 Jonathan Paolino1
Jonathan Paolino1 Barbara Degar
Barbara Degar- 1Department of Pediatric Oncology, Dana-Farber Cancer Institute, Harvard Medical School, Boston, MA, United States
- 2Division of Hematology, Brigham and Women’s Hospital, Harvard Medical School, Boston, MA, United States
Hemophagocytic lymphohistiocytosis (HLH) is a syndrome of multiorgan system dysfunction that is caused by hypercytokinemia and persistent activation of cytotoxic T lymphocytes and macrophages. A nearly ubiquitous finding and a diagnostic criterion of HLH is the presence of cytopenias in ≥ 2 cell lines. The mechanism of cytopenias in HLH is multifactorial but appears to be predominantly driven by suppression of hematopoiesis by pro-inflammatory cytokines and, to some extent, by consumptive hemophagocytosis. Recognition of cytopenias as a manifestation of HLH is an important consideration for patients with bone marrow failure of unclear etiology.
Introduction
Hemophagocytic lymphohistiocytosis (HLH) is a rare syndrome in which immune dysregulation and severe pathologic inflammation result in multiorgan dysfunction. While affected patients share certain clinical features, the inciting event, nature of external predisposing factors, and presence of underlying genetic predisposition vary widely. In HLH, the inability of natural killer (NK) and CD8+ cytotoxic T lymphocytes (CTLs) to provide critical negative feedback in response to an immunologic trigger leads to uncontrolled activation of CTLs and macrophages and initiation of a “cytokine storm” (1–3). Peripheral blood cytopenias are a universal feature of HLH and are an important diagnostic criterion for the syndrome (4–6). The causes of bone marrow failure in HLH are multifactorial, heterogeneous, and incompletely understood.
Since it was first described in 1939 by Scott and Robb-Smith, HLH has remained diagnostically challenging with highly variable clinical presentation and no single pathognomonic feature (7–10). Historically, HLH has been dichotomized as “primary” (familial or FHL) for those with a family history of HLH or predisposing genetic mutation, or “secondary” for those without an identified mutation but with an underlying infectious, rheumatologic, or malignant disease (6, 11). There is, however, considerable overlap between the two groups, with increasing understanding of the role of novel mutations and monoallelic variants in the pathogenesis of HLH (12–14). In addition, FHL may be initiated by an inciting infectious or inflammatory insult, further blurring the line between “primary” and “secondary” groups.
The constellation of clinical findings and laboratory abnormalities observed in HLH reflect the common pathway through which HLH progresses. The Histiocyte Society developed standard diagnostic criteria for the disorder and undertook the HLH94 clinical trial with the goal of improving survival of children with familial HLH, a uniformly fatal diagnosis at that time (Table 1) (17). However, the suitability of these criteria for diagnosing various forms of secondary HLH is not established. Modified criteria for patients with rheumatologic disease and underlying malignancy have been proposed (16, 18, 19). A scoring system termed the Optimized HLH inflammatory (OHI) index was recently developed to aid in the diagnosis and management of HLH in the context of hematologic malignancy (20).
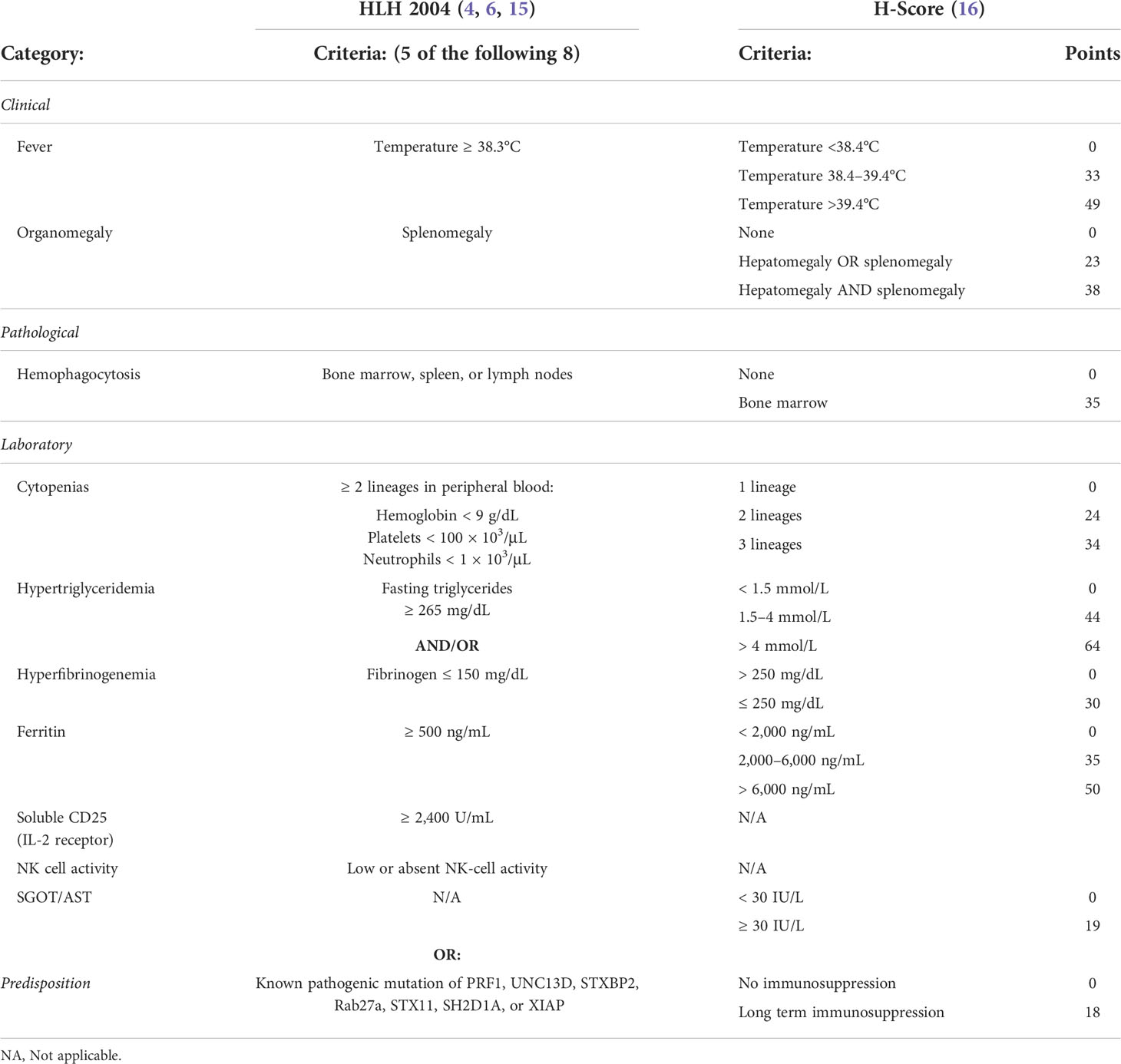
Table 1 Diagnostic criteria for HLH.
