 Daniela Criscuolo
Daniela Criscuolo Rosario Avolio
Rosario Avolio Danilo Swann Matassa
Danilo Swann Matassa Franca Esposito
Franca Esposito
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 23 November 2021
Sec. Cancer Metabolism
Volume 11 - 2021 | https://doi.org/10.3389/fonc.2021.797265
This article is part of the Research Topic Developing Combined Modality Therapy with Mitochondria-Targeting Strategy View all 8 articles
Extensive metabolic remodeling is a fundamental feature of cancer cells. Although early reports attributed such remodeling to a loss of mitochondrial functions, it is now clear that mitochondria play central roles in cancer development and progression, from energy production to synthesis of macromolecules, from redox modulation to regulation of cell death. Biosynthetic pathways are also heavily affected by the metabolic rewiring, with protein synthesis dysregulation at the hearth of cellular transformation. Accumulating evidence in multiple organisms shows that the metabolic functions of mitochondria are tightly connected to protein synthesis, being assembly and activity of respiratory complexes highly dependent on de novo synthesis of their components. In turn, protein synthesis within the organelle is tightly connected with the cytosolic process. This implies an entire network of interactions and fine-tuned regulations that build up a completely under-estimated level of complexity. We are now only preliminarily beginning to reconstitute such regulatory level in human cells, and to perceive its role in diseases. Indeed, disruption or alterations of these connections trigger conditions of proteotoxic and energetic stress that could be potentially exploited for therapeutic purposes. In this review, we summarize the available literature on the coordinated regulation of mitochondrial and cytosolic mRNA translation, and their effects on the integrity of the mitochondrial proteome and functions. Finally, we highlight the potential held by this topic for future research directions and for the development of innovative therapeutic approaches.
Mitochondria are cellular organelles with a double-membrane structure that perform several crucial functions for the homeostasis of eukaryotic cells. Their main role is to generate chemical energy through the oxidative phosphorylation (OXPHOS) system, which is composed of five multi-subunit respiratory complexes associated to the inner mitochondrial membrane (IMM). Additionally, they work as biosynthetic hubs for the synthesis of amino acids, nucleotides, lipid heme and iron-sulphur clusters (1, 2). Moreover, mitochondria control the redox homeostasis and regulate cell death pathways (3, 4).
Mammalian mitochondria originate from the endocytosis of a bacterial ancestor by a pre-eukaryotic cell (5). Although mitochondria still maintain their own genome, during evolution most of the original endosymbiont genes were lost or transferred to the nuclear genome of the host cell (6). The mitochondrial DNA (mtDNA) is found in multiple copies in the mitochondrial matrix and, in humans, consist of approximately 16,000 base pairs encoding 13 polypeptides, all of which are key subunits of the OXPHOS complexes, 2 mitochondrial ribosomal RNA (mt-rRNA) and 22 transfer RNA (mt-tRNAs), that allow the translation of the 13 mitochondrial protein-coding RNAs. Indeed, translation of these mitochondrial messenger RNAs (mt-mRNAs) requires a dedicated translation apparatus, which is located in the mitochondrial matrix and includes mt-rRNAs, mt-tRNAs, nuclear-encoded translation factors and organelle-specific ribosomes. However, most mitochondria-resident proteins, including many subunits of the OXPHOS complexes, are encoded by the nuclear genome, synthesized by cytoplasmic ribosomes and then imported into the mitochondria (7). Therefore, accurate assembly of respiratory complexes requires a tight coordination between cytosolic and mitochondrial translation and efficient protein quality control (PQC) mechanisms to monitor protein import and turnover (8, 9).
Despite early theories on the metabolic characteristics of cancer cells postulated a loss of mitochondrial functions as a key feature of cellular transformation, it is now evident that this feature is often crucial for tumor development and progression (10). Moreover, several studies have shown that different cancer cells predominantly use mitochondrial respiration to satisfy their bioenergetic and biosynthetic demands, especially when moving towards a metastatic or chemoresistant phenotype (11–13). Accordingly, upregulation of the mitochondrial translational machinery has been reported to support the energy needs of cancer cells favoring tumor progression. Therapeutic approaches that interfere with mitochondrial translation, directly, by targeting mitoribosomes, or indirectly, by altering mitochondrial PQC systems, have recently attract great attention as anticancer strategies.
Here, we review the main mechanisms affecting mitochondrial protein homeostasis. First, we provide an overview of mitochondrial translation, and we focus on how it is strictly interconnected to the cytosolic apparatus. Then, we describe the importance of mitochondrial protein quality control systems in coordinating these translational programs, and present the case of the molecular chaperone TRAP1, likely first example of a protein with dual localization that participate in the regulation of proteostasis on both sides of the mitochondrial membranes. Finally, we provide some hints about dysfunctions of mitochondrial protein homeostasis and cancer development, highlighting the most relevant therapeutic approaches proposed so far in the field.
Mitochondria have their own genome and translational machinery that allow synthesis and assembly of OXPHOS complexes, which are in turn responsible for the generation of most of the cellular energy. The mtDNA is compacted with an array of proteins in a structure called “nucleoid” that resembles the bacterial one. The protein components of the nucleoid are transcription and replication factors such as the mitochondrial transcription factor A (TFAM), mitochondrial single-strand binding protein (mtSSB), POLG, and mtRNA polymerase (POLRMT) (14). Other factors seem not to bind directly mtDNA, but are rather peripheral nucleoid proteins involved in scaffolding, helping translation and interaction with cellular signaling components (14). Among these ADAT3 (ATPase AAA domain-containing protein 3), PHB1 (Prohibitin 1), PHB2 and M19/MNF1 (mitochondrial nucleoid factor 1). The core nucleoid component POLRMT, an RNA polymerase structurally similar to the T3 and T7 bacteriophages one, in association with TFAM, is instead responsible for the transcription process. Notably, it has been recently described the first-in-class specific inhibitor of mitochondrial transcription that target the human POLRMT. This compound (IMT1) has shown relevant anti-tumor effects in mouse xenograft, with no significant toxicity in normal tissues (15). These findings represent a promising novel weapon in the fight for cancer treatment, but also a useful tool to study the role of mtDNA expression in physiology and disease.
However, to the current knowledge, mitochondrial gene expression is predominantly regulated at post-transcriptional level through the modulation of mRNA maturation and stability (16). Transcription of the mitochondrial genome by the RNA polymerase generates long polycistronic precursors containing mRNA and rRNA coding sequences flanked by tRNAs. Mitochondrial RNAs are processed by two endonucleases, RNase P and RNase Z (ELAC2), which cleave the 5’- and 3’-ends respectively, excising the tRNAs and releasing the rRNAs and mRNAs, a process known as tRNA punctuation (17). Subsequently, all mt-mRNAs, except ND6, are polyadenylated at 3’ termini by the mitochondrial polyA polymerase (mtPAP) (18). The mt-mRNA polyadenylation creates a functional stop codon, as 7 of 13 transcripts have incomplete stop codons in their coding sequence (19). Moreover, polyadenylation regulates the half-life of specific subset of mRNAs, increasing the stability of some transcripts and decreasing that of others by targeting them for degradation (20). Proteins that regulate mt-mRNA maturation and degradation determine which subsets of mitochondrial transcripts have to be translated, by affecting the availability of functional transcripts that can be engaged by the mitoribosomes.
LRPPRC (leucine-rich pentatricopeptide repeat containing), and Fas-activated serine/threonine kinase (FASTK) are protein families playing a major role in mt-mRNA stability and translation and whose dysregulation is related to diverse pathological processes, including cancer. LRPPRC, in complex with SLIRP (SRA stem loop-interacting RNA-binding protein), behaves as a mRNA chaperone, by preventing the formation of secondary structures, and affects the stability of mitochondrial transcriptome (21, 22). In particular, LRPPRC-SLIRP complex promotes mt-mRNA stability, by preventing their degradation, and polyadenylation, by simulating mtPAP activity (22). Moreover, LRPPRC-SLIRP complex has also been shown to stabilize a pool of non-translating transcripts that are not engaged with mitoribosomes (23). It is clear that LRPPRC-SLIRP complex is necessary for coordinated mitochondrial translation as its loss causes dysregulations, increasing translation of some transcripts and inhibiting translation of others (23, 24). Interestingly, human LRPPRC–SLIRP complex preferentially binds the human mt-Cyb RNA, whereas the mouse complex preferentially recognized the mouse corresponding transcript (21), and therefore that preferred locations of LRPPRC-binding sites within mitochondrial RNAs differ from mouse to humans. This testifies the importance of these proteins in dictating the local RNA structures that are critical in the lifecycles of mitochondrial RNAs. Several studies have shown that LRPPRC is upregulated in different cancer tissues and cell lines, including prostate, gastric, lung and colon cancer (25). Moreover, LRPPRC has been proposed as prognostic biomarker for gastric cancer. Indeed, a higher LRPPRC expression was found in cancer tissues compared to paired noncancerous regions and in patients with a poor survival rate (26).
FASTK family proteins are particularly expressed in the mitochondrial matrix, where they post-transcriptionally regulate the expression of different mitochondrial transcripts (27). For instance, FASTK interacts with the ND6 mRNA to prevent its degradation, by ensuring correct biogenesis of the complex I, whereas FASTKD1 negatively regulates complex I activity by destabilizing the ND3 mRNA (28, 29). Recently, a pan-cancer analysis showed that FASTK genes are frequently mutated in different cancer types highlighting the potential role of FASTK family members as therapeutic targets. In particular, gene amplification was found for FASTK and FASTKD3 in ovarian and lung cancers, respectively, while increased mRNA levels of all FASTK members were found in esophageal, stomach, liver and lung cancers (30).
A detailed discussion of the complex mechanisms involved in mitochondrial translation is beyond the scope of this article. For a comprehensive view of this topic, we recommend further reading [e.g (31)]. Hereafter, we provide a brief overview of the mitochondrial translation process, as well as of synthesis and import of the nuclear-encoded, mitochondria-destined proteins, with the aim to highlight how recent advances on this topic could open new scenarios on the mechanisms involved in metabolic remodeling in diseases, providing solid bases for future therapeutic approaches.
Translation of mt-mRNAs occurs on specialized ribosome resident in the mitochondrial matrix: the mitoribosomes. The mammalian 55S mitoribosomes are macromolecular complexes composed of two subunits, the large 39S subunit (LSU) and the small 28S subunit (SSU). The 39S subunit contains 16S mt-rRNA and 52 mitoribosomal proteins (MRPs), whereas the 28S subunits is composed of 12S mt-rRNA and 30 MRPs (32). Mitochondrial rRNAs are exclusively encoded by mtDNA, whereas MRPs are all encoded by the nuclear genome, translated in the cytosol and then imported into the mitochondrial matrix to be assembled coordinately with mt-rRNAs to form functional ribosomes. Mitoribosomes assembly takes place in close proximity to mtDNA, probably in mitochondrial RNA granules or mitochondriolus, membraneless structures containing MRPs, mitoribosome assembly factors and rRNA modifying enzymes required for post-transcriptional processing of mt-RNAs (33, 34).
Although mitoribosomes are evolutionarily derived from bacterial ribosomes, they have strongly diverged from them in terms of composition, function, and structure, by acquiring mitochondrial-specific proteins, and exhibiting differences in the number and total amount of the rRNAs. These structural changes have been accompanied by a strong functional specialization, considering that mammalian mitoribosomes exclusively synthesize membrane proteins, represented by components of the respiratory complexes, which functionally explains the acquired feature of mitochondrial ribosomes to be permanently attached to the IMM (32). The 13 proteins encoded by the mtDNA are indeed all subunits of respiratory chain complexes and, as such, are highly hydrophobic polypeptides predominantly associated with the IMM. To avoid unproductive protein aggregations, the mitochondrial translation products are cotranslationally inserted into the IMM (35). Accordingly, the mitoribosomes extensively interact with the IMM to facilitate the membrane insertion of nascent polypeptides. In particular, the LSU subunit MRPL45 anchors the mitoribosome to the IMM aligning the polypeptide exit tunnel with the insertase OXA1L, that mediates the co-translational insertion of newly synthetized proteins into the IMM (36, 37) (Figure 1).
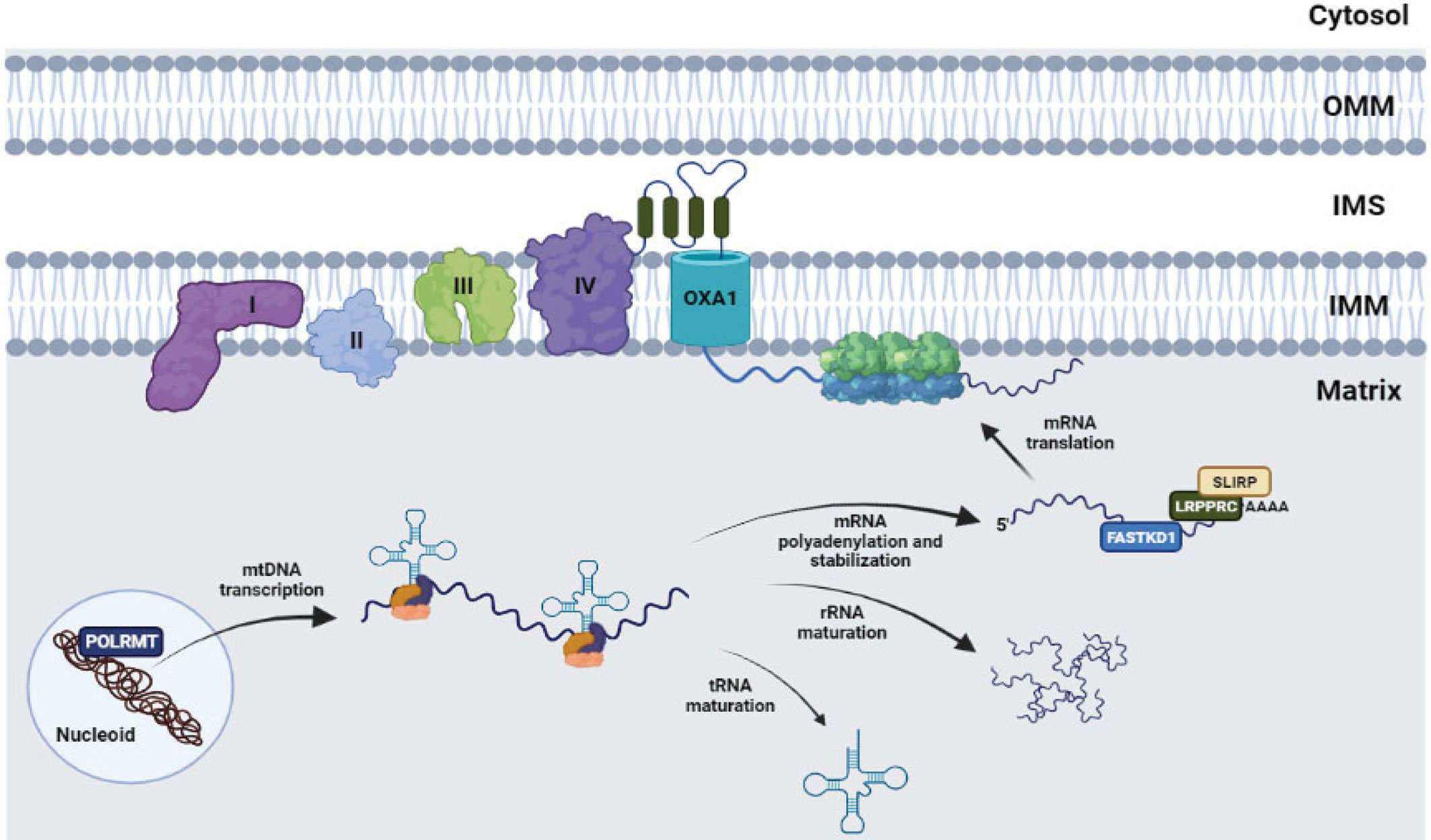
Figure 1 The mtDNA is organized in structures called “nucleoids”, in which both core and peripheral proteins contribute to organization, stability and communication of the mtDNA with additional factors. Among the nucleoid components, POLRMT plays a key role, being responsible for the transcription process. Subsequently, the original polycistronic transcript is subject to extensive maturation, yielding mitochondrial tRNAs, rRNAs and mRNAs. The latter encode 13 polypeptides, all members of the respiratory chain, can be stabilized and regulated by RNA-binding proteins such as LRPPRC, SLIRP FASTKD1, and finally translated by inner membrane-tethered mitoribosomes, to be cotranslationally assembled into the OXPHOS complexes. I, II, III, IV: respiratory complexes I-IV.
