 Ling Gu
Ling Gu Ping Liao
Ping Liao Hanmin Liu
Hanmin Liu
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 19 December 2022
Sec. Hematologic Malignancies
Volume 12 - 2022 | https://doi.org/10.3389/fonc.2022.1022979
This article is part of the Research Topic Acute Leukemias: Molecular Characterization, Leukemia-Initiating Cells, and Influence of the Microenvironment View all 18 articles
Although the prognosis for acute leukemia has greatly improved, treatment of relapsed/refractory acute leukemia (R/R AL) remains challenging. Recently, increasing evidence indicates that the bone marrow microenvironment (BMM) plays a crucial role in leukemogenesis and therapeutic resistance; therefore, BMM-targeted strategies should be a potent protocol for treating R/R AL. The targeting of cancer-associated fibroblasts (CAFs) in solid tumors has received much attention and has achieved some progress, as CAFs might act as an organizer in the tumor microenvironment. Additionally, over the last 10 years, attention has been drawn to the role of CAFs in the BMM. In spite of certain successes in preclinical and clinical studies, the heterogeneity and plasticity of CAFs mean targeting them is a big challenge. Herein, we review the heterogeneity and roles of CAFs in the BMM and highlight the challenges and opportunities associated with acute leukemia therapies that involve the targeting of CAFs.
Acute leukemia is a clonal hematopoietic cancer originating in the bone marrow (BM) and can be classified into two types: acute lymphoid leukemia (ALL) and acute myeloid leukemia (AML). With the advancement of therapies, leukemia is no longer an incurable disease. In children, the 5-year event-free survival (EFS) rate is approximately 85-90% for ALL, sometimes exceeding 90% in ALL trials in developed countries (1–4), and approximately 45-65% for AML (5, 6). However, in adults, the 5-year EFS rate for ALL is only 35-45% (7, 8). The prognosis for adult AML is better in acute promyelocytic leukemia (APL), with a 5-year EFS rate exceeding 80% (9, 10). However, only 35-40% of patients with AML manage to survive for more than 5 years (10, 11). Even in the most curable pediatric ALL, 10-15% of patients do not survive because of chemo-resistance and relapse, which is named relapsed/refractory (R/R) ALL (12, 13). The proportion of R/R AML and R/R adult ALL cases is far higher than that of R/R pediatric ALL.
Until now, the treatment of leukemia has been focused on targeting leukemic cells. While the intensity of chemoradiotherapy is limited by toxic side effects, such as pancytopenia, BM transplantation (BMT) has been drawn into the therapeutic protocol to help reconstruct hematologic and immunologic capacity following high-intensity chemotherapy and radiation treatment to eradicate leukemic cells (14–16). Even so, the prognosis for patients with R/R leukemia remains poor. The exploration of innovative approaches is crucial for patients with R/R leukemia. Immunotherapy, especially chimeric antigen receptor T (CAR-T) cell and antibody therapy, improves the response rate in patients with R/R leukemia by targeting leukemic cells (1, 8, 13–19). However, the cure rate has not been noticeably improved, especially in patients with R/R AML, which highlights an urgent need for novel and synergistic therapies.
‘Seed-and-soil’ theory is well known in cancer research and the term was coined by Dr. Stephen Paget in 1889 (20, 21). ‘Seed’ and ‘soil’ crosstalk may push cancer progression. Remodeling of the ‘soil’ will make it more difficult for cancer cells but more suitable for normal cells, thus potentially helping to cure cancer. As a matter of course, the next target should be the ‘soil’. ‘Soil’ remodeling is important for R/R leukemia patients as it may provide conditions in which cancer cells and cancer stem cells struggle to survive in (22–25). It generally accepted that the ‘soil’ of solid cancer, known as the tumor microenvironment (TME), is a target-rich environment (26–31). Cancer-associated fibroblasts (CAFs), the major players in the TME, have drawn much attention for their multiple functions, including extracellular matrix (ECM) remodeling, growth factor, cytokine, and chemokine production, angiogenesis regulation, and metabolism and immune system modulation (24, 32–39). In this review, we summarize the role of CAFs in acute leukemia and highlight the challenges and opportunities associated with CAF-targeting therapy.
First, we must understand the ‘soil’ of leukemic cells and stem cells, the BM microenvironment (BMM). BMM plays a key role in regulating normal hematopoiesis, as well as chondrogenesis and osteogenesis. Initially, BMM was identified as necessary for successful BMT to reconstruct hematopoiesis. In the 1950s, few patients with leukemia benefitted from BMT (40). After the human histocompatibility antigen system was recognized, a modern era of human BMT began. From then on, the BMM has been slowly demystified.
In 1961, Fliedner et al. (41) pointed out that the recovery of hematopoiesis in rats following 1000 cGy total body irradiation required the recovery of vasculogenesis as support. Then, in 1967, Wolf and Trentin applied the term ‘hemopoietic inductive microenvironment’ to this event in the spleen and BM (42–44). In 1978, Raymond Schofield (45) formally proposed the ‘stem cell niche’ in BM as a specialized microenvironment for stem cells in vivo. Since the 1980s, an increasing number of studies have showed that the BM niche (also called BMM) plays a crucial role in both hematopoiesis and leukemogenesis (46–51). Traditionally, the BMM was divided into endosteal and vascular niches, which may participate in different divisions of labor (52–61). Through technological breakthroughs, such as the construction of transgenic mouse models, the development of sophisticated imaging technologies, and single-cell sequencing, the atlas of BMM is becoming clearer. BMM is a continuum in which hematopoietic stem cells (HSCs) and leukemic stem cells (LSCs) may locate in their corresponding niche. The trouble is that LSCs remodel the BMM into a leukemia-permissive microenvironment while suppressing a hematopoietic-permissive microenvironment (50, 60, 62–67). Clinically, this hypothesis is best supported by donor cell leukemia, in which leukemia originates from engrafted donor cells after allogeneic HSC transplantation, i.e., the leukemia-permissive microenvironment may initiate leukemogenesis in healthy cells (68–72). Therefore, targeting of the leukemia-permissive BMM to restore hematopoietic-permissive BMM can be a useful strategy for overcoming R/R leukemia. Herein, the next issue is to dig out the potent target cells.
Initially, in the 1960s, Owen and Macapheson (73, 74) observed a group of pre-osteoblasts growing in the inner periosteal surface of the femur. In 1968, Friedenstein (75) and Tavassoli et al. (76) found that BMT could generate non-hematopoietic osteogenic cells. Then, in the 1980s, many papers reported fibroblast colonies originating from stromal osteogenic precursor cells in BM (77–81). In 1991, Caplan (82) termed precursor cells with multipotency properties as mesenchymal stem cells (MSCs). In the present day, autoradiography, BM smear and biopsy, flowcytometry, in vivo BMT, and in vitro cell culture have helped us recognize the cellular components of the BMM, including MSCs, endothelial cells (ECs), adipocytes, Cxcl12-abundant reticular (CAR) cells, osteogenic cells, macrophages, fibroblasts, Schwann cells, and possibly other stromal cells (81, 83, 84). Cre-mediated lineage tracing and deletion of molecular factors helped trace cell fate and differentiation, which were still limited in a small piece of a whole. Recently, single-cell and spatial transcriptomic technologies provided the first systematic and label-free identification of cell types of the BMM (85–90). So far, we can map the cellular composition and distribution in the BMM. Different BM resident cell types are successfully allocated to endosteal, sinusoidal, arteriolar, and non-vascular niches (90). Baryawno et al. (86) first profiled all non-hematopoietic (Ter119-/CD71-/Lin-) cells in mouse BM and gained 17 clusters spanning MSCs (Lepr+Cxcl12+), osteolineage cells (Bglap+), chondrocytes (Acan+Col2a1+), fibroblasts (S100a4+), BMECs (Cdh5+), pericytes (Acta2+), and possible transitional states. Based on single-cell and spatial transcriptomics, Baccin et al. (87), identified nine cell types in BM-resident non-hematopoietic cells and demonstrated their differential localization, including two different EC clusters (Ly6a+ arterial ECs and Emcn+ sinusoidal ECs), CAR cells (Leprhigh Adipo-CAR and osterixhigh Leprlow Osteo-CAR), three distinct fibroblast clusters (stromal, arteriolar, and endosteal localizations), myofibroblasts, Ng2+ Nestin+ MSCs, chondrocytes (Acan and Sox9), osteoblasts (Osteocalcin/Bglap and Col1a1), smooth muscle cells (Tagln and Acta2), and Schwann cells (Mog, Mag).
In the BMM, MSCs and ECs are the most abundant subsets (86), and have been fully researched, especially MSCs. Fibroblasts, myofibroblasts, and Schwann cells were found to be more abundant in crushed bones than in flushed bones (87). Therefore, these cells might be ignored during regular clinical examinations without broken bones, such as BM aspiration and biopsy, due to the limited number of cells. Baryawno et al. (86) revealed that Fibroblast-1 and -2 cells are MSC-like as they expressed the progenitor marker CD34 and MSC markers (Ly6a, Pdgfra, Thy1, and Cd44), but not BMECs or pericytes genes (Cdh5 and Acta2). While in the BMM of AML, Cxcl12, Kitl, and Angpt1 were upregulated in Fibroblast-1 cells (similar to Cxcl12-secreting CAFs). CAFs are defined as fibroblasts that are located within or adjacent to cancer cells, and have been extensively studied due to the ease with which they can be obtained and cultured in vitro from solid cancers (24, 33, 35, 91). In the past decade, CAFs have been well recognized as a promising target in the TME (25, 33, 34, 37).
Fibroblasts are defined as interstitial cells of a mesenchymal lineage that are not epithelial, endothelial, or immune cells (34, 37, 92). The origins and roles of fibroblasts in different tissues remain ambiguous, resulting in a lack of unified biomarkers to define them (36, 93). It is generally accepted that CAFs are the main participants in ECM remodeling, wound-healing responses, immune cell recruitment, inflammation, and fibrosis (32, 34–37, 93). The origins and roles of CAFs are even more complicated than fibroblasts. So far, over 10 origins of CAFs have been found in solid tumors, including tissue-resident cells (fibroblasts, myofibroblasts, fibrocytes, epithelial cells, endothelial cells, adipocytes, smooth muscle cells, and immune cells) and BM-derived cells (MSCs, circulating fibrocytes, and immune cells) (24, 35, 37). Still, the precise origins of CAFs and CAF subgroups, and the differences between CAFs and fibroblasts in normal tissues, remain elusive due to the phenotypic and functional plasticity of these cells and the lack of well-defined lineage biomarkers (34, 37). However, based on scRNA-seq and spatial transcription technology, there is a considerable understanding of the heterogeneity of CAFs in solid cancers, such as pancreatic cancer, liver cancer, gastric cancer, head and neck cancer, and breast cancer (94–102).
