
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol., 05 January 2023
Sec. Pharmacology of Anti-Cancer Drugs
Volume 12 - 2022 | https://doi.org/10.3389/fonc.2022.1026377
 Abdulrahman Theyab1,2*
Abdulrahman Theyab1,2* Khalaf F. Alsharif3*
Khalaf F. Alsharif3* Khalid J. Alzahrani3
Khalid J. Alzahrani3 Atif Abdulwahab A. Oyouni4
Atif Abdulwahab A. Oyouni4 Yousef MohammedRabaa Hawsawi2,5
Yousef MohammedRabaa Hawsawi2,5 Mohammad Algahtani1
Mohammad Algahtani1 Saad Alghamdi6
Saad Alghamdi6 Amal F. Alshammary7
Amal F. Alshammary7Over the last 20 years, granulocyte colony-stimulating factors (G-CSFs) have become the major therapeutic option for the treatment of patients with neutropenia. Most of the current G-CSFs require daily injections, which are inconvenient and expensive for patients. Increased understanding of G-CSFs’ structure, expression, and mechanism of clearance has been very instrumental in the development of new generations of long-acting G-CSFs with improved efficacy. Several approaches to reducing G-CSF clearance via conjugation techniques have been investigated. PEGylation, glycosylation, polysialylation, or conjugation with immunoglobulins or albumins have successfully increased G-CSFs’ half-lives. Pegfilgrastim (Neulasta) has been successfully approved and marketed for the treatment of patients with neutropenia. The rapidly expanding market for G-CSFs has increased demand for G-CSF biosimilars. Therefore, the importance of this review is to highlight the principle, elimination’s route, half-life, clearance, safety, benefits, and limitations of different strategies and techniques used to increase the half-life of biotherapeutic G-CSFs. Understanding these strategies will allow for a new treatment with more competitive manufacturing and lower unit costs compared with that of Neulasta.
● Most of the current G-CSFs require daily injections, which are inconvenient and expensive.
● Different strategies have been used to overcome the short half-life of the first-generation rhG-CSFs.
● Understanding of G-CSF structure, expression, and mechanism of action on neutrophils may contribute to development of safe long-acting G-CSF therapies for patients with neutropenia.
In 1991, filgrastim (FIL; NEUPOGEN®) was the first recombinant human granulocyte colony-stimulating factor (rhG-CSF) used for hematopoietic progenitor cell (HPC) mobilization. Filgrastim is a non-glycosylated G-CSF form produced in E. coli. It has a molecular weight of approximately 18.8 kDa and contains a methionine group at its N-terminus. It was the first G-CSF medicine approved by the United States Food and Drug Administration (US FDA) for treating neutropenia for various indications (1–3). Filgrastim is indicated to decrease the neutrophil recovery time and the duration of fever in patients with acute myeloid leukemia. It can be used in patients with cancer receiving myelosuppressive chemotherapy (MSC) to reduce the incidence of infection and prevent febrile neutropenia (FN). Filgrastim is also used to reduce neutropenia in patients with non-myeloid leukemia who are undergoing myeloablative chemotherapy followed by bone marrow transplantation. In addition, filgrastim has been approved to treat hematopoietic syndrome of acute radiation syndrome (the treatment plan for patients acutely exposed to myelosuppressive doses of radiation). It is the most commonly used growth factor for the mobilization of autologous HPCs into the peripheral blood (4). It has a short half-life (between 3.5 and 3.8 h on average), with filgrastim concentration and neutrophil count being the factors of clearance. The drug is cleared by the kidney (5–8).
Lenograstim is another first-generation rhG-CSF. It is produced from Chinese hamster ovary cells and has a single O-glycosylated form at position Thr-133 (9). It also has a short half-life similar to that of filgrastim, with the O-linked providing stability by shielding the cysteine-17–containing sulfhydryl group from oxidation by free radicals (5, 10). It was assumed that lenograstim may show clinical benefits over filgrastim, although an in vivo comparative study exhibited no differences between the two rhG-CSF products (11).
Tbo-filgrastim is another short-acting G-CSFs that produced by recombinant DNA technology using the E. coli K802 bacterium strain. It is a non-glycosylated recombinant methionyl human granulocyte colony-stimulating factor. It is composed of 175 amino acids and has a molecular weight of approximately 18.8 kDa. Tbo-filgrastim was approved by the European Union as a biosimilar to filgrastim in 2008. Four years later, US FDA had approved tbo-filgrastim as a biologic product with one similar indication to filgrastim (12, 13).
To overcome the short half-life of the first-generation rhG-CSFs, different strategies have been employed to increase G-CSF half-life, which include increasing the molecular weight by conjugation with another moiety such as glycosylation, polysialation, and PEGylation to overcome rapid elimination by renal filtration. In addition, using the mechanism of neonatal fragment crystallizable (Fc) receptor (FcRn) recycling through fusing several proteins with Fc portion of albumin or immunoglobulin (14–16) (Table 1). These strategies have successfully prolonged G-CSF half-life (40). This article reviews the underlying principles, elimination’s route, half-life, clearance, safety, benefits, and limitations of each of these strategies from a chemical and structural standpoint.
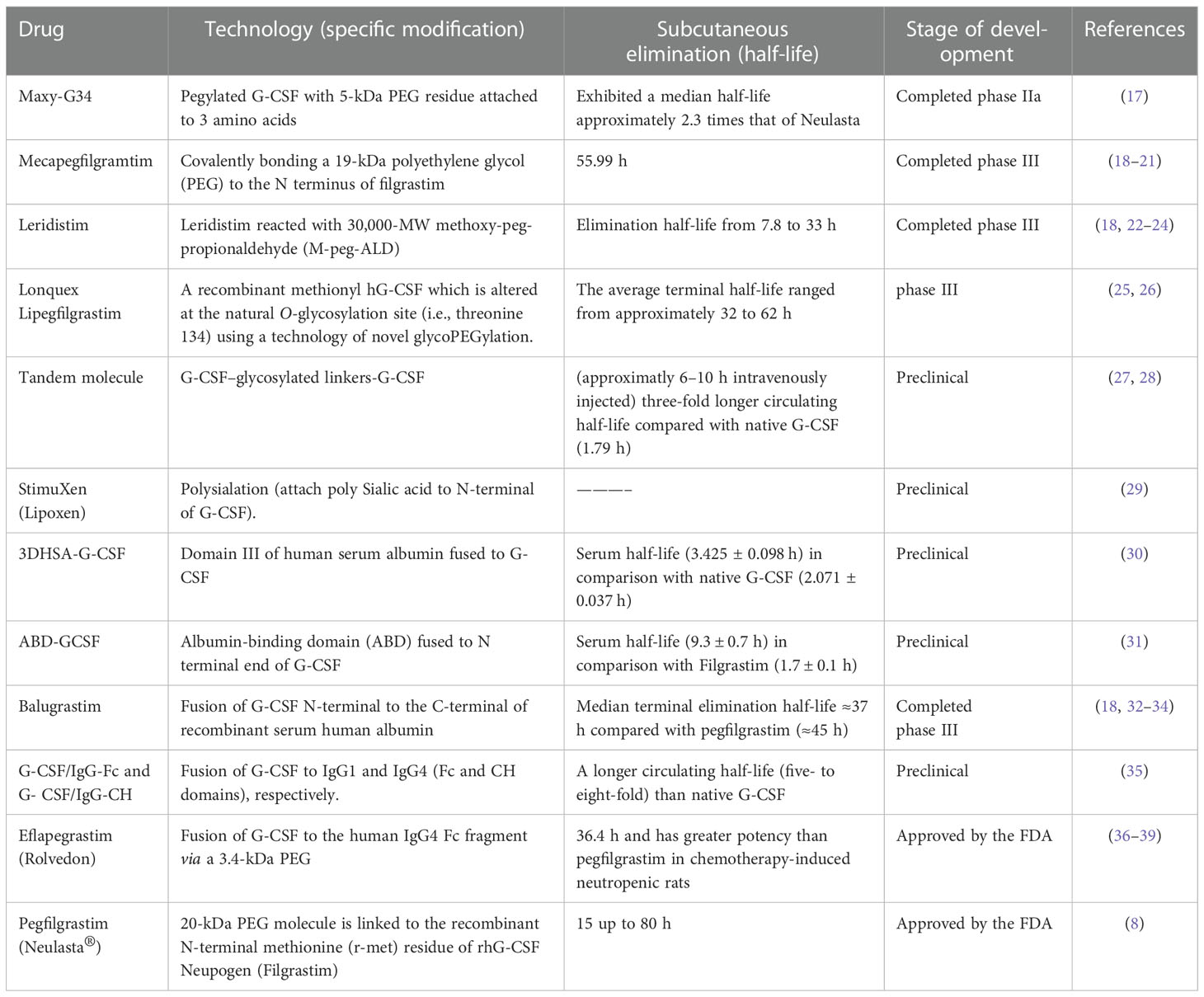
Table 1 Long-acting G-CSF formulations are considered for increasing the in vivo residence time of G-CSF compared with native G-CSF, filgrastim (rhG-CSF), and Neulasta.
Conjugation with polyethylene glycol (PEG), known as PEGylation, was first described in the 1970s by both Abuchowski and Davis, who discovered that PEGylation process may increase the longevity of different proteins and improve their immunological properties, such as albumin and bovine liver catalase (41). Consequently, studies have been performed with a view to improving the PEG process, resulting in PEGs with a wide range of molecular weights (42). In 2003, a notable example is pegvisomant (recombinant pegylated growth hormone antagonist), which was used to treat patients with acromegaly (43, 44). Pegfilgrastim (Neulasta®) is another PEGylated form of second-generation rhG-CSF, and it is the only long-acting (once weekly) G-CSF that has been approved by the FDA (Figure 1) (6, 45).
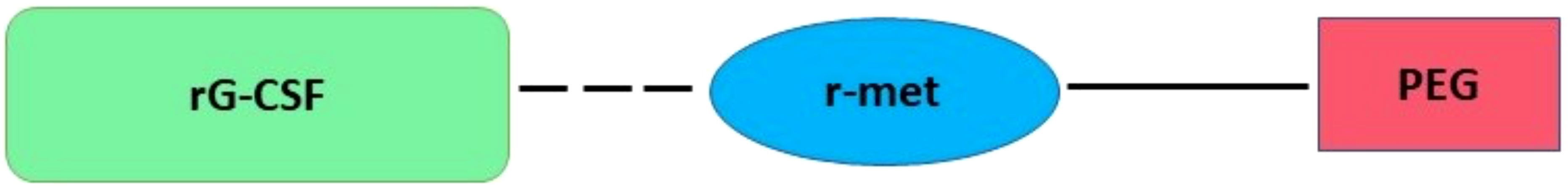
Figure 1 Plan structures of pegfilgrastim. Covalently bound of a single 20-kDa PEG molecule attached to the recombinant N-terminal methionine (r-met) residue of rG-CSF.