Mitochondrial translation begins when a mt-mRNA is loaded onto the SSU, then a start codon is recognized by the initiator tRNA carrying a formylated methionine (fMet-tRNAMet) (38). Before mRNA loading, two mitochondrial initiation factors (mIFs), mIF2 and mIF3, assemble on the SSU. Initially, mtIF3 binds SSU to prevent the premature reassociation with LSU and avoid binding of fMet-tRNAMet to the P-site in the absence of mRNA and mtIF2 (39). Subsequently, mtIF2:GTP binds the SSU and promotes the binding of fMet-tRNAMet to the P site while avoiding the association of tRNAs to the ribosomal A site. Following correct codon-anticodon interaction between fMet-tRNAMet and the start codon, LSU joins the SSU forming the monosome, mtIF2 hydrolizes GTP to GDP and the initiation factors are released from the ribosome resulting in the mature ribosome ready to enter the elongation phase (38).
Currently, how mitochondrial transcripts are loaded onto the mitoribosomes and how the start codon is selected is unclear, as human mt-mRNAs lack the Shine–Dalgarno or the Kozak sequences, the most common cis-regulatory elements located at the 5’ UTRs of the prokaryotic and eukaryotic mRNAs respectively, that help to recruit mRNA to the ribosome and to recognize the start codon during translation initiation (40).
During elongation, selected amino acids are sequentially added to the nascent polypeptide. Aminoacyl-tRNAs are delivered to the A-site of mitoribosomes by the mitochondrial elongation factor EFTu (mtEFTu) bound to GTP. Upon correct codon-anticodon interaction, GTP is hydrolyzed to GDP and mtEFTu : GDP is released from the complex. The recycling of mtEFTu needs the elongation factor Ts (mtEFTs), that exchange GDP for GTP on mtEFTu, allowing it to bind and deliver the next aminoacylated tRNA (41). After the release of mtEF-Tu, the peptidyl transferase center in the LSU catalyzes the formation of the peptide bond between the nascent peptide chain of peptidyl-tRNA in the P site and the new amino acids carried by the aminoacyl-tRNA present in the A-site leaving a deacetylated tRNA in the P-site and one residue longer peptidyl-tRNA in the A-site (42). Subsequently, the mitochondrial elongating factor G1 (mtEFG1) catalyzes the GTP hydrolysis-dependent translocation of the mitoribosome, moving the deacylated tRNA from the P to the E-site and the peptidyl-tRNA from the A-site to the P-site, hence a new codon is exposed in the A-site and the cycle can start again (43).
The elongation cycle is reiterated until the polypeptide chain is completed and a stop codon reaches the ribosomal A-site. The stop codon association is recognized by the mitochondrial release factor 1a (mtRF1a) that mediates the hydrolysis of the ester bond between the last tRNA and the completed polypeptide, resulting in the release of the newly formed protein (44). Subsequently, two mitochondrial ribosome recycling factors, mtRRF1 and mtRRF2 (also known as mtEFG2), promote the dissociation of the mitoribosomal subunits and the release of mt-mRNA and deacylated mt-tRNA (45).
Aberrant expression of mitoribosomal proteins has been associated with several types of cancer in recent years (46). In breast cancer, analysis of genome-wide transcriptional profiling data and subsequent validation by immunohistochemistry, highlighted a significant enrichment in mitoribosomal proteins among the genes upregulated in the tumor tissue compared to the adjacent stroma. This suggests a tissue organization comprising highly oxidative epithelial breast cancer cells rich in mitochondria (and mitoribosomes), and a surrounding glycolytic stroma (47). On the other hand, lactate-mediated suppression of MRPL13 expression in hepatoma cells seems to promote hepatoma cell invasiveness and hepatocellular carcinoma development (48), highlighting the importance of the metabolic context in the contribution of mitochondrial protein synthesis to pathogenesis of human cancers.
In line with the endosymbiotic theory, mitochondria originate from a respiring proteobacterium, whose genome has been transferred during evolution into the nucleus of the eukaryotic host cell. Consequently, the vast majority of mitochondria-resident proteins are encoded by the nuclear genome, synthesized by the cytosolic translational machinery and imported into the mitochondria. The nuclear-encoded mitochondrial proteins are synthesized by cytosolic ribosomes as precursors bearing specific targeting signals that direct them to different mitochondrial sub-compartments, such as the N-terminal presequence required for a localization to the IMM or the matrix (49).
According to the classical view, once their synthesis is complete, preproteins are delivered on the mitochondrial surface in an unfolded state by molecular chaperones and then are imported via translocases of the outer mitochondrial membrane (OMM) and IMM (TOM/TIM complexes) (50, 51). However, experimental evidence suggest that mitochondria-destined proteins may be synthesized by cytosolic ribosomes localized near the OMM and co-translationally imported into the mitochondria (52, 53).
Already back in the 1970s, electron microscopy analysis found that cytoplasmic ribosomes can be localized near the OMM (54). Moreover, many microarray and RNA-seq analyses of biochemically fractionated mitochondria highlight the presence of nuclear-encoded mRNAs that are co-purified with mitochondria, and fluorescent microscopy analyses confirm these observations (55–58). Importantly, it has been shown that active translation is a key part of the localization process, as disassembly of polysomes by EDTA or puromycin treatment reduces the association between mRNAs and mitochondria (55, 59). In addition, ribosome profiling analyses performed on fractions of ribosomes isolated in the proximity of mitochondria confirmed that many IMM protein coding transcripts are co-translationally targeted to mitochondria (58). Delivery of mRNAs to the mitochondrial surface requires cis-acting signals, present in the transcripts or in the encoded polypeptide, and proteins that recognize these signals. Both 3′ UTR and coding regions, primarily through mitochondrial targeting sequences (MTSs), contribute to mitochondrial localization of transcripts (60, 61). In yeast, two classes of mRNAs that are translated near the mitochondria have been identified: class I mRNAs, bearing a binding motif in the 3’UTR recognized by the RNA-binding protein Puf3, and class II mRNAs, that are localized to mitochondria in a Puf3-independent manner (57). Both mRNA groups, independently transported on the mitochondrial surface, participate in the assembly of respiratory complexes: class I mRNAs encode assembly factors, whereas class II mRNAs encode structural proteins, indicating that differential regulation of mRNA localization near mitochondria is a potential mechanism to post-transcriptionally coordinate the construction of OXPHOS complexes (57). The TOM complex also participates to mRNA localization in both yeast and mammalian cells through interaction of protein receptor Tom20 with the MTS of the nascent polypeptide as it is translated (59, 62) instead, the outer-membrane protein OM14 is a mitochondrial receptor for the ribosome nascent-chain-associated complex (NAC), which interacts with both cytosolic translating ribosomes and nascent polypeptides as they emerged from exit tunnel (63). In Drosophila ovaries, the AKAP protein MDI, in complex with the translation stimulator La-related protein (Larp), promotes site-specific translation on the OMM of mRNAs encoding for mtDNA replication factors, mitochondrial ribosomal proteins, and electron-transport chain subunits, which is crucial for mitochondrial biogenesis during oogenesis (64).
These findings support the idea of co-translational import of nuclear-encoded proteins into mitochondria (65). Thus, the localization of transcripts in proximity of mitochondria and the activity of RNA-binding proteins as trans-acting factors provide a tool for a post-transcriptional regulation of gene expression at both a temporal and spatial level, to control protein import and respiratory complex assembly (56, 57, 66).
Finally, interactions between mitochondria and mRNA/nascent-peptide complexes can be altered by the kinetics of protein synthesis, which leads to enhanced protein expression for these factors during respiratory conditions (67). In this context, the length of translation time plays an important role in mRNA localization to the mitochondria, and it has been shown that increased translation time due to a translation elongation stall caused by polyproline sequences is one way exploited by yeast cell to extend the “competent state” of a translating mRNA to be recruited to the mitochondrion; moreover other mechanisms that increase translation duration such as increased ORF length, the presence of rare codons within the transcript, and mRNA structures could likely play a similar role in mRNA localization (67).
As stated above, mitochondrial proteome is composed mostly of nuclear-encoded proteins that need to be imported into the mitochondria. Two transport mechanisms have been described, the post-translational and the co-translational translocation. Post-translational protein import implies that unfolded polypeptides, synthesized by cytosolic ribosomes, are guided to receptors of the TOM complex (Tom20, Tom22 and Tom70) by chaperones of the Hsp70, Hsp40, and Hsp90 families (50). Precursors containing the MTS are recognized by protein receptor Tom20, whereas Tom70 recognizes internal signals of hydrophobic polypeptides (68). Alternatively, ribosomes translating mitochondrial proteins can localize in the proximity of mitochondria through the action of RNA-binding proteins, such as Puf3 or NAC complex assembled on nascent polypeptides, controlling the co-translational translocation of proteins into mitochondria (63). In both cases, once transported across the OMM, mitochondrial precursors interact with the TIM23 complex, which transports preproteins into the IMM or matrix, aided by presequence translocase-associated motor (PAM) (69, 70).
TIM23 complex is emerging as a relevant factor for respiratory complex biogenesis, not only by regulating the import of presequence-carrying subunits, but also by promoting the incorporation of these subunits into the complexes. Indeed, TIM21, a subunit of TIM23 complex, is associated with complex I and complex IV assembly intermediates, where it transports the nuclear-encoded subunits for integration with the subunits encoded by the mitochondria (71). Moreover, TIM21 couples the TIM23 translocase to the cytochrome bc1-cytochrome c oxidase supercomplexes of the respiratory chain via a direct interaction with UQCR6, a subunit of complex III, supporting the import of presequence proteins under membrane potential limiting conditions, which makes it crucial for the import of protein when the membrane potential is reduced (72). Of note, it has been recently shown that integrity of complex III is crucial for the biogenesis and maturation of complex I and IV (73). Interestingly, in plants the TIM23 isoforms were found to associate with complex I (NADH dehydrogenase), too (74).
As proof of the intimate connection between respiratory chain functions and protein import, the succinate dehydrogenase (complex II) subunit Sdh3, which constitutes, along with Sdh4, a membrane integral module required for the recruitment of the catalytic subunits Sdh1 and Sdh2 to the IMM, is a moonlighting protein that participate, in partnership with Tim18, to the formation of TIM22 [reviewed in (75)]. Of note, Tim28 is also a close homolog of Shd4.
Once imported into the matrix via TIM23 complex, the precursor proteins must undergo the cleavage of their N-terminal presequences by the mitochondrial processing peptidase (MPP), which is crucial for the following folding and for the functionality of their catalytic activity. MPP consists of two homologous subunits, Mas1 and Mas2, that in turn are highly homologous to UQCRC1 and UQCRC2 core subunit of the respiratory complex III. Amazingly, it has been found in plants that Mas1 and Mas2 replace the core proteins and the MPP-activity is exclusively integrated into the complex (76).
It is noteworthy that these processes impact directly on the assembly and activity of respiratory complexes. In cancer biology, the contribution of mitochondrial metabolism to disease development and progression has long been underrated. This is due to the original hypothesis that cancer is a result of mitochondrial insufficiency, that was at the basis of Otto Warburg’s formulation of the “aerobic glycolysis” model. Although the Warburg effect remains central to our understanding of cancer cell metabolic remodeling, we now know that mitochondria play fundamental roles in several neoplasms such as sarcomas, cervical cancer, and melanomas (77), or, alternatively, in specific growth stages of the same tumor (78). Moreover, OXPHOS has been proven important to sustain survival and proliferation of chemoresistant cells (79). For these reasons, although our understanding of these phenomena in cancer cells is still at the early stages, it is reasonable to hypothesize that cancer cells must be particularly sensitive to uncoupling of mitochondrial and cytosolic translation, and to disruption of all the quality control networks highly connected to the electron transport chain functionality.
The coordinated expression and assembly of respiratory chain subunits, encoded by nuclear and mitochondrial genomes, require different PQC systems involving molecular chaperones and proteases that ensure the efficient import of nuclear encoded proteins, the correct folding of both nuclear and mitochondrial encoded proteins and the degradation of misfolded proteins or unassembled subunits (80).
Mitochondrial PQC occurs at the cytosolic side of the OMM to survey the import of nuclear-encoded proteins, in the intermembrane space and in the matrix to control their state and turnover. In this way, PQC occurs early on nascent polypeptides, which can be efficiently folded, modified and targeted to cellular membranes to avoid mis-localization, or rapidly ubiquitinated and degraded to prevent the accumulation of protein aggregates (81, 82).
The relevance of this phenomenon is testified by the recent discovery of a novel pathway of mitochondria-mediated cell death named mitochondrial Precursor Over-accumulation Stress (mPOS), that is characterized by aberrant accumulation of mitochondrial precursors in the cytosol (83). This condition is induced not only by mutations of components of the protein import machinery, but also by malfunction of the inner membrane. In keeping with these data, there is also evidence that cytosolic proteins are stabilized and mitochondrial protein import is reduced by condition of mitochondrial dysfunction (84–86). In this view, the positive effects evidenced by inhibition of mTOR on progression of mitochondrial diseases (87) could be interpreted as a result of decreases protein synthesis.
A second layer of PQC is exerted within the two sides of mitochondrial inner membrane by two ATP-dependent proteolytic complexes: the m-AAA complex, which functions at the matrix side of the membrane; and the i-AAA complex, whose role resides in the intermembrane space. The first comprises two isoenzymatic forms, the homo-oligomeric AFG3L2 subunits, implicated in the processing of Cox1 and MT-ATP6 respiratory chain subunits, and the hetero-oligomeric AFG3L2 and SPG7 subunits, involved in the degradation of the EMRE subunit of the mitochondrial calcium uniporter complex (88). The i-AAA complex plays a fundamental role in mitochondrial dynamics. Its subunit YMEL1, together with OMA1, was shown to regulate the processing of OPA1, thus affecting the process of mitochondrial fusion. Moreover, non-assembled Cox4, NDUFB6 and ND1 respiratory chain subunits, and the TIM23 subunit Tim17A, were all shown to be proteolitically processed by YMEL1 (89, 90).
Given the central role played by PQC in shaping mitochondrial functions following stressful conditions, it is not surprising that mitochondrial proteases could be relevant to the pathophysiology of some cancers. Accordingly, a recent study demonstrated how YMEL1 can rewire mitochondrial proteome to sustain the growth of pancreatic ductal adenocarcinoma (PDAC). PDAC is a solid tumor able to reprogram glutamine metabolism to overcome hypoxic and nutrient-deprived environment through the stabilization of HIF1α. Together with HIF1α stabilization, analysis of PDAC patient biopsies revealed that depletion of YMEL1 substrates represent a further mechanism encountered to optimize mitochondria metabolism rewiring and tumor progression. Indeed, depletion of YMEL1 was able to reduce both the growth of cultured PDAC cells as well as tumor formation in vivo. Conversely, the same effect was not observed for hepatocellular carcinoma, with HepG2 and Huh7 cell lines showing no differences in spheroids formation following YMEL1 silencing. These data suggest that the proteolytic rewiring by YMEL1 could strongly depend on both the metabolic needs of each tumor and the tumor microenvironment (91).
