 Xiaoxu Fang
Xiaoxu Fang Shaokun Yu†
Shaokun Yu†- Department of Oncology, The First Affiliated Hosptial of Nanjing Medicial University, Nanjing, China
Circulating tumor DNA (ctDNA) has contributed immensely to the management of hematologic malignancy and is now considered a valuable detection tool for solid tumors. ctDNA can reflect the real-time tumor burden and be utilized for analyzing specific cancer mutations via liquid biopsy which is a non-invasive procedure that can be used with a relatively high frequency. Thus, many clinicians use ctDNA to assess minimal residual disease (MRD) and it serves as a prognostic and predictive biomarker for cancer therapy, especially for non-small cell lung cancer (NSCLC). Advanced methods have been developed to detect ctDNA, and recent clinical trials have shown the rationality and feasibility of ctDNA for identifying mutations and guiding treatments in NSCLC. Here, we have reviewed recently developed ctDNA detection methods and the importance of sequence analyses of ctDNA in NSCLC.
Introduction
Lung cancer is the second most common cancer worldwide, responsible for the maximum number of cancer deaths (1). Non-small cell lung cancer (NSCLC) represents approximately 85% of diagnosed lung cancers; lung adenocarcinoma (LUAD) and lung squamous cell carcinoma (LUSC) are the two most common subtypes (2). However, the development and application of precision therapy, including targeted therapy and Immune checkpoint inhibitor (ICI) therapy, have fundamentally altered the management of NSCLC patients. Targeted therapy has shown potential in the treatment of patients with driver gene alterations such as epidermal growth factor receptor (EGFR) mutations, anaplastic lymphoma kinase (ALK) fusions, human epidermal growth factor receptor 2 (HER2) mutations, ROS1 fusions, MET amplification, BRAF mutations, and RET fusions. It is now widely used in daily clinical practice (3). ICI therapy, which suppresses programmed cell death-1 (PD-1) or programmed cell death ligand-1 (PD-L1), has also been successful in prolonging the life of patients (4).
Clinical diagnosis requires a solid biopsy in order to determine tumor histology and staging. Compared with tissue biopsy, liquid biopsy is a non-invasive way to identify patients who might response to therapy, to dynamically monitor treatment effect and to unveil resistance mechanism. Liquid biopsy could typically detect circulating tumor cells (CTCs), circulating tumor DNA (ctDNA), exosomes, microRNAs (miRNA), peripheral blood circulating RNA, tumor-educated blood platelets (TEPs), and circulating tumor vascular endothelial cells (CTECs). ctDNA is one of the most commonly detected biomarkers (5).
Circulating cell-free DNAs (cfDNA) are DNA fragments ranging from 150 to 200 base pairs in length mainly derived from apoptotic or necrotic cells (6). Tumor cells also release circulating tumor DNA (ctDNA), accounting for <0.01% of total cfDNA, which need detection techniques with high sensitivity. Besides the traditional quantitative or real-time PCR (qPCR) and next-generation sequencing (NGS), other recently introduced methods to analyze ctDNA are advanced PCR-based techniques such as digital PCR (dPCR), droplet digital PCR (ddPCR), beads emulsion amplification magnetics (BEAMing), NGS-based techniques such as tagged amplicon deep sequencing (TAM-Seq), safe-sequencing (Safe-Seq), cancer personalized profiling by deep sequencing (CAPP-Seq), and Phased variant enrichment and detection sequencing (PhasED-seq). These will be briefly explained below.
Minimal residual disease (MRD) is a disease status in patients that escapes clinical observation by radiology. In oncology, MRD represents early tumor development and tumor relapse which needs to be urgently detected and assessed (7). In MRD detection, liquid biopsy of these tumor - derived factors plays an important role in clinical application. First of all, liquid biopsy can be used for early cancer screening which lacks detectable abnormalities found by radiology approaches. Secondly, liquid biopsy could monitor micrometastatic disease to assess the risk of disease recurrence after a radical treatment. Finally, the dynamic characterization of tumor burden and disease biological changes could clarify drug resistance mechanisms and guide the treatment strategies. (Figure 1) (8). More recently, ctDNA from a liquid biopsy has shown showing their potential to be a reliable plasma-based biomarker for MRD. Quantitative characterization of ctDNA via liquid biopsy has been associated with clinical and pathologic features of cancer, including stage, tumor burden, vascularization, and response to therapy. ctDNA can help detect the mutations and activity of different tumor sub-clones,which tissue biopsy cannot because of tumor heterogeneity (9). Moreover, the short half-life of ctDNA ensures that the detection results are in real-time. The molecular precision of longitudinal tumor surveillance via serial ctDNA measurement enables the identification of mutations that drive cancer progression and treatment resistance.
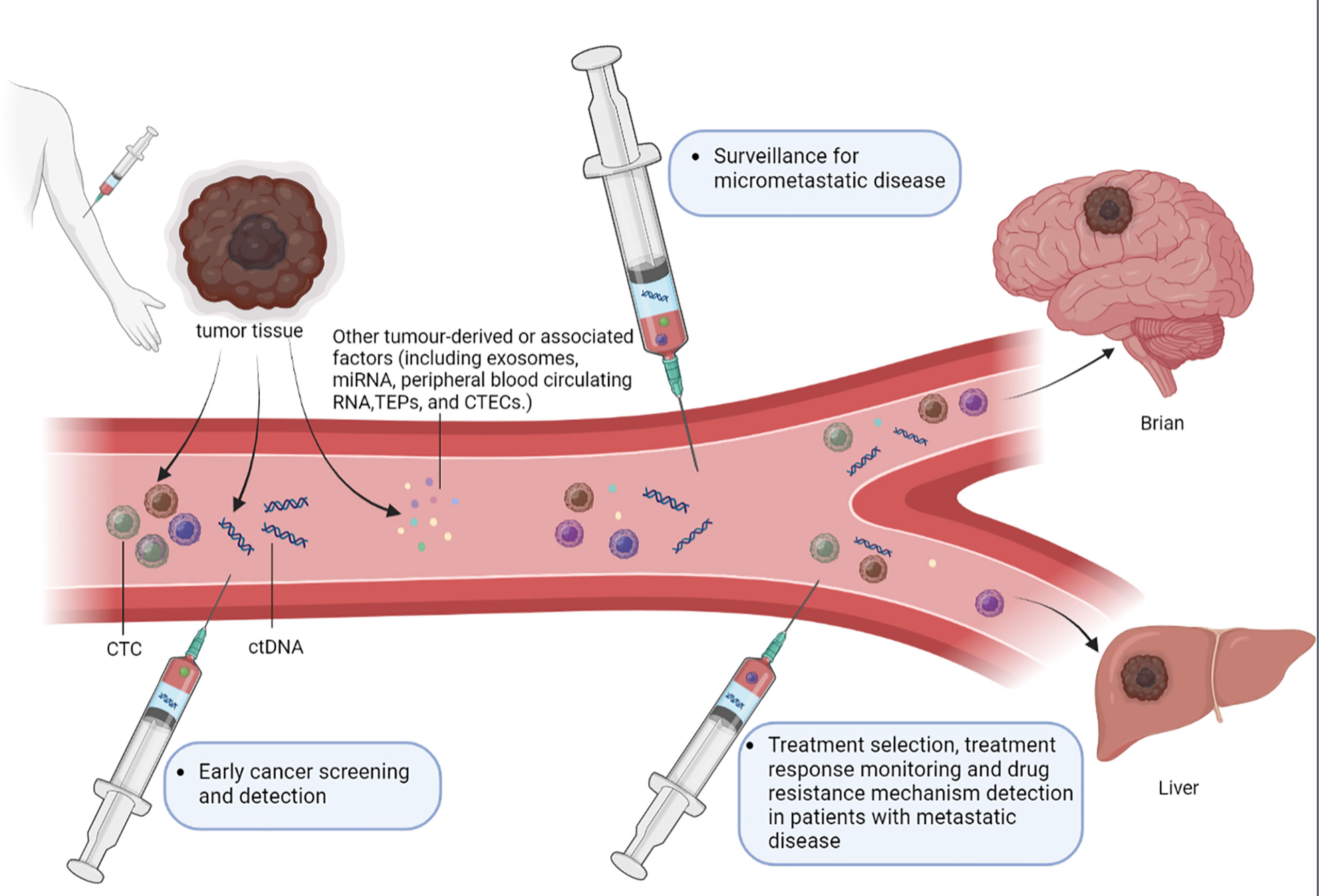
Figure 1 The role of liquid biopsy in MRD detection. Liquid biopsy typically detects circulating tumor cells (CTCs), circulating tumor DNA (ctDNA), exosomes, microRNAs (miRNA), peripheral blood circulating RNA, tumor-educated blood platelets (TEPs) and circulating tumor vascular endothelial cells (CTECs). Liquid biopsy assays of these tumor-derived factors can serve several purposes in the MRD detection. (1) Early cancer screening and detection, liquid biopsy approaches could also be used to further investigate abnormalities detected on imaging examinations. (2) Surveillance for micrometastatic disease following curative-intent treatment of a primary tumor, in order to evaluate the risk of disease recurrence and enable timely management of recurrent disease. (3) Guiding the selection of the most appropriate treatment, monitoring treatment responses and detecting the resistance mechanisms in patients with overt metastatic disease through dynamic characterization of changes in tumor burden and disease biology.
