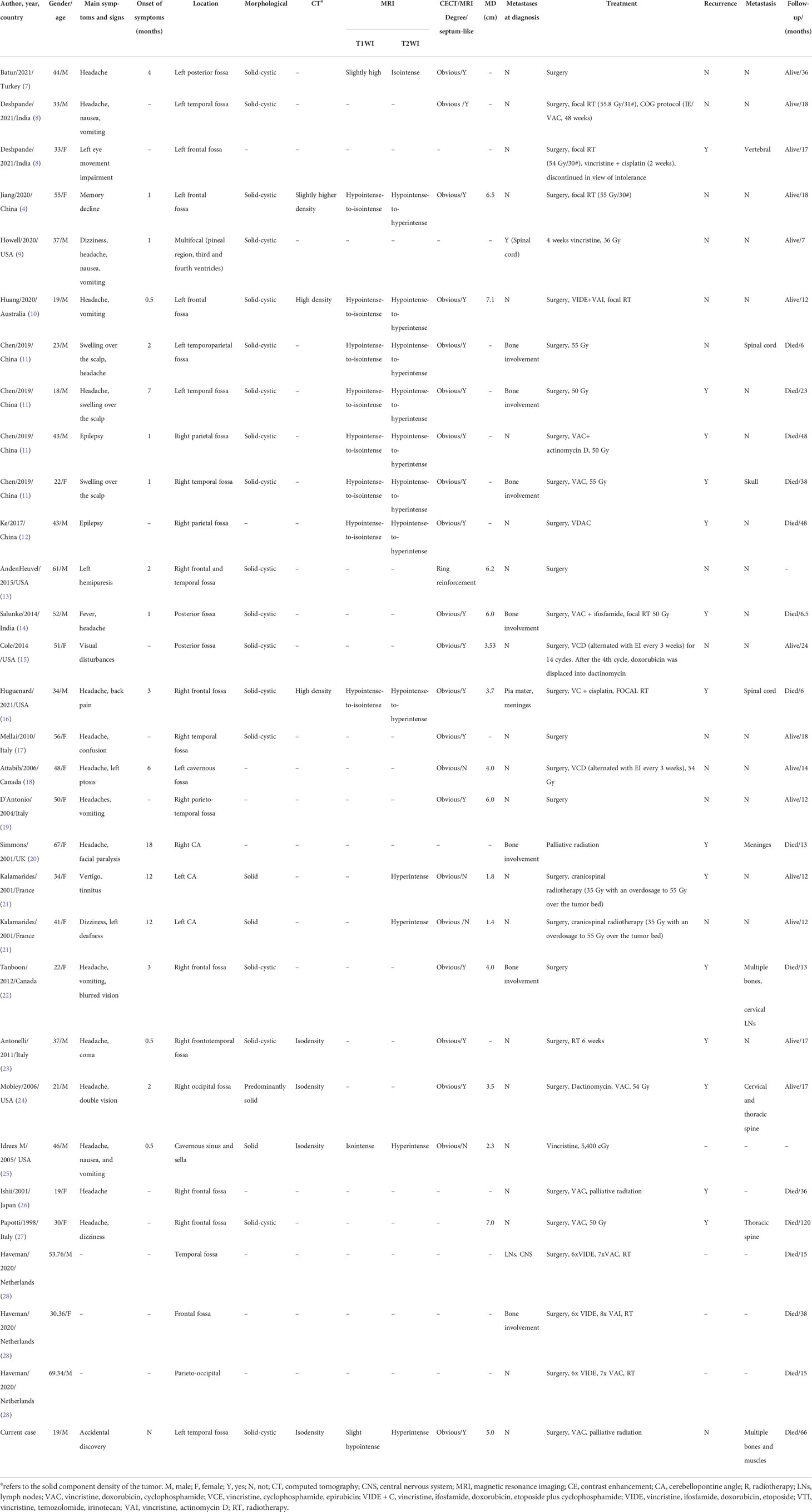
Table 1 Clinical and imaging features of the cases of primary intracranial EWs/PNETs.