A single 20-kDa PEG molecule is covalently bound to the recombinant N-terminal methionine (r-met) residue of rhG-CSF Neupogen (Filgrastim) (Figure 1) (6, 45). Structurally, each ethylene oxide of PEG can combine with two or three water particles, increasing its water solubility (more hydrophilic) and hydrodynamic radius. This increased molecule size to ~38.8 kDa, consequently decreasing renal clearance. Moreover, PEG technology generates a hydrophilic protection that shields the proteins from immunologic recognition and proteolysis (46, 47).
The addition of a PEG moiety to rhG-CSF reduces its renal clearance via glomular filtration, making neutrophil-mediated clearance the primary route of elimination (8). This elimination route is started when pegfilgrastim binds to the G-CSF receptor on the surface of neutrophil cells, causing the pegfilgrastim–receptor complex to be internalized through endocytosis and then degraded inside the cell (48). Following subcutaneous administration, the serum half-life of pegfilgrastim varies hugely depending on the absolute neutrophil counts, with a range of 15 to 80 h. Forty-two hours is the median serum half-life (8).
Pegfilgrastim has a neutrophil-induced self-regulating clearance mechanism (48, 49). The clearance is dependent on the neutrophil counts and body weights of the patients; the clearance increases with an increasing number of granulocytes and lower body weights (FDA-approved drug products). Following chemotherapy-induced neutropenia, pegfilgrastim remains in the blood until neutrophils begin to recover; as neutrophil numbers increase, pegfilgrastim’s elimination increases (49). The obvious clearance of serum is 14 ml/h/kg (Cancer Care Ontario Drug Information: Pegfilgrastim).
The highest dose of pegfilgrastim reported in clinical trials was 300 mcg/kg (50). Pegfilgrastim’s overdosage may result in bone pain and leukocytosis. In case of overdose, patients should be observed for signs and symptoms of toxicity and given general supportive care as necessary (50, 51).
The advantage of pegfilgrastim over filgrastim was its ability to avoid renal clearance. However, it is still cleared via neutrophil-mediated clearance (52), which is reliant on the number of circulating neutrophils. Hence, pegfilgrastim’s concentration remains high in serum during neutropenia and begins to clear once the neutrophil starts to normalize. Therefore, a single dose of pegfilgrastim is equal to seven daily injections of filgrastim (53).
In addition to the high cost of PEGylated proteins (54), they are usually excreted through the renal system without undergoing primary biodegradation that causes renal toxicity (55) as evidenced by the presence of PEG in bile (56) and vacuoles in renal tubules (29). It has been shown that PEGylation reduced the in vitro bioactivity of rhG-CSF to two- or three-fold, mainly due to the structural changes in the macromolecule that could attenuates its potency (57, 58). Furthermore, administration of first dose of PEGylated proteins could induce the production of anti-PEG immunoglobulin M (IgM). However, upon the administration of subsequent doses, liver Kupffer cells start to eradicate these harmful IgMs (59).
Glycosylation is a common enzymatic modification and refers to the process of adding glycans to macromolecules (60). Glycosylation increases the molecular weight of proteins, improves thermal stability, and prevents proteolytic degradation. The cell membrane surface of glycoproteins contains sialic acid, which induces the overexpression of negatively charged monosaccharides, thereby inhibiting the passage of glycoproteins via charge repulsion alongside the glomerular filtration membrane of the kidney and extending their circulatory half-life (61, 62).
Glycosylation might help to reduce the immunogenicity of polypeptides by enhancing their solubility, shielding the hydrophobic residues, and decreasing the possibility of aggregation that may result of a stationary precipitate for antibody recognition (63). An additional proposed theory suggests that sialylation (sialic acid located at the terminal of glycan) shields peptides by reducing the visible surface area that is exposed to antibody recognition sites. Darbepoetin alfa was the first to report this mechanism (a recombinant human erythropoietin analogue consisted of two N-linked glycosylation sequences) (64, 65).
Different classes of protein glycosylation have been identified, such as the addition of N-linked glycans, O-linked glycans, glycosam inoglycans, phosphorylated glycans, and formation of glycosylphosphatidylinositol anchors to peptide backbones as well as residues of C-mannosylation-tryptophan (60). However, the two major forms of glycosylation associated with G-CSF are N-linked and O-linked glycosylation (66). Understanding the forms and functions of the glycosylation to G-CSF is essential for an optimal conjugation of glycosylation moieties to G-CSF, as evidenced in lipegfilgrastim development.
O-linked glycosylation is a diverse protein glycan that is naturally attached to the oxygen atom of hydroxyl groups (-OH) of serine (Ser), threonine (Thr), or tyrosine (Tyr) residues within a protein (60, 67). Nevertheless, no specific consent sequences or motifs have been documented for this attachment. Furthermore, it is unknown why certain Ser/Thr residues are oppositely glycosylated to other residues. It is possible that the protein’s alternative structure contributes to the glycosylation site’s availability (64). The O-glycan biosynthetic pathways start in the Golgi apparatus, where polypeptide GalNAc transferase (GlcNAcT) catalyzes the transfer of the GalNAc moiety of uridine diphosphate to the hydroxyl of Ser or Thr (68). Later, a few glycosyltransferases can convert the resulting glycoprotein into different core structures linked by different α- or β-glycosidic linkages (69). O-linked glycosylation is found at Thr-134 site of rhG-CSF and recognizable as the only site altered with a single mannose, allowing glycoengineered Pichia. Pastoris is used as a possible model for biotherapeutic rhG-CSF production (70).
In N-linked glycosylation, an N-acetylglucosamine (GlcNAc) residue attaches to the amide group of an asparagine residue. It occurs in consensus sequences Asn-X-Ser/Thr, where X indicates any residue except proline (Pro) (71). N-glycosylation has also been observed at non-canonical motifs in some publications, many of which were found in the conformation Asn-X-Cys (cysteine) (72). N-glycosylation biosynthetic pathways start in endothelial reticulum (Figure 2). An early attachment of a 9-mannose glycan to the peptides of an N-linked glycan identifies it as a high mannose type. The inclusion of N-glycans is critical for the folding of newly synthesized proteins to be regulated. Following the effective folding of newly synthesized proteins, the glycoprotein migrates to the Golgi apparatus, where mannosidases remove the mannose sugar group. Later, specific glycosyltransferases assist in the binding of various monosaccharides into a developing glycan chain. This hybrid type has a high mannose content (73). The biosynthetic process in the Golgi apparatus is now complete, with a fully sialylated glycan complex containing six sugars: mannose (Man), galactose (Gal), N-acetylglucosamine (GlcNAc), fucose (Fuc), sialic acid (NeuAc), linked by different α- or β-glycosidic linkages (recognized as a complex type) (74, 75).
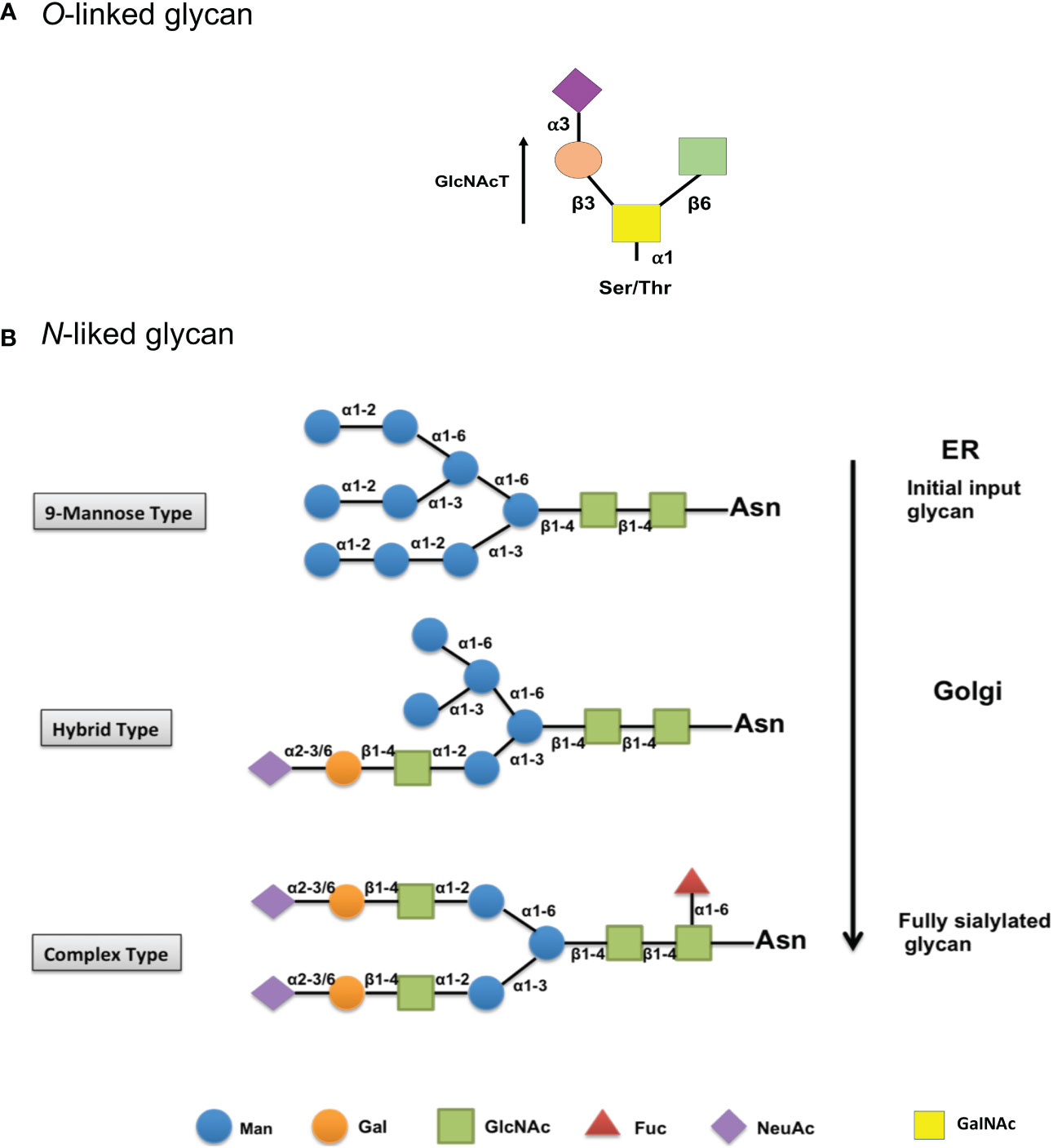
Figure 2 Biosynthesis and structure of O- and N-linked glycans. (A) Initial biosynthetic pathway of O-glycans with an O-GalNAc moiety starts in the Golgi apparatus, where a GalNAc residue is attached by different polypeptide polypeptide GalNAc transferases (GlcNAcT). Later, a few glycosyltransferases can convert the resulting glycoprotein into different core structures. (B) The biosynthetic pathway of the early input glycan (9-mannose glycan) begins in the endothelial reticulum (Top). Then, glycoprotein migrates to the Golgi, where the mannose group is removed, and other monosaccharides are added in a hybrid type process (mid). The biosynthetic process is then finished as a completely sialylated glycan complex in the Golgi (Bottom). Man, mannose; Gal, galactose; GlcNAc, N-acetylglucosamine; Fuc, fucose; NeuAc, sialic acid; GalNAc, N-acetylgalactosamine.