Peripheral blood findings
Cytopenias affecting at least two cell lines are a cardinal feature of HLH. Thrombocytopenia is almost always present (4, 21). The platelet count may initially be normal or modestly depressed; however, it often falls as the disease progresses (22). Normocytic anemia with reticulocytopenia is also common. Leukopenia and neutropenia are more variably present (22). For example, 92% of children enrolled on the HLH2004 trial demonstrated bi-cytopenia (4). Similarly, among 775 adult patients with HLH, significant cytopenias were present in the majority: platelets < 100 × 103/μL in 78% and < 10 × 103/μL in 6%; hemoglobin of < 9 g/dL in 67% and < 7 g/dL in 22%; absolute neutrophil count (ANC) < 1 × 103/μL in 42% and < 0.5 × 103/μL in 23% (21).
Bone marrow findings
In patients with HLH, the bone marrow demonstrates diffuse histiocytic infiltration, histiocyte hyperplasia, and variable numbers of cytotoxic T-cells (9, 22). Hemophagocytosis, the pathologic finding of activated macrophages engulfing erythrocytes, leukocytes, platelets, and their precursor cells, is variably present (Figure 1). This process occurs not just in the bone marrow but throughout the reticuloendothelial system, including in the spleen, liver, and lymph nodes. While the finding of hemophagocytosis in bone marrow or tissue supports the diagnosis of HLH in the proper clinical context, it is neither essential for the diagnosis nor pathognomonic. In a series of 122 children with HLH from the Histiocyte Society’s International Registry, only 75% had evidence of hemophagocytosis at diagnosis (23). Conversely, hemophagocytosis may be present in patients without HLH. In one study, among 107 adult patients who died of multiorgan failure unrelated to HLH, none of whom fulfilled standard HLH diagnostic criteria, 69 (64.5%) demonstrated histiocytic hyperplasia and hemophagocytosis on postmortem bone marrow analysis (24). Because of its non-specific nature, hemophagocytosis must be carefully considered in the context of other clinical findings.
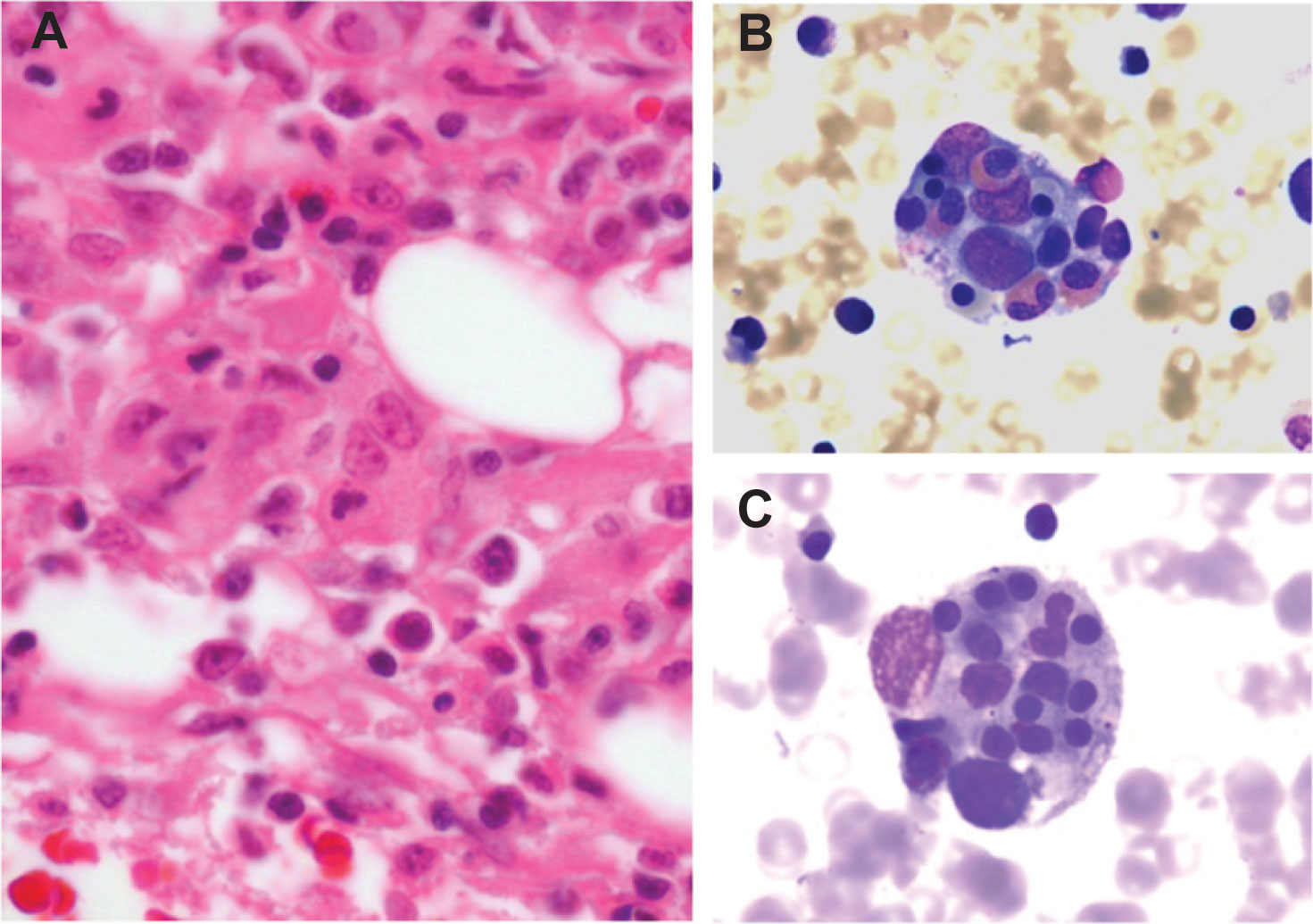
Figure 1 Histopathology of HLH (A) Histopathology of a bone marrow section in HLH demonstrates marrow infiltration by macrophages (hematoxylin and eosin stained, 400x magnification). (B, C) Bone marrow aspirate stain demonstrates macrophages engulfing hematopoietic cells, including eosinophils and erythroid precursors (Wright-Giemsa stained, 1000x magnification).
Familial HLH
Genetic linkage studies performed on families with HLH led to the discovery of bi-allelic Prf1 mutations as the cause of FHL-2 in 1999 (25). Perforin, encoded by Prf1, is a constituent of cytotoxic granules within CD8+ cytotoxic T cells and NK cells (26). Activated CTLs and NK cells form an immunologic synapse with target cells, such as virally infected or cancer cells, allowing cytotoxic granules to undergo a complex series of events through which they dock, prime, and fuse with the cytoplasmic membrane to release their contents (Figure 2) (27–31). This leads to perforin-dependent pore formation in the target cell membrane and allows serine protease granzymes to induce apoptosis of the target cell (26, 32, 33). Absent or reduced expression of functional perforin impairs effector function and clearance of the inflammatory insult, resulting in persistent activation of this pathway (25).