Through a systematic search, 30 adult primary intracranial EWs/PNETs were published prior to our present case (1, 4, 7–28). A total of 31 patients including our patient, consisted of 17 male and 14 female patients with a median age of 37 years, with the top three cases published from China (23%), USA (19%), and Italy (13%), and the detailed distribution of patients is shown in Figures 4A, B. Most patients came to the hospital for medical assistance due to short-term headache, dizziness, vomiting, and visual disturbances. Rare clinical symptoms include epilepsy, fever, memory loss, skull invasion in some patients, and local signs of scalp soft tissue swelling. The main foci originated from frontal fossa (7/31), temporal fossa (5/31), followed by posterior fossa, parietal fossa, pontine cerebellar triangle, frontotemporal fossa, frontoparietal fossa, parietal fossa junction, cavernous sinus, etc. Most of the tumor foci were solid-cystic; only four cases were solid with small diameters, ranging from 1.4 to 3.5 cm. The solid component of the tumor showed isodense or slightly high density on CT, slightly low or isointense on T1WI, and hypointense-to-hyperintense on T2WI. On contrast-enhanced CT or T1WI, most of the solid components of the tumor were significantly enhanced, and septum-like enhancement was seen in the cystic components; only one patient showed ring enhancement. Solid tumors all showed obvious uniform enhancement. Of all adults with primary intracranial EWs/PNETs, seven had cranial involvement at diagnosis, and three had meningeal or spinal metastases (as shown in Figure 4C). Most of the patients received surgical resection of the tumor combined with radiotherapy and chemotherapy. Overall, intracranial EWs/PNET has a poor prognosis, with a median survival of 38 months and a 5-year survival rate of only 18%. There was no significant difference in overall survival between male and female patients, and cranial invasion or distant metastasis at the first visit was a prognostic factor (as shown in Figure 5).
