 Juan Bautista Blaquier
Juan Bautista Blaquier Andrés Felipe Cardona
Andrés Felipe Cardona Gonzalo Recondo
Gonzalo Recondo
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Oncol., 24 December 2021
Sec. Thoracic Oncology
Volume 11 - 2021 | https://doi.org/10.3389/fonc.2021.787585
This article is part of the Research TopicTargeting KRAS in Lung CancerView all 7 articles
KRAS mutations are one of the most prevalent oncogenic alterations in cancer. Until recently, drug development targeting KRAS did not convey clinical benefits to patients. Specific KRASG12C inhibitors, such as sotorasib and adagrasib, have been designed to bind to the protein’s mutant structure and block KRASG12C in its GDP-bound inactive state. Phase 1/2 trials have shown promising anti-tumor activity, especially in pretreated non-small cell lung cancer patients. As expected, both primary and secondary resistance to KRASG12C inhibitors invariably occurs, and molecular mechanisms have been characterized in pre-clinical models and patients. Several mechanisms such as tyrosine kinase receptors (RTKs) mediated feedback reactivation of ERK-dependent signaling can result in intrinsic resistance to KRAS target therapy. Acquired resistance to KRASG12C inhibitors include novel KRAS mutations such as Y96D/C and other RAS-MAPK effector protein mutations. This review focuses on the intrinsic and acquired mechanisms of resistance to KRASG12C inhibitors in KRASG12C mutant non-small cell lung cancer and the potential clinical strategies to overcome or prevent it.
KRAS was the first described oncogene in human cancer (1) and one of the most frequently mutated genes (2). It encodes a GTPase superfamily protein that mediates the intracellular signaling of activated tyrosine kinase receptors by switching between inactive GDP-bound and active GTP-bound states. This activation induces phosphorylation of effectors of the mitogen-activated protein kinase (MAPK) pathway like RAF, MEK, and ERK leading to apoptosis inhibition and activation of transcription factors that promote cancer cell survival and metastasis (3, 4). Guanine-nucleotide-exchange factors (GEFs) catalyze the exchange of GDP for GTP, hence turning KRAS into its active state; on the other hand, GTPase activating proteins (GAPs) take part in the GTP hydrolysis reaction, inactivating KRAS (5). Single aminoacidic mutations at codons 12,13, and 61 are responsible for structural changes that prevent GAPs from hydrolyzing GTP, and therefore blocking KRAS in its GTP-bound active state (6). The hotspot transversion mutation KRASG12C is the most frequently found in lung adenocarcinoma accounting for 13% of cases in the western population (7, 8). Despite its high prevalence and importance in the development of cancer, efforts to effectively target KRAS have long been futile as a consequence of micromolar GTP concentrations in cells (~0.5 mmol/L) combined with a picomolar affinity (dissociation constant at ~10-10 mol/L) of KRAS for GTP (9). Moreover, the lack of allosteric regulatory sites needed for drug binding coined KRAS as an “undruggable” target (10). This has been modified with the discovery in the KRASG12C mutant protein of a pocket that resides beneath de switch II region, adjacent to the mutant cysteine that allows the binding of small inhibitory molecules and, as a consequence favoring the affinity of KRAS for GDP, blocking KRASG12C in its inactive state (11). This unique feature of the KRAS G12C mutation has led to the development of specific KRASG12C irreversible inhibitors such us adagrasib and sotorasib which is currently FDA approved to treat patients with KRASG12C lung cancer.
Unfortunately, primary and acquired resistance invariably occur. This review focuses on the intrinsic and acquired mechanisms of resistance to KRASG12C inhibitors, mainly in KRASG12C mutant non-small cell lung cancer, and potential clinical strategies to overcome or prevent resistance.
Particular RAS mutations may modify RAS proteins’ biochemical behavior, including their ability to bind GTP and GDP. Further studies continue reporting differences in GTP binding and intrinsic or GAP-mediated GTP hydrolysis (12). Smith et al. detected KRASG12V in the GTP-bound conformation, which was consistent with its high transforming potential. In addition, experiments in MCF10A cells transduced with different KRAS mutations revealed that KRAS wild-type (WT) and KRASG12D and KRASG13D could bind GTP with similar affinity to control cells, which only express endogenous KRAS, after EGF stimulation. In contrast, KRASG12C showed an increase in GTP-binding up to 2-fold mutant compared to control cells (13). Another study also reported that KRASG12A, G12R, Q61H, and Q61L decreased GTP hydrolysis speed approximately by 40- to 80-fold compared to KRAS wild-type, the G12C mutation had a minimal impact in this respect. Regarding this endpoint, KRASG12D, G12V, and KRASG13D mutant proteins displayed an intermediate effect (14).
Anchorage-independent growth is the ability of transformed cells to grow in suspension or unattached to any matrix (13) an associated characteristic for tumor metastasis regulated by the RAS/RAF/MAPK signaling pathway (15). Seeburg et al. reported that except for HRAS WT and HRAS G12P, all the HRAS codon 12 mutants could grow in soft agar (16), results paralleling their data on transforming potential of these mutant proteins. Immortalized human bronchial epithelial cells with specific shRNA knockdown of p53 mRNA expressing KRASG12C were able to form colonies in soft agar compared to KRASG12D easily- and KRAS WT-transfected cells (17), suggesting that the genetic background could also affect the phenotypical manifestation of mutant RAS variants.
The RAF1 serine/threonine kinase is one of the best characterized RAS effector proteins, located directly downstream of RAS in the MAPK pathway (14). Considering that point mutations at codons 12 and 61 of HRAS differ in their phenotypical properties as previously reported (18), Voice et al. hypothesized that mutant RAS proteins might activate RAF1 differentially (19) and found that KRASG12A, G12C, G13D, Q61L, and Q61H showed 1.2- to 2.3-fold decrease in relative affinity compared to WT KRAS and KRASG12D, G12R and G12V displayed an even more pronounced reduction in affinity for RAF1 (4.8-, 6.2-, and 7.3-fold, respectively).
RAS proteins also activate the PI3K/AKT/mTOR pathway to promote cell survival by activating survival factors and inhibiting apoptotic proteins (20). Therefore, different in vitro (13) and in vivo (18, 21) studies also assessed the activation of this pathway by the interaction of various RAS mutated variants with PI3K and different downstream proteins, such as AKT, 4EBP, or RPS6. The comparison of KRASG12C and G12V with WT KRAS in a panel of 67 NSCLC cell lines showed that these mutations decreased AKT activation compared to WT KRAS (17). Despite this low activation of AKT, cells expressing KRASG12C or G12V showed the same phosphorylation levels of 70S6K and 4EBP proteins compared to WT KRAS in the absence of serum. In contrast, the addition of serum to the media enabled KRASG12C and G12V to strongly phosphorylate 70S6K compared to WT KRAS (17).