In this review, we will introduce several commonly used ctDNA detection approaches and discuss the clinical application of ctDNA-based MRD evalution.
Recently developed detection techniques
PCR assay
Detection and quantitation of specific nucleic acid sequences using PCR is fundamental to a large body of research and a growing number of molecular diagnostic tests. The first generation of PCR users performed end-point analysis by gel electrophoresis to obtain qualitative results. The advent of real-time PCR spawned a second generation. rtPCR is an analogue measurement based on monitoring amplification after each cycle of PCR using fluorescence probes. The point at which the reaction fluorescence crosses an intensity threshold is called the cycle threshold(Ct). As many factors can influence PCR efficiency and hence the Ct value, the accuracy and precision of real-time PCR can vary widely (10). Vogelstein and Kinzler introduced a new form of PCR called digital PCR (dPCR) in 1999 (11). Compared with those conventional PCRs, dPCR partitions samples into multiple parallel quantitative PCR reactions within separate compartments and therefore improves sensitivity, absolute quantification, and rare allele detection (12, 13). However, the large reaction volume and the limited number of compartments to minimize the dimensions of the chip have greatly limited its possible clinical applications (10, 14), and it is believed that ddPCR might overcome these limitations.
Droplet digital PCR (ddPCR) uses aqueous droplets with volumes ranging from a few femtoliters to nanoliters dispersed in oil to compartmentalize PCR reactions, having a theoretically unlimited number of compartments (10). In addition, ddPCR needs only a single reaction tube (10). Nanoliter-sized droplet technology paired with digital PCR (ddPCR) holds promise for highly precise, absolute nucleic acid quantification. Hindson et al. compared the microRNA quantification by ddPCR and real-time PCR which revealed greater precision (coefficients of variation decreased 37–86%) and improved day-to-day reproducibility (by a factor of seven) of ddPCR but with comparable sensitivity (15). Frank Diehl et al. found another droplet-based digital PCR named BEAMing (beads, emulsion, amplification, and magnetics) in 2006. BEAMing couples oligonucleotide primers to beads and distributes beads to compartments. After amplification, every compartment contains a bead binding to thousands of copies of the initial DNA molecule. The DNA can then be released from the beads and analyzed with flow cytometry or optical scanning instruments to locate mutation (16, 17). A recent LungBEAM study demonstrated the great potential of BEAMing in optimizing treatment in patients with advanced NSCLC (18).
Presently, ddPCR and BEAMing are the two most commonly used PCR techniques in clinics to detect ctDNA, their reports must match. Ben O’Leary and his colleagues collected plasma from patients with advanced breast cancer and assessed ESR1 and PIK3CA mutations in ctDNA using both PCR techniques simultaneously. ESR1 mutation was calculated as 24.2% (88/363) with BEAMing and 25.3% (92/363) with ddPCR, (κ = 0.91; 95% CI, 0.85-0.95). The result for PIK3CA mutation was 26.2% (95/363) with BEAMing and 22.9% (83/363) with ddPCR, (κ = 0.87; 95% CI, 0.81– 0.93), showing consistency of results from BEAMing and ddPCR (19). Despite droplet-based digital PCR being highly sensitive, it can only detect known mutations and needs customed assays (20), which restrict its clinical applicability.
Targeted next generation sequencing approaches
The development of NGS including whole exome sequencing (WES) and whole genome sequencing (WGS), has facilitated cancer diagnosis over the past decade through samples from a tissue biopsy (21). However, the sensitivity of NGS for single nucleotide variants (SNV) detection is about 4% to 10% (22), which is enough for tissue samples but makes it hard to detect rare mutations in ctDNA for its extremely low percentage in cfDNA. To maintain NGS’s covering broad areas across the genome and meanwhile improve the sensitivity, targeted NGS approaches that detect specific areas of the genome were developed (23).
Forshew et al. established a technique named tagged-amplicon deep sequencing (TAm-Seq; 2012) and successfully applied it to detect ctDNA mutations in patients with metastatic breast cancers and ovary cancers (24). TAm-Seq can detect cancer mutations with allele frequencies as low as 2% and sensitivity and specificity as high as 97% (24). Kinde et al. pioneered Safe Sequencing System (Safe-SeqS; 2011), in which they tagged each template molecule with a 12- or 14- base unique identifier (UID), then amplified the tagged molecules with two cycles of amplicon-specific PCR to create UID families, and sequenced the amplified product redundantly with NGS (25). This redundant sequencing approach makes Safe-SeqS detect rare mutations with high specificity. Moreover, its unique algorithm increases the accuracy of the base calling and reduces the error rate to an average of 2×10-4 errors/bp although efficacy is still limited by artifactual mutations occurring during the PCR as well as any residual base-calling errors (25).
Newman et al. developed cancer personalized profiling by deep sequencing (CAPP-Seq; 2014) with ultrasensitive detection of ctDNA. CAPP-Seq utilizes DNA probes to hybridize and capture ctDNAs for its quantification and sequencing. These probes are designed for regions with high driver mutation frequencies in certain cancer types (26). This method can even detect 0.02% of cfDNA and ctDNA in patients with early or advanced stages of NSCLC (26). To further improve the efficiency of ctDNA detection, Newman et al. upgraded CAPP-Seq to integrate digital error suppression-enhanced CAPP-seq (iDES-enhanced CAPP-seq), which tags each template molecule with a UID just like the Safe-seqS to reach a detection limit of 0.001% and a specificity of 96% (27).
Phased variant enrichment and detection sequencing (PhasED-seq; 2021) is the most recent method that uses multiple somatic mutations in individual DNA fragments to improve the sensitivity of ctDNA detection. PhasED-seq can detect less than 0.0001% of tumor DNA, which is better than any earlier approaches (28). David et al. demonstrate that PhasED-seq can meaningfully improve detection of ctDNA in clinical samples both during therapy and before disease relapse. They analyzed serial samples from a participant with stage III lung adenocarcinoma treated with chemoradiotherapy. CAPP-seq and PhasED-seq detected similar ctDNA levels before therapy; however, three samples after treatment initiation had undetectable ctDNA by CAPP-seq before ctDNA re-emerged at the time of biopsy-confirmed recurrent disease. Using PhasED-seq, they observed molecular residual disease in 3/3 (100%) of samples that were undetected by single-nucleotide variants (SNVs), with tumor fraction as low as 0.00016% (28).
Clinical laboratories are increasingly developing and deploying NGS tests, ranging from targeted ‘hotspot’ panels to comprehensive genome-scale platforms. Ahmet et al.developed and implemented MSK-IMPACT, a hybridization capture–based NGS panel with distinct advantages over small panels for detecting all protein-coding mutations, copy number alterations (CNAs), and selected promoter mutations (29). Ivo et al. demonstrate that panel size is a critical parameter that influences confidence intervals (CIs) and cutoff values as well as important test parameters including sensitivity, specificity, and positive predictive value. Panels between 1.5 and 3 Mbp are ideally suited to estimate TMB with small CIs, whereas smaller panels tend to deliver imprecise TMB estimates for low to moderate TMB (0–30muts/Mbp) (30).
ctDNA and diagnosis of NSCLC
Early-stage diagnosis
According to the International Association for the Study of Lung Cancer (IASLC) lung cancer staging project, the 5-year survival of NSCLC diminishes rapidly as the disease stage progresses (82% for stage IA, 52% for stage IIA, 36% for stage IIIA and 6% for stage IV). Thus, the detection of early-stage NSCLC is urgent (31). However, early-stage NSCLC has few radiographic characteristics to be distinguished from benign solitary nodules, so the chances of false positives from radiology approaches are too high. Wong et al. assigned 10,061 candidates to the CANTOS (Canakinumab anti-inflammatory thrombosis outcome study), 71 of them developed lung cancer and each participant had deposited two plasma samples at two different time points during the study; one was at the baseline time point (collected at the beginning of the trial) and the other after the clinical diagnosis of lung cancer. The test of these samples indicated that patients with COSMIC (catalog of somatic mutations in Cancer) ctDNA mutations at baseline exhibited a shorter time to their lung cancer clinical diagnosis (407 days versus 837 days, P=0.011), indicating that mutations in ctDNA might predict an early-stage NSCLC to some extent (32).
A study analyzed ctDNA at different stages of NSCLC utilizing CAPP-seq and found that the diagnostic sensitivity of ctDNA was 64%, 82%, and 100% for tumor stages I, II, and III, respectively. A similar finding was reported from another study (57.9%, 66.7%, and 90% for tumor stages I, II and III, respectively), which implied a correlation between ctDNA levels and tumor volume and outlined the difficulty of early NSCLC detection (33, 34). However, Liang et al. established a method of DNA methylation profiling by high throughput DNA bisulfite sequencing that can distinguish malignant tumors from benign solitary nodules with a sensitivity of 79.5% (63.5%-90.7%) and a specificity of 85.2% (66.3% -95.8%) (35).
Detection of known mutations
The efficiency of targeted therapy depends on the precise detection of the driver gene mutations. Mack et al. tested plasma samples from 8388 patients and made a plasma-based comprehensive genomic profiling. Driver gene mutations were identified in 48% of patients, including EGFR mutations (26.4%), MET mutations (6.1%), BRAF mutations (2.8%), and fusions (ALK, RET, and ROS1; 2.3%) (36).