Although the BMM has been studied extensively since 1978, research on CAFs in hematological malignancies is falling far behind that of solid tumors. The major reason for this is that BM biopsy specimens are relatively hard to obtain. Additionally, lineage tracing of CAFs might be more difficult in BM. According to the achievements with solid tumors, we can conclude that there are abundant resident origins of CAFs in the BMM, such as MSCs, fibroblasts, myofibroblasts, fibrocytes, smooth muscle cells, endothelial-mesenchymal transformation cells, adipocyte-mesenchymal transition cells (24, 35, 103), pericyte-fibroblast transformation cells (104, 105), monocyte-fibroblast transition cells (106, 107), macrophage-mesenchymal transformation cells (108), and leukemia cells (109–111) (Figure 1). Different cell origins of CAFs might suggest different phenotypes and roles. Additionally, most of the cell origins of CAFs in BM contain populations with multipotent differentiation capacity, which may make lineage tracing of CAFs more difficult (Figure 2). For example, MSCs can differentiate into osteoblasts, chondrocytes, and adipocytes in vitro and in vivo (82, 112, 113). Adipocytes can differentiate into myofibroblasts (103) and osteoblasts (114). CD34(+) fibrocytes are BM-derived monocyte progenitor cells, which can differentiate into adipocytes, osteoblasts, and chondrocytes (115, 116). Monocytes can differentiate into fibrocytes and macrophages (117, 118). Furthermore, the cell origins of BM MSCs currently remain unclear; a mesodermal, a neuro-ectodermal, or even a dual origin have been suggested (113). The pericytes of ectodermal origin can differentiate into MSCs (113, 119, 120). BM MSCs may arise from BM or adipose tissue (121). Similarly, activated fibroblasts can transform into MSCs, adipocytes, chondrocytes, endothelial cells, ECs, and pericytes, and can even be induced to become induced pluripotent stem cells (iPSCs) (35, 122).
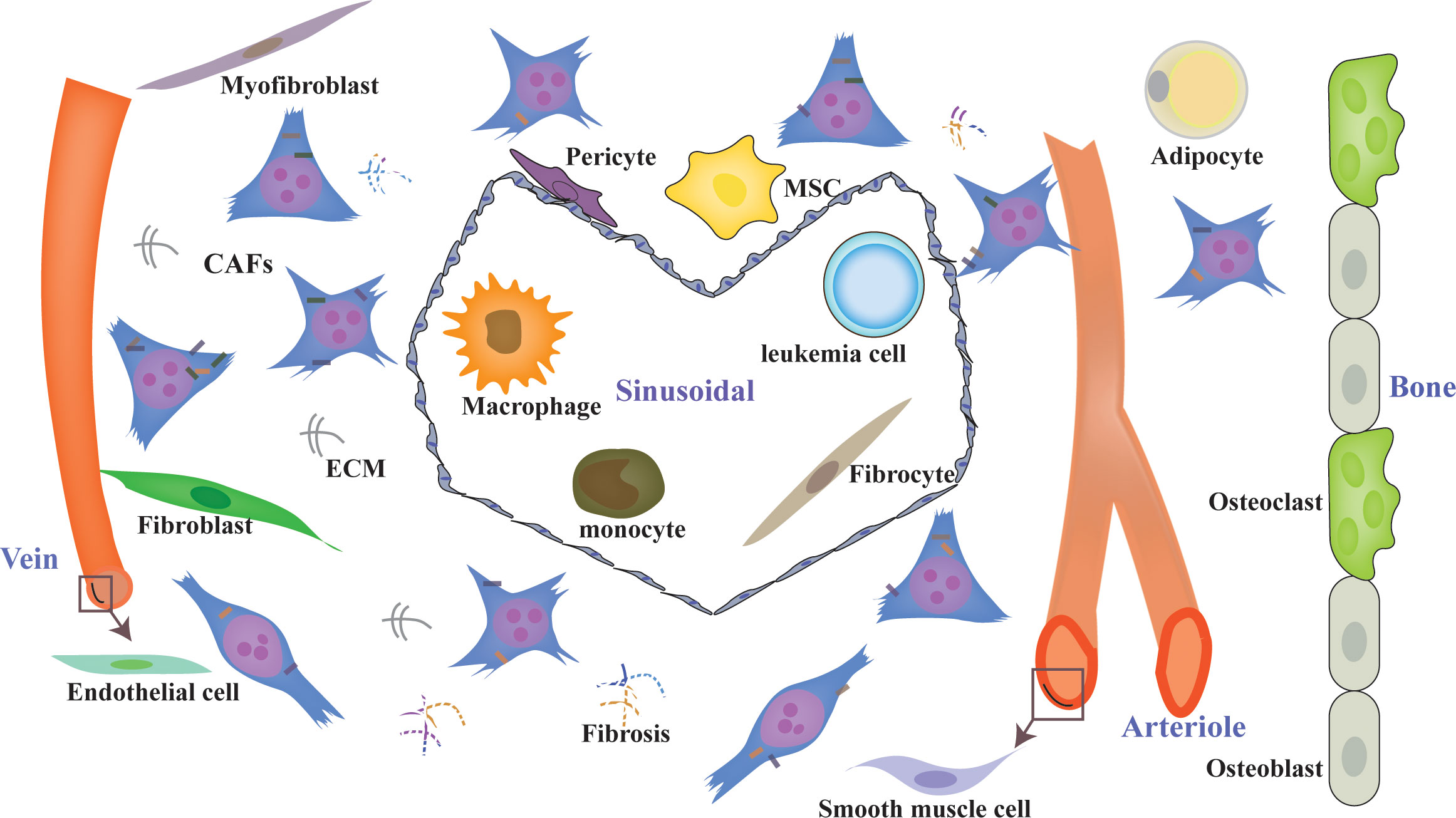
Figure 1 Diverse origins of CAFs in the BMM. CAFs can originate from diverse sources, such as MSCs, fibroblasts, myofibroblasts, fibrocytes, smooth muscle cells, endothelial cells, adipocyte pericytes, monocytes, macrophages, and leukemia cells, with different phenotypes. CAFs are a heterogeneous population with distinct functions in the BMM. ECM, extracellular matrix; CAFs, cancer-associated fibroblasts.
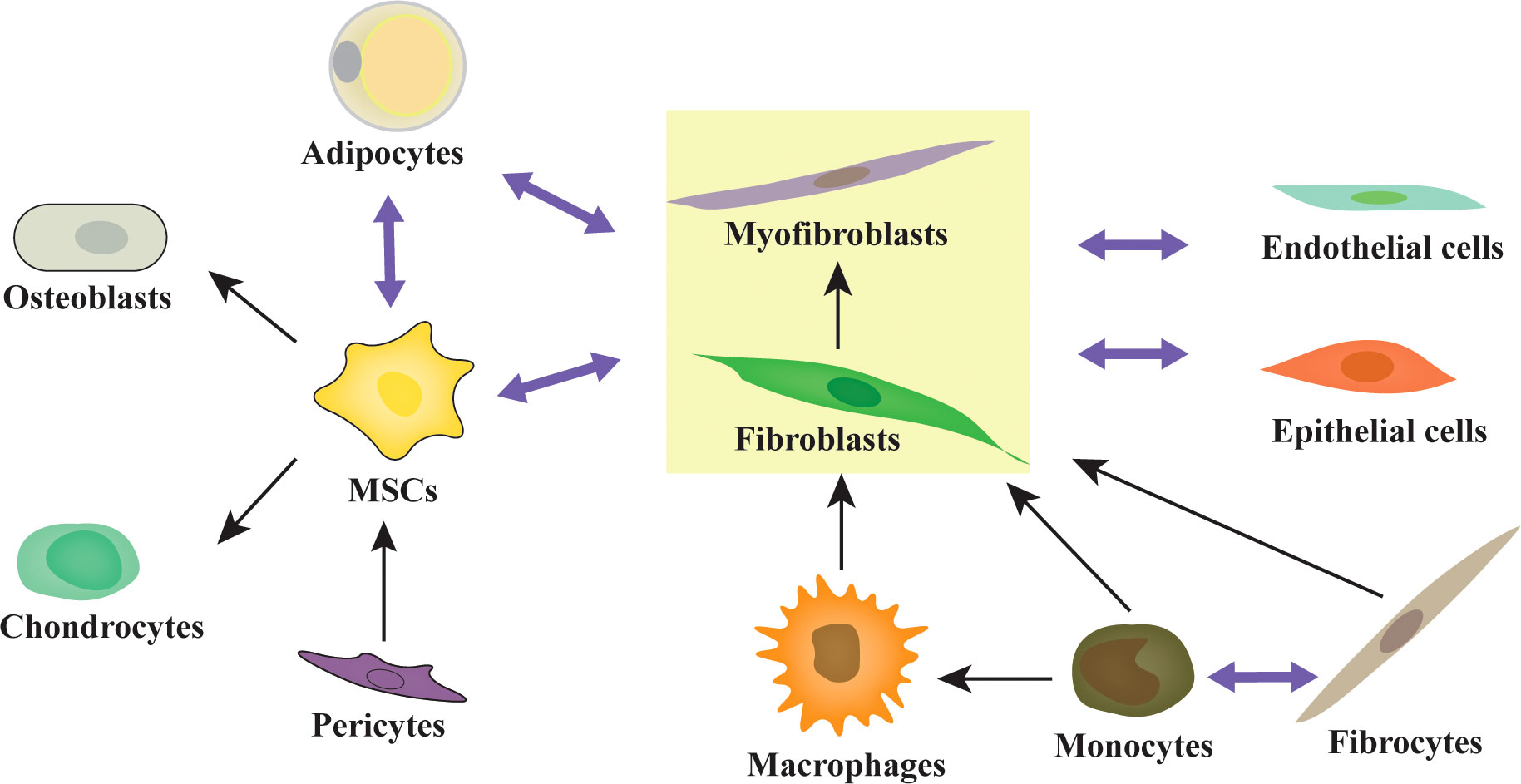
Figure 2 The multipotent differentiation capacity of origin cells of CAFs. Activated fibroblasts, MSCs, and adipocytes are highly plastic and exhibit multipotent capacity. MSCs can differentiate into osteoblasts, chondrocytes, adipocytes, and fibroblasts/myofibroblasts. Adipocytes can differentiate into fibroblasts/myofibroblasts and osteoblasts. Fibrocytes can differentiate into monocytes, fibroblasts/myofibroblasts, adipocytes, osteoblasts, and chondrocytes. Monocytes can differentiate into fibrocytes, macrophages, and fibroblasts/myofibroblasts. The pericytes and adipocytes can differentiate into MSCs. Similarly, activated fibroblasts can transform into MSCs, adipocytes, chondrocytes, endothelial cells, ECs, and pericytes.
First described in 1879, BM fibrosis with fibroblast infiltration and excessive ECM deposition (123, 124) is a typical type of BMM remodeling (125). Now, myelofibrosis (MF) is defined as a clonal hematopoietic BCR-ABL-negative myeloproliferative neoplasm characterized by BM fibrosis, extramedullary hematopoiesis, megakaryocytic hyperplasia, and constitutional symptoms (126). MF may be primary or secondary with a heterogeneous clinical course, ranging from a chronic asymptomatic state to acute leukemic transformation, and possibly a preleukemic state (126, 127). Most forms of secondary MF collaborate with myeloproliferative neoplasms (MPN) and chronic myeloid leukemia (CML). Leukemia transformation is rare in patients with non-fibrotic MPNs but common in patients with MF (128–131). Patients with acute leukemia transformed from MF have a dismal prognosis, with a median survival time of approximately 3 months (128, 129). BM fibrosis in ALL and AML was first described in 1964 (132). Although BM fibrosis may disappear and accompany the complete remission of leukemia, a higher degree of fibrosis (measured as reticulin fibrin density) may correlate with relapse and higher minimal residual disease (MRD) in ALL, especially B-ALL (133–136), and with a poor prognosis in AML (137); however, there remains controversy (138, 139). These results imply that CAFs may play a crucial role in a part of patients with acute leukemia.
