 Jordan A. Harry
Jordan A. Harry Mark L. Ormiston
Mark L. Ormiston- 1Department of Medicine, Queen’s University, Kingston, ON, Canada
- 2Department of Biomedical and Molecular Sciences, Queen’s University, Kingston, ON, Canada
- 3Department of Surgery, Queen’s University, Kingston, ON, Canada
Breast cancer is the most common cancer affecting women and is the second leading cause of cancer related death worldwide. Angiogenesis, the process of new blood vessel development from pre-existing vasculature, has been implicated in the growth, progression, and metastasis of cancer. Tumor angiogenesis has been explored as a key therapeutic target for decades, as the blockade of this process holds the potential to reduce the oxygen and nutrient supplies that are required for tumor growth. However, many existing anti-angiogenic approaches, such as those targeting Vascular Endothelial Growth Factor, Notch, and Angiopoietin signaling, have been associated with severe side-effects, limited survival advantage, and enhanced cancer regrowth rates. To address these setbacks, alternative pathways involved in the regulation of tumor angiogenesis are being explored, including those involving Bone Morphogenetic Protein-9 signaling, the Sonic Hedgehog pathway, Cyclooxygenase-2, p38-mitogen-activated protein kinase, and Chemokine Ligand 18. This review article will introduce the concept of tumor angiogenesis in the context of breast cancer, followed by an overview of current anti-angiogenic therapies, associated resistance mechanisms and novel therapeutic targets.
Introduction
Breast cancer is the most common invasive cancer affecting women. Despite an array of new therapies, diagnostic advances, and increased screening, it remains the second leading cause of cancer related death, emphasizing the critical need for new treatment options (1). The high mortality rates associated with breast cancer are due to distant metastasis from the primary breast tumor to the bones, liver, lungs, or brain (1–3). Angiogenesis, the process of new blood vessel formation from the pre-existing vasculature, is a hallmark of cancer and has been implicated in the growth, progression, and metastasis of breast cancer (4, 5). Beyond enhancing the supply of oxygen and nutrients to the tumor, angiogenesis also contributes to its metastatic potential. The fragile, tortuous, permeable and hypermalleable nature of the newly formed blood vessels allows cancer cells to enter the vasculature and travel to other tissues in the body (6, 7). These malformed vessels also contribute to a hypoxic and acidic tumor microenvironment that favours the selection of more aggressive cancer cells (8).
Initial therapeutic approaches targeting the tumor vasculature were primarily aimed at inhibiting angiogenesis, based on the mindset that preventing capillary network formation would slow tumor growth and reduce metastasis (9). However, these anti-angiogenic therapies have been limited by drug resistance and side effects, including venous thromboembolic complications, hypertension, and hemorrhaging (10–12). By reducing the blood supply to tumors, these approaches also limit the ability for chemotherapies and immune cells to infiltrate these tissues (10–12). As an alternative, therapeutic approaches based on vascular normalization have been explored, with the goal of improving tissue perfusion to provide a functional vasculature for the delivery of intravenous radiotherapy and chemotherapy (13, 14). These contrasting approaches illustrate a critical point of debate in breast cancer research; should tumor vessel formation be inhibited to prevent oxygen and nutrients from reaching primary tumors, or should the vessels be normalized to reduce metastatic spread and improve pathways for therapeutic delivery?
This review will assess the current state of both anti-angiogenic and vascular normalization strategies for the treatment of breast cancer. The various mechanisms that tumors use to develop and maintain their blood supply will be summarized, followed by a review of the pre-clinical and clinical outcomes for conventional anti-angiogenic therapies targeting well-established players in angiogenic signaling, such as Vascular Endothelial Growth Factor (VEGF), Notch signaling and the Angiopoietins (Ang). Emerging avenues for modulating tumor vascularization will also be discussed, including approaches targeting bone morphogenetic protein-9 (BMP9) signaling, Sonic Hedgehog Pathway (Shh) inhibitors, Cyclooxygenase-2 (COX-2) inhibitors, p38-mitogen-activated protein kinase (p38-MAPK) pathway inactivation and Chemokine Ligand 18 (CCL18) inhibition.
Mechanisms of Tumor Vascularization
Sprouting Angiogenesis
Sprouting angiogenesis is a primary source of new vascular growth in tumors (15). This process, which involves the growth of new capillary blood vessels from the pre-existing vasculature (Figure 1A), is highly regulated and occurs in healthy tissues through a fine balance of angiogenic and angiostatic cues (16). In tumors, changes in physiological stimuli, such as hypoxia (17), disrupt this balance and shift the process towards a pathogenic pro-angiogenic state (16). Sprouting angiogenesis is initiated by a process of growth factor-mediated endothelial cell activation, proliferation (5), loss of cell-cell junctions and decreased endothelial monolayer integrity (18). While this occurs, proteases degrade the extracellular matrix and basement membrane surrounding endothelial cells, which allows for their migration (5). Migrating endothelial cells are polarized into tip and stalk cell phenotypes, contribute to lumen formation, and eventually the establishment of immature blood vessels (5). In physiological angiogenesis, these immature vessels are then stabilized through angiostatic signals, resulting in tightened cell-cell junctions, the recruitment of mural cells and restoration of the basement membrane (19).
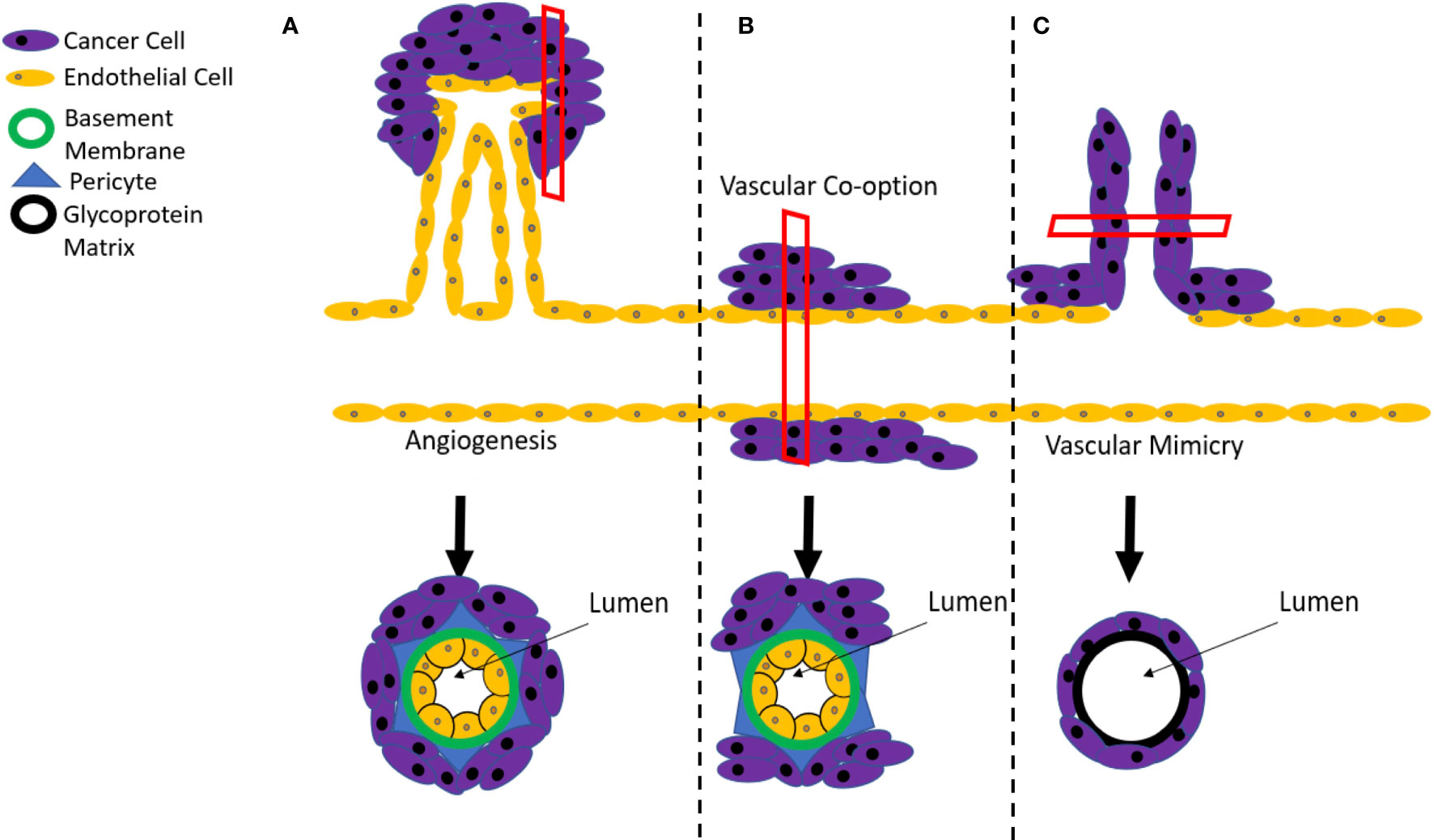
Figure 1 Mechanisms of tumor angiogenesis. (A) Tumor vascularization typically occurs by sprouting angiogenesis, involving the generation of new vessels via sprouting from the pre-existing circulation. However, resistance mechanisms to anti-angiogenic therapies can include (B) vascular co-option, where the tumor grows along existing vessels or (C) vascular mimicry where cancer stem cells create a de novo vasculature.
