
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol., 03 October 2022
Sec. Molecular and Cellular Oncology
Volume 12 - 2022 | https://doi.org/10.3389/fonc.2022.1009948
This article is part of the Research TopicReviews in Molecular and Cellular OncologyView all 47 articles
 Yangzhou Jiang1†
Yangzhou Jiang1† Yang Xiang1†Chuanchuan Lin1Weiwei Zhang1
Yang Xiang1†Chuanchuan Lin1Weiwei Zhang1 Zhenxing Yang1Lixin Xiang1Yanni Xiao1
Zhenxing Yang1Lixin Xiang1Yanni Xiao1 Li Chen1Qian Ran1*Zhongjun Li1,2*
Li Chen1Qian Ran1*Zhongjun Li1,2*Sustaining proliferative signaling and enabling replicative immortality are two important hallmarks of cancer. The complex of cyclin-dependent kinase (CDK) and its cyclin plays a decisive role in the transformation of the cell cycle and is also critical in the initiation and progression of cancer. CRIF1, a multifunctional factor, plays a pivotal role in a series of cell biological progresses such as cell cycle, cell proliferation, and energy metabolism. CRIF1 is best known as a negative regulator of the cell cycle, on account of directly binding to Gadd45 family proteins or CDK2. In addition, CRIF1 acts as a regulator of several transcription factors such as Nur77 and STAT3 and partly determines the proliferation of cancer cells. Many studies showed that the expression of CRIF1 is significantly altered in cancers and potentially regarded as a tumor suppressor. This suggests that targeting CRIF1 would enhance the selectivity and sensitivity of cancer treatment. Moreover, CRIF1 might be an indispensable part of mitoribosome and is involved in the regulation of OXPHOS capacity. Further, CRIF1 is thought to be a novel target for the underlying mechanism of diseases with mitochondrial dysfunctions. In summary, this review would conclude the latest aspects of studies about CRIF1 in cancers and mitochondria-related diseases, shed new light on targeted therapy, and provide a more comprehensive holistic view.
As an irreversible progress, the cell cycle balance of normal cells mainly depends on regulating the activities of cyclin-dependent kinases (CDKs). Cancer cells exhibit uncontrolled cell cycle progression and mitosis, due to the extraordinary proliferative ability. To sustain the capacity of rapid proliferation, cancer cells are peculiarly prone to get energy from glycolysis and display enhanced aerobic glycolysis which are referred to as the ‘Warburg effect’ (1). Researchers intend to exploit new therapeutic strategies or targets following the above two aspects to overcome the resistance encountered in cancer therapy. One strategy is to deepen the cell cycle exit, and the other one is to interfere the energy metabolism of cancer cells. However, the initial CDK inhibitors show obvious toxicities and limited efficacy, and the new class inhibitors (e.g., palbociclib, ribociclib, and abemaciclib) also display obvious tolerability (2). Recently, a novel factor (Figure 1), CR6-interacting factor 1 (CRIF1), was discovered to regulate the cell cycle progression. CRIF1 was also found to be expressed in mitochondria and to influence the OXPHOS capacity. All these suggest that CRIF1 has a considerable potential of being a new target for regulating the cell cycle and cell energy metabolism and might be applied in cancer treatment.
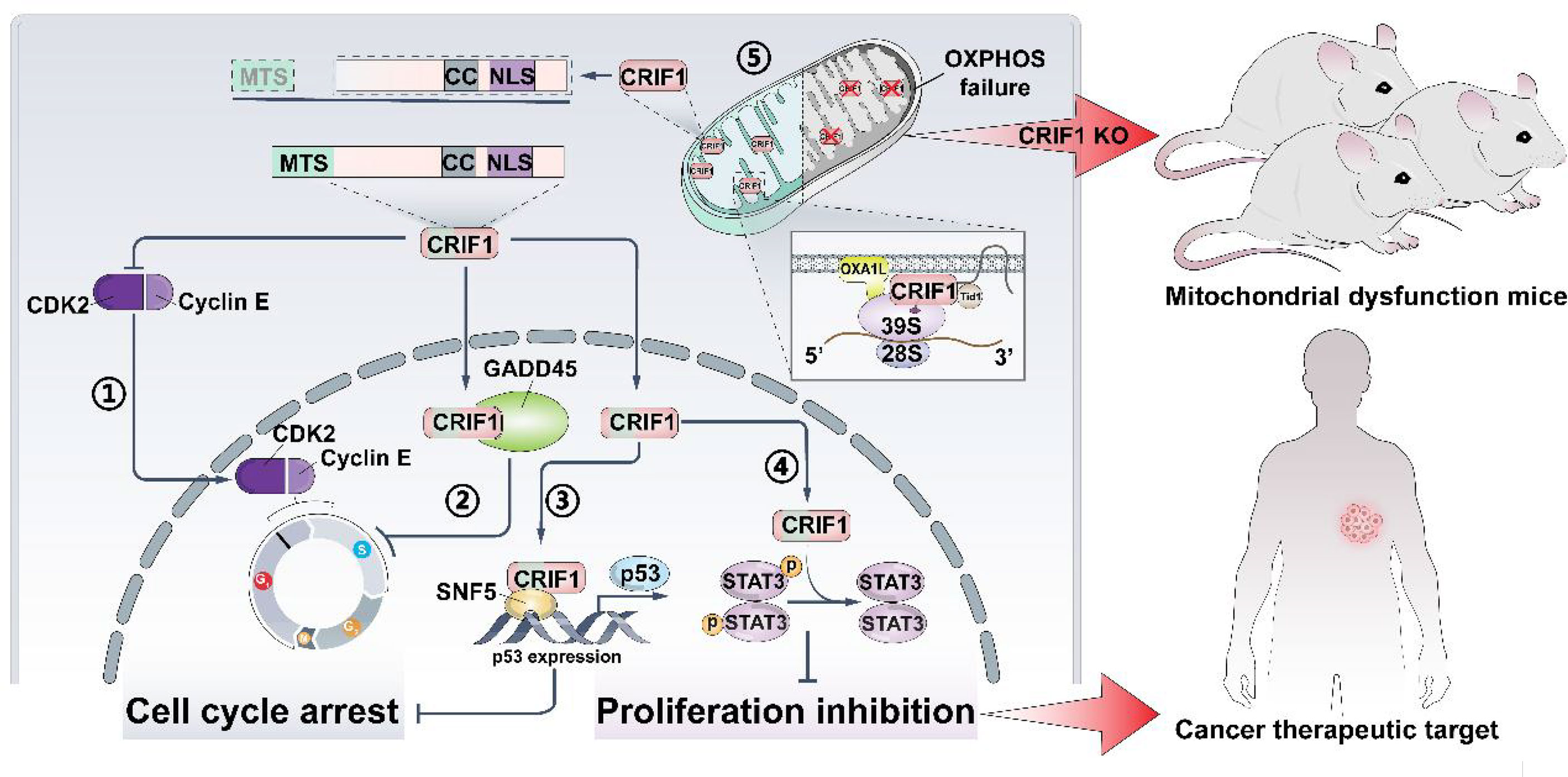
Figure 1 CRIF1 regulates cell cycle, cell proliferation, and OXPHOS capacity. It could be a cancer therapeutic target, and knocking down CRIF1 is an effective way to establish mitochondrial dysfunction in mice. CRIF1 binds to CDK2 and inhibits its activity ①. CRIF1 binds to Gadd45 family proteins and enhances its activity ②. CRIF1 increases P53 expression by coactivating with SNF5 ③. CRIF1 decreases STAT3 phosphorylation and inhibits cell proliferation ④. CRIF1 transports into the mitochondrial matrix, associates with the LSU of the mitoribosome, and regulates OXPHOS capacity ⑤.
The CRIF1 gene, which is located on chromosome 19p13.2 on the human reference genome and is known as GADD45GIP1, CKBBP2, and CKbetaBP2, holds a 669-bp open reading frame (ORF) encoding a protein of 222 amino acids. It is ubiquitously expressed in mammalian tissues, with high abundance in the heart, lymph nodes, trachea, thyroid gland, and adrenal tissues (3). In posttranslational modification of CRIF1, it is phosphorylated on serine residue at position 221 by interacting with the β subunit of CKII and promotes proliferation of cells (4). CRIF1 was originally identified as a nuclear protein which enhances the activity of Gadd45 family proteins. As the underlying role of CRIF1 further develops, it is believed that CRIF1 plays a pivotal role in cell cycle progression including the induction of cell cycle arrest through three main ways (3, 5–9). First, CRIF1 binds to the Gadd45 family proteins and consequently inhibits the activities of Cdc2. Second, CRIF1 can directly interact with CDK2 via the long alpha helical region and induces cell cycle arrest. Third, CRIF1 enhances the expression of P53 and its downstream genes including P21, which mediates P53-induced G1/S arrest. Overall, CRIF1 is an important regulator of the cell cycle and is a potential tumor suppressor. In the cancer field, it is critical to enhance the sensitivity of cancer cells to treatments (radiotherapy and drugs), while CRIF1 could be a target to turn the cancer cells resistant to sensitive and could become a novel therapeutic target due to its functions in the cell cycle.
