 Mamatha Bhat1,2,3,4*
Mamatha Bhat1,2,3,4* Elisa Pasini1Chiara Pastrello5,6Marc Angeli1Cristina Baciu1Mark Abovsky5,6Angella Coffee7
Elisa Pasini1Chiara Pastrello5,6Marc Angeli1Cristina Baciu1Mark Abovsky5,6Angella Coffee7 Oyedele Adeyi7Max Kotlyar5,6
Oyedele Adeyi7Max Kotlyar5,6 Igor Jurisica5,6,8,9,10*
Igor Jurisica5,6,8,9,10*- 1Ajmera Transplant Program, University Health Network, Toronto, ON, Canada
- 2Division of Gastroenterology & Hepatology, University of Toronto, Toronto, ON, Canada
- 3Institute of Medical Sciences, University of Toronto, Toronto, ON, Canada
- 4Toronto General Hospital Research Institute, University Health Network, Toronto, ON, Canada
- 5Osteoarthritis Research Program, Division of Orthopedic Surgery, Schroeder Arthritis Institute, University Health Network, Toronto, ON, Canada
- 6Krembil Research Institute, University Health Network, Toronto, ON, Canada
- 7Department of Pathology and University of Minnesota Medical Center, University of Minnesota, Minneapolis, MN, United States
- 8Department of Medical Biophysics, University of Toronto, Toronto, ON, Canada
- 9Department Computer Science, University of Toronto, Toronto, ON, Canada
- 10Institute of Neuroimmunology, Slovak Academy of Sciences, Bratislava, Slovakia
Background: Hepatocellular Carcinoma (HCC) is a sexually dimorphic cancer, with female sex being independently protective against HCC incidence and progression. The aim of our study was to understand the mechanism of estrogen receptor signaling in driving sex differences in hepatocarcinogenesis.
Methods: We integrated 1,268 HCC patient sample profiles from publicly available gene expression data to identify the most differentially expressed genes (DEGs). We mapped DEGs into a physical protein interaction network and performed network topology analysis to identify the most important proteins. Experimental validation was performed in vitro on HCC cell lines, in and in vivo, using HCC mouse model.
Results: We showed that the most central protein, ESR1, is HCC prognostic, as increased ESR1 expression was protective for overall survival, with HR=0.45 (95%CI 0.32-0.64, p=4.4E-06), and was more pronounced in women. Transfection of HCC cell lines with ESR1 and exposure to estradiol affected expression of genes involved in the Wnt/β-catenin signaling pathway. ER-α (protein product of ESR1) agonist treatment in a mouse model of HCC resulted in significantly longer survival and decreased tumor burden (p<0.0001), with inhibition of Wnt/β-Catenin signaling. In vitro experiments confirmed colocalization of β-catenin with ER-α, leading to inhibition of β-catenin-mediated transcription of target genes c-Myc and Cyclin D1.
Conclusion: Combined, the centrality of ESR1 and its inhibition of the Wnt/β-catenin signaling axis provide a biological rationale for protection against HCC incidence and progression in women.
Introduction
Hepatocellular carcinoma (HCC) is a high-fatality cancer that develops in the context of chronic liver disease (1, 2). It is a cancer with sexual dimorphism that arises in a non-reproductive organ (3). In fact, men are up to 8 times more likely to develop HCC and have worse prognosis than women (2). Female sex is independently protective for HCC incidence [Hazard Ratio (HR) 0.75; 95%CI 0.65-0.86] and overall survival (HR 0.83; 95%CI 0.77-0.88) (4). A case-control study of 234 post-menopausal women treated for HCC showed that estrogen replacement therapy reduced incidence (HR 0.53; 95%CI 0.32-0.88) and was associated with better survival (HR 0.55; 95%CI 0.40-0.77) (5). Other retrospective studies suggest oral contraceptive use to be protective against HCC (6–8). These studies suggest that HCC biology is subject to sex hormone modulation (3, 4, 9–16).
ESR1 has been recognized as a tumor suppressor gene, with promoter hypermethylation being predictive of tumor progression (17). Its expression has been shown to inversely correlate with HCC tumor size and disease stage in a genome-wide expression analysis (18, 19), consistent with preclinical data demonstrating that loss of ESR1 accelerates carcinogenesis. Binding of estrogen to Estrogen Receptor-alpha and -beta (ER-α and -β) induces receptor dimerization and transcriptional regulation (20, 21). Hormone receptor dimers bind DNA at their specific response elements (22–25), or interact with chromatin-associated proteins to exert transcriptional activation of proliferation, cell cycle progression and cell survival (26). These data suggest an important role for ESR1 in incidence and progression of HCC, though it is unclear how ESR1 impacts cancer signaling pathways critical to HCC progression.
To further examine the role of estrogen receptor signaling in driving sex differences in hepatocarcinogenesis, we performed a comprehensive integrative analysis of publicly available HCC patient profiles and analyzed the resulting physical protein-protein interaction network. We discovered ESR1 to be a central gene in HCC pathogenesis, independently protective for survival in HCC in both men and women. Activation of ESR1 resulted in marked inhibition of Wnt/β-catenin signaling in both male and female mice with HCC. We finally interrogated the mechanistic basis of this effect in vitro, by demonstrating that colocalization of ER-α (protein product encoded by ESR1) with the transcription factor β-catenin along the Wnt pathway results in decreased transcription of target genes cyclin D1 and c-myc. The effect of ER-α in suppressing HCC tumor growth through inhibition of Wnt signaling, the most commonly dysregulated in HCC, provides a new contributory mechanism for decreased HCC incidence and progression in women.
Materials and Methods
Data Collection, Analysis, and Database Compiling
All available high-throughput gene expression microarray datasets related to HCC samples were downloaded from published datasets. All entries on PubMed since 2002, representing the advent of high-throughput profiling, until February 2017 were considered for inclusion. A second search was performed using Gene Expression Omnibus (GEO), a public functional genomics data repository containing high-throughput array data (https://www.ncbi.nlm.nih.gov/geo) covering all HCC high-throughput gene expression profiling datasets comparing HCC to adjacent non-tumoral tissue. These datasets publicly available on GEO and published in papers were analyzed using GEO2R (https://www.ncbi.nlm.nih.gov/geo/info/geo2r.html), a web tool available on the portal, to identify genes differentially expressed between samples of HCC versus the non-tumoral portion of the liver. GEO2R compares original submitter-supplied processed data tables using the GEOquery and limma R packages from the Bioconductor project (27–29). Following instructions available online at https://www.ncbi.nlm.nih.gov/geo/info/geo2r.html, all dysregulated genes were retrieved, and only those with an adjusted p-value p<0.05 and an expression fold-change value below ≤0.5 or above ≥1.5 were collected for further analysis. We also included genes from 17 papers presenting and validating gene signatures (Supplementary Figure 1 and Supplementary Table 1).
The study workflow is illustrated and reasons for study exclusion are detailed in Supplementary Figure 1. The included papers (n=36) compared gene expression profiling in human HCC tissue versus the non-tumoral liver tissue in the same patient. These were obtained from 19 papers with datasets and 17 gene signature papers. This comprised data across 1,268 HCC patient samples and 1,402 controls (Supplementary Figure 1). Gene expression profiling had been performed using Affymetrix and Illumina microarray platforms, as detailed in Supplementary Table 1. The list of dysregulated genes for 19 datasets was obtained using GEO2R (p-value<0.05), and an integrative network analysis was performed on this data as described below. The genes from the 17 signatures and their modulations listed in the papers were collected and curated to serve as independent validation of the HCC relevance for the genes identified in the network analysis.
Available patient data, including sex, etiology of liver disease (Hepatitis C, Hepatitis B, alcohol, fatty liver disease) on the basis of which the HCC tumors developed, presence of cirrhosis, the Model for End-stage Liver Disease score (MELD score, an assessment of the severity of liver dysfunction), tumor histology, stage of cancer, alpha-fetoprotein (AFP) level, overall and recurrence-free survival following treatment were also documented.
Network Construction
A Protein interaction network was constructed using NAViGaTOR 3.0.13 (http://ophid.utoronto.ca/navigator), a visualization application, wherein networks are represented as annotated graphs and defined by nodes (for example, genes or proteins) and edges (the relationships between the proteins, such as physical protein interactions)12. In our study, nodes are proteins, annotated with Gene Ontology Molecular Function (node color), and edges represent direct physical interactions from IID database (http://ophid.utoronto.ca/iid) (30).
Calculation of Network Centrality
For all differentially expressed genes reported with a consistent modulation in at least 10 out of 19 publications, we retrieved the known interactors using the Integrated Interactions Database (http://ophid.utoronto.ca/iid) (30) version 2017-04, selecting only human experimentally-detected, physical protein-protein interactions, whose encoding genes are expressed in liver tissue. Interactors reported several times were highlighted.
Betweenness centrality was calculated in a protein-protein interaction network comprising the most frequently dysregulated genes (proteins) and their experimentally detected interaction partners annotated with human liver using the betweenness function in the igraph library version 1.0.1, in R version 3.3.1 (31). This network was visualized and analyzed in NAViGaTOR (32) ver. 3.0.13.