The Tumor Microenvironment
In tumors, pathological angiogenic sprouting is perpetuated by a local microenvironment, composed of proliferating tumor cells, blood vessels, specialized immune cells, and inflammatory cytokines (20, 21). The increased expression of angiogenic factors, inflammation, and hypoxia in the tumor microenvironment is thought to contribute to the reduced efficacy of current anti-angiogenic therapies (22, 23). As tumors grow, high levels of oxygen are consumed. This, alongside reduced nutrients and the accumulation of metabolic wastes, creates a hypoxic and acidic microenvironment that persistently activates pro-angiogenic pathways, reduces the stabilization of vessels and promotes the formation of a leaky, disorganized vascular network (24, 25). The release of mitochondrial reactive oxygen species (ROS) by tumor cells and other cellular components of the microenvironment also helps to stabilize hypoxia-inducible factor (HIF) subunits when (26–28), further promoting the expression of VEGF and upregulating angiogenesis (29, 30).
Beyond the direct actions of VEGF on the sprouting endothelium, VEGF can also serve to promote an angiogenic and immunosuppressive microenvironment through the inhibition of dendritic cell maturation and the promotion of tumor associated macrophage (TAM) recruitment and polarization in the presence of Th2 cytokines like IL-4 and IL-10 (31–33). TAMs are a major contributor to the tumor microenvironment. Their production of growth factors, chemokines and proteolytic enzymes actively promote tumor angiogenesis, while also suppressing the antitumor immune response (34–36). The recruitment and polarization of TAMs from bone marrow or tissue-derived macrophages is driven by local factors, including chemokines like CCL2 and cytokines like IL-6, as well as VEGF and hypoxia (37–39).
Vessel Co-Option
In addition to relying on sprouting angiogenesis, tumors can also be sustained through a process of vessel co-option (Figure 1B). This process involves solid tumors originating close to, and growing along, previously existing vessels (40). Vessel co-option is often distinguished from angiogenesis at the histopathological level, where tumors that achieve blood supply via co-option have a well-organized vascular network that resembles normal tissue and differs from the disorganized network that is produced by pathological sprouting angiogenesis (41). It is believed that breast cancer metastases to the brain, liver and lungs are sustained through this process, which consequently contributes to their resistance to conventional anti-angiogenic therapies (42–45). Although the process of vessel co-option has been linked to both innate and acquired resistance mechanisms to anti-angiogenic therapies (46), an increase in vessel co-option tends to proceed after angiogenic inhibition rather than precede it, indicating that it is more likely to be an acquired, rather than innate feature of the tumor (42).
Vascular Mimicry
Vascular mimicry involves the development of a vascular-like structures within tumors that contain no endothelial cells and are instead composed of a basement membrane surrounded by cancer cells (Figure 1C) (47–52). This process has been attributed to the actions of a subset of cancer stem cells that possess a high degree of differentiation plasticity and can acquire a range of endothelial markers, including cluster of differentiation 31 (CD31), VEGF receptor-2 (VEGFR2), Tie2, ephrin A2, and VE-Cadherin (53, 54). Importantly, these markers enable the incorporation of mimicked vascular structures into endothelial-based vessels at the boundaries between tumors and normal tissue (55). A blood supply model for melanoma developed by Zhang and colleagues indicated that vascular mimicry is the dominant blood supply in early tumor growth, followed by the development of conventional endothelial-based vessels to support the later stages of tumor expansion (56). In addition to supporting growth, vascular mimicry also serves to increase the proportion of tumor cells that are located adjacent to blood flow, enhancing their likelihood of infiltrating the bloodstream and metastasizing to distant sites in the body (57). As a consequence, the occurrence of vascular mimicry is linked with distant metastases, poor overall survival, and local cancer relapse (58).
Conventional Anti-Angiogenic Therapies
Several therapies targeting conventional angiogenic mediators, including the VEGF, Notch and Ang pathways, have been explored in both a pre-clinical and clinical setting, as summarized in Table 1.
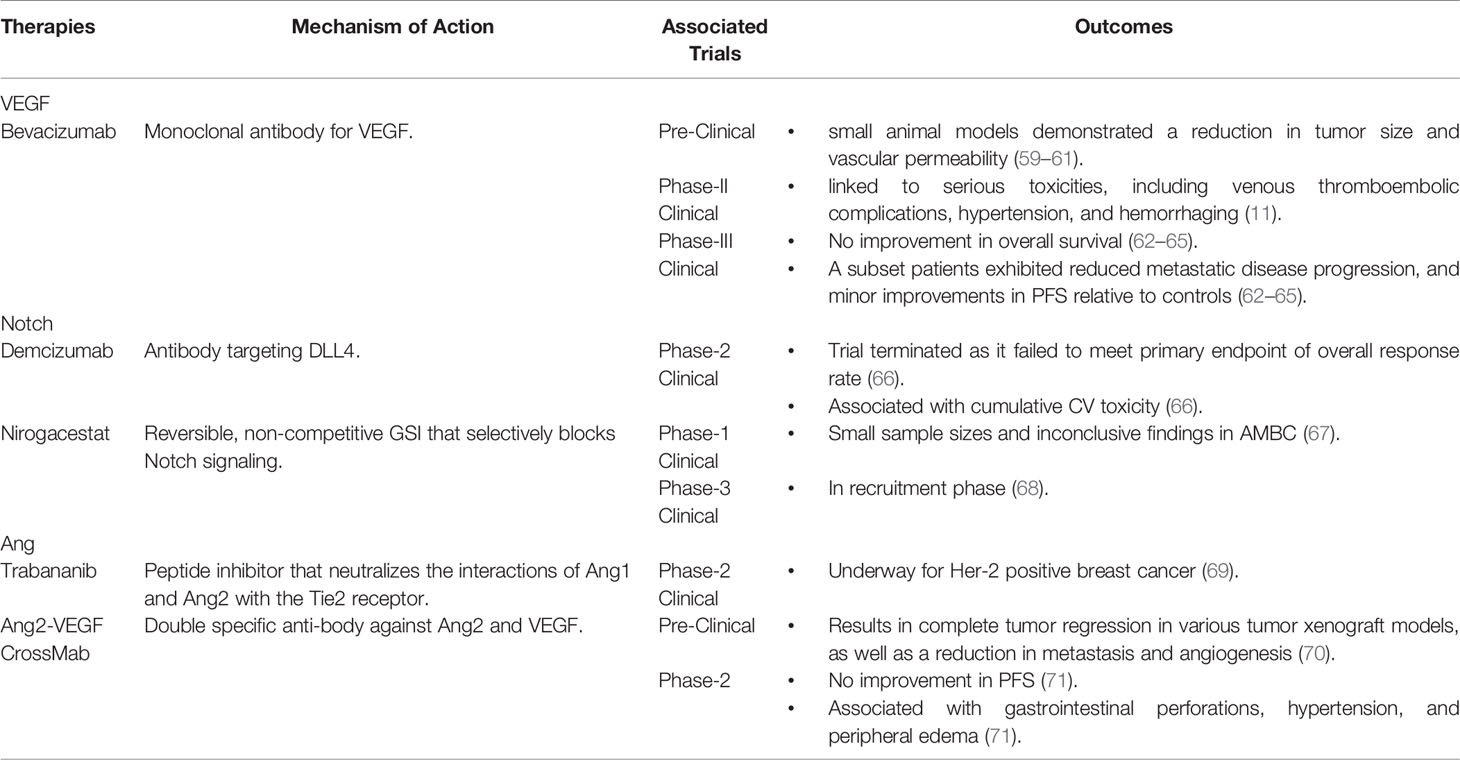
Table 1 Summary of Available Therapies with the Ability to Target Tumor Angiogenesis in Breast Cancer.
VEGF Pathway Inhibitors
In humans and other mammals, the VEGF family of growth factors includes VEGF-A, VEGF-B, VEGF-C, VEGF-D and Placental Growth Factor (PlGF) (72). While some members of this family, such as VEGF-C and VEGF-D, act primarily on the lymphatic endothelium, VEGF-A, referred to hereafter as VEGF, is the most potent direct-acting protein involved in angiogenesis (73). As such, the VEGF pathway was among the first and most broadly interrogated targets for the development of anti-angiogenic therapeutics (73). The production and release of VEGF is stimulated by a variety of factors, with the most notable being tissue hypoxia (74, 75). VEGF initiates vascular permeability, as well as endothelial cell proliferation and migration (76), through the activation of multiple tyrosine kinase receptors, including VEGFR1 and VEGFR2, with the majority of angiogenic responses being mediated by VEGFR2 activation (77).
