 Madelyn Espinosa-Cotton
Madelyn Espinosa-Cotton Nai-Kong V. Cheung*
Nai-Kong V. Cheung*- Department of Pediatrics, Memorial Sloan Kettering Cancer Center, New York, NY, United States
Desmoplastic small round cell tumor (DRSCT) is a highly aggressive primitive sarcoma that primarily affects adolescent and young adult males. The 5-year survival rate is 15-30% and few curative treatment options exist. Although there is no standard treatment for DSRCT, patients are most often treated with a combination of aggressive chemotherapy, radiation, and surgery. Targeted therapy inhibitors of PDGFA and IGF-1R, which are almost uniformly overexpressed in DSRCT, have largely failed in clinical trials. As in cancer in general, interest in immunotherapy to treat DSRCT has increased in recent years. To that end, several types of immunotherapy are now being tested clinically, including monoclonal antibodies, radionuclide-conjugated antibodies, chimeric antigen receptor T cells, checkpoint inhibitors, and bispecific antibodies (BsAbs). These types of therapies may be particularly useful in DSRCT, which is frequently characterized by widespread intraperitoneal implants, which are difficult to completely remove surgically and are the frequent cause of relapse. Successful treatment with immunotherapy or radioimmunotherapy following debulking surgery could eradiate these micrometasteses and prevent relapse. Although there has been limited success to date for immunotherapy in pediatric solid tumors, the significant improvements in survival seen in the treatment of other pediatric solid tumors, such as metastatic neuroblastoma and its CNS spread, suggest a potential of immunotherapy and specifically compartmental immunotherapy in DSRCT.
Introduction
Background
Desmoplastic small round cell tumor (DRSCT) was first described in 1989 by Gerald and Rosai as a highly aggressive primitive sarcoma characterized by nests of blue-staining tumor cells surrounded by dense stroma. Of note was the fact that these tumor cells were positive for markers of mesenchymal, neural, and epithelial lineages, suggesting that they may arise from undifferentiated progenitor cells (1, 2). A (11:22), (p13:q12) chromosomal translocation is present in all cases, resulting in an EWS-WT1 gene fusion product (3–5). This feature is considered pathognomonic and is required for its definitive diagnosis (6). DSRCT is rare, with an age-adjusted incidence rate of 0.3/million in the United States (7), and primarily arises in adolescent and young adult males, with around 80% of patients being male and an average age of 18-22 years at diagnosis (2, 8). It may disproportionately affect African Americans (7, 9). Overall 5-year survival is a dismal 15-30% (7, 10).
Clinical Presentation and Staging
Most patients initially present with abdominal pain and distension, evidence of ascites caused by extensive tumor seeding of the peritoneum (6). The vast majority (>90%) of primary tumors are found on the serosal surfaces of the abdomen, but others have been found in the thoracic cavity, the skull, or even the hand (8). Rarely, early-stage DSRCT is discovered incidentally during surgery or imaging for other indications. More commonly, however, the disease has spread extensively and is considered Stage IV at the time of diagnosis. Because of this, a new staging system for DSRCT has been proposed by Hayes-Jordan and colleagues: Stage I would include patients with one or two abdominal tumors, Stage II would include patients with extensive peritoneal spread, Stage III would include patients with peritoneal disease plus liver metastases, and Stage IV would include patients with disease that has spread outside the abdomen (6).
Treatment
There is no standard therapy for DSRCT. Treatment typically consists of multimodal neoadjuvant chemotherapy followed by surgery and radiation. Treatment with the P6 protocol – cyclophosphamide, doxorubicin and vincristine alternating with ifosfamide and etoposide – is used in the United States, whereas a slightly different protocol used to treat Ewing Sarcoma patients (vincristine, doxorubicin, and cyclophosphamide alternating with ifosfamide and etoposide) is the most common regimen in other parts of the world (11, 12). Because of the extent of disease spread within the peritoneal cavity, surgery is most effective after neoadjuvant chemotherapy, since these tumors are typically responsive to chemotherapy and either shrink or become less vascularized (6). Complete surgical resection of all visible tumors was found to be essential for survival beyond 3 years, with 58% of patients who underwent complete resection surviving to 3 years compared to 0% of patients whose tumors were not resected and instead received only chemotherapy and radiation (10). While surgical excision of all visible tumors is critical, it may be impossible to remove every tumor cell from the peritoneal cavity. Hyperthermic intraperitoneal chemotherapy (HIPEC) has been proposed as a treatment for microscopic residual disease after complete surgical resection. A recent phase II clinical trial showed that HIPEC using cisplatin is effective at improving survival, with a 3-year overall survival rate of 79% (13). However, this treatment modality does not appear effective for patients whose tumors were not able to be completely removed during surgery (14). Whole abdominopelvic irradiation has also been used to treat residual disease following surgery and appears to reduce the incidence of peritoneal relapse, however, severe gastrointestinal and hematopoietic toxicity is common (15, 16). Intensity modulated radiation therapy reduces grade 2-4 toxicities without compromising efficacy (17). Autologous stem cell transplant has been investigated and does not appear to improve outcomes for DSRCT patients (18); allogenic stem cell transplant could be an alternative (19). Several clinical trials are currently underway to test new combinations of chemotherapy and targeted therapy (Table 1). Progress has been made in extending survival, however, cures are rare.

Table 1 Current clinical trials using chemotherapy and targeted therapy for DSRCT.
Mutations, Targets, and Dysregulated Pathways
Many common mutations have been found in DSRCT (Table 2), some of which have been explored as targets for therapy. Several of these are targets of the EWS-WT1 fusion protein, which is present in every case of DSRCT and is required for positive diagnosis (6). This protein results from a chromosomal translocation involving breakage of chromosomes 11 and 22 at sites of genes known to be involved in Wilms’ tumor (WT1) and Ewing’s sarcoma (EWSR1), respectively (20). WT1 is a tumor suppressor gene encoding a transcription factor (WT1) that generally represses gene expression and was first noted for its deletion in Wilms’ tumor (21, 22). Fusion of the transcription-activating N-terminal domain of EWS to a set of zinc fingers in WT1 produces a unique transcription factor capable of upregulating a number of genes that promote tumor progression, many of which are repressed by wild-type WT1 (20).
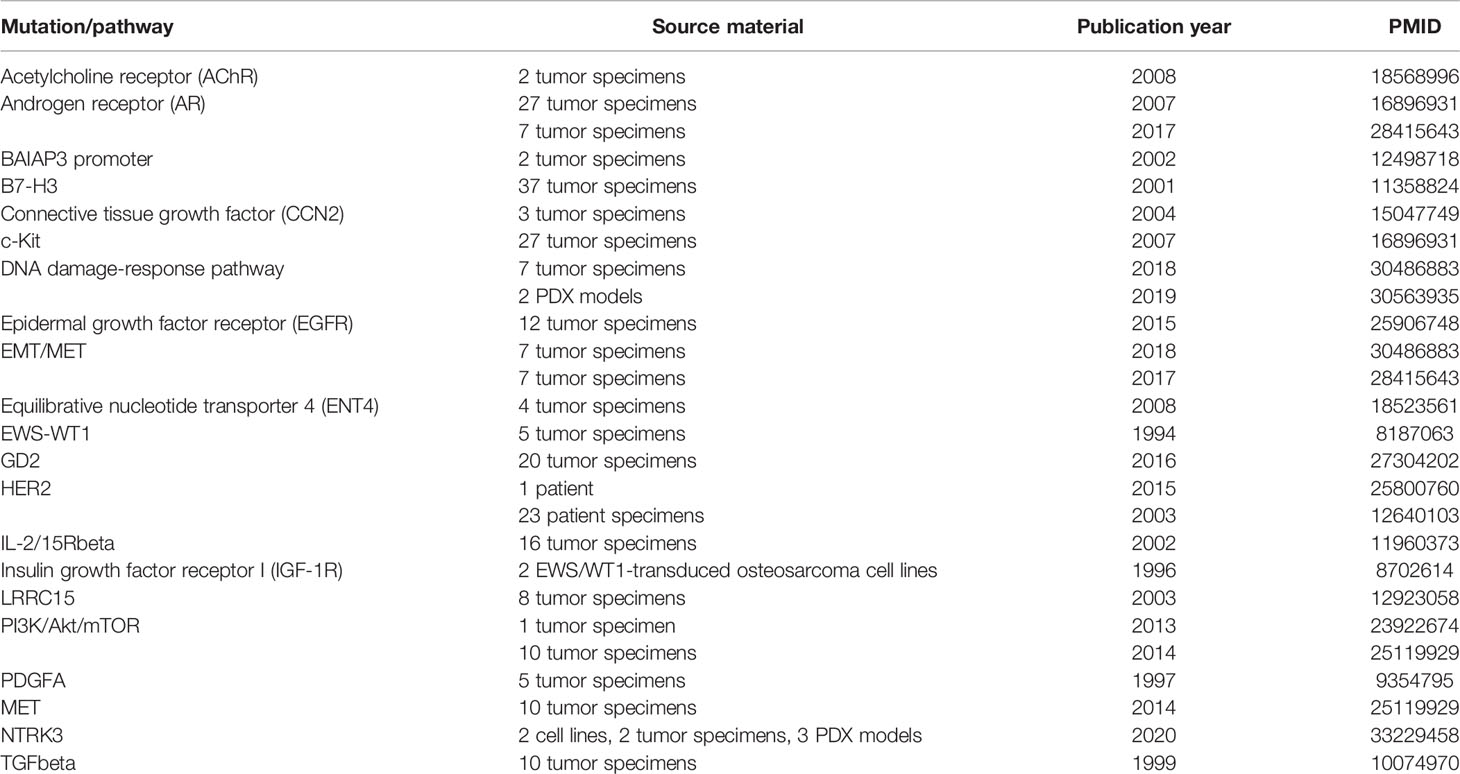
Table 2 Mutations and dysregulated pathways in DSRCT.