HCC Mouse Model
A genetic mouse model of HCC was generated through hydrodynamic tail vein injection of proto-oncogene-coding plasmids (33). Twenty µg of pT3-EF5a-G12D-mutant-K-Ras and pT3-EF5a-S45Ymutant-β-catenin-Myc-Tag along with Sleeping Beauty transposase (a kind gift from Dr. X. Chen at University of California, San Francisco) in a ratio of 25:1 was diluted in 2 ml of 0.9% NaCl, filtered through a 0.22 µm filter, and injected into the lateral tail vein of 8-10-week-old FVB/N mice [Charles River (Wilmington, MA)] in 5-7 seconds (33, 34). The mice were housed in the animal facility at the University of Toronto. The experimental protocol was approved by the Institutional Animal Care and Use Committee at the University of Toronto. Mice were fed a standard diet and monitored according to the animal committee’s regulations Prior to establishment of HCC tumors, animals were started at 2 weeks post-injection on a 5-week treatment as follows: 1) ER-a agonist Propyl pyrazole triol (PTT) subcutaneously at 1mg/kg (Sigma-Aldrich H6036) N=6; 2) ER-a antagonist methyl-piperidino-pyrazole (MPP) given subcutaneously at 200ug/kg (Sigma-Aldrich M7068) N=6; and 3) Control N=4. All animals were carefully monitored for signs of morbidity or discomfort. Animals were euthanized at 10 weeks after injection or at the earliest signs of morbidity. Body weights and liver weights were recorded. Gross images were obtained at the time of sacrifice to document macroscopic tumors. The liver was collected for histology and RNA extraction purposes. Microscopic tumor burden was assessed on H&E-stained sections. All the samples were cut across and the surface of each section occupied by tumor was estimated at low magnification (2.5X) by the pathologist who was blinded to the groups. Kaplan-Meier survival analysis was performed with GraphPad Prism ver. 8 (GraphPad Software, La Jolla, California).
Mouse Gene Expression Array
RNA was obtained from N=2 animals per group, (agonist and control both male and female) from each group using RNeasy mini kit from Qiagen.
Integrity of DNA and RNA was assessed by Agilent 2100 Bioanalyzer (Agilent, Santa Clara, CA, USA). 500 nanograms of RNA were used for analysis with Affymetrix Mouse Gene 2.0 ST platform (Thermo Fisher, Waltham, MA). Raw array data were processed and analyzed using the Affy package in R included in the Bioconductor package (version 3.6).
Transcript profiling: Gene expression data is available at Gene Expression Omnibus (GEO), (https://www.ncbi.nlm.nih.gov/geo) with accession GSE167175.
Validation in Patient Samples
To determine whether the central gene ESR1 had prognostic value, we used KMplotter on liver cancer RNA-seq data (n=364 patients), a web-based tool that enables survival analysis across multiple cancers and datasets (35). KMplotter compares two patient cohorts by plotting Kaplan-Meier curves and calculating a hazard ratio and log-rank P value. Patient samples were split into two groups according to auto selection of the best cutoff for ESR1 (RNAseq probe ID 2099), in order to assess its prognostic value. We ran multivariate overall survival analysis based on the high versus low expression of ESR1 in tumors. The two groups were compared by a Kaplan-Meier survival plot, and the hazard ratio with 95% confidence intervals and log-rank p-value were calculated.
Statistical Analysis
Data was presented as mean with standard deviation. GraphPad Prism software ver. 8 was used for all statistical calculations (GraphPad Software, La Jolla, California). Two-group comparisons were performed using unpaired two-tailed Student’s t-tests, for more than two groups comparison we used one-way ANOVA followed by Tukey’s post-hoc test. Survival curves analysis was performed using Gehan-Breslow-Wilcoxon test.
Ethics Approval: Animal studies were conducted under the approval of local Animal Care Committees (University of Toronto AUP 20012289, University Health Network AUP 6105), following Canadian Council on Animal Care (CCAC) guidelines.
Additional materials and methods used in this study are explained in the Supplementary Material.
Results
Literature Search Results
We identified 5,406 abstracts retrieved by searching PubMed for high-throughput gene expression data on human HCC samples, published between January 2002 and December 2017. The flowchart outlining the selection process is detailed in Supplementary Figure 1. Details regarding the included studies are provided in Supplementary Table 1 and made available as a database of curated cancer gene expression signatures, annotated with their demographic and clinical characteristics, at the liver page of the Cancer Data Integration Portal (CDIP) (http://ophid.utoronto.ca/CDIPLiver).
Integrative Analysis Reveals ESR1 as a Central Player in HCC Pathogenesis
Integrative network analysis of high-throughput gene expression data revealed genes most frequently altered in HCC, along with the direction of change (Supplementary Table 2). We identified the protein-protein interactions of the top genes (considering deregulation in greater than half the number of publications) using IID (36, 37), and prioritized top-genes with at least 250 interactions (n=27), due to the exponential increase in number of proteins with a degree lower than 250 (Supplementary Figure 2). Betweenness centrality was calculated using a liver-specific network comprising the 27 proteins and their experimentally-detected interaction partners (Figure 1A and Supplementary Table 3), thereby identifying the most important proteins in the HCC network. ESR1 had the highest betweenness centrality, suggesting its importance in the HCC pathology, and indicating a potentially important role in hepatocarcinogenesis.
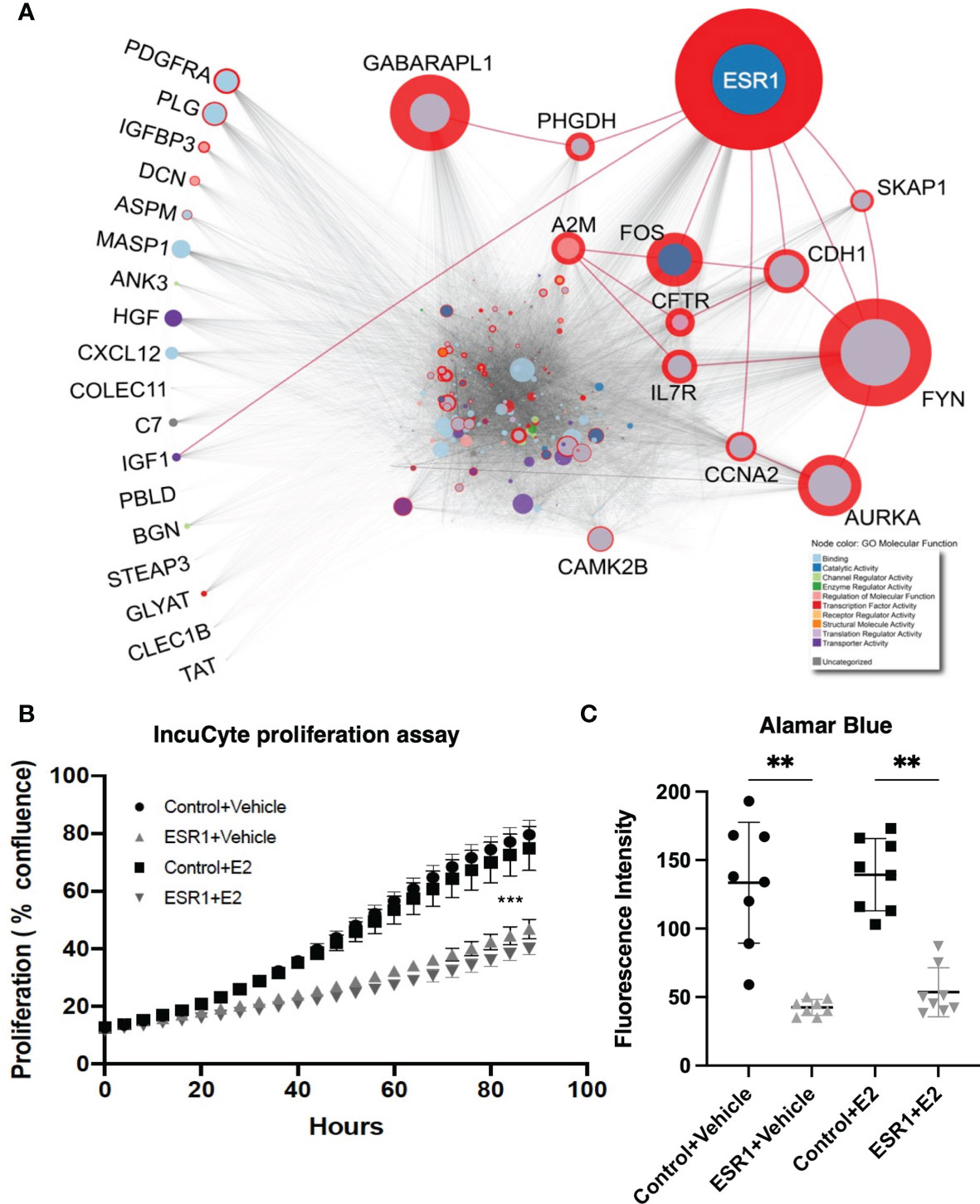
Figure 1 ESR1 is the most central node in the HCC protein-protein interaction network, and its overexpression affects HepG2 cells proliferation and viability reflecting its importance in HCC pathogenesis. (A) HCC protein-protein interaction network. The diameter of the red circle is proportional to the centrality of the protein. The size of the circles is proportional to the degree of the protein. Gene Ontology molecular function of the proteins is highlighted as node color, shown in the legend in the bottom-right corner. Red edges highlight direct interaction of ESR1 with other most central proteins. The proteins listed on the left are those dysregulated most commonly in the 19 datasets and with consistent modulation. Despite being the most commonly dysregulated, none of these are central to the HCC PPI network. (B) Results from the IncuCyte proliferation assay. ESR1-overexpressing cells proliferated significantly less than the control cells. Images were acquired every four hours for up to 90 hours. Mean with SD values from N=3 independent experiments are represented in the graph. ***p < 0.001, One-way ANOVA, Tukey correction for multiple comparisons. (C) Viability assay with Alamar Blue performed in n=3 independent experiments at 72 hours after transfection shows significant decrease of cell viability in cells transfected with ESR1 compared to the control vector Mean with SD values from N=3 independent experiments are represented in the graph. **p < 0.01, One-way ANOVA, Tukey correction for multiple comparisons. SD, standard deviation.