Mitochondria are well-known organelles which provide ATP via OXPHOS for cells. However, cancer cells are prone to obtain energy by glycolysis instead of aerobic oxidation, indicating that metabolic rewiring is important in malignant transformation for cancer cells (10). Beyond energy supplies, mitochondria are central players of metabolic signaling and are potential energy sensors and beacons (11). The mitochondrial proteins are partly encoded by their own DNA (mtDNA). In mammals, mtDNA encodes 13 essential proteins which are crucial for OXPHOS (12, 13). Although CRIF1 is not encoded by mtDNA, it is identified as a mitochondrial ribosomal protein (MRP) in the large subunit (LSU) (14). The expression of CRIF1 directly regulates mitochondrial OXPHOS capacity. Mechanically, CRIF1 is a key protein in the process of integrating OXPHOS polypeptides into the mitochondrial membrane in mammals (15). In different types of human diseases, mitochondrial dysfunction characterized by impaired OXPHOS capacity is implicated in the pathological process. Consequently, to reveal the underlying mechanisms of mitochondrial dysfunction in diseases, CRIF1 is an effective target to regulate the OXPHOS capacity.
The transcription factor P53 and the CDK–cyclin complex are two crucial factors to rule the cell cycle. P53 is a well-known tumor suppressor that upregulates numerous genes resulting in cell cycle arrest, such as Gadd45 and P21. The CDK–cyclin complex determines the phosphorylation of retinoblastoma (Rb) proteins. Phosphorylated Rb (pRb) can dissociate from the Rb–E2F complex and free E2F to the nucleus and contact the E2F transcription factor binding sites in the gene promoters to promote the cell cycle progression (16). The cell cycle machinery proteins are canonical targets in cancer therapy, and many kinase inhibitors have been investigated in preclinical/clinical trial studies or applied in clinical use, especially inhibitors for CDKs (17–20). However, with the increase in clinical application, CDK inhibitors show limitations such as drug resistance, off-target effect, and minimal effects on the kinase-independent role of CDK targets. Beyond ameliorating the structure of inhibitors, researchers attempt to overcome these shortcomings by combining inhibitors with other drugs, selecting patients with identified biomarkers or optimizing treatment sequencing (21–23). Besides, CRIF1 is another protein found to directly bind to Gadd45 and CDK2. This provides a new strategy to interfere with the activity of critical enzymes in the cell cycle; thus, CRIF1 has a considerable potential to become a therapeutic target.
CRIF1 was initially reported as a nuclear protein which can interact with Gadd45 family proteins including Gadd45α, MyD118/Gadd45β, and CR6/OIG37/Gadd45γ (3). Gadd45 family proteins have been implicated in the responses of cell cycle regulation, genomic stability, and apoptosis (24–26). In the cell cycle, Gadd45 family proteins regulate G1/S and G2/M checkpoints in different cell types. Cdc2 is a downstream gene regulated by the Rb–E2F complex. Gadd45 family proteins bind to Cdc2 kinase; at the same time, CRIF1 promotes the inhibiting effect on Cdc2 kinase in an additive manner by directly interacting with Gadd45 proteins (27–30). Overexpressed CRIF1 binds to Gadd45 and suppresses the activity of some critical enzymes in the cell cycle. The percentage of G1 cells is significantly increased (3). Gadd45 family genes were always downregulated in cancers due to promoter methylation (31). Downregulated Gadd45α promoted mammary tumor formation which is driven by Ras activation (32). Further studies revealed that Gadd45 may play different roles depending on oncogenic stress. It was regarded as a prognostic marker in breast cancer and was considered as a target in breast cancer therapy (33–35). Beyond upregulating the expression of Gadd45 family genes, increasing the CRIF1 expression is a new strategy to enhance the capacity of Gadd45 family proteins in the cell cycle. Consequently, the CRIF1 potential becomes a therapeutic target that exercises cell cycle-regulating functions through directly binding to Gadd45.
Further studies found that CRIF1 interacts with CDK2 in both nucleus and cytoplasm by co-immunoprecipitation in Jurkat cells (7). In dividing cells, CDK2 is essential for G1/S transitions and S-phase progression. CDK2 is also driven by various oncogenic signaling pathways to regulate cancers, since potent oncogenes partly enhance the activity of CDK2. Specifically inhibiting CDK2 leads to significant defects in anchorage-independent growth of cancer cells and cells with mutations in oncogenes (36). The activity of CDK2 is inhibited by CRIF1, and cells are arrested in the G0/G1 phase. In line with this, Ran et al. revealed that BMMSCs regulate the cell cycle of leukemic cells by altering the CRIF1 expression. In cocultured leukemic cells, CRIF1 directly interacts with CDK2 and acts as an inhibitor to promote leukemic cells to be arrested in the G0/G1 phase (5, 7). The long alpha helical regions of CRIF1 which include His120, Glu116, and Gln112 were considered to interact with Arg200, Arg214, and Asp210 of CDK2 to form the interaction interface of CRIF1–CDK2. More interestingly, they found that interface inhibitors observably increase ionizing radiation (IR) inhibition potential from 19.9% to 59.6% (37). In cancer treatment, CDK2 inhibitors always show exceptional anticancer activity. CDK2 inhibitors have been screened for decades, and a large number of inhibitors have been identified. Dozens of inhibitors have undergone preclinical studies, and some have been approved for clinical use (38–40). The original CDK2 inhibitors were limited by its off-target effects and high toxicity. Although some improved small-molecule CDK2 inhibitors have progressed to clinical trials or clinical use, their low selectivity is the biggest obstacle encountered in application (41, 42). Different from classical inhibitors targeting the ATP-binding site, these interface inhibitors of CRIF1–CDK2 directly bind to CRIF1 and lead to CDK2 overactivation. Overactivated CDK2 selectively promotes apoptosis and G2/M arrest in osteosarcoma (OS) cell lines (37). In other words, disrupting the complex formation of CRIF1–CDK2 by targeting CRIF1 instead of CDK2 might be a novel strategy to be used for overcoming selectivity issues and regulation of cancer cell radiosensitivity.
CRIF1 also regulates the cell cycle by altering the P53 expression. P53-P21-DREAM-E2F/CHR is an important pathway identified to induce cell cycle arrest. P21 is an important transcriptional target of P53, since it mediates P53-induced G1/S arrest by forming complexes with Cdc2, CDK2, CDK3, CDK4, and CDK6 together with specific cyclins (43). The p53 mutation is always found in ~60% of colorectal cancers. Moreover, mutated p53 activates NF-κB and promotes epithelial–mesenchymal transition (EMT) (44). Researchers found that CRIF1 enhances the expression of P53 and induces G1/S arrest in HCT116 cells. Mechanically, CRIF1 associates with chromatin remodeler SNF5 to bind to the upstream promoter sequences of the p53 gene, increasing the expression of the P53 gene. Downstream target genes such as p21 and Gadd45 are activated. P21 governs Rb phosphorylation and ultimately induces G1/S arrest in HCT116 cells (9). These advances suggest that the regulation of the cell cycle must be a result of various factors, and CRIF1 is probably involved in this progress and acts as a regulator. Depending on cancer type and stage, CRIF1 perhaps exercises its functions via different signal pathways which might exhibit different effects on tumorigenesis and progression. Due to the role of CRIF1 in cell cycle regulation, it is expected to become a target for cancer therapy and to enhance the sensitization of radiotherapy and chemotherapy.
The high capacity of proliferation is a critical characteristic of cancer cells (45). Therefore, exploiting drugs to kill cancer cells with high basal level of proliferation and regeneration is another common strategy in cancer treatment. Clinically available targeted therapies always focus on blocking the constitutive activation of signal transduction pathways and regulating hormone or receptor level (46–48). For example, the mitogen-activated protein kinase (MAPK) pathway potentially acts as the most frequently mutated signaling pathway in human cancer and plays an important role in cancer cell proliferation. Targeting the MAPK pathway has led to the clinical success of BRAF and MEK inhibitors, and ERK inhibitors exhibit potential advantages of improving cancer therapy (49). However, due to cancer heterogeneity and genomic instability, these inhibitors exhibiting a high frequency of drug resistance is the biggest obstacle encountered in clinical application. The JAK-STAT pathway is characterized by rapid membrane-to-nucleus signaling transduction, and its aberrant activation is closely associated with tumor formation (50). Moreover, some steroid hormones are regarded as cancer initiators and alter the cell genotype. For example, androgen activates androgen receptors (ARs) and promotes cell proliferation via altering gene expression (51). CRIF1 is involved in cancer cell proliferation via activating several critical signal transduction pathways, regulating hormone receptors or some transcriptional factors. This suggests that researchers could target CRIF1 to inhibit cancer cell proliferation through exercising its function of regulating cell proliferation.
CRIF1 is reported as a transcriptional coactivator of STAT3 and is important for maintaining STAT3 DNA binding activity (52). STAT3 is generally regarded as a direct transcription factor and also alters gene expression through causing epigenetic changes, such as DNA methylation and chromatin modulation. Many inhibitors targeting STAT3 are exploited for cancer treatment, including antisense oligonucleotides, STAT3 decoy oligonucleotides, and small-molecule inhibitors (53). Although the future of STAT3 inhibitors as therapies is promising, the toxicity is the most important side effect that has to be overcome. As a coactivator of STAT3, CRIF1 interacts with STAT3 via its C-terminal coiled-coil domain (CCD). STAT3 target genes such as Myc, Socs3, c-Fos, and JunB would be downregulated by CRIF1 deficiency. In mice with CRIF1 deficiency, both defective cellular proliferation and increased cell death would lead to embryonic growth deficit or death (52). Beyond this, CRIF1 negatively regulates phosphorylation of STAT3 via enhancing Socs3 activity in T cells (54). Taken together, CRIF1 is essential for STAT3 transcriptional activity. However, it also negatively regulates STAT3 phosphorylation. Exploiting new drugs targeting CRIF1 to interfere STAT3 DNA binding activity or enhancing CRIF1 activity to inhibit STAT3 phosphorylation are both potential strategies in cancer treatment.