Platelet-Derived Growth Factor A
One of the most well-characterized gene targets of EWS-WT1 is PDGFA, whose role in DSRCT was first described by Haber and colleagues in 1997, just a few years after the identification of DSRCT as a distinct malignancy (23). This group found that PDGFA was upregulated in an osteosarcoma cell line following induced expression of EWS-WT1 (23). Additionally, PDGFA expression was found in 13/14 DSRCT tumor specimens and correlated with expression of EWS-WT1 (23). These results and subsequent publications (24, 25) confirming the role of PDGFA in DSRCT laid the groundwork for clinical trials using imatinib mesylate, a tyrosine kinase inhibitor that targets abl, c-Kit, and PDGF receptor (PDGFR) and is known for its use in the treatment of chronic myelogenous leukemia (26). Unfortunately none of these trials have shown any efficacy in DSRCT (27–29).
Insulin-Like Growth Factor 1 Receptor (IGF-1R)
Another target of EWS-WT1 is IGF-1R, a tyrosine kinase receptor that is frequently upregulated in cancer cells, leading to dysregulation of the IGF pathway (30). There are several reports of DSRCT patients presenting with severe hypoglycemia as a result of an elevated IGF-II : IGF-I ratio, a consequence of IGF pathway dysregulation (31, 32). Several IGF inhibitors have been tested in DSRCT patients in early phase clinical trials. In a phase II trial of 16 DSRCT patients treated with ganitumab [monoclonal antibody (mAb) IGF-1R inhibitor], 25% (4/16) achieved clinical benefit (PR + SD ≥ 24 weeks) (33). In another study combining cixutumumab (mAb IGF-1R inhibitor) and temsirolimus (mTOR inhibitor) in 20 patients with Ewing’s sarcoma family tumors, two DSRCT patients achieved partial tumor regression and one progressed (34).
Androgen Receptor (AR)
Because DSRCT predominantly affects male patients, it has been hypothesized that androgen receptor (AR) could play a role in this disease. In one study of 27 patients with end-stage DSRCT, 10 (37%) had tumors that were positive for AR by IHC (35). Among the 10 AR-positive patients, 6 had been treated with combined androgen blockade (CAB). Three responded, with either stable disease or a reduction in tumor burden, though these responses were not durable, lasting only 3-4 months. The patients who responded to CAB had normal testosterone levels, whereas the patients who did not respond had castrate levels of testosterone, which could explain the lack of efficacy of CAB (35). A subsequent study found that AR-positive tumors were enriched for markers of stemness, which could also partially explain why the response to CAB in AR-positive patients with normal testosterone levels was short-lived (36).
In addition to the targets above, numerous other potential targets have been identified and are in various stages of pre-clinical investigation (Table 2). These include pathways common to many types of cancer (DNA damage-response pathway, epithelial-mesenchymal transition) as well as receptors that have been identified as overexpressed or activated specifically in DSRCT as targets of the EWS-WT1 transcription factor (LRRC15, NTRK3).
Targets for Immunotherapy
A suitable target for immunotherapy must be expressed consistently on tumor cells but be restricted in its normal tissue expression to prevent on-target, off-tumor toxicity. Several appropriate targets have been identified in DSRCT. B7-H3 (targeted by the monoclonal antibody 8H9) is expressed in almost all DSRCT (35/37 tumors in one study (37), 44/46 in another) (38). It is tightly regulated by microRNA-29; despite universal transcription in most normal tissues, protein expression is highly restricted (39). Its hepatic expression explained the liver sequestration after intravenous injection, forcing its clinical development into compartmental administrations (NCT00582608). A clinical trial using intraperitoneal compartmental RIT using 131I-8H9 in DSRCT is ongoing (NCT01099644). Two studies have found GD2 expression in DSRCT, albeit to varying degrees (32/36 in one study (38), 2/20 in another.) (40) A T-cell engaging bispecific antibody (BsAb) trial is currently open for GD2(+) DSRCT (NCT03860207). EGFR, which is frequently mutated or overexpressed in cancer, was found to be amplified in 2/10 (20%) DSRCT tumors (41), and an EGFR CAR T cell trial for children and young adults with refractory/recurrent solid tumors (including DSRCT) is currently underway (NCT03618381). Another phase I CAR T cell trial was carried out for patients with HER2-positive sarcomas and the one DSRCT patient included achieved stable disease for 14 months (42). In a study assessing expression of various markers in DSRCT, HER2 was found to be expressed in 7/18 tumors, albeit at a low level for the majority of the positive-staining tumors (43). The use of these targets for immunotherapy in DSRCT is discussed in more detail below.
Immunotherapy and Radioimmunotherapy for DSRCT
Although there has been a relatively recent resurgence in interest in cancer immunotherapy, the practice of using immunomodulatory agents to activate an anti-tumor immune response is over a hundred years old (44). Immunotherapy holds the potential for durable responses and even cures of metastatic disease. Immune cells activated against the tumor can seek out and destroy micrometastases and prevent recurrence. This type of treatment is particularly useful in DSRCT because of the extensive intraperitoneal seeding that typically has occurred at the time of diagnosis. While it is practically impossible to surgically remove every tumor cell when there are often dozens or hundreds of tumor implants on the peritoneum, removing all visible tumors surgically and following up with consolidation treatment to “clean up” leftover tumor cells is a viable strategy already in use for DSRCT in the form of HIPEC and abdominal radiation (14, 15, 17).
Another way in which immunotherapy is particularly well-suited for DSRCT and other cancers that affect mainly children and young adults is the lack of long-term adverse effects, or at least side effects that do not overlap with classic genotoxic chemoradiotherapy. Because these patients are treated early in life, any lasting adverse effects of treatment have the potential to impact them for decades. Furthermore, some treatments, including cranial radiation and intrathecal chemotherapy used to treat CNS metastases, have the potential to cause cognitive deficits and endocrinopathies (45). Although the wide-spread use of immunotherapy in children and young adults is relatively new, it also appears to be generally safe, though long-term follow-up will be required to identify any late adverse effects.
Because of these enormous potential benefits, various types of immunotherapy are being investigated for DSRCT. A summary of current clinical trials involving immunotherapy for DSRCT, and their molecular targets can be found in Table 3. Cellular targets that have been used for immunotherapy in other types of cancer have been identified on DSRCT tumors. For example, GD2 is reportedly expressed on the surface of DSRCT and is the target of a clinical trial (NCT00445965) employing the radionuclide-conjugated anti-GD2 antibody 131I-3F8 for patients with DSRCT and other tumors that either spread to or originate in the CNS. Neuroblastoma cells uniformly express GD2 and the use of monoclonal antibody therapy in high-risk neuroblastoma has been particularly encouraging (46).
