 Zhu Zeng
Zhu Zeng Tao Wang2
Tao Wang2 Yuehong Wang
Yuehong Wang- 1Department of Respiratory Diseases, Thoracic Disease Center, The First Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, Zhejiang, China
- 2Department of R&D, Hangzhou Repugene Technology Co., Ltd., Hangzhou, China
- 3Zhejiang Provincial Key Laboratory of Pancreatic Disease, The First Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, China
We report a case with a novel ALK-R3HDM1 and EML4-ALK dual fusion that might be a delicate mechanism for the acquired resistance of epidermal growth factor receptor (EGFR)–tyrosine kinase inhibitor (TKI). A patient with EGFR L858R lung adenocarcinoma developed disease progression after 72.7 months of gefitinib therapy; rebiopsy was done, and next-generation sequencing showed the disappearance of the previous EGFR mutations. In addition, two new ALK fusions emerged, indicating that the emergence of dual ALK rearrangement may be the underlying mechanism of gefitinib resistance. The patient exhibits an excellent response to second-line alectinib treatment with a significant clinical benefit and a high quality of life. Finally, we summarized previous studies in which ALK fusion is a required resistance mechanism to EGFR-TKI.
Introduction
Epidermal growth factor receptor (EGFR) mutations and anaplastic lymphoma kinase (ALK) fusion represent two distinct subgroups of lung cancer, which are conventionally considered mutually exclusive (1, 2). Gefitinib has been approved as a first-line tyrosine kinase inhibitor (TKI) for treating EGFR-mutant patients. Despite the efficacy of gefitinib, disease relapse or progression is inevitable (3). The most common resistance mechanism is the secondary EGFR mutation of T790M in exon 20 (4). Recently, a few cases have been reported that required ALK rearrangement as a mechanism for EGFR-TKIs (5–7). Nevertheless, the newly emerged ALK fusion always coexists with the pre-existing EGFR mutation, and the authors concluded that the regimen of EGFR-TKI combined with ALK-TKI achieved a more satisfactory efficacy than monotherapy (5). Herein, we reported the first case of a novel ALK-R3HDM1 and EML4-ALK double fusion as an acquired resistance mechanism to gefitinib with the disappearance of the original EGFR mutations and responding to alectinib. Consent for publication in print has been obtained from the patient.
Case presentation
During his routine examination, a 65-year-old, never-smoker Chinese man was found with a nodule in the right lower lobe. The patient was assessed as acceptable for surgery, and he underwent radical resection on 28th January 2010. The final diagnosis was moderately differentiated lung adenocarcinoma (papillary mixed mucinous subtype) staged as pT1N1M0 IIA. Despite three cycles of the adjuvant chemotherapy of vinorelbine combined with carboplatin (he cannot tolerate the last cycle of chemotherapy), the patient developed multiple bilateral pulmonary in February 2014. Genetic analysis was performed in the tumor tissue of surgical specimens with EGFR exon 21 L858R mutation, ALK/ROS1/KRAS/BRAF-negative (real-time PCR for EGFR/KRAS/BRAF mutation testing, IHC for ALK fusion testing, and FISH for ROS1 fusion testing). After first-line gefitinib treatment, tumors shrink and achieve partial response (PR). The patient suffered from increasing puffy face and shortness of breath after 72.7 months of treatment with gefitinib. CT scan showed multiple supraclavicular and intrapulmonary lymph node metastases (Figure 1). Meanwhile, an ultrasonographic (US) scan on superficial lymph nodes was performed for staging; the patient had supraclavicular lymphadenopathy and received a supraclavicular lymph node biopsy. Immunohistochemistry (IHC) indicated that this lymph node was metastatic lung adenocarcinoma with ALK-positive (Figure 2). Moreover, next-generation sequencing (NGS) approved that ALK-R3HDM1(A19: R21) and EML4-ALK (E6: A20, variant 3) rearrangement coexisted in specimens from the supraclavicular lymph node (Figure 3) without EGFR mutation. The novel ALK-R3HDM1 rearrangement with an abundance of 27.48% and the EML4-ALK fusion was identified at an abundance of 20%. Unfortunately, the specimens acquired by bronchoscopy cannot meet the requirements for genetic testing, and we failed to validate the genetic profile in the lung. Thereafter, the patient received oral alectinib 600 mg bid as second-line therapy from 5 May 2020, with PR as the best response (Figure 1), and continued to receive clinical benefits from treatment. There is no clinical and radiological evidence of disease progression. The duration of response (DOR) of alectinib is over 26 months (Figure 4).
