 Gaël Vermeersch
Gaël Vermeersch Michel Delforge
Michel Delforge Violaine Havelange2
Violaine Havelange2 Carlos Graux
Carlos Graux Timothy Devos
Timothy Devos
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Oncol., 07 December 2022
Sec. Hematologic Malignancies
Volume 12 - 2022 | https://doi.org/10.3389/fonc.2022.1014671
This article is part of the Research TopicCase Reports in Hematological Malignancies : 2022View all 34 articles
Chronic neutrophilic leukemia (CNL) is a rare but potentially aggressive BCR::ABL1 negative myeloproliferative neoplasm, characterized by sustained mature, neutrophilic leukocytosis. The discovery of key driver mutations in the colony-stimulating-factor-3 receptor (CSF3R) gene resulted in the updated World Health Organization (WHO) diagnostic criteria in 2016. A significant number of CNL cases have been associated with plasma cell dyscrasias, predominantly multiple myeloma (MM) and monoclonal gammopathy of unknown significance (MGUS). Compared to pure CNL, mutated CSF3R is infrequently reported in CNL cases associated with monoclonal gammopathies (MG). Until now it remains unclear whether CNL and occurring plasma cell neoplasms are clonally related or CNL is developing secondary to the underlying dyscrasia. Owing to its rarity, currently no standard of care management exists for CNL and MG-associated CNL. In this case series we report the multi-center experience of five MG-associated CNL cases with a median age of diagnosis of 69 years. Three patients (66%) showed predominance of lambda light chain expression. Four (80%) eventually evolved to MM, and one CNL-MGUS patient developed secondary acute myeloid leukemia (AML). Mutated CSF3R was present in the patient who developed AML but was absent in other cases. To assess possible associated genetic aberrations we performed recurrent analysis with next-generation sequencing (NGS). Two patients (40%) deceased with a median time of survival of 8 years after CNL diagnosis. Three (60%) are currently in follow-up with no reoccurring leukocytosis. This case series, followed by a short review, provides a long-term clinical and genetic overview of five CNL cases associated with MG.
Chronic neutrophilic leukemia (CNL) is an infrequent BCR::ABL1 negative myeloproliferative neoplasm (MPN) defined by sustained, mature neutrophilia and bone marrow hypercellularity with granulocytic hyperplasia (1, 2). Approximately 200 CNL cases have been described; however, many of these do not meet the current diagnostic criteria (3). The identification of key driver mutations in the colony-stimulating-factor-3 receptor (CSF3R) gene resulted in the updated diagnostic criteria by the World Health Organization (WHO) in 2016 (4, 5). CSF3R is mutated in up to 80% of CNL cases (6). Approximately 32% of CNL cases are associated with plasma cell dyscrasias, wherein CSF3R tends to be less frequently mutated (1, 7). It remains unclear whether both entities should be considered clonally related or neutrophilia is provoked by the plasma cell dyscrasia (1). The majority of CNL-associated paraproteinemias express lambda light chains (8). The lack of genetic markers and arbitrary WHO-diagnostic criteria, such as leukocyte count, challenges the diagnosis. This case series provides a long-term clinical and genetic overview of five CNL cases associated with monoclonal gammopathies (MG). Four patients developed multiple myeloma (MM), and one patient developed secondary acute myeloid leukemia (AML).
In 2012, a 61-year-old woman was referred due to persistent, incremental neutrophilia since 2009. Medical history included no major abnormalities. At presentation medical therapy comprised estriol cream and alprazolam. She had one sibling diagnosed with polycythemia vera (PV). Ultrasound excluded hepatosplenomegaly. Serum electrophoresis and immunofixation identified immunoglobulin G (IgG) kappa-type paraproteinemia (cfr. Table 1 for results of the full blood analysis from all patients). Bone marrow biopsy showed hypercellularity without dysplasia. Karyotyping resulted normal. Positron emission tomography-computed tomography (PET-CT) showed no bone lesions. No therapy was initiated. In 2016, MGUS evolved to MM “Revised International Staging System” stadium I (R-ISS). M-peak rose from 10 to 32 g/L. Bone marrow biopsy showed right-shifted hypercellularity (90%) with approximately 73.3% segmented neutrophils and central plasmacytosis of 35% without central blast excess. Amyloidosis was excluded by Congo Red staining. Next-generation sequencing (NGS) and fluorescence in situ hybridization (FISH) resulted normal. Initiation of bortezomib-thalidomide-dexamethasone (VTD) resulted in a very good partial response (VGPR) after six cycles. Autologous hematopoietic stem cell transplantation (HSCT) was executed in 2017. Six months later, MM progressed and FISH revealed a mono-allelic loss of 17p13/TP53 (February 2018). Daratumumab-lenalidomide-dexamethasone (DRd) was started as the patient showed intolerance towards carfilzomib-lenalidomide-dexamethasone (KRd). Six months afterwards, fourth-line therapy with bendamustine was started as PET-CT showed disease progression. The patient died due to progressive multiple myeloma, respectively 9 and 3 years after CNL and MM diagnosis. No reoccurrence of peripheral blood leukocytosis ≥25 × 109/L was observed after the initiation of VTD and following therapies.