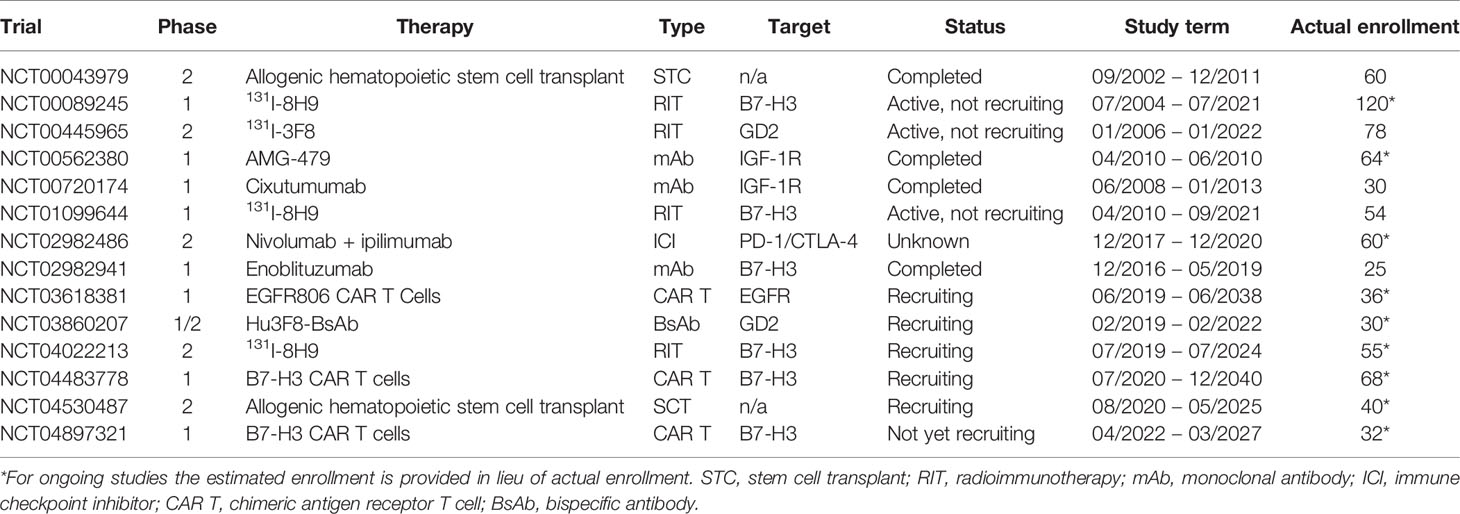
Table 3 Previous and current clinical trials using immunotherapy to treat DSRCT.
Antibody-Based Therapies
Several types of antibody-based therapies are used in the treatment of cancer, all of which rely on the specificity of an antibody-antigen interaction and require an appropriate tumor antigen to be identified that is expressed highly in tumor tissue and not in normal tissue. The most basic antibodies are monoclonal antibodies directed against a tumor antigen and designed to engage effectors cells through their Fc receptors, to mediate antibody-dependent, cell-mediated cytotoxicity (ADCC) either by natural killer cells (NK-ADCC) or by myeloid cells such as neutrophils and macrophages (myeloid-ADCC). In some tumors (e.g. neuroblastoma), complement-mediated cytotoxicity (CDC) can also play a role in the anti-tumor response. Enoblituzumab specific for B7-H3, is being tested in a phase I clinical trial in children and young adults with B7-H3-expressing relapsed/refractory solid tumors including DSRCT (NCT02982941). Enoblituzumab is also being tested in combination with checkpoint inhibitors pembrolizumab (NCT02475213) and ipilimumab (NCT02381314) among patients with B7-H3-expressing tumors.
There is also an ongoing trial to test the efficacy of a T cell-engaging BsAb in patients with advanced GD2+ tumors including DSRCT (NCT03860207). These BsAb are wide-ranging in structure, with one or more domains designed to engage T cells (usually targeting CD3) and one or more domains specific for a tumor antigen and serve as a sort of link between T cells and tumor cells, turning polyclonal T cells into specific killers. The advantage of BsAb over traditional antibody therapy is that they can initiate a robust T cell response against tumors at comparatively very low antibody concentrations and low target density. So far just one BsAb, blinatumomab, which targets CD3 and CD19, has been approved for use in cancer, in this case for ALL (47). Many others are in phase I-II trials, but only in adults (48).
Additionally, tumor antigen-specific antibodies can be used as vehicles to deliver radionuclide conjugates selectively to tumors in a process called radioimmunotherapy (RIT). This type of therapy has been successful clinically and two drugs have so far been approved for non-Hodgkin lymphoma (49). Currently, two radionuclide-conjugated antibodies are in trials for DSRCT: 131I-3F8, directed against GD2, which has previously been tested in medulloblastoma (50) and as a diagnostic tool in neuroblastoma (51), and 131I-8H9, directed against B7-H3, which has been previously tested in neuroblastoma (52). A recent Phase I trial (NCT01099644) of intraperitoneal 131I-8H9 in 48 DSRCT patients and four patients with other B7-H3-positive sarcomas found the treatment to be safe and well-tolerated with no dose-limiting toxicities (53). A Phase II trial for DSRCT is now accruing patients (NCT04022213).
CAR T Cells
In addition to using bi-specific antibodies to direct T cells to tumors, it is also possible to redirect T cells by genetically engineering them to express receptors specific for more classic antibody targets, i.e. not T cell receptor (TCR) peptide-MCH targets. These engineered cells are termed chimeric antigen receptor (CAR) T cells. In the past two years, two CAR T cell therapies have been approved by the FDA for treating CD19-expressing cancers based on clinical trials that showed they can induce complete responses in a significant proportion of patients (54, 55). Although current success with CAR T cell therapy has been largely limited to liquid tumors, clinical trials for solid tumors, including DSRCT and other sarcomas, are ongoing (56). HER2-CAR T cells were used in a phase I/II trial of sarcoma patients (including one with DSRCT) because evidence shows that HER2 is expressed at low levels by many types of sarcoma, including DSRCT (43, 57). While this low level of expression precludes immunotherapy with IgG monoclonal antibodies, CAR T cell therapy could be effective (57). This trial showed that HER2-CAR T cells delivered at low doses were safe, tolerable, and effective in maintaining stable disease in a subset of patients, with one DSRCT patient stable for >14 months (42). Phase I trials testing CAR T cells targeting EGFR (NCT03618381) and B7-H3 (NCT04483778, NCT04897321) are currently underway for patients with solid tumors including DSRCT.
Cancer Vaccines
It has been proposed that chromosomal translocation products can serve as tumor-specific antigens in sarcomas including DSRCT (58). This occurs when mutant proteins are processed by proteasomes and displayed as peptides on MHC molecules on the surface of tumor cells, as was demonstrated with mutant p53 (59, 60). A 9-amino acid epitope from the EWS-WT1 fusion protein has been described that binds to HLA-A3, providing the potential target for cytotoxic T cells (61). In one study among sarcoma patients, 2 patients with DSRCT were treated with autologous lymphocytes, tumor-pulsed dendritic cells and IL-7; unfortunately, neither had any measurable response (62). In another WT1 peptide vaccine trial among a group of 20 children with glioma, rhabdomyosarcoma, neuroblastoma, osteosarcoma, and clear cell sarcoma of the kidney (63), WT1-specific immune response was demonstrated in 4/18 (22%) evaluable patients (63). Although no DSRCT patients were included in this study, these results may provide a rationale for using peptide vaccines in DSRCT and other tumors with chromosomal translocation products that contain WT1.
Hematopoietic Stem Cell Transplantation
Allogenic stem cell transplants (SCT) have been transformative in the treatment of hematological cancers. Although intense pre-transplant chemotherapy drastically reduces tumor burden, it rarely eliminates all tumor cells from the marrow compartment (64). Rather, a graft-versus-tumor effect is probably responsible for long term tumor control (65, 66). In this way, SCT is a form of cancer immunotherapy. Allogenic SCT has been shown to increase survival in advanced DSRCT (11.4 months vs. 1.9 months) (19). Autologous SCT has also been used but does not appear to be effective in increasing survival or reducing recurrence (18, 67). Yet, one DSRCT patient treated with CD34+ selected peripheral blood stem cells (PBSC) following surgical resection and high-dose chemotherapy, has maintained remission for over 10 years (68).