Although the golden standard guiding target therapy remains gene mutations detected from a tissue biopsy, non-invasive liquid biopsy utilizing ctDNA is sometimes preferred, and ctDNA increases the chances of identifying several targetable mutations, especially EGFR mutation (37, 38). However, it is crucial to clarify whether the mutations detected from ctDNA agree with those from tumor cell lesions (Table 1). A clinical trial study (NCT01203917) aimed to assess the efficacy and tolerability of gefitinib as first-line therapy for common EGFR mutations (19del, L858R, T790M) positive patients in stage III/IV NSCLC. Researchers found EGFR mutations to be similar in tumor and plasma samples (ctDNA) with a sensitivity of 65.7% (95% CI: 55.8–74.7) and a specificity of 99.8% (95% CI: 99.0–100.0) (40), implying that plasma samples are useful to identify patients who might benefit from gefitinib when tumor tissue is unavailable. A similar comparison was designed in the FASTACT-2 study, and the sensitivity and specificity of the mutation detection were 75% and 96%, respectively (43). Cobas EGFR Mutation Test v2, a real-time PCR assay that can identify 42 different EGFR gene mutations, was the first approach approved by FDA to detect EGFR mutations in 2016 (52).
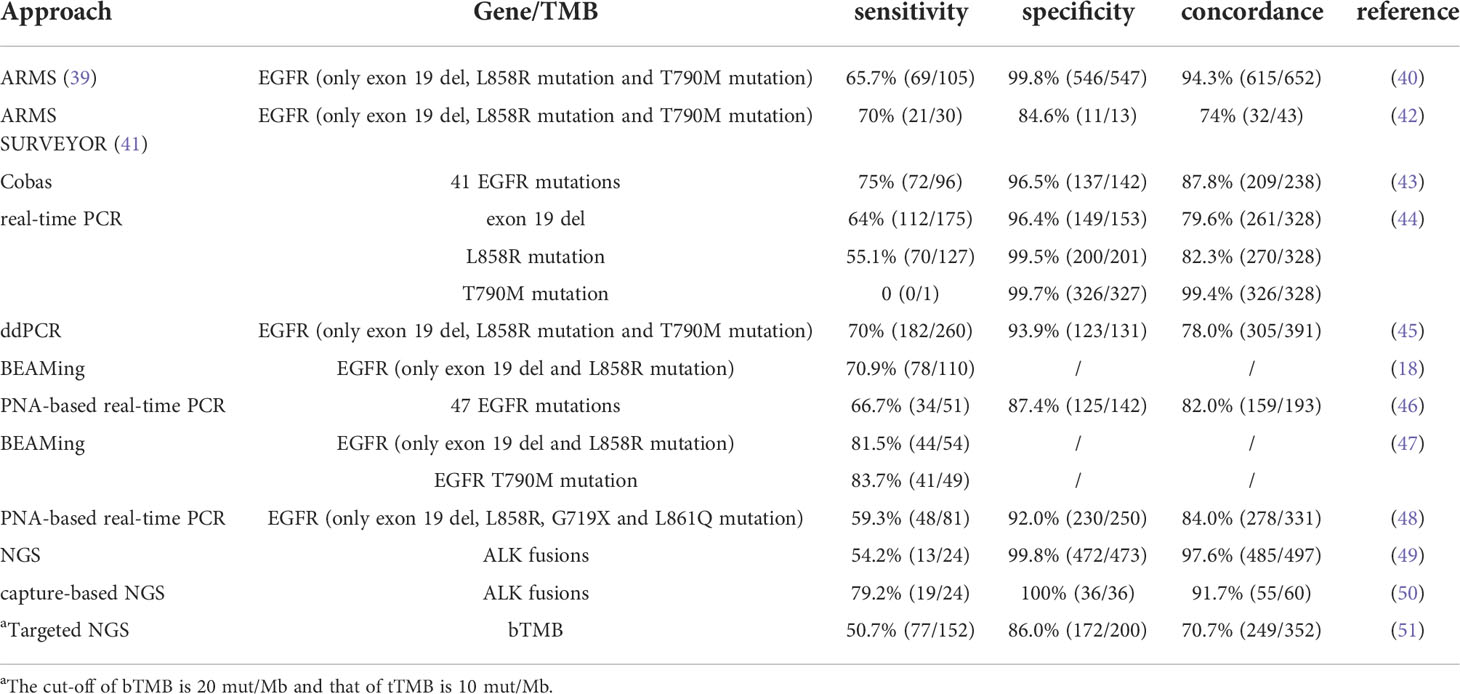
Table 1 The mutation results obtained and compared between tumor and plasma DNA samples.
Pertaining to the oncogenic fusions, ctDNA reflects a high similarity with those detected in tissue samples. Horn et al. analyzed ALK fusions in tumor and plasma samples and found a concordance of 91% (20/22) between them (53). Wang et al. also detected ALK fusions in ctDNA from 19 out of 24 patients with ALK fusions in their tumor tissue, demonstrating a sensitivity of 79.2% (95%CI: 57.9%-92.9%). They could not detect ALK fusions in ctDNA from 36 patients without ALK fusions in their tumor tissue, implying that the specificity of the method was 100% (50). Plasma ROS1 fusions analysis also showed a 100% concordance with those observed in the tissue samples (54).
Vansteenkiste et al. found that the similarity in PIK3CA mutations between tissue and ctDNA samples was 55.3%. However, the concordance was 81.8% (9 of 11 samples) between ctDNA and metastatic tissue samples, compared with 44.4% (12 of 27 samples) between ctDNA and primary tissue, implying that ctDNA PI3K pathway mutations were more correlated with metastatic lesions than with primary tumor (55).
However, despite the high sensitivity of methods for detecting mutations in ctDNA, it is wise to retest tissue samples if the result is negative (56).
It is noteworthy that MRD detection can also be confounded by clonal hematopoiesis of indeterminate potential (CHIP). CHIP arises when age-dependent mutations accumulate in hematopoietic progenitor cells, leading to the formation of a genetically distinct subpopulation that contributes disproportionately to the population of mature blood cells. These distinct subclones have driver mutations and have been implicated in hematologic diseases. In the measurement of ctDNA, CHIP can result in false-positive results due to detection of non-reference variants in the blood plasma, which is especially problematic when the ctDNA mutant allele fraction is low in the setting of MRD detection. Thus, CHIP must be properly accounted for in order to specifically measure ctDNA, such as by sequencing matched PBMCs to similar depth, especially when using ultra-sensitive assays that are capable of achieving detection of low mutant allele fraction variants (57).
ctDNA and Treatment of NSCLC
Relapse after operation
The relapse after curative-intent resection has confused surgeons for years and ctDNA might be an early predictor of it. The DYNAMIC prospective study tested plasma ctDNA collected from 36 patients that underwent curative-intent lung resections 7 times, immediately before surgery (time A), after tumor resection [time B (5 minutes), time C (30 minutes), and time D (2 hours)] and after surgery [time P1 (1 day), time P2 (3 days), and time P3 (1 month)]. A rapid decrease in the content of ctDNA was found after the curative-intent resection (the mean mutant allele fraction at times A, B, C, and D was 2.72%, 2.11%, 1.14%, and 0.17%, respectively) which implied that the half-time of ctDNA is short and there is an association between ctDNA and tumor volume. In addition, the detection of ctDNA at time P2 (278 days versus 637 days, P=0.002) and time P3 (295 days versus 662 days, P=0.003) rather than time P1 (528 days versus 543 days, P=0.657) was negatively correlated with recurrence free survival (RFS) of patients; similar correlations were observed between ctDNA detection and overall survival (OS) (58).
Xia et al. analyzed ctDNA in another prospective, multicenter study (LUNGCA-1; 2021) on NSCLC surgery patients. They found that detectable ctDNA before operation (RFS; HR=4.2, 95%CI: 2.6-6.7; P < 0.001) or at 3 days and/or 1 month after operation (RFS; HR=11.1, 95%CI: 6.5-19.0; P < 0.001) was a robust predictor for relapse in patients with stage I–III NSCLC. Moreover, ctDNA status was tightly associated with the benefit of postoperative adjuvant therapy — ctDNA-positive patients who received adjuvant therapies had improved RFS over those that did not receive (RFS; HR=0.3, 95%CI: 0.1-0.8; P=0.008), while ctDNA-negative patients receiving adjuvant therapies had impaired RFS than those that did not receive (RFS; HR=3.1, 95%CI: 1.7-5.5; P < 0.001) (7).
Chaudhuri et al. prespecified “MRD landmark” as the ctDNA status following the first phlebotomy of curative-intent resection and within 4 months from the end of therapy, progression at 36 months after the MRD landmark was 100% and 7% in patients with detectable and undetectable ctDNA MRD (HR=43.4, 95%CI=5.7–341; P < 0.001), respectively (59).