Bevacizumab, a monoclonal antibody for VEGF, is the main anti-angiogenic agent for use in breast cancer (78). It works by preventing the interaction of VEGF with its receptors, as well as through the neutralization of VEGF release from cancer cells (78). Bevacizumab is not approved as a monotherapy. However, it is approved to be given alongside chemotherapy for advanced or metastatic breast cancer (AMBC) (79, 80). In addition to the prevention of sprouting angiogenesis, Bevacizumab can also promote tumor vessel normalization by reducing vascular permeability and promoting the recruitment of pericytes that support the endothelium (81). While numerous pre-clinical studies in small animal models demonstrated a reduction in tumor size and vascular permeability with Bevacizumab (59–61), similar results have not been achieved in human trials.
In patients with AMBC, Bevacizumab did not improve disease related symptoms or overall survival (65). However, a subset of patients who received Bevacizumab did exhibit reduced metastatic disease progression, which resulted in a minor improvement in progression free survival (PFS) relative to controls (62–65). However, no improvement in overall survival has been reported (62–65). Part of Bevacizumab’s lack of efficacy may be attributable to resistance mechanisms like vessel co-option and vascular mimicry. In patients with colorectal cancer, the process of vessel co-option has been linked to poor responses to anti-angiogenic therapies like Bevacizumab (42). The trans-differentiated endothelial-like cancer cells that drive vascular mimicry have also shown limited sensitivity to anti-angiogenic therapeutics (55, 82), with tumors that are resistant to these therapies exhibiting higher levels of vascular mimicry (83).
In addition to this lack of efficacy, long term trials with Bevacizumab concluded that excessive neutralization of VEGF with high doses or prolonged exposure caused vascular regression and promoted hypoxia (84, 85), driving the selection of more invasive cancer cells that can contribute to tumor resistance and increased metastatic potential (84). Prolonged exposure to Bevacizumab did not just disrupt tumor vasculature, but also upregulated multiple angiogenic factors, including PlGF and stromal derived factor-1 (SDF-1), in addition to causing increased cancer cell migration and enhancing metastatic potential. Treatment with Bevacizumab also increased cancer regrowth rates after treatment cessation, which has been attributed to elevated tumor hypoxia levels and the upregulation of compensatory angiogenic pathways (86, 87).
Bevacizumab has also been linked to serious toxicities. Patients with colorectal cancer treated with Bevacizumab had >33% risk of developing any type of thrombosis (88). A meta-analysis of patients receiving Bevacizumab also identified a significantly increased risk of hypertension in this group (89). Hemorrhaging is also a widely reported side effect of Bevacizumab therapy, although the mechanisms underlying this and other side effects are not fully understood (11). The United States Food and Drug Administration (FDA) initially approved the use of Bevacizumab as a monotherapy for AMBC. However, this indication was later revoked due to considerable side effects, no convincing evidence of clinical benefit, and a lack of effective biomarkers to determine treatment response (90). Bevacizumab is still available on the market for prescription. However, its use in cancer is not approved by the FDA (91).
Notch Signaling Pathway
The Notch signaling pathway plays a vital role in sprouting angiogenesis through the regulation endothelial cell proliferation, VEGF receptor expression and the commitment of sprouting endothelial cells to stalk or tip cell fates (92). As a consequence, there have been over 70 clinical trials therapeutically targeting Notch signaling in tumor angiogenesis (68). In canonical Notch signaling, Notch transmembrane receptors interact with Notch ligands, such as Delta-like 4 (DLL4) and Jagged-1 (JAG1), causing them to cleave and release their intracellular domain (93). In angiogenesis, DLL4 is induced by VEGF as a negative feedback regulator that induces the expression of Hes/Hey genes in stalk cells (94). These transcriptional regulators subsequently downregulate the expression of VEGFR2, making the cells less responsive to VEGF (95) and promoting the formation of a mature vascular network through the suppression of overexuberant angiogenic sprouting (96). Conversely, JAG1 alters the balance between DLL4-Notch and VEGFRs by antagonizing DLL4-mediated Notch activation. This reduction allows tip and stalk cells to change positions, resulting in a dense and tortuous vascular network (97). JAG1 also induces the expression of VEGF-receptor 3 (VEGFR3) (97), which influences the expression of both pro- and anti-angiogenic factors via the regulation of protein kinase B (AKT) signalling (98, 99).
In cancer cells, the balance of Notch signaling is pathogenically shifted so that the angiogenic actions of JAG1 dominate over the angiostatic effects of Notch-DLL4 (97). Despite this shift, Notch-DLL4 signalling has been therapeutically targeted using various inhibitory strategies, such as anti-DLL4 antibodies, DNA vaccinations, and Notch signaling inhibitors (100–102). Although blocking a pathway that is known to serve as an angiogenic “off switch” seems counterintuitive, agents targeting DLL4 have been shown to reduce tumor growth in vivo by promoting disorganized and non-productive endothelial sprouting and poor tumor perfusion (103). An antibody targeting DLL4, Demcizumab, was one of the first treatments to selectively target the Notch pathway in clinical trials (104). Demcizumab is a humanized monoclonal antibody for DLL4 that functions by blocking Notch receptor binding (104). Unfortunately, the phase 2 clinical trial for Demcizumab in pancreatic cancer was discontinued, as clinical outcomes were superior in the placebo arm over the treatment arm (66, 105). In addition to this failure to meet the primary endpoint of overall response rate, Demcizumab was also associated with cumulative cardiovascular (CV) toxicity (66), further hampering the translational potential of this approach.
Notch signaling in cancer has also been targeted using gamma secretase inhibitors (GSIs), which block the protease complex that cleaves Notch transmembrane domains (67). Despite their initial promise, GSIs have been linked to significant gastrointestinal toxicity, caused by the inhibition of Notch signaling in the stem-cell progenitor compartment of intestinal crypts (106, 107). As GSIs also cleave membrane proteins in various other signaling pathways, their non-specific inhibition can result in systemic toxicity (40, 108). Nirogacestat, the first Notch GSI to begin phase 3 clinical trials, is a reversible, non-competitive GSI that selectively blocks the Notch signaling pathway (67). A recent clinical trial assessing the impact of Nirogacestat on AMBC had small sample sizes and inconclusive findings (67). The ongoing phase 3 trial is currently in its recruitment phase (68), but should provide definitive insights into the ultimate potential of this approach.
Alternative strategies, such as the selective upregulation of DLL4 signaling with a soluble DLL4-Fc have also been explored to treat tumor angiogenesis (109). Pre-clinical studies based on this approach have demonstrated increased tumor vessel density and mural cell recruitment, along with decreasing tumor size (110), indicating potential merit in future clinical studies.
Ang Pathway Inhibitors
Ang1 and Ang2 regulate angiogenesis and vascular remodeling through their binding to the endothelial receptor tyrosine kinase, Tie2 (111). The activation of Tie2 is associated with vascular stabilization, via increased pericyte coverage and decreased blood vessel permeability (112, 113). Consequently, Tie2 activation has been linked to increased vessel diameter, vascular density, and perfusion within tumors (112, 113). Although both Ang1 and Ang2 bind Tie2 with similar affinity, only Ang1 promotes vascular development and maturation via robust receptor activation. In contrast, Ang2 is viewed as an Ang1 antagonist (114) that only weakly activates Tie2 (112, 113). In the presence of VEGF, Ang2 promotes vascular sprouting and destabilizes blood vessels through reduced endothelial-pericyte interactions (114). In contrast, when VEGF is absent, Ang2 accelerates blood vessel regression and promotes endothelial cell death (115).
Ang2 is highly expressed in breast cancer and has been linked to tumor angiogenesis in AMBC (116), making it an exciting therapeutic target (113). Therapies targeting Ang signaling in tumor angiogenesis include Trebananib, a peptide inhibitor that can neutralize the interactions of Ang1 and Ang2 with the Tie2 receptor (69). Trebananib has been used as a combination therapy for Her2-negative breast cancer and is currently in phase 2 clinical trials for ovarian cancer and Her2-positive breast cancer (69). This combination treatment has shown promising anti-tumor activity, especially in solid tumors (117). Ang2-VEGF CrossMab is another therapy targeting angiogenesis through the Ang2 pathway. This is a double specific antibody against both Ang2 and VEGF that causes complete tumor regression in various tumor xenograft models, as well as a reduction in metastasis and angiogenesis (70). Despite pre-clinical success, clinical trials of Ang2-VEGF CrossMab did not show improved PFS and were instead associated with increased anti-angiogenic toxicity, such as gastrointestinal perforations, hypertension, and peripheral edema (71). Overall, there is a promising amount of research targeting tumor angiogenesis through the Ang pathway. However, to date, these therapies have had limited success, and have only been used in combination with other chemotherapies, or anti-angiogenic agents.