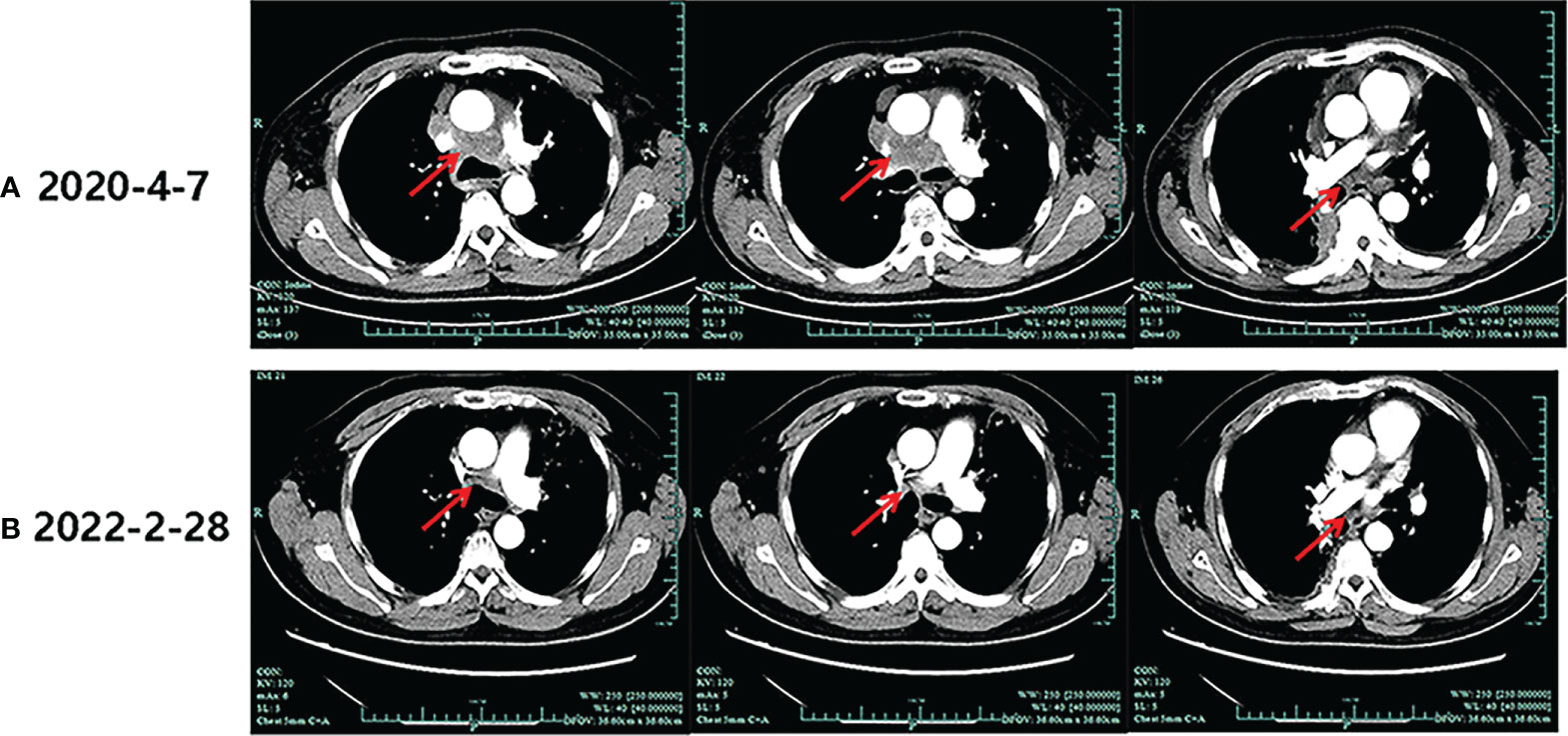
Figure 1 Imaging evolution of the patient. (A) The baseline computed tomography scan of alectinib treatment. (B) Computed tomography scan of the last follow-up of alectinib treatment.