Yilong Wu et al. elucidated the role of MRD monitoring in patients with stage I to IIIA NSCLC after definite surgical resection. Patients with undetectable MRD at landmark or longitudinal time points had better disease-free survival (DFS) than those with detectable MRD [landmark: unreached vs. 12.1 months (4.7–19.5); HR = 0.08; 95% CI, 0.02–0.33; longitudinal: unreached vs. 15.9 months (13.8–18.0); HR = 0.02; 95% CI, 0.01–0.05]. 96.8% of patients with longitudinal undetectable MRD were still disease-free at the last follow-up and had nothing to do with the clinical stage, thus it may represent the potentially cured population, which has important application value for the treatment of early lung cancer in the future. Because MRD status reflected the tumor load, adjuvant therapy was found to confer a survival benefit for patients with detectable MRD (P = 0.022; HR = 0.34; 95% CI, 0.12-0.88) (60).
Development of resistance due to targeted therapy
Driver gene mutations might exhibit changes during tumor development or treatment that can lead to resistance to the drugs, which limits the long-term use of targeted therapy. Thus, the new driver gene mutations need to be detected through a re-biopsy. Liquid biopsy can specifically detect the new gene mutations, and this can be used to predict targeted therapy resistance development in patients.
Approximately half or more NSCLC patients with EGFR mutations who develop resistance to the first- and second-generation EGFR-tyrosine kinase inhibitors (TKI) will develop a secondary EGFR T790M mutation in the tumor (61). Additionally, the ctDNA T790M mutation is more likely to be seen in patients with an initial EGFR del19 mutation compared with the EGFR L858R mutation (62). ctDNA analysis may help in predicting such resistance and directing the use of subsequent therapy such as the use of osimertinib, an oral, irreversible third-generation EGFR-TKI, approved by FDA in 2015 (42, 63). LiquidLung-O-Cohort 2 study screened ctDNA from patients with EGFR T790M mutation with a detection sensitivity of 56.8% (64). Serial monitoring of EGFR mutation in ctDNA is able to detect EGFR T790M mutation much earlier (range: 15-344 days) than clinical manifestation of the disease progression (65). However, since osimertinib is considered standard first-line therapy for NSCLC patients with EGFR mutations (66), some patients on osimertinib would inevitably develop new mutations (detected in ctDNA), including EGFR C797S mutation, MET amplification, HER2 exon 20 insertions, BRAFV600E mutation, PIK3CA mutation, and EGFR amplification and thus be resistant to the drug (67–71).
Dagogo et al. analyzed plasma and tumor samples from patients with progressed ALK-positive NSCLC treated with alectinib (2019). There was no difference in ALK mutation frequency (67% versus 63%), but ctDNA was more likely to harbor ≥2 ALK mutations (24% versus 2%, P=0.004). However, ALK L1196M, a gatekeeper mutation that leads to resistance to crizotinib, showed little prevalence between tumor DNA and ctDNA (2% versus 22%, P=0.008), which implies that ctDNA can predict ALK-TKI resistance sometimes. A similar phenomenon was found in those with lorlatinib, showing a promoted acquisition of ALK resistance mutations after sequential treatment with increasingly potent ALK-TKIs (72). Secondary ALK mutations such as ALK G1202R, ALK G1269A, and ALK L1196M were found in ctDNA through NGS, causing genetic resistance to first- and second-generation ALK-TKIs (50, 72–74).
Prognosis and treatment response after target therapy
ctDNA is commonly used to monitor the benefit of the treatment and predict progression via liquid biopsy. Several studies have found a significant association between the quantitative changes in ctDNA, the response of cancer to the targeted therapy, and the prognosis of NSCLC (Table 2).
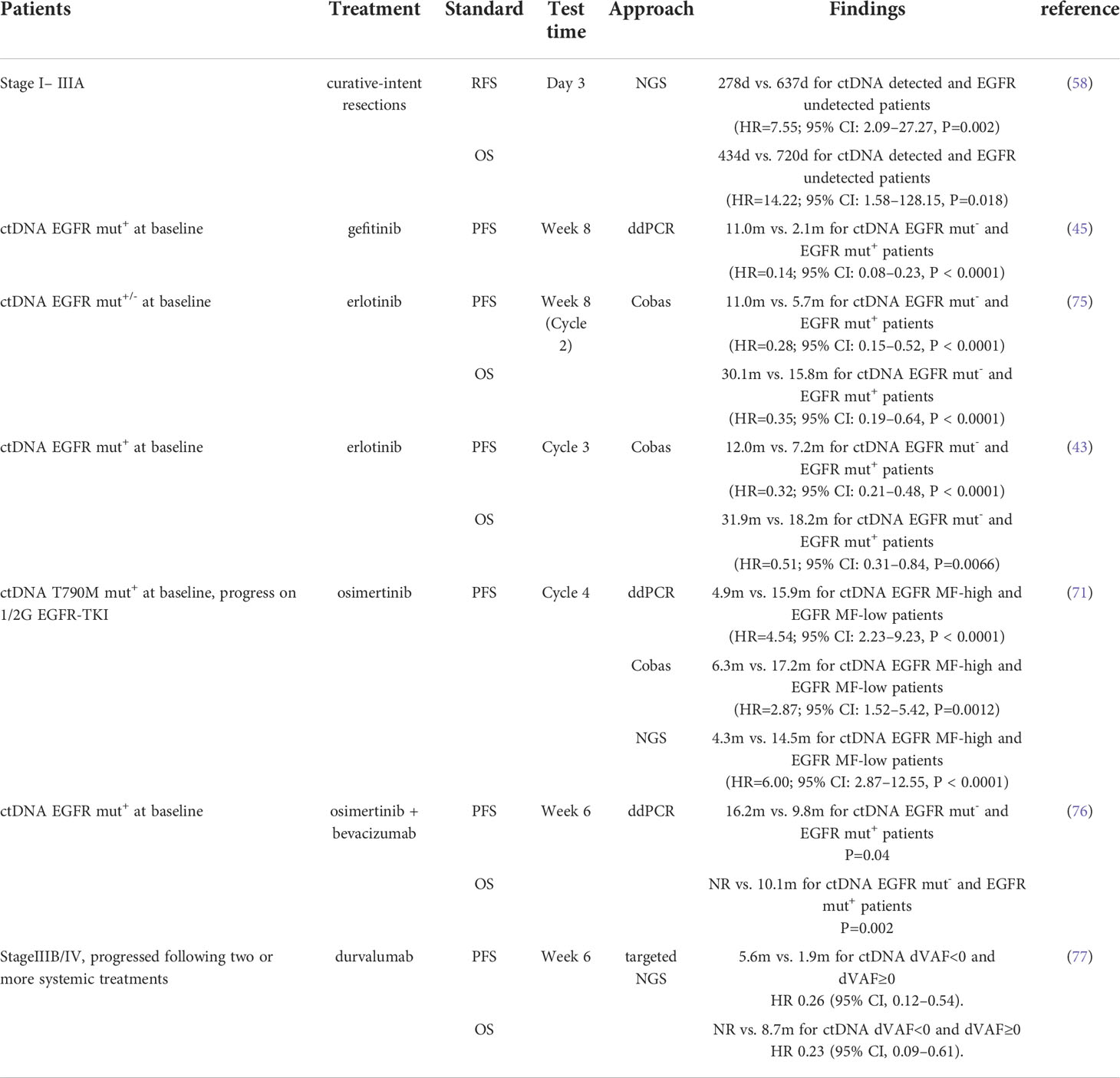
Table 2 ctDNA as a treatment response predictor.
High plasma cfDNA is associated with poor OS (16.0 months versus 28.6 months, P=0.030) and increased risk of death (HR=1.23, 95% CI: 1.01-1.50; P=0.045) (78). Bordi et al. defined a cut-off of 2200 copies/ml generated by means of ROC analysis and found a lower number of mutations (< 2200 copies/mL; at baseline) are associated with better progression free survival (PFS; 17.8 months versus 4.3 months, p=0.022) and OS (23.6 months versus 7.7 months, p= 0.016) (79).
Baseline EGFR T790M mutation detection in ctDNA might correlate with a larger baseline tumor size (56 mm for T790M (+) versus 39 mm for T790M (-); P < 0.0001) and a higher probability of extra thoracic metastasis [58% M1b for T790M (+) versus 39% M1b for T790M (-); P = 0.002] (80). Moreover, tissue T790M positive patients without detectable T790M mutation in the ctDNA had a longer PFS, which might be attributed to a lower tumor burden (80).
Identification of EGFR mutation in ctDNA before the start of the treatment procedure helps to select patients who might benefit from EGFR-TKI treatment, and monitoring ctDNA consistently for further EGFR mutation helps to predict the outcome of current treatment and the patient’s prognosis (45, 81). In patients treated with erlotinib and assessed to be stable disease (SD), undetectable ctDNA at week 8 is correlated with survival improvement (PFS: HR=0.27, 95%CI: 0.13-0.57, p<0.0001; OS: HR=0.40, 95% CI 0.20–0.80, p=0.009) (75). In NSCLC patients with progression after EGFR-TKI therapy, chest- or brain-limited disease has a significantly higher rate of ctDNA T790M mutation than the others (P<0.001). This showed that both ctDNA T790M mutation status and TKI treatment failure can predict prognosis (82). Furthermore, the persistence of EGFR mutation in ctDNA at 6 weeks in patients treated with osimertinib was associated with shorter PFS (9.8 months versus 16.2 months, P=0.04) (76), while loss of EGFR exon 19 deletion or L858R mutation post-treatment appears to correlate with longer PFS (14.7 months versus 5.5 months) (67). The monitering of ctDNA for EGFR mutations in NSCLC patients at treatment cycle 4 is optimal for predicting the treatment outcomes for patients receiving osimertinib (71).
