
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Oncol. , 27 May 2022
Sec. Gastrointestinal Cancers: Colorectal Cancer
Volume 11 - 2021 | https://doi.org/10.3389/fonc.2021.783564
This article is part of the Research Topic Prognostic and Predictive Markers in Colorectal Cancer View all 22 articles
 Dongzheng Li1Shiye Jiang1Xin Zhou1Chengshuai Si1Peng Shao1Qian Jiang1Liuqing Zhu2Lu Shen2Qi Meng2Jiani C. Yin2
Dongzheng Li1Shiye Jiang1Xin Zhou1Chengshuai Si1Peng Shao1Qian Jiang1Liuqing Zhu2Lu Shen2Qi Meng2Jiani C. Yin2 Yang Shao2,3
Yang Shao2,3 Yueming Sun4*Liu Yang1*
Yueming Sun4*Liu Yang1*Colorectal cancer (CRC) is one of the leading causes of cancer-related deaths globally. Metastasis is associated with a poor prognosis, yet the underlying molecular mechanism(s) remained largely unknown. In this study, a total of 85 CRC patients were included and the primary tumor lesions were evaluated by next-generation sequencing using a targeted panel for genetic aberrations. Patients were sub-divided according to their metastasis pattern into the non-organ metastases (Non-OM) and organ metastases (OM) groups. By comparing the genetic differences between the two groups, we found that mutations in FBXW7 and alterations in its downstream NOTCH signaling pathway were more common in the Non-OM group. Moreover, correlation analysis suggested that FBXW7 mutations were independent of other somatic alterations. The negative associations of alterations in FBXW7 and its downstream NOTCH signaling pathway with CRC organ metastasis were validated in a cohort of 230 patients in the TCGA CRC dataset. Thus, we speculated that the genomic alterations of FBXW7/NOTCH axis might be an independent negative indicator of CRC organ metastases.
Colorectal cancer (CRC) is the second most common cause for cancer-related deaths and the fourth most commonly occurring malignant tumor worldwide (1). The main reason for poor treatment outcomes in CRC patients lies in the distant spread of cancer cells and organ metastasis could be the major cause for death (2–5). Thus, early interventions preventing the initial spread and occurrence of multiple metastases are crucial to improve CRC patient survival benefits (6, 7). However, the molecular mechanisms concerning the initiation, growth, spread, and distant metastasis of colorectal cancer are particularly complicated and partially unknown (8).
Universally, most of the CRC originated with the loss of function of the reputed gatekeeper gene APC and resulting in the formation of adenomas (9, 10). As the adenomas are growing, KRAS or BRAF malfunction begins to emerge and cell reproductive capacity would be abnormally enhanced (11). Eventually, a considerable amount of genetic alterations would accumulate, namely, the mutations of PIK3CA, SMAD4 or TP53, and would enable the adenomas to transform into malignant tumors (11, 12). Moreover, several key pathways underlie the initiation and development of CRC, namely, the RTK-RAS, PI3K, and WNT pathways (13–15).
Aside from these relatively well-known primary developmental processes of CRC, there is limited evidence demonstrating some genetic features that could affect the distant metastasis. The CRC cells would eventually acquire the ability to migrate and colonize a new site, as they continuously accumulate additional genetic alterations (16, 17). Some studies have elucidated the associations between specific gene mutations and metastasis (18). For instance, it has been shown that peritoneal, distant lymphatic, and central nervous system (CNS) metastasis favor CRC patients harboring the BRAF V600E mutations (19). Abnormal activation of the cytokine receptor c-KIT, which is usually suppressed in the CRC cells, would result in an increase in the metastatic potential of the cancer cells (20, 21). In addition, chromosome 18q deletion would result in the simultaneous hepatic metastasis in CRC (22). These pieces of literature implicated a close relation of genetic alterations with CRC metastasis and could be the theoretical basis for our research.
To get a better understanding of the intrinsic molecular features relating to the organ metastasis in CRC patients, we profiled the genomic alterations of 85 patients with or without organ metastasis by using targeted next-generation sequencing (NGS) of 425 cancer-relevant genes. By comparing the molecular features among different metastatic groups, we discovered several characteristics that may serve as indicators of CRC organ metastasis. In the meantime, a parallel analysis of a TCGA CRC cohort (n = 230) was performed to validate the results we obtained (23).
From January 2020 to June 2021, a total of 85 colorectal cancer patients with or without organ metastasis who underwent surgical resections in the Jiangsu Cancer Hospital were included in this study. Surgical samples of the primary lesion were collected for genomic sequencing tests. Matched peripheral blood samples were also profiled as germline controls. The clinicopathologic features of each patient were retrospectively reviewed. All patients provided informed consents for taking part in this study.
The tissue and blood samples were tested in a CLIA- and CAP-accredited central laboratory (Nanjing Geneseeq Technology Inc., Nanjing, China). In brief, genomic DNA (gDNA) was extracted from formalin-fixed paraffin-embedded tissue samples by using QIAamp DNA FFPE Tissue Kit (Qiagen) according to the manufacturer’s protocol. Peripheral blood was centrifuged at 1,900g for 10 min and gDNA from the white blood cells was also extracted as negative germline control. DNA quality was examined using the NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific Waltham, MA, USA), and DNA quantification was performed on the Qubit 3.0 Fluorometer (Thermo Fisher Scientific, Waltham, MA, USA) with the dsDNA HS Assay Kit (Life Technologies, Carlsbad, CA, USA). The purified DNA was fragmented, and DNA libraries were constructed with the KAPA Hyper Prep kit (KAPA Biosystems) according to the manufacturer’s instruction. A panel targeting 425 cancer related genes (GeneseeqPrime™) using customized xGen lockdown probes (Integrated DNA Technologies) was used for hybridization capture. The hybridization reaction was performed with Dynabeads M-279 (Life Technologies) and xGen lockdown hybridization and wash kit (Integrated DNA Technologies). The target-enriched libraries were sequenced on the Illumina Hiseq4000 NGS platform (Illumina, San Diego, CA, USA) using the 150 bp paired-end reading according to the manufacturer’s protocol. The average coverage depth for the tumor tissue and the white blood cells were 1,000× and 60× respectively.