The Need for Alternative Pathways for Breast Cancer Therapeutics
As detailed above, there is a significant need for novel targets and approaches that improve upon the toxicities, limited survival advantage and enhanced cancer regrowth rates associated with current anti-angiogenic therapies (118). In addition to conventional angiogenic mediators like VEGF, Notch and Ang(s), the last decade has seen the rise of multiple alternative signaling pathways that could be targeted to address pathological angiogenesis in breast cancer. Approaches targeting the pathways detailed below have been summarized in Table 2 and offer the potential to improve upon current therapeutic strategies by addressing resistance mechanisms and promoting the efficient long-term stabilization of tumor blood vessels.
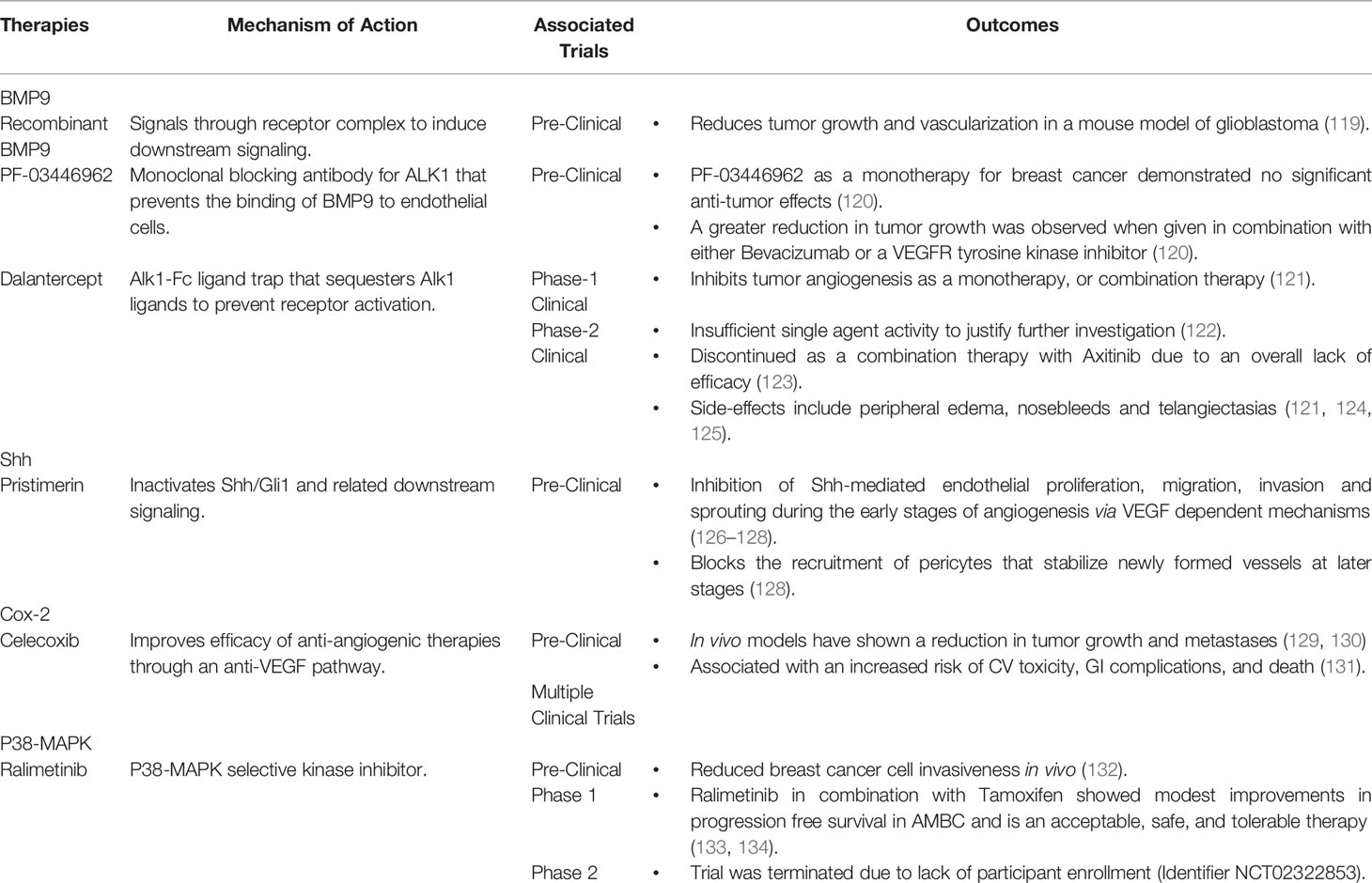
Table 2 Summary of Therapies Targeting Novel Tumor Angiogenesis Pathways in Breast Cancer.
Bone Morphogenetic Protein-9 (BMP9) Signaling
Since its discovery as a ligand for the orphan receptor, Activin receptor-like kinase 1 (Alk1), BMP9 has received significant attention as a mediator of vascular growth, stability, and integrity (135–137). BMP9 is a member of the transforming growth factor-β (TGFβ) superfamily and signals through complexes of type-I and type-II receptors (138). Alk1 is the dominant type-I BMP receptor in the endothelium and can pair with either BMPR-II or the activin type-II receptors (ActR-IIa and ActR-IIb) to form a receptor complex for BMP9, BMP10 and heterodimers of the two ligands, which are present at active concentrations in the circulation (138, 139) (Figure 2A). Receptor binding can induce both canonical signaling through phosphorylation of the SMAD 1, 5 and 9 transcriptional mediators, as well as non-canonical signaling via a variety of pathways, including Notch, Wnt, and p38 MAPK (140–143). Although recent studies have shown that the majority of BMP9 in the circulation exists as a heterodimer with a BMP10 subunit (139), most investigations into the potential of BMP9 to influence angiogenesis have explored the angiogenic potential of BMP9 homodimers alone.
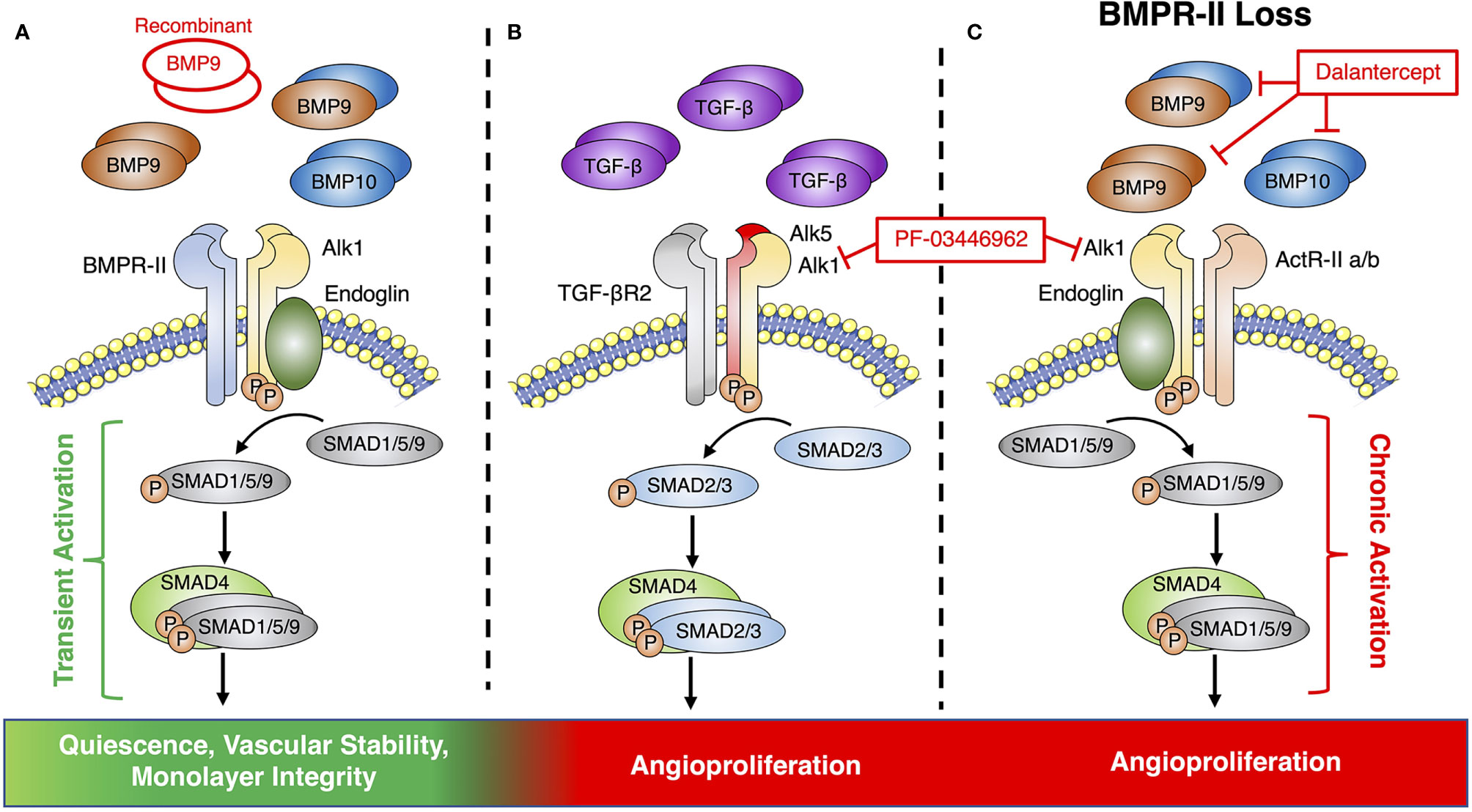
Figure 2 Endothelial BMP9 and TGFβ signaling via the Alk1 signaling axis. (A) Conventional endothelial BMP9 and BMP10 signaling via a receptor complex of Alk1 and BMPR-II leads to transient activation of downstream SMAD signaling, endothelial quiescence and vascular stability. Previous therapeutic studies have pursued vascular normalization by supplementing this pathway with recombinant BMP9. (B) Alk1 can also partner with ALK5 and TGFβR2 to form a receptor complex for TGFβ to promote angiogenesis. (C) BMPR-II loss causes signaling via alternative type-II receptors, such as ActR-IIa/b, which can promote angiogenesis via the chronic activation of canonical SMAD signaling. Therapeutic targeting of pre-angiogenic signaling of TGFβ or BMP9 via Alk1 has been pursued by receptor blockade with PF-03446962 or the Alk1-Fc ligand trap, Dalantercept.