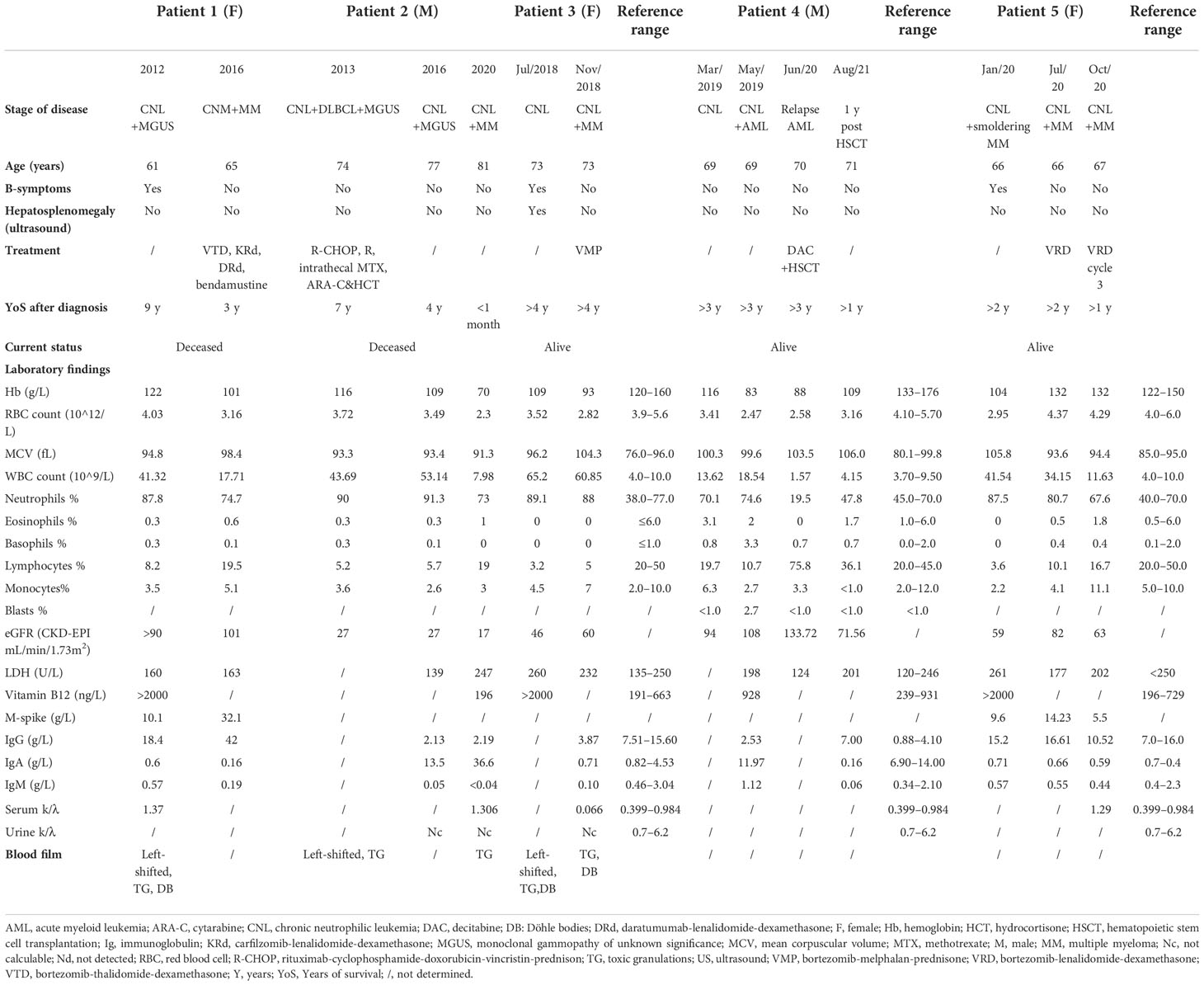
Table 1 Overview of clinical and laboratory findings at presentation and follow-up.
A 74-year-old man, known with IgA-type MGUS since 2003 and persistent neutrophilic leukocytosis since 2009, was diagnosed with spinal located diffuse large B-cell lymphoma (DLBCL) subtype ABC in 2013 (KI67 20%–25%, karyotype (46, XY[20]). Familial anamnesis did not include hemato-oncological pathologies. Medical therapy at presentation comprised nebivolol (2.5 mg QD) and spironolacton/altizide (12.5 mg/7.5 mg QD). Biochemical analysis showed leukocytosis of 43.69 × 109/L (90.7% neutrophils) with M-spikes in both β- and γ-fractions.
Bone marrow biopsy revealed hypercellularity with increased representation of the myeloid lineage (mature and immature) and local clustering (5%–10%) of monotypic plasma cells (kappa-type, CD20-). Karyotyping resulted normal. Clonality between the spinal located DLBCL and bone marrow tissue was proved by polymerase chain reaction (PCR) (IGH/IGK).
Thus, the patient was diagnosed with bone marrow involved DLBCL, CNL, and MGUS. Six cycles of rituximab-cyclophosphamide-doxorubicin-vincristin-prednisone (R-CHOP), followed by rituximab monotherapy (administered twice), were given. In addition, intrathecal chemotherapy (methotrexate, cytarabine, and hydrocortisone) was administered three times. During therapy, bone marrow biopsy showed minimal CD5+ positivity (<0.1% all nucleated cells (ANC)) and isolated loss of the Y-chromosome (45,X,-Y[3]/46,XY[7]). CSF3R mutations were absent. Complete remission (CR) of DLBCL was obtained on PET-CT scan by September 2014. Asymptomatic neutrophilic leukocytosis remained during remission (>30 × 109/L; >80% neutrophils).
In February 2016, an increasing absolute neutrophil count of 48.5 × 109/L and M-spike were observed (IgA 13.5g/L). Urine kappa FLCs were not calculable. In June 2020, MGUS progressed to MM, type IgA kappa (R-ISS III, IgA 36.60 g/L); the total number of leukocytes normalized. Cytogenetic analysis showed hyperdiploidy with gain on chromosome 1q. CSF3R-analysis was not performed at this stage. Bone marrow biopsy showed hypercellularity and right-shifted myeloid cells with normal morphology. There was plasmocytosis of 55.7% ANC. The patient preferred no further therapies and deceased shortly after, roughly 7 years after CNL diagnosis.
In 2018, a 73-year-old woman was referred due to persistent leukocytosis for 1 year. Medical therapy comprised acetylsalicylic acid (80 mg QD), bisoprolol (2.5 mg QD), and rosuvastatin (10 mg QD). Besides significant weight loss (>10% within 12 months), no B-symptoms were present. Blood film analysis confirmed the presence of left-shifted neutrophilia with toxic granulations and Döhle bodies. Bone marrow biopsy showed hypercellularity (>95%) with myeloid hyperplasia, plasmacytosis of 10%–15% (lambda positive), and bone marrow fibrosis grade 1. Karyotyping resulted normal. NGS revealed mutated ASXL1 (c.1934dup;p.Gly646Trpfs*12)(VAF 26%); CSF3R appeared normal (Table 2). The tentative diagnosis of CNL was made. No therapy was initiated.