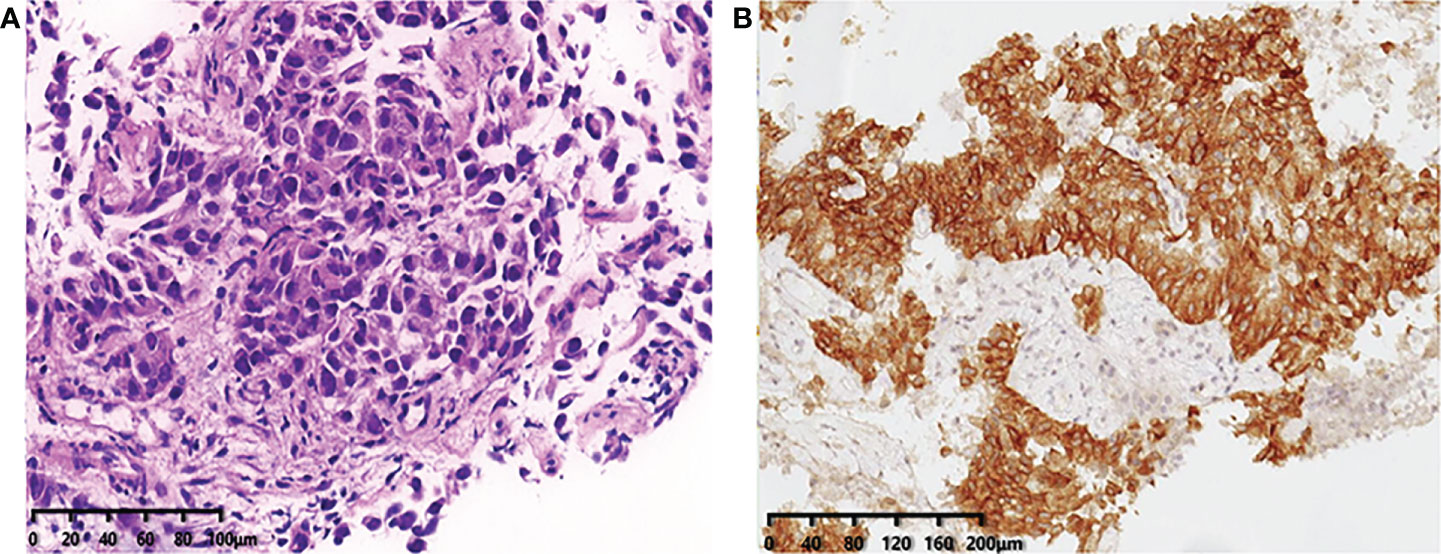
Figure 2 (A) Immunohistochemistry confirms the pathological typing of lung adenocarcinoma (×20). (B) Immunohistochemical staining of ALK expression (Ventana anti-ALK D5F3 clones, Roche-Ventana), and the pathologic assessment was positive (×20), localized in the cytoplasm. .