BMP9 was originally shown to serve as a vascular stability factor that suppresses endothelial proliferation in vitro (144, 145) and angiogenesis in vivo (136). In cancer, multiple studies have shown that BMP9 can mediate the maturation phase of angiogenesis and may contribute to blood vessel normalization through the inhibition of endothelial proliferation and migration, as well as the recruitment of pericytes (142, 146–148). Deletion of Gdf2, the gene encoding BMP9, leads to enhanced tumor growth and increased lung metastases in the syngeneic orthotopic E0771 mouse model of metastatic breast cancer (149). This finding was associated with increased tumor vascular density and reduced mural cell coverage (149). The administration of recombinant BMP9 has also been shown to reduce tumor growth and vascularization in mouse models of glioblastoma (119). This approach may be a promising option for breast cancer, as it could be given in conjunction with current chemotherapies.
While there is substantial evidence supporting this anti-angiogenic and vascular stabilizing role for BMP9 in cancer, these effects appear to be highly context dependent (150–155). Multiple in vivo studies have also reported a pro-angiogenic role for BMP9 (151, 153) in the tumor microenvironment that is mediated via a synergistic activity with TGFβ (156, 157). Interestingly, TGFβ has been shown to signal via Alk1 in some endothelial cell models through a complex containing the type-II TGFβ receptor and Alk5, which is known as the conventional type-I receptor for TGFβ (158) (Figure 2B).
PF-03446962 is a monoclonal blocking antibody for Alk1 that was developed to target the pro-angiogenic effects of TGFβ, but has also been shown to prevent the binding of BMP9 to endothelial cells (159). Although pre-clinical studies using PF-03446962 as a monotherapy for breast cancer demonstrated no significant anti-tumor effects, a greater reduction in tumor growth was observed when it was given in combination with either Bevacizumab or a VEGFR tyrosine kinase inhibitor (120). An additional anti-angiogenic therapy targeting the BMP9-Alk1 axis is Dalantercept, an Alk1-Fc ligand trap that sequesters Alk1 ligands and prevents them from activating the receptor (121). Unlike PF-03446962, Dalantercept was shown to inhibit tumor angiogenesis as either a monotherapy or in combination with other treatments (121). While Dalantercept demonstrated promising efficacy and was well-tolerated in early studies (121), later clinical trials demonstrated no benefit in endometrial cancer (122) and it has since been discontinued as a combination therapy for advanced clear cell renal cell carcinoma due to an overall lack of efficacy (123, 160). Overall, these findings call into question the ultimate merits of blocking signaling via the BMP9-Alk1 axis as a viable clinical approach for breast cancer.
In addition to the contradictory information surrounding the pro- and anti-angiogenic effects of BMP9 signaling, the therapeutic potential of targeting this pathway is further complicated by the fact that BMP9 does not act on the endothelium in isolation, but can also influence the growth of tumor cells, as well as interactions between the two cell types in the tumor microenvironment (161, 162). A study by Eleftheriou and colleagues indicated that inhibiting BMP9 decreases tumor volume, while increasing vascular branching and metastases (154), suggesting that BMP9 may cause certain cancer cells to proliferate, while also promoting vascular quiescence. Along this line, a pro-proliferative effect of BMP9 has been reported in ovarian, liver, bladder, and pancreatic cancer cell lines, but not in breast cancer (151, 153–155).
Beyond its role in cancer, the angiogenic actions of BMP9 have also been explored in other pathologies, including pulmonary arterial hypertension (PAH), a disease of occlusive pulmonary vascular remodeling that is linked to excessive endothelial cell proliferation and loss-of-function mutations in the gene encoding BMPR-II (163). This work may help to explain the contradictory actions of BMP9 in cancer by identifying BMP9 signaling as an “angiogenic switch” that can either promote or prevent angiogenesis, based on the availability of BMPR-II as its type-II receptor (164). Under this model, the loss of BMPR-II in the endothelium drives BMP9 signaling via alternative type-II receptors, such as ActR-IIa or ActR-IIb, resulting in prolonged activation of the canonical signaling pathway and a cancer-like shift of the BMP9 response towards pathological endothelial proliferation (Figure 2C).
Although a similar shift has not yet been identified in cancer, the recognition of a central role for type-II receptor utilization in regulating the balance between the pro- and anti-angiogenic effects of BMP9 highlights the value of a more nuanced understanding of BMP9 signaling in disease. Such an approach could improve on the poor efficacy of ligand traps like Dalantercept and blocking antibodies like PF-03446962, which block all BMP9-related signaling, including the beneficial effects of BMP9 on vascular growth and stability via BMPR-II. Blockade of the vascular stabilizing effects of BMP9 could also explain why Dalantercept trials demonstrated poor efficacy in human cancers, accompanied by side-effects linked to vascular instability, such as peripheral edema, nosebleeds and telangiectasias (121, 124, 125). Profiling the relative expression of type-II BMP receptors in the tumor endothelium could also help to identify breast cancer tumors that would benefit from recombinant BMP9 therapy, versus those for which this treatment may enhance disordered vascularization.
Shh Signaling
Recently, the Shh pathway has emerged as a main regulator of tumor angiogenesis and a promising therapeutic target (165). There are three different pathways involved in Hedgehog signaling, Shh, Indian-Hedgehog, and Desert-Hedgehog (166). The Shh pathway signals through active and inactive pathways (Figure 3A). Inactive signaling occurs due to the ligand independent interaction of Shh and the transmembrane protein Patched (Ptch1), whereas active signaling occurs through factors downstream of smoothened (Smo), a G protein-coupled receptor protein that is required to activate glioma associated oncogenic (Gli) proteins (167, 168) (Figure 3A). The active Shh signaling pathway is induced when Shh binds and inactivates Ptch1 (169). This binding stimulates the release of Smo, which activates the transcription factor Gli homolog 1 (Gli1) and allows for the regulation of genes involved in cell growth, differentiation, drug resistance, and angiogenesis (167, 169).