ESR1 Overexpression Affects Proliferation and Viability of HCC Cells
To evaluate the impact of ESR1 overexpression, the HepG2 cell line was transfected with the ESR1 plasmid. Transfected cells were then exposed to estradiol (E2) or vehicle and monitored for 90 hours using the IncuCyte Zoom™ live cell imaging platform. Proliferation was monitored by analyzing the area occupied by cells (percentage confluence) transfected with ESR1 or pCMV control vector and exposed to E2 or vehicle. The confluence of cells growing in normal conditions for up to 90 hours showed that ESR1 overexpression significantly reduces cell proliferation (p<0.0001, one-way ANOVA followed by Tukey’s post-hoc test). The antiproliferative effects of ESR1 were strongly evident in HepG2 cells, and the addition of the ligand further improved the antiproliferative effect (Figure 1B).
The decrease in fluorescence detected by the Alamar blue between cells overexpressing ESR1 and receiving the control vector was significant (p=0.0019, one-way ANOVA followed by Tukey’s post-hoc test), even without the addition of the natural ligand E2 (p=0.0016, one-way ANOVA followed by Tukey’s post-hoc test), confirming that ESR1 expression directly affects cell viability (Figure 1C).
Gene Expression Array
Upregulation of ESR1 resulted in significant up- or downregulated 365 genes (abs(FC)>1.5) as compared to control, of which 175 were protein-coding (Supplementary Table 4). We submitted these genes to pathDIP, in order to identify significantly enriched pathways (http://ophid.utoronto.ca/pathDIP) (38). 133 of the genes were annotated with at least one pathway. 83 pathways were significantly enriched with False Discovery Rate (FDR) < 0.05. The cancer-associated list of pathways is provided in Table 1. ESR1 upregulation significantly affected the Wnt/β-catenin signaling pathway (FDR=2.05E-06). The complete list is provided in Supplementary Table 5.

Table 1 ESR1 overexpression modulated genes associated with cancer pathways.
ER-α Agonist Exposure Increases Survival in HCC Mice
To investigate the effect of ER-α activation or inhibition in HCC, we used a well-described HCC mouse model (33). The control and antagonist mouse livers, in both male and female groups, had multiple HCC nodules infiltrating the liver as reported in Figure 2A. In contrast, the female agonist group showed smaller isolated tumor foci. ER-α activation through agonist treatment significantly improved survival (Figure 2B) by an additional 10 days for male and up to 14 days for female mice respectively (p<0.0001, Gehan-Breslow-Wilcoxon test). Mice exposed to ER-α antagonist showed decreased survival in male mice (48 days for antagonist mice compared to 52 days for the control group). The same treatment in the female mice did not shorten their lifespan (University Health Network Animal Use Protocol 6105). Control mice showed a tumor burden involving 85% of their livers, while the male mice treated with ER-α agonist showed tumor burden decreased to 78%. The decrease in tumor burden was even more pronounced (equal to 67%) in the female group mice treated with ER-a agonist (Figure 2C, p<0.05 multiple t-test).
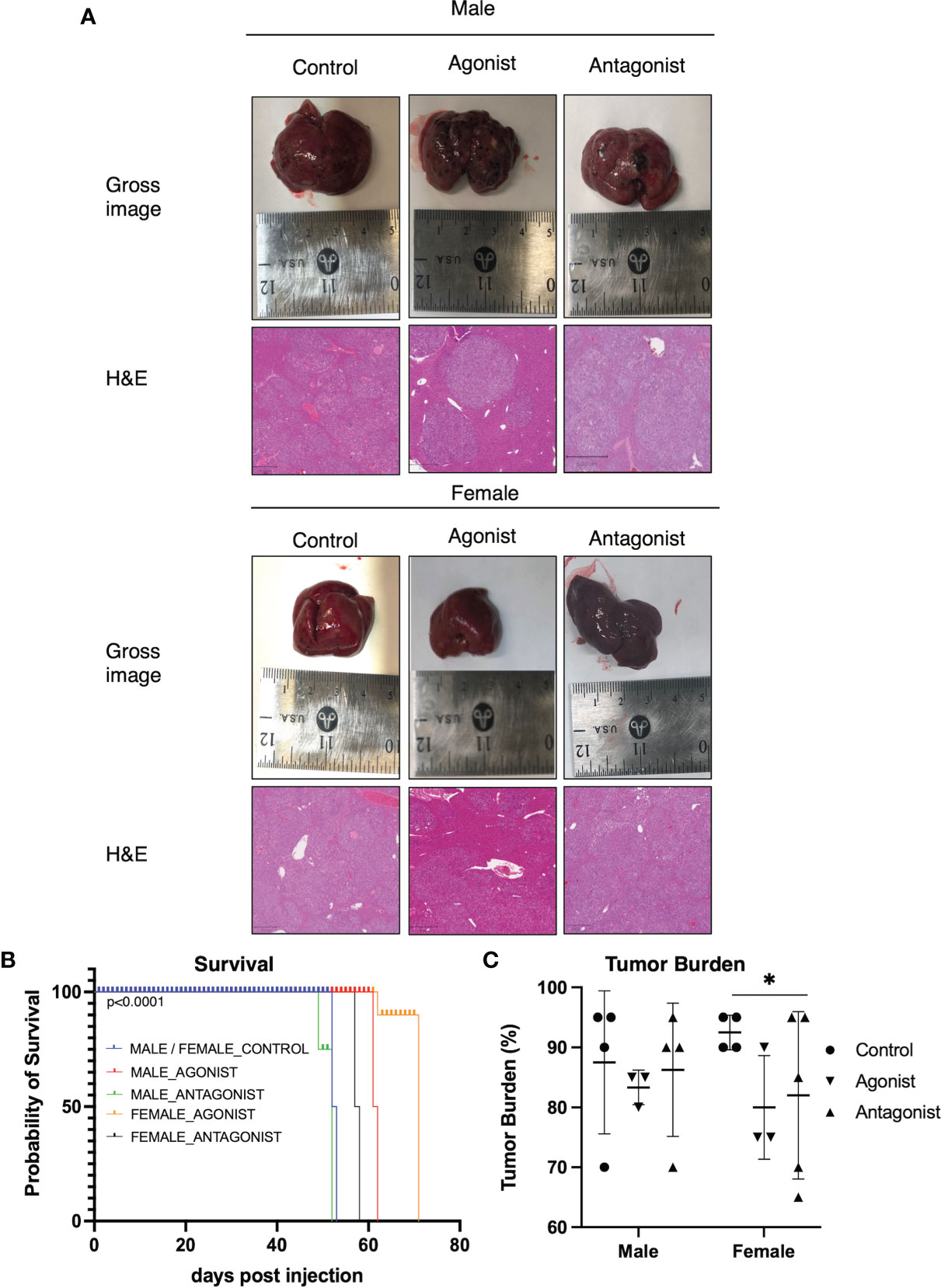
Figure 2 ER-α agonist exposure improves survival and decreases tumor burden (A) Gross images of livers from mice upon sacrifice show notable macroscopic disease in all groups. H&E staining of representative liver sections from the three groups in both male and female mice. Control group livers show more malignant nodules infiltrating the tissue. The agonist groups have smaller foci of tumor nodules (Magnification: 100X). (B) Kaplan-Meier curve comparing survival of HCC control mice (N = 4) and mice injected with ER-α agonist (N=6) or antagonist (N = 6). Agonist mice show significantly increased survival as compared to the control groups. p < 0.0001, Gehan-Breslow-Wilcoxon test. (C) Bar plot depicting tumor burden expressed as percentage of tumor cells in the liver tissue. Control groups livers show a higher percentage of infiltrating malignant cells. Mean with SD values are represented in the graph. *P < 0.05, multiple t-test. SD, standard deviation.
ER-α Agonist Treatment Inhibited Wnt/β-Catenin Signaling
Given the differences in survival and tumor burden with ER-a agonist treatment, gene expression profiling was performed to compare the agonist versus control mice. A higher number of dysregulated genes were identified in the female mice treated with ER-α agonist, with 266 upregulated and 280 downregulated genes (log2FC>1 or log2FC<-1, p value<0.05), while male mice treated with ER-α agonist had 173 upregulated and 64 downregulated genes compared to male control group. Pathway enrichment analysis was performed separately for the up- and down-modulated genes (Table 2 and Supplementary Tables 6, 7). Female mice treated with ER-α agonist had marked enrichment in the Wnt/β-catenin signaling pathway, followed by mTOR and Hippo signaling pathways (Supplementary Tables 6, 7). The male mice showed a similarly marked enrichment for downmodulated genes involving the Wnt signaling pathway, followed by MAPK.