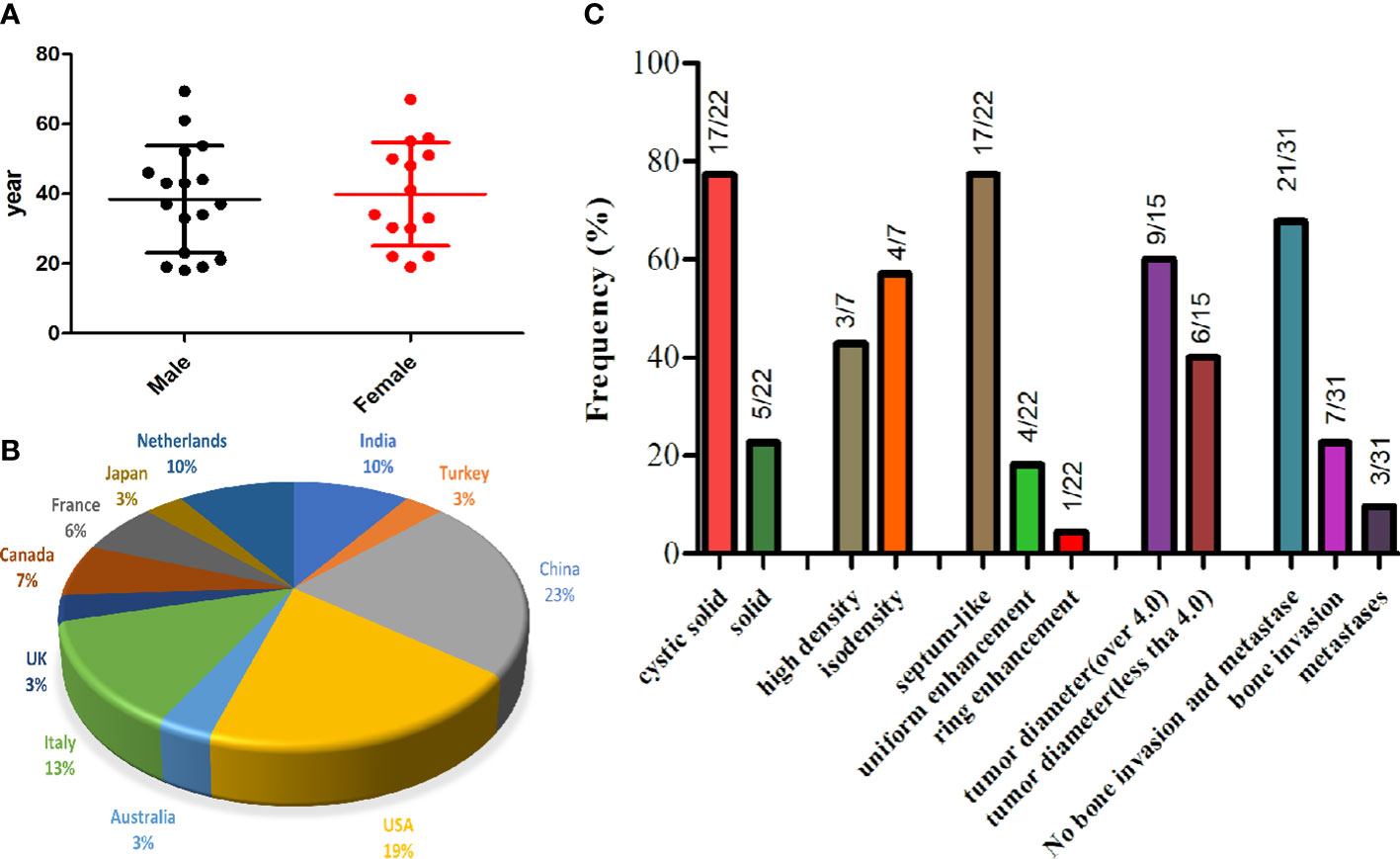
Figure 4 The gender and age distribution (A) and country proportion (B) of 31 adult patients with primary intracranial EWs/PNET, and imaging features of primary intracranial EWs/PNET (C).
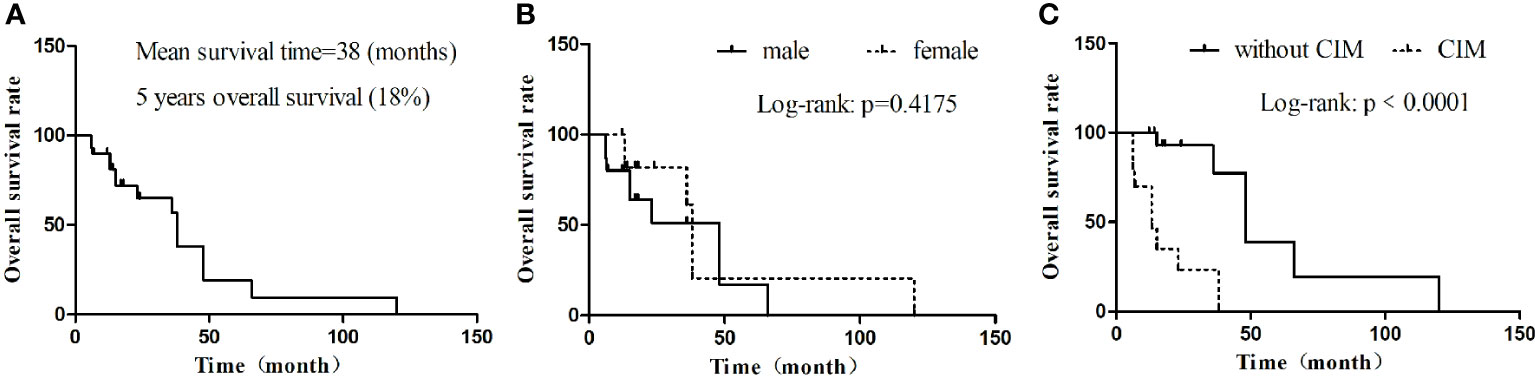
Figure 5 Kaplan–Meier OS. (A) OS in all cases; (B) OS by gender; (C) OS by whether there are cranial invasion or distant metastasis. CIM, cranial invasion or distant metastasis; MST, median survival time; OS, overall survival.
Intracranial EWs/PNETs are rare and account for only 0.03% of all intracranial tumors (29), which is more common in children and adolescents, and previous studies have shown that the median age at diagnosis of intracranial EWs/PNET is 15 years, and there are slightly more male than female patients (30). The case reported here is an intracranial EWs/PNET in the left temporo-occipital fossa of a 19-year-old male patient who was found incidentally after a trauma and had no previous clinical symptoms. In addition, we reviewed all cases of primary intracranial EWs/PNET in adults (≥18 years of age); 30 patients had been reported before our case. The oldest reported patient with primary intracranial EWs/PNET was 69 years old, and the median age at diagnosis of adult patients was 37 years. Our current study shows that the supratentorial fossa including the temporal fossa and frontal fossa are the most common sites for intracranial EWs/PNET, and the most common clinical manifestations include headache, dizziness, vomiting, and visual disturbances, without specificity.