RAS proteins can also interact and activate effectors that do not belong to the MAPK and the PI3K canonical cascades. For example, RAC, a subfamily of small GTPases of the RHO family, can interact with RAS via the RacGEF called Tiam1. The RAS/RAC signaling pathway controls several cellular functions by regulating actin cytoskeleton, including cell morphology, locomotion, and polarity (22). Another RAS downstream effector subfamily is the RAL group of proteins involved in membrane trafficking, proliferation, survival, and metastasis in many types of cancer (23). In this sense, WT KRAS and KRASG12C, but not KRASG12D, activated RALA and RALB effector proteins (17).
Brunelli et al. characterized the metabolic profile of the isogenic NCI-H1299 NSCLC cell line overexpressing WT KRAS or KRASG12C, G12D, or G12V (24). Most metabolites identified were common to all three KRAS-mutated lines (G12C, G12D, and G12V), although these mutants harbored 74, 58, and 48 unique metabolites, respectively, compared to WT. Moreover, the deregulated metabolites between WT and mutant KRAS variants were classified into biochemical groups. The two most abundant classes for KRASG12C, G12D, and G12V were glycerophospholipids and amino acids. KRASG12C and KRASG12D mainly affected phosphatidylcholines (PC) and phosphatidylinositol (PI), whereas KRASG12V influenced PI and phosphatidylserine. In addition, the report by Brunelli et al. provided further insights into the biology of the deregulated metabolites. KRASG12C, G12D, and G12V variants showed an increase of metabolites related to protein biosynthesis, glutathione, glutamate metabolism and ammonia recycling (24). Moreover, KRASG12C, G12D, and G12V had lower levels of glutamate, glutamine, asparagine, and proline, amino acids interconnected in the glutamate synthase cycle, and lower levels of NAD+, an essential coenzyme involved in many cellular metabolic pathways.
Following on these results, the group of Pastorelli continued studying the metabolic profile of KRASG12C, as it is the most representative KRAS mutation in NSCLC patients. In this work, the NCI-H1299 NSCLC cell line expressing WT or KRASG12C and xenograft tumors generated from this cell line were analyzed (25). They identified 26 and 23 deregulated metabolites in vitro and in vivo, respectively, between WT KRAS and KRASG12C. The enriched pathway analysis of these deregulated metabolites showed that KRASG12C alters the same metabolic pathways in vitro and in vivo, including pathways involved in protein biosynthesis, ammonia recycling, and urea cycle (25). Focusing on the deregulated metabolites whose abundance changed significantly in vitro and in vivo between WT KRAS and KRASG12C, 11 and 16 metabolites were significantly altered, respectively. Moreover, in both in vitro and in vivo models, KRASG12C, decreased glutamine and glutamate levels, two amino acids involved in nitrogen balance maintenance, supporting the central role of glutaminolysis and nitrogen anabolism to provide energy for cancer cell growth and proliferation. This indicates that cells expressing the KRASG12C variant use glutaminolysis as a source of energy (25). In addition, KRASG12C, mutation induced a significant increase in the levels of carnitine, acetyl-carnitine, and butyryl-carnitine, which are involved in the oxidation of fatty acids. This increase could be associated with the mitochondrial fatty acid beta-oxidation to respond to the increasing energy demand triggered by KRASG12C, to fuel cell or tumor growth and proliferation (25).
The development of specific KRASG12C inhibitors started with ARS-1620, a small covalent specific inhibitor, that showed in vitro and in vivo activity in KRASG12C mutant models (26). This was followed by the development of two KRASG12C inhibitors, sotorasib (AMG 510) and adagrasib (MRTX849), supported by robust preclinical evidence and reaching clinical development in phase I/II trials (27, 28). Both compounds have a common mechanism of action by inhibiting KRAS in its GDP-bound state, however, the binding properties of both inhibitors differ slightly. The KRASG12C cryptic pocket is formed by residues H95/Y96/Q99 is exploited by the hydrogen mediated bonding of the hydroxyl group of Y96 with the pyrimidine ring of adagrasib (29) and by the water bridges between Y96 and a carboxyl group in sotorasib (30).
The phase I trial of sotorasib CodeBreak100 included pretreated 125 patients with metastatic KRASG12C mutant solid tumors; 45.7% had non-small cell lung cancer (NSCLC) (31). The objective response was 32.2% in patients with lung cancer, and the disease control rate was 88.1%. No dose-limiting toxicities or treatment-related deaths were observed at the established phase II dose of 960mg (31). These results were recently confirmed by the phase II trial, which specifically included 122 patients with NSCLC. The objective response rate was 37.1%, with a median duration of response of 11 months and an 80.6% disease control rate. Median progression-free survival was 6.8 months, and median overall survival was 12.5 months (32). In the explorative analysis, assessing the association between response to sotorasib and the presence of previously described co-occurring mutations (STK11, KEAP1, and TP53), which define three subgroups of KRAS mutant cancers with different biology and response to treatment (33), sotorasib showed efficacy in all subgroups, with a lower percentage of response in patients with KEAP1 mutated tumors, although this was not statistically significant. Also, similar response rates were observed in PDL-1 positive or negative (score <1%) patients as well as in tumors with high (≥10 mutations per megabase) or low (<10 mutations per megabase) mutational burden. These findings led to the FDA’s accelerated approval in May 2021 of sotorasib in previously treated patients with mutant KRASG12C NSCLC. Efficacy of sotorasib versus docetaxel in previously treated patients KRASG12C mutant NSCLC is currently being tested on a phase III clinical trial (NCT04303780). Moreover, sotorasib as first-line treatment is under investigation on a phase II trial (NCT04933695).
In a similar manner, data from the phase I/II KRYSTAL-I clinical trial, that included 79 previously treated patients with advanced stage KRASG12C mutant NSCLC treated with adagrasib, showed an objective response rate of 45% and a 96% diseases control rate (34). This trial is still ongoing (NCT03785249).
Interestingly, response rates for KRASG12C mutant colorectal cancer cohorts included in both trials were considerably lower, with 7% and 17% for sotorasib and adagrasib, respectively (31, 34). This primary resistance is driven by upstream activation of EGFR, which can be overcome by combining KRASG12C inhibitors with anti EGFR monoclonal antibodies, like sotorasib and cetuximab, respectively (35).
Given the clinical evidence of the efficacy of KRASG12C inhibitors in lung cancer, a considerable proportion of patients do not experience a tumor response or have primary progression, for which treatment intensification may be required. Also, compared to other targeted therapies like EGFR, ALK, and ROS1, patients with KRAS mutant tumors that initially achieve a partial response have a shorter duration of responses, driven by acquired resistance mechanisms (36).
Despite the encouraging clinical results, about half of the patients included in clinical trials with sotorasib and adagrasib do not experience significant tumor shrinkage with specific KRASG12C inhibitors. Moreover, nearly 10% of patients will experience primary disease progression. On the other hand, as with other target therapies, all patients who initially experience an objective response or stable disease will eventually progress with KRASG12C inhibitors, given the emergence of resistance mechanisms in cancer cells. In general, biologic mechanisms of resistance can be categorized into two main groups: on-target resistance, driven by mutations or amplification in KRAS that halters drug binding, and off-target or bypass mechanisms of resistance, when KRAS is inhibited, but another effector commands oncogenic signaling. In addition, other resistance mechanisms imply histologic transformation and other events affecting epigenetic modifications and apoptosis. A summary of biologic resistance mechanisms to KRASG12C inhibitors is shown in Figure 1.