The N-linked glycan terminal site contains sialic acid (N-acetylneuraminic acid, NeuAc), characterized by a diverse group of nine carbon-containing carbohydrates with a negatively charged carboxylate (C1), and has been shown to be critical in maintaining the half-life of several glycoproteins in the bloodstream (74, 76, 77). Sialic acids can confer glomerular filtration or blood cell charge repulsion due to their negative charge and hydrophilicity, which may delay glycoproteins in the blood circulation as evidenced in human erythropoietin (78, 79).
O-linked glycosylation occurs most commonly at clustered Ser or Thr residues, making N-glycan profiling more feasible than O-glycan profiling, as evidenced by a universal endoglycosidase. Peptide-N-glycosidase F catalyzes the deglycosylation of most N-glycans and cleaves 9-high-mannose, hybrid, and complex monosaccharide chains but has not been identified for O-glycan (80, 81). Hence, N-linked glycan is preferred in several technologies of protein modification and biopharmaceutical functioning (82, 83). Recent strategies of N-linked glycan can be classified into site-directed mutagenesis and glycosylated linker.
Glycoengineering is a process of enhancing the properties of therapeutic proteins through altering their glycosylation to improve its pharmacokinetic and biological activity (84). One of the most common methods for glycoengineering is DNA mutagenesis. In vivo, additional glycosylation sites can be added to DNA via mutagenesis. This can be performed, for example, by detecting the third position of Thr/Ser residues in a protein’s sequence and mutating the first amino acid to Asn or by detecting Asn residues in a protein’s sequence and mutating the third amino acid to Thr/Ser (85). Site-directed DNA mutagenesis was used to generate new darbepoetin alfa, by mutating Ala-30, His-32 to Asn-30, Thr-32, Pro-87, Trp-88, Pro-90 to Val-87, Asn-88, and Thr-90. Alterations were found to be glycosylated, with a molecular weight increase from 35 to approximately 43 kDa while retaining biological activity (86). G-CSF has also been performed to site-directed mutagenesis by mutating Phe-140 to Asn-140, resulting in an N-linked glycosylation site on rhG-CSF. This novel mutant was exhibited to be glycosylated and had more efficiency at stimulating hematopoietic cell proliferation and differentiation than native G-CSF (16).
The use of glycosylated linker is another approach for increasing longevity and enhancing bioactivity of some therapeutic proteins in serum. Glycosylated linkers have been shown to be incorporated between two ligands of the same protein, as demonstrated in the development of recombinant human follicle-stimulating hormone (rhFSH), where either an O- or N-linked glycosylated linker was used between the α and β subunits of rhFSH. As a result, glycosylated linker increased half-life of the new tandem rhFSH by up to two-fold compared with native FSH (87, 88).
To generate a long-acting G-CSF, the advantages of glycosylated linker design have been used. Two G-CSF ligands were linked via a flexible (Gly4Ser)n linker including different glycosylation sites to form a tandem [G-CSF contains two glycosylation motifs (G-CSF2NAT): G-CSF4NAT and G-CSF8NAT] and their respective controls (QAT instead of NAT; Q = glutamine, therefore, is not recognized by cell for glycosylation). Using Western blot, the preclinical study demonstrated an increase in the molecular weight of isolated glycosylated G-CSF tandems compared with controls (Figure 3). In comparison to rhG-CSF, all G-CSF tandems exhibit increased bioactivity with two- to three-fold lower half maximal effective concentration (EC50s) (27). After intravenous injection to rats, G-CSF2NAT, G-CSF4NAT, and G-CSF8NAT, including two, four, and eight glycosylation sites, respectively, exhibited a lower rate of clearance in comparison to rhG-CSF (achieved a longer circulating half-life, nearly three-fold compared with rhG-CSF) (27). Although tandem G-CSF is still in the preclinical stage, we point out that the use of glycosylated linkers is safer than the use of site-directed mutagenesis to avoid mutating G-CSF itself, which may affect the protein’s bioactivity.
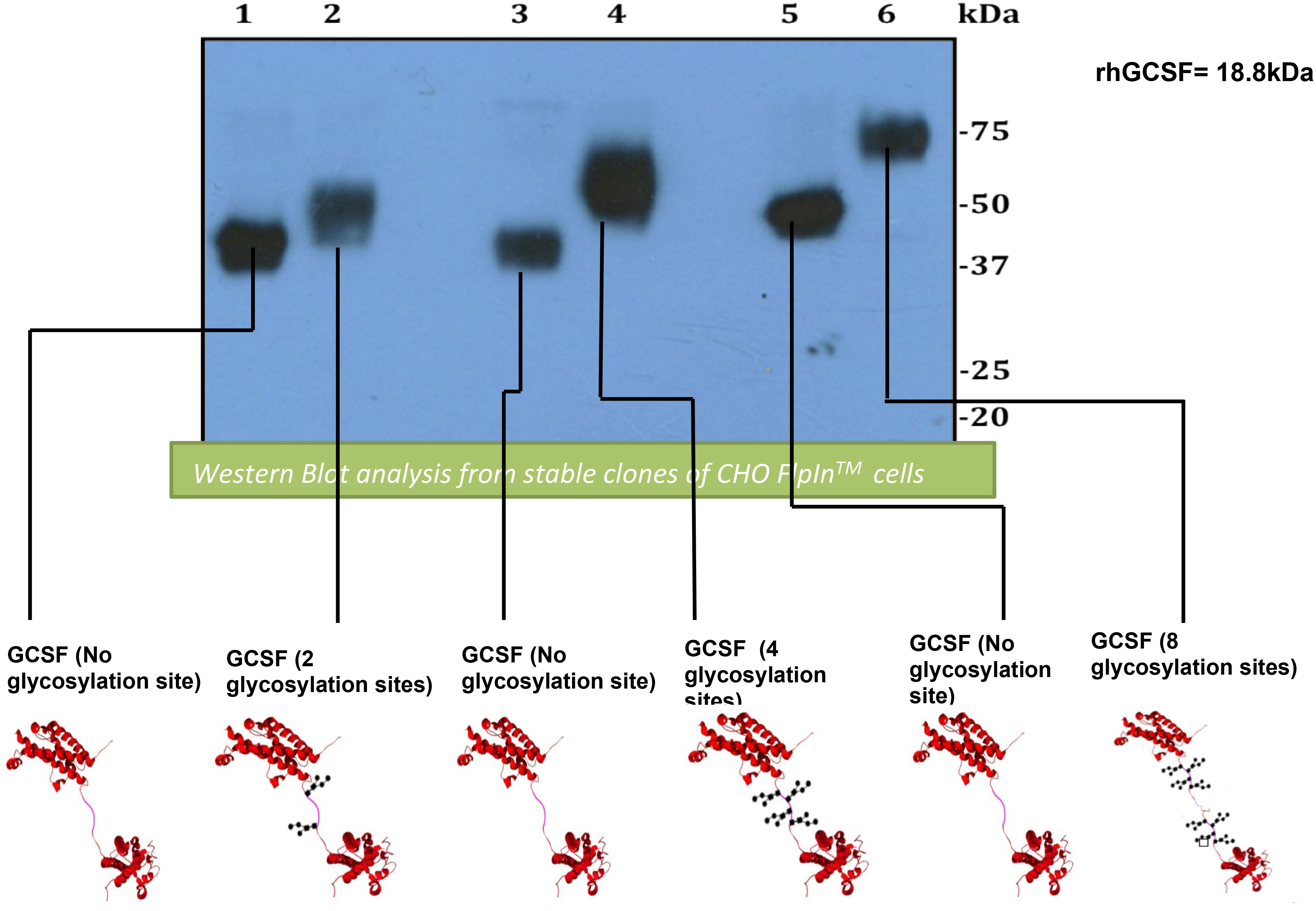
Figure 3 Analysis of Western blot expressing and comparing G-CSF tandems and their controls. First lane, GCSF2QAT_control (2 × QAT). Second lane, GCSF2NAT (2 × NAT). Third lane, GCSF4QAT_control (4 × QAT). Fourth lane, GCSF4NAT (4 × NAT). Fifth lane, GCSF8QAT_control (8 × QAT). Sixth lane, GCSF8NAT (8 × NAT). Analysis of Western blot displays an obvious increase in molecular weight for glycosylated GCSF tandems (GCSF2NAT, GCSF5NAT, and GCSF8NAT) when compared with non-glycosylated controls (GCSF2QAT, GCSF4QAT, and GCSF8QAT).