The phenotypes and roles of BM CAFs were first reported in patients and mice with multiple myeloma (MM) in 2014 (140). The same year, Duan et al. (141) found that ALL cells may induce a dynamically transient niche in the BMM with the help of chemotherapy: beginning with Nestin+ MSCs, maturating through their transition to a-SMA+ cells, and terminating with fiber residues, called the NSM niche, in mice models and patients with ALL after chemotherapy. The NSM niche was associated with additional difficulties in achieving complete remission after therapy in ALL patients, i.e., the transit of Nestin+ MSCs to a-SMA+ CAFs might correlate with BM fibrosis and poor prognosis in ALL. In 2015, Paggetti et al. (142) reported that exosomes from chronic lymphoid leukemia (CLL) may induce the transition of BM MSCs to CAFs. In 2016, a retrospective study on BM biopsies from patients with AML showed that CAFs were widespread within the BM. Furthermore, excessive reticular fibers in the BM led to a higher frequency of relapse and mortality in primary ALL patients (143). In 2019, Burt et al. (144) pinpointed that CAFs/activated MSCs are frequently presence in ALL, which could prevent ALL cell apoptosis and death from reactive oxygen species-inducing agents by mitochondrial transfer. Exposure to Ara-C or daunorubicin may generate CAFs in vitro and in ALL mice models (144). Then, Pan et al. (145, 146) found that TGF-β is a key factor for BM MSCs to obtain a CAF-like phenotype in a B-ALL microenvironment, which may interact with ALL cells through an SDF1-CXCR4 signaling axis to promote the progression of B-ALL. Using single-cell sequencing, Baryawno et al. (86) revealed a decrease in Fibroblast-5s (Sox9, Spp1, Nt5e, cspg4, and clip), an increase in Fibroblast-2s (Cd34, Ly6a, Pdgfra, Thy1, and Cd44), and a Cxcl12-secreting CAF phenotype of Fibroblast-1s (with upregulation of Cxcl12, Kitl, and Angpt1) in mice BM with AML. In 2021, our team established the first CAF tumor cell line, HXWMF-1 (α-SMA, vimentin, HSP47, S100A4/FSP1, FAP, PDGFRβ, and CD34 positive) (147), originated from the subcutaneous xenografts of HXEX-ALL1 (148), a cell line from a relapsed patient with B-ALL. The cell line provides firm evidence that leukemia cells may induce malignant transformation of CAFs (147). Malignant CAFs might remodel the BMM to form a more aggressive niche. Although the exact roles and underlying mechanisms of CAFs in BM remain elusive, it is clear that CAFs in BM may correlate with BM fibrosis, promote leukemia progression, and induce chemoresistance (Table 1) (86, 141, 143–146). Chemotherapeutic drugs, such as Ara-C and daunorubicin, may induce the generation of CAFs (Table 1) (141, 144). In general, CAFs in BM may have distinct phenotypes and play crucial roles in leukemogenesis and therapy resistance. Understanding the role of CAFs in BM and AL may have clinical significance as it may facilitate the identification of novel drug targets for BMM and immunotherapy.
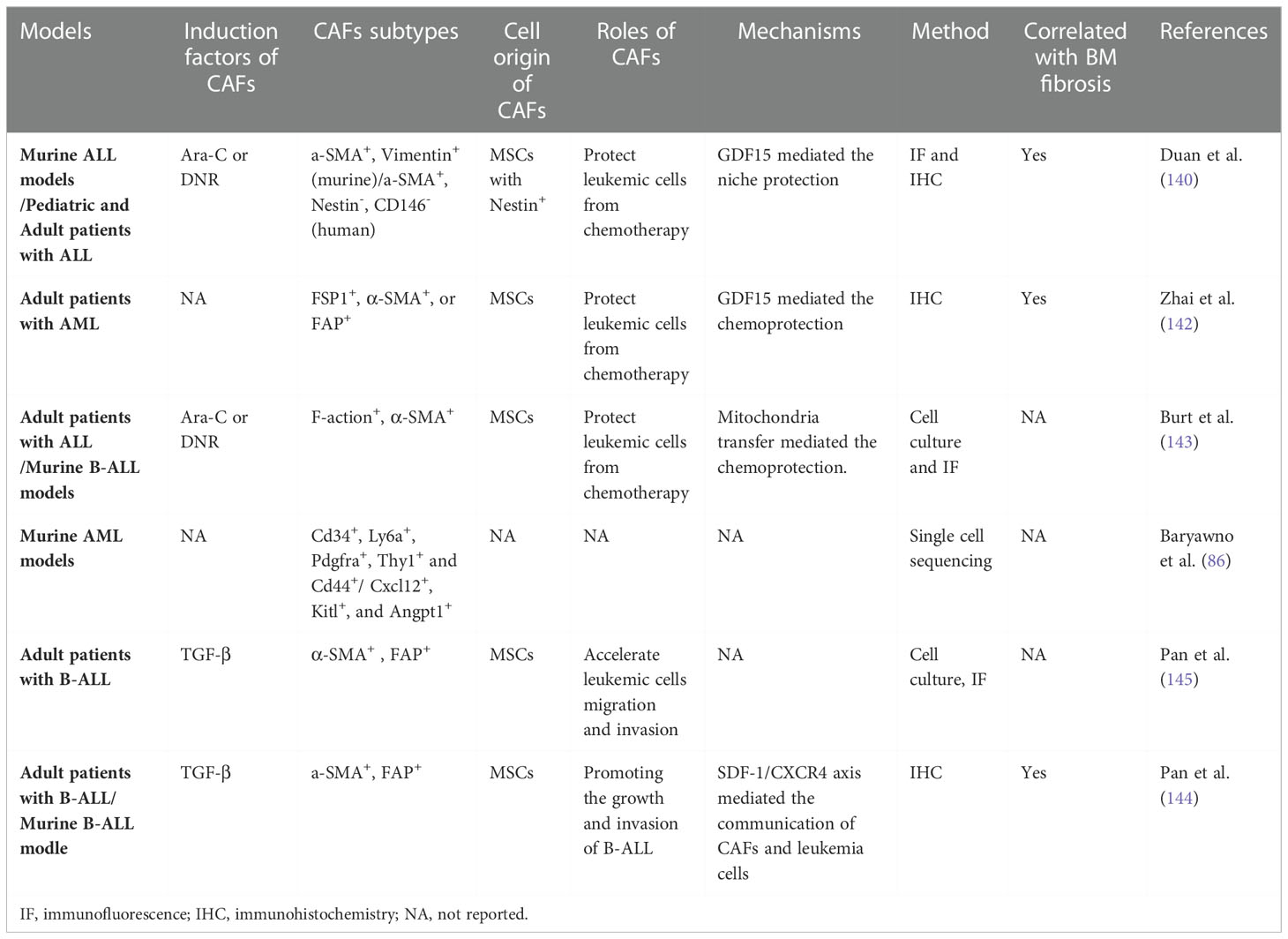
Table 1 CAFs in AL and their functions in leukemia progression.
G-banding analysis showed that HXWMF-1 cells have 60–70 chromosomes with complex structural chromosomal abnormalities (147), which raises the question of whether there are cytogenetic abnormalities in BM stromal cells in patients with acute leukemia? Some studies reported that stromal cells in the BM of MM, myelodysplastic syndrome (MDS), AML, ALL, and CML patients had numerical and structural chromosomal abnormalities, which were different from the abnormalities of leukemic cells (149–153). However, other researchers were unable to find chromosomal abnormalities in stromal cells from different hematological diseases, including MDS, AML, ALL, CLL, and CML (154–157). Gunsilius et al. (158) reported that ECs from patients with CML expressed the BCR-ABL fusion gene. Zhou et al. (159) found that clonal expansion of fibroblasts with somatic copy number alterations is prevalent in patients with colorectal cancer. The genetic profile of cancer cells can affect the surrounding stoma (160), and genetic alterations have been detected in a few stroma cells in solid tumors (161–163). In general, cytogenetic alterations could appear in stromal cells in some patients with leukemia but not all. The presence of chromosomal aberrations in BM MSCs has been associated with a bad prognosis (150).
Although, studies of the BMM of malignant hematological diseases have suggested a tumor-promoting role for CAFs (140–146, 164), studies on solid tumors revealed highly heterogeneous phenotypes in CAFs, with both tumor-promoting and restraining functions (35, 36, 38, 93, 165–169), which may partly explain the failure in clinical trials of targeting CAFs as a whole (34). The former phenotype represents most of the CAF population (38), which helps reprogram malignant ECM, increase angiogenesis and neovascurization, fuel cancer cells, direct cancer cell proliferation, metastasis, and invasion, deregulate metabolism, induce epigenetic reprogramming, unlock phenotypic plasticity, promote the stemness of cancer cells, resist cell death, shape the tumor immune microenvironment, and confer therapeutic resistance (32, 33, 36, 38, 39, 91, 93, 96, 97, 99, 168, 170, 171). Therefore, CAFs may participate in constructing almost all the fourteen hallmarks of cancer proposed by Dr. Hanahan and Dr. Weinberg (172–174), including acquiring capabilities for sustaining proliferative signaling, evading growth suppressors, resisting cell death, enabling replicative immortality, tumor-promoting inflammation, inducing/accessing vasculature, activating invasion and metastasis, reprogramming cellular metabolism, avoiding immune destruction, genome instability and mutation, unlocking phenotypic plasticity, non-mutational epigenetic reprogramming, polymorphic microbiomes, and senescent cells (172). Kochetkova and Samuel (33) reviewed the published evidence and summarized that CAF-mediated differentiation may give rise to cancer-associated immune cells, adipocytes, nerves, endothelia, and vasculature. They pointed out that CAFs are well equipped to assume the role of master organizer in the cancer by interacting with cancer cells and other stromal cells and immune cells in the TME, and producing cancer-specific ECM and secretome. Therefore, targeting CAFs to destroy cancer might be a potent therapeutic protocol for improving and perfecting cancer therapy.