Most of prostate cancers express AR throughout the course of the progression. Tan et al. revealed that CRIF1 plays a negative role in proliferation of prostate cancer cells (55). AR is a nuclear transcriptional factor regulated by ligand and drives cancer growth. AR signaling inhibitors (ARSIs) have been exploited and applied in clinical treatment for many years. However, the application of most ARSIs has been blighted by short-lived and therapy resistance (56). AR recruits CRIF1 to endogenous AR target promoters and interacts with either the N-terminal or C-terminal half-regions of CRIF1. Thus, the transactivation of AR is repressed. Interestingly, CRIF1 interacts with the C-terminal of AR and inhibits the interaction between p160 coactivator TIF2 and the AR C-terminal region. CRIF1 leads to AR N/C interaction disruption, and the activity of the AR N-terminal-DNA binding domain fragment decreased. CRIF1 outcompetes AR coactivators and represses AR transactivation. In addition, histone deacetylase 4 can be recruited by CRIF1 which also performs a negative role in transactivation of AR (55). Taken together, the CRIF1 potential acts as a natural inhibitor of AR and suppresses its DNA binding activity in prostate cancer. It is important to reveal the underlying effect of CRIF1 in AR-overexpressed cancers and try to fix the shortcomings of classic ARSIs.
Orphan nuclear receptor Nur77 is a transcription factor which is implicated in various biological processes like apoptosis and tumorigenesis (57–60). Recently, CRIF1 has been identified as a novel coregulator which interacts with Nur77 and participates in transactivation of Nur77. CRIF1 binds to the Nur77 AB domain and suppresses the transactivation of Nur77 which is mediated by TSH (61). Ultimately, CRIF1 suppresses the expression of Nur77 downstream genes and controls cell proliferation. In addition, CRIF1 function is inhibited by interacting with lymphocyte-specific protein tyrosine kinase (Lck) and leads to promotion of leukemic T-cell survival. Lck, which belongs to Src family kinases (SFKs), is universally expressed in T cells and easily detected in blood malignancies and many other solid tumors (62–64). In hepatocellular carcinoma (HCC), CRIF1 is beneficial to patient survival. These results show that CRIF1 suppresses epithelial–mesenchymal transition (EMT) via the TGFβ signaling pathway and inhibits the expression of matrix metalloproteinase-3 (MMP3) which plays an important role in tissue remodeling and metastasis (65). Although the direct role of CRIF1 in the proliferation of tumor cells has not been confirmed, it indeed participates in the proliferation of tumor cells through the interaction with STAT3, AR, NUR77, and other important cell proliferation regulation factors. Therefore, targeting CRIF1 can affect the activity of its binding factors, which is expected to go further in tumor treatment.
CRIF1 plays a critical role in cell cycle and cell proliferation in cancer and non-cancer cells. It regulates the cell cycle via directly binding to Gadd45 and CDK2. It also enhances P53 expression. In cell proliferation, CRIF1 always acts as a negative regulator in cancer cells. Although CDK inhibitors exhibit extraordinary capacity in anticancer, the toxicity, resistance, and off-target effect are ineluctable limitations encountered in clinical application. Recently, inhibitors targeting the CRIF1–CDK2 interface have exhibited a new strategy of indirectly suppressing CDK2 activity. Exploiting effective interface inhibitors are promising to try to overcome the shortcomings of other inhibitors encountered in clinical practice (8, 37). It provides an unprecedented strategy that targets CRIF1 rather than its binding proteins to overcome these shortcomings in radiotherapy and chemotherapy.
CRIF1 was originally identified only as a nuclear protein, due to binding to Gadd45 family proteins which locate in the nuclei. There is a nuclear localization sequence (NLS) in the C-terminal. To confirm the subcellular location of CRIF1, researchers constructed GFP tags exactly at its N-terminal (3). CRIF1 was then regarded as a nuclear protein for a long time. However, further studies found that CRIF1 could interact with LSUs and ICT1 near the polypeptide exit site of the mitoribosome (66–68). Due to the limitations of the LC-MS/MS method, CRIF1 was not detected as a mitoribosomal protein in the initial screening of mitoribosomal subunits (69, 70). Through protein sequence analysis, a mitochondrial targeting sequence (MTS) was predicted in the N-terminal region of CRIF1. Kim et al. identified that when GFP was tagged at the CRIF1 N-terminal region, it was only detected in the nuclei. In contrast, C-terminally tagged CRIF1 was mainly localized in mitochondria (15). This is because NLS is in the C-terminal region and MTS is in the N-terminal region. It suggests that CRIF1 might not only regulate the cell cycle in nuclei but also potentially regulate mitochondrial function.
CRIF1 was cleaved after entering mitochondria and positioned close to the peptide exit tunnel of the mitoribosome and together with MRPL23, MRPL24, and MRPL44. Although mitoribosome biogenesis is not influenced by CRIF1, it indeed interacts with the LSU of the motoribosome and potentially acts as an integral component of the mitoribosome. Some molecular chaperones, especially Tid1, interact with CRIF1 and contributes to the translational and cooperative posttranslational integration of nascent OXPHOS subunits (14, 15). CRIF1 deficiency easily causes deficit of OXPHOS capacity and increases risk of diseases related mitochondrial dysfunction. Taken together, CRIF1 is an indispensable MRP in the mitoribosome. Its deficiency significantly causes aberrant insertion of nascent OXPHOS subunits and leads to defective intersubunit assembly and decreases translation activities of mitoribosomes associated with the inner mitochondrial membrane. Thus, CRIF1 deficiency has been implicated in aberrant energy metabolism and many diseases related to mitochondrial OXPHOS dysfunction such as vascular disorders, diabetes, and neurodegenerative diseases (Figure 2).
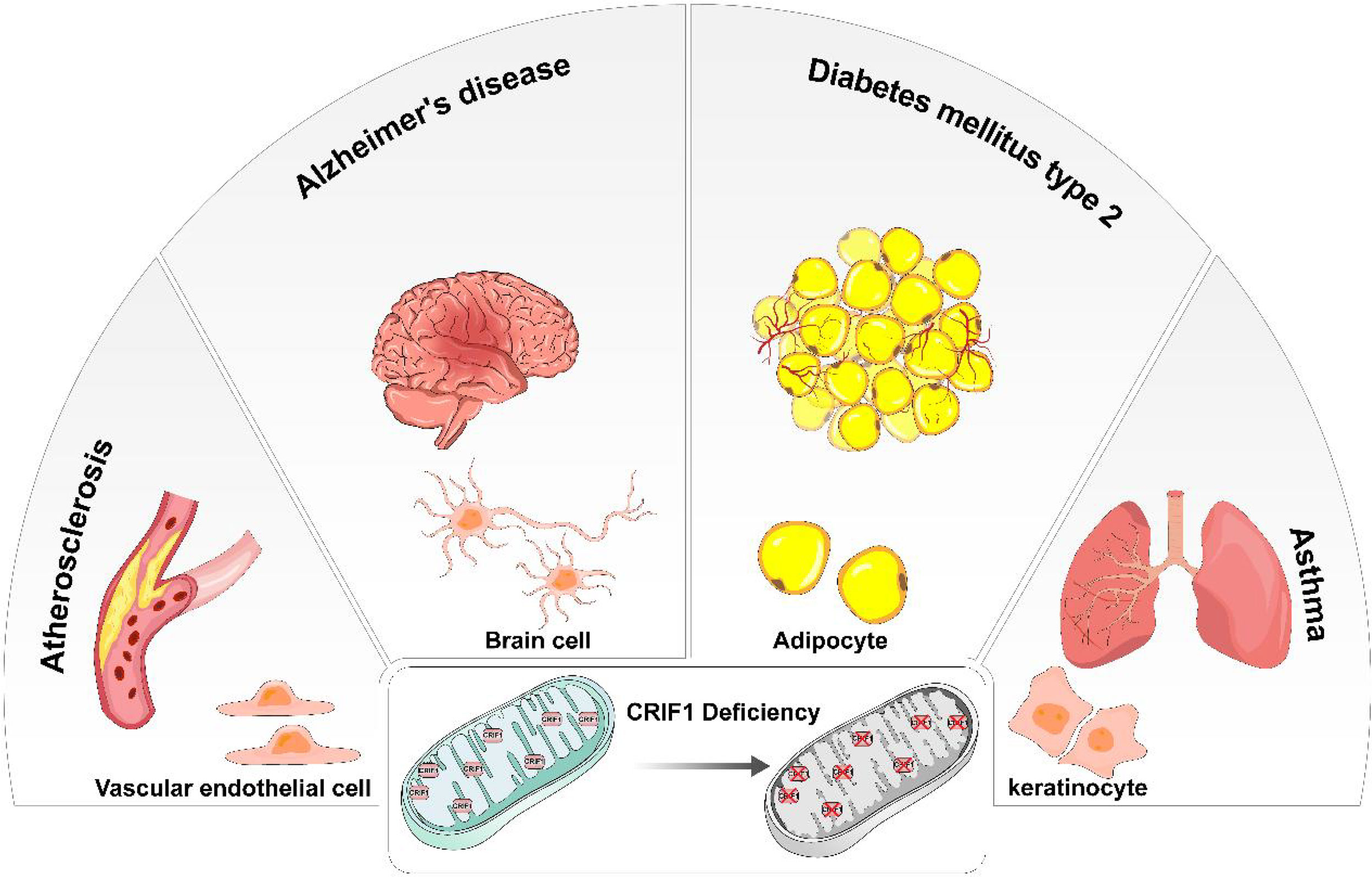
Figure 2 In different types of cells, CRIF1 deficiency induces mitochondrial dysfunction and eventually causes different diseases. In vascular endothelial cells, mitochondrial dysfunction induced by CRIF1 deficiency potentially causes atherosclerosis. In brain cells, mitochondrial dysfunction induced by CRIF1 deficiency potentially causes Alzheimer’s diseases. In adipocytes, mitochondrial dysfunction induced by CRIF1 deficiency potentially causes diabetes mellitus type 2. In keratinocytes, mitochondrial dysfunction induced by CRIF1 deficiency potentially causes asthma.