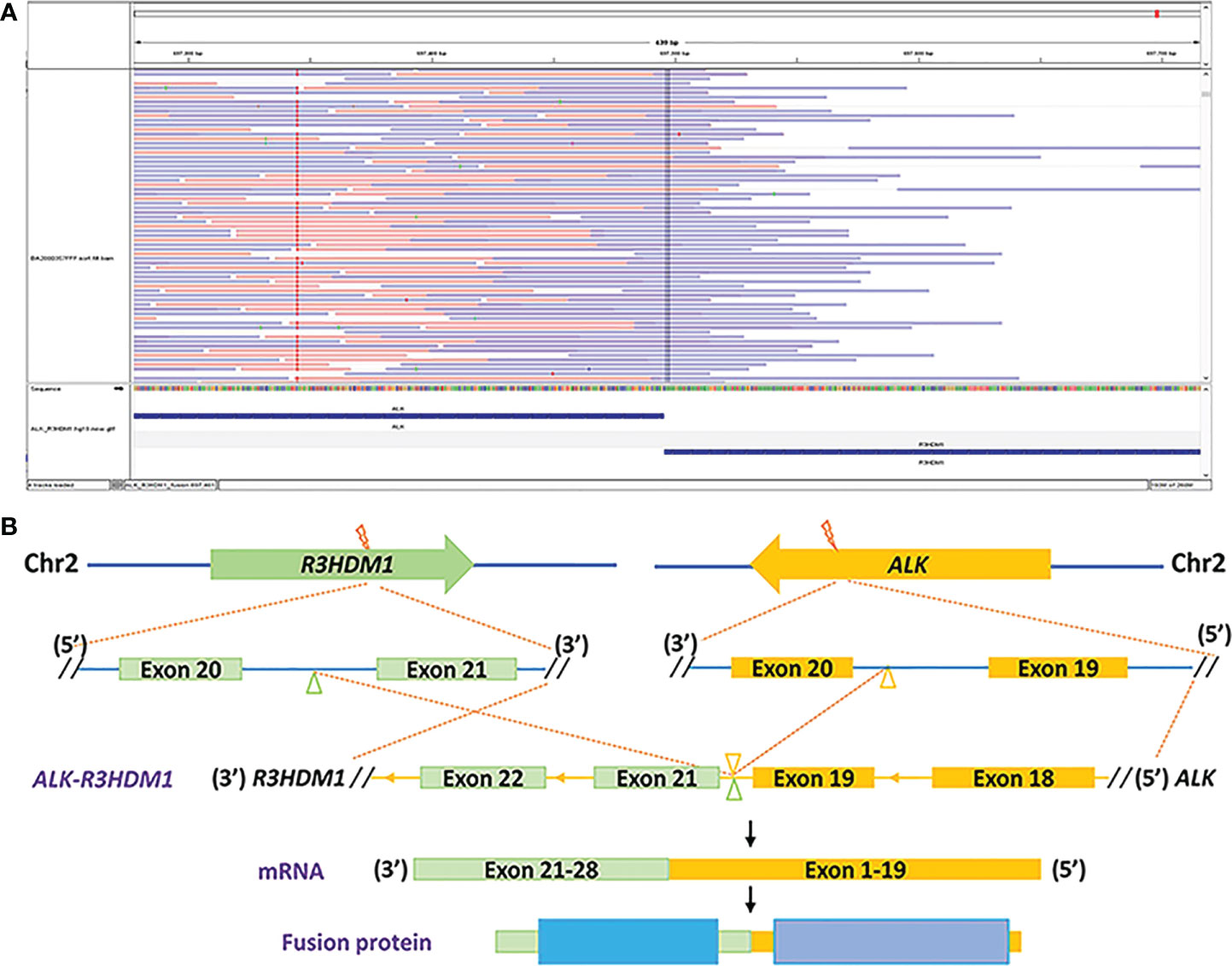
Figure 3 Identification of the ALK-R3HDM1 fusion by next-generation sequencing. (A) Integrated Genomics Viewer snapshot demonstrating the ALK-R3HDM1 translocation. (B) The schematic structure of the genomic DNA sequence shows the ALK-R3HDM1 fusion points.