The first clinical trial of targeting CAFs was reported in 1994, using iodine 131-labeled monoclonal antibody F19 (131I-mAbF19) to target FAP+CAFs in colorectal carcinoma patients with hepatic metastasis. The results prompted the diagnostic and therapeutic applications of mAbF19 (175). Then, an increasing number of preclinical and clinical trials of different targets and strategies were undertaken or are still in progress. However, in recent years, CAFs have been the focus of debate. There are numerous obstacles and challenges in targeting CAFs, such as a lack of specific CAF cell markers and signaling pathways, and the heterogeneous roles of CAFs. Increasing evidence has added further complication by indicating that the phenotypes of CAFs are dynamic and able to interconvert depending on tumor status, culture conditions, and therapeutic protocols (34, 93, 176–178). This presents a challenge and an opportunity, as modulating the phenotype of CAFs from tumor promoting to tumor restraining might be an attractive approach for cancer treatment (38, 93). Unfortunately, owing to the same difficulty, there are no definite and standardized markers to classify the functional subtypes of CAFs. Traditionally, α-smooth muscle actin (α-SMA) was identified as a marker of active CAFs and a prognostic factor in tumor patients; however, certain subtypes of CAFs are characterized by a far lower degree of α-SMA (176, 179, 180). Currently, a number of markers, such as α-SMA, FAP, PDGFRα/β, vimentin, S100A4 (FSP1), CAV1(caveolin 1), transgelin (TAGLN), periostin (POSTN), podoplanin (PDPN), integrin α11β1 (ITGA11), collagen type XI alpha I chain (COL11A1), and microfibril-associated protein 5 (MFAP5), are used to identify CAF populations and subgroups (24, 32, 38, 93, 179). Just as Dr. Song mentioned, CAFs are frequently defined by what they are not, typically using multiple biomarkers, resulting in an incomprehensive definition of a CAF (38). Recently, novel CAF-specific biomarkers were discovered in different cancers, such as CD10+GPR77+ CAFs in breast and lung cancer (25), G protein-coupled receptor 30+ CAFs in prostate cancer (181), netrin G1+ CAFs (182), neuregulin+ CAFs (183), leucine-rich-repeat-containing 15+ CAFs (91), Gli1+ CAFs (184), CD105+ CAFs in pancreatic cancer (99), and EGRhigh CAFs in adult T cell leukemia/lymphoma (185). Novel markers may help to precisely attack the tumor-promoting CAFs.
According to the target spot, there are two strategies for targeting CAFs, direct and indirect, which were recently comprehensively reviewed by Saw et al. (38). The direct targeting approach includes CAF depletion via cell markers, inhibition of CAF activation by targeting the signaling pathway, halting infiltration of CAFs, and reprogramming tumor-promoting CAFs to a quiescent state or tumor-restraining phenotype (24, 32, 38, 39). The indirect targeting approach includes targeting the TME, CAF-derived ECM, and downstream effectors (24, 32, 38, 39). However, parts of the clinical trials of targeting CAFs ended in failure, and in some cases, even accelerated cancer progression (34). Recently, there have been numerous studies on FAP-specific CAR-T cells, which can specifically attack FAP+ CAFs with concomitant antitumor efficacy and no severe toxicity (24, 186–188). CAR-T, which was first described by Gross et al. (189) in 1989, can enable T cells to recognize antigens independent of major histocompatibility complex II. The first FDA-approved CAR-T cell therapy obtained a good response in aspects of patients’ ALL (190, 191). CAR-T cell therapy is mainly performed in patients with hematological malignancies and is a revolutionary new treatment for cancer (192). However, responses are transient in patients as CAR-T cells may become exhausted/dysfunctional. Recently, Sakemura et al. (164) constructed a dual-targeting BCMA-FAP and BCMA-SLAMF CAR-T to target both malignant plasma cells and BM CAFs. The results showed that dual-targeting of CAR-T can overcome BM-CAF-mediated inhibition of BCMA-CAR-T (targeting plasma cells only) in an MM mice model. This study is a perfect preclinical attempt to target both cancer cells and the TME with immunotherapeutic strategies, and a brand-new attempt at targeting CAFs in the BMM. Encouragingly, the study suggests that FAP-CAR-T can be applied to target BM CAFs in hematologic malignancies to combat BMM-mediated therapy resistance.
Studies on CAFs are exciting and critical for leukemia treatment. The challenge is to better understand the heterogeneity and plasticity of CAFs, which may help to develop novel CAF-targeting therapeutic strategies. Compared with solid tumors, the targeting of CAFs is more challenging in hematological malignancy. First, the BM biopsy samples are harder to obtain. To complicate matters further, it is difficult to obtain enough CAFs through regular BM aspiration and biopsy, whereas fibroblasts and myofibroblasts are abundant in crushed bones (87). Second, the precursor cells in the BMM are more complex and plastic, which make lineage tracing more challenging. In general, there are still many questions about CAFs in the BMM that need to be answered, including the following:
1. What kinds of CAFs in the BMM might correlate with R/R AL? What are their cell origins? Do these CAFs have chromosomal alterations?
2. Do CAFs contribute to donor cell leukemia? What kinds of CAFs might induce donor cell leukemia? What are the underlying mechanisms?
3. What kinds of ALs might induce the malignant transformation of CAFs? What are the exact roles of malignant CAFs?
4. What are the underlying mechanisms of the transition of precursor cells to CAFs? Are there any influences of therapeutic protocols on the transition of CAFs?
LG drafted the manuscript and prepared the figures. LG and PL conceived the review. HL designed and revised the review. All authors contributed to the article and approved the submitted version.
This work was supported by the Sichuan Science and Technology Program of China (Grant no. 2021YFH0062), and Fundamental Research Funds for the Central Universities (Grant no. SCU2019C4005).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
ALL, acute lymphoid leukemia; AML, acute myeloid leukemia; APL, acute promyelocytic leukemia; BM, bone marrow; BMM, BM microenvironment; BMT, BM transplantation; CAFs, cancer associated fibroblasts; CAR, Cxcl12-abundant reticular; CAR-T, chimeric antigen receptor T; CLL, chronic lymphoid leukemia; CML, chronic myeloid leukemia; ECs, endothelial cells; ECM, extracellular matrix; EFS, event free survival; HSCs, hematopoietic stem cells; LSCs, leukemic stem cells; MSCs, mesenchymal stem cells; MDS, myelodysplastic syndrome; MM, multiple myeloma; MF, myelofibrosis; MPN, myeloproliferative neoplasms; R/R, relapsed/refractory; TME, tumor microenvironment.
1. Malard F, Mohty M. Acute lymphoblastic leukaemia. Lancet (2020) 395(10230):1146–62. doi: 10.1016/S0140-6736(19)33018-1
2. Jeha S, Pei D, Choi J, Cheng C, Sandlund JT, Coustan-Smith E, et al. Improved CNS control of childhood acute lymphoblastic leukemia without cranial irradiation: St Jude total therapy study 16. J Clin Oncol (2019) 37(35):3377–91. doi: 10.1200/JCO.19.01692
3. Inaba H, Pui CH. Advances in the diagnosis and treatment of pediatric acute lymphoblastic leukemia. J Clin Med (2021) 10(9):1926. doi: 10.3390/jcm10091926
4. Hunger SP, Mullighan CG. Acute lymphoblastic leukemia in children. N Engl J Med (2015) 373(16):1541–52. doi: 10.1056/NEJMra1400972
5. Creutzig U, van den Heuvel-Eibrink MM, Gibson B, Dworzak MN, Adachi S, de Bont E, et al. Diagnosis and management of acute myeloid leukemia in children and adolescents: recommendations from an international expert panel. Blood (2012) 120(16):3187–205. doi: 10.1182/blood-2012-03-362608
6. Conneely SE, Stevens AM. Acute myeloid leukemia in children: Emerging paradigms in genetics and new approaches to therapy. Curr Oncol Rep (2021) 23(2):16. doi: 10.1007/s11912-020-01009-3
7. Richard-Carpentier G, Kantarjian H, Jabbour E. Recent advances in adult acute lymphoblastic leukemia. Curr Hematol Malig Rep (2019) 14(2):106–18. doi: 10.1007/s11899-019-00503-1
8. Leonard J, Stock W. Progress in adult ALL: incorporation of new agents to frontline treatment. Hematol Am Soc Hematol Educ Program (2017) 2017(1):28–36. doi: 10.1182/asheducation-2017.1.28
9. Abaza Y, Kantarjian H, Garcia-Manero G, Estey E, Borthakur G, Jabbour E, et al. Long-term outcome of acute promyelocytic leukemia treated with all-trans-retinoic acid, arsenic trioxide, and gemtuzumab. Blood (2017) 129(10):1275–83. doi: 10.1182/blood-2016-09-736686
10. Kantarjian HM, Short NJ, Fathi AT, Marcucci G, Ravandi F, Tallman M, et al. Acute myeloid leukemia: Historical perspective and progress in research and therapy over 5 decades. Clin Lymphoma Myeloma Leuk (2021) 21(9):580–97. doi: 10.1016/j.clml.2021.05.016
11. Naina HV, Patnaik MM, Harris S. Anthracycline dose intensification in acute myeloid leukemia. N Engl J Med (2009) 361(26):2578. doi: 10.1056/NEJMc0910366
12. Pui CH, Yang JJ, Hunger SP, Pieters R, Schrappe M, Biondi A, et al. Childhood acute lymphoblastic leukemia: Progress through collaboration. J Clin Oncol (2015) 33(27):2938–48. doi: 10.1200/JCO.2014.59.1636
13. Hunger SP, Raetz EA. How I treat relapsed acute lymphoblastic leukemia in the pediatric population. Blood (2020) 136(16):1803–12. doi: 10.1182/blood.2019004043
14. Brown PA, Shah B, Advani A, Aoun P, Boyer MW, Burke PW, et al. Acute lymphoblastic leukemia, version 2.2021, NCCN clinical practice guidelines in oncology. J Natl Compr Canc Netw (2021) 19(9):1079–109. doi: 10.6004/jnccn.2021.0042
15. Pollyea DA, Bixby D, Perl A, Bhatt VR, Altman JK, Appelbaum FR, et al. NCCN guidelines insights: Acute myeloid leukemia, version 2.2021. J Natl Compr Canc Netw (2021) 19(1):16–27. doi: 10.6004/jnccn.2021.0002
16. Brown P, Inaba H, Annesley C, Beck J, Colace S, Dallas M, et al. Pediatric acute lymphoblastic leukemia, version 2.2020, NCCN clinical practice guidelines in oncology. J Natl Compr Canc Netw (2020) 18(1):81–112. doi: 10.6004/jnccn.2020.0001
17. Inaba H, Pui CH. Immunotherapy in pediatric acute lymphoblastic leukemia. Cancer Metastasis Rev (2019) 38(4):595–610. doi: 10.1007/s10555-019-09834-0
18. Lussana F, Gritti G, Rambaldi A. Immunotherapy of acute lymphoblastic leukemia and lymphoma with T cell-redirected bispecific antibodies. J Clin Oncol (2021) 39(5):444–55. doi: 10.1200/JCO.20.01564
19. Khaldoyanidi S, Nagorsen D, Stein A, Ossenkoppele G, Subklewe M. Immune biology of acute myeloid leukemia: Implications for immunotherapy. J Clin Oncol (2021) 39(5):419–32. doi: 10.1200/JCO.20.00475
20. Fidler IJ. The pathogenesis of cancer metastasis: the ‘seed and soil’ hypothesis revisited. Nat Rev Cancer (2003) 3(6):453–8. doi: 10.1038/nrc1098
21. Paget S. The distribution of secondary growths in cancer of the breast. 1889. Cancer Metastasis Rev (1989) 8(2):98–101.