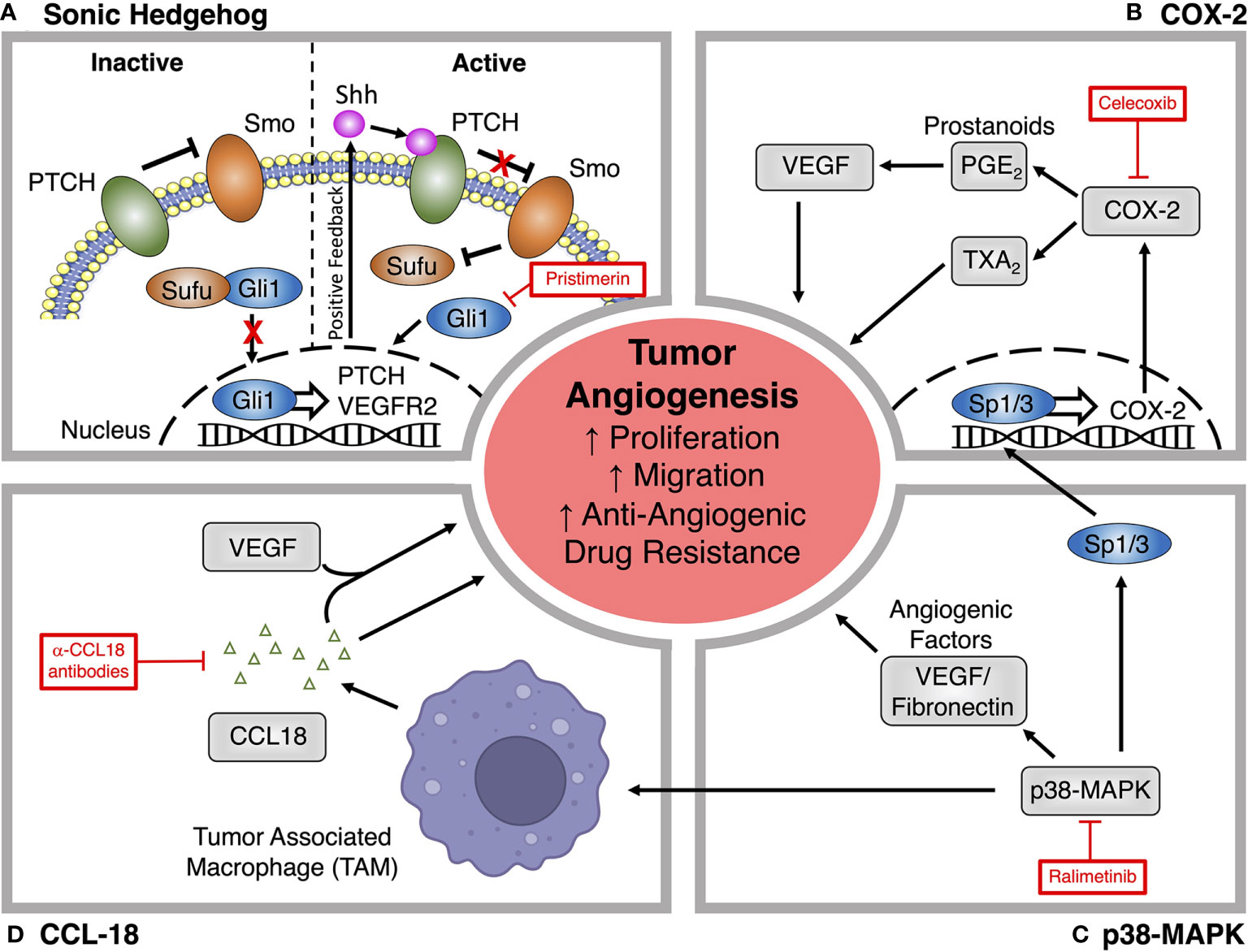
Figure 3 Alternative signaling pathways for targeting tumor angiogenesis. (A) Pristimerin inhibits the pro-angiogenic actions of Sonic hedgehog (Shh) signaling by targeting downstream effects via the transcription factor Gli1. (B) The COX-2 inhibitor Celecoxib prevents the synthesis of downstream prostanoids, which can promote tumor angiogenesis, endothelial proliferation and migration through the production of angiogenic factors like VEGF. (C) Ralimetinib blocks p38-MAPK signaling, which promotes the deposition of angiogenic factors within the tumor microenvironment and can also promote angiogenesis through actions on tumor associated macrophages (TAM) and COX-2 expression. (D) Production of CCL18 by TAMs promotes angiogenesis through VEGF-dependent and independent mechanisms. CCL18 neutralizing antibodies have been explored as a novel treatment for tumor angiogenesis.
Shh signaling can promote vascularization through its actions on both the VEGF/VEGFR2, and Ang/Tie2 signaling pathways to promote endothelial proliferation, migration, invasion, and new vessel maturation (170, 171). As the main transcription factor of the Shh pathway, Gli1 upregulates the release of multiple pro-angiogenic molecules, including VEGF and the VEGFR1 and VEGFR2 co-receptors cysteine-rich angiogenic inducer 61 (CYR61) and neuropilin 2 (NRP2) (172–175). Shh signaling can also facilitate tumor vascularization by upregulating the expression of VEGFR2 on the surface of cancer cells (165). Clinically, dysregulation of Shh signaling plays a key role in breast cancer progression and metastasis and is a measure of poor prognosis (176–178). Overexpression of Gli1 is also correlated with increased vascular density in breast cancer (165). There are numerous potential therapeutic targets in the Shh pathway that could influence tumor angiogenesis.
Pristimerin is an anti-inflammatory quinonemethide triterpenoid compound that is being explored as a potential anti-angiogenic candidate targeting Shh signaling (126). Pristimerin exerts its anti-angiogenic effects by inactivating Shh/Gli1 and related downstream signaling, resulting in the inhibition of Shh-mediated endothelial proliferation, migration, invasion and sprouting during the early stages of angiogenesis via VEGF dependent mechanisms (126–128). Pristimerin can also act at later stages to block the recruitment of pericytes that stabilize newly formed vessels (126–128). In addition to the effects of Pristimerin on angiogenesis, it can induce cancer stem cell toxicity in breast cancer (179), indicating a potential to impact the vascular mimicry-mediated resistance that limits other anti-angiogenic approaches. Overall, it appears that Pristimerin is efficacious as an anti-angiogenic therapy in cancer. However, these findings have not yet been translated into clinical success and the precise mechanisms driving these effects remain unclear (180, 181).
COX-2 Inhibition
COX is the rate-limiting enzyme for the synthesis of prostaglandins, with COX-2 being the dominant isoform driving prostaglandin production during inflammation (182). It has been previously confirmed that the expression of COX-2 in the endothelium can be induced by hypoxia-stimulated release of VEGF through a mechanism that is dependent on the activation of the p38-MAPK and c-Jun N-terminal kinase (JNK) pathways (183). P38-MAPK signaling can also upregulate COX-2 expression within tumors via the Sp1/Sp3 transcription factors (184). The prostaglandins produced by COX-2 stimulate the production of angiogenic mediators, like VEGF, in cancer and endothelial cells (Figure 3B), driving a positive feedback loop between the two cell types (185). The expression of COX-2 is 90% elevated in colorectal cancer, 70% in lung cancer, and 37% in breast cancer and has been correlated with a poor prognostic outcome (186).
A study by Xu et al., has predicted that COX-2 inhibitors targeting tumor inflammation and angiogenesis could enhance the activity of conventional anti-angiogenic therapeutics against pre-established metastases and improve the prognosis for patients with COX-2 overexpressing tumors (185). Celecoxib is a COX-2 inhibitor that has been used to treat angiogenesis in breast cancer (129, 130). Although Celecoxib may improve the efficacy of anti-angiogenic therapies through a distinct anti-VEGF pathway, this approach is also limited by dose-dependent side effects that have reduced enthusiasm for this approach (129, 130). In clinical trials, patients treated with COX-2 inhibitors have an increased risk of CV toxicity, GI complications, and death (131, 187, 188). Whilst initial results are promising, additional controlled clinical trials are needed to confirm how COX-2 inhibitors perform when combined with VEGF inhibitors for the treatment of AMBC (128, 185). Due to the abundance of clinically available COX-2 inhibitors and years of experience with these drugs in various patient populations, a demonstration of clinical efficacy from these trials could allow for the rapid incorporation of COX-2 inhibitors into conventional therapeutic regimens.
P38-MAPK Pathway
P38-MAPK is a signal transduction mediator that is involved in inflammation, the cell cycle, cell death, cellular development, differentiation, senescence and tumor development (189). A meta-analysis by Limoge et al. indicated that p38 target genes and p38-MAPK signaling are elevated in breast cancer, and have been linked to increased tumorigenesis, invasiveness, metastasis, disease recurrence, and poor outcomes (190). P38-MAPK also contributes to the formation of blood vessels in tumors by enhancing the production, and deposition of pro-angiogenic factors that alter the tumor microenvironment (Figure 3C), including pro-angiogenic cytokines and fibronectin, a component of the extracellular matrix that serves as an anchor for VEGF (190).
The p38-MAPK pathway may also contribute to angiogenesis through its effects on TAMs and neutrophils, central immune components of the tumor microenvironment. TAMs contribute to vascular sprouting by bridging tip cells, as well as by secreting a variety of angiogenic growth factors such as TGFβ, VEGF, endothelial growth factor, and a variety of chemokines (34, 191–193). The p38-MAPK pathway is also known to regulate TGFα, TGFβ, and interleukins, which are all established drivers of TAM function and play a role in the colonization of cancer cells and angiogenesis (194). In addition, p38 may also modulate VEGF-mediated endothelial migration, further contributing to angiogenesis (195).