Lipegfilgrastim is novel long-acting G-CSF, site-specific glycolpegylated r-metHu G-CSF formed by conjugation of a single 20-kDa PEG–sialic acid (Sia) to the O-linked glycan bound at the Thr-134 residue site of G-CSF, using the technology of glycopegylation (Figure 4) (25, 89). Lipegfilgrastim provides a therapeutic alternative to pegfilgrastim but has more limited global distribution (obtainable in Europe) and, therefore, less experience with its use. Clinical trials of pegfilgrastim have been shown to have a favourable competence and safety profile for prophylactic usage in patients with cancer receiving chemotherapy and at risk of severe neutropenia and may be preferred by both physicians and patients over short-acting G-CSF due to enhanced adherence and a simple once-per-cycle subcutaneous injection (90–92). Lipegfilgrastim was found to be an effective option for reducing the duration of severe chemotherapy-induced neutropenia and preventing FN in older patients with aggressive B-cell NHL receiving MSC (36). Although the current studies published for lipegfilgrastim are still limited, there is an indication that it is a promising treatment for chemotherapy-induced neutropenia.

Figure 4 Plan structures of lipegfilgrastim. Conjugation of a single 20-kDa PEG–sialic acid (Sia) to a natural O-linked glycan moiety bound at the Thr-134 residue site of rG-CSF.
Lipegfilgrastim has two different clearance pathways: a linear pathway involving proteolytic enzyme degradation and a non-linear pathway involving neutrophil-mediated clearance (93). However, at higher doses, the elimination pathway by neutrophil-mediated clearance is saturated, and its degraded fragments may undergo renal clearance (26, 94). After a single subcutaneous injection of 6 mg of lipegfilgrastim in healthy individuals, the average terminal half-life ranged from 32 to 62 h, which was 7–10 h longer for lipegfilgrastim 100 mcg/kg compared with that reported for pegfilgrastim at 100 mcg/kg (26). In phase I of a different multinational, open-label, single-arm study of pediatric patients with the Ewing family of tumors or rhabdomyosarcoma treated with MSC, the average evident clearance (CL/F) was nearly 70 ml/h for patients aged 2–6 years, 120 ml/h for patients aged 6–12 years, and 116 ml/h for patients aged 12–18 years (94).
In studies on the safety of lipegfilgrastim in dogs and rats, a single subcutaneous dose of 10 mg/kg was well tolerated. Similarly, an intravenous dose of 250 mcg/kg was well effective and tolerated in the renal excretion of rats. In a study of 139 patients, adverse events related to lipegfilgrastim occurred in 55 (39.5%) patients; bone pain and back pain were the most common (95).
It appears that glycolpegylation alters the pharmacokinetic and pharmacodynamic profiles of lipegfilgrastim. In phase I studies involving healthy volunteers, lipegfilgrastim at subcutaneous dose of 6 mg demonstrated 64% greater cumulative exposure and 36% greater peak exposure than pegfilgrastim with the same dose. In addition to pegfilgrastim, lipegfilgrastim had a longer half-life (geometric means, 32.4 h vs. 27.2 h, respectively). In a randomized, double-blind, phase III trial, it was determined that lipegfilgrastim was non-inferior to pegfilgrastim in terms of the duration of severe neutropenia in patients with breast cancer (96, 97). Back pain and bone pain were the only limitations, as mentioned previously.
One of the most common antibodies found in extracellular fluids and circulation is immunoglobulin G (IgG). Although it can directly protect the body from infections by activating its antigen binding site, IgG immune functions are mostly mediated through receptors and proteins expressed by special cell subsets that bind to a region of IgG called Fc. The neonatal FcRn belongs to a large family of Fc gamma (γ) receptors (FcγRs) and has become increasingly important on binding IgGs and albumin. FcRn is known as a recycling receptor and has been shown to bind and maintain IgGs and albumin in the blood circulation and to bidirectionally transport both ligand molecules through polarized cellular barriers (98).
In general, FcRn can attach to albumin and IgGs at the cell membrane in a pH-dependent way. Because of the presence of histidines in albumin and IgGs, the imidazole group of histidine protonates at a pH of 6.0 and binds to the FcRn receptor. The FcRn–IgG or FcRn–albumin complex interaction is then internalized and absorbed through the cell membrane, protecting albumin or IgGs from lysosomal degradation (99–101). The FcRn–albumin or FcRn-IgG complex is then recycled to the blood circulation, where it is liberated when exposed to physiological pH 7.4 (Figure 5) (102–104).
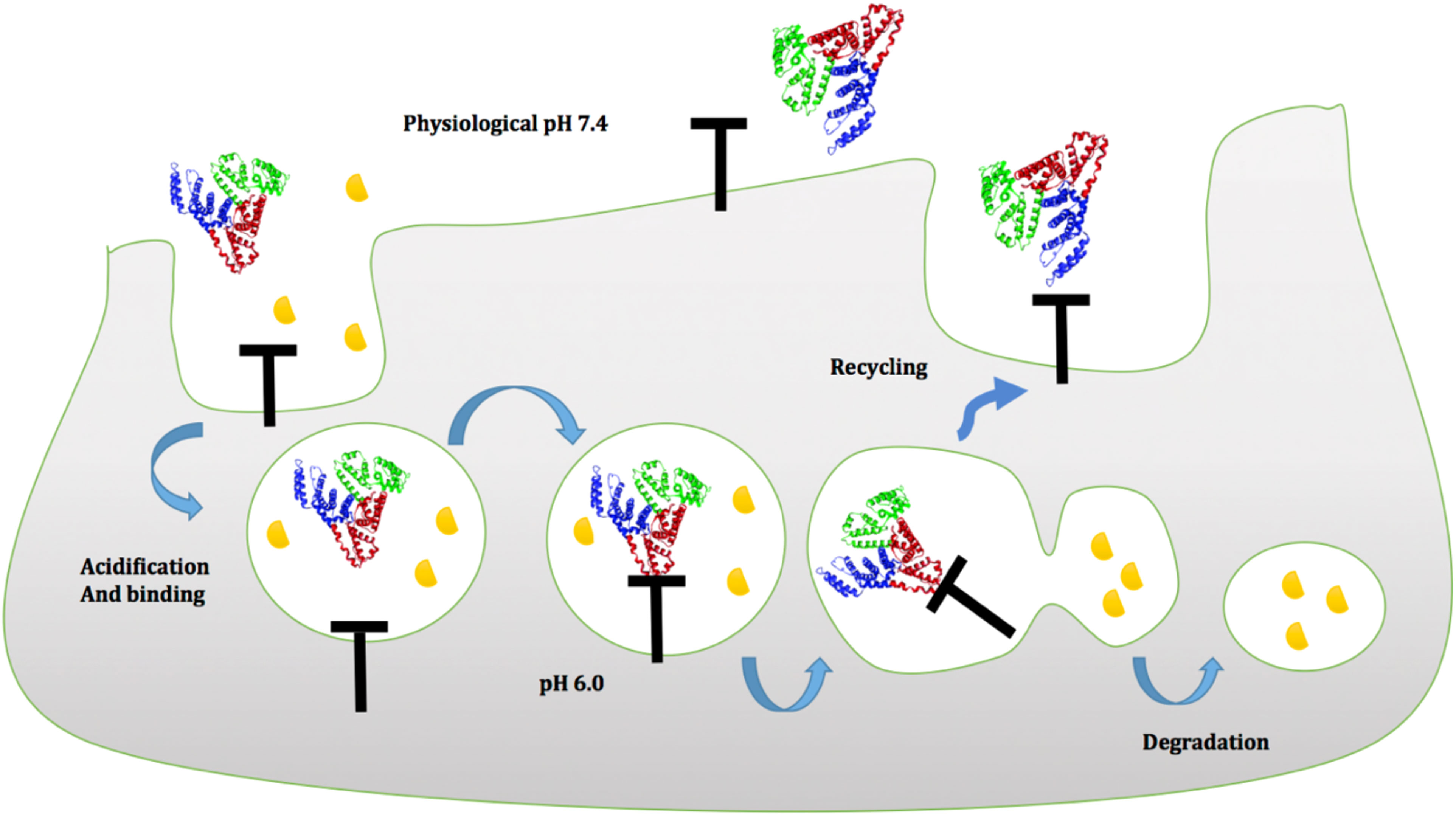
Figure 5 Proposed model of albumin to FcRn in a pH-dependent manner at the cell membrane. Green (domain I), blue (domain II), and red (domain III) indicate the three domains of albumin. Domain III albumin binds to the FcRn receptor at an acidic pH of 6.0, protecting it from lysosomal destruction, and then recycles to the blood circulation, where it is released when exposed to physiological pH 7.4.
Human serum albumin (HSA) is the main protein produced in the liver and serves a variety of physiological functions, including bilirubin, fatty acids, ion transport, and regulation of colloid blood pressure control. Because albumin is a massive protein (molecular weight of 66.5 kDa and an average half-life of 19 days), it can be used to conjugate with multiple recombinant proteins to prolong their half-lives. Consequently, these conjugated proteins will eventually acquire a molecular weight that is too large and challenging for the kidney to filter, extending the residency time of plasma proteins in the blood circulation (100, 105).
Later, the importance of carboxy-terminal domain III human serum albumin (HAS) was recognized and genetically merged with the N-terminus of rhG-CSF. As a result, pharmacokinetic and pharmacodynamic experiments revealed a longer circulating half-life and a high number of white blood cells (WBC) counts in neutropenic mice when compared with rhG-CSF (30).
IgG1 and IgG4 immunoglobulins have a circulation half-life of 23 days in serum and have been utilized to generate various long-acting fusion proteins (106). As a result, immunoglobulins were chosen as the ideal antibodies for Fc fusion proteins. They are structurally made up of two identical heavy and light chains that are linked via disulfide linkages. Both chains have two regions: the antigen-binding fragment (Fab), an antibody’s head portion, is essential for detecting immunogenicity, whereas the Fc, an antibody’s tail component that interacts with a cell surface receptor, is crucial to maintaining IgG circulation (106).
IgG immunoglobulin is composed of two fragment domains: Fc (Hinge-CH2-CH3) and CH (CH1-Hinge-CH2-CH3). The Hinge domain connects the Fc and Fab areas and allows for more flexibility, and it has been reported that various biotherapeutic proteins can be linked via the carboxy-termini of human IgGs’ Fc (Hinge-CH2-CH3) and CH (CH1-Hinge-CH2-CH3) domains (35). IgG fusion proteins are made and released from mammalian cells as disulfide-linked homodimers. This is because cysteine residues in the hinge region of IgGs form inter-chain disulfide bonds with each other. Furthermore, the dimeric structure of IgG fusion proteins increases their effective size and circulation half-life (15, 35).