PQC is carried out in the mitochondrial matrix by molecular chaperones systems and proteases of the AAA+ (ATPase associated with diverse cellular activities) family that maintain the correct protein folding and remove the unfolded or damaged proteins and unassembled OXPHOS subunits (92, 93). The AAA+ proteases of the mitochondrial matrix include CLPXP and LON. CLPXP is a complex constituted by two components, the serine protease ClpP and the chaperone ClpX, that recognizes and delivers the protein substrates to ClpP for degradation (94). Mitochondrial LON protease plays a central role in the PQC in the mitochondrial matrix by removing unfolded and oxidized proteins and promoting the folding of imported proteins through interaction with the chaperone mtHSP70 (95, 96).
In addition to the degradation of unfolded proteins in the matrix, CLPX and LON regulate mitochondrial protein synthesis, and thus the biogenesis of respiratory complexes (97, 98). The ClpXP complex regulates mitoribosome assembly through degradation of ERAL1, a putative 12S rRNA chaperone essential for SSU assembly, but whose removal is necessary to form a mature mitoribosome and for translation initiation (97, 99). Loss of ClpP or loss of ClpXP activity affects mitoribosome assembly and reduces mitochondrial translation, leading to respiratory chain dysfunction (97). LON influences mitochondrial gene expression by regulating the degradation of mitochondrial transcription factor A (TFAM), essential for mtDNA transcription initiation, MRPP3A, the RNase P subunit responsible for mtRNA processing, and FASTKD2, a factor involved in the mitoribosome biogenesis (98, 100–102). Depletion of LON in human cells reduces the levels of mtDNA, impairs mature mitoribosomes assembly and thus abolishes mitochondrial protein synthesis (100, 101).
As expected, both LON and ClpP proteases levels correlate with tumor development (103). Indeed, the RNA levels of these proteases are up-regulated in several cancers, particularly in prostate cancer (104). Indeed, LON and ClpP synergistically cooperate to promote cell growth and survival of prostate cancer cells, with patients showing a worst survival outcome when the levels of both proteases are concomitantly high. This is in agreement with a significantly marked reduction in prostate cancer cell growth and increased sensitivity to metabolic stress inducers following silencing of the two proteases (104). The mechanism behind the tumorigenic role of LON and ClpP involves the PQC exerted on the SHMT2 enzyme, whose inhibition leads to a significant reduction in cell growth with a more pronounced effect when the proteases are depleted (104).
Perturbations of mitochondrial proteostasis leads to the activation of the mitochondrial unfolded protein response (mtUPR), a retrograde signal direct to nucleus aimed at maintaining the mitochondrial proteome integrity (105). Primarly, mtUPR attempts to relieve stress by inducing the expression of chaperones and proteases that increase mitochondrial protein folding capacity (106). In addition, in order to decrease the mitochondrial folding load, mtUPR reduces protein import and decreases mitochondrial translation by impairing mt-RNA processing and inducing the degradation of mt-mRNAs and MRPs (90, 100). The activation of mtUPR is a compensatory response that can be use by cancer cells as a cytoprotective strategy supporting adaptation to unfavorable milieus (107). However, prolonged activation of this stress response pathway can result in cell death. Therefore, targeting factors that control the protein folding environment within mitochondria has been explored as anticancer strategy. In this context, the molecular chaperone TRAP1 (Tumor Necrosis Factor Receptor-Associated Protein-1), the mitochondrial paralog of the HSP90 family, is recognized as a relevant factor in the control of mitochondrial homeostasis (108). TRAP1 is a gene of monophyletic origin only present in Animalia and some Protista, mostly similar to a eukaryotic HtpG (a E. coli heat shock protein), with the addition of a N-terminal transit peptide sequence for the targeting to mitochondria, likely evolved at the base of the TRAP1 lineage. TRAP1 has therefore arisen from the ancestral eukaryotes, and was not derived from an endosymbiont of bacterial origin (109). TRAP1 is indeed the only mitochondrial member of the HSP90 family, with high homology with cytosolic HSP90 though, holding an ATPase domain and an HSP90-domain involved in client protein binding that share an overall 26% identity and 45% similarity with cytosolic HSP90 (110). TRAP1 protects the mitochondria integrity under oxidative stress by preventing the permeability transition pore opening, through binding with cyclophilin D, and acting as downstream effector of PINK1 (111, 112). Relevant for this protective function is also a regulation of mitochondrial metabolism through both direct and indirect interaction with the respiratory chain (113), with relevant effects on cancer progression and drug resistance, especially in ovarian cancer (114–116). Moreover, it has been shown that genetic silencing or pharmacological inhibition of TRAP1 in human cancer cells induces the hallmarks of mtUPR signaling, including accumulation of unfolded matrix proteins and upregulation of multiple chaperones and stress response transcription factors CHOP and C/EBPβ (117). In addition to its role in the regulation of protein folding within the mitochondria, TRAP1 contributes to maintain the mitochondrial proteostasis, also acting in the cytosol. Indeed, TRAP1 is localized to the outer face of endoplasmic reticulum, where it interacts with both the proteasome and the ribosomes to regulate co-translational degradation of mitochondria-destined proteins such as F1ATPase beta subunit and a mitochondrial isoform of Sorcin (118, 119).
All mitochondrial-encoded proteins participate in the formation of respiratory complexes together with nuclear encoding ones. Due to their dual genetic origin, the biogenesis of OXPHOS complexes requires the coordinated regulation of the mitochondrial and cytoplasmic translational machineries. The OXPHOS system subunits synthesized by mitochondrial translational machinery are: ND1-6 and ND4L for complex I (NADH dehydrogenase); cytochrome b for complex III (cytochrome c reductase); COX1-3 for complex IV (cytochrome c oxidase); ATP6 and ATP8 for complex V (ATP synthase). Hence, all the respiratory chain complexes but complex II have a dual genetic origin.
Studies performed in yeast demonstrate that expression of dual-origin OXPHOS complexes is induced upon adaptation to respiratory growth through a rapid and synchronous translation regulation across compartments, whereas OXPHOS mRNAs are not coordinately induced. Indeed, while nuclear transcripts are rapidly induced in response to a nutrient shift, the mitochondrial ones are induced more slowly, most likely reflecting the absence of mitochondrial transcription factor responsive to environmental changes (120). Synchronized translation could therefore serve to maximize the efficiency of OXPHOS complex assembly, especially in a mutable metabolic context, but also to limit nonproductive or harmful off-target interactions.
Of note, how the cytoplasmic and mitochondrial translation are synchronized in human cells is currently unknown. Conversely, studies in the yeast Saccharomyces cerevisiae extensively described feedback loops that coordinate mitochondrial translation with the availability of nuclear-encoded subunits to optimize the assembly of respiratory chain complexes. In particular, mitochondrial gene expression is regulated by several nuclear-encoded translational activators located into the IMM and in contact with the mitoribosomes (121). Translational activators tune the translation rate of specific mt-mRNAs to the import of nuclear encoded OXPHOS subunits to allow mitochondria to synthesize only those subunits that can be assembled into complexes, and thus avoiding the accumulation of unassembled subunits (121).
The best understood regulatory feedback of mitochondrial translation is the one involved in the synthesis of COX1 during the assembly of complex IV. The translational activator Mss51 binds Cox1 mRNA to start its translation, and interacts with the newly synthesized COX1 polypeptide in a pre-complex temporary assembled with the assembly factors Coa1, Coa3, Cox14 and Shy1 (122–125). In this complex, Mss51 is unable to stimulate the translation of the Cox1 transcript until the COX1 protein associates with additional subunits, imported into the mitochondria during the complex assembly, releasing Mss51, which can initiate a new round of Cox1 synthesis (121).
As for mammalian mitochondria, the only translation activator identified is TACO1 (translational activator of cytochrome oxidase I), which is necessary for the efficient translation of COX1 (126). However, a regulatory feedback exerted by cytosolic translation products on mitochondrial translation has been identified in human cells as a mechanism for the complex IV assembly (127). During translation of the Cox1 mRNA, two inner membrane proteins, C12ORF62 (COX14) and MITRAC12 (COA3) interact with the nascent polypeptide, inducing translation elongation arrest (127). Stalled mitoribosomes resume Cox1 mRNA translation only when COX4, the first nuclear-encoded subunit incorporated into the complex, is imported (127). Therefore, human mitoribosomes display a translational plasticity to coordinate their protein synthesis rate with the influx of cytosolic OXPHOS subunits and the assembly of respiratory complexes. It will be interesting to investigate whether the translational plasticity regulates the assembly of others human OXPHOS complexes with dual genetic origin.
Remarkable for the aim of this issue, several works pursuing the inhibition of mitochondrial protein synthesis as a therapeutic strategy against different tumors have shown that it leads to a decoupling of cytosolic and mitochondrial translation and consequent reduction in cell proliferation and fitness (128–130), suggesting that cancer cells could be particularly sensitive to translation uncoupling. In this view, particularly relevant is the function of the lncRNA SAMMSON, aberrantly expressed in a large fraction of melanomas and hepatocellular carcinomas (131), that has been found to concertedly stimulate rRNA biogenesis and protein synthesis in both cytosol and mitochondria of tumor cells (132). As a result, SAMMSON confers a growth advantage to immortalized cells irrespective of their tissue of origin, and is able to transform immortalized cells of melanocytic origin, allowing tumor growth in nude mice. Of note, knockdown of SAMMSON decreases melanoma viability by impairing mitochondrial translation and inducing an mPOS-like response (133), and induces apoptosis even before any effect of its depletion on ribosome biogenesis and cytosolic protein synthesis could be observed (132), supporting the importance of the coordination between both mechanisms in tumor cells.
An additional “study case” in the context of human tumors is the mitochondrial chaperone TRAP1, that is involved in the control of respiration and mitochondrial PQC, but also in the regulation of mitochondrial translation. By using two complementary approaches, it has been found that one of the functions most heavily affected by inhibition of mitochondrial HSP90 activity is mitochondrial translation, with many ribosomal proteins found aggregated and misfolded following treatment with non-cytotoxic concentrations of the Hsp90-inhibitor Gamitrinib (134). In support of this, an immunoprecipitation mass spectrometry experiment has shown that the mitochondrial translation elongation factor mtEf-Tu and several components of the mitochondrial protein import complexes TOM/TIM are, among others, TRAP1 interactors (135). Accordingly, TOM40 was also found in an independent proteomic experiment in search of TRAP1 interactors in HeLa cells (136). These pieces of evidence preliminary suggest that a single chaperone with predominant mitochondrial localization but with described functions associated to protein synthesis and co-translational PQC in the cytosolic compartment could be involved in the regulated synthesis of mitochondrial proteins on both sides of the mitochondrial membranes.
The coordination of processes that control the homeostasis of mitochondrial proteome, from cytosolic translation in proximity of mitochondria to PQC, protein import and mitochondrial translation, are schematically represented in Figure 2.
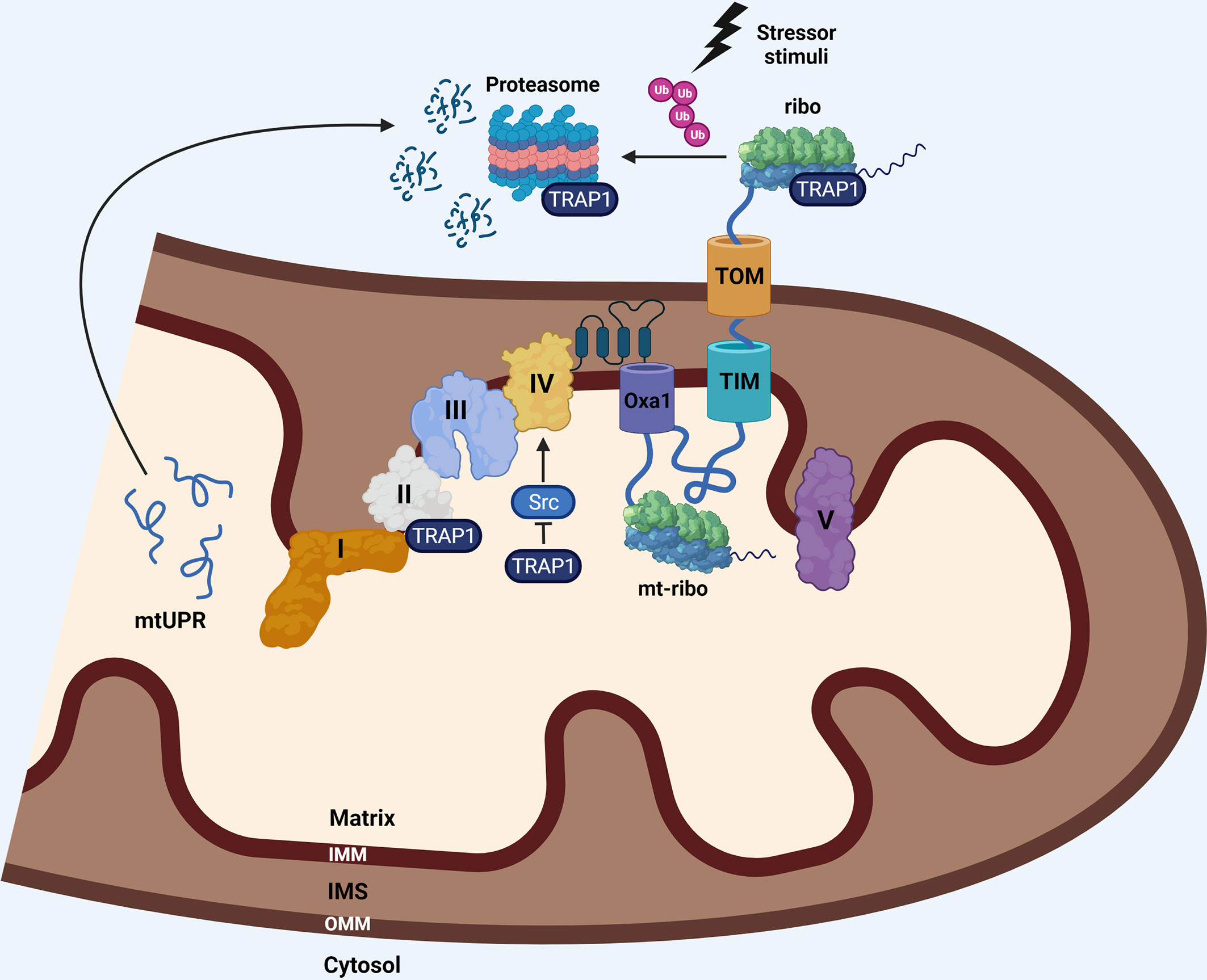
Figure 2 The nuclear-encoded mitochondrial proteins are synthesized by cytosolic ribosomes (ribo) that can be localized at the OMM, allowing co-translational import of nascent proteins into the organelle via the TOM/TIM complexes. Translating ribosomes can act as a platform for early PQC by ribosome-associated chaperones, including TRAP1, that, under stress conditions, prevents aberrant aggregation of proteins, directing them to co-translational ubiquitin-mediated proteasomal degradation. The imported proteins taking part to respiratory complexes are then assembled in supercomplexes along with the 13 components that are synthesized within the organelle by the mitochondrial ribosomes (mt-ribo). The co-translational insertion of these subunits into the IMM is mediated by OXA1, which is crucial for the assembly of functional respiratory complexes. The same molecular chaperone assisting PQC of mitochondrial proteins, TRAP1, is contemporary a regulator of respiration, through a direct binding to complex II, and an indirect regulation on complex IV, through the stabilization of the inactive form of c-Src, which is known to stimulate complex IV activity. Inhibition of TRAP1 leads to a mtUPR and related stress response. I, II, III, IV: respiratory complexes I-IV.