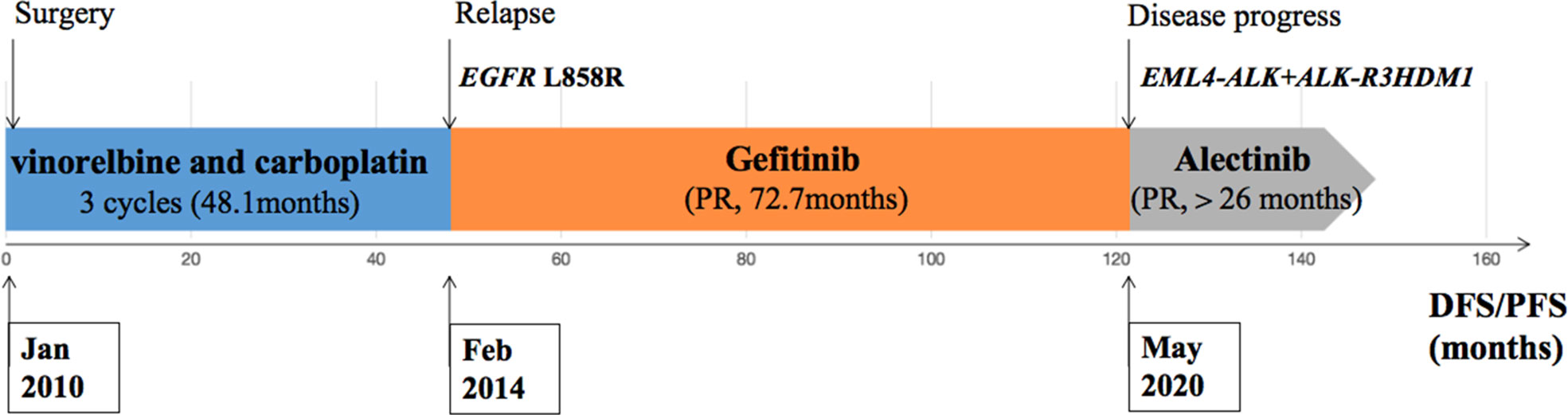
Figure 4 Description of the clinical course and pertinent molecular findings for the patient presented.
Discussion and review of the literature
Numerous researchers have demonstrated that the acquired resistance mechanisms to EGFR-TKI were highly heterogeneous. The secondary EGFR mutations, alternative pathways activation and small-cell lung cancer transformation, are associated with resistance to EGFR-TKIs (8). Recent reports reveal the emergence of de novo receptor tyrosine kinase (RTK) fusions, including ALK rearrangement, as resistance mechanisms to EGFR-TKI (7). Herein, we presented a case with ALK-R3HDM1 and EML4-ALK dual rearrangement set as a novel acquired resistance mechanism to gefitinib. To the best of our knowledge, this is the first study to report dual ALK rearrangements that participated in acquired resistance to gefitinib, with the original EGFR mutation disappearing in rebiopsy tumor tissue.
Previous studies clarified that ALK fusion is a resistance mechanism to EGFR-TKIs, but the prevalence of newly acquired ALK fusions developed during different EGFR-TKI treatments might be distinct; it could be found at a higher rate with osimertinib resistance and extremely low in first-generation EGFR-TKIs (6, 7). Table 1 summarizes 12 cases that might indicate the novel mechanism of ALK rearrangement involvement in acquired resistance to EGFR-TKI. According to NGS testing after EGFR-TKI resistance, all these patients harbored an emerging ALK rearrangement that coexists with pre-existing EGFR mutations. Among these patients, 75% (9/12) of them have detected ALK fusion after the acquired resistance to osimertinib; only one patient (8.3%, 1/12) found that ALK rearrangement may serve as a molecular mechanism underlying acquired resistance to first-line TKI (erlotinib). It is unknown whether this potential enrichment of acquired fusions is related to the more potent EGFR inhibition of osimertinib or the later-line setting after multiple lines of EGFR inhibition.