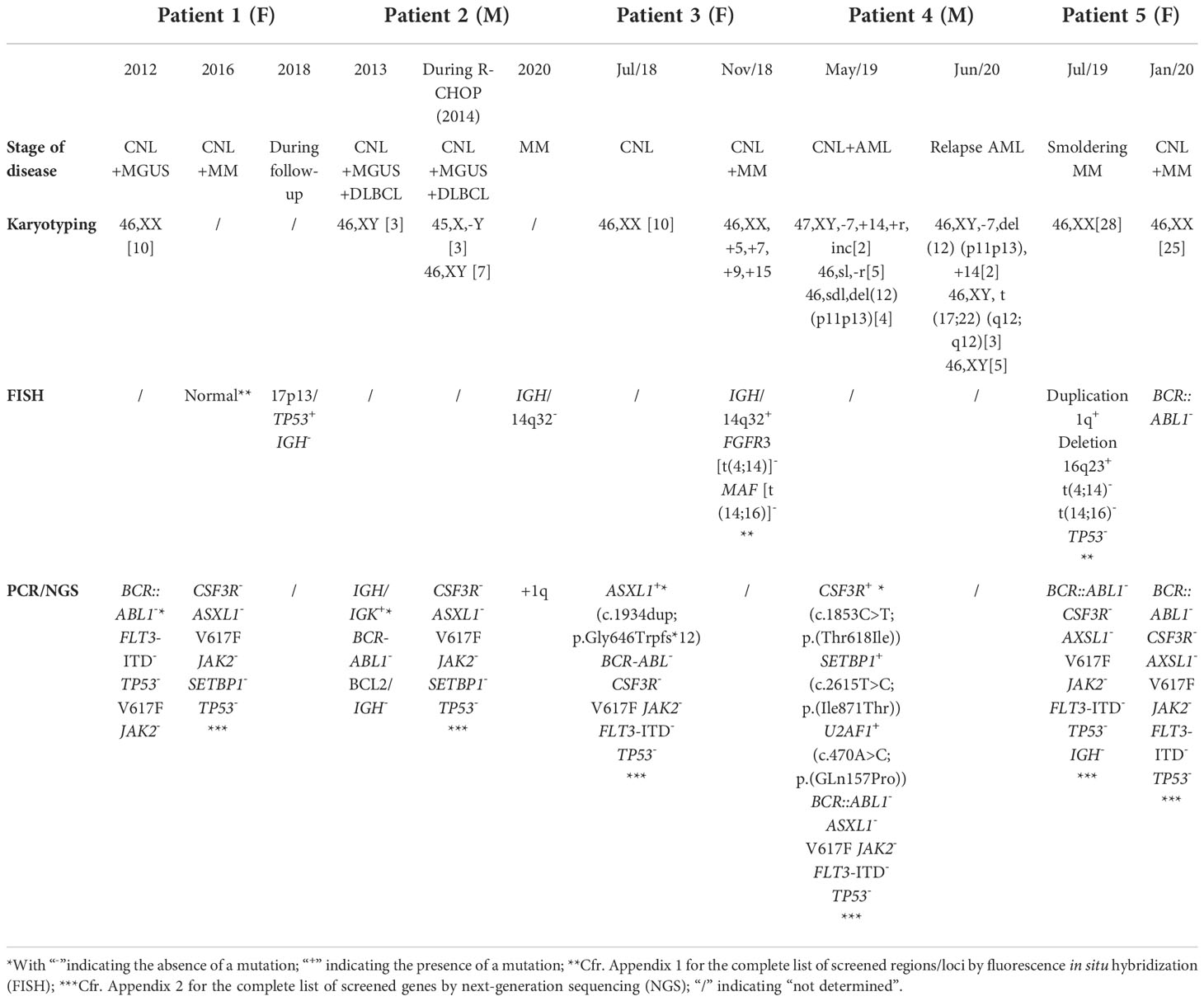
Table 2 Genetic and molecular findings at diagnosis and follow-up.
Four months later, the patient was diagnosed with lambda light chain MM (R-ISS I). Biochemical analysis revealed neutrophilic leukocytosis, hypogammaglobulinemia, and a normal β2-microglobulin concentration. Serum lambda FLCs were significantly increased (free λ: 276 mg/L (ref. 10–34) (Table 1)). There was significant proteinuria (6.79 g/24 h) with prominent lambda excretion (9816 mg/24 h). Bone marrow biopsy showed hypercellularity with segmented neutrophils (36.3% ANC) and increased central plasmocytosis (13.7% ANC) without blastosis. Congo Red staining resulted normal. Karyotyping revealed hyperdiploidy with gain on chromosomes 5, 7, 9, and 15. FISH detected IGH/14q32 rearrangement. Bortezomib-melphalan-prednisone (VMP) was initiated and halted after nine cycles as biochemical CR and minimal residual disease on PET-CT were obtained. Currently, more than 4 years after CNL and MM diagnosis, the patient is stable. Peripheral blood leukocyte count is <25 × 109/L.
In 2019, a 69-year-old man was referred due to incremental neutrophilic leukocytosis and fatigue since several months. Medical history included diabetes mellitus type 2. Medical therapy comprised sitagliptin/metformin (50 mg/1000 mg BID), rosuvastatin (10 mg QD), and allopurinol (100 mg QD). Biochemical analysis showed normocytic anemia with concurrent neutrophilic leukocytosis and 2.7% blasts. Electrophoresis and immunofixation indicated MGUS (IgG lambda type). Bone marrow biopsy showed hypercellularity with blast excess (20%). Cytogenetic analysis identified a complex karyotype with monosomy of chromosome 7, trisomy of chromosome 14, and deletion of p12 (Table 2). CSF3R (44%), SETBP1 (45%), and U2AF1 (42%) appeared mutated. The diagnosis of AML secondary to CNL was made. Complete morphologic and cytogenetic remission was obtained after eight cycles of decitabine. Planned allogeneic HSCT was postponed due to flu and the SARS-CoV-2 pandemic. The patient relapsed several months later. Cytogenetic analysis detected the reoccurrence of previous described mutations in combination with a new translocation t(17;22) (Table 2). Despite that no CR was obtained after remission-reinduction, allogeneic HSCT was performed. Bone marrow analysis 2 and 4 months afterwards showed no blast excess; mutated CSF3R was absent. The patient recovered and is currently, more than 2 years after HSCT, in CR. Paraproteinemia and neutrophilic leukocytosis did not reoccur.
In 2020, a 66-year-old woman presented with recurrent headaches, fever and generalized myalgia/arthralgia since several months. Clinical investigation was unremarkable. Familial anamnesis did not include hemato-oncological abnormalities. There was no use of medicines. Medical history included successfully treated mammary carcinoma in 2010 (surgical resection, radio- and hormonal therapy; CR at presentation). In 2019, the patient developed auto-immune aortitis, successfully treated with corticoids. A few months later, the patient was diagnosed with smoldering MM (IgG lambda). Bone marrow biopsy then showed hypercellularity with a strong representation of the granulocytic lineage in intermediary and mature stages, toxic granulations, and sea-blue histiocytes. Central plasmocytosis was 10.5%. No central blast excess was observed. Cytogenetic and molecular analysis resulted normal. Since the diagnosis of smoldering MM, there was persistent neutrophilia. Given the suspicion of CNL, the patient was shortly treated with hydroxycarbamide, which was rapidly stopped due to provoked cytopenia. Relapse of breast carcinoma was excluded and PET-CT resulted normal.