Although glycolysis has long been considered the major metabolic pathway for ATP production in cancer cells, even under aerobic conditions, several studies have now shown that some types of cancer cells choose OXPHOS for their metabolic demands (137–139). Of note, the same TRAP1 protein with roles in mitochondrial PQC within/outside mitochondria, is also considered a bona fide OXPHOS regulator, through the direct binding to SDH and an activity control exerted on complex IV, which require the modulation of c-Src phosphorylation (113). Indeed, TRAP1 appears to be upregulated in predominantly glycolytic tumors, while it is downregulated in highly respiratory ones (13, 140). This suggests the existence of gene expression programs in which genes are clustered for the activation of metabolic plans to integrate energetic and biosynthetic demands with nutrient and oxygen availability. In this context, high mitochondrial translation may be required to support the bioenergetic needs of cancer cells. Additionally, emerging evidence suggests that the mitochondrial ribosomal proteins, beside their role in mitoribosomes assembly, also exhibit moonlight functions in the regulation of cell cycle progression and apoptosis signaling (141). Therefore, it is not surprising that altered expression of mitochondrial translational machinery components has been identified in different tumor types (46, 47, 142).
Inhibition of mitochondrial functions has been explored as therapeutic strategy for cancer treatment. Owing to their prokaryotic origin, mitoribosomes are susceptible to the inhibitory effect of some antibiotics commonly used to treat bacterial infections (143, 144). In addition, due to the tight coupling between translation across compartments, PQC mechanisms and assembly of respiratory complexes, as well as mechanism of protein import and mitochondrial protein homeostasis can be also promisingly targeted for therapeutic purposes in all the systems in which mitochondrial function is key for cell survival and/or proliferation.
The main compounds developed for their capacity to target at different levels homeostasis of the mitochondrial proteome are discussed below, and listed in Table 1.
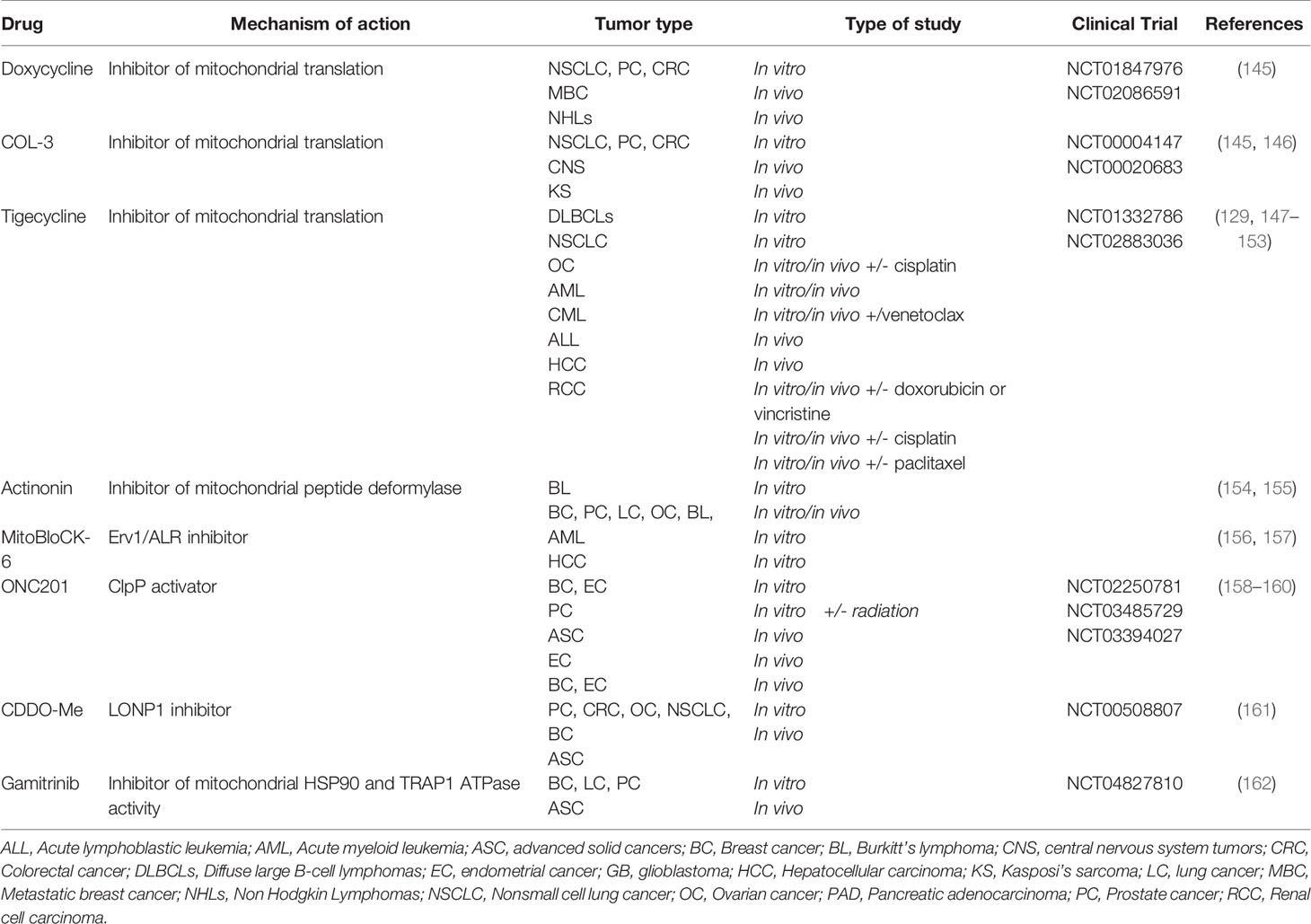
Table 1 List of mitochondrial proteostasis targeting agents used as anticancer drugs in preclinical and clinical studies.
Tetracyclines are broad spectrum antibiotics discovered in the late 1940s as natural products of Streptomyces aureofaciens strain and currently used to treat a wide variety of bacterial infections (163). The bacteriostatic activity of tetracyclines depends on their capacity to inhibit the protein synthesis by preventing the interaction of aminoacyl-tRNAs with the A-site of the ribosome and thus the peptide elongation (164). Besides being antimicrobial agents, tetracycline analogues, such as doxycycline, COL-3 and tigecycline, have shown anti-tumor effects in several human cancers in both pre-clinical and clinical studies (129, 145, 165).
The anticancer effects of doxycycline and COL-3, semisynthetic and chemically modified tetracycline, respectively, were mainly related to their inhibitory effects on the expression and activation of matrix metalloproteases (166, 167). In fact, doxycycline and COL-3 exert antiangiogenic and antimetastatic activity in different cancer cell lines including leukemias, osteosarcoma, breast, colorectal and prostate cancer (166, 168–172).
Recently, it has been show that doxycyline and COL-3 antiproliferative and pro-apoptotic effects are related to the inhibition of mitochondrial protein synthesis with a decreased OXPHOS, resulting in a significant slowdown of proliferation rate (145, 173). Moreover, the reduction of IMM potential induced by tetracyclines yields oxidative stress, bringing the cancer cells closer to the apoptotic threshold (173).
Tigecycline, a third generation tetracycline, has been identified by a chemical screening as an effective drug in reducing the viability of leukemia cell lines (129). Anti-leukemic activity of tigecycline is due to the inhibition of mitochondrial translation which significantly reduces the OXPHOS capacity of cancer cells (129). Tigecyclin inhibition is selective for mitochondrial translation as it reduces the expression levels of Cox-1 and Cox-2, subunits of respiratory complex IV translated by mitoribosomes, without changing the expression of COX-4 that is translated by cytosolic ribosomes (129, 152). As an evidence that tigecycline targets mitoribosomes, knockdown of the mitochondrial elongation factor EF-Tu mimics the effects of tigecycline (129, 153). Leukemia cells are particularly sensitive to tigecycline, being them heavily reliant on OXPHOS (174). Accordingly, experimental evidence in different human cancer cell lines support the idea that tigecycline exerts pro-apoptotic effects are more common in systems with high mitochondrial biogenesis and upregulated oxidative metabolism (130, 147–149, 151–153). Finally, tigecycline has been shown to have a synergistic effect with several chemotherapeutic drugs such as cisplatin (149, 152), paclitaxel (153), venetoclax (150), doxorubicin, vincristine (151), BRAF and MEK inhibitors (175).
Actinonin is a peptidomimetic antibiotic naturally produced by actinomyces that arrests bacterial growth by inhibiting the peptide deformylase also identified in human cells (155, 176). Human mitochondrial peptide deformylase (HsPDF) is a metalloprotease that catalyzes the co-translational removal of the formyl group from N-terminal methionine of newly synthesized proteins (155). Expression of HsPDF was found significantly increased in breast, colon, and lung cancer tissues, suggesting that this enzyme may act as an oncogene to promote cancer cell proliferation (177). Inhibition of HsPDF by actinonin-based antibiotics reduces mitochondrial translation and OXPHOS complex assembly and inhibits the proliferation of several human cancer cell lines (129, 155, 178). The antiproliferative effects of actinonin has also been attributed to the activation of a mitoribosome quality control pathway that precedes the loss of mitochondrial respiration, although the molecular mechanisms are yet to be elucidated (154, 179). According to this interpretation, actinonin blocks the mitoribosomal polypeptyde exit tunnel, probably by trapping HsPDF on the LSU, leading to the accumulation of polypeptydyl-tRNAs in the P-site, which causes mitoribosome stall (179). Stalled mitoribosomes trigger a retrograde signal to the nucleus, that causes cell proliferation arrest (179). Sustained retrograde signal mediated by actinonin induces a mitochondrial decay pathway with degradation of mt-rRNAs, mt-mRNAs and mitoribosomes, which impairs the respiratory chain function (179). This mechanism of action may explain the anticancer effect of mitochondrial translation inhibitors even in cancer cells that do not rely on the OXPHOS for their energy demand.
Carla Koehler’s research group has identified and characterized small molecules, including MitoBloCK-6 (MB-6) and MitobloCK-10 (MB-10), which interfere with mitochondrial protein import process in both yeast and mammalian cells. These two compounds attenuate precursor translocation by targeting different components of the import machineries: MB-10 binds Tim44, a component of the PAM complex, impairing the protein import into the matrix via TIM23 complex. On the other hand, MB-6 inhibits the sulfhydryl oxidase Erv1 and, in turn, Mia40 function in the import of intermembrane space proteins (180, 181). Interestingly, the human homolog of Erv1 (ALR) is found upregulated in hepatocellular carcinoma cell lines and tissues, while its silencing or inhibition through MB-6 impairs mitochondrial function and inhibits the proliferation of liver cancer cells (157, 182). MitoBloCKs can be a useful tool to study the role of mitochondrial translocation machinery in cancer, and the transcriptional and proteomic responses induced by accumulation of precursors in the cytosol, following the inhibition of protein import.
Mitochondrial protease ClpP was found overexpressed in a subset of hematological and solid tumors where it is necessary for cancer cell viability (183–185). Inhibition of ClpP has been proposed as a strategy to impair OXPHOS and induce apoptosis in leukemic cells characterized by a high reliance on mitochondrial respiration (183). Bacterial ClpP inhibitors, ß-lactones derivatives (A2-32-01) and phenyl ester compounds (TG42, TG53), cross-react with human ClpP and show anti-proliferative and pro-apoptotic effects in human cancer cell lines (183, 186). However, these drugs are mainly a chemical tool to be used for functional studies and, therefore, further efforts are needed to design more specific drugs for human ClpP and reduce off-target effects. As for the inhibition, hyperactivation of ClpP also impairs OXPHOS and induces cancer cell death by uncontrolled degradation of ClpP respiratory chain substrates (187). Indeed, the ClpP activator imipridone ONC201 has shown efficacy as a single agent or in combination with other anti-cancer therapies in several solid and hematologic tumors and it is currently being tested in clinical trials (188).
The matrix protease LONP1 is upregulated in different tumor types, including lymphomas, cervical, colorectal, bladder and non small cell lung cancer (189). Inhibition of LON by synthetic triterpenoids, 2-cyano-3, 12-dioxooleana-1,9(11)-dien-28-oic acid (CDDO) and its C-28 methyl ester derivative (CDDO-Me), show a cytotoxic effect in human cancer cell lines by impairing mitochondrial functions (190). However, these compounds exert inhibitory effects on different oncogenic factors, such as IκB kinase (IKK), ubiquitin-specific-processing protease 7 (USP7), erythroblastic oncogene B2 (ErbB2) or peroxisome proliferator activated receptor (PPAR)-γ, making the contribution of LONP1 inhibition to the anticancer effect unclear (161).
The molecular chaperone TRAP1 has been found upregulated in several cancer types, including breast, lung, prostate and colorectal cancers, where it is related to poor prognosis and advanced stages, whereas its genetic silencing induces an attenuation of cancer cells proliferation and in vivo tumor growth, providing a strong rationale for TRAP1 targeting as anticancer therapy (191).
Gamitrinibs are the first class of Hsp90 inhibitors that selectively accumulate in the mitochondrial matrix (162). Structurally, gamitrinibs are constituted by a backbone derived from 17-(allylamino)-17-demethoxygeldanamycin (17-AAG), needed to inhibit the ATPase activity of Hsp90, a linker region and a mitochondrial targeting module provided by one to four tandem repeats of cyclic guanidinium (gamitrinib-(G1-G4) or a triphenylphosphonium (gamitrinib-TPP) (162). Once in the mitochondrial matrix, gamitrinib inhibits the ATPase activity of mitochondrial HSP90 and TRAP1, inducing the accumulation of unfolded proteins with the consequent activation of the mtUPR and organelle disfunction (100, 117). Gamitrinib has been shown a “mitochondriotoxic” effect and anticancer activity in several human cancer cell lines, including squamous cell, breast, lung, prostate carcinoma and leukemia cells, and in in vivo models (162) in addition, it is recently approved to begin phase I clinical trial for advanced solid cancers.
Over the years, several efforts have been directed to designing others more selective TRAP1-targeting drugs with no effects on HSP90 activity, in order to reduce the overall cell toxicity. Rodanin et al. identified a strategy to selectively target TRAP1 ATPase domain by binding cationic appendages to the HSP90 inhibitors core (192). An alternative approach to achieve specific inhibition of TRAP1 is the identification of allosteric ligands disturbing the substructure that controls ATP hydrolysis by binding an allosteric site distal from the ATPase site (193). Highly selective small molecules targeting TRAP1 are needed to dissect the dynamics of client interaction under different conditions and thus the biochemical functions of this chaperone in cancer cells.
Metabolic reprogramming is now recognized as one of the hallmarks of cancer (194). In recent years, an impressive amount of work has been done to identify energetic pathways that could be targeted for therapy, alone or in combination with more traditional anticancer drugs. At the same time, targeting biosynthetic pathways, as protein, nucleotide and lipid biosynthesis, have also attract attention and proposed the adoption of new drugs (or the repurposing of old ones), that have sometimes entered clinical trials (195, 196). Targeting protein synthesis seems an obvious strategy, considering that the aberrantly increased protein synthesis is one of the most common features of cancer cells, and that dysregulated ribosome biogenesis has been one of the first characteristics to be identified, in the form of hypertrophic nucleoli (197). Although the modulation of gene expression has been traditionally attributed to transcription regulation, in the last few years it has become increasingly evident that many processes were regulated at translational level (198), and among those, assembly and activity of respiratory complexes, whose subunits are inserted into the nascent macromolecular units in a co-translational manner (75). As an additional level of complexity, the respiratory complexes have a dual genetic origin, and are therefore composed both by proteins synthesized into the cytosol and later imported into the organelle and proteins synthesized in the matrix by the mitochondrial translational apparatus. Brilliant research projects mainly performed in yeast have shown in recent years that the two processes are tightly connected, co-regulated and coordinated, to ensure fine-tuned responses to energetic demands and nutrient availability (120). The available evidence shows that many of these processes (schematically summarized in Figure 3) are conserved in humans, and that in human cells a large amount of mRNAs encoding mitochondrial proteins can be found in the proximity of mitochondria (66). These transcripts can be locally translated, and the nascent protein co-translationally targeted to the organelle through a protein import channel (75). This complex presents, in turn, multiple connection with the respiratory chain, where mitochondrial-encoded subunits are simultaneously inserted co-translationally, based on the availability of the nuclear-encoded subunits (127). Given the central role played by these phenomena in cancer cells, we believe that shedding light on their regulation in cancer could provide an entire new avenue of both knowledge and therapeutic opportunities. Indeed, preliminary data suggest that cancer cells are particularly sensitive to translation uncoupling, and many compounds are already available to be tested in pre-clinical models. We believe that this scenario holds great promises either in terms of research advances and of opportunities for translation to the clinic.