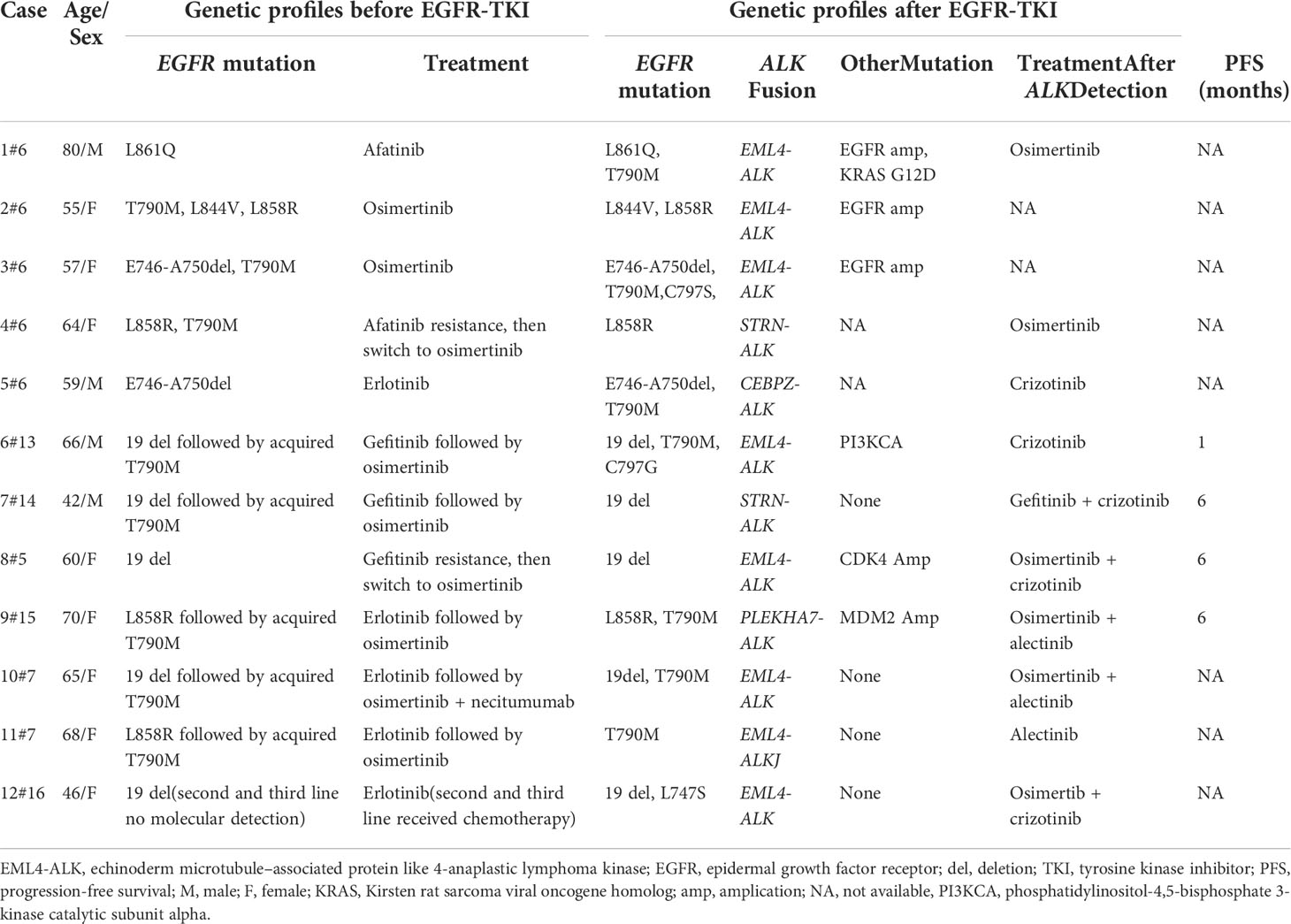
Table 1 Details of published cases of novel ALK rearrangement attributed to acquired resistance to the epidermal growth factor receptor–tyrosine kinase inhibitor.
It has been previously reported that approximately 1.3%–1.6% of non-small cell lung cancer (NSCLC) patients harbor concomitant EGFR mutations and ALK rearrangements in the baseline (9, 10). Hence, it is critical to identify truly acquired alterations from concomitant genetic abnormalities in pretreatment tissue. In this case, the initial tumor tissue from the primary surgical specimens showed only EGFR L858R mutation and negative ALK fusion, while rebiopsy from the metastatic supraclavicular lymph node at the progression of gefitinib treatment revealed the newly emerged fusions in ALK without the original EGFR mutation, indicating that the tumor progression was most likely due to the emergence of concurrent ALK- R3HDM1 and EML4-ALK fusions. Although the new ALK alteration emerged in previous studies as a resistance mechanism to EGFR-TKI, the original driver gene EGFR mutation was preserved (Table 1). Hence, previous studies indicated that dual TKI treatment might benefit EGFR-TKI-acquired resistant NSCLC patients induced by ALK rearrangement rather than the ALK inhibitor alone (5). In the current case, the absence of EGFR gene mutation in the rebiopsy tissue suggested that ALK drives oncogenesis after EGFR-TKI resistance. Hence, the patient received an exceptional response to treatment with alectinib monotherapy. Unfortunately, we only performed the NGS analysis of supraclavicular lymph nodes and could not present the detailed genetic profile by NGS in the primary lung tumor since the surgical specimens are not available.