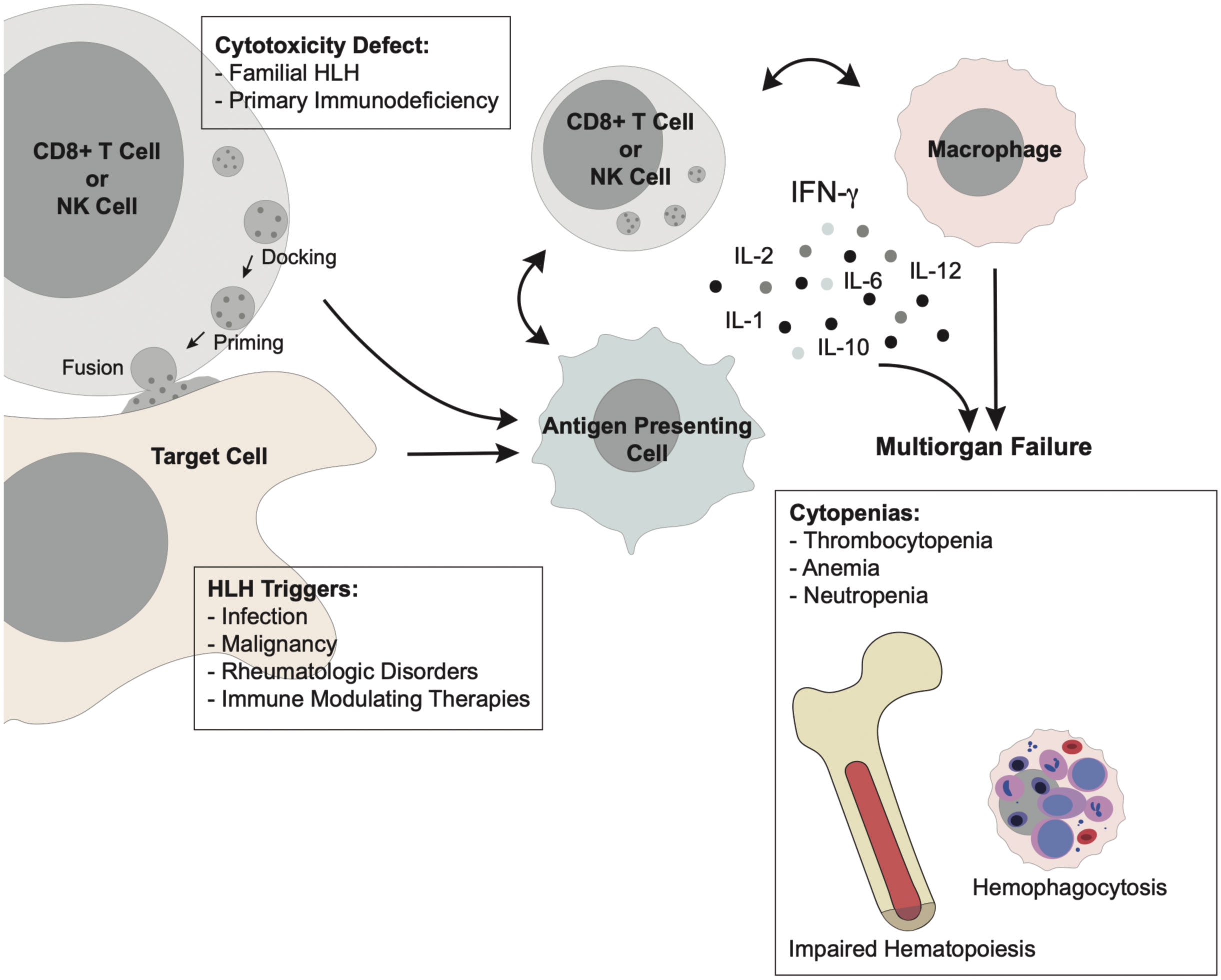
Figure 2 Pathophysiology of HLH Primary HLH arises from defective cytotoxicity in CD8+ T cells or NK cells, allowing persistence of an antigenic stimulus. Secondary HLH arises from unchecked immune activation secondary to infection, malignancy, rheumatologic disorders, or immune-modulating therapy. This immune activation acts through a common pathway in which uncontrolled stimulation of CD8+ T cells and macrophages results in hypercytokinemia which drives the clinical manifestations of multi-organ failure. Cytopenias in the form of neutropenia, anemia, and thrombocytopenia are ubiquitous in HLH downstream of excessive production of IFN- γ and other inflammatory cytokines. This “cytokine storm” suppresses normal hematopoiesis and results in diffuse hemophagocytosis by activated macrophages.
The finding of Prf1 mutations in families with FHL-2 was followed by the discovery of the genetic basis for FHL-3, FHL-4, and FHL-5, which are caused by mutations affecting expression or function of Munc13-4, Syntaxin11, and Munc18-2, respectively (Table 2) (34–36). These proteins are necessary for docking, priming, and fusion of cytotoxic granules with the T or NK cell membrane (34–36). Other genetic syndromes associated with defective trafficking of cytotoxic granules, including Chediak-Higashi syndrome, Griscelli syndrome type 2, Hermansky-Pudlak syndrome type 2, and RhoG deficiency, are also associated with HLH (Table 2) (1, 37–40, 46).
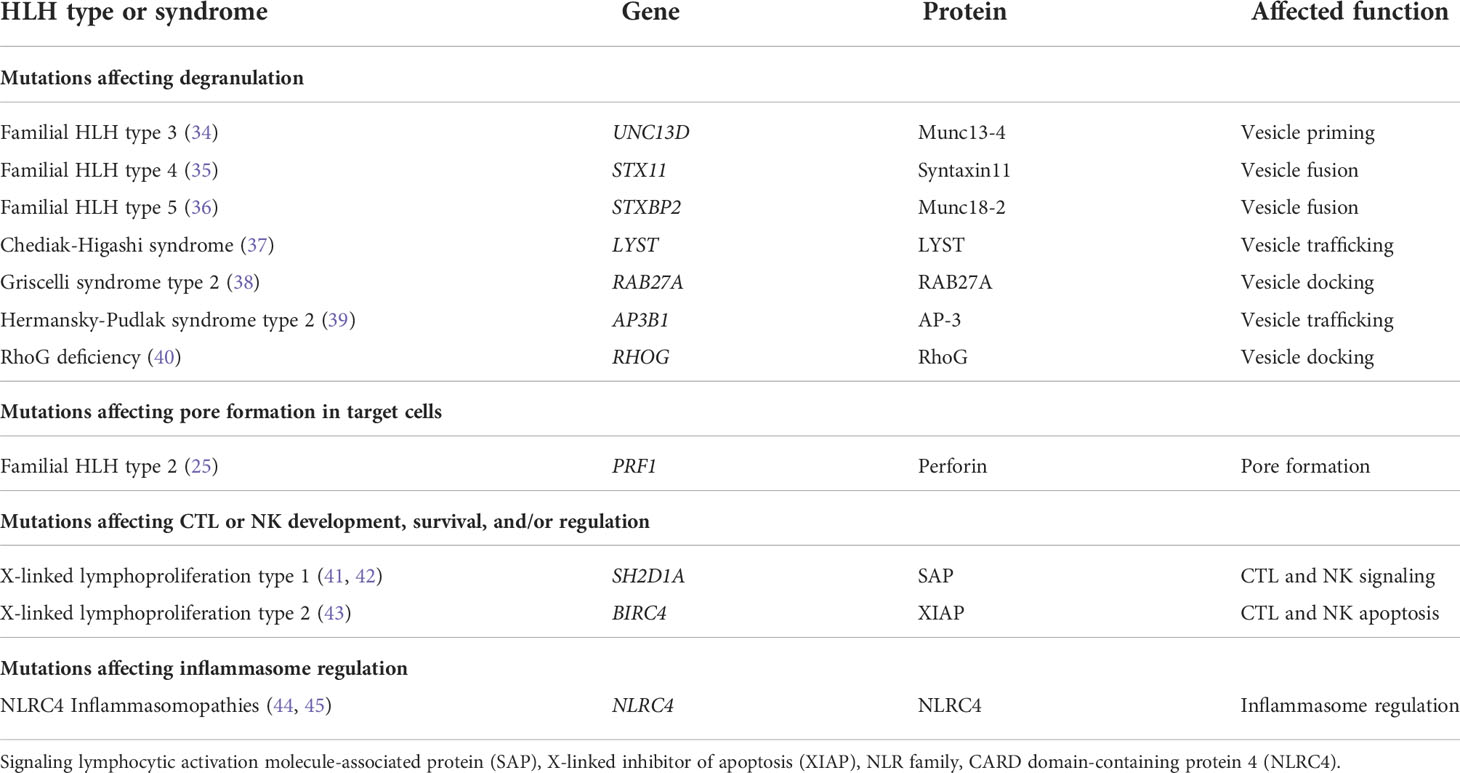
Table 2 Genetic mutations associated with primary HLH.