Patients with EML4-ALK fusion variants 1 detected in ctDNA at baseline exhibited longer PFS than those with EML4-ALK fusion variants 3 [8.2 months (95% CI: 2.1–11.7) versus 1.9 months (95% CI: 1.8-not estimable)] (53). In the ALTA-1L study, researchers found detectable baseline EML4-ALK fusion variant 3 rather than variant 1 in ctDNA, which was associated with poor PFS in patients treated with ALK TKI [crizotinib: HR: 3.42 (1.56–7.50), P=0.002; brigatinib: HR: 2.45 (1.07–5.60), P=0.033] (83).
Prognosis and treatment outcome after immunotherapy
Even though long-term positive responses have been observed in NSCLC patients receiving ICI therapy, the majority of them become refractory with an eventual unfavorable clinical outcome (84). Rapid as well as sensitive detection of dynamic changes in the ctDNA might help to identify NSCLC patients and plan appropriate immunotherapy for them (85). (Table 2)
Goldberg et al. defined ctDNA response as a >50% decrease in mutant allele fraction from its baseline (2018). In ICI therapy receiving patients with metastatic NSCLC, ctDNA response greatly agreed with the radiographic response (κ=0.753), and benefits could be assessed faster from ctDNA than radiographically (median 24.5 days versus median 72.5 days). Additionally, a ctDNA response is associated with long-term treatment benefit (205.5 days versus 69 days; P<0.001) as well as better prognosis (PFS: HR=0.29, 95%CI: 0.09–0.89, P=0.03; OS: HR=0.17, 95%CI: 0.05–0.62; P= 0.007) (86). Similar to target therapy, patients with undetectable levels of ctDNA were demonstrated to have significantly longer PFS (P=0.001) and OS (P=0.008) compared with those with no evidence of ctDNA clearance (85). Hellmann et al. tested ctDNA of 31 patients with advanced NSCLC and had achieved long-term benefit from ICI therapy (PFS≥12 months) at a median time of 26.7 months after the initiation of therapy. They found 25/27 (93%) patients with ctDNA negative remained progression-free, while in 4 patients with ctDNA positive, the desease eventually progressed (87).
Nabet et al. established an approach named DIREct-On (Durable Immunotherapy Response Estimation by immune profiling and ctDNA- On-treatment) to predict whether patients with NCSLC would show durable clinical response to ICI therapy. DIREct-On incorporated pre-treatment ctDNA and immune profiling with early on-treatment ctDNA response assessment and could get an accuracy of 92% to identify the potential patients obtaining benefit (88).
A high tumor mutation burden (TMB) and microsatellite instability (MSI) are demonstrated to correlate with a better response to immunotherapy in NSCLC (89–91).,TMB is measured from tumor tissue traditionally. Si et al. measured and compared TMB from tissue (tTMB) and plasma (bTMB) samples and found a positive correlation between bTMB (using a cutoff of 20 mut/Mb) and tTMB (using a cutoff of 10 mut/Mb) values (P<0.0001, χ2 test). They also found that higher bTMB was also associated with the clinical benefits of immunotherapy (51). When the bTMB cut-off point was set to 6, patients with higher bTMB showed superior PFS (NR versus 2.9 m; HR=0.39, 95%CI: 0.18-0.84, P=0.01) (92). Goldberg et al. found that bTMB is independently predictive of the immunotherapy outcome benefits without association with high PD-L1 expression. They further discovered that bTMB=16 mut/Mb is a clinically meaningful cut-off point in NSCLC and patients with bTMB≥16 mut/Mb benefited from a second-line immunotherapy rather than chemotherapy (PFS was 4.2m in the atezolizumab arm and 2.9m in the docetaxel arm, HR=0.57, 95%CI: 0.33–0.99; OS was 13.0m and 7.4m, HR=0.56, 95%CI: 0.31–0.99) (93). Similarly, Georgiadis et al. defined ctDNA bTMB≥10 mut/Mb of the whole exome as bTMB-high and demonstrated that bTMB-high before immunotherapy predicted a better PFS (HR=0.23, 95% CI, 0.07–0.63, P=0.003) and OS (HR=0.26, 95% CI, 0.08–0.72, P=0.008) in pan-cancer. Additionally, patients with blood MSI also had a better PFS (HR=0.21, 95% CI, 0.08–0.54, P=0.001) and OS (HR=0.41, 95% CI, 0.16–1.05, P=0.063) than those with microsatellite stability (MSS) (94).
Variant allele frequencies (VAF) in ctDNA can also predict immunotherapy response as an alteration of TMB. Patients with decreased VAF at week 6 of the treatment had a mean reduced tumor volume by 39%, while those with a high VAF had a mean increased tumor volume by 36% (77). Additionally, a decrease of VAF at week 6 of the immunotherapy implied a longer PFS and OS (77).
Discussion and limitation
Several clinical trials have demonstrated definite correlations between ctDNA levels and NSCLC patients’ medical status, including tumor sizes, recurrence post operations, choice of treatments, treatment response and prognosis. Thus, ctDNA could help to guide clinicians in selecting appropriate therapies for each patient: whether to utilize adjuvant therapies after curative-intent resections, how to select a treatment that could benefit a patient maximumly and how to evaluate treatment efficiency and diagnose drug resistance on time. The short half-life of ctDNA enables it to exhibit a real-time status of a dynamic disease and overcome temporal tumor heterogeneity. Additionally, as ctDNA could represent spatial heterogeneity better than primary tumors or metastatic lesions biopsy, ctDNA has an inherent advantage in monitoring a patient’s condition. Undoubtedly, ctDNA can serve as a predictor of MRD and can be frequent applied in the medical management of NSCLC patients. At the same time, there were a lot of research about MRD in predicting the recurrence trajectory of early lung cancer and the curative effect of consolidation immunotherapy, which show great clinical application prospect.
Based on this, we put forward some ideas. For lung cancer patients with driver gene mutations after radical resection, ctDNA-based MRD monitoring to guide the use of targeted drugs, rather than continuous drug use mode, can theoretically delay the development of drug-resistant clones of tumor targeted therapy, thereby delaying drug resistance? At the same time, the treatment burden of patients can be reduced; For patients with inoperable locally advanced NSCLC, after radical therapy, ctDNA-based MRD monitoring can be used to guide immunodrug maintenance therapy, which can not only predict the population benefiting from ICI consolidation therapy, but also reduce the treatment burden. Starting from a population of patients with advanced targeted therapy, MRD monitoring should be used to guide the use of targeted drugs after patients have achieved complete remission or local treatment for oligometrics.
However, some limitations can hamper the wide use of ctDNA. Early-stage NSCLC remains tough to be detected by ctDNA mainly because of its extremely low concentrations due to small tumor sizes. Thus, highly sensitive methods need to be developed. Moreover, most of the trials that utilize ctDNA to plan the targeted therapy and predict treatment response or prognosis focused on the most common driver gene mutation, EGFR mutations. The use of ctDNA in patients with ALK fusions, MET amplification, HER2 mutations, and other rare mutations still needs to be studied in detail.
To conclude, a transformation in the management of NSCLC patients by analyzing ctDNA is afoot.
Author contributions
XF, SY, YJ, YX, KL drafted the manuscript, conceived and designed the study, and accomplished the revision of manuscript for important intellectual content. KL obtained funding. All authors contributed to the article and approved the submitted version.