Table 2 ESR1 agonist affects genes involved in crucial HCC pathways.
ER-α Agonist Exposure Decreases Tumor Proliferation
To evaluate if the downmodulation in Wnt/β-catenin signaling identified by gene expression affected HCC cell proliferation, mice treated with ER-α agonist were evaluated for β-catenin, cyclin-D1 and Ki67 IHC staining as a reflection of tumor proliferation rate in comparison to controls. The overall decrease of β-catenin in the agonist mice compared to control (Supplementary Figure 3A), was followed by a decrease in the intensity of its target proteins cyclin-D1 and Ki67, both known markers of DNA replication and cell proliferation (Supplementary Figures 3B, C).
Further, we found that mice treated with ER-α agonist showed an overall different cellular positivity and distribution of ER-α compared to the control mice, supporting the hypothesis that activation of the receptor was responsible for its nuclear translocation (Supplementary Figure 3D). Moreover, there was increased nuclear positivity for both β-catenin (Supplementary Figure 4A) and ER-α (Supplementary Figure 4B) with concomitantly decreased nuclear positivity of cyclin-D1 (Supplementary Figure 4C) in the mice treated with ER-α agonist compared to control.
ER-a Agonist Downmodulated ESR1 Interactor Genes Involved in Wnt/β-Catenin
To better understand the molecular connection between Wnt-β-catenin and ER-a, the protein interactors of ER-α were retrieved. 66 protein members of Wnt signaling are interactors of ER-a. Among these interacting proteins was β-catenin (CNNTB1), a crucial member of the nuclear transcription machinery (Supplementary Figure 5A). Moreover, CCND1 and MYC were identified as target genes of β-catenin (Supplementary Table 8). HepG2 cells overexpressing ESR1 (FC=4.85, p<0.0001 Student’s t-test) (Supplementary Figure 5B) showed a decreased expression in β-catenin-transcribed genes like CCND1 (FC=0.31 p=0.0019 Student’s t-test) and MYC (p=0.0023, Student’s t-test) (Supplementary Figures 5C, D).
ER-α Colocalization With β-Catenin Decreases Target Gene Expression
Cytoplasmic localization of β-catenin and ER-α in HeLa and MCF7 cells by confocal microscopy was used as positive control (top left panel Figure 3A). ER-a was detected in the nucleus of MCF7 (bottom left panel Figure 3A). HepG2 transfected with the empty vector showed cytoplasmic localization of β-catenin with no concomitant ER-α (Figure 3B). HepG2 transfected with ESR1 vector and exposed 72 hours to E2 had evidence of colocalization of ER-α and β-catenin (yellow signal as indicated by the white arrows in the bottom right panel, Figure 3C). There was significantly decreased expression of the β-catenin-transcribed genes CCND1 (p<0.0001, Student’s t-test) and MYC (p<0.0001, Student’s t-test) by real-time PCR.
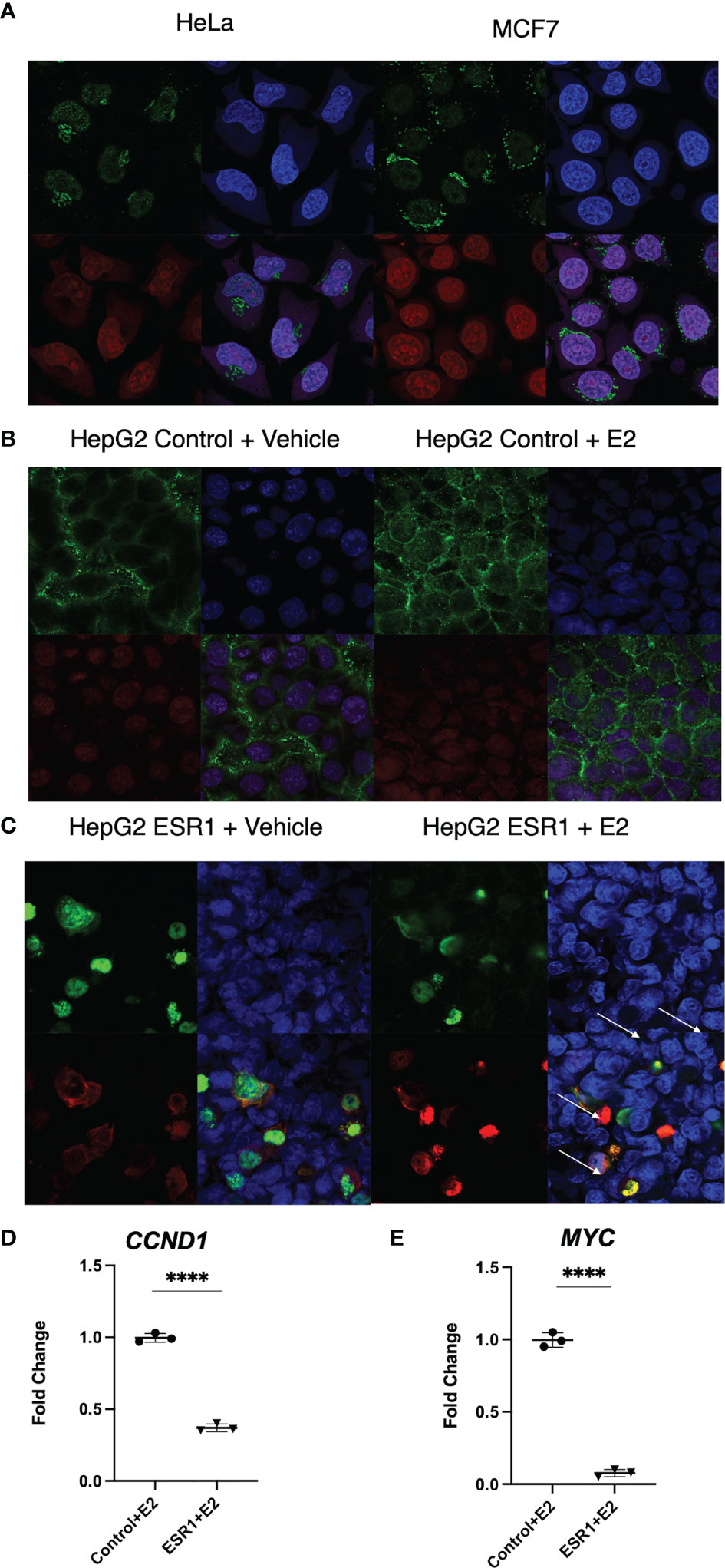
Figure 3 ER-α colocalization with β-catenin decreases β-catenin target gene expression in vitro. (A) Immunofluorescent analysis of β-catenin (top left panel) and ER-a (bottom left panel) on HeLa and MCF7 cells. Nuclei (top right panel) were stained with DAPI. Merged image (bottom right panel) showing a nuclear localization of ER-a in MCF7 and cytoplasmic localization of β-catenin in both cell lines. The images were captured at 40X magnification. (B) Immunofluorescent analysis of β-catenin (top left panel) and ER-α (bottom left panel) on HepG2 cells transfected with the control vector and exposed to vehicle or E2. Nuclei (top right panel) were stained with DAPI. Merged image (bottom right panel) showing the detection of cytoplasmic localization of β-catenin and absence of ER-α in the nucleus of HepG2 cells. (C) Immunofluorescent analysis of β-catenin (top left panel) and ER-α (bottom left panel) on HepG2 cells transfected with ESR1 coding vector and exposed to vehicle or E2. Nuclei (top right panel) were stained with DAPI. Merged image (bottom right panel) showing the concomitant detection in the nucleus of both β-catenin and ER-α in HepG2 cells represented by the yellow fluorescence as indicated by the white arrows. The images were captured at 40X magnification. (D) HepG2 cells overexpressing ESR1 expressed less CCND1 (FC = 0.3), MYC (FC = 0.065). Mean with SD values from N = 3 independent experiments are represented in the graph. ****p < 0.0001, Student’s t-test. SD, standard deviation.
ESR1 Overexpression Reduces β-Catenin Transcriptional Activity
Given that colocalization experiments revealed ESR1 translocation into the nucleus and colocalization with β-catenin, we evaluated whether this alters transcription by β-catenin of the target genes CCND1 and MYC. Thus, we evaluated if overexpression of ESR1 and exposure to E2 could downregulate β-catenin transcriptional activity compared to HepG2 cells transfected with control vector (p=0.0015). (Figure 4A). Using Real-Time PCR analysis, we determined that ESR1 overexpression in the presence of E2 effectively downregulated the mRNA expression of β-catenin target genes such as CCND1 (FC=0.02, p< 0.001, Student’s t-test) (Figure 4B) and MYC (FC=0.05, p=0.0013, Student’s t-test) (Figure 4C).