The sequence data analysis was carried out as previously described (24).In brief, the adapter reads, low quality reads and N bases were trimmed from the raw FASTQ files using Trimmomatic (25). Paired-end reads were aligned to the reference human genome (hg19). PCR deduplication was performed with Picard (http://broadinstitute.github.io/picard/). The base quality score recalibration and indel realignment were performed using the Genome Analysis Tool Kit (25). Somatic single nucleotide variants (SNVs) and small insertion/deletions (indels) were called by VarScan2 (26). Sequencing results from the tissue samples were compared with the matched white blood cell to identify the somatic mutations. SNVs and INDELs were further filtered using the following criteria: i) minimum ≥5 variant supporting reads and variant allele fraction (VAF) of ≥1%, ii) filtered out if present in >1% population frequency in the 1000 Genomes Project or ExAC database, iii) filtered out through an internal database of recurrent sequencing errors (≥3 variant reads and ≤20% VAF in at least 30 out of 2,000 normal control samples) on the same sequencing platform. Copy number variation (CNV) was screened using CNV kit. A fold change of more than 1.50 was considered to be a copy number gain and less than 0.65 was defined as copy number loss. Tumor mutational burden (TMB) was defined as the total number of base substitutions and indels in the coding region of targeted genes, namely, synonymous variants to reduce sampling noise and excluding known driver mutations as they are over-represented in the Panel, as previously described (27, 28).
MSI analysis was performed as previously described (29). The stability of a total of 52 microsatellite sites covered by the 425-cancer gene panel with a minimum of 15 bp repeats, including the classic MSI sites BAT-25, BAT-26, NR-21, NR-24, and MONO-27, was estimated and compiled into an overall MSI score. A site is considered qualified if it is covered by at least 101× depth of coverage. A sample is identified as MSI if more than 40% of the qualified sites with at least 100× coverage displayed instability.
Comparisons of proportion between groups were carried out using the Fisher’s exact test. Correlation analyses among gene alterations in different group were calculated by Spearman’s rank test. A two-sided P-value of <0.05 was considered statistically significant. All statistical analyses were performed using R (v.3.4.1).
Baseline clinicopathological features of the 85 CRC patients are summarized in Table 1. The median age of the study cohort was 55 years (range 29-78) and 50% were males. All patients were ECOG PS 0-1. Of these, 34 (40%) of patients had their primary lesions localized to the left colon, 23 (27.1%) right colon, and 12 (22.2%) rectum. The majority (77/85, 90.4%) of the cohort had adenocarcinoma, followed by the mixed histological type of six (7.1%) patients. Based on the status of metastasis, patients were subdivided into non-organ metastasis (Non-OM, n = 59) and organ-metastasis (OM, n = 26) groups. Notably, two (2.4%) cases were classified as signet-ring cell carcinoma. Both were stage IV CRC and belonged to the OM group, with metastasis to the very uncommon organ peritoneum plus bladder and peritoneum plus bone, respectively. Within the OM group, 20 (76.9%) were with liver metastasis, followed by 17 (65.4%) peritoneum metastasis, five (19.2%) lung metastasis and three other organ metastasis. Moreover, there are ten (38.5%), 13 (50%), and three (11.5%) cases possessing one, two, and three organ metastatic sites, respectively.
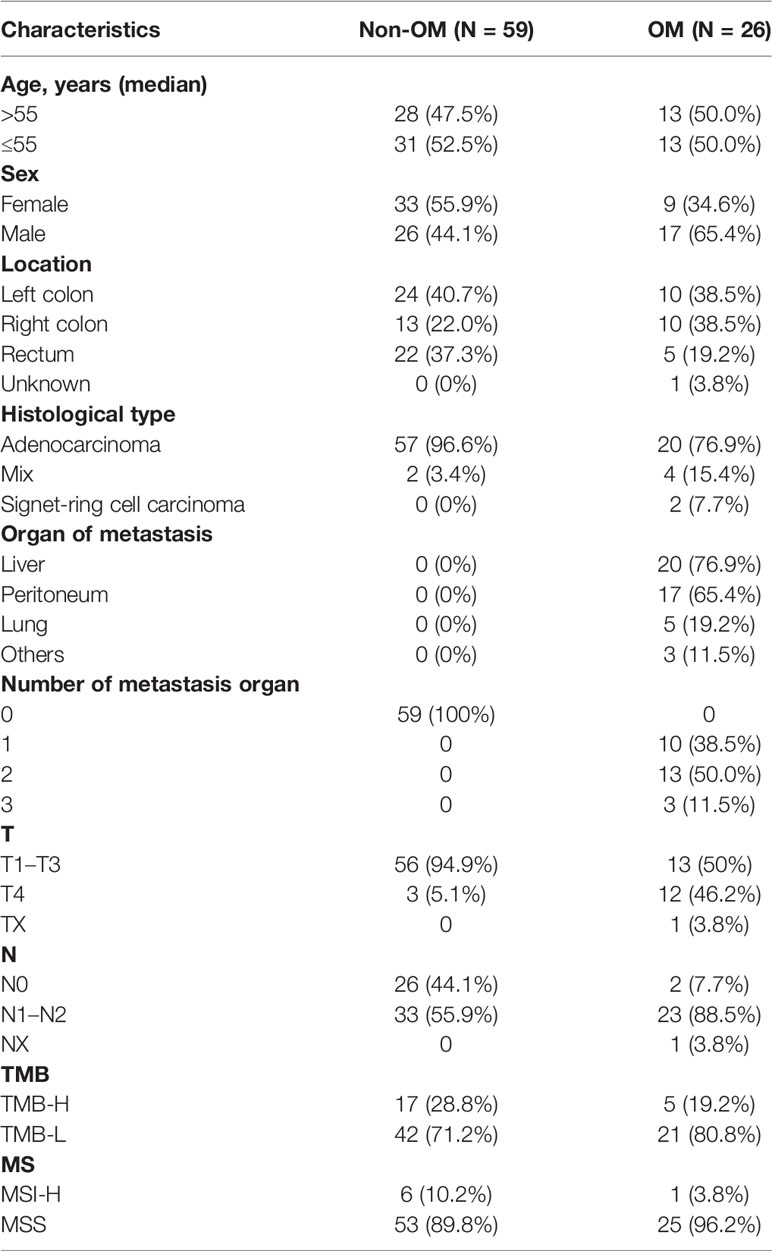
Table 1 Clinical characteristics of our cohort.