Funding
This study is supported by the National Natural Science Foundation of China (81902327, 82172708); and the Jiangsu National Natural Science Foundation (BK20191064).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin (2021) 71:209–49. doi: 10.3322/caac.21660
2. Travis WD, Brambilla E, Nicholson AG, Yatabe Y, Austin JHM, Beasley MB, et al. The 2015 world health organization classification of lung tumors: Impact of genetic, clinical and radiologic advances since the 2004 classification. J Thorac Oncol (2015) 10:1243–60. doi: 10.1097/jto.0000000000000630
3. Kris MG, Johnson BE, Berry LD, Kwiatkowski DJ, Iafrate AJ, Wistuba II, et al. Using multiplexed assays of oncogenic drivers in lung cancers to select targeted drugs. Jama (2014) 311:1998–2006. doi: 10.1001/jama.2014.3741
4. Doroshow DB, Sanmamed MF, Hastings K, Politi K, Rimm DL, Chen L, et al. Immunotherapy in non-small cell lung cancer: Facts and hopes. Clin Cancer Res (2019) 25:4592–602. doi: 10.1158/1078-0432.Ccr-18-1538
5. Li W, Liu J-B, Hou L-K, Yu F, Zhang J, Wu W, et al. Liquid biopsy in lung cancer: significance in diagnostics, prediction, and treatment monitoring. Mol Cancer (2022) 21:25. doi: 10.1186/s12943-022-01505-z
6. Corcoran RB, Chabner BA. Application of cell-free DNA analysis to cancer treatment. N Engl J Med (2018) 379:1754–65. doi: 10.1056/NEJMra1706174
7. Xia L, Mei J, Kang R, Deng S, Chen Y, Yang Y, et al. Perioperative ctDNA-based molecular residual disease detection for non-small cell lung cancer: A prospective multicenter cohort study (LUNGCA-1). Clin Cancer Res (2021) 28: 3308–17. doi: 10.1158/1078-0432.Ccr-21-3044
8. Ignatiadis M, Sledge GW, Jeffrey SS. Liquid biopsy enters the clinic - implementation issues and future challenges. Nat Rev Clin Oncol (2021) 18:297–312. doi: 10.1038/s41571-020-00457-x
9. Murtaza M, Dawson S-J, Pogrebniak K, Rueda OM, Provenzano E, Grant J, et al. Multifocal clonal evolution characterized using circulating tumour DNA in a case of metastatic breast cancer. Nat Commun (2015) 6:8760. doi: 10.1038/ncomms9760
10. Hindson BJ, Ness KD, Masquelier DA, Belgrader P, Heredia NJ, Makarewicz AJ, et al. High-throughput droplet digital PCR system for absolute quantitation of DNA copy number. Anal Chem (2011) 83:8604–10. doi: 10.1021/ac202028g
11. Vogelstein B, Kinzler KW. Digital PCR. Proc Natl Acad Sci U.S.A. (1999) 96:9236–41. doi: 10.1073/pnas.96.16.9236
12. Huggett JF, Whale A. Digital PCR as a novel technology and its potential implications for molecular diagnostics. Clin Chem (2013) 59:1691–3. doi: 10.1373/clinchem.2013.214742
13. Zonta E, Garlan F, Pécuchet N, Perez-Toralla K, Caen O, Milbury C, et al. Multiplex detection of rare mutations by picoliter droplet based digital PCR: Sensitivity and specificity considerations. PloS One (2016) 11:e0159094. doi: 10.1371/journal.pone.0159094
14. Perkins G, Lu H, Garlan F, Taly V. Droplet-based digital PCR: Application in cancer research. Adv Clin Chem (2017) 79:43–91. doi: 10.1016/bs.acc.2016.10.001
15. Hindson CM, Chevillet JR, Briggs HA, Gallichotte EN, Ruf IK, Hindson BJ, et al. Absolute quantification by droplet digital PCR versus analog real-time PCR. Nat Methods (2013) 10:1003–5. doi: 10.1038/nmeth.2633
16. Dressman D, Yan H, Traverso G, Kinzler KW, Vogelstein B. Transforming single DNA molecules into fluorescent magnetic particles for detection and enumeration of genetic variations. Proc Natl Acad Sci U.S.A. (2003) 100:8817–22. doi: 10.1073/pnas.1133470100
17. Diehl F, Li M, He Y, Kinzler KW, Vogelstein B, Dressman D. BEAMing: single-molecule PCR on microparticles in water-in-oil emulsions. Nat Methods (2006) 3:551–9. doi: 10.1038/nmeth898
18. Garrido P, Paz-Ares L, Majem M, Morán T, Trigo JM, Bosch-Barrera J, et al. LungBEAM: A prospective multicenter study to monitor stage IV NSCLC patients with EGFR mutations using BEAMing technology. Cancer Med (2021) 10:5878–88. doi: 10.1002/cam4.4135
19. O’Leary B, Hrebien S, Beaney M, Fribbens C, Garcia-Murillas I, Jiang J, et al. Comparison of BEAMing and droplet digital PCR for circulating tumor DNA analysis. Clin Chem (2019) 65:1405–13. doi: 10.1373/clinchem.2019.305805
20. Wan JCM, Massie C, Garcia-Corbacho J, Mouliere F, Brenton JD, Caldas C, et al. Liquid biopsies come of age: towards implementation of circulating tumour DNA. Nat Rev Cancer (2017) 17:223–38. doi: 10.1038/nrc.2017.7
21. Yohe S, Thyagarajan B. Review of clinical next-generation sequencing. Arch Pathol Lab Med (2017) 141:1544–57. doi: 10.5858/arpa.2016-0501-RA
22. D’Haene N, Le Mercier M, De Nève N, Blanchard O, Delaunoy M, El Housni H, et al. Clinical validation of targeted next generation sequencing for colon and lung cancers. PloS One (2015) 10:e0138245. doi: 10.1371/journal.pone.0138245
23. Larribère L, Martens UM. Advantages and challenges of using ctDNA NGS to assess the presence of minimal residual disease (MRD) in solid tumors. Cancers (Basel) (2021) 13:5698. doi: 10.3390/cancers13225698
24. Forshew T, Murtaza M, Parkinson C, Gale D, Tsui DW, Kaper F, et al. Noninvasive identification and monitoring of cancer mutations by targeted deep sequencing of plasma DNA. Sci Transl Med (2012) 4:136ra168. doi: 10.1126/scitranslmed.3003726
25. Kinde I, Wu J, Papadopoulos N, Kinzler KW, Vogelstein B. Detection and quantification of rare mutations with massively parallel sequencing. Proc Natl Acad Sci U.S.A. (2011) 108:9530–5. doi: 10.1073/pnas.1105422108
26. Newman AM, Bratman SV, To J, Wynne JF, Eclov NC, Modlin LA, et al. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat Med (2014) 20:548–54. doi: 10.1038/nm.3519
27. Newman AM, Lovejoy AF, Klass DM, Kurtz DM, Chabon JJ, Scherer F, et al. Integrated digital error suppression for improved detection of circulating tumor DNA. Nat Biotechnol (2016) 34:547–55. doi: 10.1038/nbt.3520
28. Kurtz DM, Soo J, Co Ting Keh L, Alig S, Chabon JJ, Sworder BJ, et al. Enhanced detection of minimal residual disease by targeted sequencing of phased variants in circulating tumor DNA. Nat Biotechnol (2021) 39:1537–47. doi: 10.1038/s41587-021-00981-w
29. Zehir A, Benayed R, Shah RH, Syed A, Middha S, Kim HR, et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med (2017) 23:703–13. doi: 10.1038/nm.4333
30. Buchhalter I, Rempel E, Endris V, Allgäuer M, Neumann O, Volckmar A-L, et al. Size matters: Dissecting key parameters for panel-based tumor mutational burden analysis. Int J Cancer (2019) 144:848–58. doi: 10.1002/ijc.31878
31. Goldstraw P, Chansky K, Crowley J, Rami-Porta R, Asamura H, Eberhardt WE, et al. The IASLC lung cancer staging project: Proposals for revision of the TNM stage groupings in the forthcoming (Eighth) edition of the TNM classification for lung cancer. J Thorac Oncol (2016) 11:39–51. doi: 10.1016/j.jtho.2015.09.009
32. Wong CC, Baum J, Silvestro A, Beste MT, Bharani-Dharan B, Xu S, et al. Inhibition of IL1β by canakinumab may be effective against diverse molecular subtypes of lung cancer: An exploratory analysis of the CANTOS trial. Cancer Res (2020) 80:5597–605. doi: 10.1158/0008-5472.Can-19-3176
33. Chabon JJ, Hamilton EG, Kurtz DM, Esfahani MS, Moding EJ, Stehr H, et al. Integrating genomic features for non-invasive early lung cancer detection. Nature (2020) 580:245–51. doi: 10.1038/s41586-020-2140-0
34. Chen K, Zhang J, Guan T, Yang F, Lou F, Chen W, et al. Comparison of plasma to tissue DNA mutations in surgical patients with non-small cell lung cancer. J Thorac Cardiovasc Surg (2017) 154:1123–1131.e1122. doi: 10.1016/j.jtcvs.2017.04.073
35. Liang W, Zhao Y, Huang W, Gao Y, Xu W, Tao J, et al. Non-invasive diagnosis of early-stage lung cancer using high-throughput targeted DNA methylation sequencing of circulating tumor DNA (ctDNA). Theranostics (2019) 9:2056–70. doi: 10.7150/thno.28119
36. Mack PC, Banks KC, Espenschied CR, Burich RA, Zill OA, Lee CE, et al. Spectrum of driver mutations and clinical impact of circulating tumor DNA analysis in non-small cell lung cancer: Analysis of over 8000 cases. Cancer (2020) 126:3219–28. doi: 10.1002/cncr.32876