Beyond energy supporting, the function of mitochondria in endothelial cells is signaling cellular responses to environmental cues (71). In endothelial cells, dysfunction of mitochondria is an important cause of cell senescence, angiogenesis, and adaptive responses to hypoxia, inflammation, and oxidative stress (72). CRIF1 is an indispensable component of MRP in the large subunit. Deficiency of CRIF1 easily causes mitochondrial dysfunction. It might ultimately lead to cardiovascular disorders (73).
CRIF1 deficiency would cause the downregulation of deacetylase SIRT3 and SIRT1 in endothelial cells. This is associated with endothelial inflammation and senescence. Senescence and inflammation of the vascular endothelium have been identified to easily induce vascular dysfunction (e.g., atherosclerosis and hypertension). SIRT3 is located in the mitochondrial matrix and regulates ROS production through promoting the expression of key mitochondrial metabolic and detoxification proteins (74–76). Due to lack of a stabilizing effect of CRIF1, the degradation of NRF2 and PGC1α was accelerated through proteasome-dependent ubiquitination and SIRT3 levels decreased (77). This changed the mitochondrial antioxidant mechanism and ultimately led to endothelial senescence. Similarly, downregulated SIRT1 by CRIF1 deficiency easily causes endothelial inflammation. SIRT1 was identified to exhibit an antagonistic cross talk with the NF-κB pathway and inhibit p65 translocation (78). CRIF1 deficiency downregulated SIRT1 levels and promoted transcription factor NF-κB translocation to the nucleus and decreased its inhibitor IκBα. The NF-κB pathway was then activated and continued to enhance proinflammatory cytokine expression and augment cellular inflammation (79). In a model of mice with endothelial CRIF1 deficiency, severe cardiac dysfunction and premature death could be rescued by injecting exogenously the SIRT1 activator (80). SIRT1 and SIRT3 are two important protection factors which could be downregulated by CRIF1 deficiency and potentially cause cardiovascular diseases. Thus, loss of CRIF1 expression in the mitochondria easily leads to endothelial cell dysfunction and exhibits cardiovascular diseases.
In addition, CRIF1 might also regulate endothelial function by influencing the folate cycle. Lee et al. revealed that CRIF1 deficiency promotes eNOS uncoupling by inhibiting the SIRT1-eNOS pathway and the de novo BH4 synthesis pathway, resulting in reduced NO production (81). The dihydrofolate reductase expression is reduced by CRIF1 deficiency and results in increased production of homocysteine (82). The reduced NO production and increased homocysteine production both contribute to endothelial impairment.
The redox p66shc is another targeted protein to be regulated by CRIF1 in endothelial cells. The redox p66shc, a member of the ShcA adaptor protein family, has been shown to regulate the oxidative function in endothelial cells, which contributes to endothelial dysfunction (83). Decreased CRIF1 stimulates p66shc expression, and upregulated p66shc further activates the generation of reactive oxygen species (ROS). As a result, mitophagy and endothelial activation are induced by p66shc-mediated ROS in endothelial cells (84, 85). However, the effect of mitophagy in vascular disorders is controversial and needs to be further studied. Collectively, many advances approved that CRIF1 always acts as a protective factor in vascular disorders. It is a promising target to reveal the underlying mechanism of vascular disorders with mitochondrial dysfunction.
Metabolic alteration potentially plays an important role in many pathological progresses. CRIF1, as a component of the mitoribosome, could be involved in regulating mitochondrial metabolism via various ways.
Adipose tissues support various aspects of metabolism; thus, adipogenesis is important for systematic metabolism. CRIF1 is identified to play an important role in adipogenic differentiation. In the bone marrow, adipocyte tissues derive from BMSCs. After irradiation, upregulated CRIF1 contributes to break the balance of adipogenic and osteogenic differentiation and BMSCs lean toward adipogenic differentiation (86). Mechanically, upregulated CRIF1 contributes to adipogenesis probably via the PKA-cAMP signaling pathway. The change in CRIF1 expression has no effect on PKAα cat level; however, these two proteins directly interact with each other and the downstream factors could be regulated (87). CRIF1 is involved in the regulation of adipocyte differentiation and development. Within CRIF1-deficient macrophages, impaired OXPHOS capacity leads to adipose inflammation and systematic insulin resistance (88). Moreover, CRIF1 deficiency partly affects the expression of several nuclear transcription factors such as PPARγ in adipose-derived stem cells (ADSCs). At the same time, the generation of endogenous PPARγ ligands might be influenced by CRIF1 (89). In other words, CRIF1 partly regulates adipogenic genes. In addition, different levels of CRIF1 deficiency show different results in adipocytes. Homozygotic loss of CRIF1 severely impairs the development of whit adipose tissue (WAT). Ultrastructural abnormalities of mitochondria such as swollen and distorted cristae are obvious in the adipocytes of WAT. Systematic inflammation which responds to CRIF1 deficiency is caused by macrophage infiltration. In contrast, heterozygous CRIF1 deficiency in adipocytes exhibits no difference from normal adipocytes except for OXPHOS capacity (90).
Many studies reported that the primary defects of mitochondrial electron transport chain activity protect from obesity and insulin resistance (91). CRIF1 deficiency easily induces defects of mitochondrial electron transport chain activity. Therefore, mice with CRIF1-deficient hepatocytes were used to reveal the mechanisms and factors underlying the changes in systemic energy (92). It is generally recognized that induced UPRmt exerts beneficial effects on resistance of obesity and systematic metabolism (93, 94). Growth differential factor 15 (GDF15) and fibroblast growth factor 21 (FGF21) are two main mitokines which are responsive to UPRmt. Under mitochondrial stress in CRIF1-deficient cells, the level of GDF15 is increased via the p38–CHOP pathway which could regulate UPRmt. Consistent with the change in GDF15, the expression of angiopoietin-like 6 (ANGPTL6) is increased by the reduction in mitochondrial OXPHOS capacity. Moreover, ANGPTL6 increases the expression of PPARα via activating the ERK-MAPK pathway. PPARα plays a pivotal role in stimulating the secretion of FGF21 (95, 96). Both GDF15 and FGF21 confer metabolic benefits. Taken together, CRIF1 deficiency partly stimulates UPRmt and enhances insulin sensitivity. It means that CRIF1 potential regulates insulin sensitivity and is involved in the progression of diabetes.
CRIF1 expression is also associated with diabetes. Kim et al. revealed that mitochondrial diabetes and progressive loss of beta cells were caused by disruption of CRIF1 in mouse islets (97). In hearing loss mice, which come from a mouse model of diabetes, an obvious reduction of CRIF1 could be easily detected in impaired mitochondria from the cochlea (98). Furthermore, some researchers hold the view that low-level and high-level mitochondrial dysfunction caused by CRIF1 deficiency might induce opposing metabolic effects. Severe mitochondrial dysfunction which is induced by CRIF1 homodeficiency in hypothalamic proopiomelanocortin (POMC) neurons unexpectedly leads to maturity-onset obesity, reduced energy expenditure, and glucose intolerance resembling features of human diabetes mellitus type 2 (DM2) in both male and female mice. However, mild mitochondrial dysfunction which acts as CRIF1 heterodeficiency promotes a high-turnover metabolism with enhanced thermogenesis and resistance to diet-induced obesity. Further research found that the level of Pomc transcription is upregulated by mitochondrial DNA-encoded peptide (MOTS-c) in coordination with STAT3. β-Endorphin, which potentially plays an indispensable role in mediating the communication between POMC neurons and distant adipose tissues, is markedly induced by CRIF1 heterodeficiency (99). This explains the controversial role of CRIF1 played before. In contrast to CRIF1 homodeficiency which leads to type 2 diabetes-like symptoms, mild CRIF1 deficit enhances metabolism and contributes to insulin sensitivity. Taken together, CRIF1 is partly involved in regulating adipocyte differentiation and energy metabolism changes. Besides, CRIF1 deficiency has implicated in metabolic disorder and contributes to diabetes.
Many neurodegenerative diseases such as Alzheimer’s disease (AD) and Parkinson’s disease (PD) ubiquitously exhibit mitochondrial dysfunction (100, 101). Hence, the relationship between the expression of CRIF1, an indispensable component of mitochondria, and neurodegenerative diseases has aroused the interest of many researchers. As an irreversible neurodegenerative disorder with impaired cognition, AD is the most common form of dementia characterized by aberrant accumulation of amyloid beta (Aβ) peptide, tau neurofibrillary degeneration, microglial and astrocyte responses, and blood–brain barrier disruption (102). In sporadic AD, familial AD, and toxin-induced models, mitochondrial dysfunction can be easily confirmed (103). In line with this, CRIF1 expression is decreased in AD patients and AD mouse models. The accumulation of non-functional mitochondria contributes to Aβ peptide and tau pathologies, resulting in acceleration of the process of AD (104). Increasing CRIF1 expression significantly contributes to rescue Aβ-induced disruption of mitochondrial morphology. Mechanically, accumulation of Aβ mediated the increase in ROS production. Enhanced ROS and ROS-dependent sumoylation of transcription factor specificity protein 1 (SP1) mediates the reduction of CRIF1 in the pathological region of the brain (105). This suggests that CRIF1 potentially becomes a new therapeutic target to improve symptoms via cleaning Aβ. Similarly, the role of CRIF1 in the pathological process of other neurodegenerative diseases is also a valuable researching direction.