A total of 1,981 somatic mutations in 334 genes were detected. The top frequently altered genes in this cohort included TP53 (82.4%), APC (69.4%), KRAS (55.3%), SMAD4 (25.9%), PIK3CA (22.4%), and FBXW7 (21.2%) (Figure 1). The majority of variant types were missense mutations (1,427/1,981, 72.0%), which was followed by nonsense mutations (236/1,981, 11.9%) and frame-shift mutations (205/1,981, 10.3%; Figure 2). The two most common mutated genes in CRC were analyzed. A total of 80 TP53 alterations in 70 patients were detected (Supplementary Figure S1A). The p.R175H alteration was most common and 90% (72/80) of the alterations in TP53 were oncogenic or likely oncogenic according to OncoKB annotation. Mutations in LRP1B were found in 12 (14.1%) patients, and were mutually exclusive with TP53 alterations (P <0.01; Figure 3). Meanwhile, we detected 105 APC alterations belonging to 59 patients (Supplementary Figure S1B) and 88.6% (93/105) of the mutations were oncogenic and likely oncogenic.
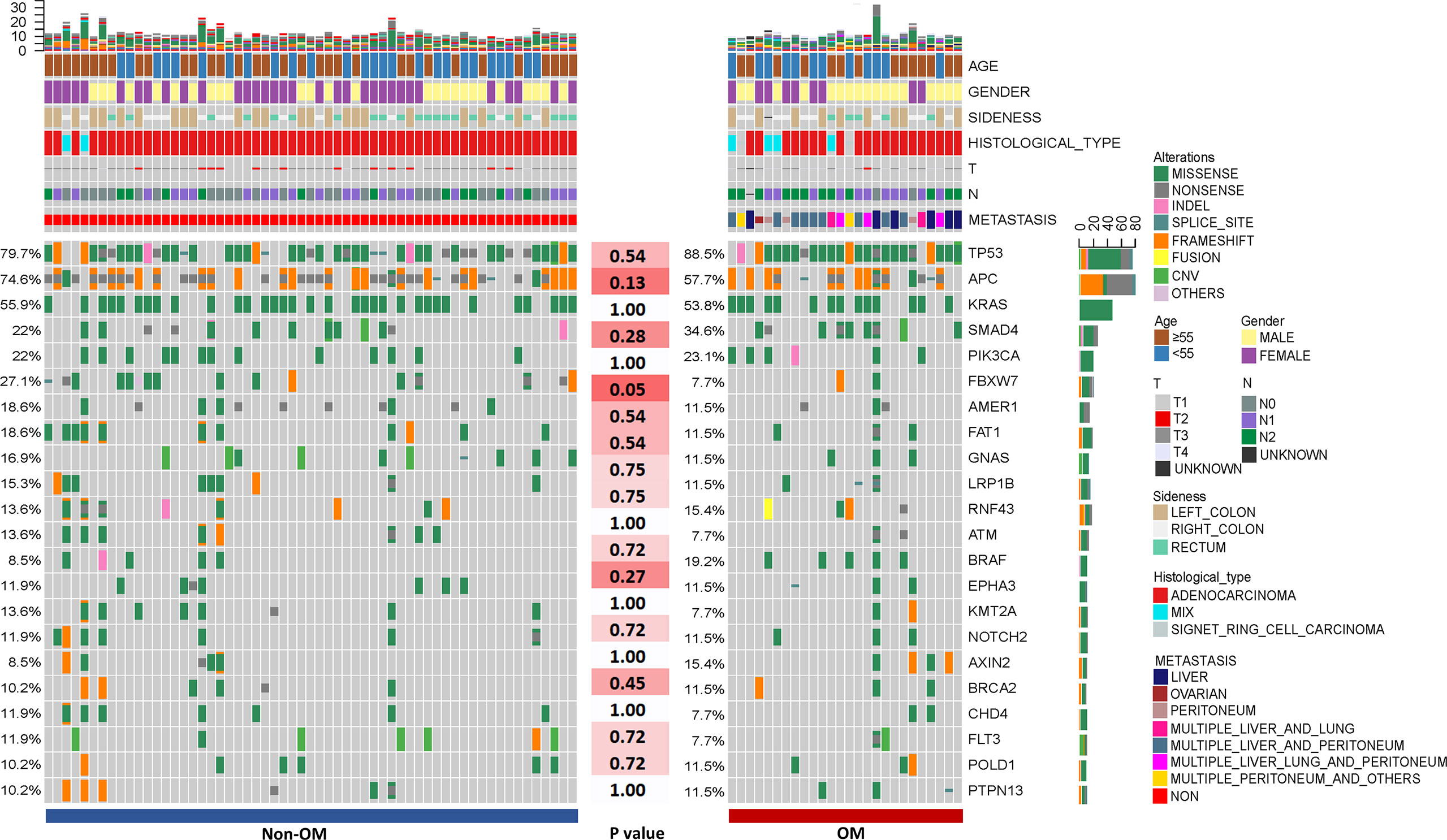
Figure 1 The clinical information and mutational spectrum of the colorectal patients in this study. Comparison of the mutational profile between the Non-OM and OM subgroups. The top 21 genes of the NGS cohort and P-values according to the Fisher’s exact were shown.

Figure 2 The somatic mutational type proportion of this cohort.