X-linked lymphoproliferative disease type 1 (XLP1) is caused by hemizygous mutations in signaling lymphocytic activation molecule-associated protein (SAP) (41, 42). SAP plays a critical role in T cell response through interactions with signaling lymphocyte activation molecules (SLAM) family receptors (47, 48). Patients with XLP1 develop severe, overwhelming immune dysregulation in the setting of Epstein-Barr virus (EBV) infection (47, 48). In X-linked lymphoproliferative disease type 2 (XLP2), mutations in X-linked inhibitor of apoptosis (XIAP) lead to reduced survival of T and NK cells (43, 49). Patients with XIAP deficiency have a high incidence of HLH (43, 49). Collectively, these disorders result in ineffective cytotoxicity of CTLs and NK cells which results in persistence of the antigenic stimulus and predisposes to the pathogenesis of HLH.
In patients with HLH, uncontrolled activation of immune cells leads to production of pro-inflammatory cytokines, which in turn act to amplify immune dysregulation in an unchecked positive feedback loop. Multiple cytokines have been implicated in the pathogenesis of HLH, including interferon-γ (IFN- γ), interleukin (IL)-1, IL-2, IL-6, IL-12, IL-18, and tumor necrosis factor-alpha (TNF-α) (50–52). In FHL, interferon-γ has emerged as a key driver of disease activity (2, 53–55). Importantly, further insight into the role of individual cytokines in HLH has potential therapeutic implications (51, 56, 57). For example, neutralization of IFN-γ with emapalumab led to its approval for treatment of FHL in patients with refractory HLH or intolerance to standard chemotherapy (55).
Secondary HLH
Unlike FHL, which typically presents in infancy or early childhood, secondary HLH is far more prevalent in the older child and adult population. Secondary HLH arises in the setting of an immune stimulus associated with a malignancy, infection, rheumatologic disorder, primary immunodeficiency syndrome, and/or immune-modulating treatment, as outlined in Table 3 (1, 6, 58). The two most common triggers of secondary HLH are infection and malignancy (1, 21, 66–68). Epstein-Barr virus and other herpes viruses are especially frequent; in one study, herpes viruses were identified in 62% of virus-associated HLH cases in adults (21, 59). Lymphoma and leukemia underlie the vast majority of cases of malignancy-associated HLH (19, 58, 69). In addition, several newly developed cancer therapies, such as immune checkpoint inhibitors, monoclonal antibodies, and chimeric antigen receptor T-cells, may also lead to hyperinflammation and/or cytokine release syndrome that may resemble HLH (58).

Table 3 Secondary HLH.
Patterns of T cell activation differ between primary and secondary HLH, reflecting potential differences in the underlying pathogenesis (70). In one study of T cell activation patterns, patients with primary or virus-associated secondary HLH were found to have significantly higher expression of HLA-DR in CD8 + T cells, a marker of T-cell activation, when compared to those with secondary HLH without a viral trigger (64.4% and 61.5% vs. 21%, respectively) (70). Cytokine profiles are frequently similar but may vary based on the underlying inflammatory trigger (51, 71). IFN- γ, TNF-α, IL-10, and IL-18 are commonly elevated in secondary HLH (72–75). Despite the etiologic heterogeneity of secondary HLH, the pathophysiology follows a similar common pathway in which persistent antigenic stimulation leads to an amplified and unchecked immune response, excessive cytokine production, and the resultant development of hallmark features of HLH (1, 50, 57).
Cytopenias in HLH
Many factors contribute to the development of cytopenias in patients with HLH, including impairment of hematopoiesis mediated through the action of pro-inflammatory cytokines, consumption of hematopoietic progenitors due to hemophagocytosis throughout the reticuloendothelial system, shortened survival of blood cells due to hepatosplenomegaly and/or disseminated intravascular coagulopathy, co-existing viral infection, marrow invasion by cancer, and treatment-related myelosuppression (21, 51, 76–79). Despite the complex interactions of these factors, two distinct drivers are attributable to the phenomenon of HLH itself: impaired hematopoiesis as a result of hypercytokinemia and consumptive hemophagocytosis by activated macrophages.
Impaired hematopoiesis
Suppression of hematopoiesis by interferon-γ and other inflammatory cytokines is well described (79–82). Indeed, IFN-γ has been implicated in the pathogenesis of aplastic anemia (83, 84). In the setting of inflammation, interferon-γ exhibits both stimulating and suppressive effects on hematopoietic precursors in a lineage-dependent manner (80, 85). IFN-γ plays a key role in myelopoiesis, directing differentiation to either monocyte or neutrophil populations (86). In contrast, IFN-γ exhibits a predominantly suppressive effect on hematopoietic erythroid progenitors and disrupts thrombopoietin signaling in hematopoietic stem cell precursors (87–89). Interestingly, interferon-γ and TNF-α, two cytokines frequently elevated in patients with HLH, have demonstrated the potential to synergistically suppress bone marrow erythroid and multipotential progenitor cells (79). The role of inflammatory cytokines as an etiology of bone marrow failure has substantial implications in the context of the hypercytokinemia observed in patients with HLH.