Genetic inactivation of p38 has been shown to reduce tumor angiogenesis (132, 190), offering promise that anti-p38 drugs could be a new therapeutic option for treating AMBC, as well as breast cancer in general. The pharmacological inhibition of p38 significantly reduces tumor growth, angiogenesis, and lung metastasis (196). P38 inhibition can also help to improve the efficacy of current anti-angiogenic therapies, as TAMs have been shown to contribute to the resistance of VEGF-targeted anti-angiogenics (197). Ralimetinib, a p38-MAPK selective kinase inhibitor, has been shown to reduce breast cancer cell invasiveness in vivo (132). A phase 1 clinical trial assessing Ralimetinib in combination with Tamoxifen demonstrated that it could provide modest improvements in progression free survival in AMBC and is an acceptable, safe, and tolerable therapy (133, 134). A phase 2 clinical trial of Ralimetinib was initiated to address the efficacy and safety of p38-MAPK inhibition in AMBC; however, the trial was terminated due to lack of participant enrollment (identifier NCT02322853). Despite the early promising findings, a major concern associated with this therapeutic approach is the fact that p38 is a well-recognized tumor suppressor (198), suggesting the need to balance any beneficial effects on tumor angiogenesis against potentially damaging actions on the tumor itself. The time of administration is also an important factor in this therapeutic approach, as p38 inhibition is impacted by circadian rhythms (199).
CCL18 in Tumor Angiogenesis
Several chemokines are currently being investigated for their association with angiogenesis However, CCL18 appears to be the most promising. CCL18 is the most abundant and specific chemokine released from TAMs, and serves the function of recruiting B-cells, T-cells, and natural killer (NK) cells (200). It is also essential for the promotion of tumor angiogenesis and endothelial cell survival (201) and is linked to poor prognosis in breast cancer (200). CCL18 can either work synergistically with VEGF, or through VEGF-independent mechanisms (Figure 3D), to promote migratory and angiogenic effects both in vitro and in vivo (202). Beyond its pro-angiogenic activity, CCL18 can also promote endothelial to mesenchymal transition (EndMT) in the tumor microvasculature, which can lead to loss of cell-to-cell junctions, and enhanced invasion and migration (202).
Therapeutically, targeting CCL18 represents a promising treatment for patients with resistance to anti-VEGF therapies. Neutralizing antibodies towards CCL18 do not only block its effects on tumor angiogenesis though VEGF-independent mechanisms, but may also improve the response to other anti-angiogenic therapies via TAM-driven processes (202). CCL18 should be further assessed for biological activities using mechanistic studies and the pre-clinical and clinical evaluation of novel inhibitors (203).
In conclusion, drug resistance, severe side-effects, limited survival advantage, and enhanced cancer regrowth rates associated with current anti-angiogenic therapies highlight the critical need for novel targets and therapies targeting this aspect of tumor growth and metastasis. Beyond the novel therapies discussed in this review, non-coding RNAs, including long non-coding and circular RNAs are being investigated as potential master regulators of tumor angiogenesis. These therapies are advantageous as they have multiple targets and broad affects are difficult to achieve through approaches targeting individual pathways or ligands. Increased understanding of the factors that stimulate tumor angiogenesis and the interactions between these novel pathways and more established mediators may also allow for a refinement of therapeutic approaches, without the need for new drugs. Together these advances offer the promise of new treatment options that will improve the prognosis for AMBC patients.
Author Contributions
JH contributed to the conception and design of the review article. JH wrote each section of the manuscript and prepared the figures. MO provided feedback on the conception and contributed to the revision of the manuscript and figures. All authors contributed to the article and approved the submitted version.
Funding
This work is funded by the Canadian Institutes of Health Research. Grant #: PJT-178078.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We acknowledge Dr. Peter Greer for providing editorial contributions to the manuscript.
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global Cancer Statistics 2018: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin (2018) 68:394–424. doi: 10.3322/caac.21492
2. Sihto H, Lundin J, Lundin M, Lehtimaki T, Ristimaki A, Holli K, et al. Breast Cancer Biological Subtypes and Protein Expression Predict for the Preferential Distant Metastasis Sites: A Nationwide Cohort Study. Breast Cancer Res (2011) 13:R87. doi: 10.1186/bcr2944
3. Sopik V, Narod SA. The Relationship Between Tumour Size, Nodal Status and Distant Metastases: On the Origins of Breast Cancer. Breast Cancer Res Treat (2018) 170:647–56. doi: 10.1007/s10549-018-4796-9
4. Hanahan D, Weinberg RA. Hallmarks of Cancer: The Next Generation. Cell (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
5. Davis G, Speichinger K, Norden P, Kim D, Bowers S. Endothelial Cell Polarization During Lumen Formation, Tubulogenesis, and Vessel Maturation in 3D Extracellular Matrices. Cell Polarity (2015) 1:205–20. doi: 10.1007/978-3-319-14463-4_9
6. Hashizume H, Baluk P, Morikawa S, Mclean JW, Thurston G, Roberge S, et al. Openings Between Defective Endothelial Cells Explain Tumor Vessel Leakiness. Am J Pathol (2000) 156:1363–80. doi: 10.1016/S0002-9440(10)65006-7
7. Inai T, Mancuso M, Hashizume H, Baffert F, Haskell A, Baluk P, et al. Inhibition of Vascular Endothelial Growth Factor (VEGF) Signaling in Cancer Causes Loss of Endothelial Fenestrations, Regression of Tumor Vessels, and Appearance of Basement Membrane Ghosts. Am J Pathol (2004) 165:35–52. doi: 10.1016/S0002-9440(10)63273-7
8. Li H, Fan X, Houghton J. Tumor Microenvironment: The Role of the Tumor Stroma in Cancer. J Cell Biochem (2007) 101:805–15. doi: 10.1002/jcb.21159
9. Folkman J. Role of Angiogenesis in Tumor Growth and Metastasis. Semin Oncol (2002) 29:15–8. doi: 10.1053/sonc.2002.37263
10. Arriaga Y, Becerra CR. Adverse Effects of Bevacizumab and Their Management in Solid Tumors. Support Cancer Ther (2006) 3:247–50. doi: 10.3816/SCT.2006.n.023
11. Widakowich C, De Castro G Jr., De Azambuja E, Dinh P, Awada A. Review: Side Effects of Approved Molecular Targeted Therapies in Solid Cancers. Oncologist (2007) 12:1443–55. doi: 10.1634/theoncologist.12-12-1443
12. Elice F, Rodeghiero F. Side Effects of Anti-Angiogenic Drugs. Thromb Res (2012) 129 Suppl1:S50–53. doi: 10.1016/S0049-3848(12)70016-6
13. Hamzah J, Jugold M, Kiessling F, Rigby P, Manzur M, Marti HH, et al. Vascular Normalization in Rgs5-Deficient Tumours Promotes Immune Destruction. Nature (2008) 453:410–4. doi: 10.1038/nature06868
14. Mazzone M, Dettori D, De Oliveira RL, Loges S, Schmidt T, Jonckx B, et al. Heterozygous Deficiency of PHD2 Restores Tumor Oxygenation and Inhibits Metastasis via Endothelial Normalization. Cell (2009) 136:839–51. doi: 10.1016/j.cell.2009.01.020
15. Hanahan D, Weinberg RA. The Hallmarks of Cancer. Cell (2000) 100:57–70. doi: 10.1016/S0092-8674(00)81683-9
16. Hanahan D, Folkman J. Patterns and Emerging Mechanisms of the Angiogenic Switch During Tumorigenesis. Cell (1996) 86:353–64. doi: 10.1016/S0092-8674(00)80108-7
17. Dor Y, Porat R, Keshet E. Vascular Endothelial Growth Factor and Vascular Adjustments to Perturbations in Oxygen Homeostasis. Am J Physiol Cell Physiol (2001) 280:C1367–1374. doi: 10.1152/ajpcell.2001.280.6.C1367