A few months later, smoldering MM evolved into MM (R-ISS II). Treatment with bortezomib-lenalidomide and dexamethasone (VRD) was initiated and resulted in partial response (PR). The patient refused intensification with autologous HSCT. VRD was stopped after three cycles due to neurological complications. According to patient requirement, low-dose lenalidomide (5 mg) was continued in monotherapy and PR was maintained. Under lenalidomide, the patient progressively developed nephrotic syndrome. Renal biopsy showed glomerulonephritis with mesangial IgA deposits, probably not related to the hematologic condition. Currently, the patient is in follow-up; leukocyte count remains normal.
Five patients diagnosed with CNL between 2012 and 2020 were included in this study. In retrospect, CNL diagnosis was based on the 2016 WHO diagnostic criteria (4). Cytogenetic and molecular analysis of patients 1–3 was performed at the University Hospitals Leuven, Belgium. Cytogenetic analysis of patient 4 was performed at Université Catholique de Louvain, CHU UCL Namur, Belgium. Molecular analysis of patient 4 was performed at the University Hospitals Leuven. Cytogenetic analysis of patient 5 was performed at Université Catholique de Louvain Saint-Luc, Woluwe-Saint-Lambert, Belgium. Molecular analysis of patient 5 was performed at Institut de Pathologie et de Génétique (IPG), Charleroi, Belgium. We report cytogenetic and molecular analyses at diagnosis and during follow-up. Used probes and screened genes by FISH and NGS are clarified in the appendices. Patients 1–3 are treated at the University Hospitals Leuven. Patient 4 is treated at Université Catholique de Louvain, CHU UCL Namur, and patient 5, at Université Catholique de Louvain Saint-Luc, Woluwe-Saint-Lambert.
In this series the median age of CNL diagnosis is 69 years, 40% of patients (n = 2) are men; 20% (n = 1) of patients carried mutated CSF3R. B-symptoms and splenomegaly were present in 40% (n = 2) and 20% (n = 1) of patients, respectively. Three patients (66%) showed a predominance of lambda light chain expression. Four patients (80%) evolved to multiple myeloma. Hydroxycarbamide was shortly initiated in one patient. All patients received treatments focusing on associated malignancies such as AML and MM, among these 40% (n = 2) underwent HSCT. Two patients died after a median time of survival of 8 years after CNL diagnosis. Three patients (66%) are currently in follow-up (approximately 3 years after diagnosis) and show no signs of reoccurring leukocytosis.
In 1920, CNL was first described as “polymorphonuclear neutrophil hyperleukocytosis” (9). Roughly 200 CNL cases are currently reported in literature; however, many cases may not meet the WHO-defined diagnostic criteria (1, 3). The lack of cytogenetic markers has complicated accurate diagnosis in the past. Recent identification of oncogenic driver mutations in CSF3R resulted in the updated WHO diagnostic criteria in 2016 (1, 4, 5).
Besides the presence of mutated CSF3R, the diagnostic criteria include a peripheral blood leukocytosis of ≥25 × 109/L with ≥80% neutrophils (segmented and band), <10% peripheral neutrophil precursors, absence of monocytosis/eosinophilia/basophilia, and rarely observed peripheral myeloblasts. Arber et al. provide a complete list of the current diagnostic criteria (4, 7, 10). Levels of vitamin B12 and lactate dehydrogenase (LDH) are frequently elevated but are no solid criteria. Neutrophil morphology typically presents with non-specific characteristics such as toxic granulations and Döhle bodies (8, 11). Bone marrow biopsy shows hypercellularity with increased numbers of normal maturating neutrophilic granulocytes. Chronic myeloid leukemia and other BCR::ABL1 negative MPNs must be excluded; rearrangement of PDGFRA, PDGFRB, FGR, or PCM1::JAK2 should be absent. Without mutated CSF3R, the diagnosis is possible if persistent non-reactive neutrophilia (≥3 months) is present. In this context, demonstrated myeloid clonality by cytogenetic or molecular analysis is preferred (4, 10).
When strictly applied, only cases 1–3 and 5 meet the CNL criteria concerning leukocytosis. In case 4, even before the diagnosis of AML, peripheral blood leukocytosis was <25 × 109/L with <80% neutrophils. However, the persistence of leukocytosis (>3 months) without an identifiable cause of reactive neutrophilia combined with mutated CSF3R strongly supports the CNL diagnosis. In AML, the incidence of mutated CSF3R is approximately 1%. This case emphasizes the risk of an arbitrary leukocyte number as an absolute criterion for diagnosis (5, 12).
Elliott et al., analyzing 40 WHO-defined CNL cases, reported a median age of 66 years (range 16–86). The majority of these cases were men (56%) (3). In our series two of five patients (40%) were male and the median age of diagnosis was 69 years. Splenomegaly, which is removed as a solid criterion in the current WHO criteria, was present in one of five (20%).
CSF3R is the gene coding for the receptor of colony-stimulating-factor-3 (CSF3), a primary neutrophil growth factor. Mutated CSF3R is considered as a solid CNL-defining criterion. In CNL, the mutational frequency of CSF3R is approximately 60%–80%, with CSF3RT618I as the most frequent reported mutation (7, 13). CSF3RT618I occurs in the extracellular or transmembrane domain of the receptor and results in the activation of the JAK-STAT pathway; which explains the marked clinical improvement after the initiation of ruxolitinib in some patients. Other mutations preferentially activate the SRC tyrosine kinase. We refer to Maxson et al. for qualitative information about CSF3R (5, 6, 13). In MG-associated CNL, the incidence of mutated CSF3R appears to be significantly lower; however, data is scarce (7).
According to the WHO diagnostic criteria, plasma cell neoplasms should be excluded in the absence of mutated CSF3R (4). Nevertheless, up to 32% of CNL cases report concurrent plasma cell dyscrasias, with the predominance of lambda light chain expression (66% or 3/5 in our series) (1). All patients showed sign of MG at the time of CNL diagnosis; 80% (4/5) eventually evolved into MM. Larger data sets are necessary to evaluate whether patients with CNL-associated MGUS carry an inherent higher risk to evolve into MM.
It is unclear whether MG-associated CNL has to be considered as a genuine myeloproliferative neoplasm or neutrophilic reaction, for example, secondary to production of plasma cell-derived cytokines such as G-CSF (1). Quantification of G-CSF serum concentration was not performed in our series but could be useful. The lower frequency of mutated CSF3R, combined with the longer median time of survival in MG-associated CNL, may indicate different etiopathogenesis (1, 7). Only patient 4 carried mutated CSF3R. MG-associated CNL shows a median time of survival of approximately 5 years, compared to 24 months in true CNL (3, 7, 14). Patients 1 and 2 show a median time of survival of 8 years after CNL diagnosis. Patients 3, 4, and 5 are in follow-up, roughly 3 years after CNL diagnosis.