The carboxy-terminus of rhG-CSF has been fused to the amino termini of the Fc (Hinge-CH2-CH3) and CH (CH1-Hinge-CH2-CH3) domains of human IgG4 and IgG1 immunoglobulins, which are connected by a 7–amino acid flexible linker (Ser-Gly-Gly-Ser-Gly-Gly-Ser) (Figure 6). Fusions of rhG-CSF to the IgG domain resulted in homodimers with a massive molecular weight, longer circulation half-lives (approximately five- to eight-fold longer than reported for native G-CSF), and a high number of neutrophil counts in vivo, without affecting G-CSF bioactivity in vitro (15, 35).
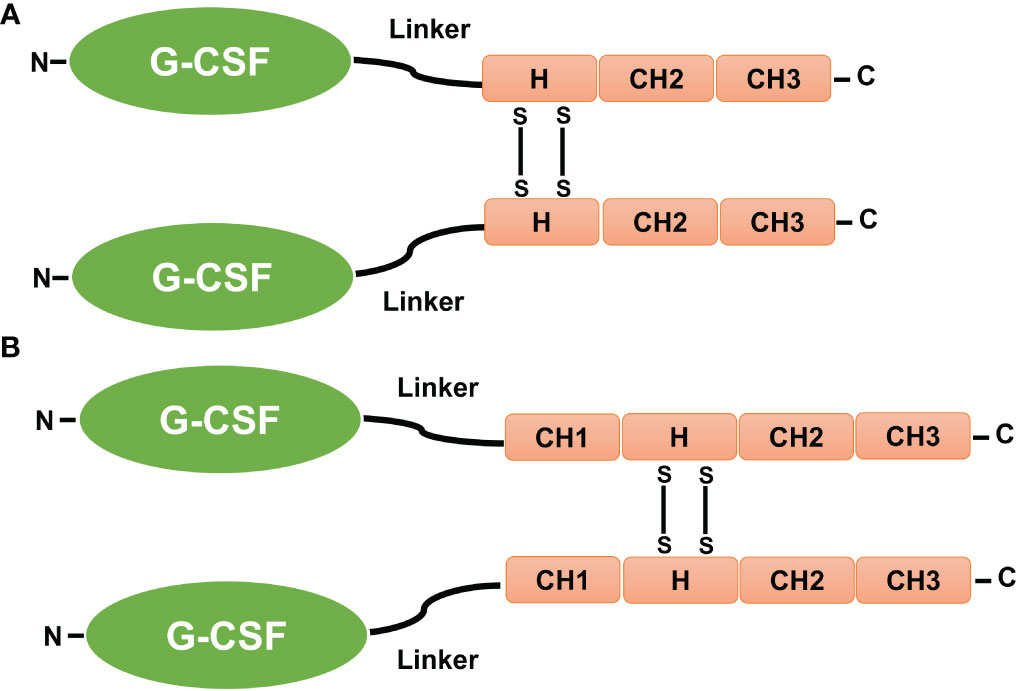
Figure 6 Fusion of G-CSF to Fc and CH domains. (A) The carboxy-terminus of human GCSF is fused to the amino termini of the IgG-Fc and IgG-CH domains by a 7–amino acid fixable linker (L). (B) The CH1, CH2, and CH3 sections of the IgG domains, as well as the hinge (H), are also shown. The presence of disulfide bonds (SS) that occur between cysteine residues causes fusion proteins in the IgG hinge region to be dimeric (15, 35).
Eflapegrastim (Rolvedon), a novel long-acting rhG-CSF, is created by fusing the rhG-CSF to the Fc fragment of human IgG4 via a PEG linker to increase G-CSF half-life (38, 107). The findings suggested that the human IgG4 Fc fragment of eflapegrastim interacts with FcRn, which is expressed on various tissues including bone marrow, and thus reduces eflapegrastim renal clearance, protects it from lysosomes, and prolongs its retention in bone marrow (Figure 7) (108). Preclinical studies of phase I and II pharmacokinetic and pharmacodynamic data exhibited an increased potency for neutrophil count for eflapegrastim compared with pegflgrastim (40, 108, 109). Phase III results showed noninferiority and analogous safety for eflapegrastim at a lower dose of G-CSF compared with pegflgrastim. Therefore, eflapegrastim has a high potential to provide improved clinical benefit and permit more clinical studies in patients at higher risk for contrast-induced nephropathy (37, 38). A recent study by Jeon et al. (2022) showed that eflapegrastim has greater potency than pegfilgrastim in chemotherapy-induced neutropenic rats (39, 110). In September 2022, the US FDA approved eflapegrastim as a prophylactic against infection, as evidenced by FN, in patients receiving certain myelosuppressive anti-cancer drugs.
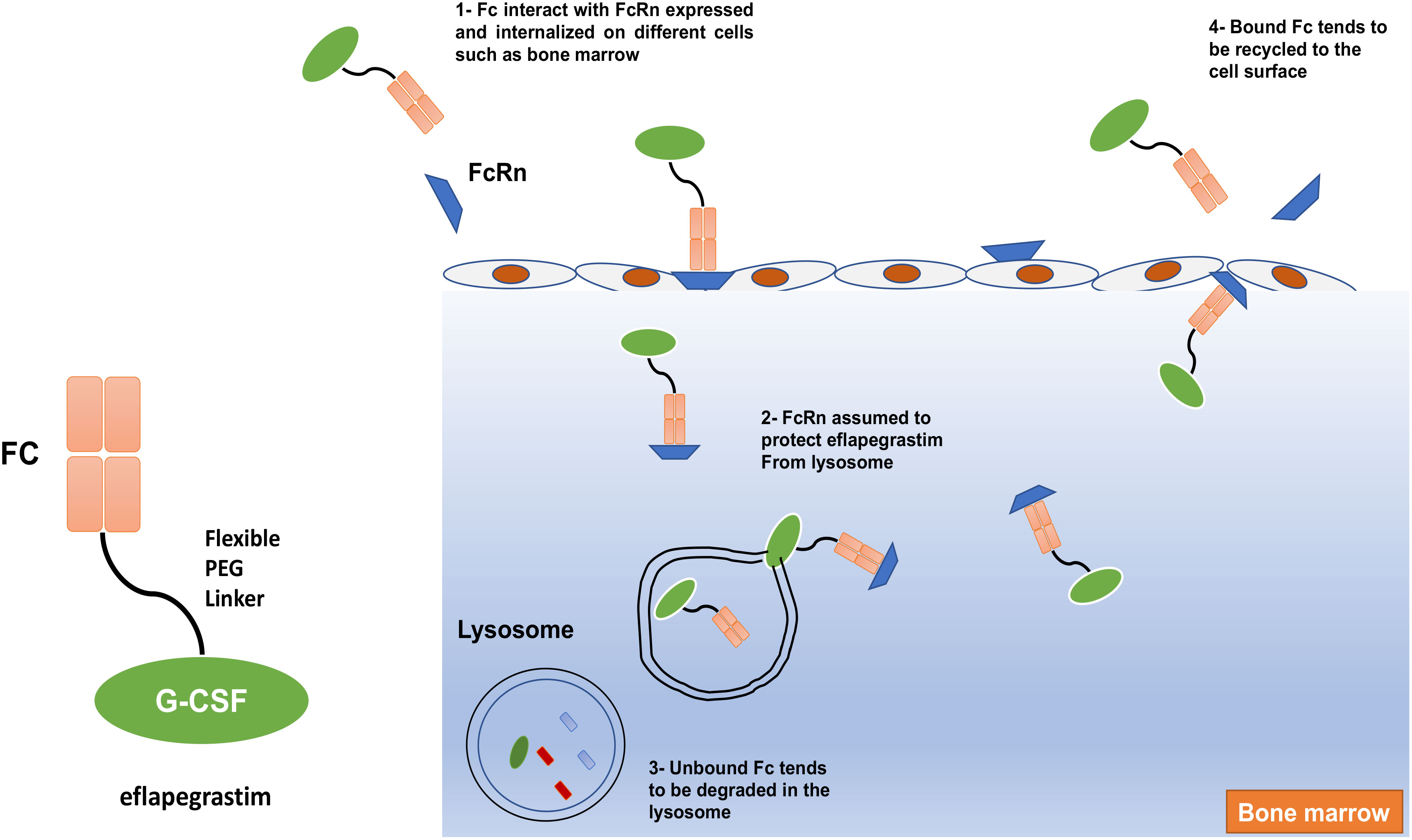
Figure 7 The mechanism of action of eflapegrastim. The human IgG4 Fc fragment of Eflapegrastim interacts with FcRn, which is expressed and internalized on different cells such as bone marrow and assumed to protect eflapegrastim from lysosome leads to elongate its retention in bone marrow.
Eflapegrastim is not detectable in the urine after subcutaneous dose administration (109). In patients with breast cancer, eflapagrastim has a median half-life of 36.4 h. Eflapegrastim clearance decreased with increasing dose, indicating target-mediated clearance by neutrophils. Clearance appears to increase with repeated dosing, possibly due to the subsequent increase of neutrophils in the circulation (FDA-approved drug products).
Leukocytosis and bone pain may result from an eflapegrastim overdose. In this case, patients must be monitored for these adverse effects, and general supportive measures should be taken as required (FDA-approved drug products).
A study in rat models showed that eflapegrastim improved clinical benefit and was associated with higher bone marrow and serum concentrations than pegfilgrastim, which resulted in a significantly shorter duration of neutropenia when administered 24 h after chemotherapy compared with pegfilgrastim (111). Leukocytosis and bone pain due to eflapegrastim overdose are considered the only limitations, as mentioned previously (FDA-approved drug products).
All the long-acting G-CSFs in development rely on pegylation, glycopegylation, conjugation to IgG fragments, or serum human albumin (Table 1). Nonetheless, understanding G-CSF structure, expression, and mechanism of action on neutrophils may contribute to the development of a safe long-acting G-CSF therapy for patients with neutropenia that maintains the pharmacodynamic and pharmacokinetic of pegfilgrastim (Neulasta), but with more competitive manufacturing and lower unit costs compared with Neulasta (the cost for Neulasta is $6,417.99* per dose as of 18 August 2021).