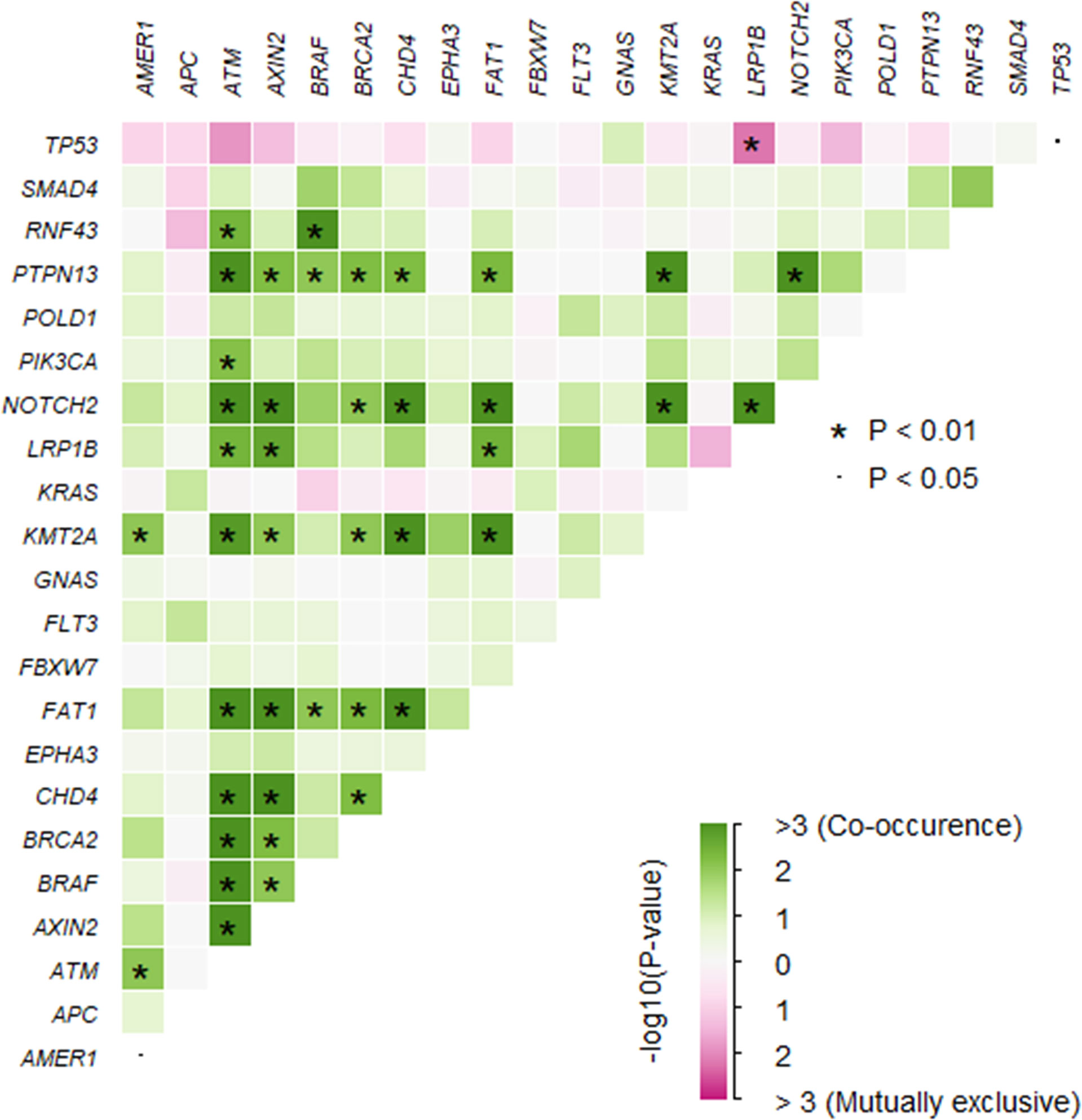
Figure 3 The somatic interactions of the study. Significant exclusive or co-occurrent variants were analyzed with Fisher’s exact test.
Next, we examined the frequencies of oncogenic driver mutations in our cohort. Oncogenic mutations in KRAS, NRAS, and BRAF were detected in 55.3% (47/85), 7.1% (6/85), and 11.8% (10/85) of the cohort. KRAS G12 (82%, 41/50) was the most commonly mutated codon, and included G12D (30%, 15/50), G12V (16%, 8/50), G12R (8%, 4/85), G12C (6%, 3/50), G12A (4%, 2/50), and G12S (4%, 2/50) variants. The second most frequently altered codon was G13, consisting of G13D (14%, 7/50) and G13R (2%, 1/50). Furthermore, we also detected another eight (16%) types of KRAS variants located in the 2nd, 3rd, and 4th exons (Supplementary Figure S1C).
We also detected relatively high frequencies of alterations in EGFR (4.7%, 4/85), ALK (8.2%, 7/85), MET (2.4%, 2/85), ROS1 (5.9%, 5/85), ERBB2 (7.1%, 6/85), and NTRK1/2/3 (10.1%, 10/85), which may potentially serve as targets for targeted therapies. None of these mutations were known oncogenic/targetable mutations, except for an irregular SPTBN1~ALK fusion gene that retained the complete kinase domain of ALK. The presence of ALK fusion suggested that CRC might benefit from ALK-targeted therapies.
In addition, we performed mutational analysis comparing patients with various clinical characteristics. As compared to T1-3, T4 tumors had significantly higher mutational frequency in three genes, BRAF (5/15 vs. 5/69, p = 0.01), IFNE (2/15 vs. 0/69, p = 0.03), and RNF43 (5/69 vs. 7/69, p = 0.03). By comparing the patients with and without lymph metastasis, we found that alteration frequencies of 79 genes were significantly different between two groups, of those six genes had mutational frequencies more than 10% in this cohort (Supplementary Table 1). Specifically, mutations of five genes favored non-lymph metastasis, namely ATM (8/28 vs. 2/56, p <0.01), CHD4 (7/28 vs. 2/56, p <0.01), AMER1 (9/28 vs. 5/56, p = 0.01), KMT2A (7/28 vs. 3/56, p = 0.01), and NOTCH2 (7/28 vs. 3/56, p = 0.01). On the other hand, TP53 mutations were enriched in the lymph metastasis group (19/28 vs. 51/56, p = 0.01). Moreover, a comparison between patients older or younger than median age was performed and showed that mutations in KDR (7/44 vs. 0/41, p = 0.01) were more common in the younger patients while mutations in ARAF (0/44 vs. 5/41, p = 0.02), AXL (0/44 vs. 5/41, p = 0.02) and APC (26/44 vs. 33/41, p = 0.04) were more common in the older patients. These results could potentially help to reveal the pathogenesis of CRC patients with diverse clinical features.