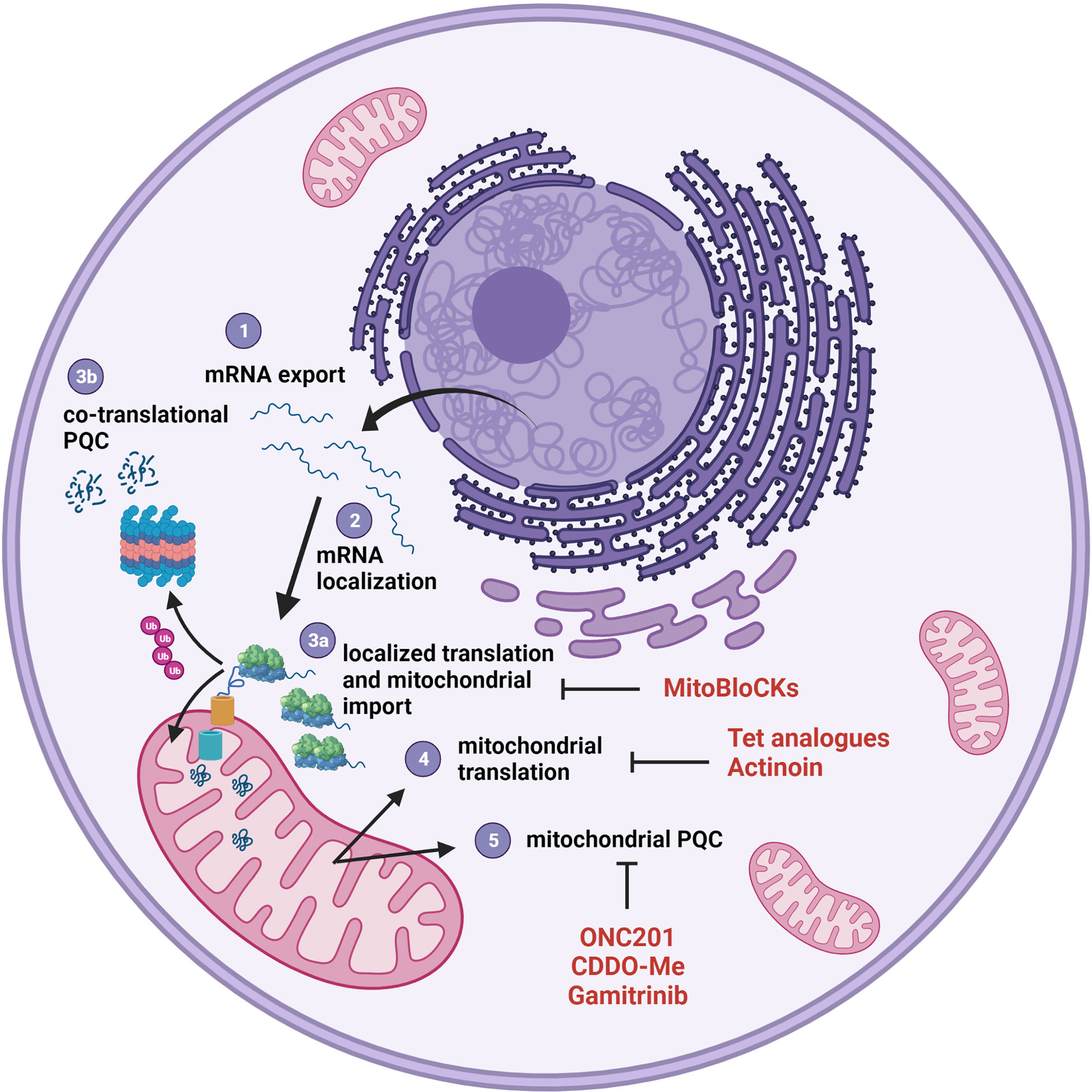
Figure 3 The mitochondrial proteome is controlled at several levels. The vast majority of mitochondrial proteins is encoded by the nuclear genome; therefore, the transcribed mRNAs must be exported from the nucleus (1) to be translated into the cytosol. This translation process can be compartmentalized through a localization of transcript to the organelle (2) with the contribution of the protein synthesis machinery. Proteins synthesized on the surface of mitochondria can be imported via TOM/TIM (3a) or discarded and degraded if they don’t pass the PQC step (3b). mRNA translation (4) and associated PQC (5) also occur in the mitochondrial matrix. All these steps can be potentially targeted with compound listed in Table 1 and represented here in red.
DC, RA, DM, and FE wrote the manuscript. FE critically revised the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by POR CAMPANIA FESR 2014/2020 [project “SATIN” (Sviluppo di Approcci Terapeutici INnovativi per patologie neoplastiche resistenti ai trattamenti)] and FRA (Finanziamento della Ricerca in Ateneo) 2020 grant to DM.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Spinelli JB, Haigis MC. The Multifaceted Contributions of Mitochondria to Cellular Metabolism. Nat Cell Biol (2018) 20(7):745–54. doi: 10.1038/s41556-018-0124-1
2. Lill R, Hoffmann B, Molik S, Pierik AJ, Rietzschel N, Stehling O, et al. The Role of Mitochondria in Cellular Iron–Sulfur Protein Biogenesis and Iron Metabolism. Biochim Biophys Acta (BBA) - Mol Cell Res (2012) 1823(9):1491–508. doi: 10.1016/j.bbamcr.2012.05.009
3. Lopez J, Tait SWG. Mitochondrial Apoptosis: Killing Cancer Using the Enemy Within. Br J Cancer (2015) 112(6):957–62. doi: 10.1038/bjc.2015.85
4. Wang C, Youle RJ. The Role of Mitochondria in Apoptosis. Annu Rev Genet (2009) 43(1):95–118. doi: 10.1146/annurev-genet-102108-134850
5. Zimorski V, Ku C, Martin WF, Gould SB. Endosymbiotic Theory for Organelle Origins. Curr Opin Microbiol (2014) 22:38–48. doi: 10.1016/j.mib.2014.09.008
6. Timmis JN, Ayliffe MA, Huang CY, Martin W. Endosymbiotic Gene Transfer: Organelle Genomes Forge Eukaryotic Chromosomes. Nat Rev Genet (2004) 5(2):123–35. doi: 10.1038/nrg1271
7. Chacinska A, Koehler CM, Milenkovic D, Lithgow T, Pfanner N. Importing Mitochondrial Proteins: Machineries and Mechanisms. Cell (2009) 138(4):628–44. doi: 10.1016/j.cell.2009.08.005
8. Ott M, Amunts A, Brown A. Organization and Regulation of Mitochondrial Protein Synthesis. Annu Rev Biochem (2016) 85(1):77–101. doi: 10.1146/annurev-biochem-060815-014334
9. Vazquez-Calvo C, Suhm T, Büttner S, Ott M. The Basic Machineries for Mitochondrial Protein Quality Control. Mitochondrion (2020) 50:121–31. doi: 10.1016/j.mito.2019.10.003
10. Porporato PE, Filigheddu N, Pedro JMB-S, Kroemer G, Galluzzi L. Mitochondrial Metabolism and Cancer. Cell Res (2018) 28(3):265–80. doi: 10.1038/cr.2017.155
11. LeBleu VS, O’Connell JT, Gonzalez Herrera KN, Wikman H, Pantel K, Haigis MC, et al. PGC-1α Mediates Mitochondrial Biogenesis and Oxidative Phosphorylation in Cancer Cells to Promote Metastasis. Nat Cell Biol (2014) 16(10):992–1003. doi: 10.1038/ncb3039
12. Bosc C, Selak MA, Sarry J-E. Resistance Is Futile: Targeting Mitochondrial Energetics and Metabolism to Overcome Drug Resistance in Cancer Treatment. Cell Metab (2017) 26(5):705–7. doi: 10.1016/j.cmet.2017.10.013
13. Matassa DS, Amoroso MR, Lu H, Avolio R, Arzeni D, Procaccini C, et al. Oxidative Metabolism Drives Inflammation-Induced Platinum Resistance in Human Ovarian Cancer. Cell Death Differ (2016) 23(9):1542–54. doi: 10.1038/cdd.2016.39
14. Gilkerson R, Bravo L, Garcia I, Gaytan N, Herrera A, Maldonado A, et al. The Mitochondrial Nucleoid: Integrating Mitochondrial DNA Into Cellular Homeostasis. Cold Spring Harbor Perspect Biol (2013) 5(5):a011080–a011080. doi: 10.1101/cshperspect.a011080
15. Bonekamp NA, Peter B, Hillen HS, Felser A, Bergbrede T, Choidas A, et al. Small-Molecule Inhibitors of Human Mitochondrial DNA Transcription. Nat (2020) 588(7839):712–6. doi: 10.1038/s41586-020-03048-z
16. Barchiesi A, Vascotto C. Transcription, Processing, and Decay of Mitochondrial RNA in Health and Disease. IJMS (2019) 20(9):2221. doi: 10.3390/ijms20092221
17. Ojala D, Montoya J, Attardi G. tRNA Punctuation Model of RNA Processing in Human Mitochondria. Nat (1981) 290(5806):470–4. doi: 10.1038/290470a0
18. Levy S, Schuster G. Polyadenylation and Degradation of RNA in the Mitochondria. Biochem Soc Trans (2016) 44(5):1475–82. doi: 10.1042/BST20160126
19. Anderson S, Bankier AT, Barrell BG, de Bruijn MHL, Coulson AR, Drouin J, et al. Sequence and Organization of the Human Mitochondrial Genome. Nat (1981) 290(5806):457–65. doi: 10.1038/290457a0
20. Slomovic S, Laufer D, Geiger D, Schuster G. Polyadenylation and Degradation of Human Mitochondrial RNA: The Prokaryotic Past Leaves Its Mark. Mol Cell Biol (2005) 25(15):6427–35. doi: 10.1128/MCB.25.15.6427-6435.2005
21. Siira SJ, Spåhr H, Shearwood A-MJ, Ruzzenente B, Larsson N-G, Rackham O, et al. LRPPRC-Mediated Folding of the Mitochondrial Transcriptome. Nat Commun (2017) 8(1):1532. doi: 10.1038/s41467-017-01221-z
22. Chujo T, Ohira T, Sakaguchi Y, Goshima N, Nomura N, Nagao A, et al. LRPPRC/SLIRP Suppresses PNPase-Mediated mRNA Decay and Promotes Polyadenylation in Human Mitochondria. Nucleic Acids Res (2012) 40(16):8033–47. doi: 10.1093/nar/gks506
23. Ruzzenente B, Metodiev MD, Wredenberg A, Bratic A, Park CB, Cámara Y, et al. LRPPRC Is Necessary for Polyadenylation and Coordination of Translation of Mitochondrial mRNAs: LRPPRC Regulates Mitochondrial Translation. EMBO J (2012) 31(2):443–56. doi: 10.1038/emboj.2011.392
24. Lagouge M, Mourier A, Lee HJ, Spåhr H, Wai T, Kukat C, et al. SLIRP Regulates the Rate of Mitochondrial Protein Synthesis and Protects LRPPRC From Degradation. Barsh GS editor PloS Genet (2015) 11(8):e1005423. doi: 10.1371/journal.pgen.1005423
25. Cui J, Wang L, Ren X, Zhang Y, Zhang H. LRPPRC: A Multifunctional Protein Involved in Energy Metabolism and Human Disease. Front Physiol (2019) 10:595. doi: 10.3389/fphys.2019.00595
26. Li X, Lv L, Zheng J, Zhou J, Liu B, Chen H, et al. The Significance of LRPPRC Overexpression in Gastric Cancer. Med Oncol (2014) 31(2):818. doi: 10.1007/s12032-013-0818-y
27. Jourdain AA, Popow J, de la Fuente MA, Martinou J-C, Anderson P, Simarro M. The FASTK Family of Proteins: Emerging Regulators of Mitochondrial RNA Biology. Nucleic Acids Res (2017) 45(19):10941–7. doi: 10.1093/nar/gkx772
28. Jourdain AA, Koppen M, Rodley CD, Maundrell K, Gueguen N, Reynier P, et al. A Mitochondria-Specific Isoform of FASTK Is Present in Mitochondrial RNA Granules and Regulates Gene Expression and Function. Cell Rep (2015) 10(7):1110–21. doi: 10.1016/j.celrep.2015.01.063
29. Boehm E, Zaganelli S, Maundrell K, Jourdain AA, Thore S, Martinou J-C. FASTKD1 and FASTKD4 Have Opposite Effects on Expression of Specific Mitochondrial RNAs, Depending Upon Their Endonuclease-Like RAP Domain. Nucleic Acids Res (2017) 45(10):6135–46. doi: 10.1093/nar/gkx164
30. Magraner-Pardo L, Gobelli D, de la Fuente MA, Pons T, Simarro M. Systematic Analysis of FASTK Gene Family Alterations in Cancer. IJMS (2021) 22(21):11337. doi: 10.3390/ijms222111337
31. Kummer E, Ban N. Mechanisms and Regulation of Protein Synthesis in Mitochondria. Nat Rev Mol Cell Biol (2021) 22(5):307–25. doi: 10.1038/s41580-021-00332-2
32. Greber BJ, Ban N. Structure and Function of the Mitochondrial Ribosome. Annu Rev Biochem (2016) 85(1):103–32. doi: 10.1146/annurev-biochem-060815-014343
33. Antonicka H, Shoubridge EA. Mitochondrial RNA Granules Are Centers for Posttranscriptional RNA Processing and Ribosome Biogenesis. Cell Rep (2015) 10(6):920–32. doi: 10.1016/j.celrep.2015.01.030
34. Bogenhagen DF, Martin DW, Koller A. Initial Steps in RNA Processing and Ribosome Assembly Occur at Mitochondrial DNA Nucleoids. Cell Metab (2014) 19(4):618–29. doi: 10.1016/j.cmet.2014.03.013
35. Ott M, Herrmann JM. Co-Translational Membrane Insertion of Mitochondrially Encoded Proteins. Biochim Biophys Acta (BBA) - Mol Cell Res (2010) 1803(6):767–75. doi: 10.1016/j.bbamcr.2009.11.010
36. Greber BJ, Boehringer D, Leibundgut M, Bieri P, Leitner A, Schmitz N, et al. The Complete Structure of the Large Subunit of the Mammalian Mitochondrial Ribosome. Nat (2014) 515(7526):283–6. doi: 10.1038/nature13895
37. Stiburek L, Fornuskova D, Wenchich L, Pejznochova M, Hansikova H, Zeman J. Knockdown of Human Oxa1l Impairs the Biogenesis of F1Fo-ATP Synthase and NADH:Ubiquinone Oxidoreductase. J Mol Biol (2007) 374(2):506–16. doi: 10.1016/j.jmb.2007.09.044
38. Christian BE, Spremulli LL. Mechanism of Protein Biosynthesis in Mammalian Mitochondria. Biochim Biophys Acta (BBA) - Gene Regul Mech (2012) 1819(9–10):1035–54. doi: 10.1016/j.bbagrm.2011.11.009
39. Koc EC, Spremulli LL. Identification of Mammalian Mitochondrial Translational Initiation Factor 3 and Examination of Its Role in Initiation Complex Formation With Natural mRNAs. J Biol Chem (2002) 277(38):35541–9. doi: 10.1074/jbc.M202498200
40. Temperley RJ, Wydro M, Lightowlers RN, Chrzanowska-Lightowlers ZM. Human Mitochondrial mRNAs—Like Members of All Families, Similar But Different. Biochim Biophys Acta (BBA) - Bioenerg (2010) 1797(6–7):1081–5. doi: 10.1016/j.bbabio.2010.02.036
41. Cai Y-C, Bullard JM, Thompson NL, Spremulli LL. Interaction of Mitochondrial Elongation Factor Tu With Aminoacyl-tRNA and Elongation Factor Ts. J Biol Chem (2000) 275(27):20308–14. doi: 10.1074/jbc.M001899200
42. Nissen P. The Structural Basis of Ribosome Activity in Peptide Bond Synthesis. Sci (2000) 289(5481):920–30. doi: 10.1126/science.289.5481.920
43. Bhargava K, Templeton P, Spremulli LL. Expression and Characterization of Isoform 1 of Human Mitochondrial Elongation Factor G. Protein Expression Purif (2004) 37(2):368–76. doi: 10.1016/j.pep.2004.06.030
44. Soleimanpour-Lichaei HR, Kühl I, Gaisne M, Passos JF, Wydro M, Rorbach J, et al. Mtrf1a Is a Human Mitochondrial Translation Release Factor Decoding the Major Termination Codons UAA and UAG. Mol Cell (2007) 27(5):745–57. doi: 10.1016/j.molcel.2007.06.031
45. Rorbach J, Richter R, Wessels HJ, Wydro M, Pekalski M, Farhoud M, et al. The Human Mitochondrial Ribosome Recycling Factor is Essential for Cell Viability. Nucleic Acids Res (2008) 36(18):5787–99. doi: 10.1093/nar/gkn576
46. Huang G, Li H, Zhang H. Abnormal Expression of Mitochondrial Ribosomal Proteins and Their Encoding Genes With Cell Apoptosis and Diseases. IJMS (2020) 21(22):8879. doi: 10.3390/ijms21228879
47. Sotgia F, Whitaker-Menezes D, Martinez-Outschoorn UE, Salem AF, Tsirigos A, Lamb R, et al. Mitochondria “Fuel” Breast Cancer Metabolism: Fifteen Markers of Mitochondrial Biogenesis Label Epithelial Cancer Cells, But Are Excluded From Adjacent Stromal Cells. Cell Cycle (2012) 11(23):4390–401. doi: 10.4161/cc.22777
48. Lee Y-K, Lim JJ, Jeoun U, Min S, Lee E, Kwon SM, et al. Lactate-Mediated Mitoribosomal Defects Impair Mitochondrial Oxidative Phosphorylation and Promote Hepatoma Cell Invasiveness. J Biol Chem (2017) 292(49):20208–17. doi: 10.1074/jbc.M117.809012
49. Abe Y, Shodai T, Muto T, Mihara K, Torii H, Nishikawa S, et al. Structural Basis of Presequence Recognition by the Mitochondrial Protein Import Receptor Tom20. Cell (2000) 100(5):551–60. doi: 10.1016/S0092-8674(00)80691-1