Checkpoint Inhibitors
The field of immunotherapy was revolutionized by the advent of immune checkpoint inhibitors, drugs meant to release the “brakes” on T cells to reboot or to recruit T cells in their anti-tumor immune responses. Drugs targeting CTLA4 (ipilimumab) and PD-1 (programmed cell death protein 1) (nivolumab and pembrolizumab) have proven effective in treating melanoma, and, most excitingly, many of the patients who respond are alive years after treatment, suggesting that their remission is durable (69). Unfortunately, DSRCT, like sarcomas in general, do not respond well to checkpoint inhibition (36, 70). Typical among pediatric cancers driven by gene fusion, DSRCT has low tumor mutation burden, and hence few neoepitopes and insufficient anti-tumor T cell clonal frequencies (71). Tumor mutation burden, which often also correlates with a low degree of lymphocyte infiltration, is a predictor of response to checkpoint inhibitors (72). In contrast to melanoma and other tumors that are responsive to checkpoint inhibition, DSRCT appears to be an immunologically “cold” tumor, that is to say it is not heavily populated by tumor infiltrating lymphocytes (TILs) (73). DSCRT has low or no expression of PD-1/PD-L1 (programmed death ligand 1), suggesting that signaling along this axis is uncommon (41, 74). Put simply, removing the “brakes” from T cells is only effective if the host already has T cells capable of recognizing tumor epitopes just waiting to be rebooted, or if the naive T cells can be recruited to go after neoepitopes. Despite their failure as monotherapies in DSRCT, they could still hold promise when combined with other forms of immunotherapy, such as CAR T cells, cancer vaccines, and BsAb (75–77).
Challenges in Immunotherapy for DSRCT
Low mutation load is now a well-recognized Achilles heel for most solid tumors in children and adolescents/young adults (71). This partly explained the paucity of TILs, which is made worse by the upfront use of aggressive chemotherapy, adopted as the standard of care from the time of diagnosis (6, 78). While this chemotherapy often succeeds at reducing tumor burden, it also depletes the patient’s immune cells, which may lead to depletion of effector cells, and especially T cells, thereby reducing the efficacy of immunotherapies (78).
Another challenge lies in achieving sufficient concentrations of antibody or large proteins in the tumor. When delivered intravenously, it may be difficult to achieve therapeutic doses of antibodies without on-target, off-tumor toxicity and other adverse effects. One solution for this is to deliver the immunotherapeutic agent regionally or to a specific biological compartment (56). Currently, a clinical trial (NCT00445965) is underway to test the efficacy of intrathecal delivery of the radiolabeled antibody 131I-3F8 to DSRCT and other tumors that have spread to or originated in the brain. Another clinical trial (NCT04022213) is in progress to determine the efficacy of 131I-8H9 delivered intraperitoneally for DSRCT and other solid tumors which metastasize within the peritoneum. Regional delivery has also been tested in radioimmunotherapy of neuroblastoma that has spread to the CNS, and even CAR T cells in ovarian cancer (52, 56, 79). These delivery methods aim to allow the treatment to reach sufficient levels in and around the tumor while protecting other normal tissues from the deleterious effects of high concentrations of these immunotherapeutic agents. Given the peripatetic presence across all tissues, T cells have the capability to penetrate deep into tissues unlike antibodies. Hence BsAbs that arm T cells either in vivo or ex vivo may be able to overcome penetration hurdles.
Although speculative, regional or compartmental delivery of BsAb may reduce on-target, off-tumor effects, which can be life-threatening. The most common adverse effect experienced by patients in these T cell-based therapy is cytokine release syndrome, which occurs when large numbers of T cells become activated and release cytokines such as IFN-y, GM-CSF, IL-10, and IL-6 (80). This occurs because of immediate contact between active T cells with targets in the hematogenous compartment (e.g. CD19), or direct activation of T cells even before arriving at the tumor site. The effects of this “cytokine storm” range from mild to severe, and can cause fever, fatigue, pain, nausea, hypotension, and organ failure (80). One patient treated with 1 × 1010 HER2 CAR T cells experienced respiratory distress within minutes of the infusion and died 5 days later (81). It is thought that the CAR T cells localized to her lungs and became active upon recognition of low levels of HER2 expressed in lung tissue (81). However, patients have safely been treated with HER2 CAR T cells at much lower doses, including one DSRCT patient treated with 1 × 107/m2 cells (42). Such life-threatening complications can potentially be avoided if the BsAbs are first administered into a “cold” compartment to bind to tumors before TILs arrive.
Finally, it is well known that tumors evolve resistance to treatments by downregulating expression of target molecules, and by upregulating expression of immune checkpoint molecules. In the context of immunotherapy, this is called immune escape (82). It has been demonstrated that tumors upregulate PD-1 and CD47 in response to treatment with BsAbs (77, 83). It has also been shown that tumors are capable of downregulating the targets of CARs, evidenced by the fact that 11% of B-cell malignancy patients who initially responded to treatment with CD19 CAR T cells eventually relapsed with CD19- tumors (84). One potential strategy to combat this phenomenon is to develop treatments that target multiple tumor antigens, including bispecific CAR T cells and T cell engaging antibodies with more than one tumor-targeting domain (82).
Discussion
Overall cancer death rates have steadily declined over the past several decades in the United States, owing to prevention, early detection, and advances in treatment (85). Deaths from childhood cancers have also declined, though this is primarily due to the availability of better treatments, since childhood cancers (which are rarely caused by infection or exposure to carcinogens) can only rarely be prevented (86, 87). Furthermore, while the adult cancer death rate has been improved by efforts to regularly screen at-risk populations (through cervical cancer screening, mammograms, and colonoscopies, for example), for childhood cancers including DSRCT it is not feasible to implement any screening programs because no at-risk populations have been identified and no suitable screening tests exist to implement on a population-wide scale (88–90). For these reasons, efforts to reduce deaths from DSRCT and other childhood cancers can only be focused on improving treatments. While some novel approaches, such as the use of HIPEC, could delay progression, few have yielded few long-term DSRCT survivors who can be considered cured (6, 14). Immunotherapy and radioimmunotherapy are attractive options for DSRCT because they have the potential to produce durable responses without long-term side effects.
So far most of the reports on immunotherapy success in DSRCT are anecdotal. However, it is useful to learn from the successful implementation of immunotherapy in other pediatric solid tumors, like high-risk metastatic neuroblastoma which was incurable three decades ago. As mentioned previously, GD2-targeted immunotherapy has revolutionized treatment of high-risk neuroblastoma even when recurrent (46). Beginning with murine monoclonal antibodies, anti-GD2 treatment has evolved into chimeric and humanized monoclonal antibodies, and trials are underway testing CAR T cells (NCT03294954), an antibody vaccine(NCT00911560), and BsAb therapy (NCT03860207) (46). Though decades behind neuroblastoma, DSRCT could benefit from similar types of immunotherapies.
While DSRCT and other immunologically “cold” tumors don’t have de novo anti-tumor immune responses, passive immunotherapy, such as monoclonal antibodies, may still be effective. The success of monoclonal antibody therapy in neuroblastoma and leukemia, even in patients who are immunocompromised by prior chemoradiotherapy, suggests that these passive immunotherapeutic approaches could be successfully implemented even after receiving dose-intensive therapies among DSRCT patients. In fact, a monoclonal antibody against B7-H3 (NCT02982941) and two radionuclide-labeled monoclonal antibodies against B7-H3 (NCT04022213) and GD2 (NCT00445965) are currently being tested in clinical trials open to patients with DSRCT.
Immunotherapy may also be made more efficacious for DSRCT by compartmental delivery to the peritoneum. While DSRCT is often advanced at diagnosis, in a substantial proportion of patients their disease is contained within the peritoneum. By delivering the immunotherapeutic agent intraperitoneally, the concentration of the agent in and around the tumor could be substantially higher compared to systemic delivery. The likelihood of adverse on-target, off-tumor effects would also likely be reduced for tissues expressing the target but located outside the peritoneal cavity. In this way, the impact of the agent on the tumors would be maximized while the impact of the agent on normal tissue would be minimized. Intraperitoneal delivery of CAR T cells has proven superior to systemic delivery in an animal model of metastatic peritoneal colorectal cancer (91) and this strategy is being implemented in clinical trials for ovarian cancer (NCT02498912), peritoneal mesothelioma (NCT03608618) and advanced gastric cancer (NCT03563326) with peritoneal spread. When DSRCT does spread beyond the peritoneum, it often metastasizes to the CNS, which is walled off by the blood brain barrier from most treatments that are delivered parenterally. To reach these tumors treatment must be delivered intrathecally or intraparenchymally using catheters connected to an Ommaya reservoir. A clinical trial testing intrathecal 131I-3F8 (NCT00445965) is ongoing for patients with CNS tumors, including metastatic DSRCT.