In vivo preclinical models of HLH support the role of IFN-γ in the development of impaired hematopoiesis. In one study, IFN-γ knockout abrogated the development of anemia in a murine model of toll-like receptor 9 (TLR9)-induced fulminant macrophage activating syndrome (90). Interestingly, in this study, both IFN-γ wild-type and knockout mice developed an MAS/HLH-like syndrome following exposure to a TLR9 agonist, suggesting that the clinical phenotype was not mediated by IFN-γ alone. The IFN- γ knockout mice, however, did not develop anemia despite the presence of hemophagocytosis. These mice were found to have compensatory splenic erythroid precursor production, leading authors to conclude that dyserythropoiesis, not hemophagocytosis, was primarily responsible for anemia in this model (90). Further supporting the role of interferon-γ in the development of cytopenias are the findings that anti-interferon-γ antibodies correct peripheral blood cytopenias and histiocytic infiltration of the marrow, liver, and spleen in perforin and Rab27a-deficient mice (2, 53).
Consumptive hemophagocytosis
Hemophagocytosis is frequently observed in HLH, although it is neither specific nor required for the diagnosis. The role of hemophagocytosis in the development of pancytopenia in HLH is uncertain. In one animal model, sustained exposure to IFN- γ in wild-type mice induced the development of dose-dependent normocytic anemia and compensatory reticulocytosis (54). In this model, mice were infused with IFN- γ over a five-day period, during which anemia became apparent within 48 hours. There was no change in red blood cell morphology or evidence of a significant hemolytic process (54). Anemia was accompanied by thrombocytopenia and leukopenia, and was associated with diffuse hemophagocytosis. The temporal relationship between the start of IFN- γ infusion and the development of anemia, as well as the accompanying reticulocytosis, led the authors to conclude that the cytopenias observed in this model were likely to be predominantly the result of a consumptive process secondary to acute inflammation rather than suppression of hematopoiesis (54). These findings further implicate IFN- γ as a key driver of the cytopenias observed in patients with HLH.
Conclusions
Hemophagocytic lymphohistiocytosis (HLH) is a hyperinflammatory syndrome that results from persistent activation of cytotoxic T lymphocytes and macrophages. The underlying causes of HLH are heterogeneous; however, peripheral blood cytopenias are almost universally present. The mechanism of cytopenias is multifactorial and may be exacerbated by concomitant effects by factors such as infection of hematopoietic progenitors, bone marrow infiltration, and myelosuppressive therapy. Inflammatory cytokines, especially interferon-γ, play a significant role in suppressing hematopoiesis in HLH, leading to cytopenias in animal models and patients. To a variable extent, consumptive hemophagocytosis throughout the reticuloendothelial system by macrophages contributes. Importantly, recognition of cytopenias as a manifestation of HLH is essential for patients with bone marrow failure of unclear etiology.
Author contributions
The manuscript was written by JP and extensively edited by NB and BD. All authors contributed to the article and approved the submitted version.
Funding
JP was supported by NIH 5T32HL007574-38.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Al-Samkari H, Berliner N. Hemophagocytic lymphohistiocytosis. Annu Rev Pathol (2018) 13:27–49. doi: 10.1146/annurev-pathol-020117-043625
2. Jordan MB, Hildeman D, Kappler J, Marrack P. An animal model of hemophagocytic lymphohistiocytosis (HLH): CD8+ T cells and interferon gamma are essential for the disorder. Blood (2004) 104(3):735–43. doi: 10.1182/blood-2003-10-3413
3. Fajgenbaum DC, June CH. Cytokine storm. New Engl J Med (2020) 383(23):2255–73. doi: 10.1056/NEJMra2026131
4. Bergsten E, Horne A, Aricó M, Astigarraga I, Egeler RM, Filipovich AH, et al. Confirmed efficacy of etoposide and dexamethasone in HLH treatment: Long-term results of the cooperative HLH-2004 study. Blood (2017) 130(25):2728–38. doi: 10.1182/blood-2017-06-788349
5. Trottestam H, Horne A, Aricò M, Egeler RM, Filipovich AH, Gadner H, et al. Chemoimmunotherapy for hemophagocytic lymphohistiocytosis: Long-term results of the HLH-94 treatment protocol. Blood (2011) 118(17):4577–84. doi: 10.1182/blood-2011-06-356261
6. Jordan MB, Allen CE, Greenberg J, Henry M, Hermiston ML, Kumar A, et al. Challenges in the diagnosis of hemophagocytic lymphohistiocytosis: Recommendations from the north American consortium for histiocytosis (NACHO). Pediatr Blood Canc (2019) 66(11):e27929. doi: 10.1002/pbc.27929
7. Scott R R-SA.Histiocytic medullary reticulocytosis. Lancet (1939) 2:194−8. doi: 10.1016/S0140-6736(00)61951-7
8. Rosado FG, Rinker EB, Plummer WD, Dupont WD, Spradlin NM, Reichard KK, et al. The diagnosis of adult-onset haemophagocytic lymphohistiocytosis: Lessons learned from a review of 29 cases of bone marrow haemophagocytosis in two large academic institutions. J Clin Pathol (2016) 69(9):805–9. doi: 10.1136/jclinpath-2015-203577
9. Kikuchi A, Singh K, Gars E, Ohgami RS. Pathology updates and diagnostic approaches to haemophagocytic lymphohistiocytosis. Histopathology (2022) 80(4):616–26. doi: 10.1111/his.14591
10. Nikiforow S, Berliner N. The unique aspects of presentation and diagnosis of hemophagocytic lymphohistiocytosis in adults. Hematol Am Soc Hematol Educ Program (2015) 2015:183–9. doi: 10.1182/asheducation-2015.1.183
11. Emile JF, Abla O, Fraitag S, Horne A, Haroche J, Donadieu J, et al. Revised classification of histiocytoses and neoplasms of the macrophage-dendritic cell lineages. Blood (2016) 127(22):2672–81. doi: 10.1182/blood-2016-01-690636
12. Zhang K, Jordan MB, Marsh RA, Johnson JA, Kissell D, Meller J, et al. Hypomorphic mutations in PRF1, MUNC13-4, and STXBP2 are associated with adult-onset familial HLH. Blood (2011) 118(22):5794–8. doi: 10.1182/blood-2011-07-370148
13. Kaufman KM, Linghu B, Szustakowski JD, Husami A, Yang F, Zhang K, et al. Whole-exome sequencing reveals overlap between macrophage activation syndrome in systemic juvenile idiopathic arthritis and familial hemophagocytic lymphohistiocytosis. Arthritis Rheumatol (2014) 66(12):3486–95. doi: 10.1002/art.38793
14. Carvelli J, Piperoglou C, Farnarier C, Vely F, Mazodier K, Audonnet S, et al. Functional and genetic testing in adults with HLH reveals an inflammatory profile rather than a cytotoxicity defect. Blood (2020) 136(5):542–52. doi: 10.1182/blood.2019003664
15. Henter JI, Horne A, Aricó M, Egeler RM, Filipovich AH, Imashuku S, et al. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Canc (2007) 48(2):124–31. doi: 10.1002/pbc.21039
16. Fardet L, Galicier L, Lambotte O, Marzac C, Aumont C, Chahwan D, et al. Development and validation of the HScore, a score for the diagnosis of reactive hemophagocytic syndrome. Arthritis Rheumatol (2014) 66(9):2613–20. doi: 10.1002/art.38690
17. Henter JI, Elinder G, Ost A. Diagnostic guidelines for hemophagocytic lymphohistiocytosis. FHL Study Group Histiocyte Society Semin Oncol (1991) 18(1):29–33.