50. Hansen KG, Herrmann JM. Transport of Proteins Into Mitochondria. Protein J (2019) 38(3):330–42. doi: 10.1007/s10930-019-09819-6
51. Wiedemann N, Pfanner N. Mitochondrial Machineries for Protein Import and Assembly. Annu Rev Biochem (2017) 86(1):685–714. doi: 10.1146/annurev-biochem-060815-014352
52. Vardi-Oknin D, Arava Y. Characterization of Factors Involved in Localized Translation Near Mitochondria by Ribosome-Proximity Labeling. Front Cell Dev Biol (2019) 7:305. doi: 10.3389/fcell.2019.00305
53. Gold VA, Chroscicki P, Bragoszewski P, Chacinska A. Visualization of Cytosolic Ribosomes on the Surface of Mitochondria by Electron Cryo-Tomography. EMBO Rep (2017) 18(10):1786–800. doi: 10.15252/embr.201744261
54. Kellems RE, Allison VF, Butow RA. Cytoplasmic Type 80 S Ribosomes Associated With Yeast Mitochondria. J Biol Chem (1974) 249(10):3297–303. doi: 10.1016/S0021-9258(19)42672-0
55. Fazal FM, Han S, Parker KR, Kaewsapsak P, Xu J, Boettiger AN, et al. Atlas of Subcellular RNA Localization Revealed by APEX-Seq. Cell (2019) 178(2):473–90.e26. doi: 10.1016/j.cell.2019.05.027
56. Garcia M, Darzacq X, Delaveau T, Jourdren L, Singer RH, Jacq C. Mitochondria-Associated Yeast mRNAs and the Biogenesis of Molecular Complexes. Fox T, Editor. MBoC (2007) 18(2):362–8. doi: 10.1091/mbc.e06-09-0827
57. Saint-Georges Y, Garcia M, Delaveau T, Jourdren L, Le Crom S, Lemoine S, et al. Yeast Mitochondrial Biogenesis: A Role for the PUF RNA-Binding Protein Puf3p in mRNA Localization. Bähler J, Editor. PloS One (2008) 3(6):e2293. doi: 10.1371/journal.pone.0002293
58. Williams CC, Jan CH, Weissman JS. Targeting and Plasticity of Mitochondrial Proteins Revealed by Proximity-Specific Ribosome Profiling. Sci (2014) 346(6210):748–51. doi: 10.1126/science.1257522
59. Eliyahu E, Pnueli L, Melamed D, Scherrer T, Gerber AP, Pines O, et al. Tom20 Mediates Localization of mRNAs to Mitochondria in a Translation-Dependent Manner. Mol Cell Biol (2010) 30(1):284–94. doi: 10.1128/MCB.00651-09
60. Sylvestre J, Margeot A, Jacq C, Dujardin G, Corral-Debrinski M. The Role of the 3′ Untranslated Region in mRNA Sorting to the Vicinity of Mitochondria Is Conserved From Yeast to Human Cells. MBoC (2003) 14(9):3848–56. doi: 10.1091/mbc.e03-02-0074
61. Garcia M, Delaveau T, Goussard S, Jacq C. Mitochondrial Presequence and Open Reading Frame Mediate Asymmetric Localization of Messenger RNA. EMBO Rep (2010) 11(4):285–91. doi: 10.1038/embor.2010.17
62. Lesnik C, Golani-Armon A, Arava Y. Localized Translation Near the Mitochondrial Outer Membrane: An Update. RNA Biol (2015) 12(8):801–9. doi: 10.1080/15476286.2015.1058686
63. Gamerdinger M, Kobayashi K, Wallisch A, Kreft SG, Sailer C, Schlömer R, et al. Early Scanning of Nascent Polypeptides Inside the Ribosomal Tunnel by NAC. Mol Cell (2019) 75(5):996–1006.e8. doi: 10.1016/j.molcel.2019.06.030
64. Zhang Y, Chen Y, Gucek M, Xu H. The Mitochondrial Outer Membrane Protein MDI Promotes Local Protein Synthesis and Mt DNA Replication. EMBO J (2016) 35(10):1045–57. doi: 10.15252/embj.201592994
65. Lesnik C, Cohen Y, Atir-Lande A, Schuldiner M, Arava Y. OM14 Is a Mitochondrial Receptor for Cytosolic Ribosomes That Supports Co-Translational Import Into Mitochondria. Nat Commun (2014) 5(1):5711. doi: 10.1038/ncomms6711
66. Martin KC, Ephrussi A. mRNA Localization: Gene Expression in the Spatial Dimension. Cell (2009) 136(4):719–30. doi: 10.1016/j.cell.2009.01.044
67. Tsuboi T, Viana MP, Xu F, Yu J, Chanchani R, Arceo XG, et al. Mitochondrial Volume Fraction and Translation Duration Impact Mitochondrial mRNA Localization and Protein Synthesis. eLife (2020) 9:e57814. doi: 10.7554/eLife.57814
68. Stan T. Recognition of Preproteins by the Isolated TOM Complex of Mitochondria. EMBO J (2000) 19(18):4895–902. doi: 10.1093/emboj/19.18.4895
69. van der Laan M, Schrempp SG, Pfanner N. Voltage-Coupled Conformational Dynamics of Mitochondrial Protein-Import Channel. Nat Struct Mol Biol (2013) 20(8):915–7. doi: 10.1038/nsmb.2643
70. Kang P-J, Ostermann J, Shilling J, Neupert W, Craig EA, Pfanner N. Requirement for Hsp70 in the Mitochondrial Matrix for Translocation and Folding of Precursor Proteins. Nat (1990) 348(6297):137–43. doi: 10.1038/348137a0
71. Mick DU, Dennerlein S, Wiese H, Reinhold R, Pacheu-Grau D, Lorenzi I, et al. MITRAC Links Mitochondrial Protein Translocation to Respiratory-Chain Assembly and Translational Regulation. Cell (2012) 151(7):1528–41. doi: 10.1016/j.cell.2012.11.053
72. van der Laan M, Wiedemann N, Mick DU, Guiard B, Rehling P, Pfanner N. A Role for Tim21 in Membrane-Potential-Dependent Preprotein Sorting in Mitochondria. Curr Biol (2006) 16(22):2271–6. doi: 10.1016/j.cub.2006.10.025
73. Protasoni M, Pérez-Pérez R, Lobo-Jarne T, Harbour ME, Ding S, Peñas A, et al. Respiratory Supercomplexes Act as a Platform for Complex III -Mediated Maturation of Human Mitochondrial Complexes I and IV. EMBO J (2020) 39(3):e102817. doi: 10.15252/embj.2019102817
74. Wang Y, Carrie C, Giraud E, Elhafez D, Narsai R, Duncan O, et al. Dual Location of the Mitochondrial Preprotein Transporters B14.7 and Tim23-2 in Complex I and the TIM17:23 Complex in Arabidopsis Links Mitochondrial Activity and Biogenesis. Plant Cell (2012) 24(6):2675–95. doi: 10.1105/tpc.112.098731
75. Kulawiak B, Höpker J, Gebert M, Guiard B, Wiedemann N, Gebert N. The Mitochondrial Protein Import Machinery has Multiple Connections to the Respiratory Chain. Biochim Biophys Acta (BBA) - Bioenerg (2013) 1827(5):612–26. doi: 10.1016/j.bbabio.2012.12.004
76. Braun HP, Emmermann M, Kruft V, Schmitz UK. The General Mitochondrial Processing Peptidase From Potato Is an Integral Part of Cytochrome C Reductase of the Respiratory Chain. EMBO J (1992) 11(9):3219–27. doi: 10.1002/j.1460-2075.1992.tb05399.x
77. Moreno-Sánchez R, Rodríguez-Enríquez S, Marín-Hernández A, Saavedra E. Energy Metabolism in Tumor Cells: Glycolytic and Mitochondrial Metabolism of Tumor Cells. FEBS J (2007) 274(6):1393–418. doi: 10.1111/j.1742-4658.2007.05686.x
78. Weber GF. Time and Circumstances: Cancer Cell Metabolism at Various Stages of Disease Progression. Front Oncol (2016) 6:257/full. doi: 10.3389/fonc.2016.00257/full
79. Viale A, Corti D, Draetta GF. Tumors and Mitochondrial Respiration: A Neglected Connection. Cancer Res (2015) 75(18):3687–91. doi: 10.1158/0008-5472.CAN-15-0491
80. Song J, Herrmann JM, Becker T. Quality Control of the Mitochondrial Proteome. Nat Rev Mol Cell Biol (2021) 22(1):54–70. doi: 10.1038/s41580-020-00300-2
81. Hsieh H-H, Lee JH, Chandrasekar S, Shan S. A Ribosome-Associated Chaperone Enables Substrate Triage in a Cotranslational Protein Targeting Complex. Nat Commun (2020) 11(1):5840. doi: 10.1038/s41467-020-19548-5
82. Gandin V, Topisirovic I. Co-Translational Mechanisms of Quality Control of Newly Synthesized Polypeptides. Translation (2014) 2(1):e28109. doi: 10.4161/trla.28109
83. Wang X, Chen XJ. A Cytosolic Network Suppressing Mitochondria-Mediated Proteostatic Stress and Cell Death. Nat (2015) 524(7566):481–4. doi: 10.1038/nature14859
84. Merkwirth C, Martinelli P, Korwitz A, Morbin M, Brönneke HS, Jordan SD, et al. Loss of Prohibitin Membrane Scaffolds Impairs Mitochondrial Architecture and Leads to Tau Hyperphosphorylation and Neurodegeneration. PloS Genet (2012) 8(11):e1003021. doi: 10.1371/journal.pgen.1003021
85. Segref A, Kevei É, Pokrzywa W, Schmeisser K, Mansfeld J, Livnat-Levanon N, et al. Pathogenesis of Human Mitochondrial Diseases is Modulated by Reduced Activity of the Ubiquitin/Proteasome System. Cell Metab (2014) 19(4):642–52. doi: 10.1016/j.cmet.2014.01.016
86. Nargund AM, Pellegrino MW, Fiorese CJ, Baker BM, Haynes CM. Mitochondrial Import Efficiency of ATFS-1 Regulates Mitochondrial UPR Activation. Sci (2012) 337(6094):587–90. doi: 10.1126/science.1223560
87. Johnson SC, Yanos ME, Kayser E-B, Quintana A, Sangesland M, Castanza A, et al. mTOR Inhibition Alleviates Mitochondrial Disease in a Mouse Model of Leigh Syndrome. Sci (2013) 342(6165):1524–8. doi: 10.1126/science.1244360
88. Levytskyy RM, Bohovych I, Khalimonchuk O. Metalloproteases of the Inner Mitochondrial Membrane. Biochem (2017) 56(36):4737–46. doi: 10.1021/acs.biochem.7b00663
89. Stiburek L, Cesnekova J, Kostkova O, Fornuskova D, Vinsova K, Wenchich L, et al. YME1L Controls the Accumulation of Respiratory Chain Subunits and Is Required for Apoptotic Resistance, Cristae Morphogenesis, and Cell Proliferation. Mol Biol Cell (2012) 23(6):1010–23. doi: 10.1091/mbc.e11-08-0674
90. Rainbolt TK, Atanassova N, Genereux JC, Wiseman RL. Stress-Regulated Translational Attenuation Adapts Mitochondrial Protein Import Through Tim17A Degradation. Cell Metab (2013) 18(6):908–19. doi: 10.1016/j.cmet.2013.11.006
91. MacVicar T, Ohba Y, Nolte H, Mayer FC, Tatsuta T, Sprenger H-G, et al. Lipid Signalling Drives Proteolytic Rewiring of Mitochondria by YME1L. Nat (2019) 575(7782):361–5. doi: 10.1038/s41586-019-1738-6
92. Kim YE, Hipp MS, Bracher A, Hayer-Hartl M, Ulrich Hartl F. Molecular Chaperone Functions in Protein Folding and Proteostasis. Annu Rev Biochem (2013) 82(1):323–55. doi: 10.1146/annurev-biochem-060208-092442
93. Truscott KN, Lowth BR, Strack PR, Dougan DA. Diverse Functions of Mitochondrial AAA+ Proteins: Protein Activation, Disaggregation, and Degradationthis Paper Is One of a Selection of Papers Published in This Special Issue Entitled 8th International Conference on AAA Proteins and has Undergone the Journal’s Usual Peer Review Process. Biochem Cell Biol (2010) 88(1):97–108. doi: 10.1139/O09-167
94. Sauer RT, Baker TA. AAA+ Proteases: ATP-Fueled Machines of Protein Destruction. Annu Rev Biochem (2011) 80(1):587–612. doi: 10.1146/annurev-biochem-060408-172623
95. Bota DA, Davies KJA. Lon Protease Preferentially Degrades Oxidized Mitochondrial Aconitase by an ATP-Stimulated Mechanism. Nat Cell Biol (2002) 4(9):674–80. doi: 10.1038/ncb836
96. Shin C-S, Meng S, Garbis SD, Moradian A, Taylor RW, Sweredoski MJ, et al. LONP1 and Mthsp70 Cooperate to Promote Mitochondrial Protein Folding. Nat Commun (2021) 12(1):265. doi: 10.1038/s41467-020-20597-z
97. Szczepanowska K, Maiti P, Kukat A, Hofsetz E, Nolte H, Senft K, et al. CLPP Coordinates Mitoribosomal Assembly Through the Regulation of ERAL 1 Levels. EMBO J (2016) 35(23):2566–83. doi: 10.15252/embj.201694253
98. Matsushima Y, Goto Y-I, Kaguni LS. Mitochondrial Lon Protease Regulates Mitochondrial DNA Copy Number and Transcription by Selective Degradation of Mitochondrial Transcription Factor A (TFAM). Proc Natl Acad Sci (2010) 107(43):18410–5. doi: 10.1073/pnas.1008924107
99. Dennerlein S, Rozanska A, Wydro M, Chrzanowska-Lightowlers ZMA, Lightowlers RN. Human ERAL1 is a Mitochondrial RNA Chaperone Involved in the Assembly of the 28S Small Mitochondrial Ribosomal Subunit. Biochem J (2010) 430(3):551–8. doi: 10.1042/BJ20100757
100. Münch C, Harper JW. Mitochondrial Unfolded Protein Response Controls Matrix Pre-RNA Processing and Translation. Nat (2016) 534(7609):710–3. doi: 10.1038/nature18302
101. Zurita Rendón O, Shoubridge EA. LONP1 Is Required for Maturation of a Subset of Mitochondrial Proteins, and Its Loss Elicits an Integrated Stress Response. Mol Cell Biol (2018) 38(20):e00412-17. doi: 10.1128/MCB.00412-17