Recently, numerous ALK fusion partners have been identified in NSCLC patients, and the response is significantly varied by the fusion partner (11). Herein, we first report a novel fusion partner RH3D1, in an NSCLC patient with EML4-ALK simultaneously. Previous studies defined these fusions as non-reciprocal/reciprocal ALK, harboring concurrent ALK fusions with at least one 3′-ALK fusion and one 5′-ALK, which may not affect inhibitor response (12). However, Zhang et al. reported that non-reciprocal/reciprocal ALK fusion is a negative prognostic factor for NSCLC patients harboring ALK fusion and a negative predictive factor for crizotinib treatment (12). This is the first case to assess the response of ALK-TKI in acquired non-reciprocal/reciprocal ALK fusion to EGFR-TKI. According to a previous study, the median PFS of patients with non-reciprocal/reciprocal ALK fusion who received first-line crizotinib was 6.1 months(n = 23, 95%CI: 5.0–11.0 months). However, our patient exhibits an excellent response to second-line alectinib treatment; the duration of response (DOR) is more than 26 months with a significant clinical benefit and a high quality of life. There are two possible reasons for this: firstly, all dual fusions reported by Yang and his colleagues were primary driver genes, whereas fusions in our patients were required after EGFR-TKI resistance, which may be attributed to a more complex molecular mechanism after drug resistance; secondly, alectinib may be more effective in the non-reciprocal/reciprocal ALK fusion than crizotinib.
It would be interesting to know how these dual ALK rearrangements contribute to the excellent response of alectinib in this patient. Further mechanistic studies are warranted to elucidate their intracellular function. Our report is also limited by the lack of tumor tissue to differentiate second primary lung cancer from intrapulmonary metastases lung cancer. A more comprehensive analysis of the primary and metastatic lesions in the genome may provide more clues to the above questions. However, owing to the lack of tumor specimens for further analyses, some crucial information was not available.
Conclusion
In summary, we report that an NSCLC patient who experienced gefitinib resistance and was shown to harbor that the emerging non-reciprocal/reciprocal ALK rearrangements achieved satisfactory efficacy with alectinib monotherapy. The finding of this new ALK-R3HDM1 fusion may provide a more molecular profiling of patients with non-reciprocal/reciprocal ALK fusions to optimize the therapeutic strategy. Furthermore, the regimen of alectinib may achieve a more satisfactory efficacy than crizotinib in patients with non-reciprocal/reciprocal ALK fusions.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
Ethics statement
This study was approved by the Ethics Committee of the First Affiliated Hospital, Zhejiang University School of Medicine. The patients/participants provided their written informed consent to participate in this study.
Author contributions
YW designed the case report, ZZ drafted the manuscript, TW and JH are responsible for data analysisand graphing. All authors contributed to the article and approved the submitted version.
Funding
This research was supported by National Natural Science Foundation of China under Grant Number 82002411 and the key projects of Natural Science Foundation of Zhejiang Province under Grant Number LZ22H160011.
Acknowledgments
The authors would like to thank all study participants and their families and are grateful for the collaboration of The First Affiliated Hospital, College of Medicine, Zhejiang University, and their staff.