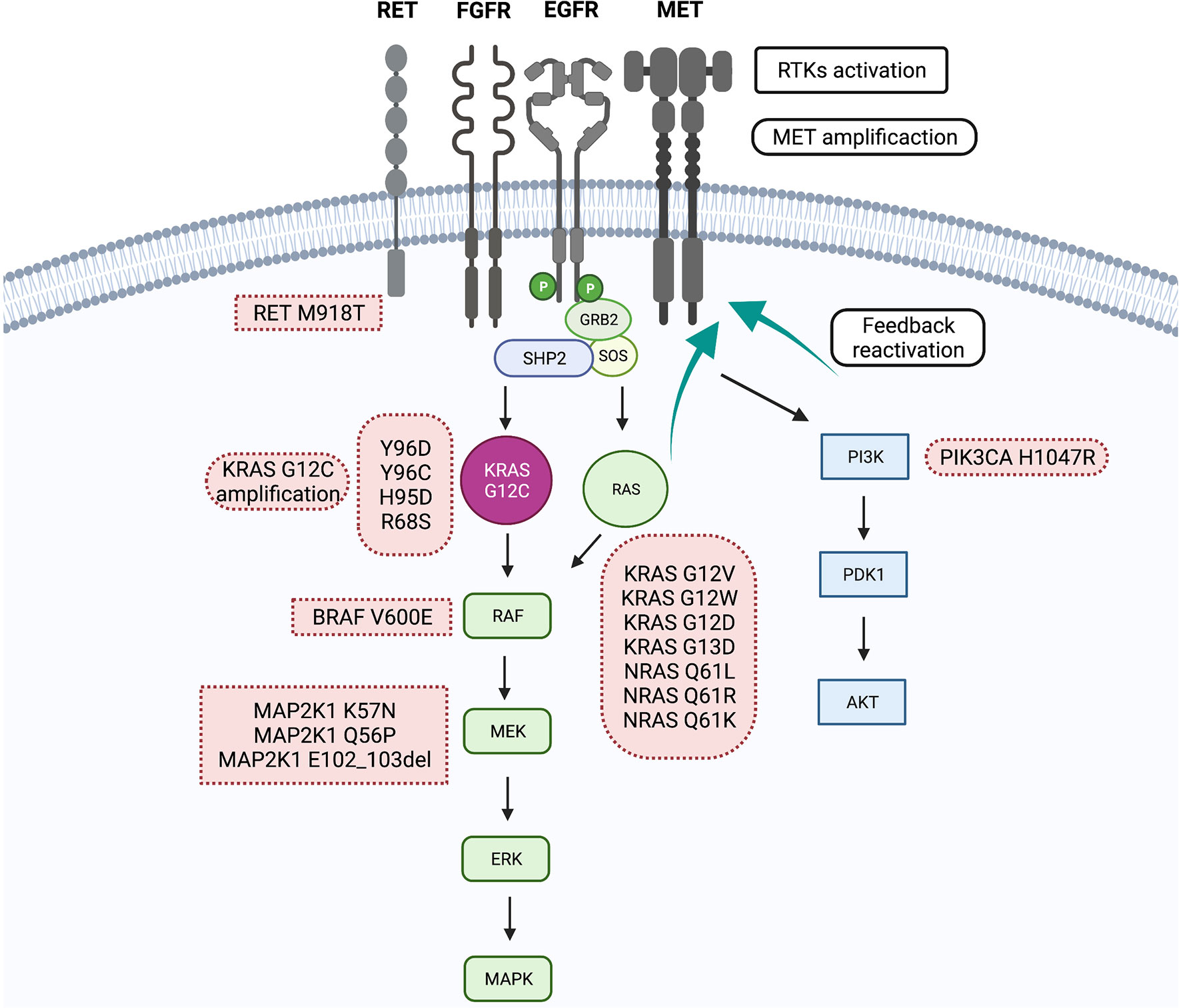
Figure 1 Summery primary and acquired resistance mechanisms to KRASG12C inhibitors.
A lack of response to targeted therapies can be in part due to intrinsic resistance in cancer cells. One potential cause of the lack of efficacy of KRASG12C inhibitors is that not all KRAS mutant cells depend on KRAS activation to maintain their viability. In an attempt to identify a gene expression signature that correlates with the KRAS dependency KRAS mutant cells, Singh et al. found a subgroup of lung cancer-derived cellular lines that maintain their viability despite ablation of the KRAS mutant protein. Moreover, activation of the two main downstream effectors (ERK and AKT) was not suppressed after KRAS knockdown (37). For example, alternative PIK3-AKT pathway activation can be due to oncogenic mutations in AKT1, AKT2, PIK3CA, or PTEN (20, 38), therefore maintaining activated pathway singling to cell proliferation and survival, independently of the status of KRAS. Lessons learned from efforts to inhibit the RAS-RAF-MEK pathway in other tumor types have revealed that a major mechanism of resistance is the adaptive feedback resistance, in which the loss of downstream signaling by the blocked mutant target, leads to reactivation of receptor tyrosine kinase (RTK) mediated signaling through wild type RAS and RAF (39). This adaptive feedback resistance was evaluated in KRASG12C inhibited cell lines using ARS-1610 and AMG-510, finding a rapid and consistent reactivation of signaling by a rebound activation of downstream ERK and RSK in addition to increase levels of active GTP-bound NRAS and HRAS wild type. Also, increased EGFR, HER2, FGFR, and c-MET was observed (39), suggesting that RTKs upstream activation is one of the critical resistance mechanisms KRASG12C inhibitors. In another study addressing the heterogeneity of initial response in KRASG12C mutant lung cancer cells, Xue and colleagues described that tumor cells initially adopt a quiescent state, in which some cells will die. Still, other cells will rapidly evade inhibition by the synthesis of new KRASG12C that is quickly converted to its active state due to upstream stimuli mediated by epithelial growth factor receptor (EGFR) and Aurora Kinase A (AURKA) (40). Hence, primary resistance relies on that as a canonical and fundamental pathway for cell survival, RAS-RAF-ERK has multiple independent mechanisms that can maintain signaling in its active state while being selectively targeted.
As stated before, eventually all patients treated with target therapies will eventually develop progressive disease. In some cases, progression will occur after an initial response as a consequence of acquired resistance mechanisms triggered by the selective pressure of target therapies (41, 42).