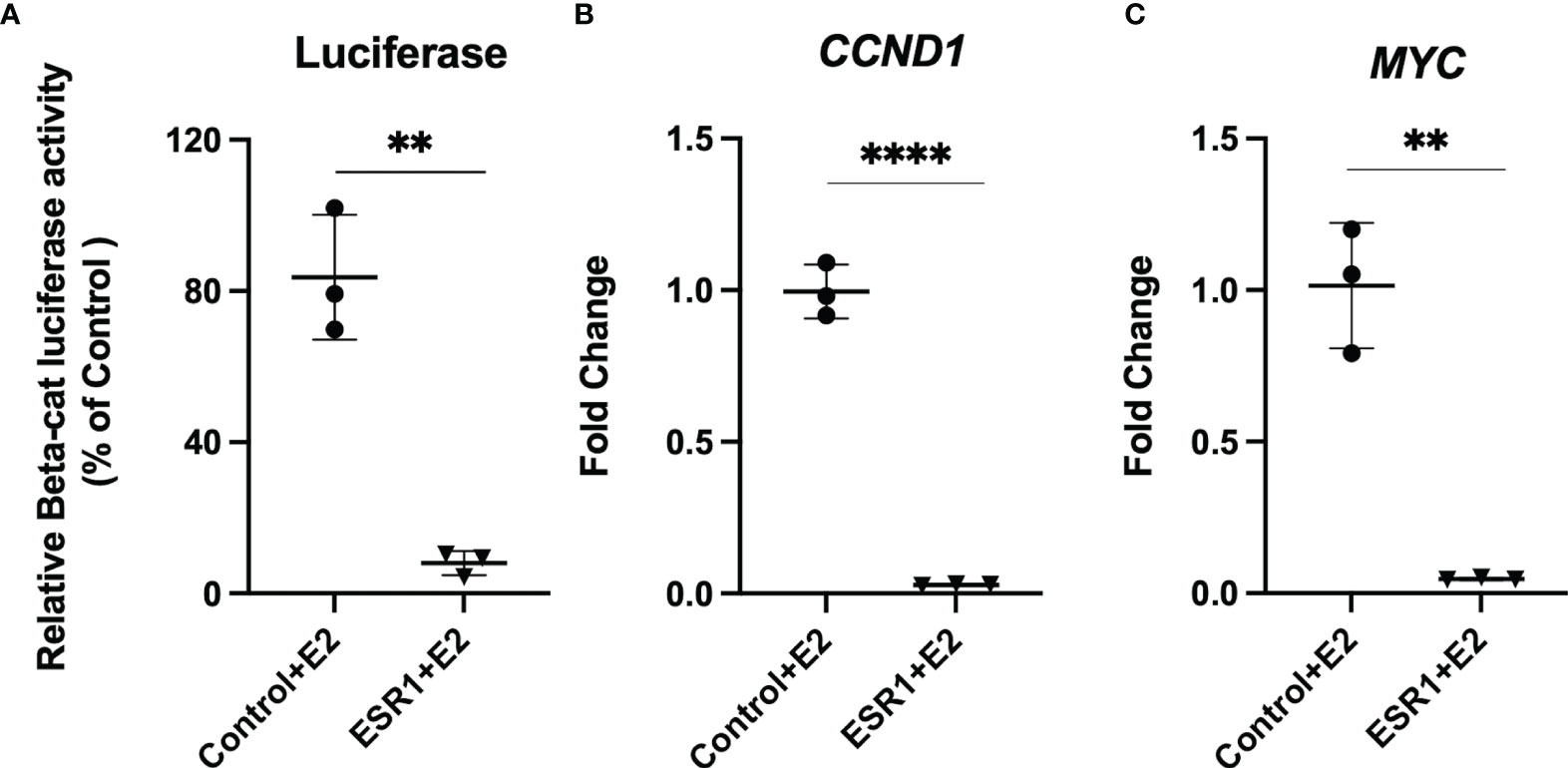
Figure 4 ESR1 overexpression decreases transcriptional activity and gene expression of β-catenin and Wnt/β-catenin signaling targets. (A) Representative bar graphs showing reduction of β-catenin transcriptional activity, as measured by luciferase assay in HepG2 cells following ESR1 co-transfection or control vector and exposure to E2 or vehicle. (B) Representative bar graphs showing reduction of mRNA expression of CCND1 in HepG2 cells as measured by Real-Time PCR. (C) Representative bar graphs showing reduction of mRNA expression of MYC in HepG2 cells as measured by Real-Time PCR. Mean with SD values from N = 3 independent experiments are represented in the graph. **p < 0.01, ****p < 0.0001 Student’s t-test. SD, standard deviation.
ESR1 Is Prognostic of HCC Survival, Independent of Sex and Race
Overall survival analysis was evaluated using The Cancer Genome Atlas (TCGA) HCC RNAseq data normalized with DESeq. ESR1 was significantly predictive of overall survival in 364 HCC patients with a significantly decreased hazard ratio of 0.45 (95%CI 0.32-0.64, log-rank p=4.4E-06), as shown in Figure 5A. Patients with high ESR1-expressing tumors had a median survival of 81.9 months, as compared to the cohort with low ESR1-expressing tumors at 27.6 months. This finding was independent of sex: overall survival was significantly improved in both males (HR 0.43 (95%CI 0.27-0.67, log-rank p=0.0001) (Figure 5B) and females (HR 0.39 (95%CI 0.22-0.68, log-rank p=0.0007) when ESR1 was upregulated (Figure 5C). Furthermore, we identified ESR1 to be significantly predictive of recurrence-free survival (HR 0.54 (95%CI 0.43-0.79, log-rank p=0.00044)) and progression-free survival (HR 0.4 (95%CI 0.25-0.63, log-rank p=3.6E-05)) (Supplementary Figure 6). Separation of patients by ESR1 expression in Kaplan-Meier plotter was based on the best cutoff computed from all the possible cutoff values between the lower and upper quartile (39).
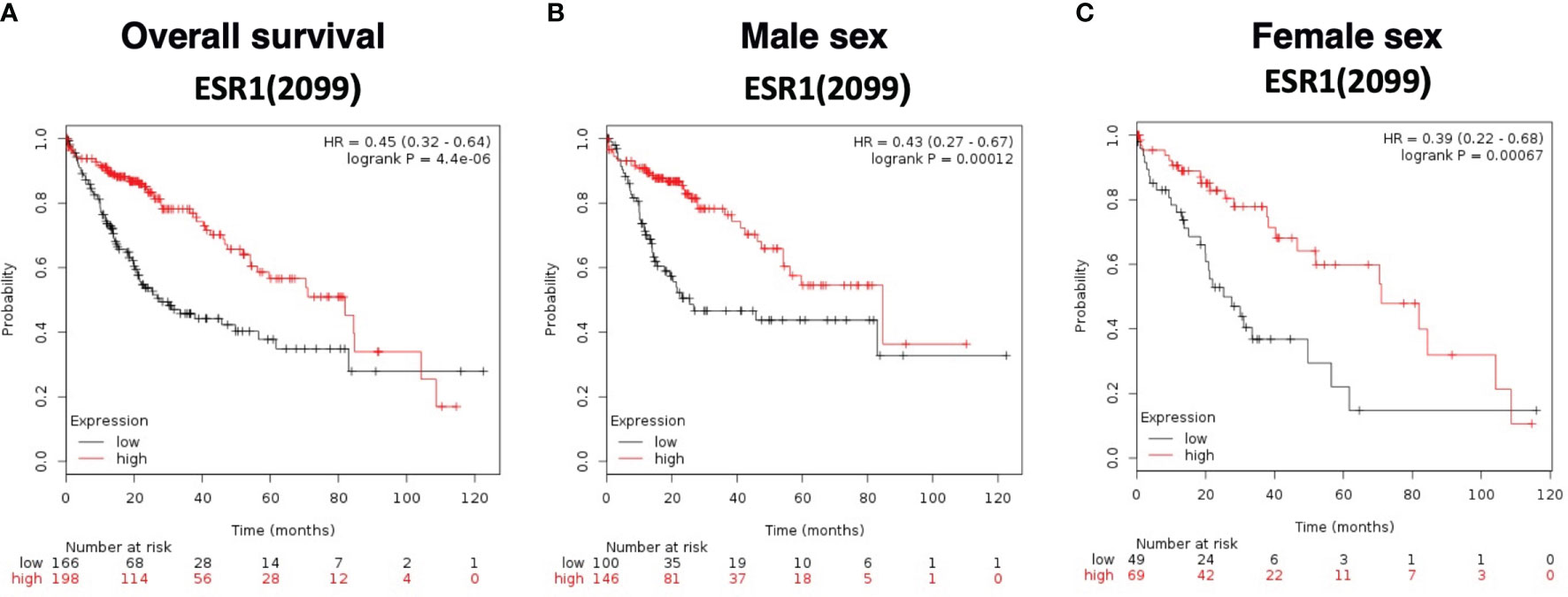
Figure 5 Survival curves based on expression of ESR1 (RNAseq ID 2099) in the TCGA dataset. (A) Overall survival according to ESR1 status based on auto selected best cutoff value (79). (B) Survival according to ESR1 status and further stratified by Male sex, based on auto selected best cutoff value (79) (C) Survival according to ESR1 status and further stratified by Female sex based on auto selected best cutoff value (43). Separation of patients by ESR1 expression in Kaplan-Meier plotter was based on the best cutoff computed from all the possible cutoff values between the lower and upper quartile (39).