Imaging tests including CT and MRI play a significant role in the diagnosis of central nervous system diseases, especially MRI. On imaging, primary intracranial EWs/PNET is mostly a mass that grows with the dura mater as a wide base, which can grow across the cranial fossa and midline of the brain. Large cystic necrotic areas can be seen in most tumors, and hemorrhage can be seen in some patients, but few calcifications appear. The adjacent skull may be compressed and thinned or invaded to cause bone destruction. Our study showed that intracranial EWs/PNETs were mostly solid-cystic masses, in which the solid components were mostly isodense or slightly hypodense on CT, and only a few small masses showed uniform density. On the T1WI sequence of MRI, the solid component of the tumor was slightly hypointense or isointense, and T2WI was hypo-to-hyperintense, while the cystic part was hypointense on T1WI sequence and hyperintense on T2WI. On contrast-enhanced CT or T1WI, the solid part of the tumor in most patients was significantly enhanced, and the adjacent dura mater was significantly enhanced; only a patient showed primary ring-enhancing cystic lesions in all published cases (13). Notably, on contrast-enhanced scans, septum-like enhancement was seen in cystic regions of tumors in our patient and most of the published literature, which we believe is different from other tumors. Intracranial EWs/PNET including our patient and the published cases were misdiagnosed as meningioma or hemangiopericytoma (HPC) preoperatively; thus, these two tumors are the most important imaging differential diagnoses for intracranial EWs/PNET. Meningiomas are common adult central nervous system tumors, mostly benign. Meningiomas on CT are mostly isodense or high-density spherical or hemispherical masses, with a higher probability of calcification, but with less low-density cystic necrosis, and even if present, the volume of cystic necrosis is small (31). On MRI, the signal of meningiomas is relatively uniform, and the T1WI and T2WI sequences are iso-to-hyperintense, and hyperintensity is more common on DWI, while our patient was hypointense on DWI, which is different from it. Angiography can clearly show the internal and external carotid arteries supplying blood to the meningioma. Typical signs include the tumor body compressing adjacent blood vessels, but the tumor body and its interface are still smooth, showing a “holding ball” change (32). HPC is a malignant tumor with abundant blood vessels, which often leads to low-density necrotic foci due to insufficient local blood supply to the tumor, and the morphology of the tumor is irregular, most of which is lobulated (33), while the shape of EWs/PNET is more regular. On contrast-enhanced scans, HPC was more enhanced and intratumoral vascular flow void signals were more common, and angiography showed multiple tortuous and thickened vascular shadows in the mass (34), which is different from EWs/PNET. Moreover, malignant triton tumor (MTT), namely, malignant schwannoma with rhabdomyoma differentiation, another rare intracranial tumor, is often misdiagnosed as meningioma in previous literature reports; thus, it should also be considered as one of the differential diagnoses of EWs/PNET. MTT is usually equal and slightly hyperdense on CT, and a small volume of cystic necrosis is often seen. Tumors on MRI are mostly low signal on T1WI and high signal on T2WI, and arc or linear low-signal shadows appear in the high-signal tumor tissue, which is relatively specific for the diagnosis of MTT (35).
The diagnosis of EWs/PNET mainly depends on pathology and immunohistochemistry. Under the microscope, the tumors are composed of small round cells or elliptic cells, with hyperchromatic nuclei, and mildly basophilic cytoplasm and Homer–Wright chrysanthemum or Flexner–Weinsteiner chrysanthemum structures can be seen in some tumors (2, 29). Immunohistochemistry showed that almost all tumor cells of intracranial EWs/PNET patients consistently and diffusely expressed CD99, but there was no specificity (36). In addition, tumor cells expressed Vimentin, NSE, Syn, and membrane protein FLI-1 to varying degrees, and molecular detection to find EWSR1 gene rearrangement is the current gold standard for diagnosing EWs/PNETs (2, 12).