To identify potential genetic risk factors of organ metastasis, we analyzed the genetic features between the Non-OM and OM subgroups. To examine if the results were reliable, a TCGA CRC dataset (23) was used as a validation cohort.
To assess the influences of single gene aberrations on organ metastasis, we compared the genetic mutations (with mutational frequencies >10%) between the two groups. We observed a higher frequency of FBXW7 alterations in Non-OM than that in the OM group (27.1% vs. 7.7%; p = 0.05; Figure 1), which was validated in the TCGA CRC dataset (Non-OM, 19.3%; OM, 0%; p <0.01; Supplementary Figure S2A). To further examine if FBXW7 mutations act independently of other genes, we performed a mutual exclusivity or co-occurrence analysis. Notably, the FBXW7 mutations occurred independently from mutations in other genes (Figure 3). Functional annotations of all the FBXW7 variants with OncoKB revealed that the oncogenic or likely oncogenic FBXW7 mutations were significantly enriched in the Non-OM group than that in the OM group (20.3% vs. 3.8%; p = 0.05; Supplementary Figures S2B, C; Supplementary Table 2). We also evaluated the functional variants in various oncogenic signaling pathways that might be related to CRC organ metastasis. Interestingly, the NOTCH pathway locating downstream of FBXW7 was more frequently altered in the Non-OM than in the OM group in both our cohort (22% vs. 3.8%, p = 0.05) and the TCGA cohort (10.9% vs. 0, p = 0.05). Thus, we speculated that FBXW7 and its downstream NOTCH pathway might play an independent and suppressive role in CRC organ metastasis.
In addition, we studied the distribution of all the altered genes in both our and the TCGA cohorts and found that there are 54 genes (all with mutational frequencies less than 10%) only mutated in the Non-OM groups of both cohorts but not in the OM groups. A pathway analysis implicated that these mutations were significantly enriched in several important cancer-related pathways, including the p38 MAPK cascade, response to cAMP, and leukocyte activation, etc. (Supplementary Figure S3). Thus, these pathways might play vital roles in the regulation/suppression of CRC organ metastases.
Apart from the feature of single genes and pathways, distinct mutational features were identified among different groups. We analyzed the mutational type proportion between the two groups. The results suggested that in both groups, the top common mutational type were missense, frame-shift and nonsense variants, with minor trends of differences between the Non-OM subgroup [68.4% (793/1,160), 13.7% (159/1,160), and 11.7% (136/1,160) respectively] and the OM subgroup [77.2% (634/821), 5.6% (46/821), and 12.2% (100/821) (Supplementary Figure S4A). Similar results were observed in the TCGA cohort (Supplementary Figure S4B). Moreover, the numbers of the somatic mutations of each patient between the groups were also investigated, though no significant difference was observed in either our cohort or the validation cohort (Supplementary Figures S5A, B).
Since there are evidences that microsatellite (MS) instable CRC and tumor mutation burden (TMB) high CRC may possess disparate biological characters (30, 31), we subsequently inspected the associations of TMB and MS status with respect to the organ metastasis status (Table 1). In our cohort, 22 patients had high TMB (TMB ≥10 mut/Mb), of which seven patients were MSI-H (Supplementary Figure S6). Given the moderate sample size, both the TMB-H (n = 22, 25.9%, p = 0.43) and MSI-H (n = 7, 8.2%, p = 0.43) patients showed no statistically significant associations with OM vs. Non-OM compared with the TMB-L and MSS patients, respectively (Supplementary Figures S7A, B). In FBXW7-mutated tumors, there was a trend towards higher TMB compared with those with wild-type FBXW7 (p = 0.12; Supplementary Figure S8). In line with the previous result, within the TMB-L population, FBXW7 mutations more commonly occurred in the Non-OM group than that of OM group (26.2% vs. 4.8%, p = 0.05). Interestingly, within the TMB-H population, it was CTNNB1, but not FBXW7, mutations that significantly differed between the non-OM and OM groups, with the frequency of 23.5% (4/17) in the Non-OM and 80% (4/5) in the OM (p = 0.04) groups. This finding suggested that TMB-H CRC and TMB-L CRC might be driven by distinct organ metastatic mechanisms. Due to the low number of the MSI-H cases, we could not perform the similar analysis.
In the past decades, efforts have been made to discover the molecular mechanisms underlying the organ metastasis in CRC. By comparing the genomic aberrations in different metastatic groups, previous studies have discovered a number of key genes and pathways that function to regulate the spread and colonization of CRC cells to the distant organs. In this work, we investigated 425 cancer-related genes in a cohort of 85 CRC patients and an independent validation cohort of 230 patients in the TCGA CRC dataset. By comparing the genetic features of Non-OM and OM group, we discovered that the mutations in FBXW7, as well as its downstream NOTCH signaling pathway genes, were significantly enriched in the CRC patients without organ metastasis, which was validated in the TCGA cohort. In accordance with our finding, FBXW7 mutations have been reported to exhibit a tendency more commonly appeared in early-stage CRC patients (32, 33).
The frequency of FBXW7 mutations in CRC is around 6 to 10% (32). FBXW7 is a key participant in the ubiquitin-proteosome system and negatively regulates the downstream cancer-related genes (34). Mounting studies have demonstrated that FBXW7 plays critical roles in tumor initiation, cell proliferation, cell differentiation and angiogenesis (35–37). In line with this, it has been reported that some missense mutations in FBXW7 could enhance cell proliferation, migration and invasion in cervical cancer cells (38). Previous reports have suggested that FBXW7 mutations could improve cancer-initiating cell activities through the NOTCH signaling pathways (39, 40). In our study, we found that not only FBXW7 mutations but also alterations in its downstream NOTCH pathway were significantly enriched in the Non-OM group. This phenomenon was also validated in the TCGA CRC cohort. Therefore, the FBXW7/NOTCH regulating axis might play an important role in the organ metastasis of CRC. In summary, we speculate that FBXW7 and its downstream NOTCH pathway could be the indicators for CRC organ metastasis.