Discussion
It has been well established that women are protected against HCC incidence and progression in comparison to men, independent of environmental exposures (40). This significant impact of sex on HCC incidence and survival has been ascribed to hormones, though the mechanistic basis has not been well delineated. In this study, we substantiate these sex-specific differences in HCC by demonstrating that ESR1, and therefore, the estrogen-mediated effect is an important contributor to sex differences in HCC. We employed the betweenness centrality measure on the HCC protein-protein interaction network obtained through a comprehensive integrative analysis of all publicly available gene expression data in HCC. In vivo validation confirmed that ER-α agonism decreases HCC tumor burden and enhances overall survival. This was accompanied by significant inhibition of the Wnt/β-catenin signaling cascade, a critical pathway in hepatocarcinogenesis. Additionally, we demonstrated through knockdown in vitro experiments that the mechanistic basis of ESR1’s effect is through its colocalization with β-catenin, thereby preventing transcription of the pro-oncogenic genes c-myc and cyclin D1. Thus, using a network analysis approach, we demonstrate ESR1 as being important to HCC pathogenesis through inhibition of Wnt signaling, and more specifically through inhibition of β-catenin transcriptional activity, thereby providing a molecular explanation for women being protected against HCC incidence and progression.
These findings are in line with previous literature on female sex being independently protective for HCC incidence in patients aged 18 to 44 years (HR 0.75; 95%CI 0.65-0.86, p<0.001) (17). Overall survival in women with HCC was also significantly higher, with a hazard ratio of 0.83 (95%CI 0.77-0.88) (17). This male predominance of HCC has led to questioning the importance of sex hormones in HCC pathogenesis. A case-control study of 234 female patients treated for HCC showed that estrogen replacement therapy reduced risk (HR 0.53; 95%CI 0.32-0.88) and decreased the risk of death from HCC (HR 0.55; 95%CI 0.40-0.77, p=0.01) (41). Median survival was also significantly higher in estrogen users (33.5 months) as compared to non-users (24.1 months, p=0.008) (5). A retrospective study of over 3,000 patients determined that oral contraceptive use was associated with increased survival in women with HCC (5).
Susceptibility to HCC has also been demonstrated with specific ESR1 polymorphisms, as evidenced by a study of 248 patients with HCC compared to controls. There was a greater than two-fold increase in susceptibility to HCC when one of three ESR1 polymorphisms were found (42). ESR1 has been identified as a tumor suppressor protein whose expression inversely correlates with tumor size and disease stage, based on genome-wide expression and microRNA analyses (18). An epigenetic study of HCC also confirmed ESR1 as a tumor suppressor gene, with up to 83% of HCC patient tumors demonstrating ESR1 promoter hypermethylation (5) and predicting tumor progression (17). Variant ESR1 expression is predominant in male patients with hepatitis B-associated HCC and is predictive of worse survival (18, 19, 41).
ESR1 is a ligand-dependent transcription factor that, upon binding to estrogen, recruits coregulatory proteins and binds regulatory DNA sites containing an estrogen responsive element. This alters transcription of genes relevant to tumorigenesis and the immune response (43, 44). In vitro and in vivo data have demonstrated that estrogen inhibits HCC growth through upregulation of the JAK-STAT pathway (16), suppression of Interleukin-6 secretion, decreased hepatocyte growth factor production (13, 45), suppression of PPARa-associated hepatocarcinogenesis (46, 47), and inhibition of NF-KB (48). Specific effects of estrogen on the immune cells within the HCC tumor microenvironment have also been shown in vivo (49, 50). Estrogen repressed HCC growth through inhibition of tumor-associated macrophages and the NLRP3 inflammasome (49). Conversely, in a preclinical model of diethylnitrosamine-induced HCC, loss of ESR1 was shown to accelerate hepatocarcinogenesis (51).
Our in vivo validation revealed that ER-α particularly inhibits the Wnt signaling cascade. In terms of processes, ESR1 significantly impacted transcription regulation, histone methylation, DNA repair, cell cycle and senescence. The effect of ESR1 on these processes is compatible with the literature on the impact of ESR1 in other cancers (52–60), with impact on transcriptional regulation (61), cell cycle, and tumor suppression (62). Interestingly, ER-α antagonist did not produce the opposite effect, but rather resulted in tumor burden similar to the control conditions. This again suggests that the positive effects of ER-α agonism are through inhibition of β-catenin transcriptional activity, without any graded effect once ER-α is inactivated.
Although Wnt signaling is the dominant pathway dysregulated in 40% of HCC tumors (15, 63), the current first-line therapies of lenvatinib or atezolizumab and bevacizumab target other less crucial pathways such as Ras/Raf/MAPK (64). The Wnt signaling pathway has remained elusive to therapeutic targeting, due to issues with protein-binding sites (β-Catenin) or significant side effects (65, 66). Beyond consideration for signaling pathways, sex differences have been noted in treatment response. Women have better outcomes than men, by virtue of slow-growing HCC that presents early enough for curative therapy (67, 68). Our studies could inform a potentially synergistic targeting of Wnt signaling, under sex hormone receptor modulation, alongside the current first-line therapy targeting other less dysregulated pathways in HCC.
The current study also represents a large integration of publicly available gene expression data in HCC, covering 36 studies and 1,268 patient sample profiles. We integrated gene expression data from PubMed and the Gene Expression Omnibus enabled the identification of the most consistently deregulated genes involved in hepatocarcinogenesis as detected by transcriptomics. These systematically curated data are made available in our Cancer Data Integration Portal (http://ophid.utoronto.ca/CDIPLiver) and will serve as a public resource for analyzing integrated HCC high-throughput gene expression data.
One key study limitation was that most HCC samples in this integrative analysis came from patients who had undergone hepatectomy, which we discovered was the nature of most publicly available gene expression data. The molecular features of different HCC stages could potentially differ, and ESR1 may not be central in those contexts.
Additionally, estrogen receptor signaling likely has an impact on other aspects of HCC biology, but we chose to focus on Wnt/B-catenin given the impact of ER-a agonist treatment on this critical HCC pathway.
In conclusion, we provide a mechanism by which ESR1 plays an important role in the sexual dimorphism of HCC, by inhibiting Wnt/β-catenin signaling, the most commonly dysregulated pathway in HCC. Our results suggest that ER-α colocalizes with the transcription factor β-catenin to inhibit transcription of pro-oncogenic genes. Our study findings serve to enhance our understanding of the mechanistic basis for protection against HCC incidence and progression in women as compared to men.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Ethics Statement
The animal study was reviewed and approved by University of Toronto AUP 20012289, University Health Network AUP 6105.
Author Contributions
Conceptualization: MB and IJ. Methodology: MB, EP, CP, CB, MK, OA, and IJ. Investigation: MB, EP, CP, MAn, MAb, MK, OA, and IJ. Visualization: MB and IJ. Funding acquisition: MB and IJ. Project administration: MB, EP, and IJ. Supervision: MB and IJ. Writing – original draft: MB, EP, CP, and IJ. Writing – review and editing: MB, EP, CP, MAn, CB, MK, and IJ.
Funding
Canadian Institutes for Health Research Fellowship (MB),Toronto General and Western Hospital Research Foundation (MB), Canadian Liver Foundation (MB), Natural Sciences Research Council (IJ), Canada Foundation for Innovation (IJ), Ontario Research Fund (IJ), IBM and Ian Lawson van Toch Fund (IJ).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors acknowledge the assistance of the animal facilities at the University of Toronto and Toronto General Hospital Research Institute.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2021.777834/full#supplementary-material
References
1. Wong CR, Nguyen MH, Lim JK. Hepatocellular Carcinoma in Patients With non-Alcoholic Fatty Liver Disease. World J Gastroenterol (2016) 22(37):8294–303. doi: 10.3748/wjg.v22.i37.8294
2. Ha NB, Ha NB, Ahmed A, Ayoub W, Daugherty TJ, Chang ET, et al. Risk Factors for Hepatocellular Carcinoma in Patients With Chronic Liver Disease: A Case-Control Study. Cancer Causes Control (2012) 23(3):455–62. doi: 10.1007/s10552-012-9895-z
3. El-Serag HB, Rudolph KL. Hepatocellular Carcinoma: Epidemiology and Molecular Carcinogenesis. Gastroenterology (2007) 132(7):2557–76. doi: 10.1053/j.gastro.2007.04.061
4. Ma WL, Hsu CL, Yeh CC, Wu MH, Huang CK, Jeng LB, et al. Hepatic Androgen Receptor Suppresses Hepatocellular Carcinoma Metastasis Through Modulation of Cell Migration and Anoikis. Hepatology (2012) 56(1):176–85. doi: 10.1002/hep.25644
5. Hassan MM, Botrus G, Abdel-Wahab R, Wolff RA, Li D, Tweardy D, et al. Estrogen Replacement Reduces Risk and Increases Survival Times of Women With Hepatocellular Carcinoma. Clin Gastroenterol Hepatol (2017) 15(11):1791–9. doi: 10.1016/j.cgh.2017.05.036
6. Lam CM, Yong JL, Chan AO, Ng KK, Poon RT, Liu CL, et al. Better Survival in Female Patients With Hepatocellular Carcinoma: Oral Contraceptive Pills Related? J Clin Gastroenterol (2005) 39(6):533–9. doi: 10.1097/01.mcg.0000165670.25272.46
7. Chang KK, Mo LR, Wang CH, Hsieh CC, Hsu HY, Tseng YC, et al. Long-Term Effects of Hormone Replacement Therapy on Hepatocellular Carcinoma Risk and Overall Survival Rate in Women With Chronic Hepatitis C: A Population-Based Cohort Study in Taiwan. Adv Dig Med (2021) 1–9. doi: 10.1002/aid2.13250
8. Yu MW, Chang HC, Chang SC, Liaw YF, Lin SM, Liu CJ, et al. Role of Reproductive Factors in Hepatocellular Carcinoma: Impact on Hepatitis B- and C-Related Risk. Hepatology (2003) 38(6):1393–400. doi: 10.1016/j.hep.2003.09.041
9. Chiu CM, Yeh SH, Chen PJ, Kuo TJ, Chang CJ, Chen PJ, et al. Hepatitis B Virus X Protein Enhances Androgen Receptor-Responsive Gene Expression Depending on Androgen Level. Proc Natl Acad Sci USA (2007) 104(8):2571–8. doi: 10.1073/pnas.0609498104
10. Firminger HI, Reuber MD. Influence of Adrenocortical, Androgenic, and Anabolic Hormones on the Development of Carcinoma and Cirrhosis of the Liver in A X C Rats Fed N-2-Fluorenyldicetamide. J Natl Cancer Inst (1961) 27:559–95.