Due to its rarity, the standard treatment regimen for intracranial EWs/PNET has not been established, and surgery for the purpose of total tumor resection is currently the main treatment method. In our review of the published literature, most patients were treated with a combination of radiotherapy and chemotherapy including vincristine, cyclophosphamide, doxorubicin, ifosfamide, etoposide, doxorubicin, and radiomycin D and other drugs after surgical resection (30). Our study including 31 adults with intracranial EWs/PNET showed that the prognosis was poor, and the longest follow-up was 10 years in a patient who eventually metastasized to the thoracic spine, progressed, and died (27). Among these patients, their median survival was only 38 months, and the 5-year survival rate was less than 20%.
In conclusion, adult patients with primary intracranial EWs/PNET are rare, whose imaging findings should be considered as one of the differential diagnoses of meningioma, HPC, and MTT. There are large cystic lesions in the tumor, and septum-like enhancement on contrast-enhanced scan may be a relatively specific feature of EWs/PNET, whereas the rarity of the disease limits the clinical application of our findings, and more cases and long-term follow-up studies are needed in the future to fully understand EWs/PNET in the adult population.
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding authors.
Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
JC and PW: funding acquisition; DL: investigation; QH and JW: methodology; XH: writing—original draft; XH, PW, and JC: writing—review and editing. All authors contributed to the article and approved the submitted version.
This study was funded by the National Natural Science Foundation of the People’s Republic of China, NSFC (grant numbers: 81571712), Zunyi Medical College Research Start Fund 2018ZYFY03, and QianKeHe platform talents [2017] (Grant No. 5733-035).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. de Alava E. Ewing Sarcoma, an update on molecular pathology with therapeutic implications. Surg Pathol Clin (2017) 10:575–85. doi: 10.1016/j.path.2017.04.001
2. Applebaum MA, Worch J, Matthay KK, Goldsby R, Neuhaus J, West DC, et al. Clinical features and outcomes in patients with extraskeletal Ewing sarcoma. Cancer (2011) 117(13):3027–32. doi: 10.1002/cncr.25840
3. Grünewald T, Cidre-Aranaz F, Surdez D, Tomazou EM, de Álava E, Kovar H, et al. Ewing Sarcoma. Nat Rev Dis Primers (2018) 4:5. doi: 10.1038/s41572-018-0003-x
4. Jiang Y, Zhao L, Wang Y, Liu X, Wu X, Li Y. Primary intracranial Ewing Sarcoma/Peripheral primitive neuroectodermal tumor mimicking meningioma: A case report and literature review. Front Oncol (2020) 10:528073. doi: 10.3389/fonc.2020.528073
5. Bacci G, Longhi A, Ferrari S, Mercuri M, Versari M, Bertoni F. Prognostic factors in non-metastatic ewing's sarcoma tumor of bone: an analysis of 579 patients treated at a single institution with adjuvant or neoadjuvant chemotherapy between 1972 and 1998. Acta Oncol (2006) 45:469–75. doi: 10.1080/02841860500519760
6. Ladenstein R, Pötschger U, Le Deley MC, Whelan J, Paulussen M, Oberlin O, et al. Primary disseminated multifocal Ewing sarcoma: results of the Euro-EWING 99 trial. J Clin Oncol (2010) 28:3284–91. doi: 10.1200/JCO.2009.22.9864
7. Ishii N, Hiraga H, Sawamura Y, Shinohe Y, Nagashima K. Alternative EWS-FLI1 fusion gene and MIC2 expression in peripheral and central primitive neuroectodermal tumors. Neuropathology (2001) 21(1):40–4. doi: 10.1046/j.1440-1789.2001.00367.x
8. Deshpande G, Epari S, Gupta C, Shetty O, Gurav M, Chinnaswamy G, et al. Primary intracranial Ewing sarcoma/ peripheral primitive neuroectodermal tumor, an entity of unacquaintance: a series of 8 cases. Childs Nerv Syst (2021) 37:839–49. doi: 10.1007/s00381-020-04850-w
9. Howell SG, Lyon KA, Garrett D Jr, Huang JH, Fonkem E. Optimum treatment for primary intracranial Ewing sarcoma. Proc (Bayl Univ Med Cent) (2020) 33:430–2. doi: 10.1080/08998280.2020.1755199
10. Huang J, Ghent F, Levingston R, Scholsem M. Intracranial Ewing sarcoma - a case report. Surg Neurol Int (2020) 11:134. doi: 10.25259/SNI_178_2020
11. Chen J, Jiang Q, Zhang Y, Yu Y, Zheng Y, Chen J, et al. Clinical features and long-term outcome of primary intracranial Ewing Sarcoma/Peripheral primitive neuroectodermal tumors: 14 cases from a single institution. World Neurosurg (2019) 122:e1606–1606e1614. doi: 10.1016/j.wneu.2018.11.151
12. Ch K, Duan Q, Yang H, Zhu F, Yan M, Xu SP, et al. Meningeal Ewing Sarcoma/Peripheral PNET: Clinicopathological, immunohistochemical and FISH study of four cases. Neuropathology (2017) 37:35–44. doi: 10.1111/neup.12325
13. VandenHeuvel KA, Al-Rohil RN, Stevenson ME, Qian J, Gross NL, McNall-Knapp R, et al. Primary intracranial ewing's sarcoma with unusual features. Int J Clin Exp Pathol (2015) 8:260–74.