Another challenge to consider which may inform the future direction of immunotherapy in DSRCT is antigen heterogeneity and antigen loss, a process in which tumors lose expression of antigens targeted by immunotherapy and thereby “escape” immune surveillance. This is now a well-known mechanism of resistance in CD19 CAR T cells, where recurrent leukemia cells no longer express CD19 following treatment. A proposed solution for this problem is to target two antigens instead of one, since the probability of two distinct mutations arising in the tumor that would downregulate both targets and allow for antigen escape during treatment is statistically unlikely (92). Since DSRCT expresses several tumor-associated targets, they should be suitable for this dual targeting approach. Targeting multiple antigens would also increase efficacy of immunotherapy in tumors where the expression of targets is heterogenous, where no single target is expressed uniformly on every tumor cell. Radionuclide-conjugated antibodies offer a similar benefit, as they can kill not only the tumor cells that express their antibody targets but other tumor cells in the vicinity. This killing is achieved by way of cross-firing during radioactive decay by high energy particles such as electrons and positrons that travel millimeter distances, and alpha particles that travel micron distances. By combining antibody formats that target multiple targets with novel radioimmunotherapy platforms, the potential exists for designing a radioimmunotherapy strategy for DSRCT, which is well-known to be radiation-responsive.
In addition to the scientific challenges confronting immunotherapy approaches, it is important to take into consideration the logistical hurdles, including the cost of these therapies and the accessibility for the general public world-wide. These issues have surfaced quickly in the field of CAR T cells, which must be tailor-made for each patient using ex vivo T cell expansion and viral transduction. In the case of Kymriah and Yescarta, the two FDA-approved CAR T cell therapies, the drug cost is currently $475,000 and $373,000 per patient, respectively (93). These prices make them inaccessible for many patients, even those in developed countries. In addition to the cost, the treatment can take several weeks to prepare for each individual patient. Although these therapies have undoubtedly saved lives, the fact remains that they are time-consuming and expensive to produce. While antibody therapy is less personalized and therefore does not have to be tailored made for each patient, it is still expensive in the current market. In the United States, a course of treatment with dinutuximab, an anti-GD2 monoclonal antibody, for example, can cost upwards of $150,000 for the drug only, and the only BsAb approved for cancer, blinatumomab, costs over $170,000 per year. The cost of these revolutionary treatments needs to be considered when evaluating the success of immunotherapy, considering that patients cannot benefit from drugs they cannot access.
Until recently, the improvements in survival for DSRCT have been incremental, owing mostly to the use of multi-agent chemotherapy. These combinations of drugs are highly toxic, have debilitating side effects, and only serve to prolong survival by a few months or years, at best. Today, there are numerous ongoing trials of various types of immunotherapy for DSRCT patients, including a BsAb, radionuclide-conjugated antibodies, a monoclonal antibody, CAR T cells, and checkpoint inhibitors (Table 3). New clinical studies aiming to collate information on clinical attributes (NCT04690374) and immune characteristics (NCT03967834) of patients with DSRCT will improve our knowledge this rare disease. Immunotherapy offers the promise of eradicating chemotherapy-resistant, microscopic tumors that cause relapse and eventually death in a majority of DSRCT patients, without the same adverse long-term effects encountered by genotoxic therapies. It is instructive to look at the successes of immunotherapy in other diseases, such as neuroblastoma and leukemia, which have seen children with relapsed tumors cured for the first time by antibody therapy and CAR T cells. At the same time, it is important to learn from the challenges experienced in the development and evolution of these treatments so that they may be implemented most efficiently and effectively in DSRCT.
Author Contributions
N-KC and ME-C both wrote and edited this manuscript. All authors contributed to the article and approved the submitted version.
Funding
The authors acknowledge support of the NCI Cancer Center Support Grant P30 CA008748, NCI Predoctoral to Postdoctoral Fellow Transition Award K00 CA223062, and support from the following donors and organizations: Runway Heroes, Renaissance Charitable Foundation, Tay-Bandz, Inc., The Steven Vanover Foundation, and Brenda D. Vanover.
Conflict of Interest
N-KC reports receiving commercial research grants from Y-mAbs Therapeutics and Abpro-Labs Inc.; holding ownership interest/equity/options in Y-mAbs Therapeutics Inc., and in Abpro-Labs, and owning stock options in Eureka Therapeutics. N-KC is the inventor of pending and issued patents filed by MSK, including hu3F8 and 8H9 licensed to Y-mAbs Therapeutics, beta-glucan to Biotec Pharmacon, and HER2 bispecific antibody to Abpro-labs. N-KC is an advisory board member for Abpro-Labs and Eureka Therapeutics.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Gerald WL, Rosai J. Case 2. Desmoplastic Small Cell Tumor With Divergent Differentiation. Pediatr Pathol (1989) 9:177–83. doi: 10.3109/15513818909022347
2. Gerald WL, Miller HK, Battifora H, Miettinen M, Silva EG, Rosai J. Intra-Abdominal Desmoplastic Small Round-Cell Tumor. Report of 19 Cases of a Distinctive Type of High-Grade Polyphenotypic Malignancy Affecting Young Individuals. Am J Surg Pathol (1991) 15:499–513. doi: 10.1097/00000478-199106000-00001
3. Sawyer JR, Tryka AF, Lewis JM. A Novel Reciprocal Chromosome Translocation T(11;22)(P13;Q12) in an Intraabdominal Desmoplastic Small Round-Cell Tumor. Am J Surg Pathol (1992) 16:411–6. doi: 10.1097/00000478-199204000-00010
4. Rodriguez E, Sreekantaiah C, Gerald W, Reuter VE, Motzer RJ, Chaganti RS. A Recurring Translocation, T(11;22)(P13;Q11.2), Characterizes Intra-Abdominal Desmoplastic Small Round-Cell Tumors. Cancer Genet Cytogenet (1993) 69:17–21. doi: 10.1016/0165-4608(93)90105-U
5. Ladanyi M, Gerald W. Fusion of the EWS and WT1 Genes in the Desmoplastic Small Round Cell Tumor. Cancer Res (1994) 54:2837–40.