22. Plaks V, Kong N, Werb Z. The cancer stem cell niche: how essential is the niche in regulating stemness of tumor cells. Cell Stem Cell (2015) 16(3):225–38. doi: 10.1016/j.stem.2015.02.015
23. Najafi M, Mortezaee K, Majidpoor J. Cancer stem cell (CSC) resistance drivers. Life Sci (2019) 234:116781. doi: 10.1016/j.lfs.2019.116781
24. Chen X, Song E. Turning foes to friends: targeting cancer-associated fibroblasts. Nat Rev Drug Discovery (2019) 18(2):99–115. doi: 10.1038/s41573-018-0004-1
25. Su S, Chen J, Yao H, Liu J, Yu S, Lao L, et al. CD10+GPR77+ cancer-associated fibroblasts promote cancer formation and chemoresistance by sustaining cancer stemness. Cell (2018) 172(4):841–56.e16. doi: 10.1016/j.cell.2018.01.009
26. Xiao Y, Yu D. Tumor microenvironment as a therapeutic target in cancer. Pharmacol Ther (2021) 221:107753. doi: 10.1016/j.pharmthera.2020.107753
27. Roma-Rodrigues C, Mendes R, Baptista PV, Fernandes AR. Targeting tumor microenvironment for cancer therapy. Int J Mol Sci (2019) 20(4):840. doi: 10.3390/ijms20040840
28. Kaymak I, Williams KS, Cantor JR, Jones RG. Immunometabolic interplay in the tumor microenvironment. Cancer Cell (2021) 39(1):28–37. doi: 10.1016/j.ccell.2020.09.004
29. Duan Q, Zhang H, Zheng J, Zhang L. Turning cold into hot: Firing up the tumor microenvironment. Trends Cancer (2020) 6(7):605–18. doi: 10.1016/j.trecan.2020.02.022
30. Peng J, Yang Q, Shi K, Xiao Y, Wei X, Qian Z. Intratumoral fate of functional nanoparticles in response to microenvironment factor: Implications on cancer diagnosis and therapy. Adv Drug Delivery Rev (2019) 143:37–67. doi: 10.1016/j.addr.2019.06.007
31. Chen Q, Liu G, Liu S, Su H, Wang Y, Li J, et al. Remodeling the tumor microenvironment with emerging nanotherapeutics. Trends Pharmacol Sci (2018) 39(1):59–74. doi: 10.1016/j.tips.2017.10.009
32. Mhaidly R, Mechta-Grigoriou F. Role of cancer-associated fibroblast subpopulations in immune infiltration, as a new means of treatment in cancer. Immunol Rev (2021) 302(1):259–72. doi: 10.1111/imr.12978
33. Kochetkova M, Samuel MS. Differentiation of the tumor microenvironment: are CAFs the organizer. Trends Cell Biol (2022) 32(4):285–94. doi: 10.1016/j.tcb.2021.11.008
34. Chen Y, McAndrews KM, Kalluri R. Clinical and therapeutic relevance of cancer-associated fibroblasts. Nat Rev Clin Oncol (2021) 18(12):792–804. doi: 10.1038/s41571-021-00546-5
35. Kalluri R. The biology and function of fibroblasts in cancer. Nat Rev Cancer (2016) 16(9):582–98. doi: 10.1038/nrc.2016.73
36. Pradhan RN, Krishnamurty AT, Fletcher AL, Turley SJ, Müller S. A bird’s eye view of fibroblast heterogeneity: A pan-disease, pan-cancer perspective. Immunol Rev (2021) 302(1):299–320. doi: 10.1111/imr.12990
37. Sahai E, Astsaturov I, Cukierman E, DeNardo DG, Egeblad M, Evans RM, et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat Rev Cancer (2020) 20(3):174–86. doi: 10.1038/s41568-019-0238-1
38. Saw PE, Chen J, Song E. Targeting CAFs to overcome anticancer therapeutic resistance. Trends Cancer (2022) 8(7):527–55. doi: 10.1016/j.trecan.2022.03.001
39. Desbois M, Wang Y. Cancer-associated fibroblasts: Key players in shaping the tumor immune microenvironment. Immunol Rev (2021) 302(1):241–58. doi: 10.1111/imr.12982
40. Thomas ED, Lochte HL, Lu WC, Ferrebee JW. Intravenous infusion of bone marrow in patients receiving radiation and chemotherapy. N Engl J Med (1957) 257(11):491–6. doi: 10.1056/NEJM195709122571102
41. Fliedner TM, Bond VP, Cronkite EP. Structural, cytologic and autoradiographic (H3-thymidine) changes in the bone marrow following total body irradiation. Am J Pathol (1961) 38:599–623.
43. Curry JL, Trentin JJ, Wolf N. Hemopoietic spleen colony studies. II. erythropoiesis. J Exp Med (1967) 125(4):703–20. doi: 10.1084/jem.125.4.703
44. Curry JL, Trentin JJ, Cheng V. Hemopoietic spleen colony studies. 3. hemopoietic nature of spleen colonies induced by lymph node or thymus cells, with or without phytohemagglutinin. J Immunol (1967) 99(5):907–16.
45. Schofield R. The relationship between the spleen colony-forming cell and the haemopoietic stem cell. Blood Cells (1978) 4(1-2):7–25.
46. Bentley SA. Bone marrow connective tissue and the haemopoietic microenvironment. Br J Haematol (1982) 50(1):1–6. doi: 10.1111/j.1365-2141.1982.tb01884.x
47. Dorshkind K. Regulation of hemopoiesis by bone marrow stromal cells and their products. Annu Rev Immunol (1990) 8:111–37. doi: 10.1146/annurev.iy.08.040190.000551
48. Testa NG, Dexter TM. Long-term hematopoietic damage: concepts, approaches, and results relevant to the study of environmental toxins. Environ Health Perspect (1989) 82:51–6. doi: 10.1289/ehp.898251
49. Ayala F, Dewar R, Kieran M, Kalluri R. Contribution of bone microenvironment to leukemogenesis and leukemia progression. Leukemia (2009) 23(12):2233–41. doi: 10.1038/leu.2009.175
50. Le PM, Andreeff M, Battula VL. Osteogenic niche in the regulation of normal hematopoiesis and leukemogenesis. Haematologica (2018) 103(12):1945–55. doi: 10.3324/haematol.2018.197004
51. Medyouf H. The microenvironment in human myeloid malignancies: emerging concepts and therapeutic implications. Blood (2017) 129(12):1617–26. doi: 10.1182/blood-2016-11-696070
52. Acar M, Kocherlakota KS, Murphy MM, Peyer JG, Oguro H, Inra CN, et al. Deep imaging of bone marrow shows non-dividing stem cells are mainly perisinusoidal. Nature (2015) 526(7571):126–30. doi: 10.1038/nature15250
53. Chen JY, Miyanishi M, Wang SK, Yamazaki S, Sinha R, Kao KS, et al. Hoxb5 marks long-term haematopoietic stem cells and reveals a homogenous perivascular niche. Nature (2016) 530(7589):223–7. doi: 10.1038/nature16943
54. Bruns I, Lucas D, Pinho S, Ahmed J, Lambert MP, Kunisaki Y, et al. Megakaryocytes regulate hematopoietic stem cell quiescence through CXCL4 secretion. Nat Med (2014) 20(11):1315–20. doi: 10.1038/nm.3707
55. Itkin T, Gur-Cohen S, Spencer JA, Schajnovitz A, Ramasamy SK, Kusumbe AP, et al. Distinct bone marrow blood vessels differentially regulate haematopoiesis. Nature (2016) 532(7599):323–8. doi: 10.1038/nature17624
56. Goncalves KA, Silberstein L, Li S, Severe N, Hu MG, Yang H, et al. Angiogenin promotes hematopoietic regeneration by dichotomously regulating quiescence of stem and progenitor cells. Cell (2016) 166(4):894–906. doi: 10.1016/j.cell.2016.06.042
57. Silberstein L, Goncalves KA, Kharchenko PV, Turcotte R, Kfoury Y, Mercier F, et al. Proximity-based differential single-cell analysis of the niche to identify Stem/Progenitor cell regulators. Cell Stem Cell (2016) 19(4):530–43. doi: 10.1016/j.stem.2016.07.004
58. Ding L, Saunders TL, Enikolopov G, Morrison SJ. Endothelial and perivascular cells maintain haematopoietic stem cells. Nature (2012) 481(7382):457–62. doi: 10.1038/nature10783
59. Mendelson A, Frenette PS. Hematopoietic stem cell niche maintenance during homeostasis and regeneration. Nat Med (2014) 20(8):833–46. doi: 10.1038/nm.3647
60. Ghiaur G, Wroblewski M, Loges S. Acute myelogenous leukemia and its microenvironment: A molecular conversation. Semin Hematol (2015) 52(3):200–6. doi: 10.1053/j.seminhematol.2015.03.003
61. Comazzetto S, Shen B, Morrison SJ. Niches that regulate stem cells and hematopoiesis in adult bone marrow. Dev Cell (2021) 56(13):1848–60. doi: 10.1016/j.devcel.2021.05.018
62. Behrmann L, Wellbrock J, Fiedler W. Acute myeloid leukemia and the bone marrow niche-take a closer look. Front Oncol (2018) 8:444. doi: 10.3389/fonc.2018.00444
63. Ladikou EE, Sivaloganathan H, Pepper A, Chevassut T. Acute myeloid leukaemia in its niche: the bone marrow microenvironment in acute myeloid leukaemia. Curr Oncol Rep (2020) 22(3):27. doi: 10.1007/s11912-020-0885-0
64. Duarte D, Hawkins ED, Lo Celso C. The interplay of leukemia cells and the bone marrow microenvironment. Blood (2018) 131(14):1507–11. doi: 10.1182/blood-2017-12-784132
65. Tettamanti S, Pievani A, Biondi A, Dotti G, Serafini M. Catch me if you can: how AML and its niche escape immunotherapy. Leukemia (2022) 36(1):13–22. doi: 10.1038/s41375-021-01350-x
66. Wang X, Huang S, Chen JL. Understanding of leukemic stem cells and their clinical implications. Mol Cancer (2017) 16(1):2. doi: 10.1186/s12943-016-0574-7
67. Schepers K, Campbell TB, Passegué E. Normal and leukemic stem cell niches: insights and therapeutic opportunities. Cell Stem Cell (2015) 16(3):254–67. doi: 10.1016/j.stem.2015.02.014
68. Flynn CM, Kaufman DS. Donor cell leukemia: insight into cancer stem cells and the stem cell niche. Blood (2007) 109(7):2688–92. doi: 10.1182/blood-2006-07-021980
69. Williams L, Doucette K, Karp JE, Lai C. Genetics of donor cell leukemia in acute myelogenous leukemia and myelodysplastic syndrome. Bone Marrow Transplant (2021) 56(7):1535–49. doi: 10.1038/s41409-021-01214-z
70. Shahar Gabay T, Chapal-Ilani N, Moskovitz Y, Biezuner T, Oron B, Brilon Y, et al. Donor cell leukemia: reappearance of gene mutations in donor cells - more than an incidental phenomenon. Haematologica (2020) 105(12):2861–3. doi: 10.3324/haematol.2019.242347
71. Scheuermann A, Moskop A, Hopp A, Bone K, Drendel HM, Talano J, et al. Pediatric donor cell acute lymphoblastic leukemia following bone marrow transplant for GATA2 mutation. J Pediatr Hematol Oncol (2022) 44(5):268–70. doi: 10.1097/MPH.0000000000002437
72. Wiseman DH. Donor cell leukemia: a review. Biol Blood Marrow Transplant (2011) 17(6):771–89. doi: 10.1016/j.bbmt.2010.10.010
73. Owen M, Macpherson S. Cell population kinetics of an osteogenic tissue. II. J Cell Biol (1963) 19:33–44. doi: 10.1083/jcb.19.1.33
74. Owen M. Cell population kinetics of an osteogenic tissue. I. J Cell Biol (1963) 19:19–32. doi: 10.1083/jcb.19.1.19
75. Friedenstein AJ, Petrakova KV, Kurolesova AI, Frolova GP. Heterotopic of bone marrow. analysis of precursor cells for osteogenic and hematopoietic tissues. Transplantation (1968) 6(2):230–47.