14. Salunke P, Sharma M, Gupta K. Ewing Sarcoma of the occipital bone in an?elderly patient. World Neurosurg (2014) 81:e10–2. doi: 10.1016/j.wneu.2010.12.050
15. Cole M, Parajuli S, Laske D, Goldstein L, Morrison T, Mukherjee A, et al. Peripheral primitive neuroectodermal tumor of the dura in a 51-year-old woman following intensive treatment for breast cancer. Am J Case Rep (2014) 15:294–9. doi: 10.12659/AJCR.890656
16. Huguenard AL, Li YD, Sharifai N, Perkins SM, Dahiya S, Chicoine MR. Multifocal primary central nervous system Ewing sarcoma presenting with intracranial hemorrhage and leptomeningeal dissemination: illustrative case. J Neurosurg Case Lessons (2021) 1:CASE2042. doi: 10.3171/CASE2042
17. Mellai M, Caldera V, Comino A, Fortunato M, Bernucci C, Schiffer D. PNET/ESFT of the cranial vault: a case report. Clin Neuropathol (2010) 29:372–7. doi: 10.5414/npp29372
18. Attabib NA, West M, Rhodes RH. Peripheral primitive neuroectodermal tumor of the cavernous sinus: case report. Neurosurgery (2006) 58:E992. doi: 10.1227/01.NEU.0000210215.73374.EE
19. D'Antonio A, Caleo A, Garcia JF, Marsilia GM, De Dominicis G, Boscaino A. Primary peripheral PNET/Ewing's sarcoma of the dura with FISH analysis. Histopathology (2004) 45:651–4. doi: 10.1111/j.1365-2559.2004.01961.x
20. Simmons MA, Luff DA, Banerjee SS, Ramsden RT. Peripheral primitive neuroectodermal tumour (pPNET) of the cerebellopontine angle presenting in adult life. J Laryngol Otol (2001) 115:848–52. doi: 10.1258/0022215011909161
21. Kalamarides M, Dewolf E, Couvelard A, Shahidi A, Bouccara D, Cyna-Gorse F, et al. Extraaxial primitive neuroectodermal tumor mimicking a vestibular schwannoma: diagnostic and therapeutic difficulties. Rep two cases J Neurosurg (2001) 94:612–6. doi: 10.3171/jns.2001.94.4.0612
22. Tanboon J, Sitthinamsuwan B, Paruang T, Marrano P, Thorner PS. Primary intracranial Ewing sarcoma with an unusually aggressive course: a case report and review of the literature. Neuropathology (2012) 32:293–300. doi: 10.1111/j.1440-1789.2011.01258.x
23. Antonelli M, Caltabiano R, Chiappetta C, Oliva MA, Giangaspero F, Lanzafame S. Primary peripheral PNET/Ewing's sarcoma arising in the meninges, confirmed by the presence of the rare translocation t (21,22) (q22;q12). Neuropathology (2011) 31:549–55. doi: 10.1111/j.1440-1789.2010.01196.x
24. Mobley BC, Roulston D, Shah GV, Bijwaard KE, McKeever PE. Peripheral primitive neuroectodermal tumor/Ewing's sarcoma of the craniospinal vault: case reports and review. Hum Pathol (2006) 37:845–53. doi: 10.1016/j.humpath.2006.02.011
25. Idrees M, Gandhi C, Betchen S, Strauchen J, King W, Wolfe D. Intracranial peripheral primitive neuroectodermal tumors of the cavernous sinus: a diagnostic peculiarity. Arch Pathol Lab Med (2005) 129:e11–5. doi: 10.5858/2005-129-e11-IPPNTO