6. Hayes-Jordan A, LaQuaglia MP, Modak S. Management of Desmoplastic Small Round Cell Tumor. Semin Pediatr Surg (2016) 25:299–304. doi: 10.1053/j.sempedsurg.2016.09.005
7. Lettieri CK, Garcia-Filion P, Hingorani P. Incidence and Outcomes of Desmoplastic Small Round Cell Tumor: Results From the Surveillance, Epidemiology, and End Results Database. J Cancer Epidemiol (2014) 2014:680126. doi: 10.1155/2014/680126
8. Gerald WL, Ladanyi M, de Alava E, Cuatrecasas M, Kushner BH, LaQuaglia MP, et al. Clinical, Pathologic, and Molecular Spectrum of Tumors Associated With T(11;22)(P13;Q12): Desmoplastic Small Round-Cell Tumor and its Variants. J Clin Oncol (1998) 16:3028–36. doi: 10.1200/jco.1998.16.9.3028
9. Worch J, Cyrus J, Goldsby R, Matthay KK, Neuhaus J, DuBois SG. Racial Differences in the Incidence of Mesenchymal Tumors Associated With EWSR1 Translocation. Cancer Epidemiol Biomarkers Prev (2011) 20:449–53. doi: 10.1158/1055-9965.Epi-10-1170
10. Lal DR, Su WT, Wolden SL, Loh KC, Modak S, La Quaglia MP. Results of Multimodal Treatment for Desmoplastic Small Round Cell Tumors. J Pediatr Surg (2005) 40:251–5. doi: 10.1016/j.jpedsurg.2004.09.046
11. Kushner BH, LaQuaglia MP, Wollner N, Meyers PA, Lindsley KL, Ghavimi F, et al. Desmoplastic Small Round-Cell Tumor: Prolonged Progression-Free Survival With Aggressive Multimodality Therapy. J Clin Oncol (1996) 14:1526–31. doi: 10.1200/JCO.1996.14.5.1526
12. Whelan J, Khan A, Sharma A, Rothermundt C, Dileo P, Michelagnoli M, et al. Interval Compressed Vincristine, Doxorubicin, Cyclophosphamide Alternating With Ifosfamide, Etoposide in Patients With Advanced Ewing's and Other Small Round Cell Sarcomas. Clin Sarcoma Res (2012) 2:12. doi: 10.1186/2045-3329-2-12
13. Hayes-Jordan AA, Coakley BA, Green HL, Xiao L, Fournier KF, Herzog CE, et al. Desmoplastic Small Round Cell Tumor Treated With Cytoreductive Surgery and Hyperthermic Intraperitoneal Chemotherapy: Results of a Phase 2 Trial. Ann Surg Oncol (2018) 25:872–7. doi: 10.1245/s10434-018-6333-9
14. Hayes-Jordan A, Green HL, Lin H, Owusu-Agyemang P, Fitzgerald N, Arunkumar R, et al. Complete Cytoreduction and HIPEC Improves Survival in Desmoplastic Small Round Cell Tumor. Ann Surg Oncol (2014) 21:220–4. doi: 10.1245/s10434-013-3269-y
15. Goodman KA, Wolden SL, La Quaglia MP, Kushner BH. Whole Abdominopelvic Radiotherapy for Desmoplastic Small Round-Cell Tumor. Int J Radiat Oncol Biol Phys (2002) 54:170–6. doi: 10.1016/S0360-3016(02)02871-7
16. Honore C, Atallah V, Mir O, Orbach D, Ferron G, LePechoux C, et al. Abdominal Desmoplastic Small Round Cell Tumor Without Extraperitoneal Metastases: Is There a Benefit for HIPEC After Macroscopically Complete Cytoreductive Surgery? PloS One (2017) 12:e0171639. doi: 10.1371/journal.pone.0171639
17. Desai NB, Stein NF, LaQuaglia MP, Alektiar KM, Kushner BH, Modak S, et al. Reduced Toxicity With Intensity Modulated Radiation Therapy (Imrt) for Desmoplastic Small Round Cell Tumor (Dsrct): An Update on the Whole Abdominopelvic Radiation Therapy (Wap-Rt) Experience. Int J Radiat Oncol Biol Phys (2013) 85:e67–72. doi: 10.1016/j.ijrobp.2012.09.005
18. Forlenza CJ, Kushner BH, Kernan N, Boulad F, Magnan H, Wexler L, et al. Myeloablative Chemotherapy With Autologous Stem Cell Transplant for Desmoplastic Small Round Cell Tumor. Sarcoma (2015) 2015:269197. doi: 10.1155/2015/269197
19. Baird K, Fry TJ, Steinberg SM, Bishop MR, Fowler DH, Delbrook CP, et al. Reducedintensity Allogeneic Stem Cell Transplantation in Children and Young Adults With Ultrahigh-Risk Pediatric Sarcomas. Biol Blood Marrow Transplant (2012) 18:698–707. doi: 10.1016/j.bbmt.2011.08.020
20. Gerald WL, Haber DA. The EWS-WT1 Gene Fusion in Desmoplastic Small Round Cell Tumor. Semin Cancer Biol (2005) 15:197–205. doi: 10.1016/j.semcancer.2005.01.005
21. Gessler M, Poustka A, Cavenee W, Neve RL, Orkin SH, Bruns GA. Homozygous Deletion in Wilms Tumours of a Zinc-Finger Gene Identified by Chromosome Jumping. Nature (1990) 343:774–8. doi: 10.1038/343774a0
22. Rauscher FJ 3rd. The Wt1 Wilms Tumor Gene Product: A Developmentally Regulated Transcription Factor in the Kidney That Functions as a Tumor Suppressor. FASEB J (1993) 7:896–903. doi: 10.1096/fasebj.7.10.8393820
23. Lee SB, Kolquist KA, Nichols K, Englert C, Maheswaran S, Ladanyi M, et al. The EWSWT1 Translocation Product Induces PDGFA in Desmoplastic Small Round-Cell Tumour. Nat Genet (1997) 17:309–13. doi: 10.1038/ng1197-309
24. Froberg K, Brown RE, Gaylord H, Manivel C. Intra-Abdominal Desmoplastic Small Round Cell Tumor: Immunohistochemical Evidence for Up-Regulation of Autocrine and Paracrine Growth Factors. Ann Clin Lab Sci (1999) 29:78–85.
25. Zhang PJ, Goldblum JR, Pawel BR, Pasha TL, Fisher C, Barr FG. PDGF-a, Pdgfrbeta, Tgfbeta3 and Bone Morphogenic Protein-4 in Desmoplastic Small Round Cell Tumors With EWS-WT1 Gene Fusion Product and Their Role in Stromal Desmoplasia: An Immunohistochemical Study. Mod Pathol (2005) 18:382–7. doi: 10.1038/modpathol.3800264
26. Druker BJ, Talpaz M, Resta DJ, Peng B, Buchdunger E, Ford JM, et al. Efficacy and Safety of a Specific Inhibitor of the BCR-ABL Tyrosine Kinase in Chronic Myeloid Leukemia. N Engl J Med (2001) 344:1031–7. doi: 10.1056/nejm200104053441401
27. Bond M, Bernstein ML, Pappo A, Schultz KR, Krailo M, Blaney SM, et al. A Phase II Study of Imatinib Mesylate in Children With Refractory or Relapsed Solid Tumors: A Children's Oncology Group Study. Pediatr Blood Cancer (2008) 50:254–8. doi: 10.1002/pbc.21132
28. Chao J, Budd GT, Chu P, Frankel P, Garcia D, Junqueira M, et al. Phase II Clinical Trial of Imatinib Mesylate in Therapy of KIT and/or Pdgfralpha-Expressing Ewing Sarcoma Family of Tumors and Desmoplastic Small Round Cell Tumors. Anticancer Res (2010) 30:547–52.
29. De Sanctis R, Bertuzzi A, Bisogno G, Carli M, Ferrari A, Comandone A, et al. Imatinib Mesylate in Desmoplastic Small Round Cell Tumors. Future Oncol (2017) 13:1233–7. doi: 10.2217/fon-2016-0305
30. Werner H, LeRoith D. The Role of the Insulin-Like Growth Factor System in Human Cancer. Adv Cancer Res (1996) 68:183–223. doi: 10.1016/S0065-230X(08)60354-1
31. Barra WF, Castro G, Hoff AO, Siqueira SA, Hoff PM. Symptomatic Hypoglycemia Related to Inappropriately High Igf-Ii Serum Levels in a Patient With Desmoplastic Small Round Cell Tumor. Case Rep Med (2010) 2010:684045. doi: 10.1155/2010/684045
32. Almaghraby A, Brickman WJ, Goldstein JA, Habiby RL. Refractory Hypoglycemia in a Pediatric Patient With Desmoplastic Small Round Cell Tumor. J Pediatr Endocrinol Metab JPEM (2018) 31:947–50. doi: 10.1515/jpem-2018-0107
33. Tap WD, Demetri G, Barnette P, Desai J, Kavan P, Tozer R, et al. Phase II Study of Ganitumab, a Fully Human Anti-Type-1 Insulin-Like Growth Factor Receptor Antibody, in Patients With Metastatic Ewing Family Tumors or Desmoplastic Small Round Cell Tumors. J Clin Oncol (2012) 30:1849–56. doi: 10.1200/JCO.2011.37.2359
34. Naing A, LoRusso P, Fu S, Hong DS, Anderson P, Benjamin RS, et al. Insulin Growth Factor-Receptor (IGF-1R) Antibody Cixutumumab Combined With the Mtor Inhibitor Temsirolimus in Patients With Refractory Ewing's Sarcoma Family Tumors. Clin Cancer Res (2012) 18:2625–31. doi: 10.1158/1078-0432.CCR-12-0061
35. Fine RL, Shah SS, Moulton TA, Yu IR, Fogelman DR, Richardson M, et al. Androgen and C-Kit Receptors in Desmoplastic Small Round Cell Tumors Resistant to Chemotherapy: Novel Targets for Therapy. Cancer Chemother Pharmacol (2007) 59:429–37. doi: 10.1007/s00280-006-0280-z
36. Negri T, Brich S, Bozzi F, Volpi CV, Gualeni AV, Stacchiotti S, et al. New Transcriptionalbased Insights Into the Pathogenesis of Desmoplastic Small Round Cell Tumors (Dsrcts). Oncotarget (2017) 8:32492–504. doi: 10.18632/oncotarget.16477
37. Modak S, Kramer K, Gultekin SH, Guo HF, Cheung NK. Monoclonal Antibody 8H9 Targets a Novel Cell Surface Antigen Expressed by a Wide Spectrum of Human Solid Tumors. Cancer Res (2001) 61:4048–54.