All authors contributed equally to this publication. Conception and design: AT and KFA. Drafting: ATA and KFA. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Frampton JE, Lee CR, Faulds D. Filgrastim - a review of its pharmacological properties and therapeutic efficacy in neutropenia. Drugs (1994) 48(5):731–60. doi: 10.2165/00003495-199448050-00007
2. Welte K, Gabrilove J, Bronchud MH, Platzer E, Morstyn G. Filgrastim (r-metHuG-CSF): the first 10 years. Blood (1996) 88(6):1907–29. doi: 10.1182/blood.V88.6.1907.bloodjournal8861907
3. Yang BB, Savin MA, Green M. Prevention of chemotherapy-induced neutropenia with pegfilgrastim: pharmacokinetics and patient outcomes. Chemotherapy (2012) 58(5):387–98. doi: 10.1159/000345626
4. Inc. A. Neulasta (pegfilgrastim) prescribing information. Initial U.S. Approval. (California: Amgen Inc. One Amgen Center Drive Thousand Oaks) (2002) 91320-1799 U.S. License No. 1080.
5. Cooper KL, Madan J, Whyte S, Stevenson MD, Akehurst RL. Granulocyte colony-stimulating factors for febrile neutropenia prophylaxis following chemotherapy: systematic review and meta-analysis. BMC Cancer (2011) 11:404. doi: 10.1186/1471-2407-11-404
6. Hoggatt J, Pelus LM. New G-CSF agonists for neutropenia therapy. Expert Opin Investig Drugs (2014) 23(1):21–35. doi: 10.1517/13543784.2013.838558
7. Petros WP, Rabinowitz J, Stuart A, Petters WP. Clinical pharmacology of filgrastim following high-dose chemotherapy and autologous bone marrow transplantation. Clin Cancer Res (1997) 3(5):705–11.
8. Molineux G. The design and development of pegfilgrastim (PEG-rmetHuG-CSF, neulasta). Curr Pharm Des (2004) 10(11):1235–44. doi: 10.2174/1381612043452613
9. Nagata S, Tsuchiya M, Asano S, Kaziro Y, Yamazaki T, Yamamoto O, et al. Molecular cloning and expression of cDNA for human granulocyte colony-stimulating factor. Nature (1986) 319(6052):415–8. doi: 10.1038/319415a0
10. Hasegawa M. A thermodynamic model for denaturation of granulocyte colony-stimulating factor: O-linked sugar chain suppresses not the triggering deprotonation but the succeeding denaturation. Biochim Biophys Acta (1993) 1203(2):295–7. doi: 10.1016/0167-4838(93)90097-B
11. Ataergin S, Arpaci F, Turan M, Solchaga L, Cetin T, Ozturk M, et al. Reduced dose of lenograstim is as efficacious as standard dose of filgrastim for peripheral blood stem cell mobilization and transplantation: a randomized study in patients undergoing autologous peripheral stem cell transplantation. Am J Hematol (2008) 83(8):644–8. doi: 10.1002/ajh.21206
12. Agboola F, Reddy P. Conversion from filgrastim to tbo-filgrastim: Experience of a Large health care system. J Manag Care Spec Pharm (2017) 23(12):1214–8. doi: 10.18553/jmcp.2017.23.12.1214
13. Singh VK, Newman VL, Seed TM. Colony-stimulating factors for the treatment of the hematopoietic component of the acute radiation syndrome (H-ARS): A review. Cytokine (2015) 71(1):22–37. doi: 10.1016/j.cyto.2014.08.003
14. Natalello A, Ami D, Collini M, DÁlfonso L, Chirico G, Tonon G, et al. Biophysical characterization of met-G-CSF: effects of different site-specific mono-pegylations on protein stability and aggregation. PloS One (2012) 7(8):e42511. doi: 10.1371/journal.pone.0042511
15. Cox GN, Chlipala EA, Smith DJ, Carlson SJ, Bell SJ, Doherty DH, et al. Hematopoietic properties of granulocyte colony-stimulating factor/immunoglobulin (G-CSF/IgG-Fc) fusion proteins in normal and neutropenic rodents. PloS One (2014) 9(3):e91990. doi: 10.1371/journal.pone.0091990
16. Chung HK, Kim SW, Byun SJ, Ko EM, Chung HJ, Woo JS, et al. Enhanced biological effects of Phe140Asn, a novel human granulocyte colony-stimulating factor mutant, on HL60 cells. BMB Rep (2011) 44(10):686–91. doi: 10.5483/BMBRep.2011.44.10.686
17. Jevsevar S, Kunstelj M, Porekar VG. PEGylation of therapeutic proteins. Biotechnol J (2010) 5(1):113–28. doi: 10.1002/biot.200900218
18. Wang Y, Chen L, Liu F, Zhao N, Xu L, Fu B, et al. Efficacy and tolerability of granulocyte colony-stimulating factors in cancer patients after chemotherapy: A systematic review and Bayesian network meta-analysis. Sci Rep (2019) 9(1):15374. doi: 10.1038/s41598-019-51982-4
19. Yan B, Zhang W, Lu F, Chen ZL, Han BH, Jiang LY, et al. Safety of polyethylene glycol recombinant human granulocyte colony-stimulating factor in treating non-small cell lung cancer patients at I b stage. Asian Pac J Trop Med (2013) 6(11):912–5. doi: 10.1016/S1995-7645(13)60163-7
20. Zhou C, Huang Y, Wang D, An C, Zhou F, Li Y, et al. A randomized multicenter phase III study of single administration of mecapegfilgrastim (HHPG-19K), a pegfilgrastim biosimilar, for prophylaxis of chemotherapy-induced neutropenia in patients with advanced non-Small-Cell lung cancer (NSCLC). Clin Lung Cancer (2016) 17(2):119–27. doi: 10.1016/j.cllc.2015.12.002
21. Xu F, Zhang Y, Miao Z, Zeng X, Wu B, Cai L, et al. Efficacy and safety of mecapegfilgrastim for prophylaxis of chemotherapy-induced neutropenia in patients with breast cancer: a randomized, multicenter, active-controlled phase III trial. Ann Transl Med (2019) 7(18):482. doi: 10.21037/atm.2019.07.95
22. Farese AM, Casey DB, Smith WG, Vignulle RM, McKearn JP, MacVittie TJ, et al. Leridistim, a chimeric dual G-CSF and IL-3 receptor agonist, enhances multilineage hematopoietic recovery in a nonhuman primate model of radiation-induced myelosuppression: effect of schedule, dose, and route of administration. Stem Cells (2001) 19(6):522–33. doi: 10.1634/stemcells.19-6-522
23. Farese AM, Casey DB, Vignulle RM, Siegel NR, Finn RF, Klover JA, et al. A single dose of pegylated leridistim significantly improves neutrophil recovery in sublethally irradiated rhesus macaques. Stem Cells (2001) 19(6):514–21. doi: 10.1634/stemcells.19-6-514
24. Nabholtz JM, Cantin J, Chang J, Guevin R, Patel R, Tkaczuk K, et al. Phase III trial comparing granulocyte colony-stimulating factor to leridistim in the prevention of neutropenic complications in breast cancer patients treated with docetaxel/doxorubicin/cyclophosphamide: results of the BCIRG 004 trial. Clin Breast Cancer (2002) 3(4):268–75. doi: 10.3816/CBC.2002.n.030
25. Abdolzade-Bavil A, Cooksey BA, Scheckermann C, Lammerich A, Krasney P, Allgaier H, et al. Pegylated versus glycopegylated G-CSFs and their biochemical and physiological properties. Blood (2013) 122(21):4851–1. doi: 10.1182/blood.V122.21.4851.4851
26. Hoggatt J, Tate TA, Pelus LM. Role of lipegfilgrastim in the management of chemotherapy-induced neutropenia. Int J Nanomed (2015) 10:2647–52. doi: 10.2147/IJN.S55796
27. Alshehri A, Ross R, Wilkinson I. Generation of a long acting GCSF for treatment of neutropenia and stem cell harvest. Endocr Abstr (2014). doi: 10.1530/endoabs.34.P37
28. Tanaka H, Ishikawa RS, Ishikawa M, Matsuki M, Asano K. Pharmacokinetics of recombinant human granulocyte colony-stimulating factor conjugated to polyethylene glycol in rats. Cancer Res (1991) 51(14):3710–4.