It is worth pointing out that all patients in the Non-OM group presented with early stage CRC at the time of diagnosis. As a consequence, due to the retrospective nature of our study, we cannot fully differentiate whether the association of FBXW7 mutations were OM-specific or could be related to tumor stage at disease presentation. In addition, while our findings likely support the positive prognostic value of FBXW7 mutations in CRC patients, future investigations are needed to demonstrate the association between FBXW7 mutation and CRC prognosis.
A discrepancy of the result is that FBXW7 is a cancer suppressor gene and there are proofs that mutation of FBXW7 can increase the degree of malignancy of tumor cells (36, 38, 41). However, in our cohort, the TCGA cohort and in other pieces of literature the alterations in FBXW7 more frequently occurred in early stage CRC patients (32, 33). We think that it might be because that FBXW7 is a central regulator in cancer biology mediating a diverse array of oncogenic processes. As a consequence, alterations in FBXW7 might have distinct consequences under different biological context. For example, it was reported that FBXW7 mutation in a single allele would diminish the activity of FBXW7 more severely than the bi-allelic or the homozygous mutation in the intestine of mice (33). Furthermore, FBXW7 heterozygous mutations have been reported to exhibit a higher tumorigenic rate than homozygous mutations in mice (42). Thus, future studies are required to explain this seemingly paradoxical phenomenon.
It is well known that TMB-H and TMB-L CRC share highly distinct pathogenesis and would exhibit differential responses to various treatments (30, 43, 44). In this study, we also noticed that another gene CTNNB1 but not FBXW7 may be a more useful indicator for CRC metastasis in the TMB-H population. The CTNNB1 variants were significantly enriched in the TMB-H OM group, compared with that of TMB-H Non-OM group. In addition, mutations in CTNNB1 could irregularly activate the WNT pathway without the ligands and subsequently result in the abnormal growth of cells and development of cancer (45, 46). Thus, our data suggested that CTNNB1 mutations could be an indicator of CRC metastasis in the TMB-H population.
In conclusion, by comparing the molecular differences among CRC patients with different metastasis statuses, we discovered that FBXW7 and its downstream NOTCH pathway were more commonly mutated in the Non-OM group than in the OM group. Therefore, FBXW7/NOTCH regulating axis may play important roles in organ metastasis in CRC. Our study contributes to the understanding of the molecular mechanisms of CRC metastastic processes.
The original contributions presented in the study are publicly available. This data can be found here: https://ngdc.cncb.ac.cn/gsa-human/, HRA002412.
The study has been approved by the Ethics committee of Jiangsu Cancer Hospital. All patients have provided the written consent forms.
Study concept and design: DL and YuS. Manuscript writing and revision: DL, LY, QM, and JY. NGS testing and analysis: LZ, LS, QM, JY, YaS. Clinical information collection: DL, SJ, XZ, LY. Pathological evaluation: CS, PS, QJ. All authors contributed to the article and approved the submitted version.
This study was supported by the National Natural Science Foundation of China (81602145 and 82072704, to L.Y.), Jiangsu Primary Research & Development Plan (SBE2021740280), Jiangsu Provincial Natural Science Foundation (BK20171509 to L.Y.), Jiangsu Provincial Medical Youth Talent, The Project of Invigorating Health Care through Science, Technology Education (QNRC2016649 to L.Y.), the China Postdoctoral Science Foundation (2018M632265 to L.Y.), The “333 Talents” Program of Jiangsu Province (BRA2020390 to L.Y.) and The Talents Program of Jiangsu Cancer Hospital (2017-33 to L.Y.).
LZ, LS, QM, JY, and YS are the employees of Nanjing Geneseeq Technology Inc.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank all the patients who participated in this study. We are grateful to Nanjing Geneseeq Technology Inc. for the NGS tests.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2021.783564/full#supplementary-material
Supplementary Figure 1 | The distribution of the detected somatic variants in this study. (A), TP53; (B), APC and (C), KRAS.
Supplementary Figure 2 | (A) The mutational spectrum of the colorectal patients in TCGA cohort. Comparison of the mutational profile between the Non-OM and OM subgroup and the P values according to the Fisher’s exact test was shown. The distribution of the detected FBXW7 variants in the non-OM (B) and OM (C) groups.
Supplementary Figure 3 | The distribution of the mutated genes in both this study and TCGA cohort with or without organ metastasis (A). The GO pathway analysis of the 54 genes that only distributed in the OM groups (B).
Supplementary Figure 4 | The mutational type proportion of Non-OM and OM groups in this study (A) and TCGA cohort (B).
Supplementary Figure 5 | The numbers of the somatic mutations of each patient in the Non-OM and OM groups of this study (A) and TCGA cohort (B).
Supplementary Figure 6 | The interaction of TMB and MS status in this study.
Supplementary Figure 7 | The TMB (A) and MSI (B) distribution in the Non-OM and OM groups of this study.
Supplementary Figure 8 | The comparison of TMB between FBXW7 mutated and WT.