The main mechanism of on-target resistance to kinase inhibitors is the acquisition of mutations in key regions within the driver protein that impedes adequate drug binding and target inhibition. In vitro modeling of on-target resistance, mechanisms can be performed using N-ethyl-N-nitrosourea (ENU) mutagenesis screens, which can predict a broad spectrum of resistance that can eventually occur in patients. In a preclinical study using this technique by exposing KRASG12C mutant Ba/F3 cells to sotorasib and adagrasib, 142 acquired resistant clones were identified, of which 124 (87%) harbored secondary KRAS mutations (Y96D, A59T, A59S, R68M, R68M, M721, V8E, G13D, Q61L, Q99L, and H358). The Y96D mutation, as previously stated, occurs at a relevant position for sotorasib and adagrasib binding, leading to resistance to high concentrations (>1000nM) of both specific inhibitors (43). These mutations were also found in patients, as recently reported by Awad and colleagues, by performing a translational biomarker study of a patient treated with adagrasib in the KRYSTAL-1 study who had acquired resistance (44). Acquired resistance was defined as the disease progression after 12 weeks of stable disease or after partial or complete response. This study used next-generation sequencing (NGS) on tissue samples or circulating tumor DNA (ctDNA). A total of 38 patients were included (27 NSCLC), from whom resistance mechanisms were identify in 17 (45%), 10 (26%) corresponding to patients with NSCLC. In four patients with NSCLC, on-target mechanisms were identified, including acquired secondary KRAS mutations associated with the switch II pocket to which KRAS inhibitors bind, Y96C, R86S, and H95D mutations. In addition, acquired activating KRAS mutations other than G12C, including G12D, G12V, and G12W occurring in trans, were found, alone or concomitantly with acquired mutations in the switch II pocket. This complex resistance reveals that some clones may acquire resistance by impeding drug binding in KRASG12C cells, while others acquire mutations that are not targetable by specific G12C inhibitors. In addition, target amplification in the KRASG12C allele has been identified as an independent resistance mechanism, as previously observed with crizotinib in ALK-rearranged lung cancer (45). On target, mechanisms are summarized in Table 1. The same research group performed a preclinical analysis to better characterize the role of the secondary KRAS mutations at the Switch II region in NSCLC patients; double-mutant alleles including Y96C, H95D, and R68S Ba/F3 cells were tested with adagrasib and sotorasib (44).. Tanaka and colleagues also described multiple co-occurring acquired resistance mechanisms in a patient treated with adagrasib using cell-free DNA next-generation sequencing (NGS including two KRAS activating mutations (G12D and G12D) in trans and a Y96D mutation affecting the cryptic Switch II pocket (29). As stated before, the residue Y96 is critical in forming hydrogen bonds with sotorasib and adagrasib; the Y96D mutation disrupts the bonds between adagrasib and sotorasib ARS-1620. Double mutant KRASG12C/Y96D Ba/F cells are resistant to all three KRASG12C inhibitors (29), nevertheless other mutations at the cryptic pocket such as R68S and H95D, that confer resistance to adagrasib remain sensitive to sotorasib (44). Hence, these findings could support the use of sotorasib in certain clinical situations were adagrasib resistance mutations retina sensitivity to sotorasib. However, clinical evidence of activity in this situation is needed. On-target mechanisms include novel acquired mutations in key residues including Y96, H95, and R68 which directly affect the switch II region, impeding KRASG12C inhibitors to bind allosterically: the acquisition of activating mutations different to KRASG12C occurring in the wild type KRAS allele (in trans) and KRASG12C amplification. Strategies to overcome these resistances mechanisms will be further discussed.
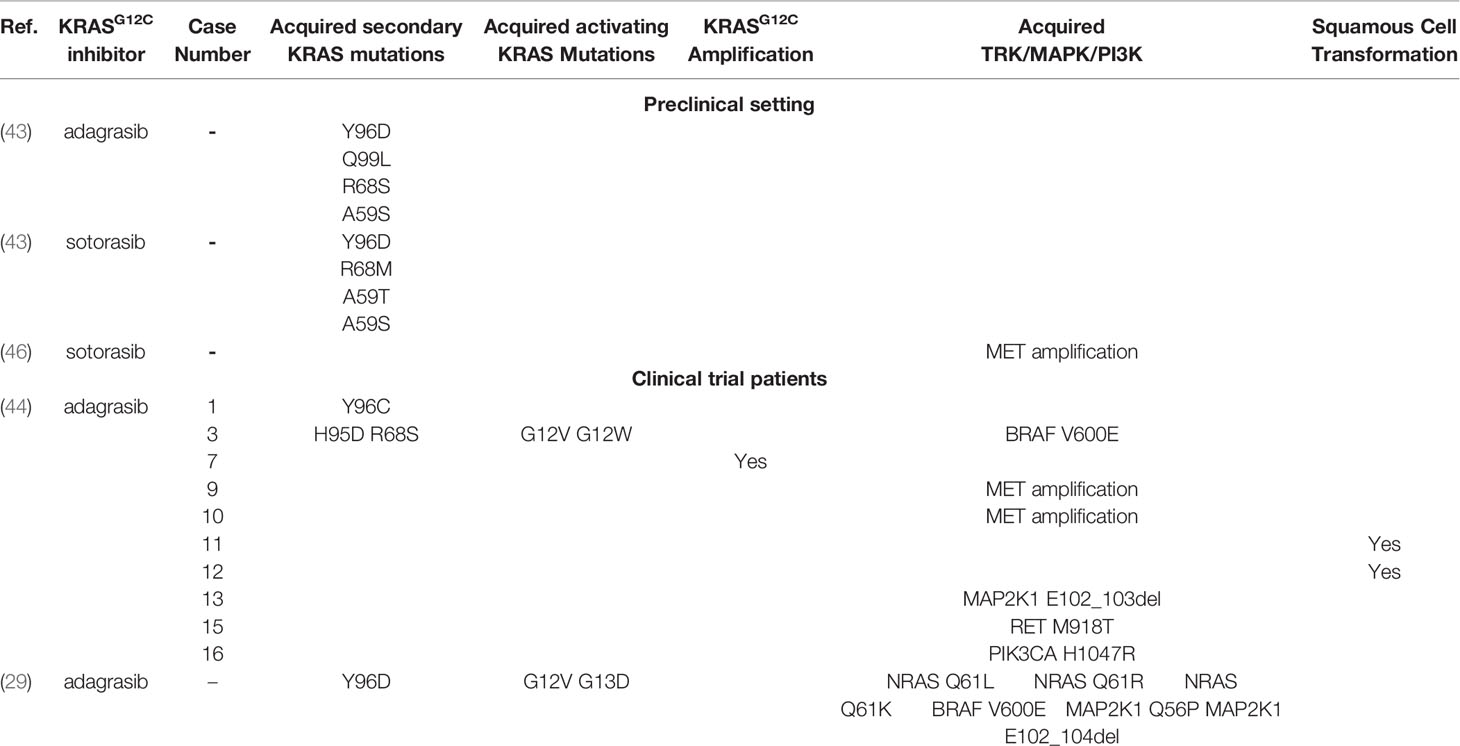
Table 1 Acquired resistance mechanisms to KRASG12C inhibitors in the preclinical setting and clinical trial patients.