38. Modak S, Gerald W, Cheung NK. Disialoganglioside GD2 and a Novel Tumor Antigen: Potential Targets for Immunotherapy of Desmoplastic Small Round Cell Tumor. Med Pediatr Oncol (2002) 39:547–51. doi: 10.1002/mpo.10151
39. Xu H, Cheung IY, Guo HF, Cheung NK. Microrna Mir-29 Modulates Expression of Immunoinhibitory Molecule B7-H3: Potential Implications for Immune Based Therapy of Human Solid Tumors. Cancer Res (2009) 69:6275–81. doi: 10.1158/0008-5472.CAN-08-4517
40. Dobrenkov K, Ostrovnaya I, Gu J, Cheung IY, Cheung NK. Oncotargets GD2 and GD3 are Highly Expressed in Sarcomas of Children, Adolescents, and Young Adults. Pediatr Blood Cancer (2016) 63:1780–5. doi: 10.1002/pbc.26097
41. Movva S, Wen W, Chen W, Millis SZ, Gatalica Z, Reddy S, et al. Multi-Platform Profiling of Over 2000 Sarcomas: Identification of Biomarkers and Novel Therapeutic Targets. Oncotarget (2015) 6:12234–47. doi: 10.18632/oncotarget.3498
42. Ahmed N, Brawley VS, Hegde M, Robertson C, Ghazi A, Gerken C, et al. Human Epidermal Growth Factor Receptor 2 (HER2) -Specific Chimeric Antigen Receptor-Modified T Cells for the Immunotherapy of HER2-Positive Sarcoma. J Clin Oncol (2015) 33:1688–96. doi: 10.1200/jco.2014.58.0225
43. Zhang PJ, Goldblum JR, Pawel BR, Fisher C, Pasha TL, Barr FG. Immunophenotype of Desmoplastic Small Round Cell Tumors as Detected in Cases With EWS-WT1 Gene Fusion Product. Mod Pathol (2003) 16:229–35. doi: 10.1097/01.Mp.0000056630.76035.F3
44. Decker WK, da Silva RF, Sanabria MH, Angelo LS, Guimaraes F, Burt BM, et al. Cancer Immunotherapy: Historical Perspective of a Clinical Revolution and Emerging Preclinical Animal Models. Front Immunol (2017) 8:829. doi: 10.3389/fimmu.2017.00829
45. Duffner PK. Long-Term Effects of Radiation Therapy on Cognitive and Endocrine Function in Children With Leukemia and Brain Tumors. Neurol (2004) 10:293–310. doi: 10.1097/01.nrl.0000144287.35993.96
46. Keyel ME, Reynolds CP. Spotlight on Dinutuximab in the Treatment of High-Risk Neuroblastoma: Development and Place in Therapy. Biologics (2019) 13:1–12. doi: 10.2147/btt.S114530
47. Curran E, Stock W. Taking a "Bite Out of ALL" - Blinatumomab Approval for MRD Positive ALL. Blood (2019) 133:1715–9. doi: 10.1182/blood-2018-12-852376
48. Sedykh SE, Prinz VV, Buneva VN, Nevinsky GA. Bispecific Antibodies: Design, Therapy, Perspectives. Drug Design Dev Ther (2018) 12:195–208. doi: 10.2147/dddt.S151282
49. Kawashima H. Radioimmunotherapy: A Specific Treatment Protocol for Cancer by Cytotoxic Radioisotopes Conjugated to Antibodies. Sci World J (2014) 2014:492061. doi: 10.1155/2014/492061
50. Kramer K, Pandit-Taskar N, Humm JL, Zanzonico PB, Haque S, Dunkel IJ, et al. A Phase II Study of Radioimmunotherapy With Intraventricular (131) I-3F8 for Medulloblastoma. Pediatr Blood Cancer (2018) 65. doi: 10.1002/pbc.26754
51. Yeh SD, Larson SM, Burch L, Kushner BH, Laquaglia M, Finn R, et al. Radioimmunodetection of Neuroblastoma With Iodine-131-3F8: Correlation With Biopsy, Iodine- 131-Metaiodobenzylguanidine and Standard Diagnostic Modalities. J Nucl Med (1991) 32:769–76.
52. Kramer K, Kushner BH, Modak S, Pandit-Taskar N, Smith-Jones P, Zanzonico P, et al. Compartmental Intrathecal Radioimmunotherapy: Results for Treatment for Metastatic Cns Neuroblastoma. J Neurooncol (2010) 97:409–18. doi: 10.1007/s11060-009-0038-7
53. Modak S, Zanzonico P, Grkovski M, Slotkin EK, Carrasquillo JA, Lyashchenko SK, et al. B7H3-Directed Intraperitoneal Radioimmunotherapy With Radioiodinated Omburtamab for Desmoplastic Small Round Cell Tumor and Other Peritoneal Tumors: Results of a Phase I Study. J Clin Oncol (2020) 38:4283–91. doi: 10.1200/jco.20.01974
54. Schuster SJ, Bishop MR, Tam CS, Waller EK, Borchmann P, McGuirk JP, et al. Primary Analysis of Juliet: A Global, Pivotal, Phase 2 Trial of CTL019 in Adult Patients With Relapsed or Refractory Diffuse Large B-Cell Lymphoma. Blood (2017) 130:577–7.
55. Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in Children and Young Adults With B-Cell Lymphoblastic Leukemia. N Engl J Med (2018) 378:439–48. doi: 10.1056/NEJMoa1709866
56. Schmidts A, Maus MV. Making CAR T Cells a Solid Option for Solid Tumors. Front Immunol (2018) 9:2593. doi: 10.3389/fimmu.2018.02593
57. Ahmed N, Salsman VS, Yvon E, Louis CU, Perlaky L, Wels WS, et al. Immunotherapy for Osteosarcoma: Genetic Modification of T Cells Overcomes Low Levels of Tumor Antigen Expression. Mol Ther (2009) 17:1779–87. doi: 10.1038/mt.2009.133
58. Goletz TJ, Mackall CL, Berzofsky JA, Helman LJ. Molecular Alterations in Pediatric Sarcomas: Potential Targets for Immunotherapy. Sarcoma (1998) 2:77–87. doi: 10.1080/13577149878037
59. Houbiers JG, Nijman HW, van der Burg SH, Drijfhout JW, Kenemans P, van de Velde CJ, et al. In Vitro Induction of Human Cytotoxic T Lymphocyte Responses Against Peptides of Mutant and Wild-Type P53. Eur J Immunol (1993) 23:2072–7. doi: 10.1002/eji.1830230905
60. Yanuck M, Carbone DP, Pendleton CD, Tsukui T, Winter SF, Minna JD, et al. A Mutant P53 Tumor Suppressor Protein is a Target for Peptide-Induced CD8+ Cytotoxic T-Cells. Cancer Res (1993) 53:3257–61.
61. Worley BS, van den Broeke LT, Goletz TJ, Pendleton CD, Daschbach EM, Thomas EK, et al. Antigenicity of Fusion Proteins From Sarcoma-Associated Chromosomal Translocations. Cancer Res (2001) 61:6868–75.