76. Tavassoli M, Crosby WH. Transplantation of marrow to extramedullary sites. Science (1968) 161(3836):54–6. doi: 10.1126/science.161.3836.54
77. Friedenstein AJ, Chailakhyan RK, Gerasimov UV. Bone marrow osteogenic stem cells: in vitro cultivation and transplantation in diffusion chambers. Cell Tissue Kinet (1987) 20(3):263–72. doi: 10.1111/j.1365-2184.1987.tb01309.x
78. Castro-Malaspina H, Gay RE, Resnick G, Kapoor N, Meyers P, Chiarieri D, et al. Characterization of human bone marrow fibroblast colony-forming cells (CFU-f) and their progeny. Blood (1980) 56(2):289–301. doi: 10.1182/blood.V56.2.289.289
79. Howlett CR, Cavé J, Williamson M, Farmer J, Ali SY, Bab I, et al. Mineralization in in vitro cultures of rabbit marrow stromal cells. Clin Orthop Relat Res (1986) 213):251–63. doi: 10.1097/00003086-198612000-00037
80. Mardon HJ, Bee J, von der Mark K, Owen ME. Development of osteogenic tissue in diffusion chambers from early precursor cells in bone marrow of adult rats. Cell Tissue Res (1987) 250(1):157–65. doi: 10.1007/BF00214667
81. Owen M, Friedenstein AJ. Stromal stem cells: marrow-derived osteogenic precursors. Ciba Found Symp (1988) 136:42–60. doi: 10.1002/9780470513637.ch4
83. Crane GM, Jeffery E, Morrison SJ. Adult haematopoietic stem cell niches. Nat Rev Immunol (2017) 17(9):573–90. doi: 10.1038/nri.2017.53
84. Mayani H, Guilbert LJ, Janowska-Wieczorek A. Biology of the hemopoietic microenvironment. Eur J Haematol (1992) 49(5):225–33. doi: 10.1111/j.1600-0609.1992.tb00053.x
85. Al-Sabah J, Baccin C, Haas S. Single-cell and spatial transcriptomics approaches of the bone marrow microenvironment. Curr Opin Oncol (2020) 32(2):146–53. doi: 10.1097/CCO.0000000000000602
86. Baryawno N, Przybylski D, Kowalczyk MS, Kfoury Y, Severe N, Gustafsson K, et al. A cellular taxonomy of the bone marrow stroma in homeostasis and leukemia. Cell (2019) 177(7):1915–32.e16. doi: 10.1016/j.cell.2019.04.040
87. Baccin C, Al-Sabah J, Velten L, Helbling PM, Grünschläger F, Hernández-Malmierca P, et al. Combined single-cell and spatial transcriptomics reveal the molecular, cellular and spatial bone marrow niche organization. Nat Cell Biol (2020) 22(1):38–48. doi: 10.1038/s41556-019-0439-6
88. Severe N, Karabacak NM, Gustafsson K, Baryawno N, Courties G, Kfoury Y, et al. Stress-induced changes in bone marrow stromal cell populations revealed through single-cell protein expression mapping. Cell Stem Cell (2019) 25(4):570–83.e7. doi: 10.1016/j.stem.2019.06.003
89. Wolock SL, Krishnan I, Tenen DE, Matkins V, Camacho V, Patel S, et al. Mapping distinct bone marrow niche populations and their differentiation paths. Cell Rep (2019) 28(2):302–11.e5. doi: 10.1016/j.celrep.2019.06.031
90. Zhang P, Li X, Pan C, Zheng X, Hu B, Xie R, et al. Single-cell RNA sequencing to track novel perspectives in HSC heterogeneity. Stem Cell Res Ther (2022) 13(1):39. doi: 10.1186/s13287-022-02718-1
91. Dominguez CX, Müller S, Keerthivasan S, Koeppen H, Hung J, Gierke S, et al. Single-cell RNA sequencing reveals stromal evolution into LRRC15+ myofibroblasts as a determinant of patient response to cancer immunotherapy. Cancer Discovery (2020) 10(2):232–53. doi: 10.1158/2159-8290.CD-19-0644
92. Buechler MB, Pradhan RN, Krishnamurty AT, Cox C, Calviello AK, Wang AW, et al. Cross-tissue organization of the fibroblast lineage. Nature (2021) 593(7860):575–9. doi: 10.1038/s41586-021-03549-5
93. Biffi G, Tuveson DA. Diversity and biology of cancer-associated fibroblasts. Physiol Rev (2021) 101(1):147–76. doi: 10.1152/physrev.00048.2019
94. Mannarapu M, Dariya B, Bandapalli OR. Application of single-cell sequencing technologies in pancreatic cancer. Mol Cell Biochem (2021) 476(6):2429–37. doi: 10.1007/s11010-021-04095-4
95. Hu B, Wu C, Mao H, Gu H, Dong H, Yan J, et al. Subpopulations of cancer-associated fibroblasts link the prognosis and metabolic features of pancreatic ductal adenocarcinoma. Ann Transl Med (2022) 10(5):262. doi: 10.21037/atm-22-407
96. Affo S, Nair A, Brundu F, Ravichandra A, Bhattacharjee S, Matsuda M, et al. Promotion of cholangiocarcinoma growth by diverse cancer-associated fibroblast subpopulations. Cancer Cell (2021) 39(6):866–82.e11. doi: 10.1016/j.ccell.2021.03.012
97. Li X, Sun Z, Peng G, Xiao Y, Guo J, Wu B, et al. Single-cell RNA sequencing reveals a pro-invasive cancer-associated fibroblast subgroup associated with poor clinical outcomes in patients with gastric cancer. Theranostics (2022) 12(2):620–38. doi: 10.7150/thno.60540
98. Kumar V, Ramnarayanan K, Sundar R, Padmanabhan N, Srivastava S, Koiwa M, et al. Single-cell atlas of lineage states, tumor microenvironment, and subtype-specific expression programs in gastric cancer. Cancer Discovery (2022) 12(3):670–91. doi: 10.1158/2159-8290.CD-21-0683
99. Hutton C, Heider F, Blanco-Gomez A, Banyard A, Kononov A, Zhang X, et al. Single-cell analysis defines a pancreatic fibroblast lineage that supports anti-tumor immunity. Cancer Cell (2021) 39(9):1227–44.e20. doi: 10.1016/j.ccell.2021.06.017
100. Obradovic A, Graves D, Korrer M, Wang Y, Roy S, Naveed A, et al. Immunostimulatory cancer-associated fibroblast subpopulations can predict immunotherapy response in head and neck cancer. Clin Cancer Res (2022) 28(10):2094–109. doi: 10.1158/1078-0432.CCR-21-3570
101. Busch S, Andersson D, Bom E, Walsh C, Ståhlberg A, Landberg G. Cellular organization and molecular differentiation model of breast cancer-associated fibroblasts. Mol Cancer (2017) 16(1):73. doi: 10.1186/s12943-017-0642-7
102. Wu SZ, Roden DL, Wang C, Holliday H, Harvey K, Cazet AS, et al. Stromal cell diversity associated with immune evasion in human triple-negative breast cancer. EMBO J (2020) 39(19):e104063. doi: 10.15252/embj.2019104063
103. Marangoni RG, Korman B, Varga J. Adipocytic progenitor cells give rise to pathogenic myofibroblasts: Adipocyte-to-Mesenchymal transition and its emerging role in fibrosis in multiple organs. Curr Rheumatol Rep (2020) 22(11):79. doi: 10.1007/s11926-020-00957-w
104. Hosaka K, Yang Y, Seki T, Fischer C, Dubey O, Fredlund E, et al. Pericyte-fibroblast transition promotes tumor growth and metastasis. Proc Natl Acad Sci U.S.A. (2016) 113(38):E5618–27. doi: 10.1073/pnas.1608384113
105. Feng F, Feng X, Zhang D, Li Q, Yao L. Matrix stiffness induces pericyte-fibroblast transition through YAP activation. Front Pharmacol (2021) 12:698275. doi: 10.3389/fphar.2021.698275
106. Haudek SB, Trial J, Xia Y, Gupta D, Pilling D, Entman ML. Fc receptor engagement mediates differentiation of cardiac fibroblast precursor cells. Proc Natl Acad Sci U.S.A. (2008) 105(29):10179–84. doi: 10.1073/pnas.0804910105
107. Dong Y, Yang M, Zhang J, Peng X, Cheng J, Cui T, et al. Depletion of CD8+ T cells exacerbates CD4+ T cell-induced monocyte-to-Fibroblast transition in renal fibrosis. J Immunol (2016) 196(4):1874–81. doi: 10.4049/jimmunol.1501232
108. Iwamoto C, Ohuchida K, Shinkawa T, Okuda S, Otsubo Y, Okumura T, et al. Bone marrow-derived macrophages converted into cancer-associated fibroblast-like cells promote pancreatic cancer progression. Cancer Lett (2021) 512:15–27. doi: 10.1016/j.canlet.2021.04.013
109. Shirasaki R, Tashiro H, Oka Y, Matsuo T, Yamamoto T, Sugao T, et al. Chronic myelogenous leukemia cells contribute to the stromal myofibroblasts in leukemic NOD/SCID mouse in vivo. J Oncol (2012) 2012:901783. doi: 10.1155/2012/901783
110. Tashiro H, Mizutani-Noguchi M, Shirasaki R, Shirafuji N. Acute myelogenous leukemia cells with the MLL-ELL translocation convert morphologically and functionally into adherent myofibroblasts. Biochem Biophys Res Commun (2010) 391(1):592–7. doi: 10.1016/j.bbrc.2009.11.104
111. Shirasaki R, Tashiro H, Mizutani-Noguchi M, Kawasugi K, Shirafuji N. Chronic myelogenous leukemia cells convert to myofibroblasts in vitro: effect of vascular endothelial growth factor on development of the microenvironment. Leuk Res (2011) 35(5):663–9. doi: 10.1016/j.leukres.2010.09.019
112. Pittenger MF, Discher DE, Péault BM, Phinney DG, Hare JM, Caplan AI. Mesenchymal stem cell perspective: cell biology to clinical progress. NPJ Regener Med (2019) 4:22. doi: 10.1038/s41536-019-0083-6
113. Andrzejewska A, Lukomska B, Janowski M. Concise review: Mesenchymal stem cells: From roots to boost. Stem Cells (2019) 37(7):855–64. doi: 10.1002/stem.3016
114. Lin D, Dass CR. Transdifferentiation of adipocytes to osteoblasts: potential for orthopaedic treatment. J Pharm Pharmacol (2018) 70(3):307–19. doi: 10.1111/jphp.12862
115. Hong KM, Burdick MD, Phillips RJ, Heber D, Strieter RM. Characterization of human fibrocytes as circulating adipocyte progenitors and the formation of human adipose tissue in SCID mice. FASEB J (2005) 19(14):2029–31. doi: 10.1096/fj.05-4295fje
116. Choi YH, Burdick MD, Strieter RM. Human circulating fibrocytes have the capacity to differentiate osteoblasts and chondrocytes. Int J Biochem Cell Biol (2010) 42(5):662–71. doi: 10.1016/j.biocel.2009.12.011
117. Niedermeier M, Reich B, Rodriguez Gomez M, Denzel A, Schmidbauer K, Göbel N, et al. CD4+ T cells control the differentiation of Gr1+ monocytes into fibrocytes. Proc Natl Acad Sci U.S.A. (2009) 106(42):17892–7. doi: 10.1073/pnas.0906070106
118. Reilkoff RA, Bucala R, Herzog EL. Fibrocytes: emerging effector cells in chronic inflammation. Nat Rev Immunol (2011) 11(6):427–35. doi: 10.1038/nri2990
119. Mangialardi G, Cordaro A, Madeddu P. The bone marrow pericyte: an orchestrator of vascular niche. Regener Med (2016) 11(8):883–95. doi: 10.2217/rme-2016-0121
120. Crisan M, Yap S, Casteilla L, Chen CW, Corselli M, Park TS, et al. A perivascular origin for mesenchymal stem cells in multiple human organs. Cell Stem Cell (2008) 3(3):301–13. doi: 10.1016/j.stem.2008.07.003
121. Zhang J, Liu Y, Yin W, Hu X. Adipose-derived stromal cells in regulation of hematopoiesis. Cell Mol Biol Lett (2020) 25:16. doi: 10.1186/s11658-020-00209-w