Due to CRIF1 deficiency, the differentiation of keratinocyte and hair follicle stem cells (HFSCs) is both impaired. Abnormal keratinocyte differentiation is perhaps related to compromising skin barrier and antimicrobial function, eventually resulting in epithelial dysregulation in asthma, eosinophilic esophagitis, and allergic rhinosinusitis (106). Mechanically, CRIF1 deficiency could also reduce epidermal proliferation and increase apoptosis, resulting in disrupted skin homeostasis. In addition, the hair growth cycle is significantly retarded via the Wnt/β-catenin signaling pathway which is downregulated by deletion of CRIF1 (107, 108).
A recent study shows that, disrupting the electron transport chain (ETC) by deleting CRIF1, mice exhibit upregulated resistance to sepsis. It can be a reason to explain why mitoribosome–target antibiotics have beneficial effects on disease tolerance which are irrelevant to their antibacterial activity (109). It suggests that CRIF1 is an important part of ETC and CRIF1 deficiency-caused ETC mild perturbation is beneficial to induce resistance of diseases such as sepsis. Moreover, CRIF1 could be a downstream target of lymphocyte expansion molecule (LEM) and benefit to activate cytotoxic CD8+ T cells (CTL) (110).
Although CRIF1 does not control mitoribosome biogenesis, it is indeed an indispensable component of large subunits of the mitoribosome. CRIF1 deficiency easily causes defects of OXPHOS capacity and leads to mitochondrial dysfunction. Many studies establish a mitochondrial dysfunction model by knocking down CRIF1 for investigating related diseases. CRIF1 deficiency occurs in many diseases, but there are no drugs or therapy targeting CRIF1 to try to cure these diseases. With the underlying mechanism of CRIF1 further developing, it is expected that exploiting drugs is based on fixing CRIF1 deficit to apply in treatment of diseases with mitochondrial dysfunction. Besides, Warburg proposed that persistent and irreversible injury of mitochondria is a key reason that turns normal cells into cancer cells (111). Moreover, cancer cells get energy depending on glycolysis, which has been identified as a biomarker of cancer progression (112, 113). Mitochondrial dysfunction has been identified to regulate normal cell or cancer cell growth via initiating several signaling pathways (114–116). Rapidly growing cancer cells can become glycolytic and be unable to upregulate OXPHOS indicating partial mitochondrial dysfunction (117–119). However, the role of CRIF1 playing between mitochondrial dysfunction and cancer cell proliferation is still unclear and needs further study.
Since CRIF1 distributes both in cell nuclei and in mitochondria, it might regulate cancer cell progression through genomic and non-genomic ways. Moreover, CRIF1 plays a pivotal role in regulating OXPHOS capacity. Moreover, it can generally be regarded as a molecular target to establish a mitochondrial dysfunction model for studying mitochondria-related diseases. Whether CRIF1 affects mitochondrial energy supplication and simultaneously affects cell proliferation should be a further discussion. With the increasing obstacles encountered in cancer therapy, the studies of CRIF1 and its mechanism in regulating cell cycle and cell proliferation might be a promising direction of cancer therapy in the future. Moreover, exploiting therapies and drugs based on targeting CRIF1 exhibits considerable potential in cancer treatment. This review would provide a fascinating snapshot of many of these roles in cancer cell progression, targeted cancer therapy, and treatment of diseases related to mitochondrial dysfunction, providing a more comprehensive holistic view of CRIF1.
ZL and QR contributed to conception and design of the review. YJ and YX wrote the first draft of the manuscript. WZ, ZY and LX wrote sections of the manuscript. CL drew the figure. YNX and LC revised the manuscript. All authors contributed to manuscript revision, read and approved the submitted version.
This work was supported by projects of the International Cooperation and Exchanges NSFC (No. 82020108025), National Natural Science Foundation of China (No. 81972973), and Chongqing Science and Technology Commission (cstc2021jcyj-jqX0004).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Icard P, Fournel L, Wu Z, Alifano M, Lincet H. Interconnection between metabolism and cell cycle in cancer. Trends Biochem Sci (2019) 44(6):490–501. doi: 10.1016/j.tibs.2018.12.007
2. Ingham M, Schwartz GK. Cell-cycle therapeutics come of age. J Clin Oncol (2017) 35(25):2949–59. doi: 10.1200/JCO.2016.69.0032
3. Chung HK, Yi YW, Jung NC, Kim D, Suh JM, Kim H, et al. CR6-interacting factor 1 interacts with Gadd45 family proteins and modulates the cell cycle. J Biol Chem (2003) 278(30):28079–88. doi: 10.1074/jbc.M212835200
4. Oh NS, Yoon SH, Lee WK, Choi JY, Min do S, Bae YS. Phosphorylation of CKBBP2/CRIF1 by protein kinase CKII promotes cell proliferation. Gene (2007) 386(1-2):147–53. doi: 10.1016/j.gene.2006.08.023
5. Ran Q, Hao P, Xiao Y, Xiang L, Ye X, Deng X, et al. CRIF1 interacting with CDK2 regulates bone marrow microenvironment-induced G0/G1 arrest of leukemia cells. PloS One (2014) 9(2):e85328. doi: 10.1371/journal.pone.0085328
6. Ran Q, Jin F, Xiang Y, Xiang L, Wang Q, Li F, et al. CRIF1 as a potential target to improve the radiosensitivity of osteosarcoma. Proc Natl Acad Sci U S A. (2019) 116(41):20511–6. doi: 10.1073/pnas.1906578116
7. Zhang X, Ran Q, Li Z, Liu Y, Liang X, Chen X. Cell cycle arrest of jurkat cells by leukemic bone marrow stromal cells: possible mechanisms and involvement of CRIF1. Transplant Proc (2011) 43(7):2770–3. doi: 10.1016/j.transproceed.2011.05.048
8. Sang X, Belmessabih N, Wang R, Stephen P, Lin SX. CRIF1-CDK2 interface inhibitors enhance taxol inhibition of the lethal triple-negative breast cancer. Cancers (Basel). (2022) 14(4):989. doi: 10.3390/cancers14040989
9. Yan HX, Zhang YJ, Zhang Y, Ren X, Shen YF, Cheng MB, et al. CRIF1 enhances p53 activity via the chromatin remodeler SNF5 in the HCT116 colon cancer cell lines. Biochim Biophys Acta Gene Regul Mech (2017) 1860(4):516–22. doi: 10.1016/j.bbagrm.2017.02.006
10. Porporato PE, Filigheddu N, Pedro JMB, Kroemer G, Galluzzi L. Mitochondrial metabolism and cancer. Cell Res (2018) 28(3):265–80. doi: 10.1038/cr.2017.155
11. Nunnari J, Suomalainen A. Mitochondria: in sickness and in health. Cell (2012) 148(6):1145–59. doi: 10.1016/j.cell.2012.02.035
12. Annesley SJ, Fisher PR. Mitochondria in health and disease. Cells (2019) 8(7):680. doi: 10.3390/cells8070680
13. Schmidt O, Pfanner N, Meisinger C. Mitochondrial protein import: from proteomics to functional mechanisms. Nat Rev Mol Cell Biol (2010) 11(9):655–67. doi: 10.1038/nrm2959
14. Koc EC, Cimen H, Kumcuoglu B, Abu N, Akpinar G, Haque ME, et al. Identification and characterization of CHCHD1, AURKAIP1, and CRIF1 as new members of the mammalian mitochondrial ribosome. Front Physiol (2013) 4:183. doi: 10.3389/fphys.2013.00183
15. Kim SJ, Kwon MC, Ryu MJ, Chung HK, Tadi S, Kim YK, et al. CRIF1 is essential for the synthesis and insertion of oxidative phosphorylation polypeptides in the mammalian mitochondrial membrane. Cell Metab (2012) 16(2):274–83. doi: 10.1016/j.cmet.2012.06.012
16. Giacinti C, Giordano A. RB and cell cycle progression. Oncogene (2006) 25(38):5220–7. doi: 10.1038/sj.onc.1209615
17. O’Leary B, Finn RS, Turner NC. Treating cancer with selective CDK4/6 inhibitors. Nat Rev Clin Oncol (2016) 13(7):417–30. doi: 10.1038/nrclinonc.2016.26
18. Pack LR, Daigh LH, Chung M, Meyer T. Clinical CDK4/6 inhibitors induce selective and immediate dissociation of p21 from cyclin d-CDK4 to inhibit CDK2. Nat Commun (2021) 12(1):3356. doi: 10.1038/s41467-021-23612-z