Some authors stress the few reported chromosomal abnormalities and better survival as evidence for a provoked neutrophilic reaction in MG-associated CNL (14, 15). However, Nedeljkovic et al. reported the presence of a homozygous JAK2V617F mutation in a patient with CNL associated with plasma cell myeloma, demonstrating molecular evidence of clonality in the absence of mutated CSF3R (16). Mutated ASXL1 in patient 3 at the time of CNL diagnosis is a sign of clonality in the absence of mutated CSF3R.
Y-chromosome loss, as in patient 2, is frequently reported in patients with hematological disorders. It remains unknown whether Y-chromosome loss should be considered as an age-related phenomenon or cytogenetic marker for malignancy (17). Copy number alterations, resulting in gain of chromosomes such as +5, +7, +9, and +15 in patient 3, are one of the most prominent perturbations in MM. The gain on chromosomes 9 and 15 may play an important role in MGUS transformation (18). Abnormalities of chromosome 1 are observed in approximately 25% of patients at MM diagnosis. The gain on 1q, as in patients 2 and 5, is associated with inferior survival (19).
At CNL diagnosis, patient 4 carried monosomy 7, trisomy 14 and 12p-deletion. This latter is associated with progression to post-MPN AML (20). Trisomy 14 is a rare cytogenetic abnormality in myeloid neoplasms such as AML. Isolated trisomy 14 is indicated as an early event in leukemogenesis; however, more research on its clinicopathological features is needed. Monosomy 7 is frequently reported in myeloid malignancies and is detected in previous CNL-cases who developed secondary AML (3, 21, 22). The translocation t(17;22)(q12;q12), which occurred in patient 4 during AML relapse, is associated with myelodysplastic syndrome (MDS) (23).
Specific mutations may indicate a negative prognosis in CNL, even if, by our knowledge, currently no mutations are validated as prognostic markers. Mutated ASXL1 as in patient 3, resulting in disrupted epigenetic regulation, is associated with a negative prognosis in CNL. Mutational frequency is varying from 30% to 81% (1, 22).
Mutated TP53 is characterized as a negative prognostic factor in myeloid and lymphoid malignancies (24, 25). Monoallelic loss of TP53 was detected in patient 1 during follow-up; the patient deceased 2 years afterwards. The relevance of mutated TP53 in MG-associated CNL is unclear.
Rearrangements involving the IGH locus on chromosome 14q32 frequently occur in MM and are associated with standard risk (26). Translocations in 14q32 less frequently co-occur with hyperdiploid chromosomes such as in patient 3 (27).
SETBP1, considered as a driver oncogene, is mutated in approximately 33% of CNL cases (7). It is associated with poor prognosis in various myeloproliferative phenotypes, including secondary AML as in patient 4. Whether mutated SETBP1 is associated with poor survival in CNL is unknown (22, 28).
A limited number of cases report the presence of JAK2V617F mutations in CNL; little is known about its true prevalence in WHO-defined CNL cases (7, 29–34). JAK2V617F and CSF3RT618I appear to be mutually exclusive (7, 10, 35). None of the patients in our series carried JAK2V617F.
Mutated spliceosome-associated genes such as SRSF2 and U2AF1 have been reported in CNL. Meggendorfer et al. report a SRSF2 mutational frequency of 21% (4/14 cases); others report a frequency of 0% (0/10) (35–37). Patient 4 carried the Glutamin(Gln)157pro mutation in the U2AF1 gene at the moment of CNL and AML diagnosis. Mutated U2AF1 results in predisposition to AML (38, 39). Other CNL-associated mutations, such as TET2 and RUNX1, were absent in our cases. TET2 mutations have an estimated mutational frequency of 29% in CNL (1, 36). Previous authors postulated that cooperating RUNX1 and CSF3R mutations in CNL may result in disease progression, resistance to ruxolitnib and may act as an early marker of AML transformation (40, 41).
Currently, no standard of care management exists for CNL. Historically, splenic irradiation and splenectomy were performed to reduce tumor bulk and abdominal discomfort. Nevertheless, splenectomy is associated with worsening of neutrophilia in CNL. The only potentially curative treatment is HSCT (1). Allogeneic HSCT showed a one-year overall survival rate of 40% in patients with CNL. However, data regarding clinical outcomes and the most optimal regimens of HSCT are scarce (3, 10, 42, 43). In our series, autologous and allogeneic HSCTs were performed in patient 1 due to progressive MM and in patient 4 due to AML, not CNL specifically.
The use of “7+3” induction chemotherapy in CNL has not been able to induce hematological remission. One report describes a young patient in the blast phase who attained a second chronic phase following induction chemotherapy (anthracycline and cytarabine) (44).
Cytoreductive agents, such as hydroxyurea and interferon-alpha (IFN-α), have demonstrated efficacy in controlling leukocytosis and splenomegaly. Currently, hydroxyurea is the most frequently used first-line agent; nonetheless, 25% of patients appear to be refractory (3, 35). Few reports mention durable remission and good tolerability after IFN-α initiation (45–47). No IFN-α is used in our series as it is not reimbursed in our country.
Promising targeted therapies are being investigated since the identification of mutated CSF3R. Depending on the mutation downstream signaling pathways through JAK-STAT or SRC tyrosine kinase are activated, these may be inhibited by ruxolitinib and dasatinib, respectively (5, 10, 42). An overall response rate of 32% was observed in patients with CSF3R-mutated CNL treated with ruxolitinib; patients harboring CSF3RT618I were most likely to respond (48). None of these agents were used in our series.
As in CNL, there is no standard of care management for MG-associated CNL (1). No specific anti-myeloma regimen can be recommended, but conventional chemotherapy, immunomodulatory drugs, proteasome inhibitors, and anti-CD38 monoclonal antibodies may reduce neutrophil counts to a variable extent. However, it is difficult to discriminate a direct effect on the CNL clone from an indirect reduction by the suppression of the malignant plasma cell clone. Keeping in mind the more favorable prognosis of MG-associated CNL, treatment strategies in our series were primarily focused on associated plasma cell dyscrasias and AML.
In patient 1, autologous HSCT was performed after VTD therapy. Subsequently, the patient received therapy existing out of KRd, DRd, and bendamustine due to MM progression. No reoccurrence of neutrophilia was observed after the initiation of VTD and following therapies. However, as various therapies were administered in a short time frame, no conclusion on the effectivity of individual therapies in CNL can be made. As mentioned, HSCT in patient 1 and 4 was performed due to progressive MM and AML respectively, not CNL specifically.
Decitabine, a demethylating agent, was initiated in patient 4 and resulted in complete morphologic and cytogenetic remission after eight cycles. Previous authors reported complete remission and suggested decitabine as a potential effective therapeutic agent in patients with secondary AML; nonetheless, data is scarce (49).