62. Merchant MS, Bernstein D, Amoako M, Baird K, Fleisher TA, Morre M, et al. Adjuvant Immunotherapy to Improve Outcome in High-Risk Pediatric Sarcomas. Clin Cancer Res (2016) 22:3182–91. doi: 10.1158/1078-0432.CCR-15-2550
63. Hirabayashi K, Yanagisawa R, Saito S, Higuchi Y, Koya T, Sano K, et al. Feasibility and Immune Response of WT1 Peptide Vaccination in Combination With OK-432 for Paediatric Solid Tumors. Anticancer Res (2018) 38:2227–34. doi: 10.21873/anticanres.12465
64. Srinivasan R, Barrett J, Childs R. Allogeneic Stem Cell Transplantation as Immunotherapy for Nonhematological Cancers. Semin Oncol (2004) 31:47–55. doi: 10.1053/j.seminoncol.2003.11.002
65. Horowitz MM, Gale RP, Sondel PM, Goldman JM, Kersey J, Kolb HJ, et al. Graftversus- Leukemia Reactions After Bone Marrow Transplantation. Blood (1990) 75:555–62. doi: 10.1182/blood.V75.3.555.555
66. Weiden PL, Flournoy N, Thomas ED, Prentice R, Fefer A, Buckner CD, et al. Antileukemic Effect of Graft-Versus-Host Disease in Human Recipients of Allogeneic-Marrow Grafts. N Engl J Med (1979) 300:1068–73. doi: 10.1056/nejm197905103001902
67. Bertuzzi A, Castagna L, Quagliuolo V, Ginanni V, Compasso S, Magagnoli M, et al. Prospective Study of High-Dose Chemotherapy and Autologous Peripheral Stem Cell Transplantation in Adult Patients With Advanced Desmoplastic Small Round-Cell Tumour. Br J Cancer (2003) 89:1159–61. doi: 10.1038/sj.bjc.6601304
68. Houet L, Moller I, Engelhardt M, Kohler G, Schmidt H, Herchenbach D, et al. Long-Term Remission After CD34+-Selected PBSCT in a Patient With Advanced Intra-Abdominal Desmoplastic Small Round-Cell Tumor. Bone Marrow Transplant (2010) 45:793–5. doi: 10.1038/bmt.2009.226
69. Lugowska I, Teterycz P, Rutkowski P. Immunotherapy of Melanoma. Contemp Oncol (Poznan Poland) (2018) 22:61–7. doi: 10.5114/wo.2018.73889
70. Tawbi HA, Burgess M, Bolejack V, Van Tine BA, Schuetze SM, Hu J, et al. Pembrolizumab in Advanced Soft-Tissue Sarcoma and Bone Sarcoma (SARC028): A Multicentre, Two-Cohort, Single-Arm, Open-Label, Phase 2 Trial. Lancet Oncol (2017) 18:1493–501. doi: 10.1016/s1470-2045(17)30624-1
71. Watson IR, Takahashi K, Futreal PA, Chin L. Emerging Patterns of Somatic Mutations in Cancer. Nat Rev Genet (2013) 14:703–18. doi: 10.1038/nrg3539
72. Chan TA, Yarchoan M, Jaffee E, Swanton C, Quezada SA, Stenzinger A, et al. Development of Tumor Mutation Burden as an Immunotherapy Biomarker: Utility for the Oncology Clinic. Ann Oncol (2019) 30:44–56. doi: 10.1093/annonc/mdy495
73. Maleki Vareki S. High and Low Mutational Burden Tumors Versus Immunologically Hot and Cold Tumors and Response to Immune Checkpoint Inhibitors. J Immunother Cancer (2018) 6:157. doi: 10.1186/s40425-018-0479-7
74. Inaguma S, Wang Z, Lasota J, Sarlomo-Rikala M, McCue PA, Ikeda H, et al. Comprehensive Immunohistochemical Study of Programmed Cell Death Ligand 1 (Pd-L1): Analysis in 5536 Cases Revealed Consistent Expression in Trophoblastic Tumors. Am J Surg Pathol (2016) 40:1133–42. doi: 10.1097/pas.0000000000000653
75. Rosewell Shaw A, Porter CE, Watanabe N, Tanoue K, Sikora A, Gottschalk S, et al. Adenovirotherapy Delivering Cytokine and Checkpoint Inhibitor Augments CAR T Cells Against Metastatic Head and Neck Cancer. Mol Ther 25:2440–51. doi: 10.1016/j.ymthe.2017.09.010 (2017
76. Kim H, Khanna V, Kucaba TA, Zhang W, Ferguson DM, Griffith TS, et al. Combination of Sunitinib and PD-L1 Blockade Enhances Anticancer Efficacy of TLR7/8 Agonist-Based Nanovaccine. Mol Pharm (2019) 16:1200–10. doi: 10.1021/acs.molpharmaceut.8b01165
77. Krupka C, Kufer P, Kischel R, Zugmaier G, Lichtenegger FS, Kohnke T, et al. Blockade of the PD-1/PD-L1 Axis Augments Lysis of AML Cells by the CD33/CD3 Bite Antibody Construct AMG 330: Reversing a T-Cell-Induced Immune Escape Mechanism. Leukemia (2016) 30:484–91. doi: 10.1038/leu.2015.214
78. Das RK, Vernau L, Grupp SA, Barrett DM. Naive T-Cell Deficits at Diagnosis and After Chemotherapy Impair Cell Therapy Potential in Pediatric Cancers. Cancer Discovery (2019) 9:492–9. doi: 10.1158/2159-8290.Cd-18-1314
79. Heiss MM, Strohlein MA, Jager M, Kimmig R, Burges A, Schoberth A, et al. Immunotherapy of Malignant Ascites With Trifunctional Antibodies. Int J Cancer (2005) 117:435–43. doi: 10.1002/ijc.21165
80. Bonifant CL, Jackson HJ, Brentjens RJ, Curran KJ. Toxicity and Management in CAR Tcell Therapy. Mol Ther Oncolytics (2016) 3:16011. doi: 10.1038/mto.2016.11
81. Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case Report of a Serious Adverse Event Following the Administration of T Cells Transduced With a Chimeric Antigen Receptor Recognizing ERBB2. Mol Ther (2010) 18:843–51. doi: 10.1038/mt.2010.24
82. Zhang X, Yang Y, Fan D, Xiong D. The Development of Bispecific Antibodies and Their Applications in Tumor Immune Escape. Exp Hematol Oncol (2017) 6:12. doi: 10.1186/s40164-017-0072-7
83. Piccione EC, Juarez S, Liu J, Tseng S, Ryan CE, Narayanan C, et al. A Bispecific Antibody Targeting CD47 and CD20 Selectively Binds and Eliminates Dual Antigen Expressing Lymphoma Cells. MAbs (2015) 7:946–56. doi: 10.1080/19420862.2015.1062192
84. Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric Antigen Receptor T Cells for Sustained Remissions in Leukemia. N Engl J Med (2014) 371:1507–17. doi: 10.1056/NEJMoa1407222
85. Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2018, Vol. 68. CA Cancer J Clin (2018). pp. 7–30. doi: 10.3322/caac.21442.
86. Smith MA, Seibel NL, Altekruse SF, Ries LA, Melbert DL, O'Leary M, et al. Outcomes for Children and Adolescents With Cancer: Challenges for the Twenty-First Century. J Clin Oncol (2010) 28:2625–34. doi: 10.1200/jco.2009.27.0421
87. Islami F, Goding Sauer A, Miller KD, Siegel RL, Fedewa SA, Jacobs EJ, et al. Proportion and Number of Cancer Cases and Deaths Attributable to Potentially Modifiable Risk Factors in the United States. CA Cancer J Clin (2018) 68:31–54. doi: 10.3322/caac.21440
88. Wiegering A, Ackermann S, Riegel J, Dietz UA, Gotze O, Germer CT, et al. Improved Survival of Patients With Colon Cancer Detected by Screening Colonoscopy. Int J Colorectal Dis (2016) 31:1039–45. doi: 10.1007/s00384-015-2501-6
89. Kaplan HG, Malmgren JA, Atwood MK, Calip GS. Effect of Treatment and Mammography Detection on Breast Cancer Survival Over Time: 1990-2007. Cancer (2015) 121:2553–61. doi: 10.1002/cncr.29371
90. Holmquist ND. Revisiting the Effect of the Pap Test on Cervical Cancer. Am J Public Health (2000) 90:620–3. doi: 10.2105/AJPH.90.4.620
91. Katz SC, Point GR, Cunetta M, Thorn M, Guha P, Espat NJ, et al. Regional CAR-T Cell Infusions for Peritoneal Carcinomatosis are Superior to Systemic Delivery. Cancer Gene Ther (2016) 23:142–8. doi: 10.1038/cgt.2016.14
92. Jackson HJ, Brentjens RJ. Overcoming Antigen Escape With CAR T-Cell Therapy. Cancer Discovery (2015) 5:1238–40. doi: 10.1158/2159-8290.Cd-15-1275
Keywords: DSRCT = desmoplastic small round cell tumor, antibodies, immunotherapy, targeted therapy, radioimmunotherapy, CAR T cell
Citation: Espinosa-Cotton M and Cheung N-KV (2021) Immunotherapy and Radioimmunotherapy for Desmoplastic Small Round Cell Tumor. Front. Oncol. 11:772862. doi: 10.3389/fonc.2021.772862
Received: 08 September 2021; Accepted: 02 November 2021;
Published: 19 November 2021.
Edited by:
Pouya Faridi, Monash University, AustraliaReviewed by:
Pingping Chen, University of Miami, United StatesTomas Sjöberg Bexelius, Karolinska Institutet (KI), Sweden
Copyright © 2021 Espinosa-Cotton and Cheung. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nai-Kong V. Cheung, Y2hldW5nbkBtc2tjYy5vcmc=