37. Aggarwal C, Thompson JC, Black TA, Katz SI, Fan R, Yee SS, et al. Clinical implications of plasma-based genotyping with the delivery of personalized therapy in metastatic non-small cell lung cancer. JAMA Oncol (2019) 5:173–80. doi: 10.1001/jamaoncol.2018.4305
38. Leighl NB, Page RD, Raymond VM, Daniel DB, Divers SG, Reckamp KL, et al. Clinical utility of comprehensive cell-free DNA analysis to identify genomic biomarkers in patients with newly diagnosed metastatic non-small cell lung cancer. Clin Cancer Res (2019) 25:4691–700. doi: 10.1158/1078-0432.Ccr-19-0624
39. Kimura H, Suminoe M, Kasahara K, Sone T, Araya T, Tamori S, et al. Evaluation of epidermal growth factor receptor mutation status in serum DNA as a predictor of response to gefitinib (IRESSA). Br J Cancer (2007) 97:778–84. doi: 10.1038/sj.bjc.6603949
40. Douillard JY, Ostoros G, Cobo M, Ciuleanu T, McCormack R, Webster A, et al. First-line gefitinib in Caucasian EGFR mutation-positive NSCLC patients: a phase-IV, open-label, single-arm study. Br J Cancer (2014) 110:55–62. doi: 10.1038/bjc.2013.721
41. Jänne PA, Borras AM, Kuang Y, Rogers AM, Joshi VA, Liyanage H, et al. A rapid and sensitive enzymatic method for epidermal growth factor receptor mutation screening. Clin Cancer Res (2006) 12:751–8. doi: 10.1158/1078-0432.Ccr-05-2047
42. Kuang Y, Rogers A, Yeap BY, Wang L, Makrigiorgos M, Vetrand K, et al. Noninvasive detection of EGFR T790M in gefitinib or erlotinib resistant non-small cell lung cancer. Clin Cancer Res (2009) 15:2630–6. doi: 10.1158/1078-0432.Ccr-08-2592
43. Mok T, Wu YL, Lee JS, Yu CJ, Sriuranpong V, Sandoval-Tan J, et al. Detection and dynamic changes of EGFR mutations from circulating tumor DNA as a predictor of survival outcomes in NSCLC patients treated with first-line intercalated erlotinib and chemotherapy. Clin Cancer Res (2015) 21:3196–203. doi: 10.1158/1078-0432.Ccr-14-2594
44. Wu YL, Sequist LV, Hu CP, Feng J, Lu S, Huang Y, et al. EGFR mutation detection in circulating cell-free DNA of lung adenocarcinoma patients: analysis of LUX-lung 3 and 6. Br J Cancer (2017) 116:175–85. doi: 10.1038/bjc.2016.420
45. Wang Z, Cheng Y, An T, Gao H, Wang K, Zhou Q, et al. Detection of EGFR mutations in plasma circulating tumour DNA as a selection criterion for first-line gefitinib treatment in patients with advanced lung adenocarcinoma (BENEFIT): a phase 2, single-arm, multicentre clinical trial. Lancet Respir Med (2018) 6:681–90. doi: 10.1016/s2213-2600(18)30264-9
46. Han JY, Choi JJ, Kim JY, Han YL, Lee GK. PNA clamping-assisted fluorescence melting curve analysis for detecting EGFR and KRAS mutations in the circulating tumor DNA of patients with advanced non-small cell lung cancer. BMC Cancer (2016) 16:627. doi: 10.1186/s12885-016-2678-2
47. Krug AK, Enderle D, Karlovich C, Priewasser T, Bentink S, Spiel A, et al. Improved EGFR mutation detection using combined exosomal RNA and circulating tumor DNA in NSCLC patient plasma. Ann Oncol (2018) 29:700–6. doi: 10.1093/annonc/mdx765
48. Park CK, Lee SY, Lee JC, Choi CM, Lee SY, Jang TW, et al. Phase II open-label multicenter study to assess the antitumor activity of afatinib in lung cancer patients with activating epidermal growth factor receptor mutation from circulating tumor DNA: Liquid-Lung-A. Thorac Cancer (2021) 12:444–52. doi: 10.1111/1759-7714.13763
49. Mondaca S, Lebow ES, Namakydoust A, Razavi P, Reis-Filho JS, Shen R, et al. Clinical utility of next-generation sequencing-based ctDNA testing for common and novel ALK fusions. Lung Cancer (2021) 159:66–73. doi: 10.1016/j.lungcan.2021.06.018
50. Wang Y, Tian PW, Wang WY, Wang K, Zhang Z, Chen BJ, et al. Noninvasive genotyping and monitoring of anaplastic lymphoma kinase (ALK) rearranged non-small cell lung cancer by capture-based next-generation sequencing. Oncotarget (2016) 7:65208–17. doi: 10.18632/oncotarget.11569
51. Si H, Kuziora M, Quinn KJ, Helman E, Ye J, Liu F, et al. A blood-based assay for assessment of tumor mutational burden in first-line metastatic NSCLC treatment: Results from the MYSTIC study. Clin Cancer Res (2021) 27:1631–40. doi: 10.1158/1078-0432.Ccr-20-3771
52. Kwapisz D. The first liquid biopsy test approved. is it a new era of mutation testing for non-small cell lung cancer? Ann Transl Med (2017) 5:46. doi: 10.21037/atm.2017.01.32
53. Horn L, Whisenant JG, Wakelee H, Reckamp KL, Qiao H, Leal TA, et al. Monitoring therapeutic response and resistance: Analysis of circulating tumor DNA in patients with ALK+ lung cancer. J Thorac Oncol (2019) 14:1901–11. doi: 10.1016/j.jtho.2019.08.003
54. Dagogo-Jack I, Rooney M, Nagy RJ, Lin JJ, Chin E, Ferris LA, et al. Molecular analysis of plasma from patients with ROS1-positive NSCLC. J Thorac Oncol (2019) 14:816–24. doi: 10.1016/j.jtho.2019.01.009
55. Vansteenkiste JF, Canon JL, De Braud F, Grossi F, De Pas T, Gray JE, et al. Safety and efficacy of buparlisib (BKM120) in patients with PI3K pathway-activated non-small cell lung cancer: Results from the phase II BASALT-1 study. J Thorac Oncol (2015) 10:1319–27. doi: 10.1097/jto.0000000000000607
56. Goto K, Ichinose Y, Ohe Y, Yamamoto N, Negoro S, Nishio K, et al. Epidermal growth factor receptor mutation status in circulating free DNA in serum: from IPASS, a phase III study of gefitinib or carboplatin/paclitaxel in non-small cell lung cancer. J Thorac Oncol (2012) 7:115–21. doi: 10.1097/JTO.0b013e3182307f98
57. Chin R-I, Chen K, Usmani A, Chua C, Harris PK, Binkley MS, et al. Detection of solid tumor molecular residual disease (MRD) using circulating tumor DNA (ctDNA). Mol Diagn Ther (2019) 23:311–31. doi: 10.1007/s40291-019-00390-5
58. Chen K, Zhao H, Shi Y, Yang F, Wang LT, Kang G, et al. Perioperative dynamic changes in circulating tumor DNA in patients with lung cancer (DYNAMIC). Clin Cancer Res (2019) 25:7058–67. doi: 10.1158/1078-0432.Ccr-19-1213
59. Chaudhuri AA, Chabon JJ, Lovejoy AF, Newman AM, Stehr H, Azad TD, et al. Early detection of molecular residual disease in localized lung cancer by circulating tumor DNA profiling. Cancer Discov (2017) 7:1394–403. doi: 10.1158/2159-8290.Cd-17-0716
60. Zhang JT, Liu SY, Gao W, Liu SM, Yan HH, Ji L, et al. Longitudinal undetectable molecular residual disease defines potentially cured population in localized non-small cell lung cancer. Cancer Discov (2022) 12:1690–701. doi: 10.1158/2159-8290.Cd-21-1486
61. Balak MN, Gong Y, Riely GJ, Somwar R, Li AR, Zakowski MF, et al. Novel D761Y and common secondary T790M mutations in epidermal growth factor receptor-mutant lung adenocarcinomas with acquired resistance to kinase inhibitors. Clin Cancer Res (2006) 12:6494–501. doi: 10.1158/1078-0432.Ccr-06-1570
62. Remon J, Caramella C, Jovelet C, Lacroix L, Lawson A, Smalley S, et al. Osimertinib benefit in EGFR-mutant NSCLC patients with T790M-mutation detected by circulating tumour DNA. Ann Oncol (2017) 28:784–90. doi: 10.1093/annonc/mdx017
63. Yang JC, Ahn MJ, Kim DW, Ramalingam SS, Sequist LV, Su WC, et al. Osimertinib in pretreated T790M-positive advanced non-Small-Cell lung cancer: AURA study phase II extension component. J Clin Oncol (2017) 35:1288–96. doi: 10.1200/jco.2016.70.3223
64. Park CK, Cho HJ, Choi YD, Oh IJ, Kim YCA. Phase II trial of osimertinib in the second-line treatment of non-small cell lung cancer with the EGFR T790M mutation, detected from circulating tumor DNA: LiquidLung-O-Cohort 2. Cancer Res Treat (2019) 51:777–87. doi: 10.4143/crt.2018.387
65. Sorensen BS, Wu L, Wei W, Tsai J, Weber B, Nexo E, et al. Monitoring of epidermal growth factor receptor tyrosine kinase inhibitor-sensitizing and resistance mutations in the plasma DNA of patients with advanced non-small cell lung cancer during treatment with erlotinib. Cancer (2014) 120:3896–901. doi: 10.1002/cncr.28964
66. Ramalingam SS, Vansteenkiste J, Planchard D, Cho BC, Gray JE, Ohe Y, et al. Overall survival with osimertinib in untreated, EGFR-mutated advanced NSCLC. N Engl J Med (2020) 382:41–50. doi: 10.1056/NEJMoa1913662