Bortezomib, a proteasome inhibitor, was administered in patients 1, 3, and 5 for the treatment of MM. To our knowledge, only one previous case report describes the use of bortezomib in CNL-MM, resulting in the complete resolution of both leukocytosis and MG (50). Reduced neutrophil count is a common observation in the use of bortezomib. Bortezomib could act directly through the effect on proteasomes in neutrophils, or indirectly through its influence on cytokine concentration. The administration of bortezomib in patients 1 and 3 resulted in a VGPR, while in patient 5 a partial response was achieved. Data of immunomodulatory drugs in MG-associated CNL, such as thalidomide and lenalidomide in patients 1 and 5 of our series, are scarce. The number of leukocytes normalized in all of these patients.
Chronic neutrophilic leukemia is a rare but potentially aggressive myeloproliferative neoplasm with a median survival of approximately 24 months. A non-negligible number of CNL cases are diagnosed with plasma cell dyscrasias; these patients show a more favorable prognosis with a median survival of 5 years. The discovery of oncogenic CSF3R driver mutations raised the diagnostic accuracy of CNL and resulted in the updated WHO diagnostic criteria in 2016. CSF3R tends to be less frequently mutated in CNL associated with monoclonal gammopathies (MG), challenging the diagnosis. Better survival and lower CSF3R mutational frequency may suggest a different etiopathogenesis between pure CNL and MG-associated CNL. Owing to the rarity of CNL, there is no defined standard of care. Currently, hematopoietic stem cell transplantation (HSCT) is the only curative treatment. This series provides an overview of cytogenetic evolution and treatment in MG-associated CNL. More knowledge about occurring mutations and the order of acquisition will hopefully result in better therapeutic approaches and outcomes.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
GV: data curation (equal), formal analysis (equal), visualization (equal), writing-original draft (lead), writing-review and editing (supporting). MD: data curation (equal), formal analysis (equal), visualization (equal), writing-original draft (supporting), writingreview and editing (supporting). VH: data curation (equal), formal analysis (equal), visualization (equal), writing-original draft (supporting), writing-review and editing (supporting). CG: data curation (equal), formal analysis (equal), visualization (equal), writingoriginal draft (supporting), writing-review and editing (supporting). LM: data curation (equal), formal analysis (equal), visualization (equal), writing-original draft (supporting), writing-review and editing (supporting). TD: data curation (equal), formal analysis (equal), visualization (equal), writing-original draft (lead), writing-review and editing (lead).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Szuber N, Elliott M, Tefferi A. Chronic neutrophilic leukemia: 2022 update on diagnosis, genomic landscape, prognosis, and management. Am J Hematol (2022) 97:491–505. doi: 10.1002/ajh.26481
2. Thomopoulos TP, Symeonidis A, Kourakli A, Papageorgiou SG, Pappa V. Case report: Chronic neutrophilic leukemia associated with monoclonal gammopathies: Case series and review of genetic characteristics and practical management. Front Oncol (2022) 12:891961/BIBTEX. doi: 10.3389/FONC.2022.891961/BIBTEX
3. Elliott MA, Hanson CA, Dewald GW, Smoley SA, Lasho TL, Tefferi A. WHO-defined chronic neutrophilic leukemia: A long-term analysis of 12 cases and a critical review of the literature. Leukemia (2005) 19:313–7. doi: 10.1038/sj.leu.2403562
4. Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, et al. The 2016 revision to the world health organization classification of myeloid neoplasms and acute leukemia. Blood (2016) 127:2391–405. doi: 10.1182/blood-2016-03-643544
5. Maxson JE, Gotlib J, Pollyea DA, Fleischman AG, Agarwal A, Eide CA, et al. Oncogenic CSF3R mutations in chronic neutrophilic leukemia and atypical CML. N Engl J Med (2013) 368:1781–90. doi: 10.1056/NEJMoa1214514
6. Maxson JE, Tyner JW. Genomics of chronic neutrophilic leukemia. Blood (2017) 129:715–22. doi: 10.1182/blood-2016-10-695981
7. Pardanani A, Lasho TL, Laborde RR, Elliott M, Hanson CA, Knudson RA, et al. CSF3R T618I is a highly prevalent and specific mutation in chronic neutrophilic leukemia. Leukemia (2013) 27:1870–3. doi: 10.1038/leu.2013.122
8. Bain BJ, Ahmad S. Chronic neutrophilic leukaemia and plasma cell-related neutrophilic leukaemoid reactions. Br J Haematol (2015) 171:400–10. doi: 10.1111/bjh.13600
9. Tuohy E. A case of splenomegaly with polymorphonuclear neutrophil hyperleukocytosis. Am J Med Sci (1920), 160:18–25. doi: 10.1097/00000441-192007000-00003
10. Elliott MA, Tefferi A. Chronic neutrophilic leukemia: 2018 update on diagnosis, molecular genetics and management. Am J Hematol (2018) 93:578–87. doi: 10.1002/ajh.24983
11. Elliott MA, Dewald GW, Tefferi A, Hanson CA. Chronic neutrophilic leukemia (CNL): A clinical, pathologic and cytogenetic study. Leukemia (2001) 15:35–40. doi: 10.1038/sj.leu.2401993
12. Dao KHT, Tyner JW, Gotlib J. Recent progress in chronic neutrophilic leukemia and atypical chronic myeloid leukemia. Curr Hematol Malig Rep (2017) 12:432–41. doi: 10.1007/s11899-017-0413-y
13. Maxson JE, Luty SB, MacManiman JD, Paik JC, Gotlib J, Greenberg P, et al. The colony-stimulating factor 3 receptor T640N mutation is oncogenic, sensitive toJAKInhibition, and mimics T618i. Clin Cancer Res (2016) 22:757–64. doi: 10.1158/1078-0432.CCR-14-3100