Resistance mechanisms due to biological alterations independent of KRASG12C inhibition have also been described and characterized. As previously mentioned, GTP-bound KRAS activates two main canonical pathways, BRAF-MEK-ERK and PI3K-AKT-mTOR (3). As seen in EGRF mutant NSCLC, off-target mechanisms such as MET amplification confer resistance to tyrosine kinase inhibitors (TKIs) by activating the HGF/MET pathway that leads to AKT and ERK activation bypassing RAS (47). This mechanism was successfully targeted in EGFR-mutant NSCLC by combining the EGFR inhibitor osimertinib and a MET inhibitor savolitinib (48). In the preclinical setting, Suzuki et al. exposed mutant KRASG12C cells to sotorsaib finding MET amplifications in resistant cells. To further confirm this as the resistance mechanisms, they demonstrated thar after MET knockdown using siRNA, cells recovered sensitivity to sotorasib (46). Moreover, they found that MET amplified cells had higher levels of GTP-bound (active) RAS, mainly in other isoforms such as NRAS, suggesting that MET amplification could confer resistance by the reactivation of RAS-BRAF-MEK-ERK pathway by direct activation of other RAS isoforms. Similar to the findings in EGFR resistant NSCLC, MET amplification was found in 2 patients (7%) at the time of progression with adagrasib (44). Similar to MET amplification, RET kinase domain activation leads to the oncogenic signaling of BRAF-MEK-ERK, PI3K-AKT-mTOR and JNKs pathways (49), favoring cell survival and cancer development. Rearrangements in RET are found in 1%-2% of all NSCLC (50); moreover, RET point mutations are not frequently found in lung cancer but play a key role in hereditary neuroendocrine syndromes such as MEN2B (49). In the same study by Awad et al., a CCDC6-RET fusion was detected in one patient, and a RET M918T was found in another patient; interestingly M918T is highly associated with MEN2B and is found in about 50% of sporadic medullary thyroid carcinomas (51, 52). Despite these findings, RET mutations in lung cancer are extremely rare, but RET fusions have also been previously described to emerge as a bypass resistance mechanism in EGFR mutant NSCLC. In addition, other fusions have been found at the time of resistance to adagrasib like EML4-ALK and FGFR3−TACC3 which are known to translate in oncogenic fusion proteins. Several fusions involving MAPK pathway effectors have also been described: AKAP9−BRAF, NRF1−BRAF, RAF1−CCDC176, RAF1−TRAK1.
The RAS family genes encode four proteins (KRAS4A, KRAS4B, HRAS, and NRAS), being KRAS the most frequently mutated isoform in NSCLC (20-40%). Mutations in NRAS are rare in this tumor type (3, 53). However, three different activating NRAS mutations (NRAS Q61L, NRAS Q61R, NRAS Q61K) were described in the same patient (29), suggesting the polyclonal activation of other RAS isoforms could play a key role in the acquisition of resistance to KRASG12C inhibitors.
In addition to parallel bypass activation, other genomic alterations in downstream effectors of KRAS, like point mutations and deletions in MAPK pathway effectors, confer resistance to KRASG12C inhibitors MAP2K1 mutations (MAP2K1 K57N, MAP2K1 Q56P and two MAP2K1 E102_103del), BRAF V600E driver mutation. The BRAF V600E mutation confers monomeric activation and signaling and is found in about 2-4% of NSCLC and can be targeted with dabrafenib (BRAF inhibitor) and trametinib (MEK inhibitor) (54). Moreover, activating mutations in the PIK3-AKT-MTOR pathway like PIK3CA H1047R, PIK3CA H1047R were found, confirming bypass activation of oncogenic signaling. Other mutations activating the same pathway were found in colorectal cancer (PIK3R1 S361fs, PTEN N48K, and PTEN G209V).
Finally, as seen with other TKIs, histologic transformation also occurs in KRASG12C mutant lung adenocarcinoma, given the squamous cell carcinoma transformation report as a resistance mechanism to adagrasib (44, 55).
The complex and diverse resistance mechanisms to KRASG12C inhibitors pose a challenge to design clinical strategies to overcome or prevent resistance. A main mechanism of primary resistance is feedback reactivation mediated by multiple RTKs, leading to preclinical research aiming to combine the inhibition of KRASG12C and other RTKs such as EGFR, FGFR, and MET (56). In this setting the combination of sotorasib and MET inhibitors such as crizotinib and capmatinib has shown to overcome resistance to KRASG12C inhibitors in vitro in cancer cells with MET amplification (46). One of the main focuses of current development is targeting transduction molecules situated downstream from RTKs such as SHP2 and SOS1 which after phosphorylation favor de GTP-bound state thus activating the ERK pathway (57). Consequently, targeting SHP2 and SOS1 can synergize with KRASG12C inhibitors given in combination. Preclinical research showed that the combination of a KRASG12C inhibition (ARS-1620) with a SHP2 inhibitor (SHP099) conveyed a more profound suppression of KRAS-GTP and total RAS-GTP in vitro and enhance tumor regression in xenograft models compared to ARS-1620 and SHP099 alone (39). In the same manner the combination of sotorasib and BI-3406, a SOS1 inhibitor, resulted in diminished activity of the ERK pathway in KRASG12C mutant cells (46). Multiple clinical trials are ongoing to test the efficacy of the combination of KRASG12C and SHP2 inhibitors including: the phase 1/2 KRYSTAL 2 trial evaluating the combination of MRTX849 and TNO155 in patients with solid tumor with KRASG12C mutation (NCT04330664) and a phase Ib/II evaluating JDQ443 (KRASG12C inhibitor) and TNO155 (NCT04699188). Other strategies to overcome primary resistance are being evaluated in preclinical models including combination of KRASG12C inhibitors with: PI3K/AKT inhibitors, chemotherapy, CDK4/6 inhibitors targeting alterative pathways, cell cycle and DNA damage (56). RM-108 is a specific KRASG12C inhibitor which binds to a chaperone protein forming a “tri-complex” that prevents the association to the downstream effector proteins (58). This has shown to be active in KRASG12C/Y96D resistant BA/F3 models (29). These strategies represent multiple opportunities to overcome primary and acquired resistance, which need validation in clinical trials.
In addition to the efficacy of single targeted therapies and combinations, cell death induced by KRASG12C inhibitors induce a pro-inflammatory microenvironment that set the bases for the observed synergism between KRASG12C and anti-PD1 immune checkpoint inhibitors in murine models (27, 59); this combination strategy is being evaluated in the multi-arm CodeBreak 101 trial (NCT04185883) with an estimated enrollment of 1273 participants. This trial includes both an experimental arm of sotorasib plus pembrolizumab in dose exploration and dose expansion cohorts and a sotorasib plus atezolizumab cohort. Other arms in this trial include the combination of sotorasib plus: afatinib, palbociclib, chemotherapy, everolimus, among others. In a similar manner the phase 2 KRYSTAL-7 trial is designed to include 250 patients with treatment naïve advanced KRASG12C mutant NSCLC to evaluate the clinical activity of adagrasib in combination with pembrolizumab in (NCT04613596).
Given the recent clinical approval of specific KRASG12C inhibitors and the ongoing development of other strategies to target other KRAS mutations, understanding the biological mechanisms that confer primary and acquired resistance is necessary to improve patients’ clinical outcomes to the response rates and progression free survival.