11. Moriya K, Fujie H, Shintani Y, Yotsuyanagi H, Tsutsumi T, Ishibashi K, et al. The Core Protein of Hepatitis C Virus Induces Hepatocellular Carcinoma in Transgenic Mice. Nat Med (1998) 4(9):1065–7. doi: 10.1038/2053
12. Wu EM, Wong LL, Hernandez BY, Ji JF, Jia W, Kwee SA, et al. Gender Differences in Hepatocellular Cancer: Disparities in Nonalcoholic Fatty Liver Disease/Steatohepatitis and Liver Transplantation. Hepatoma Res (2018) 4:66. doi: 10.20517/2394-5079.2018.87
13. Naugler WE, Sakurai T, Kim S, Maeda S, Kim K, Elsharkawy AM, et al. Gender Disparity in Liver Cancer Due to Sex Differences in MyD88-Dependent IL-6 Production. Science (2007) 317(5834):121–4. doi: 10.1126/science.1140485
14. Kohi MP. Gender-Related Differences in Hepatocellular Carcinoma: Does Sex Matter? J Vasc Interv Radiol (2016) 27(9):1338–41. doi: 10.1016/j.jvir.2016.06.035
15. Medavaram S, Zhang Y. Emerging Therapies in Advanced Hepatocellular Carcinoma. Exp Hematol Oncol (2018) 7:17. doi: 10.1186/s40164-018-0109-6
16. Hou J, Xu J, Jiang R, Wang Y, Chen C, Deng L, et al. Estrogen-Sensitive PTPRO Expression Represses Hepatocellular Carcinoma Progression by Control of STAT3. Hepatology (2013) 57(2):678–88. doi: 10.1002/hep.25980
17. Liang J, Lv J, Liu Z. Identification of Dysfunctional Biological Pathways and Their Synergistic Mechanism in Hepatocellular Carcinoma Process. Exp Mol Pathol (2015) 98(3):540–5. doi: 10.1016/j.yexmp.2015.03.028
18. Hishida M, Nomoto S, Inokawa Y, Hayashi M, Kanda M, Okamura Y, et al. Estrogen Receptor 1 Gene as a Tumor Suppressor Gene in Hepatocellular Carcinoma Detected by Triple-Combination Array Analysis. Int J Oncol (2013) 43(1):88–94. doi: 10.3892/ijo.2013.1951
19. Villa E, Moles A, Ferretti I, Buttafoco P, Grottola A, Del Buono M, et al. Natural History of Inoperable Hepatocellular Carcinoma: Estrogen Receptors' Status in the Tumor is the Strongest Prognostic Factor for Survival. Hepatology (2000) 32(2):233–8. doi: 10.1053/jhep.2000.9603
20. Kocanova S, Mazaheri M, Caze-Subra S, Bystricky K. Ligands Specify Estrogen Receptor Alpha Nuclear Localization and Degradation. BMC Cell Biol (2010) 11:98. doi: 10.1186/1471-2121-11-98
21. Ochiai I, Matsuda K, Nishi M, Ozawa H, Kawata M. Imaging Analysis of Subcellular Correlation of Androgen Receptor and Estrogen Receptor Alpha in Single Living Cells Using Green Fluorescent Protein Color Variants. Mol Endocrinol (2004) 18(1):26–42. doi: 10.1210/me.2002-0262
22. Helsen C, Kerkhofs S, Clinckemalie L, Spans L, Laurent M, Boonen S, et al. Structural Basis for Nuclear Hormone Receptor DNA Binding. Mol Cell Endocrinol (2012) 348(2):411–7. doi: 10.1016/j.mce.2011.07.025
23. Lonard DM, O'Malley BW. The Expanding Cosmos of Nuclear Receptor Coactivators. Cell (2006) 125(3):411–4. doi: 10.1016/j.cell.2006.04.021
24. Gadaleta RM, Magnani L. Nuclear Receptors and Chromatin: An Inducible Couple. J Mol Endocrinol (2014) 52(2):R137–49. doi: 10.1530/JME-13-0170
25. Kininis M, Kraus WL. A Global View of Transcriptional Regulation by Nuclear Receptors: Gene Expression, Factor Localization, and DNA Sequence Analysis. Nucl Recept Signal (2008) 6:e005. doi: 10.1621/nrs.06005
26. Clocchiatti A, Cora E, Zhang Y, Dotto GP. Sexual Dimorphism in Cancer. Nat Rev Cancer (2016) 16(5):330–9. doi: 10.1038/nrc.2016.30
27. Smyth GK. Linear Models and Empirical Bayes Methods for Assessing Differential Expression in Microarray Experiments. Stat Appl Genet Mol Biol (2004) 3 Article3:1544–6115. doi: 10.2202/1544-6115.1027
28. Limma GKS. Linear Models for Microarray Data. In: Gentleman R, Carey VJ, Huber W, Irizarry RA, Dudoit S, editors. Bioinformatics and Computational Biology Solutions Using R and Bioconductor Statistics for Biology and Health. New York, NY: Springer (2005). p. 397–420.
29. Davis S, Meltzer PS. GEOquery: A Bridge Between the Gene Expression Omnibus (GEO) and BioConductor. Bioinformatics (2007) 23(14):1846–7. doi: 10.1093/bioinformatics/btm254
30. Bruix J, Sherman M, American Association for the Study of Liver Diseases. Management of Hepatocellular Carcinoma: An Update. Hepatology (2011) 53(3):1020–2. doi: 10.1002/hep.24199
31. Csardi G, Nepusz T. The Igraph Software Package For Complex Network Research. IJ Comp Syss (2006) 53(3):1020–2. doi: 10.1002/hep.24199
32. Brown KR, Otasek D, Ali M, McGuffin MJ, Xie W, Devani B, et al. NAViGaTOR: Network Analysis, Visualization and Graphing Toronto. Bioinformatics (2009) 25(24):3327–9. doi: 10.1093/bioinformatics/btp595
33. Tao J, Zhang R, Singh S, Poddar M, Xu E, Oertel M, et al. Targeting Beta-Catenin in Hepatocellular Cancers Induced by Coexpression of Mutant Beta-Catenin and K-Ras in Mice. Hepatology (2017) 65(5):1581–99. doi: 10.1002/hep.28975
34. Chen X, Calvisi DF. Hydrodynamic Transfection for Generation of Novel Mouse Models for Liver Cancer Research. Am J Pathol (2014) 184(4):912–23. doi: 10.1016/j.ajpath.2013.12.002
35. Lanczky A, Nagy A, Bottai G, Munkacsy G, Szabo A, Santarpia L, et al. Mirpower: A Web-Tool to Validate Survival-Associated miRNAs Utilizing Expression Data From 2178 Breast Cancer Patients. Breast Cancer Res Treat (2016) 160(3):439–46. doi: 10.1007/s10549-016-4013-7
36. Kotlyar M, Pastrello C, Malik Z, Jurisica I. IID 2018 Update: Context-Specific Physical Protein-Protein Interactions in Human, Model Organisms and Domesticated Species. Nucleic Acids Res (2019) 47(D1):D581–9. doi: 10.1093/nar/gky1037
37. Fortney K, Griesman J, Kotlyar M, Pastrello C, Angeli M, Sound-Tsao M, et al. Prioritizing Therapeutics for Lung Cancer: An Integrative Meta-Analysis of Cancer Gene Signatures and Chemogenomic Data. PloS Comput Biol (2015) 11(3):e1004068. doi: 10.1371/journal.pcbi.1004068
38. Rahmati S, Abovsky M, Pastrello C, Jurisica I. pathDIP: An Annotated Resource for Known and Predicted Human Gene-Pathway Associations and Pathway Enrichment Analysis. Nucleic Acids Res (2017) 45(D1):D419–26. doi: 10.1093/nar/gkw1082
39. Nagy Á, Munkácsy G, Győrffy B. Pancancer Survival Analysis of Cancer Hallmark Genes. Sci Rep (2021) 11(1):6047. doi: 10.1038/s41598-021-84787-5
40. Ananthakrishnan A, Gogineni V, Saeian K. Epidemiology of Primary and Secondary Liver Cancers. Semin Intervent Radiol (2006) 23(1):47–63. doi: 10.1055/s-2006-939841
41. Villa E, Grottola A, Colantoni A, De Maria N, Buttafoco P, Ferretti I, et al. Hepatocellular Carcinoma: Role of Estrogen Receptors in the Liver. Ann N Y Acad Sci (2002) 963:37–45. doi: 10.1111/j.1749-6632.2002.tb04092.x
42. Zhai Y, Zhou G, Deng G, Xie W, Dong X, Zhang X, et al. Estrogen Receptor Alpha Polymorphisms Associated With Susceptibility to Hepatocellular Carcinoma in Hepatitis B Virus Carriers. Gastroenterology (2006) 130(7):2001–9. doi: 10.1053/j.gastro.2006.02.030