18. Ravelli A, Minoia F, Davì S, Horne A, Bovis F, Pistorio A, et al. 2016 Classification criteria for macrophage activation syndrome complicating systemic juvenile idiopathic arthritis: A European league against Rheumatism/American college of Rheumatology/Paediatric rheumatology international trials organisation collaborative initiative. Arthritis Rheumatol (2016) 68(3):566–76. doi: 10.1002/art.39332
19. Daver N, McClain K, Allen CE, Parikh SA, Otrock Z, Rojas-Hernandez C, et al. A consensus review on malignancy-associated hemophagocytic lymphohistiocytosis in adults. Cancer (2017) 123(17):3229–40. doi: 10.1002/cncr.30826
20. Zoref-Lorenz A, Murakami J, Hofstetter L, Iyer S, Alotaibi AS, Mohamed SF, et al. An improved index for diagnosis and mortality prediction in malignancy-associated hemophagocytic lymphohistiocytosis. Blood (2022) 139(7):1098–110. doi: 10.1182/blood.2021012764
21. Ramos-Casals M, Brito-Zerón P, López-Guillermo A, Khamashta MA, Bosch X. Adult haemophagocytic syndrome. Lancet (9927) 2014:1503–16:383. doi: 10.1016/S0140-6736(13)61048X
22. Orkin SH. Nathan And oski’s hematology and oncology of infancy and childhood. Eighth edition. Philadelphia, PA: Elsevier Saunders (2015).
23. Aricò M, Janka G, Fischer A, Henter JI, Blanche S, Elinder G, et al. Hemophagocytic lymphohistiocytosis. report of 122 children from the international registry. FHL study group of the histiocyte society. Leukemia (1996) 10(2):197–203.
24. Strauss R, Neureiter D, Westenburger B, Wehler M, Kirchner T, Hahn EG. Multifactorial risk analysis of bone marrow histiocytic hyperplasia with hemophagocytosis in critically ill medical patients–a postmortem clinicopathologic analysis. Crit Care Med (2004) 32(6):1316–21. doi: 10.1097/01.CCM.0000127779.24232.15
25. Stepp SE, Dufourcq-Lagelouse R, Le Deist F, Bhawan S, Certain S, Mathew PA, et al. Perforin gene defects in familial hemophagocytic lymphohistiocytosis. Science (1999) 286(5446):1957–9. doi: 10.1126/science.286.5446.1957
26. Voskoboinik I, Whisstock JC, Trapani JA. Perforin and granzymes: function, dysfunction and human pathology. Nat Rev Immunol (2015) 15(6):388–400. doi: 10.1038/nri3839
27. Russell JH, Ley TJ. Lymphocyte-mediated cytotoxicity. Annu Rev Immunol (2002) 20(1):323–70. doi: 10.1146/annurev.immunol.20.100201.131730
28. Biassoni R, Cantoni C, Pende D, Sivori S, Parolini S, Vitale M, et al. Human natural killer cell receptors and co-receptors. Immunol Rev (2001) 181:203–14. doi: 10.1034/j.1600-065X.2001.1810117.x
29. Basu R, Whitlock BM, Husson J, Le Floc'h A, Jin W, Oyler-Yaniv A, et al. Cytotoxic T cells use mechanical force to potentiate target cell killing. Cell (2016) 165(1):100–10. doi: 10.1016/j.cell.2016.01.021
30. Orange JS. Formation and function of the lytic NK-cell immunological synapse. Nat Rev Immunol (2008) 8(9):713–25. doi: 10.1038/nri2381
31. Stinchcombe JC, Barral DC, Mules EH, Booth S, Hume AN, Machesky LM, et al. Rab27a is required for regulated secretion in cytotoxic T lymphocytes. J Cell Biol (2001) 152(4):825–34. doi: 10.1083/jcb.152.4.825
32. Brennan AJ, Chia J, Trapani JA, Voskoboinik I. Perforin deficiency and susceptibility to cancer. Cell Death Differ (2010) 17(4):607–15. doi: 10.1038/cdd.2009.212
33. Ewen CL, Kane KP, Bleackley RC. A quarter century of granzymes. Cell Death Differ (2012) 19(1):28–35. doi: 10.1038/cdd.2011.153
34. Feldmann J, Callebaut I, Raposo G, Certain S, Bacq D, Dumont C, et al. Munc13-4 is essential for cytolytic granules fusion and is mutated in a form of familial hemophagocytic lymphohistiocytosis (FHL3). Cell (2003) 115(4):461–73. doi: 10.1016/S0092-8674(03)00855-9
35. zur Stadt U, Schmidt S, Kasper B, Beutel K, Diler AS, Henter JI, et al. Linkage of familial hemophagocytic lymphohistiocytosis (FHL) type-4 to chromosome 6q24 and identification of mutations in syntaxin 11. Hum Mol Genet (2005) 14(6):827–34. doi: 10.1093/hmg/ddi076
36. Pagel J, Beutel K, Lehmberg K, Koch F, Maul-Pavicic A, Rohlfs A-K, et al. Distinct mutations in STXBP2 are associated with variable clinical presentations in patients with familial hemophagocytic lymphohistiocytosis type 5 (FHL5). Blood (2012) 119(25):6016–24. doi: 10.1182/blood-2011-12-398958
37. Nagle DL, Karim MA, Woolf EA, Holmgren L, Bork P, Misumi DJ, et al. Identification and mutation analysis of the complete gene for chediak-higashi syndrome. Nat Genet (1996) 14(3):307–11. doi: 10.1038/ng1196-307
38. Ménasché G, Pastural E, Feldmann J, Certain S, Ersoy F, Dupuis S, et al. Mutations in RAB27A cause griscelli syndrome associated with haemophagocytic syndrome. Nat Genet (2000) 25(2):173–6. doi: 10.1038/76024
39. Dell'Acqua F, Saettini F, Castelli I, Badolato R, Notarangelo LD, Rizzari C. Hermansky-pudlak syndrome type II and lethal hemophagocytic lymphohistiocytosis: Case description and review of the literature. J Allergy Clin Immunol Pract (2019) 7(7):2476–8.e5. doi: 10.1016/j.jaip.2019.04.001
40. Kalinichenko A, Perinetti Casoni G, Dupré L, Trotta L, Huemer J, Galgano D, et al. RhoG deficiency abrogates cytotoxicity of human lymphocytes and causes hemophagocytic lymphohistiocytosis. Blood (2021) 137(15):2033–45. doi: 10.1182/blood.2020008738
41. Coffey AJ, Brooksbank RA, Brandau O, Oohashi T, Howell GR, Bye JM, et al. Host response to EBV infection in X-linked lymphoproliferative disease results from mutations in an SH2-domain encoding gene. Nat Genet (1998) 20(2):129–35. doi: 10.1038/2424
42. Nichols KE, Hom J, Gong SY, Ganguly A, Ma CS, Cannons JL, et al. Regulation of NKT cell development by SAP, the protein defective in XLP. Nat Med (2005) 11(3):340–5. doi: 10.1038/nm1189
43. Rigaud S, Fondanèche MC, Lambert N, Pasquier B, Mateo V, Soulas P, et al. XIAP deficiency in humans causes an X-linked lymphoproliferative syndrome. Nature (2006) 444(7115):110–4. doi: 10.1038/nature05257
44. Romberg N, Al Moussawi K, Nelson-Williams C, Stiegler AL, Loring E, Choi M, et al. Mutation of NLRC4 causes a syndrome of enterocolitis and autoinflammation. Nat Genet (2014) 46(10):1135–9. doi: 10.1038/ng.3066
45. Canna SW, de Jesus AA, Gouni S, Brooks SR, Marrero B, Liu Y, et al. An activating NLRC4 inflammasome mutation causes autoinflammation with recurrent macrophage activation syndrome. Nat Genet (2014) 46(10):1140–6. doi: 10.1038/ng.3089