14. Ito T, Kojima H, Otani K, Komeno T, Mitsuhashi S, Hasegawa Y, et al. Chronic neutrophilic leukemia associated with monoclonal gammopathy of undetermined significance. Acta Haematol (1996) 95:140–3. doi: 10.1159/000203863
15. Standen GR, Jasani B, Wagstaff M, Wardrop CAJ. Chronic neutrophilic leukemia and multiple myeloma: An association with λ light chain expression. Cancer (1990) 66:162–6. doi: 10.1002/1097-0142(19900701)66:1<162
16. Nedeljkovic M, He S, Szer J, Juneja S. Chronic neutrophilia associated with myeloma: Is it clonal? Leuk Lymph (2014) 55:439–40. doi: 10.3109/10428194.2013.809080
17. Forsberg LA. Loss of chromosome y (LOY) in blood cells is associated with increased risk for disease and mortality in aging men. Hum Genet (2017) 136:657–63. doi: 10.1007/s00439-017-1799-2
18. Aktas Samur A, Minvielle S, Shammas M, Fulciniti M, Magrangeas F, Richardson PG, et al. Deciphering the chronology of copy number alterations in multiple myeloma. Blood Cancer J (2019) 9:39. doi: 10.1038/s41408-019-0199-3
19. Giri S, Huntington SF, Wang R, Zeidan AM, Podoltsev N, Gore SD, et al. Chromosome 1 abnormalities and survival of patients with multiple myeloma in the era of novel agents. Blood Adv (2020) 4:2245–53. doi: 10.1182/bloodadvances.2019001425
20. Klampfl T, Harutyunyan A, Berg T, Gisslinger B, Schalling M, Bagienski K, et al. Genome integrity of myeloproliferative neoplasms in chronic phase and during disease progression. Blood (2011) 118:167–76. doi: 10.1182/blood-2011-01-331678
21. Inaba T, Honda H, Matsui H. The enigma of monosomy 7. Blood (2018) 131:2891–8. doi: 10.1182/blood-2017-12-822262
22. Elliott MA, Pardanani A, Hanson CA, Lasho TL, Finke CM, Belachew AA, et al. ASXL1 mutations are frequent and prognostically detrimental in CSF3R -mutated chronic neutrophilic leukemia. Am J Hematol (2015) 90:653–6. doi: 10.1002/ajh.24031
23. Antic D, Impera L, Fekete MD, Djordjevic V, Storlazzi CT, Elezovic I. Novel chromosomal translocation (17;22)(q12;q12) in a case of myelodisplastic syndrome characterized with signs of hemolytic anemia at presentation. Gene (2012) 493:161–4. doi: 10.1016/j.gene.2011.11.002
24. Xu-Monette ZY, Jeffrey Medeiros L, Li Y, Orlowski RZ, Andreeff M, Bueso-Ramos CE, et al. Dysfunction of the TP53 tumor suppressor gene in lymphoid malignancies. Blood (2012) 119:3668–83. doi: 10.1182/blood-2011-11-366062
25. Cumbo C, Tota G, Anelli L, Zagaria A, Specchia G, Albano F. TP53 in myelodysplastic syndromes: Recent biological and clinical findings. Int J Mol Sci (2020) 21:3432. doi: 10.3390/ijms21103432
26. Rajkumar SV. Multiple myeloma: 2018 update on diagnosis, risk-stratification, and management. Am J Hematol (2018) 93:1091–110. doi: 10.1002/ajh.25117
27. Smadja NV, Leroux D, Soulier J, Dumont S, Arnould C, Taviaux S, et al. Further cytogenetic characterization of multiple myeloma confirms that 14q32 translocations are a very rare event in hyperdiploid cases. Genes Chromosomes Cancer (2003) 38:234–9. doi: 10.1002/gcc.10275
28. Shou LH, Cao D, Dong XH, Fang Q, Wu Y, Zhang Y, et al. Prognostic significance of SETBP1 mutations in myelodysplastic syndromes, chronic myelomonocytic leukemia, and chronic neutrophilic leukemia: A meta-analysis. PloS One (2017) 12:e0171608. doi: 10.1371/journal.pone.0171608
29. Gajendra S, Gupta R, Chandgothia M, Kumar L, Gupta R, Chavan SM. Chronic neutrophilic leukemia with V617F JAK2 mutation. Indian J Hematol Blood Transfus (2014) 30:139–42. doi: 10.1007/s12288-012-0203-6
30. Imashuku S, Kudo N, Kubo K, Saigo K, Okuno N, Tohyama K. Rituximab for managing acquired hemophilia a in a case of chronic neutrophilic leukemia with the JAK2 kinase V617F mutation. J Blood Med (2012) 3:157. doi: 10.2147/jbm.s37631
31. Kako S, Kanda Y, Sato T, Goyama S, Noda N, Shoda E, et al. Early relapse of JAK2 V617F-positive chronic neutrophilic leukemia with central nervous system infiltration after unrelated bone marrow transplantation. Am J Hematol (2007) 82:386–90. doi: 10.1002/ajh.20805
32. Lea NC, Lim Z, Westwood NB, Arno MJ, Gäken J, Mohamedali A, et al. Presence of JAK2 V617F tyrosine kinase mutation as a myeloid-lineage-specific mutation in chronic neutrophilic leukaemia. Leukemia (2006) 20:1324–6. doi: 10.1038/sj.leu.2404240
33. Mc Lornan DP, Percy MJ, Jones AV, Cross NCP, Mc Mullin MF. Chronic neutrophilic leukemia with an associated V617F JAK2 tyrosine kinase mutation. Haematologica (2005) 90:1696–7. doi: 10.3324/%x
34. Steensma DP, Dewald GW, Lasho TL, Powell HL, McClure RF, Levine RL, et al. The JAK2 V617F activating tyrosine kinase mutation is an infrequent event in both “atypical” myeloproliferative disorders and myelodysplastic syndromes. Blood (2005) 106:1207–9. doi: 10.1182/blood-2005-03-1183
35. Szuber N, Tefferi A. Chronic neutrophilic leukemia: New science and new diagnostic criteria. Blood Cancer J (2018) 8:19. doi: 10.1038/s41408-018-0049-8
36. Meggendorfer M, Haferlach T, Alpermann T, Jeromin S, Haferlach C, Kern W, et al. Specific molecular mutation patterns delineate chronic neutrophilic leukemia, atypical chronic myeloid leukemia, and chronic myelomonocytic leukemia. Haematologica (2014) 99:e244–6. doi: 10.3324/haematol.2014.113159
37. Ouyang Y, Qiao C, Chen Y, Zhang SJ. Clinical significance of CSF3R, SRSF2 and SETBP1 mutations in chronic neutrophilic leukemia and chronic myelomonocytic leukemia. Oncotarget (2017) 8:20834–41. doi: 10.18632/oncotarget.15355