Resistance to KRASG12C is still not fully understood. However initial reports show that these mechanisms are complex, heterogeneous, and can emerge concomitantly during treatment. In contrast with other targets like EGFR-mutant NSCLC in which a single the T790M mutation is responsible for approximately half of the progressions after first- or second-line specific inhibitors (60), progressions after treatment with KRASG12C inhibitors are caused by both single and combined mechanisms, including: on-target mutations affecting the docking site of this inhibitors; other activating KRAS mutations; activation of the canonical pathways by mutations or activation of upstream and downstream effectors and also activation of non-mutant RAS isoforms. These early reports may be selecting patients with tumors that have more complex biology; hence there is a need to further explore resistance mechanisms in patients that experience a more extended benefit with KRASG12C inhibitors. In the clinical setting, liquid biopsy NGS has proven to be a useful tool in identifying the heterogeneity of resistance mechanisms occurring in a single patient (61). The identification and understanding of acquired resistance mutations to test and develop new therapies to target them.
JB, AC, and GR: drafted and critically reviewed the work. All authors contributed to the article and approved the submitted version.
JB reports educational grants from Amgen, GR reports grants from Amgen, personal fees from Roche, Bayer, BMS, MSD, Pfizer, Takeda, and Amgen. AC disclosed financial research support from MSD, Boehringer Ingelheim, Roche, BMS, and The Foundation for Clinical and Applied Cancer Research - FICMAC. Additionally, he was linked and received honoraria as an advisor, participated in speakers’ bureau, and gave expert testimony to MSD, Boehringer Ingelheim, Roche, BMS, Pfizer, Novartis, Celldex Therapeutics, Foundation Medicine, Eli Lilly, and Foundation for Clinical and Applied Cancer Research - FICMAC.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Figure 1: created with BioRender.com.
1. Tsuchida N, Ryder T, Ohtsubo E. Nucleotide Sequence of the Oncogene Encoding the P21 Transforming Protein of Kirsten Murine Sarcoma Virus. Science (1982) 217(4563):937–9. doi: 10.1126/science.6287573
2. Merz V, Gaule M, Zecchetto C, Cavaliere A, Casalino S, Pesoni C, et al. Targeting KRAS: The Elephant in the Room of Epithelial Cancers. Front Oncol (2021) 11:638360. doi: 10.3389/fonc.2021.638360
3. Campbell SL, Khosravi-Far R, Rossman KL, Clark GJ, Der CJ. Increasing Complexity of Ras Signaling. Oncogene (1998) 17(11 Reviews):1395–413. doi: 10.1038/sj.onc.1202174
4. Tomasini P, Walia P, Labbe C, Jao K, Leighl NB. Targeting the KRAS Pathway in Non-Small Cell Lung Cancer. Oncologist (2016) 21(12):1450–60. doi: 10.1634/theoncologist.2015-0084
5. Hennig A, Markwart R, Esparza-Franco MA, Ladds G, Rubio I. Ras Activation Revisited: Role of GEF and GAP Systems. Biol Chem (2015) 396(8):831–48. doi: 10.1515/hsz-2014-0257
6. Scheffzek K, Ahmadian MR, Kabsch W, Wiesmüller L, Lautwein A, Schmitz F, et al. The Ras-RasGAP Complex: Structural Basis for GTPase Activation and its Loss in Oncogenic Ras Mutants. Science (1997) 277(5324):333–8. doi: 10.1126/science.277.5324.333
7. Biernacka A, Tsongalis PD, Peterson JD, de Abreu FB, Black CC, Gutmann EJ, et al. The Potential Utility of Re-Mining Results of Somatic Mutation Testing: KRAS Status In Lung Adenocarcinoma. Cancer Genet (2016) 209(5):195–8. doi: 10.1016/j.cancergen.2016.03.001
8. Nassar AH, Adib E, Kwiatkowski DJ. Distribution of KRAS (G12C) Somatic Mutations Across Race, Sex, and Cancer Type. New Engl J Med United States; (2021) 384:185–7. doi: 10.1056/NEJMc2030638
9. Liu P, Wang Y, Li X. Targeting the Untargetable KRAS in Cancer Therapy. Acta Pharm Sin B (2019) 9(5):871–9. doi: 10.1016/j.apsb.2019.03.002
10. Cox AD, Fesik SW, Kimmelman AC, Luo J, Der CJ. Drugging the Undruggable RAS: Mission Possible? Nat Rev Drug Discov (2014) 13(11):828–51. doi: 10.1038/nrd4389
11. Ostrem JM, Peters U, Sos ML, Wells JA, Shokat KM. K-Ras(G12C) Inhibitors Allosterically Control GTP Affinity and Effector Interactions. Nature (2013) 503(7477):548–51. doi: 10.1038/nature12796
12. Smith G, Bounds R, Wolf H, Steele RJC, Carey FA, Wolf CR. Activating K-Ras Mutations Outwith ‘Hotspot’ Codons in Sporadic Colorectal Tumours – Implications for Personalised Cancer Medicine. Br J Cancer (2010) 102(4):693–703. doi: 10.1038/sj.bjc.6605534
13. Stolze B, Reinhart S, Bulllinger L, Fröhling S, Scholl C. Comparative Analysis of KRAS Codon 12, 13, 18, 61, and 117 Mutations Using Human MCF10A Isogenic Cell Lines. Sci Rep (2015) 5:8535. doi: 10.1038/srep08535
14. Hunter JC, Manandhar A, Carrasco MA, Gurbani D, Gondi S, Westover KD. Biochemical and Structural Analysis of Common Cancer-Associated KRAS Mutations. Mol Cancer Res (2015) 13(9):1325 LP – 1335. doi: 10.1158/1541-7786.MCR-15-0203
15. Molina JR, Adjei AA. The Ras/Raf/MAPK Pathway. J Thorac Oncol Off Publ Int Assoc Study Lung Cancer (2006) 1(1):7–9. doi: 10.1016/S1556-0864(15)31506-9
16. Seeburg PH, Colby WW, Capon DJ, Goeddel DV, Levinson AD. Biological Properties of Human C-Ha-Ras1 Genes Mutated at Codon 12. Nature (1984) 312(5989):71–5. doi: 10.1038/312071a0