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global Cancer Statistics 2018: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin (2018) 68:394–424. doi: 10.3322/caac.21492
2. Hanahan D, Weinberg RA. Hallmarks of Cancer: The Next Generation. Cell (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
3. Valastyan S, Weinberg RA. Tumor Metastasis: Molecular Insights and Evolving Paradigms. Cell (2011) 147:275–92. doi: 10.1016/j.cell.2011.09.024
4. Croner RS, Fortsch T, Bruckl WM, Rodel F, Rodel C, Papadopoulos T, et al. Molecular Signature for Lymphatic Metastasis in Colorectal Carcinomas. Ann Surg (2008) 247:803–10. doi: 10.1097/SLA.0b013e31816bcd49
5. Pretzsch E, Bosch F, Neumann J, Ganschow P, Bazhin A, Guba M, et al. Mechanisms of Metastasis in Colorectal Cancer and Metastatic Organotropism: Hematogenous Versus Peritoneal Spread. J Oncol (2019) 2019:7407190. doi: 10.1155/2019/7407190
6. Duan L, Yang W, Wang X, Zhou W, Zhang Y, Liu J, et al. Advances in Prognostic Markers for Colorectal Cancer(). Expert Rev Mol Diagnostics (2019) 19:313–24. doi: 10.1080/14737159.2019.1592679
7. Walther A, Johnstone E, Swanton C, Midgley R, Tomlinson I, Kerr D. Genetic Prognostic and Predictive Markers in Colorectal Cancer. Nat Rev Cancer (2009) 9:489–99. doi: 10.1038/nrc2645
8. Pappas-Gogos G, Baltagiannis EG, Kyrochristos ID, Ziogas DE, Goussia A, Mitsis M, et al. Predictive and Patient-Monitoring Biomarkers: Precision in the Management of Colorectal Cancer. Biomarkers Med (2020) 14:335–9. doi: 10.2217/bmm-2020-0025
9. Kinzler KW, Vogelstein B. Cancer-Susceptibility Genes. Gatekeepers and Caretakers. Nature (1997) 386:761, 763. doi: 10.1038/386761a0
10. Aghabozorgi AS, Bahreyni A, Soleimani A, Bahrami A, Khazaei M, Ferns GA, et al. Role of Adenomatous Polyposis Coli (APC) Gene Mutations in the Pathogenesis of Colorectal Cancer; Current Status and Perspectives. Biochimie (2019) 157:64–71. doi: 10.1016/j.biochi.2018.11.003
11. Li ZN, Zhao L, Yu LF, Wei MJ. BRAF and KRAS Mutations in Metastatic Colorectal Cancer: Future Perspectives for Personalized Therapy. Gastroenterol Rep (2020) 8:192–205. doi: 10.1093/gastro/goaa022
12. Fearon ER, Vogelstein B. A Genetic Model for Colorectal Tumorigenesis. Cell (1990) 61:759–67. doi: 10.1016/0092-8674(90)90186-i
14. Bailey MH, Tokheim C, Porta-Pardo E, Sengupta S, Bertrand D, Weerasinghe A, et al. Comprehensive Characterization of Cancer Driver Genes and Mutations. Cell (2018) 173:371–385 e318. doi: 10.1016/j.cell.2018.02.060
15. Nie X, Liu H, Liu L, Wang YD, Chen WD. Emerging Roles of Wnt Ligands in Human Colorectal Cancer. Front Oncol (2020) 10:1341. doi: 10.3389/fonc.2020.01341
16. Vermaat JS, Nijman IJ, Koudijs MJ, Gerritse FL, Scherer SJ, Mokry M, et al. Primary Colorectal Cancers and Their Subsequent Hepatic Metastases Are Genetically Different: Implications for Selection of Patients for Targeted Treatment. Clin Cancer Res (2012) 18:688–99. doi: 10.1158/1078-0432.CCR-11-1965
17. Massague J, Obenauf AC. Metastatic Colonization by Circulating Tumour Cells. Nature (2016) 529:298–306. doi: 10.1038/nature17038
18. Tan IB, Malik S, Ramnarayanan K, McPherson JR, Ho DL, Suzuki Y, et al. High-Depth Sequencing of Over 750 Genes Supports Linear Progression of Primary Tumors and Metastases in Most Patients With Liver-Limited Metastatic Colorectal Cancer. Genome Biol (2015) 16:32. doi: 10.1186/s13059-015-0589-1
19. Morris V, Overman MJ, Jiang ZQ, Garrett C, Agarwal S, Eng C, et al. Progression-Free Survival Remains Poor Over Sequential Lines of Systemic Therapy in Patients With BRAF-Mutated Colorectal Cancer. Clin Colorectal Cancer (2014) 13:164–71. doi: 10.1016/j.clcc.2014.06.001
20. Ishaque N, Abba ML, Hauser C, Patil N, Paramasivam N, Huebschmann D, et al. Whole Genome Sequencing Puts Forward Hypotheses on Metastasis Evolution and Therapy in Colorectal Cancer. Nat Commun (2018) 9:4782. doi: 10.1038/s41467-018-07041-z
21. Gavert N, Shvab A, Sheffer M, Ben-Shmuel A, Haase G, Bakos E, et al. C-Kit Is Suppressed in Human Colon Cancer Tissue and Contributes to L1-Mediated Metastasis. Cancer Res (2013) 73:5754–63. doi: 10.1158/0008-5472.CAN-13-0576
22. Tanaka T, Watanabe T, Kitayama J, Kanazawa T, Kazama Y, Tanaka J, et al. Chromosome 18q Deletion as a Novel Molecular Predictor for Colorectal Cancer With Simultaneous Hepatic Metastasis. Diagn Mol Pathol Am J Surg Pathol Part B (2009) 18:219–25. doi: 10.1097/PDM.0b013e3181910f17
23. The Cancer Genome Atlas Network. Comprehensive Molecular Characterization of Human Colon and Rectal Cancer. Nature (2012) 487:330–7. doi: 10.1038/nature11252
24. Zhou Z, Liu Z, Ou Q, Wu X, Wang X, Shao Y, et al. Targeting FGFR in Non-Small Cell Lung Cancer: Implications From the Landscape of Clinically Actionable Aberrations of FGFR Kinases. Cancer Biol Med (2021) 18:490–501. doi: 10.20892/j.issn.2095-3941.2020.0120
25. McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The Genome Analysis Toolkit: A MapReduce Framework for Analyzing Next-Generation DNA Sequencing Data. Genome Res (2010) 20:1297–303. doi: 10.1101/gr.107524.110
26. Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, et al. VarScan 2: Somatic Mutation and Copy Number Alteration Discovery in Cancer by Exome Sequencing. Genome Res (2012) 22:568–76. doi: 10.1101/gr.129684.111
27. Fang W, Ma Y, Yin JC, Hong S, Zhou H, Wang A, et al. Comprehensive Genomic Profiling Identifies Novel Genetic Predictors of Response to Anti-PD-(L)1 Therapies in Non-Small Cell Lung Cancer. Clin Cancer Res (2019) 25:5015–26. doi: 10.1158/1078-0432.CCR-19-0585
28. Chalmers ZR, Connelly CF, Fabrizio D, Gay L, Ali SM, Ennis R, et al. Analysis of 100,000 Human Cancer Genomes Reveals the Landscape of Tumor Mutational Burden. Genome Med (2017) 9:34. doi: 10.1186/s13073-017-0424-2
29. Ying J, Yang L, Yin JC, Xia G, Xing M, Chen X, et al. Additive Effects of Variants of Unknown Significance in Replication Repair-Associated DNA Polymerase Genes on Mutational Burden and Prognosis Across Diverse Cancers. J Immunother Cancer (2021) 9. doi: 10.1136/jitc-2021-002336
30. Schrock AB, Ouyang C, Sandhu J, Sokol E, Jin D, Ross JS, et al. Tumor Mutational Burden Is Predictive of Response to Immune Checkpoint Inhibitors in MSI-High Metastatic Colorectal Cancer. Ann Oncol (2019) 30:1096–103. doi: 10.1093/annonc/mdz134
31. Lizardo DY, Kuang C, Hao S, Yu J, Huang Y, Zhang L. Immunotherapy Efficacy on Mismatch Repair-Deficient Colorectal Cancer: From Bench to Bedside. Biochim Biophys Acta Rev Cancer (2020) 1874:188447. doi: 10.1016/j.bbcan.2020.188447
32. Davis RJ, Welcker M, Clurman BE. Tumor Suppression by the Fbw7 Ubiquitin Ligase: Mechanisms and Opportunities. Cancer Cell (2014) 26:455–64. doi: 10.1016/j.ccell.2014.09.013
33. Davis H, Lewis A, Behrens A, Tomlinson I. Investigation of the Atypical FBXW7 Mutation Spectrum in Human Tumours by Conditional Expression of a Heterozygous Propellor Tip Missense Allele in the Mouse Intestines. Gut (2014) 63:792–9. doi: 10.1136/gutjnl-2013-304719
34. Li Z, Xiao J, Hu K, Wang G, Li M, Zhang J, et al. FBXW7 Acts as an Independent Prognostic Marker and Inhibits Tumor Growth in Human Osteosarcoma. Int J Mol Sci (2015) 16:2294–306. doi: 10.3390/ijms16022294
35. Wang Z, Inuzuka H, Zhong J, Wan L, Fukushima H, Sarkar FH, et al. Tumor Suppressor Functions of FBW7 in Cancer Development and Progression. FEBS Lett (2012) 586:1409–18. doi: 10.1016/j.febslet.2012.03.017
36. Izumi N, Helker C, Ehling M, Behrens A, Herzog W, Adams RH. Fbxw7 Controls Angiogenesis by Regulating Endothelial Notch Activity. PLoS One (2012) 7:e41116. doi: 10.1371/journal.pone.0041116
37. Tu K, Yang W, Li C, Zheng X, Lu Z, Guo C, et al. Fbxw7 Is an Independent Prognostic Marker and Induces Apoptosis and Growth Arrest by Regulating YAP Abundance in Hepatocellular Carcinoma. Mol Cancer (2014) 13:110. doi: 10.1186/1476-4598-13-110
38. Liu F, Zou Y, Wang F, Yang B, Zhang Z, Luo Y, et al. FBXW7 Mutations Promote Cell Proliferation, Migration, and Invasion in Cervical Cancer. Genet Test Mol Biomarkers (2019) 23:409–17. doi: 10.1089/gtmb.2018.0278
39. Weng AP, Ferrando AA, Lee W, Morris JP, Silverman LB, Sanchez-Irizarry C, et al. Activating Mutations of NOTCH1 in Human T Cell Acute Lymphoblastic Leukemia. Science (2004) 306:269–71. doi: 10.1126/science.1102160
40. Hodis E, Watson IR, Kryukov GV, Arold ST, Imielinski M, Theurillat JP, et al. A Landscape of Driver Mutations in Melanoma. Cell (2012) 150:251–63. doi: 10.1016/j.cell.2012.06.024
41. Akhoondi S, Sun D, von der Lehr N, Apostolidou S, Klotz K, Maljukova A, et al. FBXW7/hCDC4 Is a General Tumor Suppressor in Human Cancer. Cancer Res (2007) 67:9006–12. doi: 10.1158/0008-5472.CAN-07-1320
42. Mao JH, Perez-Losada J, Wu D, Delrosario R, Tsunematsu R, Nakayama KI, et al. Fbxw7/Cdc4 Is a P53-Dependent, Haploinsufficient Tumour Suppressor Gene. Nature (2004) 432:775–9. doi: 10.1038/nature03155
43. Boland CR, Goel A. Microsatellite Instability in Colorectal Cancer. Gastroenterology (2010) 138:2073–2087.e2073. doi: 10.1053/j.gastro.2009.12.064
44. Lin A, Zhang J, Luo P. Crosstalk Between the MSI Status and Tumor Microenvironment in Colorectal Cancer. Front Immunol (2020) 11:2039. doi: 10.3389/fimmu.2020.02039
45. Zhang L, Dong X, Yan B, Yu W, Shan L. CircAGFG1 Drives Metastasis and Stemness in Colorectal Cancer by Modulating YY1/CTNNB1. Cell Death Dis (2020) 11:542. doi: 10.1038/s41419-020-2707-6
Keywords: colorectal cancer, organ metastasis, FBXW7, NOTCH, NGS
Citation: Li D, Jiang S, Zhou X, Si C, Shao P, Jiang Q, Zhu L, Shen L, Meng Q, Yin JC, Shao Y, Sun Y and Yang L (2022) FBXW7 and Its Downstream NOTCH Pathway Could be Potential Indicators of Organ-Free Metastasis in Colorectal Cancer. Front. Oncol. 11:783564. doi: 10.3389/fonc.2021.783564
Received: 26 September 2021; Accepted: 07 December 2021;
Published: 27 May 2022.
Edited by:
Cornelis F. M. Sier, Leiden University, NetherlandsReviewed by:
Hisato Kawakami, Kindai University Hospital, JapanCopyright © 2022 Li, Jiang, Zhou, Si, Shao, Jiang, Zhu, Shen, Meng, Yin, Shao, Sun and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Liu Yang, eWFuZ2xpdUBuam11LmVkdS5jbg==; Yueming Sun, c3VueXVlbWluZ0Buam11LmVkdS5jbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.