26. Ishii N, Hiraga H, Sawamura Y, Shinohe Y, Nagashima K. Alternative EWS-FLI1 fusion gene and MIC2 expression in peripheral and central primitive neuroectodermal tumors. Neuropathology (2001) 21:40–4. doi: 10.1046/j.1440-1789.2001.00367.x
27. Papotti M, Abbona G, Pagani A, Monga G, Bussolati G. Primitive neuroectodermal tumor of the meninges: An histological, immunohistochemical, ultrastructural, and cytogenetic study. Endocr Pathol (1998) 9:275–80. doi: 10.1007/BF02739968
28. Haveman LM, Ranft A, Berg H, Klco-Brosius S, Ladenstein R, Paulussen M, et al. Primary and metastatic intracranial Ewing sarcoma at diagnosis: Retrospective international study and systematic review. Cancers (Basel) (2020) 12(6):1675. doi: 10.3390/cancers12061675
29. Yim J, Lee WS, Kim SK, Kang HJ, Bae J, Park SH. Intracranial Ewing sarcoma with whole genome study. Childs Nerv Syst (2019) 35:547–52. doi: 10.1007/s00381-018-3997-1
30. Cherif El Asri A, Benzagmout M, Chakour K, Chaoui MF, Laaguili J, Chahdi H, et al. Primary intracranial pPNET/Ewing sarcoma: Diagnosis, management, and prognostic factors dilemma-a systematic review of the literature. World Neurosurg (2018) 115:346–56. doi: 10.1016/j.wneu.2018.04.164
31. Shijo M, Honda H, Koyama S, Ishitsuka K, Maeda K, Kuroda J, et al. Dura mater graft-associated Creutzfeldt-Jakob disease with 30-year incubation period. Neuropathology (2017) 37:275–81. doi: 10.1111/neup.12359
32. Dabus G, Linfante I, McDermott MW. Angiography and embolization of meningiomas. Handb Clin Neurol (2020) 169:193–202. doi: 10.1016/B978-0-12-804280-9.00013-5
33. Wang X, Zhang X, Zhou Q, Zhou X, Chen L, Huang G, et al. Hemangiopericytoma arose from the site of meningioma resection. J Craniofac Surg (2017) 28:e329–329e330. doi: 10.1097/SCS.0000000000003576
34. Sibtain NA, Butt S, Connor SE. Imaging features of central nervous system haemangiopericytomas. Eur Radiol (2007) 17:1685–93. doi: 10.1007/s00330-006-0471-3
35. Hu X, He X, Wang P, Zou Y, Li D, Cai J, et al. Intracranial malignant triton tumor in children: A case report. Iran J Radiol (2021) 18(1):e106103. doi: 10.1097/MD.0000000000016797
Keywords: Ewing sarcomas, primitive neuroectodermal tumors, EWs/PNET, intracranial, magnetic resonance imaging, PET/CT
Citation: Hu X, Huang Q, Wang J, Li D, Wang P and Cai J (2022) Case report: Primary intracranial EWs/PNET in adults: Clinical experience and literature review. Front. Oncol. 12:1035800. doi: 10.3389/fonc.2022.1035800
Received: 03 September 2022; Accepted: 23 September 2022;
Published: 13 October 2022.
Edited by:
Andrea Ciarmiello, Sant’Andrea University Hospital, ItalyReviewed by:
Chunhong Li, Suining Central Hospital, ChinaCopyright © 2022 Hu, Huang, Wang, Li, Wang and Cai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jiong Cai, amlvbmdfY2FpQDE2My5jb20=; Pan Wang, MTI5ODE3ODgyOEBxcS5jb20=; Dandan Li, ODA3NDQyMDAzQHFxLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.