17. Ihle NT, Byers LA, Kim ES, Saintigny P, Lee JJ, Blumenschein GR, et al. Effect of KRAS Oncogene Substitutions on Protein Behavior: Implications for Signaling and Clinical Outcome. J Natl Cancer Inst (2012) 104(3):228–39. doi: 10.1093/jnci/djr523
18. Céspedes MV, Sancho FJ, Guerrero S, Parreño M, Casanova I, Pavón MA, et al. K-Ras Asp12 Mutant Neither Interacts With Raf, Nor Signals Through Erk and is Less Tumorigenic Than K-Ras Val12. Carcinogenesis (2006) 27(11):2190–200. doi: 10.1093/carcin/bgl063
19. Voice JK, Klemke RL, Le A, Jackson JH. Four Human Ras Homologs Differ in Their Abilities to Activate Raf-1, Induce Transformation, and Stimulate Cell Motility. J Biol Chem (1999) 274(24):17164–70. doi: 10.1074/jbc.274.24.17164
20. Khan AQ, Kuttikrishnan S, Siveen KS, Prabhu KS, Shanmugakonar M, Al-Naemi HA, et al. RAS-Mediated Oncogenic Signaling Pathways in Human Malignancies. Semin Cancer Biol (2019) 54:1–13. doi: 10.1016/j.semcancer.2018.03.001
21. Burd CE, Liu W, Huynh MV, Waqas MA, Gillahan JE, Clark KS, et al. Mutation-Specific RAS Oncogenicity Explains NRAS Codon 61 Selection in Melanoma. Cancer Discov (2014) 4(12):1418–29. doi: 10.1158/2159-8290.CD-14-0729
22. Baker NM, Yee Chow H, Chernoff J, Der CJ. Molecular Pathways: Targeting RAC-P21-Activated Serine-Threonine Kinase Signaling in RAS-Driven Cancers. Clin Cancer Res Off J Am Assoc Cancer Res (2014) 20(18):4740–6. doi: 10.1158/1078-0432.CCR-13-1727
23. Yan C, Theodorescu D. RAL GTPases: Biology and Potential as Therapeutic Targets in Cancer. Pharmacol Rev (2018) 70(1):1–11. doi: 10.1124/pr.117.014415
24. Brunelli L, Caiola E, Marabese M, Broggini M, Pastorelli R. Capturing the Metabolomic Diversity of KRAS Mutants in non-Small-Cell Lung Cancer Cells. Oncotarget (2014) 5(13):4722–31. doi: 10.18632/oncotarget.1958
25. Brunelli L, Caiola E, Marabese M, Broggini M, Pastorelli R. Comparative Metabolomics Profiling of Isogenic KRAS Wild Type and Mutant NSCLC Cells In Vitro and In Vivo. Sci Rep (2016) 6(1):28398. doi: 10.1038/srep28398
26. Janes MR, Zhang J, Li L-S, Hansen R, Peters U, Guo X, et al. Targeting KRAS Mutant Cancers With a Covalent G12C-Specific Inhibitor. Cell (2018) 172(3):578–89.e17. doi: 10.1016/j.cell.2018.01.006
27. Canon J, Rex K, Saiki AY, Mohr C, Cooke K, Bagal D, et al. The Clinical KRAS(G12C) Inhibitor AMG 510 Drives Anti-Tumour Immunity. Nature (2019) 575(7781):217–23. doi: 10.1038/s41586-019-1694-1
28. Hallin J, Engstrom LD, Hargis L, Calinisan A, Aranda R, Briere DM, et al. The KRAS(G12C) Inhibitor MRTX849 Provides Insight Toward Therapeutic Susceptibility of KRAS-Mutant Cancers in Mouse Models and Patients. Cancer Discovery (2020) 10(1):54–71. doi: 10.1158/2159-8290.CD-19-1167
29. Tanaka N, Lin JJ, Li C, Ryan MB, Zhang J, Kiedrowski LA, et al. Clinical Acquired Resistance to KRAS(G12C) Inhibition Through a Novel KRAS Switch-II Pocket Mutation and Polyclonal Alterations Converging on RAS-MAPK Reactivation. Cancer Discov (2021) 11(8)1913–22. doi: 10.1158/2159-8290.CD-21-0365
30. Pantsar T. KRAS(G12C)–AMG 510 Interaction Dynamics Revealed by All-Atom Molecular Dynamics Simulations. Sci Rep (2020) 10(1):11992. doi: 10.1038/s41598-020-68950-y
31. Hong DS, Fakih MG, Strickler JH, Desai J, Durm GA, Shapiro GI, et al. KRAS(G12C) Inhibition With Sotorasib in Advanced Solid Tumors. N Engl J Med (2020) 383(13):1207–17. doi: 10.1056/NEJMoa1917239
32. Skoulidis F, Li BT, Dy GK, Price TJ, Falchook GS, Wolf J, et al. Sotorasib for Lung Cancers With KRAS P.G12C Mutation. N Engl J Med (2021) 384(25):2371–81. doi: 10.1056/NEJMoa2103695
33. Skoulidis F, Byers LA, Diao L, Papadimitrakopoulou VA, Tong P, Izzo J, et al. Co-Occurring Genomic Alterations Define Major Subsets of ≪Em<KRAS</em<-Mutant Lung Adenocarcinoma With Distinct Biology, Immune Profiles, and Therapeutic Vulnerabilities. Cancer Discov (2015) 5(8):860 LP–877. doi: 10.1158/2159-8290.CD-14-1236
34. Jänne PA, Rybkin II, Spira AI, Riely GJ, Papadopoulos KP, Sabari JK, et al. KRYSTAL-1: Activity and Safety of Adagrasib (MRTX849) in Advanced/ Metastatic Non–Small-Cell Lung Cancer (NSCLC) Harboring KRAS G12C Mutation. Eur J Cancer (2020) 138:S1–2. doi: 10.1016/S0959-8049(20)31076-5
35. Weiss J, Yaeger RD, Johnson ML, Spira A, Klempner SJ, Barve MA, et al. LBA6 KRYSTAL-1: Adagrasib (MRTX849) as Monotherapy or Combined With Cetuximab (Cetux) in Patients (Pts) With Colorectal Cancer (CRC) Harboring a KRASG12C Mutation. Ann Oncol (2021) 32:S1294. doi: 10.1016/j.annonc.2021.08.2093
36. Hata AN, Shaw AT. Resistance Looms for KRAS(G12C) Inhibitors. Nat Med (2020) 26(2):169–70. doi: 10.1038/s41591-020-0765-z
37. Singh A, Greninger P, Rhodes D, Koopman L, Violette S, Bardeesy N, et al. A Gene Expression Signature Associated With “K-Ras Addiction” Reveals Regulators of EMT and Tumor Cell Survival. Cancer Cell (2009) 15(6):489–500. doi: 10.1016/j.ccr.2009.03.022
38. Franke TF. PI3K/Akt: Getting it Right Matters. Oncogene (2008) 27(50):6473–88. doi: 10.1038/onc.2008.313
39. Ryan MB, Fece de la Cruz F, Phat S, Myers DT, Wong E, Shahzade HA, et al. Vertical Pathway Inhibition Overcomes Adaptive Feedback Resistance to KRAS(G12C) Inhibition. Clin Cancer Res Off J Am Assoc Cancer Res (2020) 26(7):1633–43. doi: 10.1158/1078-0432.CCR-19-3523
40. Xue JY, Zhao Y, Aronowitz J, Mai TT, Vides A, Qeriqi B, et al. Rapid non-Uniform Adaptation to Conformation-Specific KRAS(G12C) Inhibition. Nature (2020) 577(7790):421–5. doi: 10.1038/s41586-019-1884-x