19. Xu XQ, Pan XH, Wang TT, Wang J, Yang B, He QJ, et al. Intrinsic and acquired resistance to CDK4/6 inhibitors and potential overcoming strategies. Acta Pharmacol Sin (2021) 42(2):171–8. doi: 10.1038/s41401-020-0416-4
20. Liu -Q, Xiang -Z, Zhu -H. The transcription factor E2F2 regulates tumor development. Blood&Genomics (2020) 4(1):45. doi: 10.46701/BG.2020012020102
21. Freeman-Cook K, Hoffman RL, Miller N, Almaden J, Chionis J, Zhang Q, et al. Expanding control of the tumor cell cycle with a CDK2/4/6 inhibitor. Cancer Cell (2021) 39(10):1404–21.e11. doi: 10.1016/j.ccell.2021.08.009
22. Braal CL, Jongbloed EM, Wilting SM, Mathijssen RHJ, Koolen SLW, Jager A. Inhibiting CDK4/6 in breast cancer with palbociclib, ribociclib, and abemaciclib: Similarities and differences. Drugs (2021) 81(3):317–31. doi: 10.1007/s40265-020-01461-2
23. Spring LM, Wander SA, Andre F, Moy B, Turner NC, Bardia A. Cyclin-dependent kinase 4 and 6 inhibitors for hormone receptor-positive breast cancer: past, present, and future. Lancet (2020) 395(10226):817–27. doi: 10.1016/S0140-6736(20)30165-3
24. Wang XW, Zhan Q, Coursen JD, Khan MA, Kontny HU, Yu L, et al. GADD45 induction of a G2/M cell cycle checkpoint. Proc Natl Acad Sci U S A. (1999) 96(7):3706–11. doi: 10.1073/pnas.96.7.3706
25. Tamura RE, de Vasconcellos JF, Sarkar D, Libermann TA, Fisher PB, Zerbini LF. GADD45 proteins: central players in tumorigenesis. Curr Mol Med (2012) 12(5):634–51. doi: 10.2174/156652412800619978
26. Salvador JM, Brown-Clay JD, Fornace AJ Jr. Gadd45 in stress signaling, cell cycle control, and apoptosis. Adv Exp Med Biol (2013) 793:1–19. doi: 10.1007/978-1-4614-8289-5_1
27. Fan W, Richter G, Cereseto A, Beadling C, Smith KA. Cytokine response gene 6 induces p21 and regulates both cell growth and arrest. Oncogene (1999) 18(47):6573–82. doi: 10.1038/sj.onc.1203054
28. Jin S, Tong T, Fan W, Fan F, Antinore MJ, Zhu X, et al. GADD45-induced cell cycle G2-m arrest associates with altered subcellular distribution of cyclin B1 and is independent of p38 kinase activity. Oncogene (2002) 21(57):8696–704. doi: 10.1038/sj.onc.1206034
29. Yang Q, Manicone A, Coursen JD, Linke SP, Nagashima M, Forgues M, et al. Identification of a functional domain in a GADD45-mediated G2/M checkpoint. J Biol Chem (2000) 275(47):36892–8. doi: 10.1074/jbc.M005319200
30. Jin S, Antinore MJ, Lung FD, Dong X, Zhao H, Fan F, et al. The GADD45 inhibition of Cdc2 kinase correlates with GADD45-mediated growth suppression. J Biol Chem (2000) 275(22):16602–8. doi: 10.1074/jbc.M000284200
31. Zerbini LF, Libermann TA. GADD45 deregulation in cancer: frequently methylated tumor suppressors and potential therapeutic targets. Clin Cancer Res (2005) 11(18):6409–13. doi: 10.1158/1078-0432.CCR-05-1475
32. Tront JS, Hoffman B, Liebermann DA. Gadd45a suppresses ras-driven mammary tumorigenesis by activation of c-jun NH2-terminal kinase and p38 stress signaling resulting in apoptosis and senescence. Cancer Res (2006) 66(17):8448–54. doi: 10.1158/0008-5472.CAN-06-2013
33. Tront JS, Huang Y, Fornace AJ Jr., Hoffman B, Liebermann DA. Gadd45a functions as a promoter or suppressor of breast cancer dependent on the oncogenic stress. Cancer Res (2010) 70(23):9671–81. doi: 10.1158/0008-5472.CAN-10-2177
34. Tront JS, Willis A, Huang Y, Hoffman B, Liebermann DA. Gadd45a levels in human breast cancer are hormone receptor dependent. J Transl Med (2013) 11:131. doi: 10.1186/1479-5876-11-131
35. Yu KD, Di GH, Li WF, Rao NY, Fan L, Yuan WT, et al. Genetic contribution of GADD45A to susceptibility to sporadic and non-BRCA1/2 familial breast cancers: a systematic evaluation in Chinese populations. Breast Cancer Res Treat (2010) 121(1):157–67. doi: 10.1007/s10549-009-0516-9
36. Horiuchi D, Huskey NE, Kusdra L, Wohlbold L, Merrick KA, Zhang C, et al. Chemical-genetic analysis of cyclin dependent kinase 2 function reveals an important role in cellular transformation by multiple oncogenic pathways. Proc Natl Acad Sci U S A. (2012) 109(17):e1019-27. doi: 10.1073/pnas.1111317109
37. Ran Q, Xiang Y, Stephen P, Wu C, Li T, Lin SX, et al. CRIF1-CDK2 interface inhibitors: An unprecedented strategy for modulation of cell radiosensitivity. J Am Chem Soc (2019) 141(4):1420–4. doi: 10.1021/jacs.8b10207
38. Coxon CR, Anscombe E, Harnor SJ, Martin MP, Carbain B, Golding BT, et al. Cyclin-dependent kinase (CDK) inhibitors: Structure-activity relationships and insights into the CDK-2 selectivity of 6-substituted 2-arylaminopurines. J Med Chem (2017) 60(5):1746–67. doi: 10.1021/acs.jmedchem.6b01254
39. Cheng W, Yang Z, Wang S, Li Y, Wei H, Tian X, et al. Recent development of CDK inhibitors: An overview of CDK/inhibitor co-crystal structures. Eur J Med Chem (2019) 164:615–39. doi: 10.1016/j.ejmech.2019.01.003
40. Lin T, Li J, Liu L, Li Y, Jiang H, Chen K, et al. Design, synthesis, and biological evaluation of 4-benzoylamino-1H-pyrazole-3-carboxamide derivatives as potent CDK2 inhibitors. Eur J Med Chem (2021) 215:113281. doi: 10.1016/j.ejmech.2021.113281
41. Tadesse S, Caldon EC, Tilley W, Wang S. Cyclin-dependent kinase 2 inhibitors in cancer therapy: An update. J Med Chem (2019) 62(9):4233–51. doi: 10.1021/acs.jmedchem.8b01469
42. Liu J, Peng Y, Wei W. Cell cycle on the crossroad of tumorigenesis and cancer therapy. Trends Cell Biol (2022) 32(1):30–44. doi: 10.1016/j.tcb.2021.07.001
43. Engeland K. Cell cycle regulation: p53-p21-RB signaling. Cell Death Differ (2022) 29(5):946–60. doi: 10.1038/s41418-022-00988-z
44. Nakayama M, Oshima M. Mutant p53 in colon cancer. J Mol Cell Biol (2019) 11(4):267–76. doi: 10.1093/jmcb/mjy075
45. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144(5):646–74. doi: 10.1016/j.cell.2011.02.013
46. Clara JA, Monge C, Yang Y, Takebe N. Targeting signalling pathways and the immune microenvironment of cancer stem cells - a clinical update. Nat Rev Clin Oncol (2020) 17(4):204–32. doi: 10.1038/s41571-019-0293-2
47. Lupulescu AP. Hormones, vitamins, and growth factors in cancer treatment and prevention. A Crit appraisal. Cancer. (1996) 78(11):2264–80. doi: 10.1002/(SICI)1097-0142(19961201)78:11<2264::AIDCNCR2>3.0.CO;2-N
48. Patel HK, Bihani T. Selective estrogen receptor modulators (SERMs) and selective estrogen receptor degraders (SERDs) in cancer treatment. Pharmacol Ther (2018) 186:1–24. doi: 10.1016/j.pharmthera.2017.12.012
49. Liu F, Yang X, Geng M, Huang M. Targeting ERK, an achilles’ heel of the MAPK pathway, in cancer therapy. Acta Pharm Sin B (2018) 8(4):552–62. doi: 10.1016/j.apsb.2018.01.008
50. Hu X, Li J, Fu M, Zhao X, Wang W. The JAK/STAT signaling pathway: from bench to clinic. Signal Transduct Target Ther (2021) 6(1):402. doi: 10.1038/s41392-021-00791-1
51. Feitelson MA, Arzumanyan A, Kulathinal RJ, Blain SW, Holcombe RF, Mahajna J, et al. Sustained proliferation in cancer: Mechanisms and novel therapeutic targets. Semin Cancer Biol (2015) 35(Suppl):S25–s54. doi: 10.1016/j.semcancer.2015.02.006
52. Kwon MC, Koo BK, Moon JS, Kim YY, Park KC, Kim NS, et al. Crif1 is a novel transcriptional coactivator of STAT3. EMBO J (2008) 27(4):642–53. doi: 10.1038/sj.emboj.7601986
53. Bharadwaj U, Kasembeli MM, Robinson P, Tweardy DJ. Targeting janus kinases and signal transducer and activator of transcription 3 to treat inflammation, fibrosis, and cancer: Rationale, progress, and caution. Pharmacol Rev (2020) 72(2):486–526. doi: 10.1124/pr.119.018440
54. Park JS, Choi SY, Hwang SH, Kim SM, Choi J, Jung KA, et al. CR6-interacting factor 1 controls autoimmune arthritis by regulation of signal transducer and activator of transcription 3 pathway and T helper type 17 cells. Immunology (2019) 156(4):413–21. doi: 10.1111/imm.13042