102. Popow J, Alleaume A-M, Curk T, Schwarzl T, Sauer S, Hentze MW. FASTKD2 is an RNA-Binding Protein Required for Mitochondrial RNA Processing and Translation. RNA (2015) 21(11):1873–84. doi: 10.1261/rna.052365.115
103. Goard CA, Schimmer AD. Mitochondrial Matrix Proteases as Novel Therapeutic Targets in Malignancy. Oncogene (2014) 33(21):2690–9. doi: 10.1038/onc.2013.228
104. Lee YG, Kim HW, Nam Y, Shin KJ, Lee YJ, Park DH, et al. LONP1 and ClpP Cooperatively Regulate Mitochondrial Proteostasis for Cancer Cell Survival. Oncogenesis (2021) 10(2):18. doi: 10.1038/s41389-021-00306-1
105. Cagin U, Enriquez JA. The Complex Crosstalk Between Mitochondria and the Nucleus: What Goes in Between? Int J Biochem Cell Biol (2015) 63:10–5. doi: 10.1016/j.biocel.2015.01.026
106. Haynes CM, Ron D. The Mitochondrial UPR – Protecting Organelle Protein Homeostasis. J Cell Sci (2010) 123(22):3849–55. doi: 10.1242/jcs.075119
107. Kenny TC, Manfredi G, Germain D. The Mitochondrial Unfolded Protein Response as a Non-Oncogene Addiction to Support Adaptation to Stress During Transformation in Cancer and Beyond. Front Oncol (2017) 7:159. doi: 10.3389/fonc.2017.00159
108. Altieri DC, Stein GS, Lian JB, Languino LR. TRAP-1, the Mitochondrial Hsp90. Biochim Biophys Acta (BBA) - Mol Cell Res (2012) 1823(3):767–73. doi: 10.1016/j.bbamcr.2011.08.007
109. Chen B, Zhong D, Monteiro A. Comparative Genomics and Evolution of the HSP90 Family of Genes Across All Kingdoms of Organisms. BMC Genomics (2006) 7(1):156. doi: 10.1186/1471-2164-7-156
110. Amoroso MR, Matassa DS, Sisinni L, Lettini G, Landriscina M, Esposito F. TRAP1 Revisited: Novel Localizations and Functions of a “Next-Generation” Biomarker (Review). Int J Oncol (2014) 45(3):969–77. doi: 10.3892/ijo.2014.2530
111. Kang BH, Plescia J, Dohi T, Rosa J, Doxsey SJ, Altieri DC. Regulation of Tumor Cell Mitochondrial Homeostasis by an Organelle-Specific Hsp90 Chaperone Network. Cell (2007) 131(2):257–70. doi: 10.1016/j.cell.2007.08.028
112. Pridgeon JW, Olzmann JA, Chin L-S, Li L. PINK1 Protects Against Oxidative Stress by Phosphorylating Mitochondrial Chaperone TRAP1. Zoghbi HY, Editor. PloS Biol (2007) 5(7):e172. doi: 10.1371/journal.pbio.0050172
113. Rasola A, Neckers L, Picard D. Mitochondrial Oxidative Phosphorylation TRAP(1)ped in Tumor Cells. Trends Cell Biol (2014) 24(8):455–63. doi: 10.1016/j.tcb.2014.03.005
114. Amoroso MR, Matassa DS, Agliarulo I, Avolio R, Maddalena F, Condelli V, et al. Stress-Adaptive Response in Ovarian Cancer Drug Resistance: Role of TRAP1 in Oxidative Metabolism-Driven Inflammation. Adv Protein Chem Struct Biol (2017) 108:163–98. doi: 10.1016/bs.apcsb.2017.01.004
115. Criscuolo D, Avolio R, Calice G, Laezza C, Paladino S, Navarra G, et al. Cholesterol Homeostasis Modulates Platinum Sensitivity in Human Ovarian Cancer. Cells (2020) 9(4):828. doi: 10.3390/cells9040828
116. Amoroso MR, Matassa DS, Agliarulo I, Avolio R, Lu H, Sisinni L, et al. TRAP1 Downregulation in Human Ovarian Cancer Enhances Invasion and Epithelial–Mesenchymal Transition. Cell Death Dis (2016) 7(12):e2522–2. doi: 10.1038/cddis.2016.400
117. Siegelin MD, Dohi T, Raskett CM, Orlowski GM, Powers CM, Gilbert CA, et al. Exploiting the Mitochondrial Unfolded Protein Response for Cancer Therapy in Mice and Human Cells. J Clin Invest (2011) 121(4):1349–60. doi: 10.1172/JCI44855
118. Amoroso MR, Matassa DS, Laudiero G, Egorova AV, Polishchuk RS, Maddalena F, et al. TRAP1 and the Proteasome Regulatory Particle TBP7/Rpt3 Interact in the Endoplasmic Reticulum and Control Cellular Ubiquitination of Specific Mitochondrial Proteins. Cell Death Differ (2012) 19(4):592–604. doi: 10.1038/cdd.2011.128
119. Landriscina M, Laudiero G, Maddalena F, Amoroso MR, Piscazzi A, Cozzolino F, et al. Mitochondrial Chaperone Trap1 and the Calcium Binding Protein Sorcin Interact and Protect Cells Against Apoptosis Induced by Antiblastic Agents. Cancer Res (2010) 70(16):6577–86. doi: 10.1158/0008-5472.CAN-10-1256
120. Couvillion MT, Soto IC, Shipkovenska G, Churchman LS. Synchronized Mitochondrial and Cytosolic Translation Programs. Nat (2016) 533(7604):499–503. doi: 10.1038/nature18015
121. Herrmann JM, Woellhaf MW, Bonnefoy N. Control of Protein Synthesis in Yeast Mitochondria: The Concept of Translational Activators. Biochim Biophys Acta (BBA) - Mol Cell Res (2013) 1833(2):286–94. doi: 10.1016/j.bbamcr.2012.03.007
122. Zambrano A, Fontanesi F, Solans A, de Oliveira RL, Fox TD, Tzagoloff A, et al. Aberrant Translation of Cytochrome C Oxidase Subunit 1 mRNA Species in the Absence of Mss51p in the Yeast Saccharomyces Cerevisiae. Walter P, Editor. MBoC (2007) 18(2):523–35. doi: 10.1091/mbc.e06-09-0803
123. Pierrel F, Bestwick ML, Cobine PA, Khalimonchuk O, Cricco JA, Winge DR. Coa1 Links the Mss51 Post-Translational Function to Cox1 Cofactor Insertion in Cytochrome C Oxidase Assembly. EMBO J (2007) 26(20):4335–46. doi: 10.1038/sj.emboj.7601861
124. Mick DU, Vukotic M, Piechura H, Meyer HE, Warscheid B, Deckers M, et al. Coa3 and Cox14 are Essential for Negative Feedback Regulation of COX1 Translation in Mitochondria. J Cell Biol (2010) 191(1):141–54. doi: 10.1083/jcb.201007026
125. Mick DU, Wagner K, van der Laan M, Frazier AE, Perschil I, Pawlas M, et al. Shy1 Couples Cox1 Translational Regulation to Cytochrome C Oxidase Assembly. EMBO J (2007) 26(20):4347–58. doi: 10.1038/sj.emboj.7601862
126. Weraarpachai W, Antonicka H, Sasarman F, Seeger J, Schrank B, Kolesar JE, et al. Mutation in TACO1, Encoding a Translational Activator of COX I, Results in Cytochrome C Oxidase Deficiency and Late-Onset Leigh Syndrome. Nat Genet (2009) 41(7):833–7. doi: 10.1038/ng.390
127. Richter-Dennerlein R, Oeljeklaus S, Lorenzi I, Ronsör C, Bareth B, Schendzielorz AB, et al. Mitochondrial Protein Synthesis Adapts to Influx of Nuclear-Encoded Protein. Cell (2016) 167(2):471–483.e10. doi: 10.1016/j.cell.2016.09.003
128. Fogal V, Richardson AD, Karmali PP, Scheffler IE, Smith JW, Ruoslahti E. Mitochondrial P32 Protein Is a Critical Regulator of Tumor Metabolism via Maintenance of Oxidative Phosphorylation. Mol Cell Biol (2010) 30(6):1303–18. doi: 10.1128/MCB.01101-09
129. Škrtić M, Sriskanthadevan S, Jhas B, Gebbia M, Wang X, Wang Z, et al. Inhibition of Mitochondrial Translation as a Therapeutic Strategy for Human Acute Myeloid Leukemia. Cancer Cell (2011) 20(5):674–88. doi: 10.1016/j.ccr.2011.10.015
130. D’Andrea A, Gritti I, Nicoli P, Giorgio M, Doni M, Conti A, et al. The Mitochondrial Translation Machinery as a Therapeutic Target in Myc-Driven Lymphomas. Oncotarget (2016) 7(45):72415–30. doi: 10.18632/oncotarget.11719
131. Yang S, Cai H, Hu B, Tu J. LncRNA SAMMSON Negatively Regulates miR-9-3p in Hepatocellular Carcinoma Cells and has Prognostic Values. Biosci Rep (2019) 39(7):BSR20190615. doi: 10.1042/BSR20190615
132. Vendramin R, Verheyden Y, Ishikawa H, Goedert L, Nicolas E, Saraf K, et al. SAMMSON Fosters Cancer Cell Fitness by Concertedly Enhancing Mitochondrial and Cytosolic Translation. Nat Struct Mol Biol (2018) 25(11):1035–46. doi: 10.1038/s41594-018-0143-4
133. Leucci E, Vendramin R, Spinazzi M, Laurette P, Fiers M, Wouters J, et al. Melanoma Addiction to the Long Non-Coding RNA SAMMSON. Nat (2016) 531(7595):518–22. doi: 10.1038/nature17161
134. Chae YC, Angelin A, Lisanti S, Kossenkov AV, Speicher KD, Wang H, et al. Landscape of the Mitochondrial Hsp90 Metabolome in Tumours. Nat Commun (2013) 4(1):2139. doi: 10.1038/ncomms3139
135. Joshi A, Dai L, Liu Y, Lee J, Ghahhari NM, Segala G, et al. The Mitochondrial HSP90 Paralog TRAP1 Forms an OXPHOS-Regulated Tetramer and Is Involved in Mitochondrial Metabolic Homeostasis. BMC Biol (2020) 18(1):10. doi: 10.1186/s12915-020-0740-7
136. Avolio R, Järvelin AI, Mohammed S, Agliarulo I, Condelli V, Zoppoli P, et al. Protein Syndesmos is a Novel RNA-Binding Protein That Regulates Primary Cilia Formation. Nucleic Acids Res (2018) 46(22):12067–86. doi: 10.1093/nar/gky873/5107578
137. Funes JM, Quintero M, Henderson S, Martinez D, Qureshi U, Westwood C, et al. Transformation of Human Mesenchymal Stem Cells Increases Their Dependency on Oxidative Phosphorylation for Energy Production. Proc Natl Acad Sci (2007) 104(15):6223–8. doi: 10.1073/pnas.0700690104
138. Vyas S, Zaganjor E, Haigis MC. Mitochondria and Cancer. Cell (2016) 166(3):555–66. doi: 10.1016/j.cell.2016.07.002
139. Ghosh P, Vidal C, Dey S, Zhang L. Mitochondria Targeting as an Effective Strategy for Cancer Therapy. IJMS (2020) 21(9):3363. doi: 10.3390/ijms21093363
140. Yoshida S, Tsutsumi S, Muhlebach G, Sourbier C, Lee M-J, Lee S, et al. Molecular Chaperone TRAP1 Regulates a Metabolic Switch Between Mitochondrial Respiration and Aerobic Glycolysis. Proc Natl Acad Sci (2013) 110(17):E1604–12. doi: 10.1073/pnas.1220659110
141. Pecoraro A, Pagano M, Russo G, Russo A. Ribosome Biogenesis and Cancer: Overview on Ribosomal Proteins. IJMS (2021) 22(11):5496. doi: 10.3390/ijms22115496
142. Kim H-J, Maiti P, Barrientos A. Mitochondrial Ribosomes in Cancer. Semin Cancer Biol (2017) 47:67–81. doi: 10.1016/j.semcancer.2017.04.004
143. Zhang L, Ging NC, Komoda T, Hanada T, Suzuki T, Watanabe K. Antibiotic Susceptibility of Mammalian Mitochondrial Translation. FEBS Lett (2005) 579(28):6423–7. doi: 10.1016/j.febslet.2005.09.103
144. Lamb R, Ozsvari B, Lisanti CL, Tanowitz HB, Howell A, Martinez-Outschoorn UE, et al. Antibiotics That Target Mitochondria Effectively Eradicate Cancer Stem Cells, Across Multiple Tumor Types: Treating Cancer Like an Infectious Disease. Oncotarget (2015) 6(7):4569–84. doi: 10.18632/oncotarget.3174
145. Protasoni M, Kroon AM, Taanman J-W. Mitochondria as Oncotarget: A Comparison Between the Tetracycline Analogs Doxycycline and COL-3. Oncotarget (2018) 9(73):33818–31. doi: 10.18632/oncotarget.26107
146. Rudek MA, New P, Mikkelsen T, Phuphanich S, Alavi JB, Nabors LB, et al. Phase I and Pharmacokinetic Study of COL-3 in Patients With Recurrent High-Grade Gliomas. J Neurooncol (2011) 105(2):375–81. doi: 10.1007/s11060-011-0602-9
147. Norberg E, Lako A, Chen P-H, Stanley IA, Zhou F, Ficarro SB, et al. Differential Contribution of the Mitochondrial Translation Pathway to the Survival of Diffuse Large B-Cell Lymphoma Subsets. Cell Death Differ (2017) 24(2):251–62. doi: 10.1038/cdd.2016.116
148. Jia X, Gu Z, Chen W, Jiao J. Tigecycline Targets Nonsmall Cell Lung Cancer Through Inhibition of Mitochondrial Function. Fundam Clin Pharmacol (2016) 30(4):297–306. doi: 10.1111/fcp.12199
149. Hu B, Guo Y. Inhibition of Mitochondrial Translation as a Therapeutic Strategy for Human Ovarian Cancer to Overcome Chemoresistance. Biochem Biophys Res Commun (2019) 509(2):373–8. doi: 10.1016/j.bbrc.2018.12.127
150. Sharon D, Cathelin S, Mirali S, Di Trani JM, Yanofsky DJ, Keon KA, et al. Inhibition of Mitochondrial Translation Overcomes Venetoclax Resistance in AML Through Activation of the Integrated Stress Response. Sci Transl Med (2019) 11(516):eaax2863. doi: 10.1126/scitranslmed.aax2863
151. Fu X, Liu W, Huang Q, Wang Y, Li H, Xiong Y. Targeting Mitochondrial Respiration Selectively Sensitizes Pediatric Acute Lymphoblastic Leukemia Cell Lines and Patient Samples to Standard Chemotherapy. Am J Cancer Res (2017) 7(12):2395–405.