Conflict of interest
Author TW was employed by Hangzhou Repugene Technology Co., Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Lin JJ, Ritterhouse LL, Ali SM, Bailey M, Schrock AB, Gainor JF, et al. ROS1 fusions rarely overlap with other oncogenic drivers in non-small cell lung cancer. J Thorac Oncol (2017) 12(5):872–7. doi: 10.1016/j.jtho.2017.01.004
2. Shaw AT, Yeap BY, Mino-Kenudson M, Digumarthy SR, Costa DB, Heist RS, et al. Clinical features and outcome of patients with non-small-cell lung cancer who harbor EML4-ALK. J Clin Oncol (2009) 27(26):4247–53. doi: 10.1200/JCO.2009.22.6993
3. Tsubata Y, Tanino R, Isobe T. Current therapeutic strategies and prospects for EGFR mutation-positive lung cancer based on the mechanisms underlying drug resistance. Cells (2021) 10(11). doi: 10.3390/cells10113192
4. Gao J, Li HR, Jin C, Jiang JH, Ding JY. Strategies to overcome acquired resistance to EGFR TKI in the treatment of non-small cell lung cancer. Clin Transl Oncol (2019) 21(10):1287–301. doi: 10.1007/s12094-019-02075-1
5. Hou H, Sun D, Zhang C, Liu D, Zhang X. ALK rearrangements as mechanisms of acquired resistance to osimertinib in EGFR mutant non-small cell lung cancer. Thorac Canc (2021) 12(6):962–9. doi: 10.1111/1759-7714.13817
6. Xu H, Shen J, Xiang J, Li H, Li B, Zhang T, et al. Characterization of acquired receptor tyrosine-kinase fusions as mechanisms of resistance to EGFR tyrosine-kinase inhibitors. Cancer Manag Res (2019) 11:6343–51. doi: 10.2147/CMAR.S197337
7. Offin M, Somwar R, Rekhtman N, Benayed R, Chang JC, Plodkowski A, et al. Acquired ALK and RET gene fusions as mechanisms of resistance to osimertinib in EGFR-mutant lung cancers. JCO Precis Oncol (2018) 2. doi: 10.1200/PO.18.00126
8. Ortiz-Cuaran S, Scheffler M, Plenker D, Dahmen L, Scheel AH, Fernandez-Cuesta L, et al. Heterogeneous mechanisms of primary and acquired resistance to third-generation EGFR inhibitors. Clin Cancer Res (2016) 22(19):4837–47. doi: 10.1158/1078-0432.CCR-15-1915
9. Ulivi P, Chiadini E, Dazzi C, Dubini A, Costantini M, Medri L, et al. Nonsquamous, non-Small-Cell lung cancer patients who carry a double mutation of EGFR, EML4-ALK or KRAS: Frequency, clinical-pathological characteristics, and response to therapy. Clin Lung Canc (2016) 17(5):384–90. doi: 10.1016/j.cllc.2015.11.004
10. Yang JJ, Zhang XC, Su J, Xu CR, Zhou Q, Tian HX, et al. Lung cancers with concomitant EGFR mutations and ALK rearrangements: diverse responses to EGFR-TKI and crizotinib in relation to diverse receptors phosphorylation. Clin Cancer Res (2014) 20(5):1383–92. doi: 10.1158/1078-0432.CCR-13-0699
11. Yoshida T, Oya Y, Tanaka K, Shimizu J, Horio Y, Kuroda H, et al. Differential crizotinib response duration among ALK fusion variants in ALK-positive non-Small-Cell lung cancer. J Clin Oncol (2016) 34(28):3383–9. doi: 10.1200/JCO.2015.65.8732
Keywords: alectinib, ALK fusion, EGFR-TKI resistance, lung cancer, non-reciprocal/reciprocal ALK
Citation: Zeng Z, Wang T, He J and Wang Y (2022) ALK-R3HDM1 and EML4-ALK fusion as a mechanism of acquired resistance to gefitinib: A case report and literature review. Front. Oncol. 12:1010084. doi: 10.3389/fonc.2022.1010084
Received: 02 August 2022; Accepted: 12 October 2022;
Published: 31 October 2022.
Edited by:
Jill Kolesar, University of Kentucky, United StatesReviewed by:
Kyoung-Ho Pyo, Yonsei University, South KoreaJames C. M. Ho, The University of Hong Kong, Hong Kong SAR, China
Copyright © 2022 Zeng, Wang, He and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yuehong Wang, eXVlaG9uZ3dAemp1LmVkdS5jbg==