55. Tan JA, Bai S, Grossman G, Titus MA, Harris Ford O, Pop EA, et al. Mechanism of androgen receptor corepression by CKβBP2/CRIF1, a multifunctional transcription factor coregulator expressed in prostate cancer. Mol Cell Endocrinol (2014) 382(1):302–13. doi: 10.1016/j.mce.2013.09.036
56. Jamroze A, Chatta G, Tang DG. Androgen receptor (AR) heterogeneity in prostate cancer and therapy resistance. Cancer Lett (2021) 518:1–9. doi: 10.1016/j.canlet.2021.06.006
57. Zhan Y, Du X, Chen H, Liu J, Zhao B, Huang D, et al. Cytosporone b is an agonist for nuclear orphan receptor Nur77. Nat Chem Biol (2008) 4(9):548–56. doi: 10.1038/nchembio.106
58. Hu M, Luo Q, Alitongbieke G, Chong S, Xu C, Xie L, et al. Celastrol-induced Nur77 interaction with TRAF2 alleviates inflammation by promoting mitochondrial ubiquitination and autophagy. Mol Cell (2017) 66(1):141–53.e6. doi: 10.1016/j.molcel.2017.03.008
59. Yang PB, Hou PP, Liu FY, Hong WB, Chen HZ, Sun XY, et al. Blocking PPARγ interaction facilitates Nur77 interdiction of fatty acid uptake and suppresses breast cancer progression. Proc Natl Acad Sci U S A. (2020) 117(44):27412–22. doi: 10.1073/pnas.2002997117
60. Niu B, Liu J, Lv B, Lin J, Li X, Wu C, et al. Interplay between transforming growth factor-β and Nur77 in dual regulations of inhibitor of differentiation 1 for colonic tumorigenesis. Nat Commun (2021) 12(1):2809. doi: 10.1038/s41467-021-23048-5
61. Park KC, Song KH, Chung HK, Kim H, Kim DW, Song JH, et al. CR6-interacting factor 1 interacts with orphan nuclear receptor Nur77 and inhibits its transactivation. Mol Endocrinol (2005) 19(1):12–24. doi: 10.1210/me.2004-0107
62. Zepecki JP, Snyder KM, Moreno MM, Fajardo E, Fiser A, Ness J, et al. Regulation of human glioma cell migration, tumor growth, and stemness gene expression using a lck targeted inhibitor. Oncogene (2019) 38(10):1734–50. doi: 10.1038/s41388-018-0546-z
63. Zheng X, Liao X, Nie L, Lin T, Xu H, Yang L, et al. LCK and CD3E orchestrate the tumor microenvironment and promote immunotherapy response and survival of muscle-invasive bladder cancer patients. Front Cell Dev Biol (2021) 9:748280. doi: 10.3389/fcell.2021.748280
64. Weiße J, Rosemann J, Müller L, Kappler M, Eckert AW, Glaß M, et al. Identification of lymphocyte cell-specific protein-tyrosine kinase (LCK) as a driver for invasion and migration of oral cancer by tumor heterogeneity exploitation. Mol Cancer. (2021) 20(1):88. doi: 10.1186/s12943-021-01384-w
65. Zhuang R, Lu D, Zhuo J, Zhang X, Wang K, Wei X, et al. CR6-interacting factor 1 inhibits invasiveness by suppressing TGF-β-mediated epithelial-mesenchymal transition in hepatocellular carcinoma. Oncotarget (2017) 8(55):94759–68. doi: 10.18632/oncotarget.21925
66. Kramer G, Rauch T, Rist W, Vorderwülbecke S, Patzelt H, Schulze-Specking A, et al. L23 protein functions as a chaperone docking site on the ribosome. Nature (2002) 419(6903):171–4. doi: 10.1038/nature01047
67. Sharma MR, Koc EC, Datta PP, Booth TM, Spremulli LL, Agrawal RK. Structure of the mammalian mitochondrial ribosome reveals an expanded functional role for its component proteins. Cell (2003) 115(1):97–108. doi: 10.1016/S0092-8674(03)00762-1
68. Richter R, Rorbach J, Pajak A, Smith PM, Wessels HJ, Huynen MA, et al. A functional peptidyl-tRNA hydrolase, ICT1, has been recruited into the human mitochondrial ribosome. EMBO J (2010) 29(6):1116–25. doi: 10.1038/emboj.2010.14
69. Koc EC, Burkhart W, Blackburn K, Moyer MB, Schlatzer DM, Moseley A, et al. The large subunit of the mammalian mitochondrial ribosome. analysis of the complement of ribosomal proteins present. J Biol Chem (2001) 276(47):43958–69. doi: 10.1074/jbc.M106510200
70. Suzuki T, Terasaki M, Takemoto-Hori C, Hanada T, Ueda T, Wada A, et al. Structural compensation for the deficit of rRNA with proteins in the mammalian mitochondrial ribosome. systematic analysis of protein components of the large ribosomal subunit from mammalian mitochondria. J Biol Chem (2001) 276(24):21724–36. doi: 10.1074/jbc.M100432200
71. Kluge MA, Fetterman JL, Vita JA. Mitochondria and endothelial function. Circ Res (2013) 112(8):1171–88. doi: 10.1161/CIRCRESAHA.111.300233
72. Morris G, Puri BK, Olive L, Carvalho A, Berk M, Walder K, et al. Endothelial dysfunction in neuroprogressive disorders-causes and suggested treatments. BMC Med (2020) 18(1):305. doi: 10.1186/s12916-020-01749-w
73. Davidson SM. Endothelial mitochondria and heart disease. Cardiovasc Res (2010) 88(1):58–66. doi: 10.1093/cvr/cvq195
74. Lombard DB, Alt FW, Cheng HL, Bunkenborg J, Streeper RS, Mostoslavsky R, et al. Mammalian Sir2 homolog SIRT3 regulates global mitochondrial lysine acetylation. Mol Cell Biol (2007) 27(24):8807–14. doi: 10.1128/MCB.01636-07
75. Kincaid B, Bossy-Wetzel E. Forever young: SIRT3 a shield against mitochondrial meltdown, aging, and neurodegeneration. Front Aging Neurosci (2013) 5:48. doi: 10.3389/fnagi.2013.00048
76. Yu W, Dittenhafer-Reed KE, Denu JM. SIRT3 protein deacetylates isocitrate dehydrogenase 2 (IDH2) and regulates mitochondrial redox status. J Biol Chem (2012) 287(17):14078–86. doi: 10.1074/jbc.M112.355206
77. Kim S, Piao S, Lee I, Nagar H, Choi SJ, Shin N, et al. CR6 interacting factor 1 deficiency induces premature senescence via SIRT3 inhibition in endothelial cells. Free Radic Biol Med (2020) 150:161–71. doi: 10.1016/j.freeradbiomed.2020.02.017
78. Yang XD, Tajkhorshid E, Chen LF. Functional interplay between acetylation and methylation of the RelA subunit of NF-kappaB. Mol Cell Biol (2010) 30(9):2170–80. doi: 10.1128/MCB.01343-09
79. Piao S, Lee JW, Nagar H, Jung SB, Choi S, Kim S, et al. CR6 interacting factor 1 deficiency promotes endothelial inflammation by SIRT1 downregulation. PloS One (2018) 13(2):e0192693. doi: 10.1371/journal.pone.0192693
80. Piao S, Lee I, Jin SA, Kim S, Nagar H, Choi SJ, et al. SIRT1 activation attenuates the cardiac dysfunction induced by endothelial cell-specific deletion of CRIF1. Biomedicines (2021) 9(1):52. doi: 10.3390/biomedicines9010052
81. Lee I, Kim S, Nagar H, Choi SJ, Jeon BH, Piao S, et al. CR6-interacting factor 1 deficiency reduces endothelial nitric oxide synthase activity by inhibiting biosynthesis of tetrahydrobiopterin. Sci Rep (2020) 10(1):842. doi: 10.1038/s41598-020-57673-9
82. Lee I, Piao S, Kim S, Nagar H, Choi SJ, Jeon BH, et al. CRIF1 deficiency increased homocysteine production by disrupting dihydrofolate reductase expression in vascular endothelial cells. Antioxidants (Basel) (2021) 10(11):1645. doi: 10.3390/antiox10111645
83. Kumar S. P66Shc and vascular endothelial function. Biosci Rep (2019) 39(4):2134. doi: 10.1042/BSR20182134
84. Piao S, Nagar H, Kim S, Lee I, Choi SJ, Kim T, et al. CRIF1 deficiency induced mitophagy via p66shc-regulated ROS in endothelial cells. Biochem Biophys Res Commun (2020) 522(4):869–75. doi: 10.1016/j.bbrc.2019.11.109
85. Nagar H, Jung SB, Kwon SK, Park JB, Shong M, Song HJ, et al. CRIF1 deficiency induces p66shc-mediated oxidative stress and endothelial activation. PloS One (2014) 9(6):e98670. doi: 10.1371/journal.pone.0098670
86. Xiang LX, Ran Q, Chen L, Xiang Y, Li FJ, Zhang XM, et al. CR6-interacting factor-1 contributes to osteoclastogenesis by inducing receptor activator of nuclear factor κB ligand after radiation. World J Stem Cells (2020) 12(3):222–40. doi: 10.4252/wjsc.v12.i3.222
87. Zhang X, Xiang L, Ran Q, Liu Y, Xiang Y, Xiao Y, et al. Crif1 promotes adipogenic differentiation of bone marrow mesenchymal stem cells after irradiation by modulating the PKA/CREB signaling pathway. Stem Cells (2015) 33(6):1915–26. doi: 10.1002/stem.2019