122. Raab S, Klingenstein M, Liebau S, Linta L. A comparative view on human somatic cell sources for iPSC generation. Stem Cells Int (2014) 2014:768391. doi: 10.1155/2014/768391
123. Greenberg BR, Woo L, Veomett IC, Payne CM, Ahmann FR. Cytogenetics of bone marrow fibroblastic cells in idiopathic chronic myelofibrosis. Br J Haematol (1987) 66(4):487–90. doi: 10.1111/j.1365-2141.1987.tb01332.x
124. Tomuleasa C, Selicean S, Gafencu G, Petrushev B, Pop L, Berce C, et al. Fibroblast dynamics as an in vitro screening platform for anti-fibrotic drugs in primary myelofibrosis. J Cell Physiol (2018) 233(1):422–33. doi: 10.1002/jcp.25902
125. Tefferi A. Pathogenesis of myelofibrosis with myeloid metaplasia. J Clin Oncol (2005) 23(33):8520–30. doi: 10.1200/JCO.2004.00.9316
126. Venugopal S, Mascarenhas J. Current clinical investigations in myelofibrosis. Hematol Oncol Clin North Am (2021) 35(2):353–73. doi: 10.1016/j.hoc.2020.12.003
127. Song IC, Yeon SH, Lee MW, Ryu H, Lee HJ, Yun HJ, et al. Myelofibrotic and leukemic transformation in 2016 WHO-defined Philadelphia-negative myeloproliferative neoplasm. Blood Res (2022) 57(1):59–68. doi: 10.5045/br.2021.2021209
128. Mughal TI, Vaddi K, Sarlis NJ, Verstovsek S. Myelofibrosis-associated complications: pathogenesis, clinical manifestations, and effects on outcomes. Int J Gen Med (2014) 7:89–101. doi: 10.2147/IJGM.S51800
129. Mesa RA, Li CY, Ketterling RP, Schroeder GS, Knudson RA, Tefferi A. Leukemic transformation in myelofibrosis with myeloid metaplasia: a single-institution experience with 91 cases. Blood (2005) 105(3):973–7. doi: 10.1182/blood-2004-07-2864
130. Kundranda MN, Tibes R, Mesa RA. Transformation of a chronic myeloproliferative neoplasm to acute myelogenous leukemia: does anything work. Curr Hematol Malig Rep (2012) 7(1):78–86. doi: 10.1007/s11899-011-0107-9
131. Tefferi A, Lasho TL, Jimma T, Finke CM, Gangat N, Vaidya R, et al. One thousand patients with primary myelofibrosis: the mayo clinic experience. Mayo Clin Proc (2012) 87(1):25–33. doi: 10.1016/j.mayocp.2011.11.001
132. Kundel DW, Brecher G, Bodey GP, Brittin GM. Reticulin fibrosis and bone infarction in acute leukemia. implications for prognosis. Blood (1964) 23:526–44. doi: 10.1182/blood.V23.4.526.526
133. Norén-Nyström U, Heyman M, Frisk P, Golovleva I, Sundström C, Porwit A, et al. Vascular density in childhood acute lymphoblastic leukaemia correlates to biological factors and outcome. Br J Haematol (2009) 146(5):521–30. doi: 10.1111/j.1365-2141.2009.07796.x
134. Norén-Nyström U, Roos G, Bergh A, Botling J, Lönnerholm G, Porwit A, et al. Bone marrow fibrosis in childhood acute lymphoblastic leukemia correlates to biological factors, treatment response and outcome. Leukemia (2008) 22(3):504–10. doi: 10.1038/sj.leu.2405072
135. Wallis JP, Reid MM. Bone marrow fibrosis in childhood acute lymphoblastic leukaemia. J Clin Pathol (1989) 42(12):1253–4. doi: 10.1136/jcp.42.12.1253
136. Nath SV, Nicholson I, Tapp H, Zola H, Zannettino AC, Revesz T. Reticulin fibres anchor leukaemic blasts in the marrow of patients with acute lymphoblastic leukaemia. Med Hypotheses (2011) 77(3):333–5. doi: 10.1016/j.mehy.2011.05.007
137. Zhang X, Wang F, Yu J, Jiang Z. Significance of bone marrow fibrosis in acute myeloid leukemia for survival in the real-world. Front Oncol (2022) 12:971082. doi: 10.3389/fonc.2022.971082
138. Bharos A, Jong AJ, Manton N, Venn N, Story C, Hodge G, et al. Bone marrow fibrosis and vascular density lack prognostic significance in childhood acute lymphoblastic leukaemia. Leukemia (2010) 24(8):1537–8. doi: 10.1038/leu.2010.134
139. Kuter DJ, Bain B, Mufti G, Bagg A, Hasserjian RP. Bone marrow fibrosis: pathophysiology and clinical significance of increased bone marrow stromal fibres. Br J Haematol (2007) 139(3):351–62. doi: 10.1111/j.1365-2141.2007.06807.x
140. Frassanito MA, Rao L, Moschetta M, Ria R, Di Marzo L, De Luisi A, et al. Bone marrow fibroblasts parallel multiple myeloma progression in patients and mice: in vitro and in vivo studies. Leukemia (2014) 28(4):904–16. doi: 10.1038/leu.2013.254
141. Duan CW, Shi J, Chen J, Wang B, Yu YH, Qin X, et al. Leukemia propagating cells rebuild an evolving niche in response to therapy. Cancer Cell (2014) 25(6):778–93. doi: 10.1016/j.ccr.2014.04.015
142. Paggetti J, Haderk F, Seiffert M, Janji B, Distler U, Ammerlaan W, et al. Exosomes released by chronic lymphocytic leukemia cells induce the transition of stromal cells into cancer-associated fibroblasts. Blood (2015) 126(9):1106–17. doi: 10.1182/blood-2014-12-618025
143. Zhai Y, Zhang J, Wang H, Lu W, Liu S, Yu Y, et al. Growth differentiation factor 15 contributes to cancer-associated fibroblasts-mediated chemo-protection of AML cells. J Exp Clin Cancer Res (2016) 35(1):147. doi: 10.1186/s13046-016-0405-0
144. Burt R, Dey A, Aref S, Aguiar M, Akarca A, Bailey K, et al. Activated stromal cells transfer mitochondria to rescue acute lymphoblastic leukemia cells from oxidative stress. Blood (2019) 134(17):1415–29. doi: 10.1182/blood.2019001398
145. Pan C, Fang Q, Liu P, Ma D, Cao S, Zhang L, et al. Mesenchymal stem cells with cancer-associated fibroblast-like phenotype stimulate SDF-1/CXCR4 axis to enhance the growth and invasion of b-cell acute lymphoblastic leukemia cells through cell-to-Cell communication. Front Cell Dev Biol (2021) 9:708513. doi: 10.3389/fcell.2021.708513
146. Pan C, Liu P, Ma D, Zhang S, Ni M, Fang Q, et al. Bone marrow mesenchymal stem cells in microenvironment transform into cancer-associated fibroblasts to promote the progression of b-cell acute lymphoblastic leukemia. BioMed Pharmacother (2020) 130:110610. doi: 10.1016/j.biopha.2020.110610
147. Li Y, Gu L. Establishment and characterization of HXWMF-1: the first mouse fibroblastic tumor cell line derived from leukemia-associated fibroblasts. Cancer Cell Int (2021) 21(1):177. doi: 10.1186/s12935-021-01870-7
148. Zhu Y, Yang R, Gao J, Zhang Y, Zhang G, Gu L. Establishment and characterization of a novel childhood acute lymphoblastic leukemia cell line, HXEX-ALL1, with chromosome 9p and 17p deletions. Cancer Cell Int (2019) 19:113. doi: 10.1186/s12935-019-0834-x
149. Kim Y, Jekarl DW, Kim J, Kwon A, Choi H, Lee S, et al. Genetic and epigenetic alterations of bone marrow stromal cells in myelodysplastic syndrome and acute myeloid leukemia patients. Stem Cell Res (2015) 14(2):177–84. doi: 10.1016/j.scr.2015.01.004
150. Blau O, Baldus CD, Hofmann WK, Thiel G, Nolte F, Burmeister T, et al. Mesenchymal stromal cells of myelodysplastic syndrome and acute myeloid leukemia patients have distinct genetic abnormalities compared with leukemic blasts. Blood (2011) 118(20):5583–92. doi: 10.1182/blood-2011-03-343467
151. Blau O, Hofmann WK, Baldus CD, Thiel G, Serbent V, Schümann E, et al. Chromosomal aberrations in bone marrow mesenchymal stroma cells from patients with myelodysplastic syndrome and acute myeloblastic leukemia. Exp Hematol (2007) 35(2):221–9. doi: 10.1016/j.exphem.2006.10.012
152. Yeh SP, Lo WJ, Lin CL, Liao YM, Lin CY, Bai LY, et al. Anti-leukemic therapies induce cytogenetic changes of human bone marrow-derived mesenchymal stem cells. Ann Hematol (2012) 91(2):163–72. doi: 10.1007/s00277-011-1254-8
153. Huang JC, Basu SK, Zhao X, Chien S, Fang M, Oehler VG, et al. Mesenchymal stromal cells derived from acute myeloid leukemia bone marrow exhibit aberrant cytogenetics and cytokine elaboration. Blood Cancer J (2015) 5:e302. doi: 10.1038/bcj.2015.17
154. Campioni D, Bardi MA, Cavazzini F, Tammiso E, Pezzolo E, Pregnolato E, et al. Cytogenetic and molecular cytogenetic profile of bone marrow-derived mesenchymal stromal cells in chronic and acute lymphoproliferative disorders. Ann Hematol (2012) 91(10):1563–77. doi: 10.1007/s00277-012-1500-8
155. Diaz de la Guardia R, Lopez-Millan B, Lavoie JR, Bueno C, Castaño J, Gómez-Casares M, et al. Detailed characterization of mesenchymal Stem/Stromal cells from a Large cohort of AML patients demonstrates a definitive link to treatment outcomes. Stem Cell Rep (2017) 8(6):1573–86. doi: 10.1016/j.stemcr.2017.04.019
156. Flores-Figueroa E, Montesinos JJ, Flores-Guzmán P, Gutiérrez-Espíndola G, Arana-Trejo RM, Castillo-Medina S, et al. Functional analysis of myelodysplastic syndromes-derived mesenchymal stem cells. Leuk Res (2008) 32(9):1407–16. doi: 10.1016/j.leukres.2008.02.013
157. Xie J, Chen J, Wang B, He X, Huang H. Bone mesenchymal stromal cells exhibit functional inhibition but no chromosomal aberrations in chronic myelogenous leukemia. Oncol Lett (2019) 17(1):999–1007. doi: 10.3892/ol.2018.9681
158. Gunsilius E, Duba HC, Petzer AL, Kähler CM, Grünewald K, Stockhammer G, et al. Evidence from a leukaemia model for maintenance of vascular endothelium by bone-marrow-derived endothelial cells. Lancet (2000) 355(9216):1688–91. doi: 10.1016/S0140-6736(00)02241-8
159. Zhou Y, Bian S, Zhou X, Cui Y, Wang W, Wen L, et al. Single-cell multiomics sequencing reveals prevalent genomic alterations in tumor stromal cells of human colorectal cancer. Cancer Cell (2020) 38(6):818–28.e5. doi: 10.1016/j.ccell.2020.09.015
160. Novo D, Heath N, Mitchell L, Caligiuri G, MacFarlane A, Reijmer D, et al. Mutant p53s generate pro-invasive niches by influencing exosome podocalyxin levels. Nat Commun (2018) 9(1):5069. doi: 10.1038/s41467-018-07339-y
161. Tuhkanen H, Anttila M, Kosma VM, Heinonen S, Juhola M, Helisalmi S, et al. Frequent gene dosage alterations in stromal cells of epithelial ovarian carcinomas. Int J Cancer (2006) 119(6):1345–53. doi: 10.1002/ijc.21785
162. Patocs A, Zhang L, Xu Y, Weber F, Caldes T, Mutter GL, et al. Breast-cancer stromal cells with TP53 mutations and nodal metastases. N Engl J Med (2007) 357(25):2543–51. doi: 10.1056/NEJMoa071825
163. Moinfar F, Man YG, Arnould L, Bratthauer GL, Ratschek M, Tavassoli FA. Concurrent and independent genetic alterations in the stromal and epithelial cells of mammary carcinoma: implications for tumorigenesis. Cancer Res (2000) 60(9):2562–6.