38. Dao KHT, Tyner JW. What’s different about atypical CML and chronic neutrophilic leukemia? Hematology (2015) 2015:264–71. doi: 10.1182/asheducation-2015.1.264
39. Yoshida K, Sanada M, Shiraishi Y, Nowak D, Nagata Y, Yamamoto R, et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature (2011) 478:64–9. doi: 10.1038/nature10496
40. Nooruddin Z, Miltgen N, Wei Q, Schowinsky J, Pan Z, Tobin J, et al. Changes in allele frequencies of CSF3R and SETBP1 mutations and evidence of clonal evolution in a chronic neutrophilic leukemia patient treated with ruxolitinib. Haematologica (2017) 102:e207–9. doi: 10.3324/haematol.2016.163089
41. Stoner RC, Press RD, Maxson JE, Tyner JW, Dao KHT. Insights on mechanisms of clonal evolution in chronic neutrophilic leukemia on ruxolitinib therapy. Leukemia (2019) 34:1684–8. doi: 10.1038/s41375-019-0688-1
42. Venugopal S, Mascarenhas J. Chronic neutrophilic leukemia: Current and future perspectives. Clin Lymph Myeloma Leuk (2019) 19:129–34. doi: 10.1016/j.clml.2018.11.012
43. Itonaga H, Ota S, Ikeda T, Taji H, Amano I, Hasegawa Y, et al. Allogeneic hematopoietic stem cell transplantation for the treatment of BCR-ABL1-negative atypical chronic myeloid leukemia and chronic neutrophil leukemia: A retrospective nationwide study in Japan. Leuk Res (2018) 75:50–7. doi: 10.1016/J.LEUKRES.2018.11.003
44. Hasle H, Olesen G, Kerndrup G, Philip P, Jacobsen N. Chronic neutrophil leukaemia in adolescence and young adulthood. Br J Haematol (1996) 94:628–30. doi: 10.1046/J.1365-2141.1996.7082329.X
45. Zhang X, Pan J, Guo J. Presence of the JAK2 V617F mutation in a patient with chronic neutrophilic leukemia and effective response to interferon alfa-2b. Acta Haematol (2013) 130:44–6. doi: 10.1159/000345851
46. Yassin MA, Kohla S, Al-Sabbagh A, Soliman AT, Yousif A, Moustafa A, et al. A case of chronic neutrophilic leukemia successfully treated with pegylated interferon alpha-2a. Clin Med Insights: Case Rep (2015) 8:33–6. doi: 10.4137/CCRep.s22820
47. Meyer S, Feremans W, Cantiniaux B, Capel P, Huygen K, Dicato M. Successful alpha-2b-interferon therapy for chronic neutrophilic leukemia. Am J Hematol (1993) 43:307–9. doi: 10.1002/ajh.2830430416
48. Dao KHT, Gotlib J, Deininger MMN, Oh ST, Cortes JE, Collins RH, et al. Efficacy of ruxolitinib in patients with chronic neutrophilic leukemia and atypical chronic myeloid leukemia. J Clin Oncol (2020) 38:1006–18. doi: 10.1200/JCO.19.00895
49. Shi J, Ni Y, Li J, Qiu H, Miao K. Concurrent chronic neutrophilic leukemia blast crisis and multiple myeloma: A case report and literature review. Oncol Lett (2015) 9:2208. doi: 10.3892/OL.2015.3043
50. Taiwo E, Wang H, Lewis R. Treatment of coexisting chronic neutrophilic leukemia and light chain multiple myeloma with hydroxyurea, bortezomib, and dexamethasone. Case Rep Hematol (2014) 2014:1–3. doi: 10.1155/2014/869395
A. List of screened regions/genes by Fluorescent In Situ Hybridization (FISH) in patient 1
LSI CDKN2A(9p21)(SO)/CEP9(SG) [9p21/9p11.1-9q11.1, Abbott], LSI TP53(SO)/CEP17(SG) [17p13/17p11.1-17q11.1, Abbott], LSI IGH(DC BA) [14q32, Abbott], CEP9(SO) [9p11.1-q11.1, Abbott], CKS1B(SG)/CEP1(SO) [1q21/1p11.1-1q11.1, CME + Abbott]
B. List of screened regions/genes by Fluorescent In Situ Hybridization (FISH) in patient 2
XL IGH(DC BA) [14q32, Metasystems]
C. List of screened regions/genes by Fluorescent In Situ Hybridization (FISH) in patient 3
LSI IGH(DC BA) [14q32, Vysis], LSI IGH(SG)/CMAF(SO)(DC DF) [14q32/16q23, Vysis], LSI IGH(SG)/CCND1-XT(SO) (DC DF) [14q32/11q13, Vysis], LSI IGH(SG)/FGFR3(SO) (DC DF) [14q32/4p16, Vysis]
D. List of screened regions/genes by Fluorescent In Situ Hybridization (FISH) in patient 5
LSI 4q12 (FIP1L1-PDGFRA) [Vysis tri-color rearrangement probe], LSI PDGFRB [Vysis], LSI JAK2 [Kreatech], LSI FGFR1 [Zytovision], LSI BCR/ABL(ES) [9q34.1/22q11.23, Vysis], TPI 17p13.1/CEP17(D17Z1) [Metasystems], IGH/FGFR3 (DC DF) [14q32/4p16, Metasystems], IGH/MAF (DC DF) [14q32/16q23, Metasystems]
A. List of screened genes by Next-Generation Sequencing (NGS) in patient 1,2, 3 & 4.
ABL1, ASXL1, ATRX, BCOR, BCORL1, BRAF, CALR, CBL, CBLB, CBLC, CDKN2A, CSF3R, CUX1, DNMT3A, ETV6, EZH2, FBXW7, FLT3, GATA1, GATA2, GNAS, IDH1, IDH2, IKZF1, JAK2, KMD6A, KIT, KRAS, MPL, MYD88, NOTCH1, NPM1, NRAS, PDGFRA, PHF6, PTEN, PTPN11, RAD21, RUNX1, SETBP1, SF3B1, SMC1A, SMC3, SRSF2, STAG2, TET2, TP53, U2AF1, WT1, ZRSR2
B. List of screened genes by Next-Generation Sequencing (NGS) in patient 5
ASXL1, CEBPA, DNMT3A, FLT3, IDH1, IDH2, cKIT, NPM1, RUNX1, TET2, TP53, WT1, SF3B1, SRSF2, U2AF1, JAK2, MPL, CALR, EZH2, SETBP1, CSF3R
Keywords: myeloproliferative disorders, chronic neutrophilic leukemia, monoclonal gammopathy, multiple myeloma, myeloid malignancy, myeloproliferative neoplasm
Citation: Vermeersch G, Delforge M, Havelange V, Graux C, Michaux L and Devos T (2022) Case report: Chronic neutrophilic leukemia associated with monoclonal gammopathies. A case series and review of genetic characteristics and practical management. Front. Oncol. 12:1014671. doi: 10.3389/fonc.2022.1014671
Received: 10 August 2022; Accepted: 04 November 2022;
Published: 07 December 2022.
Edited by:
Ahmad Antar, Almoosa Specialist Hospital, Saudi ArabiaReviewed by:
Guangsheng He, Nanjing Medical University, ChinaCopyright © 2022 Vermeersch, Delforge, Havelange, Graux, Michaux and Devos. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gaël Vermeersch, Z2FlbC52ZXJtZWVyc2NoQHV6bGV1dmVuLmJl
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.