29. Zhang F, Liu MR, Wan HT. Discussion about several potential drawbacks of PEGylated therapeutic proteins. Biol Pharm Bull (2014) 37(3):335–9. doi: 10.1248/bpb.b13-00661
30. Zhao S, Zhang Y, Tian H, Chen X, Cai D, Yao W, et al. Extending the serum half-life of G-CSF via fusion with the domain III of human serum albumin. BioMed Res Int (2013) 2013:107238. doi: 10.1155/2013/107238
31. Nikravesh FY, Shirkhani S, Bayat E, Talebkhan Y, Mirabzadeh E, Sabzalinejad M, et al. Extension of human GCSF serum half-life by the fusion of albumin binding domain. Sci Rep (2022) 12(1):667. doi: 10.1038/s41598-021-04560-6
32. Halpern W, Riccobene T, Agostini H, Baker K, Stolow D, Gu ML, et al. Albugranin, a recombinant human granulocyte colony stimulating factor (G-CSF) genetically fused to recombinant human albumin induces prolonged myelopoietic effects in mice and monkeys. Pharm Res (2002) 19(11):1720–9. doi: 10.1023/A:1020917732218
33. Gladkov O, Moieseyenko V, Brondarenko I, Shparyk Y, Barash S, Adar L, et al. A phase III study of balugrastim versus pegfilgrastim in breast cancer patients receiving chemotherapy with doxorubicin and docetaxel. Oncologist (2016) 21(1):7–15. doi: 10.1634/theoncologist.2015-0152
34. Ghidini M, Hahne JC, Trevisani F, Panni S, Ratti M, Toppo L, et al. New developments in the treatment of chemotherapy-induced neutropenia: focus on balugrastim. Ther Clin Risk Manage (2016) 12:1009–15. doi: 10.2147/TCRM.S80732
35. Cox GN, Smith DJ, Carlson SJ, Bendele AM, Chlipala EA, Doherty DH, et al. Enhanced circulating half-life and hematopoietic properties of a human granulocyte colony-stimulating factor/immunoglobulin fusion protein. Exp Hematol (2004) 32(5):441–9. doi: 10.1016/j.exphem.2004.01.012
36. Link H, Illerhaus G, Marens UM, Salar A, Depenbusch R, Kohler A, et al. Efficacy and safety of lipegfilgrastim versus pegfilgrastim in elderly patients with aggressive b cell non-Hodgkin lymphoma (B-NHL): results of the randomized, open-label, non-inferiority AVOID neutropenia study. Support Care Cancer (2021) 29(5):2519–27. doi: 10.1007/s00520-020-05711-7
37. Cobb P, Moon YW, Mezei K, Lang I, Bhat G, Chawla S, et al. A comparison of eflapegrastim to pegfilgrastim in the management of chemotherapy-induced neutropenia in patients with early-stage breast cancer undergoing cytotoxic chemotherapy (RECOVER): A phase 3 study. Cancer Med (2020) 9:(17):6234–43. doi: 10.1002/cam4.3227
38. Schwartzberg LS, Bhat G, Peguero J, Agajanian R, Bharadwaj JS, Restrepo A, et al. Eflapegrastim, a long-acting granulocyte-colony stimulating factor for the management of chemotherapy-induced neutropenia: Results of a phase III trial. Oncologist (2020) 25(8):e1233–41. doi: 10.1634/theoncologist.2020-0105
39. Jeon Y, Lee N, Baek S, Choi J, Jhee S, Lee H, et al. A randomized, double-blind, placebo- and active-controlled, escalating single-dose study to evaluate the safety, tolerability, pharmacokinetic, and pharmacodynamic profiles of subcutaneous eflapegrastim in healthy Japanese and Caucasian subjects. Drugs R&D (2022) 22(1):71–87. doi: 10.1007/s40268-021-00379-8
40. Vacirca JL, Chan A, Mezei K, Adoo CS, Papai Z, Mcgregor K, et al. An open-label, dose-ranging study of rolontis, a novel long-acting myeloid growth factor, in breast cancer. Cancer Med (2018) 7(5):1660–9. doi: 10.1002/cam4.1388
41. Abuchowski A, Es T, Palczuk NC, Davis FF. Alteration of immunological properties of bovine serum albumin by covalent attachment of polyethylene glycol. J Biol Chem (1977) 252(11):3578–81. doi: 10.1016/S0021-9258(17)40291-2
42. Jain A, Jain SK. PEGylation: an approach for drug delivery. a review. Crit Rev Ther Drug Carrier Syst (2008) 25(5):403–47. doi: 10.1615/CritRevTherDrugCarrierSyst.v25.i5.10
43. Trainer PJ, Drake WM, Katznelson L, Freda PU, Herman-Bonert V, Lely AJ, et al. Treatment of acromegaly with the growth hormone–receptor antagonist pegvisomant. New Engl J Med (2000) 342(16):1171–7. doi: 10.1056/NEJM200004203421604
44. Hamidi M, Azadi A, Rafiei P. Pharmacokinetic consequences of pegylation. Drug Deliv (2006) 13(6):399–409. doi: 10.1080/10717540600814402
45. Lord BI, Woolford LB, Molineux G. Kinetics of neutrophil production in normal and neutropenic animals during the response to filgrastim (r-metHu G-CSF) or filgrastim SD/01 (PEG-r-metHu G-CSF). Clin Cancer Res (2001) 7(7):2085–90.
46. Bailon P, Won CY. PEG-modified biopharmaceuticals. Expert Opin Drug Delivery (2009) 6(1):1–16. doi: 10.1517/17425240802650568
47. Milla P, Dosio F, Cattel L. PEGylation of proteins and liposomes: a powerful and flexible strategy to improve the drug delivery. Curr Drug Metab (2012) 13(1):105–19. doi: 10.2174/138920012798356934
48. Yang BB, Kido A. Pharmacokinetics and pharmacodynamics of pegfilgrastim. Clin Pharmacokinet (2011) 50(5):295–306. doi: 10.2165/11586040-000000000-00000
49. Arvedson T, O'Kelly J, Yang BB. Design rationale and development approach for pegfilgrastim as a long-acting granulocyte colony-stimulating factor. BioDrugs (2015) 29(3):185–98. doi: 10.1007/s40259-015-0127-4
50. Parker SD, King N, Jacobs TF. Pegfilgrastim. In: StatPearls. Treasure Island (FL: StatPearls Publishing LLC (2022).
51. Selby C, Peyton-Thomas B, Eslami P. Pegfilgrastim biosimilars: Where are we now? J Adv Pract Oncol (2021) 12(5):541–7. doi: 10.6004/jadpro.2021.12.5.9
52. Yowell SL, Blackwell S. Novel effects with polyethylene glycol modified pharmaceuticals. Cancer Treat Rev (2002) 28:3–6. doi: 10.1016/S0305-7372(02)80002-0
53. Curran MP, Goa KL. Pegfilgrastim. Drugs (2002) 62(8):1207–13. doi: 10.2165/00003495-200262080-00012
54. Pisal DS, Kosloski MP, Balu-Iyer SV. Delivery of therapeutic proteins. J Pharm Sci (2010) 99(6):2557–75. doi: 10.1002/jps.22054
55. Patel A, Cholkar K, Mitra AK. Recent developments in protein and peptide parenteral delivery approaches. Ther Delivery (2014) 5(3):337–65. doi: 10.4155/tde.14.5
56. Caliceti P, Veronese FM. Pharmacokinetic and biodistribution properties of poly(ethylene glycol)-protein conjugates. Adv Drug Delivery Rev (2003) 55(10):1261–77. doi: 10.1016/S0169-409X(03)00108-X
57. Kinstler OB, Brems DN, Lauren SL, Paige AG, Hamburger JB, Treuheit MJ, et al. Characterization and stability of n-terminally PEGylated rhG-CSF. Pharm Res (1996) 13(7):996–1002. doi: 10.1023/A:1016042220817
58. Gaertner HF, Offord RE. Site-specific attachment of functionalized poly(ethylene glycol) to the amino terminus of proteins. Bioconjug Chem (1996) 7(1):38–44. doi: 10.1021/bc950074d
59. Hoang Thi TT, Pilkington EH, Nguyen DH, Lee JS, Park KD, Truong NP, et al. The importance of poly(ethylene glycol) alternatives for overcoming PEG immunogenicity in drug delivery and bioconjugation. Polymers (2020) 12(2):298. doi: 10.3390/polym12020298
60. Reily C, Stewart TJ, Benfrow MB, Novak J. Glycosylation in health and disease. Nat Rev Nephrol (2019) 15(6):346–66. doi: 10.1038/s41581-019-0129-4
61. Varki A. Sialic acids in human health and disease. Trends Mol Med (2008) 14(8):351–60. doi: 10.1016/j.molmed.2008.06.002
62. Li Q, Xie Y, Xu G, Lebrilla C. Identification of potential sialic acid binding proteins on cell membranes by proximity chemical labeling. Chem Sci (2019) 10(24):6199–209. doi: 10.1039/C9SC01360A
63. Walsh G, Jefferis R. Post-translational modifications in the context of therapeutic proteins. Nat Biotechnol (2006) 24(10):1241–52. doi: 10.1038/nbt1252
64. Sinclair AM, Elliott S. Glycoengineering: the effect of glycosylation on the properties of therapeutic proteins. J Pharm Sci (2005) 94(8):1626–35. doi: 10.1002/jps.20319
65. Byrne B, Donohoe GG, O'Kennedy R. Sialic acids: carbohydrate moieties that influence the biological and physical properties of biopharmaceutical proteins and living cells. Drug Discovery Today (2007) 12(7-8):319–26. doi: 10.1016/j.drudis.2007.02.010
66. Saint-Jore-Dupas C, Faye L, Gomord V. From planta to pharma with glycosylation in the toolbox. Trends Biotechnol (2007) 25(7):317–23. doi: 10.1016/j.tibtech.2007.04.008
67. Wongtrakul-Kish K, Kolarich D, Pascovici D, Joss JL, Deane E, Packer NH, et al. Characterization of n- and O-linked glycosylation changes in milk of the tammar wallaby (Macropus eugenii) over lactation. Glycoconj J (2012) 30(5):523–36. doi: 10.1007/s10719-012-9452-8
68. Wolfert MA, Boons GJ. Adaptive immune activation: glycosylation does matter. Nat Chem Biol (2013) 9(12):776–84. doi: 10.1038/nchembio.1403
69. Brockhausen I, Stanley P, Varki A, Cummings RD, Esko JD, Stanley P. O-GalNAc glycans. In: Varki A, et al, editors. Essentials of glycobiology. Cold Spring Harbor (NY: Cold Spring Harbor Laboratory Press (2015).