46. Chandrakasan S, Filipovich AH. Hemophagocytic lymphohistiocytosis: advances in pathophysiology, diagnosis, and treatment. J Pediatr (2013) 163(5):1253–9. doi: 10.1016/j.jpeds.2013.06.053
47. Latour S, Veillette A. Molecular and immunological basis of X-linked lymphoproliferative disease. Immunol Rev (2003) 192:212–24. doi: 10.1034/j.1600-065X.2003.00023.x
48. Sharifi R, Sinclair JC, Gilmour KC, Arkwright PD, Kinnon C, Thrasher AJ, et al. SAP mediates specific cytotoxic T-cell functions in X-linked lymphoproliferative disease. Blood (2004) 103(10):3821–7. doi: 10.1182/blood-2003-09-3359
49. Marsh RA, Madden L, Kitchen BJ, Mody R, McClimon B, Jordan MB, et al. XIAP deficiency: A unique primary immunodeficiency best classified as X-linked familial hemophagocytic lymphohistiocytosis and not as X-linked lymphoproliferative disease. Blood (2010) 116(7):1079–82. doi: 10.1182/blood-2010-01-256099
50. Osugi Y, Hara J, Tagawa S, Takai K, Hosoi G, Matsuda Y, et al. Cytokine production regulating Th1 and Th2 cytokines in hemophagocytic lymphohistiocytosis. Blood (1997) 89(11):4100–3. doi: 10.1182/blood.V89.11.4100
51. Fujiwara F, Hibi S, Imashuku S. Hypercytokinemia in hemophagocytic syndrome. Am J Pediatr Hematol Oncol (1993) 15(1):92–8. doi: 10.1097/00043426-199302000-00012
52. Takada H, Nomura A, Ohga S, Hara T. Interleukin-18 in hemophagocytic lymphohistiocytosis. Leuk Lymphoma (2001) 42(1-2):21–8. doi: 10.3109/10428190109097673
53. Pachlopnik Schmid J, Ho CH, Chrétien F, Lefebvre JM, Pivert G, Kosco-Vilbois M, et al. Neutralization of IFNgamma defeats haemophagocytosis in LCMV-infected perforin- and Rab27a-deficient mice. EMBO Mol Med (2009) 1(2):112–24. doi: 10.1002/emmm.200900009
54. Zoller EE, Lykens JE, Terrell CE, Aliberti J, Filipovich AH, Henson PM, et al. Hemophagocytosis causes a consumptive anemia of inflammation. J Exp Med (2011) 208(6):1203–14. doi: 10.1084/jem.20102538
55. Locatelli F, Jordan MB, Allen C, Cesaro S, Rizzari C, Rao A, et al. Emapalumab in children with primary hemophagocytic lymphohistiocytosis. N Engl J Med (2020) 382(19):1811–22. doi: 10.1056/NEJMoa1911326
56. Tang Y, Xu X, Song H, Yang S, Shi S, Wei J, et al. Early diagnostic and prognostic significance of a specific Th1/Th2 cytokine pattern in children with haemophagocytic syndrome. Br J Haematol (2008) 143(1):84–91. doi: 10.1111/j.1365-2141.2008.07298.x
57. Henter JI, Elinder G, Söder O, Hansson M, Andersson B, Andersson U. Hypercytokinemia in familial hemophagocytic lymphohistiocytosis. Blood (1991) 78(11):2918–22. doi: 10.1182/blood.V78.11.2918.2918
58. Setiadi A, Zoref-Lorenz A, Lee CY, Jordan MB, Chen LYC. Malignancy-associated haemophagocytic lymphohistiocytosis. Lancet Haematol (2022) 9(3):e217–e27. doi: 10.1016/S2352-3026(21)00366-5
59. Marsh RA. Epstein-Barr Virus and hemophagocytic lymphohistiocytosis. Front Immunol (2017) 8:1902. doi: 10.3389/fimmu.2017.01902
60. La Rosée P, Horne A, Hines M, von Bahr Greenwood T, Machowicz R, Berliner N, et al. Recommendations for the management of hemophagocytic lymphohistiocytosis in adults. Blood (2019) 133(23):2465–77. doi: 10.1182/blood.2018894618
61. El-Mallawany NK, Curry CV, Allen CE. Haemophagocytic lymphohistiocytosis and Epstein-Barr virus: a complex relationship with diverse origins, expression and outcomes. Br J Haematol (2022) 196(1):31–44. doi: 10.1111/bjh.17638
62. Canna SW, Marsh RA. Pediatric hemophagocytic lymphohistiocytosis. Blood (2020) 135(16):1332–43. doi: 10.1182/blood.2019000936
63. Henderson LA, Cron RQ. Macrophage activation syndrome and secondary hemophagocytic lymphohistiocytosis in childhood inflammatory disorders: Diagnosis and management. Paediatr Drugs (2020) 22(1):29–44. doi: 10.1007/s40272-019-00367-1
64. Tangye SG, Latour S. Primary immunodeficiencies reveal the molecular requirements for effective host defense against EBV infection. Blood. (2020) 135(9):644–55. doi: 10.1182/blood.2019000928
65. Dupré A, Michot JM, Schoeffler A, Frumholtz L, Baroudjian B, Delyon J, et al. Haemophagocytic lymphohistiocytosis associated with immune checkpoint inhibitors: a descriptive case study and literature review. Br J Haematol (2020) 189(5):985–92. doi: 10.1111/bjh.16630
66. Risdall RJ, McKenna RW, Nesbit ME, Krivit W, Balfour HH Jr., Simmons RL, et al. Virus-associated hemophagocytic syndrome: a benign histiocytic proliferation distinct from malignant histiocytosis. Cancer (1979) 44(3):993–1002. doi: 10.1002/1097-0142(197909)44:3<993::AID-CNCR2820440329>3.0.CO;2-5
67. Lehmberg K, Sprekels B, Nichols KE, Woessmann W, Müller I, Suttorp M, et al. Malignancy-associated haemophagocytic lymphohistiocytosis in children and adolescents. Br J Haematol (2015) 170(4):539–49. doi: 10.1111/bjh.13462
68. Ishii E, Ohga S, Imashuku S, Yasukawa M, Tsuda H, Miura I, et al. Nationwide survey of hemophagocytic lymphohistiocytosis in Japan. Int J Hematol (2007) 86(1):58–65. doi: 10.1532/IJH97.07012
69. Tamamyan GN, Kantarjian HM, Ning J, Jain P, Sasaki K, McClain KL, et al. Malignancy-associated hemophagocytic lymphohistiocytosis in adults: Relation to hemophagocytosis, characteristics, and outcomes. Cancer (2016) 122(18):2857–66. doi: 10.1002/cncr.30084
70. Ammann S, Lehmberg K, Zur Stadt U, Janka G, Rensing-Ehl A, Klemann C, et al. Primary and secondary hemophagocytic lymphohistiocytosis have different patterns of T-cell activation, differentiation and repertoire. Eur J Immunol (2017) 47(2):364–73. doi: 10.1002/eji.201646686
71. Shimizu M, Yokoyama T, Yamada K, Kaneda H, Wada H, Wada T, et al. Distinct cytokine profiles of systemic-onset juvenile idiopathic arthritis-associated macrophage activation syndrome with particular emphasis on the role of interleukin-18 in its pathogenesis. Rheumatology (2010) 49(9):1645–53. doi: 10.1093/rheumatology/keq133
72. Hayden A, Park S, Giustini D, Lee AY, Chen LY. Hemophagocytic syndromes (HPSs) including hemophagocytic lymphohistiocytosis (HLH) in adults: A systematic scoping review. Blood Rev (2016) 30(6):411–20. doi: 10.1016/j.blre.2016.05.001
73. Ohga S, Matsuzaki A, Nishizaki M, Nagashima T, Kai T, Suda M, et al. Inflammatory cytokines in virus-associated hemophagocytic syndrome. interferon-gamma as a sensitive indicator of disease activity. Am J Pediatr Hematol Oncol (1993) 15(3):291–8.