41. Floros KV, Hata AN, Faber AC. Investigating New Mechanisms of Acquired Resistance to Targeted Therapies: If You Hit Them Harder, Do They Get Up Differently? Cancer Res (2020) 80(1):25 LP – 26. doi: 10.1158/0008-5472.CAN-19-3405
42. Neel DS, Bivona TG. Resistance is Futile: Overcoming Resistance to Targeted Therapies in Lung Adenocarcinoma. NPJ Precis Oncol (2017) 1(1):3. doi: 10.1038/s41698-017-0007-0
43. Koga T, Suda K, Fujino T, Ohara S, Hamada A, Nishino M, et al. KRAS Secondary Mutations That Confer Acquired Resistance to KRAS G12C Inhibitors, Sotorasib and Adagrasib, and Overcoming Strategies: Insights From In Vitro Experiments. J Thorac Oncol Off Publ Int Assoc Study Lung Cancer (2021) 16(8):1321–32. doi: 10.1016/j.jtho.2021.04.015
44. Awad MM, Liu S, Rybkin II, Arbour KC, Dilly J, Zhu VW, et al. Acquired Resistance to KRASG12C Inhibition in Cancer. N Engl J Med (2021) 384(25):2382–93. doi: 10.1056/NEJMoa2105281
45. Katayama R, Shaw AT, Khan TM, Mino-Kenudson M, Solomon BJ, Halmos B, et al. Mechanisms of Acquired Crizotinib Resistance in ALK-Rearranged Lung Cancers. Sci Transl Med (2012) 4(120):120ra17. doi: 10.1126/scitranslmed.3003316
46. Suzuki S, Yonesaka K, Teramura T, Takehara T, Kato R, Sakai H, et al. KRAS Inhibitor Resistance in MET-Amplified KRAS (G12C) Non-Small Cell Lung Cancer Induced By RAS- and Non-RAS-Mediated Cell Signaling Mechanisms. Clin Cancer Res Off J Am Assoc Cancer Res (2021) 27(20):5697–707. doi: 10.1158/1078-0432.CCR-21-0856
47. Wang Q, Yang S, Wang K, Sun S-Y. MET Inhibitors for Targeted Therapy of EGFR TKI-Resistant Lung Cancer. J Hematol Oncol (2019) 12(1):63. doi: 10.1186/s13045-019-0759-9
48. Sequist LV, Han J-Y, Ahn M-J, Cho BC, Yu H, Kim S-W, et al. Osimertinib Plus Savolitinib in Patients With EGFR Mutation-Positive, MET-Amplified, non-Small-Cell Lung Cancer After Progression on EGFR Tyrosine Kinase Inhibitors: Interim Results From a Multicentre, Open-Label, Phase 1b Study. Lancet Oncol (2020) 21(3):373–86. doi: 10.1016/S1470-2045(19)30785-5
49. Arighi E, Borrello MG, Sariola H. RET Tyrosine Kinase Signaling in Development and Cancer. Cytokine Growth Factor Rev (2005) 16(4):441–67. doi: 10.1016/j.cytogfr.2005.05.010
50. Network CGAR. Comprehensive Molecular Profiling of Lung Adenocarcinoma. Nature (2014) 511(7511):543–50. doi: 10.1038/nature13385
51. Rudin CM, Drilon A, Poirier JT. RET Mutations in Neuroendocrine Tumors: Including Small-Cell Lung Cancer. J Thorac Oncol (2014) 9(9):1240–2. doi: 10.1097/JTO.0000000000000301
52. Dabir S, Babakoohi S, Kluge A, Morrow JJ, Kresak A, Yang M, et al. RET Mutation and Expression in Small-Cell Lung Cancer. J Thorac Oncol Off Publ Int Assoc Study Lung Cancer (2014) 9(9):1316–23. doi: 10.1097/JTO.0000000000000234
53. Adderley H, Blackhall FH, Lindsay CR. KRAS-Mutant non-Small Cell Lung Cancer: Converging Small Molecules and Immune Checkpoint Inhibition. EBioMedicine (2019) 41:711–6. doi: 10.1016/j.ebiom.2019.02.049
54. Planchard D, Smit EF, Groen HJM, Mazieres J, Besse B, Helland Å, et al. Dabrafenib Plus Trametinib in Patients With Previously Untreated BRAF(V600E)-Mutant Metastatic non-Small-Cell Lung Cancer: An Open-Label, Phase 2 Trial. Lancet Oncol (2017) 18(10):1307–16. doi: 10.1016/S1470-2045(17)30679-4
55. Schoenfeld AJ, Chan JM, Kubota D, Sato H, Rizvi H, Daneshbod Y, et al. Tumor Analyses Reveal Squamous Transformation and Off-Target Alterations As Early Resistance Mechanisms to First-Line Osimertinib in ≪Em<EGFR</em<-Mutant Lung Cancer. Clin Cancer Res (2020) 26(11):2654 LP–2663. doi: 10.1158/1078-0432.CCR-19-3563
56. Jiao D, Yang S. Overcoming Resistance to Drugs Targeting KRAS(G12C) Mutation. Innov (New York NY) (2020) 1(2):1–11. doi: 10.1016/j.xinn.2020.100035
57. Chen D, Waters SB, Holt KH, Pessin JE. SOS Phosphorylation and Disassociation of the Grb2-SOS Complex by the ERK and JNK Signaling Pathways. J Biol Chem (1996) 271(11):6328–32. doi: 10.1074/jbc.271.11.6328
58. Nichols R, Schulze C, Bermingham A, Choy T, Cregg J, Kiss G, et al. A06 Tri-Complex Inhibitors of the Oncogenic, GTP-Bound Form of KRASG12C Overcome RTK-Mediated Escape Mechanisms and Drive Tumor Regressions in Preclinical Models of NSCLC. J Thorac Oncol (2020) 15(2):S13–4. doi: 10.1016/j.jtho.2019.12.035
59. Briere DM, Li S, Calinisan A, Sudhakar N, Aranda R, Hargis L, et al. The KRASG12C Inhibitor MRTX849 Reconditions the Tumor Immune Microenvironment and Sensitizes Tumors to Checkpoint Inhibitor Therapy. Mol Cancer Ther (2021) 20(6):975–85. doi: 10.1158/1535-7163.MCT-20-0462
60. Mok TS, Wu Y-L, Ahn M-J, Garassino MC, Kim HR, Ramalingam SS, et al. Osimertinib or Platinum-Pemetrexed in EGFR T790M-Positive Lung Cancer. N Engl J Med (2017) 376(7):629–40. doi: 10.1056/NEJMoa1612674
Keywords: KRASG12C, resistance mechanisms, NSCLC, target therapy, Y96D
Citation: Blaquier JB, Cardona AF and Recondo G (2021) Resistance to KRASG12C Inhibitors in Non-Small Cell Lung Cancer. Front. Oncol. 11:787585. doi: 10.3389/fonc.2021.787585
Received: 30 September 2021; Accepted: 09 December 2021;
Published: 24 December 2021.
Edited by:
Matthias Scheffler, University Hospital of Cologne, GermanyReviewed by:
Haruhiko Sugimura, Hamamatsu University School of Medicine, JapanCopyright © 2021 Blaquier, Cardona and Recondo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gonzalo Recondo, Z3JlY29uZG9oQGNlbWljLmVkdS5hcg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.