70. Gong B, Burnina I, Lynaugh H, Li H. O-Linked glycosylation analysis of recombinant human granulocyte colony-stimulating factor produced in glycoengineered pichia pastoris by liquid chromatography and mass spectrometry. J Chromatogr B Analyt Technol BioMed Life Sci (2014) 945-946:135–40. doi: 10.1016/j.jchromb.2013.11.031
71. Kornfeld R, Kornfeld S. Assembly of asparagine-linked oligosaccharides. Annu Rev Biochem (1985) 54:631–64. doi: 10.1146/annurev.bi.54.070185.003215
72. Lowenthal MS, Davis KS, Formolo T, Kilpatrick LE, Phinney KW, et al. Identification of novel n-glycosylation sites at noncanonical protein consensus motifs. J Proteome Res (2016) 15(7):2087–101. doi: 10.1021/acs.jproteome.5b00733
73. Chao Q, Ding Y, Chen ZH, Xiang MH, Wang N, Gao X-D, et al. Recent progress in chemo-enzymatic methods for the synthesis of n-glycans. Front Chem (2020) 8(513). doi: 10.3389/fchem.2020.00513
74. Kim PJ, Lee DY, Jeong H. Centralized modularity of n-linked glycosylation pathways in mammalian cells. PloS One (2009) 4(10):e7317. doi: 10.1371/journal.pone.0007317
75. Butler M, Spearman M. The choice of mammalian cell host and possibilities for glycosylation engineering. Curr Opin Biotechnol (2014) 30:107–12. doi: 10.1016/j.copbio.2014.06.010
76. Sola RJ, Griebenow K. Effects of glycosylation on the stability of protein pharmaceuticals. J Pharm Sci (2009) 98(4):1223–45. doi: 10.1002/jps.21504
77. D’Addio M, Frey J, Otto VI. The manifold roles of sialic acid for the biological functions of endothelial glycoproteins. Glycobiology (2020) 30(8):490–9. doi: 10.1093/glycob/cwaa008
78. Ghosh S. Sialic acid and biology of life: An introduction. Sialic Acids Sialoglycoconjugates Biol Life Health Dis (2020) p:1–61. doi: 10.1016/B978-0-12-816126-5.00001-9
79. Darling RJ, Kuchibhotla U, Glaesner W, Micanovic R, Witcher DR, Beals JM, et al. Glycosylation of erythropoietin affects receptor binding kinetics: role of electrostatic interactions. Biochemistry (2002) 41(49):14524–31. doi: 10.1021/bi0265022
80. Maley F, Trimble RB, Tarentino AL, Plummer TH Jr. Characterization of glycoproteins and their associated oligosaccharides through the use of endoglycosidases. Anal Biochem (1989) 180(2):195–204. doi: 10.1016/0003-2697(89)90115-2
81. Tokhtaeva E, Mareninova OA, Gukovskaya AS, Vagin O. Analysis of n- and O-glycosylation of lysosomal glycoproteins. Methods Mol Biol (Clifton N.J.) (2017) 1594:35–42. doi: 10.1007/978-1-4939-6934-0_3
82. Spiro RG. Protein glycosylation: nature, distribution, enzymatic formation, and disease implications of glycopeptide bonds. Glycobiology (2002) 12(4):43R–56R. doi: 10.1093/glycob/12.4.43R
83. Behnke J, Cohen AM, LaRoche J. N-linked glycosylation enzymes in the diatom thalassiosira oceanica exhibit a diel cycle in transcript abundance and favor for NXT-type sites. Sci Rep (2021) 11(1):3227. doi: 10.1038/s41598-021-82545-1
84. Sola RJ, Griebenow K. Glycosylation of therapeutic proteins: an effective strategy to optimize efficacy. BioDrugs (2010) 24(1):9–21. doi: 10.2165/11530550-000000000-00000
85. Ma B, Guan X, Li Y, Shang S, Li J, Tan Z, et al. Protein glycoengineering: An approach for improving protein properties. Front Chem (2020) 8(622). doi: 10.3389/fchem.2020.00622
86. Elliott S, Lorenzini T, Asher S, Aoki K, Brankow D, Buck L, et al. Enhancement of therapeutic protein in vivo activities through glycoengineering. Nat Biotechnol (2003) 21(4):414–21. doi: 10.1038/nbt799
87. Weenen C, Pena JE, Pollak SV, Klein J, Lobel L, Trousdale RK, et al. Long-acting follicle-stimulating hormone analogs containing n-linked glycosylation exhibited increased bioactivity compared with o-linked analogs in female rats. J Clin Endocrinol Metab (2004) 89(10):5204–12. doi: 10.1210/jc.2004-0425
88. Trousdale RK, Yu B, Pollak SV, Husami N, Vidali A, Lustbader JW, et al. Efficacy of native and hyperglycosylated follicle-stimulating hormone analogs for promoting fertility in female mice. Fertil Steril (2009) 91(1):265–70. doi: 10.1016/j.fertnstert.2007.11.013
89. Guariglia R, Martorelli MC, Lerose R, Telesca D, Milella MR, Musto P, et al. Lipegfilgrastim in the management of chemotherapy-induced neutropenia of cancer patients. Biol: Targets Ther (2016) 10:1–8. doi: 10.2147/BTT.S58597
90. Vogel CL, Wojtukiewicz MZ, Carroll RR, Tjulandin SA, Barajas-Figueroa LJ, Wiens B, et al. First and subsequent cycle use of pegfilgrastim prevents febrile neutropenia in patients with breast cancer: a multicenter, double-blind, placebo-controlled phase III study. J Clin Oncol (2005) 23(6):1178–84. doi: 10.1200/JCO.2005.09.102
91. Pro B, Fayad L, Mclaughlin P, Romaguera J, Hagemeister FB, Rodriguez MA, et al. Pegfilgrastim administered in a single fixed dose is effective in inducing neutrophil count recovery after paclitaxel and topotecan chemotherapy in patients with relapsed aggressive non-hodgkin's lymphoma. Leuk Lymphoma (2006) 47(3):481–5. doi: 10.1080/10428190500305802
92. Aapro M, Boccia R, Leonard R, Camps C, Campone M, Choquet S, et al. Refining the role of pegfilgrastim (a long-acting G-CSF) for prevention of chemotherapy-induced febrile neutropenia: consensus guidance recommendations. Support Care Cancer (2017) 25(11):3295–304. doi: 10.1007/s00520-017-3842-1
93. Gálffy G. [Lipegfilgrastim - long acting G-CSF in prevention of chemotherapy-induced neutropenia]. Magy Onkol (2018) 62(3):195–200.
94. Belogurova M, Kizyma ZP, Garami M, Csoka M, Lamson MJ, Buchner A, et al. A pharmacokinetic study of lipegfilgrastim in children with Ewing family of tumors or rhabdomyosarcoma. Cancer Chemother Pharmacol (2017) 79:155–64. doi: 10.1007/s00280-016-3216-2
95. Fontaine C, Claes N, Graas MP, Samani KK, Vuylsteke P, Vulsteke C, et al. Effect of lipegfilgrastim administration as prophylaxis of chemotherapy-induced neutropenia on dose modification and incidence of neutropenic events: real-world evidence from a non-interventional study in Belgium and Luxembourg. Acta Clin Belg (2021) 76(1):10–5. doi: 10.1080/17843286.2019.1646539
96. Mahlert F, Schmidt K, Allgaier H, Liu P, Muller U, Shen WD, et al. Rational development of lipegfilgrastim, a novel long-acting granulocyte colony-stimulating factor, using glycopegylation technology. Blood (2013) 122(21):4853. doi: 10.1182/blood.V122.21.4853.4853
97. Bondarenko I, Gladkov OA, Elsaesser R, Buchner A, Bias P, et al. Efficacy and safety of lipegfilgrastim versus pegfilgrastim: a randomized, multicenter, active-control phase 3 trial in patients with breast cancer receiving doxorubicin/docetaxel chemotherapy. BMC Cancer (2013) 13:386. doi: 10.1186/1471-2407-13-386
98. Pyzik M, Sand KMK, Hubbard J, Andersen JT, Sandlie I, Blumberg RS, et al. The neonatal fc receptor (FcRn): A misnomer? Front Immunol (2019) 10(1540). doi: 10.3389/fimmu.2019.01540
99. Chaudhury C, Brooks CL, Carter DC, Robinson JM, Anderson CL. Albumin binding to FcRn: distinct from the FcRn-IgG interaction. Biochemistry (2006) 45(15):4983–90. doi: 10.1021/bi052628y
100. Andersen JT, Sandlie I. The versatile MHC class I-related FcRn protects IgG and albumin from degradation: implications for development of new diagnostics and therapeutics. Drug Metab Pharmacokinet (2009) 24(4):318–32. doi: 10.2133/dmpk.24.318
101. Dumont JA, Low SC, Peters RT, Bitonti AJ. Monomeric fc fusions: impact on pharmacokinetic and biological activity of protein therapeutics. BioDrugs (2006) 20(3):151–60. doi: 10.2165/00063030-200620030-00002
102. Ober RJ, Matinez C, Lai X, Zhou J, Ward ES. Exocytosis of IgG as mediated by the receptor, FcRn: an analysis at the single-molecule level. Proc Natl Acad Sci U.S.A. (2004) 101(30):11076–81. doi: 10.1073/pnas.0402970101
103. Andersen JT, Dee Qian J, Sandlie I. The conserved histidine 166 residue of the human neonatal fc receptor heavy chain is critical for the pH-dependent binding to albumin. Eur J Immunol (2006) 36(11):3044–51. doi: 10.1002/eji.200636556
104. Mackness BC, Jaworski JA, Boudanova E, Park A, Valente D, Mauriac C, et al. Antibody fc engineering for enhanced neonatal fc receptor binding and prolonged circulation half-life. MAbs (2019) 11(7):1276–88. doi: 10.1080/19420862.2019.1633883
105. Dennis MS, Zhang M, Meng YG, Kadkhodayan M, Kirchhofer D, Combs, et al. Albumin binding as a general strategy for improving the pharmacokinetics of proteins. J Biol Chem (2002) 277(38):35035–43. doi: 10.1074/jbc.M205854200
106. Gaberc-Porekar V, Zore I, Po B, Me V. Obstacles and pitfalls in the PEGylation of therapeutic proteins. Curr Opin Drug Discovery Devel (2008) 11(2):242–50.
107. Pechtner V, Karanikas CA, García-Pérez LE, Glaesner W. A new approach to drug therapy: Fc-fusion technology. Prim Health Care Open Access (2017) 07. doi: 10.4172/2167-1079.1000255
108. Kim YH, Chol I, Kolli P, Reddy G. Abstract 1347: In vivo efficacy of eflapegrastim in rats with chemotherapy-induced neutropenia. Cancer Res (2017) 77(13 Supplement):1347–7. doi: 10.1158/1538-7445.AM2017-1347
109. Shin KH, Kim TE, Lim KS, Yoon SH, Cho JY, Kim SE, et al. Pharmacokinetic and pharmacodynamic properties of a new long-acting granulocyte colony-stimulating factor (HM10460A) in healthy volunteers. BioDrugs (2013) 27(2):149–58. doi: 10.1007/s40259-013-0010-0
110. Vacirca JL, Papai Z, Horvath Z, Makharadze R, Reddy G, Song T, et al. Abstract P5-11-07: Pharmacokinetics of eflapegrastim in a phase 2 open-label dose-ranging study in breast cancer patients receiving TC regimen. Cancer Res (2017) 77(4_Supplement):P5–11-07-P5-11-07. doi: 10.1158/1538-7445.SABCS16-P5-11-07
Keywords: G-CSF, long-acting, neutropenia, strategy, therapy
Citation: Theyab A, Alsharif KF, Alzahrani K, Oyouni AAA, Hawsawi YM, Algahtani M, Alghamdi S and Alshammary AF (2023) New insight into strategies used to develop long-acting G-CSF biologics for neutropenia therapy. Front. Oncol. 12:1026377. doi: 10.3389/fonc.2022.1026377
Received: 23 August 2022; Accepted: 08 December 2022;
Published: 05 January 2023.
Edited by:
Daiqing Liao, University of Florida, United StatesReviewed by:
Noriko Satake, UC Davis Medical Center, United StatesCopyright © 2023 Theyab, Alsharif, Alzahrani, Oyouni, Hawsawi, Algahtani, Alghamdi and Alshammary. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Abdulrahman Theyab, Ym9zZWl0QGhvdG1haWwuY29t; Khalaf F. Alsharif, QWxzaGFyaWZAdHUuZWR1LnNh
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.