67. Yu HA, Paz-Ares LG, Yang JC, Lee KH, Garrido P, Park K, et al. Phase I study of the efficacy and safety of ramucirumab in combination with osimertinib in advanced T790M-positive EGFR-mutant non-small cell lung cancer. Clin Cancer Res (2021) 27:992–1002. doi: 10.1158/1078-0432.Ccr-20-1690
68. Thress KS, Paweletz CP, Felip E, Cho BC, Stetson D, Dougherty B, et al. Acquired EGFR C797S mutation mediates resistance to AZD9291 in non-small cell lung cancer harboring EGFR T790M. Nat Med (2015) 21:560–2. doi: 10.1038/nm.3854
69. Guibert N, Hu Y, Feeney N, Kuang Y, Plagnol V, Jones G, et al. Amplicon-based next-generation sequencing of plasma cell-free DNA for detection of driver and resistance mutations in advanced non-small cell lung cancer. Ann Oncol (2018) 29:1049–55. doi: 10.1093/annonc/mdy005
70. Ramalingam SS, Yang JC, Lee CK, Kurata T, Kim DW, John T, et al. Osimertinib as first-line treatment of EGFR mutation-positive advanced non-Small-Cell lung cancer. J Clin Oncol (2018) 36:841–9. doi: 10.1200/jco.2017.74.7576
71. Sakai K, Takahama T, Shimokawa M, Azuma K, Takeda M, Kato T, et al. Predicting osimertinib-treatment outcomes through EGFR mutant-fraction monitoring in the circulating tumor DNA of EGFR T790M-positive patients with non-small cell lung cancer (WJOG8815L). Mol Oncol (2021) 15:126–37. doi: 10.1002/1878-0261.12841
72. Dagogo-Jack I, Rooney M, Lin JJ, Nagy RJ, Yeap BY, Hubbeling H, et al. Treatment with next-generation ALK inhibitors fuels plasma ALK mutation diversity. Clin Cancer Res (2019) 25:6662–70. doi: 10.1158/1078-0432.Ccr-19-1436
73. Sánchez-Herrero E, Serna-Blasco R, Ivanchuk V, García-Campelo R, Dómine Gómez M, Sánchez JM, et al. NGS-based liquid biopsy profiling identifies mechanisms of resistance to ALK inhibitors: a step toward personalized NSCLC treatment. Mol Oncol (2021) 15:2363–76. doi: 10.1002/1878-0261.13033
74. Lin YT, Chiang CL, Hung JY, Lee MH, Su WC, Wu SY, et al. Resistance profiles of anaplastic lymphoma kinase tyrosine kinase inhibitors in advanced non-small-cell lung cancer: a multicenter study using targeted next-generation sequencing. Eur J Cancer (2021) 156:1–11. doi: 10.1016/j.ejca.2021.06.043
75. Kok PS, Lee K, Lord S, John T, Marschner I, Wu YL, et al. Incorporating circulating tumor DNA detection to radiographic assessment for treatment response in advanced EGFR-mutant lung cancer. Lung Cancer (2022) 163:14–8. doi: 10.1016/j.lungcan.2021.11.010
76. Yu HA, Schoenfeld AJ, Makhnin A, Kim R, Rizvi H, Tsui D, et al. Effect of osimertinib and bevacizumab on progression-free survival for patients with metastatic EGFR-mutant lung cancers: A phase 1/2 single-group open-label trial. JAMA Oncol (2020) 6:1048–54. doi: 10.1001/jamaoncol.2020.1260
77. Raja R, Kuziora M, Brohawn PZ, Higgs BW, Gupta A, Dennis PA, et al. Early reduction in ctDNA predicts survival in patients with lung and bladder cancer treated with durvalumab. Clin Cancer Res (2018) 24:6212–22. doi: 10.1158/1078-0432.Ccr-18-0386
78. Lee YJ, Yoon KA, Han JY, Kim HT, Yun T, Lee GK, et al. Circulating cell-free DNA in plasma of never smokers with advanced lung adenocarcinoma receiving gefitinib or standard chemotherapy as first-line therapy. Clin Cancer Res (2011) 17:5179–87. doi: 10.1158/1078-0432.Ccr-11-0400
79. Bordi P, Del Re M, Minari R, Rofi E, Buti S, Restante G, et al. From the beginning to resistance: Study of plasma monitoring and resistance mechanisms in a cohort of patients treated with osimertinib for advanced T790M-positive NSCLC. Lung Cancer (2019) 131:78–85. doi: 10.1016/j.lungcan.2019.03.017
80. Papadimitrakopoulou VA, Han JY, Ahn MJ, Ramalingam SS, Delmonte A, Hsia TC, et al. Epidermal growth factor receptor mutation analysis in tissue and plasma from the AURA3 trial: Osimertinib versus platinum-pemetrexed for T790M mutation-positive advanced non-small cell lung cancer. Cancer (2020) 126:373–80. doi: 10.1002/cncr.32503
81. Fukuhara T, Saito H, Furuya N, Watanabe K, Sugawara S, Iwasawa S, et al. Evaluation of plasma EGFR mutation as an early predictor of response of erlotinib plus bevacizumab treatment in the NEJ026 study. EBioMedicine (2020) 57:102861. doi: 10.1016/j.ebiom.2020.102861
82. Zhang S, Zhu L, Xia B, Chen E, Zhao Q, Zhang X, et al. Epidermal growth factor receptor (EGFR) T790M mutation identified in plasma indicates failure sites and predicts clinical prognosis in non-small cell lung cancer progression during first-generation tyrosine kinase inhibitor therapy: a prospective observational study. Cancer Commun (Lond) (2018) 38:28. doi: 10.1186/s40880-018-0303-2
83. Camidge DR, Kim HR, Ahn MJ, Yang JCH, Han JY, Hochmair MJ, et al. Brigatinib versus crizotinib in ALK inhibitor-naive advanced ALK-positive NSCLC: Final results of phase 3 ALTA-1L trial. J Thorac Oncol (2021) 16:2091–108. doi: 10.1016/j.jtho.2021.07.035
84. Horn L, Spigel DR, Vokes EE, Holgado E, Ready N, Steins M, et al. Nivolumab versus docetaxel in previously treated patients with advanced non-Small-Cell lung cancer: Two-year outcomes from two randomized, open-label, phase III trials (CheckMate 017 and CheckMate 057). J Clin Oncol (2017) 35:3924–33. doi: 10.1200/jco.2017.74.3062
85. Anagnostou V, Forde PM, White JR, Niknafs N, Hruban C, Naidoo J, et al. Dynamics of tumor and immune responses during immune checkpoint blockade in non-small cell lung cancer. Cancer Res (2019) 79:1214–25. doi: 10.1158/0008-5472.Can-18-1127
86. Goldberg SB, Narayan A, Kole AJ, Decker RH, Teysir J, Carriero NJ, et al. Early assessment of lung cancer immunotherapy response via circulating tumor DNA. Clin Cancer Res (2018) 24:1872–80. doi: 10.1158/1078-0432.Ccr-17-1341
87. Hellmann MD, Nabet BY, Rizvi H, Chaudhuri AA, Wells DK, Dunphy MPS, et al. Circulating tumor DNA analysis to assess risk of progression after long-term response to PD-(L)1 blockade in NSCLC. Clin Cancer Res (2020) 26:2849–58. doi: 10.1158/1078-0432.Ccr-19-3418
88. Nabet BY, Esfahani MS, Moding EJ, Hamilton EG, Chabon JJ, Rizvi H, et al. Noninvasive early identification of therapeutic benefit from immune checkpoint inhibition. Cell (2020) 183:363–376.e313. doi: 10.1016/j.cell.2020.09.001
89. Samstein RM, Lee CH, Shoushtari AN, Hellmann MD, Shen R, Janjigian YY, et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat Genet (2019) 51:202–6. doi: 10.1038/s41588-018-0312-8
90. Chan TA, Yarchoan M, Jaffee E, Swanton C, Quezada SA, Stenzinger A, et al. Development of tumor mutation burden as an immunotherapy biomarker: utility for the oncology clinic. Ann Oncol (2019) 30:44–56. doi: 10.1093/annonc/mdy495
91. Dudley JC, Lin MT, Le DT, Eshleman JR. Microsatellite instability as a biomarker for PD-1 blockade. Clin Cancer Res (2016) 22:813–20. doi: 10.1158/1078-0432.Ccr-15-1678
92. Wang Z, Duan J, Cai S, Han M, Dong H, Zhao J, et al. Assessment of blood tumor mutational burden as a potential biomarker for immunotherapy in patients with non-small cell lung cancer with use of a next-generation sequencing cancer gene panel. JAMA Oncol (2019) 5:696–702. doi: 10.1001/jamaoncol.2018.7098
93. Gandara DR, Paul SM, Kowanetz M, Schleifman E, Zou W, Li Y, et al. Blood-based tumor mutational burden as a predictor of clinical benefit in non-small-cell lung cancer patients treated with atezolizumab. Nat Med (2018) 24:1441–8. doi: 10.1038/s41591-018-0134-3
Keywords: ctDNA, liquid biopsy, NSCLC, therapy monitoring, minimal residual disease (MRD)
Citation: Fang X, Yu S, Jiang Y, Xiang Y and Lu K (2022) Circulating tumor DNA detection in MRD assessment and diagnosis and treatment of non-small cell lung cancer. Front. Oncol. 12:1027664. doi: 10.3389/fonc.2022.1027664
Received: 25 August 2022; Accepted: 11 October 2022;
Published: 27 October 2022.
Edited by:
Qingqing Zhu, The First Affiliated Hospital of Soochow University, ChinaReviewed by:
Francesco Pepe, University of Naples Federico II, ItalyYoshiko Hashii, Osaka University, Japan
Copyright © 2022 Fang, Yu, Jiang, Xiang and Lu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kaihua Lu, bHVrYWlodWFAbmptdS5lZHUuY24=
†These authors have contributed equally to this work