164. Sakemura R, Hefazi M, Siegler EL, Cox MJ, Larson DP, Hansen MJ, et al. Targeting cancer-associated fibroblasts in the bone marrow prevents resistance to CART-cell therapy in multiple myeloma. Blood (2022) 139(26):3708–21. doi: 10.1182/blood.2021012811
165. Özdemir BC, Pentcheva-Hoang T, Carstens JL, Zheng X, Wu CC, Simpson TR, et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell (2014) 25(6):719–34. doi: 10.1016/j.ccr.2014.04.005
166. Rhim AD, Oberstein PE, Thomas DH, Mirek ET, Palermo CF, Sastra SA, et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell (2014) 25(6):735–47. doi: 10.1016/j.ccr.2014.04.021
167. Zhang Y, Lazarus J, Steele NG, Yan W, Lee HJ, Nwosu ZC, et al. Regulatory T-cell depletion alters the tumor microenvironment and accelerates pancreatic carcinogenesis. Cancer Discovery (2020) 10(3):422–39. doi: 10.1158/2159-8290.CD-19-0958
168. Paulsson J, Micke P. Prognostic relevance of cancer-associated fibroblasts in human cancer. Semin Cancer Biol (2014) 25:61–8. doi: 10.1016/j.semcancer.2014.02.006
169. Brechbuhl HM, Finlay-Schultz J, Yamamoto TM, Gillen AE, Cittelly DM, Tan AC, et al. Fibroblast subtypes regulate responsiveness of luminal breast cancer to estrogen. Clin Cancer Res (2017) 23(7):1710–21. doi: 10.1158/1078-0432.CCR-15-2851
170. Li C, Teixeira AF, Zhu HJ, Ten Dijke P. Cancer associated-fibroblast-derived exosomes in cancer progression. Mol Cancer (2021) 20(1):154. doi: 10.1186/s12943-021-01463-y
171. Kadel D, Zhang Y, Sun HR, Zhao Y, Dong QZ, Qin LX. Current perspectives of cancer-associated fibroblast in therapeutic resistance: potential mechanism and future strategy. Cell Biol Toxicol (2019) 35(5):407–21. doi: 10.1007/s10565-019-09461-z
172. Hanahan D. Hallmarks of cancer: New dimensions. Cancer Discovery (2022) 12(1):31–46. doi: 10.1158/2159-8290.CD-21-1059
173. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144(5):646–74. doi: 10.1016/j.cell.2011.02.013
174. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell (2000) 100(1):57–70. doi: 10.1016/s0092-8674(00)81683-9
175. Welt S, Divgi CR, Scott AM, Garin-Chesa P, Finn RD, Graham M, et al. Antibody targeting in metastatic colon cancer: a phase I study of monoclonal antibody F19 against a cell-surface protein of reactive tumor stromal fibroblasts. J Clin Oncol (1994) 12(6):1193–203. doi: 10.1200/JCO.1994.12.6.1193
176. Öhlund D, Handly-Santana A, Biffi G, Elyada E, Almeida AS, Ponz-Sarvise M, et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J Exp Med (2017) 214(3):579–96. doi: 10.1084/jem.20162024
177. Biffi G, Oni TE, Spielman B, Hao Y, Elyada E, Park Y, et al. IL1-induced JAK/STAT signaling is antagonized by TGFβ to shape CAF heterogeneity in pancreatic ductal adenocarcinoma. Cancer Discovery (2019) 9(2):282–301. doi: 10.1158/2159-8290.CD-18-0710
178. Friedman G, Levi-Galibov O, David E, Bornstein C, Giladi A, Dadiani M, et al. Cancer-associated fibroblast compositions change with breast cancer progression linking the ratio of S100A4(+) and PDPN(+) CAFs to clinical outcome. Nat Cancer (2020) 1(7):692–708. doi: 10.1038/s43018-020-0082-y
179. Nurmik M, Ullmann P, Rodriguez F, Haan S, Letellier E. In search of definitions: Cancer-associated fibroblasts and their markers. Int J Cancer (2020) 146(4):895–905. doi: 10.1002/ijc.32193
180. Li H, Courtois ET, Sengupta D, Tan Y, Chen KH, Goh J, et al. Reference component analysis of single-cell transcriptomes elucidates cellular heterogeneity in human colorectal tumors. Nat Genet (2017) 49(5):708–18. doi: 10.1038/ng.3818
181. Zhang R, Zong J, Peng Y, Shi J, Du X, Liu H, et al. GPR30 knockdown weakens the capacity of CAF in promoting prostate cancer cell invasion via reducing macrophage infiltration and M2 polarization. J Cell Biochem (2021). doi: 10.1002/jcb.29938
182. Francescone R, Barbosa Vendramini-Costa D, Franco-Barraza J, Wagner J, Muir A, Lau AN, et al. Netrin G1 promotes pancreatic tumorigenesis through cancer-associated fibroblast-driven nutritional support and immunosuppression. Cancer Discovery (2021) 11(2):446–79. doi: 10.1158/2159-8290.CD-20-0775
183. Ogier C, Colombo PE, Bousquet C, Canterel-Thouennon L, Sicard P, Garambois V, et al. Targeting the NRG1/HER3 pathway in tumor cells and cancer-associated fibroblasts with an anti-neuregulin 1 antibody inhibits tumor growth in pre-clinical models of pancreatic cancer. Cancer Lett (2018) 432:227–36. doi: 10.1016/j.canlet.2018.06.023
184. Garcia PE, Adoumie M, Kim EC, Zhang Y, Scales MK, El-Tawil YS, et al. Differential contribution of pancreatic fibroblast subsets to the pancreatic cancer stroma. Cell Mol Gastroenterol Hepatol (2020) 10(3):581–99. doi: 10.1016/j.jcmgh.2020.05.004
185. Joo EH, Bae JH, Park J, Bang YJ, Han J, Gulati N, et al. Deconvolution of adult T-cell Leukemia/Lymphoma with single-cell RNA-seq using frozen archived skin tissue reveals new subset of cancer-associated fibroblast. Front Immunol (2022) 13:856363. doi: 10.3389/fimmu.2022.856363
186. Wang LC, Lo A, Scholler J, Sun J, Majumdar RS, Kapoor V, et al. Targeting fibroblast activation protein in tumor stroma with chimeric antigen receptor T cells can inhibit tumor growth and augment host immunity without severe toxicity. Cancer Immunol Res (2014) 2(2):154–66. doi: 10.1158/2326-6066.CIR-13-0027
187. Bughda R, Dimou P, D’Souza RR, Klampatsa A. Fibroblast activation protein (FAP)-targeted CAR-T cells: Launching an attack on tumor stroma. Immunotargets Ther (2021) 10:313–23. doi: 10.2147/ITT.S291767
188. Hiltbrunner S, Britschgi C, Schuberth P, Bankel L, Nguyen-Kim T, Gulati P, et al. Local delivery of CAR T cells targeting fibroblast activation protein is safe in patients with pleural mesothelioma: first report of FAPME, a phase I clinical trial. Ann Oncol (2021) 32(1):120–1. doi: 10.1016/j.annonc.2020.10.474
189. Gross G, Waks T, Eshhar Z. Expression of immunoglobulin-t-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc Natl Acad Sci U.S.A. (1989) 86(24):10024–8. doi: 10.1073/pnas.86.24.10024
190. Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in children and young adults with b-cell lymphoblastic leukemia. N Engl J Med (2018) 378(5):439–48. doi: 10.1056/NEJMoa1709866
191. Halford Z, Anderson MK, Bennett LL, Moody J. Tisagenlecleucel in acute lymphoblastic leukemia: A review of the literature and practical considerations. Ann Pharmacother (2021) 55(4):466–79. doi: 10.1177/1060028020948165
Keywords: bone marrow, tumor microenvironment, leukemia, relapsed/refractory, cancer associated fibroblasts
Citation: Gu L, Liao P and Liu H (2022) Cancer-associated fibroblasts in acute leukemia. Front. Oncol. 12:1022979. doi: 10.3389/fonc.2022.1022979
Received: 19 August 2022; Accepted: 01 December 2022;
Published: 19 December 2022.
Edited by:
Spiros Vlahopoulos, University of Athens, GreeceReviewed by:
Chao Chen, Emory University, United StatesCopyright © 2022 Gu, Liao and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ling Gu, Z3VsaW5nQHNjdS5lZHUuY24=; Ping Liao, cGluZ19saWFvQG5uaS5jb20uc2c=; Hanmin Liu, bGl1aG1Ac2N1LmVkdS5jbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.