74. Xu XJ, Tang YM, Song H, Yang SL, Xu WQ, Zhao N, et al. Diagnostic accuracy of a specific cytokine pattern in hemophagocytic lymphohistiocytosis in children. J Pediatr (2012) 160(6):984–90.e1. doi: 10.1016/j.jpeds.2011.11.046
75. Mazodier K, Marin V, Novick D, Farnarier C, Robitail S, Schleinitz N, et al. Severe imbalance of IL-18/IL-18BP in patients with secondary hemophagocytic syndrome. Blood (2005) 106(10):3483–9. doi: 10.1182/blood-2005-05-1980
76. Baranski B, Armstrong G, Truman JT, Quinnan GV Jr., Straus SE, Young NS. Epstein-Barr Virus in the bone marrow of patients with aplastic anemia. Ann Intern Med (1988) 109(9):695–704. doi: 10.7326/0003-4819-109-9-695
77. Stéphan F, Thiolière B, Verdy E, Tulliez M. Role of hemophagocytic histiocytosis in the etiology of thrombocytopenia in patients with sepsis syndrome or septic shock. Clin Infect Dis (1997) 25(5):1159–64. doi: 10.1086/516086
78. Henter JI, Aricò M, Elinder G, Imashuku S, Janka G. Familial hemophagocytic lymphohistiocytosis. primary hemophagocytic lymphohistiocytosis. Hematol Oncol Clin North Am (1998) 12(2):417–33. doi: 10.1016/S0889-8588(05)70520-7
79. Broxmeyer HE, Williams DE, Lu L, Cooper S, Anderson SL, Beyer GS, et al. The suppressive influences of human tumor necrosis factors on bone marrow hematopoietic progenitor cells from normal donors and patients with leukemia: Synergism of tumor necrosis factor and interferon-gamma. J Immunol (1986) 136(12):4487–95.
80. de Bruin AM, Voermans C, Nolte MA. Impact of interferon-γ on hematopoiesis. Blood (2014) 124(16):2479–86. doi: 10.1182/blood-2014-04-568451
81. Yang L, Dybedal I, Bryder D, Nilsson L, Sitnicka E, Sasaki Y, et al. IFN-gamma negatively modulates self-renewal of repopulating human hemopoietic stem cells. J Immunol (2005) 174(2):752–7. doi: 10.4049/jimmunol.174.2.752
82. Snoeck HW, Van Bockstaele DR, Nys G, Lenjou M, Lardon F, Haenen L, et al. Interferon gamma selectively inhibits very primitive CD342+CD38- and not more mature CD34+CD38+ human hematopoietic progenitor cells. J Exp Med (1994) 180(3):1177–82. doi: 10.1084/jem.180.3.1177
83. Young NS, Calado RT, Scheinberg P. Current concepts in the pathophysiology and treatment of aplastic anemia. Blood (2006) 108(8):2509–19. doi: 10.1182/blood-2006-03-010777
84. Young NS, Scheinberg P, Calado RT. Aplastic anemia. Curr Opin Hematol (2008) 15(3):162–8. doi: 10.1097/MOH.0b013e3282fa7470
85. Baldridge MT, King KY, Boles NC, Weksberg DC, Goodell MA. Quiescent haematopoietic stem cells are activated by IFN-gamma in response to chronic infection. Nature (2010) 465(7299):793–7. doi: 10.1038/nature09135
86. de Bruin AM, Libregts SF, Valkhof M, Boon L, Touw IP, Nolte MA. IFNγ induces monopoiesis and inhibits neutrophil development during inflammation. Blood (2012) 119(6):1543–54. doi: 10.1182/blood-2011-07-367706
87. Alvarado LJ, Huntsman HD, Cheng H, Townsley DM, Winkler T, Feng X, et al. Eltrombopag maintains human hematopoietic stem and progenitor cells under inflammatory conditions mediated by IFN-γ. Blood (2019) 133(19):2043–55. doi: 10.1182/blood-2018-11-884486
88. Raefsky EL, Platanias LC, Zoumbos NC, Young NS. Studies of interferon as a regulator of hematopoietic cell proliferation. J Immunol (1985) 135(4):2507–12.
89. Wang CQ, Udupa KB, Lipschitz DA. Interferon-gamma exerts its negative regulatory effect primarily on the earliest stages of murine erythroid progenitor cell development. J Cell Physiol (1995) 162(1):134–8. doi: 10.1002/jcp.1041620116
Keywords: hemophagocytic lymphohistiocytosis, hypercytokinaemia, familial hemophagocytic lymphohistiocytosis (FHL), macrophage activation syndrome (MAS), cytopenia, bone marrow failure (BMF)
Citation: Paolino J, Berliner N and Degar B (2022) Hemophagocytic lymphohistiocytosis as an etiology of bone marrow failure. Front. Oncol. 12:1016318. doi: 10.3389/fonc.2022.1016318
Received: 10 August 2022; Accepted: 27 September 2022;
Published: 27 October 2022.
Edited by:
Panagiotis Diamantopoulos, Laiko General Hospital of Athens, GreeceReviewed by:
Robert Ohgami, The University of Utah, United StatesJulien Carvelli, Assistance Publique Hôpitaux de Marseille, France
Copyright © 2022 Paolino, Berliner and Degar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Barbara Degar, YmFyYmFyYV9kZWdhckBkZmNpLmhhcnZhcmQuZWR1