43. Nagasue N, Ito A, Yukaya H, Ogawa Y. Androgen Receptors in Hepatocellular Carcinoma and Surrounding Parenchyma. Gastroenterology (1985) 89(3):643–7. doi: 10.1016/0016-5085(85)90463-9
44. Nagasue N, Ito A, Yukaya H, Ogawa Y. Estrogen Receptors in Hepatocellular Carcinoma. Cancer (1986) 57(1):87–91. doi: 10.1002/1097-0142(19860101)57:1<87::AID-CNCR2820570118>3.0.CO;2-K
45. Wang YC, Xu GL, Jia WD, Han SJ, Ren WH, Wang W, et al. Estrogen Suppresses Metastasis in Rat Hepatocellular Carcinoma Through Decreasing Interleukin-6 and Hepatocyte Growth Factor Expression. Inflammation (2012) 35(1):143–9. doi: 10.1007/s10753-011-9299-3
46. Chang-Lee SN, Hsu HH, Shibu MA, Ho TJ, Tsai CH, Chen MC, et al. E2/ERbeta Inhibits PPARalpha to Regulate Cell-Proliferation and Enhance Apoptosis in Hep3B-Hepatocellular Carcinoma. Pathol Oncol Res (2017) 23(3):477–85. doi: 10.1007/s12253-016-0136-8
47. Wu H, Yao S, Zhang S, Wang JR, Guo PD, Li XM, et al. Elevated Expression of Erbin Destabilizes ERalpha Protein and Promotes Tumorigenesis in Hepatocellular Carcinoma. J Hepatol (2017) 66(6):1193–204. doi: 10.1016/j.jhep.2017.01.030
48. Xu H, Wei Y, Zhang Y, Xu Y, Li F, Liu J, et al. Oestrogen Attenuates Tumour Progression in Hepatocellular Carcinoma. J Pathol (2012) 228(2):216–29. doi: 10.1002/path.4009
49. Yang W, Lu Y, Xu Y, Xu L, Zheng W, Wu Y, et al. Estrogen Represses Hepatocellular Carcinoma (HCC) Growth via Inhibiting Alternative Activation of Tumor-Associated Macrophages (TAMs). J Biol Chem (2012) 287(48):40140–9. doi: 10.1074/jbc.M112.348763
50. Wei Q, Guo P, Mu K, Zhang Y, Zhao W, Huai W, et al. Estrogen Suppresses Hepatocellular Carcinoma Cells Through ERbeta-Mediated Upregulation of the NLRP3 Inflammasome. Lab Invest (2015) 95(7):804–16. doi: 10.1038/labinvest.2015.63
51. Hong EJ, Levasseur MP, Dufour CR, Perry MC, Giguere V. Loss of Estrogen-Related Receptor Alpha Promotes Hepatocarcinogenesis Development via Metabolic and Inflammatory Disturbances. Proc Natl Acad Sci USA (2013) 110(44):17975–80. doi: 10.1073/pnas.1315319110
52. Breu A, Sprinzing B, Merkl K, Bechmann V, Kujat R, Jenei-Lanzl Z, et al. Estrogen Reduces Cellular Aging in Human Mesenchymal Stem Cells and Chondrocytes. J Orthop Res (2011) 29(10):1563–71. doi: 10.1002/jor.21424
53. Pettapiece-Phillips R, Kotlyar M, Chehade R, Salmena L, Narod SA, Akbari M, et al. Uninterrupted Sedentary Behavior Downregulates BRCA1 Gene Expression. Cancer Prev Res (Phila) (2016) 9(1):83–8. doi: 10.1158/1940-6207.CAPR-15-0291
54. Wang X, Yan Z, Fulciniti M, Li Y, Gkotzamanidou M, Amin SB, et al. Transcription Factor-Pathway Coexpression Analysis Reveals Cooperation Between SP1 and ESR1 on Dysregulating Cell Cycle Arrest in Non-Hyperdiploid Multiple Myeloma. Leukemia (2014) 28(4):894–903. doi: 10.1038/leu.2013.233
55. Hartman J, Edvardsson K, Lindberg K, Zhao C, Williams C, Strom A, et al. Tumor Repressive Functions of Estrogen Receptor Beta in SW480 Colon Cancer Cells. Cancer Res (2009) 69(15):6100–6. doi: 10.1158/0008-5472.CAN-09-0506
56. Lattrich C, Stegerer A, Haring J, Schuler S, Ortmann O, Treeck O. Estrogen Receptor Beta Agonists Affect Growth and Gene Expression of Human Breast Cancer Cell Lines. Steroids (2013) 78(2):195–202. doi: 10.1016/j.steroids.2012.10.014
57. Lu W, Katzenellenbogen BS. Estrogen Receptor-Beta Modulation of the ERalpha-P53 Loop Regulating Gene Expression, Proliferation, and Apoptosis in Breast Cancer. Horm Cancer (2017) 8(4):230–42. doi: 10.1007/s12672-017-0298-1
58. Sareddy GR, Nair BC, Gonugunta VK, Zhang QG, Brenner A, Brann DW, et al. Therapeutic Significance of Estrogen Receptor Beta Agonists in Gliomas. Mol Cancer Ther (2012) 11(5):1174–82. doi: 10.1158/1535-7163.MCT-11-0960
59. Warner M, Huang B, Gustafsson JA. Estrogen Receptor Beta as a Pharmaceutical Target. Trends Pharmacol Sci (2017) 38(1):92–9. doi: 10.1016/j.tips.2016.10.006
60. Liu J, Sareddy GR, Zhou M, Viswanadhapalli S, Li X, Lai Z, et al. Differential Effects of Estrogen Receptor Beta Isoforms on Glioblastoma Progression. Cancer Res (2018) 78(12):3176–89. doi: 10.1158/0008-5472.CAN-17-3470
61. Lee MT, Ouyang B, Ho SM, Leung YK. Differential Expression of Estrogen Receptor Beta Isoforms in Prostate Cancer Through Interplay Between Transcriptional and Translational Regulation. Mol Cell Endocrinol (2013) 376(1-2):125–35. doi: 10.1016/j.mce.2013.06.023
62. Dey P, Barros RP, Warner M, Strom A, Gustafsson JA. Insight Into the Mechanisms of Action of Estrogen Receptor Beta in the Breast, Prostate, Colon, and CNS. J Mol Endocrinol (2013) 51(3):T61–74. doi: 10.1530/JME-13-0150
63. Cancer Genome Atlas Research Network. Comprehensive and Integrative Genomic Characterization of Hepatocellular Carcinoma. Cell (2017) 169(7):1327–41.e23. doi: 10.1016/j.cell.2017.05.046
64. Finn RS, Qin S, Ikeda M, Galle PR, Ducreux M, Kim TY, et al. Atezolizumab Plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N Engl J Med (2020) 382(20):1894–905. doi: 10.1056/NEJMoa1915745
65. Jung YS, Park JI. Wnt Signaling in Cancer: Therapeutic Targeting of Wnt Signaling Beyond Beta-Catenin and the Destruction Complex. Exp Mol Med (2020) 52(2):183–91. doi: 10.1038/s12276-020-0380-6
66. Voronkov A, Krauss S. Wnt/beta-Catenin Signaling and Small Molecule Inhibitors. Curr Pharm Des (2013) 19(4):634–64. doi: 10.2174/138161213804581837
67. Sobotka L, Hinton A, Conteh L. Women Receive More Inpatient Resections and Ablations for Hepatocellular Carcinoma Than Men. World J Hepatol (2017) 9(36):1346–51. doi: 10.4254/wjh.v9.i36.1346
Keywords: estrogen, hepatocellular carcinoma, high-throughput, network analysis, Wnt/b-catenin, ESR1, interactome, PPIs
Citation: Bhat M, Pasini E, Pastrello C, Angeli M, Baciu C, Abovsky M, Coffee A, Adeyi O, Kotlyar M and Jurisica I (2021) Estrogen Receptor 1 Inhibition of Wnt/β-Catenin Signaling Contributes to Sex Differences in Hepatocarcinogenesis. Front. Oncol. 11:777834. doi: 10.3389/fonc.2021.777834
Received: 16 September 2021; Accepted: 01 November 2021;
Published: 22 November 2021.
Edited by:
Shilpa S. Dhar, University of Texas MD Anderson Cancer Center, United StatesReviewed by:
Davod Javanmard, Birjand University of Medical Sciences, IranConcetta Saponaro, Istituto Nazionale dei Tumori (IRCCS), Italy
Copyright © 2021 Bhat, Pasini, Pastrello, Angeli, Baciu, Abovsky, Coffee, Adeyi, Kotlyar and Jurisica. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mamatha Bhat, TWFtYXRoYS5CaGF0QHVobi5jYQ==; Igor Jurisica, anVyaXNAYWkudXRvcm9udG8uY2E=