152. Tan J, Song M, Zhou M, Hu Y. Antibiotic Tigecycline Enhances Cisplatin Activity Against Human Hepatocellular Carcinoma Through Inducing Mitochondrial Dysfunction and Oxidative Damage. Biochem Biophys Res Commun (2017) 483(1):17–23. doi: 10.1016/j.bbrc.2017.01.021
153. Wang B, Ao J, Yu D, Rao T, Ruan Y, Yao X. Inhibition of Mitochondrial Translation Effectively Sensitizes Renal Cell Carcinoma to Chemotherapy. Biochem Biophys Res Commun (2017) 490(3):767–73. doi: 10.1016/j.bbrc.2017.06.115
154. Sheth A, Escobar-Alvarez S, Gardner J, Ran L, Heaney ML, Scheinberg DA. Inhibition of Human Mitochondrial Peptide Deformylase Causes Apoptosis in C-Myc-Overexpressing Hematopoietic Cancers. Cell Death Dis (2014) 5(3):e1152–2. doi: 10.1038/cddis.2014.112
155. Lee MD, She Y, Soskis MJ, Borella CP, Gardner JR, Hayes PA, et al. Human Mitochondrial Peptide Deformylase, a New Anticancer Target of Actinonin-Based Antibiotics. J Clin Invest (2004) 114(8):1107–16. doi: 10.1172/JCI200422269
156. Singh RP, Jeyaraju DV, Voisin V, Hurren R, Xu C, Hawley JR, et al. Disrupting Mitochondrial Copper Distribution Inhibits Leukemic Stem Cell Self-Renewal. Cell Stem Cell (2020) 26(6):926–37.e10. doi: 10.1016/j.stem.2020.04.010
157. Kabiri Y, Fuhrmann A, Becker A, Jedermann L, Eberhagen C, König A-C, et al. Mitochondrial Impairment by MitoBloCK-6 Inhibits Liver Cancer Cell Proliferation. Front Cell Dev Biol (2021) 9:725474. doi: 10.3389/fcell.2021.725474
158. Greer YE, Porat-Shliom N, Nagashima K, Stuelten C, Crooks D, Koparde VN, et al. ONC201 Kills Breast Cancer Cells In Vitro by Targeting Mitochondria. Oncotarget (2018) 9(26):18454–79. doi: 10.18632/oncotarget.24862
159. Amoroso F, Glass K, Singh R, Liberal F, Steele RE, Maguire S, et al. Modulating the Unfolded Protein Response With ONC201 to Impact on Radiation Response in Prostate Cancer Cells. Sci Rep (2021) 11(1):4252. doi: 10.1038/s41598-021-83215-y
160. Stein MN, Bertino JR, Kaufman HL, Mayer T, Moss R, Silk A, et al. First-In-Human Clinical Trial of Oral ONC201 in Patients With Refractory Solid Tumors. Clin Cancer Res (2017) 23(15):4163–9. doi: 10.1158/1078-0432.CCR-16-2658
161. Borella R, Forti L, Gibellini L, De Gaetano A, De Biasi S, Nasi M, et al. Synthesis and Anticancer Activity of CDDO and CDDO-Me, Two Derivatives of Natural Triterpenoids. Molecules (2019) 24(22):4097. doi: 10.3390/molecules24224097
162. Kang BH, Plescia J, Song HY, Meli M, Colombo G, Beebe K, et al. Combinatorial Drug Design Targeting Multiple Cancer Signaling Networks Controlled by Mitochondrial Hsp90. J Clin Invest (2009) 119(3):454–64. doi: 10.1172/JCI37613
163. Nelson ML, Levy SB. The History of the Tetracyclines: The History of the Tetracyclines. Ann New York Acad Sci (2011) 1241(1):17–32. doi: 10.1111/j.1749-6632.2011.06354.x
164. Chopra I, Roberts M. Tetracycline Antibiotics: Mode of Action, Applications, Molecular Biology, and Epidemiology of Bacterial Resistance. Microbiol Mol Biol Rev (2001) 65(2):232–60. doi: 10.1128/MMBR.65.2.232-260.2001
165. Richards C, Pantanowitz L, Dezube BJ. Antimicrobial and Non-Antimicrobial Tetracyclines in Human Cancer Trials. Pharmacol Res (2011) 63(2):151–6. doi: 10.1016/j.phrs.2010.10.008
166. Hadjimichael AC, Foukas AF, Savvidou OD, Mavrogenis AF, Psyrri AK, Papagelopoulos PJ. The Anti-Neoplastic Effect of Doxycycline in Osteosarcoma as a Metalloproteinase (MMP) Inhibitor: A Systematic Review. Clin Sarcoma Res (2020) 10(1):7. doi: 10.1186/s13569-020-00128-6
167. Lokeshwar BL. Chemically Modified Non-Antimicrobial Tetracyclines are Multifunctional Drugs Against Advanced Cancers. Pharmacol Res (2011) 63(2):146–50. doi: 10.1016/j.phrs.2010.11.003
168. Iwasaki H, Inoue H, Mitsuke Y, Badran A, Ikegaya S, Ueda T. Doxycycline Induces Apoptosis by Way of Caspase-3 Activation With Inhibition of Matrix Metalloproteinase in Human T-Lymphoblastic Leukemia CCRF-CEM Cells. J Lab Clin Med (2002) 140(6):382–6. doi: 10.1067/mlc.2002.129308
169. Song H, Fares M, Maguire KR, Sidén Å, Potácová Z. Cytotoxic Effects of Tetracycline Analogues (Doxycycline, Minocycline and COL-3) in Acute Myeloid Leukemia HL-60 Cells. Wang Z, Editor. PloS One (2014) 9(12):e114457. doi: 10.1371/journal.pone.0114457
170. Onoda T, Ono T, Dhar DK, Yamanoi A, Fujii T, Nagasue N. Doxycycline Inhibits Cell Proliferation and Invasive Potential: Combination Therapy With Cyclooxygenase-2 Inhibitor in Human Colorectal Cancer Cells. J Lab Clin Med (2004) 143(4):207–16. doi: 10.1016/j.lab.2003.12.012
171. Duivenvoorden WCM, Popović SV, Lhoták S, Seidlitz E, Hirte HW, Tozer RG, et al. Doxycycline Decreases Tumor Burden in a Bone Metastasis Model of Human Breast Cancer. Cancer Res (2002) 62(6):1588–91.
172. Lokeshwar BL, Selzer MG, Zhu B-Q, Block NL, Golub LM. Inhibition of Cell Proliferation, Invasion, Tumor Growth and Metastasis by an Oral Non-Antimicrobial Tetracycline Analog (COL-3) in a Metastatic Prostate Cancer Model. Int J Cancer (2002) 98(2):297–309. doi: 10.1002/ijc.10168
173. Dijk SN, Protasoni M, Elpidorou M, Kroon AM, Taanman J-W. Mitochondria as Target to Inhibit Proliferation and Induce Apoptosis of Cancer Cells: The Effects of Doxycycline and Gemcitabine. Sci Rep (2020) 10(1):4363. doi: 10.1038/s41598-020-61381-9
174. Nelson MA, McLaughlin KL, Hagen JT, Coalson HS, Schmidt C, Kassai M, et al. Intrinsic OXPHOS Limitations Underlie Cellular Bioenergetics in Leukemia. eLife (2021) 10:e63104. doi: 10.7554/eLife.63104
175. Martin TD, Cook DR, Choi MY, Li MZ, Haigis KM, Elledge SJ. A Role for Mitochondrial Translation in Promotion of Viability in K-Ras Mutant Cells. Cell Rep (2017) 20(2):427–38. doi: 10.1016/j.celrep.2017.06.061
176. Chen DZ, Patel DV, Hackbarth CJ, Wang W, Dreyer G, Young DC, et al. Actinonin, a Naturally Occurring Antibacterial Agent, Is a Potent Deformylase Inhibitor. Biochem (2000) 39(6):1256–62. doi: 10.1021/bi992245y
177. Randhawa H, Chikara S, Gehring D, Yildirim T, Menon J, Reindl KM. Overexpression of Peptide Deformylase in Breast, Colon, and Lung Cancers. BMC Cancer (2013) 13(1):321. doi: 10.1186/1471-2407-13-321
178. Escobar-Alvarez S, Gardner J, Sheth A, Manfredi G, Yang G, Ouerfelli O, et al. Inhibition of Human Peptide Deformylase Disrupts Mitochondrial Function. Mol Cell Biol (2010) 30(21):5099–109. doi: 10.1128/MCB.00469-10
179. Richter U, Lahtinen T, Marttinen P, Myöhänen M, Greco D, Cannino G, et al. A Mitochondrial Ribosomal and RNA Decay Pathway Blocks Cell Proliferation. Curr Biol (2013) 23(6):535–41. doi: 10.1016/j.cub.2013.02.019
180. Miyata N, Tang Z, Conti MA, Johnson ME, Douglas CJ, Hasson SA, et al. Adaptation of a Genetic Screen Reveals an Inhibitor for Mitochondrial Protein Import Component Tim44. J Biol Chem (2017) 292(13):5429–42. doi: 10.1074/jbc.M116.770131
181. Dabir DV, Hasson SA, Setoguchi K, Johnson ME, Wongkongkathep P, Douglas CJ, et al. A Small Molecule Inhibitor of Redox-Regulated Protein Translocation Into Mitochondria. Dev Cell (2013) 25(1):81–92. doi: 10.1016/j.devcel.2013.03.006
182. Nguyen KH, Nguyen AH, Dabir DV. Clinical Implications of Augmenter of Liver Regeneration in Cancer: A Systematic Review. Anticancer Res (2017) 37(7):3379–83. doi: 10.21873/anticanres.11704
183. Cole A, Wang Z, Coyaud E, Voisin V, Gronda M, Jitkova Y, et al. Inhibition of the Mitochondrial Protease ClpP as a Therapeutic Strategy for Human Acute Myeloid Leukemia. Cancer Cell (2015) 27(6):864–76. doi: 10.1016/j.ccell.2015.05.004
184. Seo JH, Rivadeneira DB, Caino MC, Chae YC, Speicher DW, Tang H-Y, et al. The Mitochondrial Unfoldase-Peptidase Complex ClpXP Controls Bioenergetics Stress and Metastasis. Christofk H, Editor. PloS Biol (2016) 14(7):e1002507. doi: 10.1371/journal.pbio.1002507
185. Luo J, Zeng B, Tao C, Lu M, Ren G. ClpP Regulates Breast Cancer Cell Proliferation, Invasion and Apoptosis by Modulating the Src/PI3K/Akt Signaling Pathway. PeerJ (2020) 8:e8754. doi: 10.7717/peerj.8754
186. Hackl MW, Lakemeyer M, Dahmen M, Glaser M, Pahl A, Lorenz-Baath K, et al. Phenyl Esters Are Potent Inhibitors of Caseinolytic Protease P and Reveal a Stereogenic Switch for Deoligomerization. J Am Chem Soc (2015) 137(26):8475–83. doi: 10.1021/jacs.5b03084
187. Ishizawa J, Zarabi SF, Davis RE, Halgas O, Nii T, Jitkova Y, et al. Mitochondrial ClpP-Mediated Proteolysis Induces Selective Cancer Cell Lethality. Cancer Cell (2019) 35(5):721–37.e9. doi: 10.1016/j.ccell.2019.03.014
188. Prabhu VV, Morrow S, Rahman Kawakibi A, Zhou L, Ralff M, Ray J, et al. ONC201 and Imipridones: Anti-Cancer Compounds With Clinical Efficacy. Neoplasia (2020) 22(12):725–44. doi: 10.1016/j.neo.2020.09.005
189. Cheng C-W, Kuo C-Y, Fan C-C, Fang W-C, Jiang SS, Lo Y-K, et al. Overexpression of Lon Contributes to Survival and Aggressive Phenotype of Cancer Cells Through Mitochondrial Complex I-Mediated Generation of Reactive Oxygen Species. Cell Death Dis (2013) 4(6):e681–1. doi: 10.1038/cddis.2013.204
190. Gibellini L, Pinti M, Bartolomeo R, De Biasi S, Cormio A, Musicco C, et al. Inhibition of Lon Protease by Triterpenoids Alters Mitochondria and is Associated to Cell Death in Human Cancer Cells. Oncotarget (2015) 6(28):25466–83. doi: 10.18632/oncotarget.4510
191. Lettini G, Maddalena F, Sisinni L, Condelli V, Matassa DS, Costi MP, et al. TRAP1: A Viable Therapeutic Target for Future Cancer Treatments? Expert Opin Ther Targets (2017) 21(8):805–15. doi: 10.1080/14728222.2017.1349755
192. Rondanin R, Lettini G, Oliva P, Baruchello R, Costantini C, Trapella C, et al. New TRAP1 and Hsp90 Chaperone Inhibitors With Cationic Components: Preliminary Studies on Mitochondrial Targeting. Bioorg Med Chem Lett (2018) 28(13):2289–93. doi: 10.1016/j.bmcl.2018.05.031
193. Sanchez-Martin C, Moroni E, Ferraro M, Laquatra C, Cannino G, Masgras I, et al. Rational Design of Allosteric and Selective Inhibitors of the Molecular Chaperone Trap1. Cell Rep (2020) 31(3):107531. doi: 10.1016/j.celrep.2020.107531
194. Hanahan D, Weinberg RA. Hallmarks of Cancer: The Next Generation. Cell (2011) 144(5):646–74. doi: 10.1016/j.cell.2011.02.013
195. Blagden SP, Willis AE. The Biological and Therapeutic Relevance of mRNA Translation in Cancer. Nat Rev Clin Oncol (2011) 8(5):280–91. doi: 10.1038/nrclinonc.2011.16
196. Longo J, van Leeuwen JE, Elbaz M, Branchard E, Penn LZ. Statins as Anticancer Agents in the Era of Precision Medicine. Clin Cancer Res (2020) 26(22):5791–800. doi: 10.1158/1078-0432.CCR-20-1967
197. Ruggero D. Revisiting the Nucleolus: From Marker to Dynamic Integrator of Cancer Signaling. Sci Signal (2012) 5(241):pe38. doi: 10.1126/scisignal.2003477
Keywords: mitochondrial translation, protein synthesis, inter-organelle coordinated translation regulation, mitochondrial protein import, mitochondrial protein quality control (mtPQC)
Citation: Criscuolo D, Avolio R, Matassa DS and Esposito F (2021) Targeting Mitochondrial Protein Expression as a Future Approach for Cancer Therapy. Front. Oncol. 11:797265. doi: 10.3389/fonc.2021.797265
Received: 18 October 2021; Accepted: 03 November 2021;
Published: 23 November 2021.
Edited by:
Yong Teng, Emory University, United StatesReviewed by:
Yoav Arava, Technion Israel Institute of Technology, IsraelCopyright © 2021 Criscuolo, Avolio, Matassa and Esposito. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Franca Esposito, ZnJhbmNhLmVzcG9zaXRvQHVuaW5hLml0
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.