88. Jung SB, Choi MJ, Ryu D, Yi HS, Lee SE, Chang JY, et al. Reduced oxidative capacity in macrophages results in systemic insulin resistance. Nat Commun (2018) 9(1):1551. doi: 10.1038/s41467-018-03998-z
89. Ryu MJ, Kim SJ, Choi MJ, Kim YK, Lee MH, Lee SE, et al. Mitochondrial oxidative phosphorylation reserve is required for hormone- and PPARγ agonist-induced adipogenesis. Mol Cells (2013) 35(2):134–41. doi: 10.1007/s10059-012-2257-1
90. Ryu MJ, Kim SJ, Kim YK, Choi MJ, Tadi S, Lee MH, et al. Crif1 deficiency reduces adipose OXPHOS capacity and triggers inflammation and insulin resistance in mice. PloS Genet (2013) 9(3):e1003356. doi: 10.1371/journal.pgen.1003356
91. Wredenberg A, Freyer C, Sandström ME, Katz A, Wibom R, Westerblad H, et al. Respiratory chain dysfunction in skeletal muscle does not cause insulin resistance. Biochem Biophys Res Commun (2006) 350(1):202–7. doi: 10.1016/j.bbrc.2006.09.029
92. Kang SG, Choi MJ, Jung SB, Chung HK, Chang JY, Kim JT, et al. Differential roles of GDF15 and FGF21 in systemic metabolic adaptation to the mitochondrial integrated stress response. iScience (2021) 24(3):102181. doi: 10.1016/j.isci.2021.102181
93. Kenny TC, Gomez ML, Germain D. Mitohormesis, UPR(mt), and the complexity of mitochondrial DNA landscapes in cancer. Cancer Res (2019) 79(24):6057–66. doi: 10.1158/0008-5472.CAN-19-1395
94. Deng P, Haynes CM. Mitochondrial dysfunction in cancer: Potential roles of ATF5 and the mitochondrial UPR. Semin Cancer Biol (2017) 47:43–9. doi: 10.1016/j.semcancer.2017.05.002
95. Chung HK, Ryu D, Kim KS, Chang JY, Kim YK, Yi HS, et al. Growth differentiation factor 15 is a myomitokine governing systemic energy homeostasis. J Cell Biol (2017) 216(1):149–65. doi: 10.1083/jcb.201607110
96. Kang SG, Yi HS, Choi MJ, Ryu MJ, Jung S, Chung HK, et al. ANGPTL6 expression is coupled with mitochondrial OXPHOS function to regulate adipose FGF21. J Endocrinol (2017) 233(1):105–18. doi: 10.1530/JOE-16-0549
97. Kim YK, Joung KH, Ryu MJ, Kim SJ, Kim H, Chung HK, et al. Disruption of CR6-interacting factor-1 (CRIF1) in mouse islet beta cells leads to mitochondrial diabetes with progressive beta cell failure. Diabetologia (2015) 58(4):771–80. doi: 10.1007/s00125-015-3506-y
98. Lyu AR, Kim TH, Shin SA, Kim EH, Yu Y, Gajbhiye A, et al. Hearing impairment in a mouse model of diabetes is associated with mitochondrial dysfunction, synaptopathy, and activation of the intrinsic apoptosis pathway. Int J Mol Sci (2021) 22(16):8807. doi: 10.3390/ijms22168807
99. Kang GM, Min SH, Lee CH, Kim JY, Lim HS, Choi MJ, et al. Mitohormesis in hypothalamic POMC neurons mediates regular exercise-induced high-turnover metabolism. Cell Metab (2021) 33(2):334–49.e6. doi: 10.1016/j.cmet.2021.01.003
100. Swerdlow RH. Mitochondria and cell bioenergetics: increasingly recognized components and a possible etiologic cause of alzheimer’s disease. Antioxid Redox Signal (2012) 16(12):1434–55. doi: 10.1089/ars.2011.4149
101. Shoshan-Barmatz V, Nahon-Crystal E, Shteinfer-Kuzmine A, Gupta R. VDAC1, mitochondrial dysfunction, and alzheimer’s disease. Pharmacol Res (2018) 131:87–101. doi: 10.1016/j.phrs.2018.03.010
102. Serrano-Pozo A, Das S, Hyman BT. APOE and alzheimer’s disease: advances in genetics, pathophysiology, and therapeutic approaches. Lancet Neurol (2021) 20(1):68–80. doi: 10.1016/S1474-4422(20)30412-9
103. Macdonald R, Barnes K, Hastings C, Mortiboys H. Mitochondrial abnormalities in parkinson’s disease and alzheimer’s disease: can mitochondria be targeted therapeutically? Biochem Soc Trans (2018) 46(4):891–909. doi: 10.1042/BST20170501
104. Kerr JS, Adriaanse BA, Greig NH, Mattson MP, Cader MZ, Bohr VA, et al. Mitophagy and alzheimer’s disease: Cellular and molecular mechanisms. Trends Neurosci (2017) 40(3):151–66. doi: 10.1016/j.tins.2017.01.002
105. Byun J, Son SM, Cha MY, Shong M, Hwang YJ, Kim Y, et al. CR6-interacting factor 1 is a key regulator in aβ-induced mitochondrial disruption and pathogenesis of alzheimer’s disease. Cell Death Differ (2015) 22(6):959–73. doi: 10.1038/cdd.2014.184
106. Goleva E, Berdyshev E, Leung DY. Epithelial barrier repair and prevention of allergy. J Clin Invest. (2019) 129(4):1463–74. doi: 10.1172/JCI124608
107. Shin JM, Choi DK, Sohn KC, Kim JY, Im M, Lee Y, et al. Targeted deletion of Crif1 in mouse epidermis impairs skin homeostasis and hair morphogenesis. Sci Rep (2017) 7:44828. doi: 10.1038/srep44828
108. Shin JM, Ko JW, Choi CW, Lee Y, Seo YJ, Lee JH, et al. Deficiency of Crif1 in hair follicle stem cells retards hair growth cycle in adult mice. PloS One (2020) 15(4):e0232206. doi: 10.1371/journal.pone.0232206
109. Colaço HG, Barros A, Neves-Costa A, Seixas E, Pedroso D, Velho T, et al. Tetracycline antibiotics induce host-dependent disease tolerance to infection. Immunity (2021) 54(1):53–67.e7. doi: 10.1016/j.immuni.2020.09.011
110. Okoye I, Wang L, Pallmer K, Richter K, Ichimura T, Haas R, et al. T Cell metabolism. the protein LEM promotes CD8⁺ T cell immunity through effects on mitochondrial respiration. Science (2015) 348(6238):995–1001. doi: 10.1126/science.aaa7516
111. Warburg O. On the origin of cancer cells. Science (1956) 123(3191):309–14. doi: 10.1126/science.123.3191.309
112. Cuezva JM, Krajewska M, de Heredia ML, Krajewski S, Santamaría G, Kim H, et al. The bioenergetic signature of cancer: a marker of tumor progression. Cancer Res (2002) 62(22):6674–81. doi: 10.1002/cncr.10960
113. Bando H, Atsumi T, Nishio T, Niwa H, Mishima S, Shimizu C, et al. Phosphorylation of the 6-phosphofructo-2-kinase/fructose 2,6-bisphosphatase/PFKFB3 family of glycolytic regulators in human cancer. Clin Cancer Res (2005) 11(16):5784–92. doi: 10.1158/1078-0432.CCR-05-0149
114. Rustin P. Mitochondria, from cell death to proliferation. Nat Genet (2002) 30(4):352–3. doi: 10.1038/ng0402-352
115. Arnould T, Vankoningsloo S, Renard P, Houbion A, Ninane N, Demazy C, et al. CREB activation induced by mitochondrial dysfunction is a new signaling pathway that impairs cell proliferation. EMBO J (2002) 21(1-2):53–63. doi: 10.1093/emboj/21.1.53
116. Elstrom RL, Bauer DE, Buzzai M, Karnauskas R, Harris MH, Plas DR, et al. Akt stimulates aerobic glycolysis in cancer cells. Cancer Res (2004) 64(11):3892–9. doi: 10.1158/0008-5472.CAN-03-2904
117. Wu M, Neilson A, Swift AL, Moran R, Tamagnine J, Parslow D, et al. Multiparameter metabolic analysis reveals a close link between attenuated mitochondrial bioenergetic function and enhanced glycolysis dependency in human tumor cells. Am J Physiol Cell Physiol (2007) 292(1):C125–36. doi: 10.1152/ajpcell.00247.2006
118. Wang GL, Semenza GL. General involvement of hypoxia-inducible factor 1 in transcriptional response to hypoxia. Proc Natl Acad Sci U S A. (1993) 90(9):4304–8. doi: 10.1073/pnas.90.9.4304
Keywords: CR6-interacting factor 1, cell cycle regulation, cell proliferation regulation, cancer treatment, mitochondrial ribosomal protein, mitochondrial dysfunction
Citation: Jiang Y, Xiang Y, Lin C, Zhang W, Yang Z, Xiang L, Xiao Y, Chen L, Ran Q and Li Z (2022) Multifunctions of CRIF1 in cancers and mitochondrial dysfunction. Front. Oncol. 12:1009948. doi: 10.3389/fonc.2022.1009948
Received: 02 August 2022; Accepted: 14 September 2022;
Published: 03 October 2022.
Edited by:
Kevin Ni, St George Hospital Cancer Care Centre, AustraliaReviewed by:
Lijian Shao, University of Illinois at Chicago, United StatesCopyright © 2022 Jiang, Xiang, Lin, Zhang, Yang, Xiang, Xiao, Chen, Ran and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qian Ran, bG91aXNlLXItcUAxNjMuY29t; Zhongjun Li, am9obm5leXVzY0BnbWFpbC5jb20=
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.