18. Cao J, Ehling M, März S, Seebach J, Tarbashevich K, Sixta T, et al. Polarized Actin and VE-Cadherin Dynamics Regulate Junctional Remodelling and Cell Migration During Sprouting Angiogenesis. Nat Commun (2017) 8:1. doi: 10.1038/s41467-017-02373-8
19. Hillen F, Griffioen AW. Tumour Vascularization: Sprouting Angiogenesis and Beyond. Cancer Metastasis Rev (2007) 26:489–502. doi: 10.1007/s10555-007-9094-7
20. Whiteside TL. The Tumor Microenvironment and its Role in Promoting Tumor Growth. Oncogene (2008) 27:5904–12. doi: 10.1038/onc.2008.271
21. Fridman WH, Zitvogel L, Sautes-Fridman C, Kroemer G. The Immune Contexture in Cancer Prognosis and Treatment. Nat Rev Clin Oncol (2017) 14:717–34. doi: 10.1038/nrclinonc.2017.101
22. Borsi E, Terragna C, Brioli A, Tacchetti P, Martello M, Cavo M. Therapeutic Targeting of Hypoxia and Hypoxia-Inducible Factor 1 Alpha in Multiple Myeloma. Transl Res (2015) 165:641–50. doi: 10.1016/j.trsl.2014.12.001
23. Viallard C, Larrivee B. Tumor Angiogenesis and Vascular Normalization: Alternative Therapeutic Targets. Angiogenesis (2017) 20:409–26. doi: 10.1007/s10456-017-9562-9
24. Azzi S, Hebda JK, Gavard J. Vascular Permeability and Drug Delivery in Cancers. Front Oncol (2013) 3:211. doi: 10.3389/fonc.2013.00211
25. Weis S, Cui J, Barnes L, Cheresh D. Endothelial Barrier Disruption by VEGF-Mediated Src Activity Potentiates Tumor Cell Extravasation and Metastasis. J Cell Biol (2004) 167:223–9. doi: 10.1083/jcb.200408130
26. Klimova T, Chandel NS. Mitochondrial Complex III Regulates Hypoxic Activation of HIF. Cell Death Differ (2008) 15:660–6. doi: 10.1038/sj.cdd.4402307
27. Weinberg F, Hamanaka R, Wheaton WW, Weinberg S, Joseph J, Lopez M, et al. Mitochondrial Metabolism and ROS Generation Are Essential for Kras-Mediated Tumorigenicity. Proc Natl Acad Sci USA (2010) 107:8788–93. doi: 10.1073/pnas.1003428107
28. Costa A, Scholer-Dahirel A, Mechta-Grigoriou F. The Role of Reactive Oxygen Species and Metabolism on Cancer Cells and Their Microenvironment. Semin Cancer Biol (2014) 25:23–32. doi: 10.1016/j.semcancer.2013.12.007
29. Chandel NS, Maltepe E, Goldwasser E, Mathieu CE, Simon MC, Schumacker PT. Mitochondrial Reactive Oxygen Species Trigger Hypoxia-Induced Transcription. Proc Natl Acad Sci USA (1998) 95:11715–20. doi: 10.1073/pnas.95.20.11715
30. Chandel NS, Mcclintock DS, Feliciano CE, Wood TM, Melendez JA, Rodriguez AM, et al. Reactive Oxygen Species Generated at Mitochondrial Complex III Stabilize Hypoxia-Inducible Factor-1alpha During Hypoxia: A Mechanism of O2 Sensing. J Biol Chem (2000) 275:25130–8. doi: 10.1074/jbc.M001914200
31. Linde N, Lederle W, Depner S, Van Rooijen N, Gutschalk CM, Mueller MM. Vascular Endothelial Growth Factor-Induced Skin Carcinogenesis Depends on Recruitment and Alternative Activation of Macrophages. J Pathol (2012) 227:17–28. doi: 10.1002/path.3989
32. Lanitis E, Irving M, Coukos G. Targeting the Tumor Vasculature to Enhance T Cell Activity. Curr Opin Immunol (2015) 33:55–63. doi: 10.1016/j.coi.2015.01.011
33. Lapeyre-Prost A, Terme M, Pernot S, Pointet AL, Voron T, Tartour E, et al. Immunomodulatory Activity of VEGF in Cancer. Int Rev Cell Mol Biol (2017) 330:295–342. doi: 10.1016/bs.ircmb.2016.09.007
34. Lin EY, Li JF, Gnatovskiy L, Deng Y, Zhu L, Grzesik DA, et al. Macrophages Regulate the Angiogenic Switch in a Mouse Model of Breast Cancer. Cancer Res (2006) 66:11238–46. doi: 10.1158/0008-5472.CAN-06-1278
35. Vasiljeva O, Papazoglou A, Kruger A, Brodoefel H, Korovin M, Deussing J, et al. Tumor Cell-Derived and Macrophage-Derived Cathepsin B Promotes Progression and Lung Metastasis of Mammary Cancer. Cancer Res (2006) 66:5242–50. doi: 10.1158/0008-5472.CAN-05-4463
36. Yang L, Zhang Y. Tumor-Associated Macrophages: From Basic Research to Clinical Application. J Hematol Oncol (2017) 10:58. doi: 10.1186/s13045-017-0430-2
37. Jeannin P, Duluc D, Delneste Y. IL-6 and Leukemia-Inhibitory Factor Are Involved in the Generation of Tumor-Associated Macrophage: Regulation by IFN-Gamma. Immunotherapy (2011) 3:23–6. doi: 10.2217/imt.11.30
38. Qian BZ, Li J, Zhang H, Kitamura T, Zhang J, Campion LR, et al. CCL2 Recruits Inflammatory Monocytes to Facilitate Breast-Tumour Metastasis. Nature (2011) 475:222–5. doi: 10.1038/nature10138
39. Henze AT, Mazzone M. The Impact of Hypoxia on Tumor-Associated Macrophages. J Clin Invest (2016) 126:3672–9. doi: 10.1172/JCI84427
40. Beel AJ, Sanders CR. Substrate Specificity of Gamma-Secretase and Other Intramembrane Proteases. Cell Mol Life Sci (2008) 65:1311–34. doi: 10.1007/s00018-008-7462-2
41. Di Tomaso E, Snuderl M, Kamoun WS, Duda DG, Auluck PK, Fazlollahi L, et al. Glioblastoma Recurrence After Cediranib Therapy in Patients: Lack of "Rebound" Revascularization as Mode of Escape. Cancer Res (2011) 71:19–28. doi: 10.1158/0008-5472.CAN-10-2602
42. Frentzas S, Simoneau E, Bridgeman VL, Vermeulen PB, Foo S, Kostaras E, et al. Vessel Co-Option Mediates Resistance to Anti-Angiogenic Therapy in Liver Metastases. Nat Med (2016) 22:1294–302. doi: 10.1038/nm.4197
43. Kienast Y, Von Baumgarten L, Fuhrmann M, Klinkert WE, Goldbrunner R, Herms J, et al. Real-Time Imaging Reveals the Single Steps of Brain Metastasis Formation. Nat Med (2010) 16:116–22. doi: 10.1038/nm.2072
44. Kuczynski EA, Yin M, Bar-Zion A, Lee CR, Butz H, Man S, et al. Co-Option of Liver Vessels and Not Sprouting Angiogenesis Drives Acquired Sorafenib Resistance in Hepatocellular Carcinoma. J Natl Cancer Inst (2016) 108(8). doi: 10.1093/jnci/djw030
45. Pereira ER, Kedrin D, Seano G, Gautier O, Meijer EFJ, Jones D, et al. Lymph Node Metastases can Invade Local Blood Vessels, Exit the Node, and Colonize Distant Organs in Mice. Science (2018) 359:1403–7. doi: 10.1126/science.aal3622
46. Rada M, Lazaris A, Kapelanski-Lamoureux A, Mayer TZ, Metrakos P. Tumor Microenvironment Conditions That Favor Vessel Co-Option in Colorectal Cancer Liver Metastases: A Theoretical Model. Semin Cancer Biol (2020) 71:52–64. doi: 10.1016/j.semcancer.2020.09.001
47. Shirakawa K, Tsuda H, Heike Y, Kato K, Asada R, Inomata M, et al. Absence of Endothelial Cells, Central Necrosis, and Fibrosis Are Associated With Aggressive Inflammatory Breast Cancer. Cancer Res (2001) 61:445–51 https://cancerres.aacrjournals.org/content/61/2/445.long.
48. El Hallani S, Boisselier B, Peglion F, Rousseau A, Colin C, Idbaih A, et al. A New Alternative Mechanism in Glioblastoma Vascularization: Tubular Vasculogenic Mimicry. Brain (2010) 133:973–82. doi: 10.1093/brain/awq044
49. Kirschmann DA, Seftor EA, Hardy KM, Seftor RE, Hendrix MJ. Molecular Pathways: Vasculogenic Mimicry in Tumor Cells: Diagnostic and Therapeutic Implications. Clin Cancer Res (2012) 18:2726–32. doi: 10.1158/1078-0432.CCR-11-3237
50. Liu TJ, Sun BC, Zhao XL, Zhao XM, Sun T, Gu Q, et al. CD133+ Cells With Cancer Stem Cell Characteristics Associates With Vasculogenic Mimicry in Triple-Negative Breast Cancer. Oncogene (2013) 32:544–53. doi: 10.1038/onc.2012.85
51. Plantamura I, Casalini P, Dugnani E, Sasso M, D’ippolito E, Tortoreto M, et al. PDGFRbeta and FGFR2 Mediate Endothelial Cell Differentiation Capability of Triple Negative Breast Carcinoma Cells. Mol Oncol (2014) 8:968–81. doi: 10.1016/j.molonc.2014.03.015
52. Wagenblast E, Soto M, Gutierrez-Angel S, Hartl CA, Gable AL, Maceli AR, et al. A Model of Breast Cancer Heterogeneity Reveals Vascular Mimicry as a Driver of Metastasis. Nature (2015) 520:358–62. doi: 10.1038/nature14403
53. Maniotis AJ, Folberg R, Hess A, Seftor EA, Gardner LM, Pe’er J, et al. Vascular Channel Formation by Human Melanoma Cells In Vivo and In Vitro: Vasculogenic Mimicry. Am J Pathol (1999) 155:739–52. doi: 10.1016/S0002-9440(10)65173-5
54. Ge H, Luo H. Overview of Advances in Vasculogenic Mimicry - a Potential Target for Tumor Therapy. Cancer Manag Res (2018) 10:2429–37. doi: 10.2147/CMAR.S164675
55. Sun B, Zhang D, Zhang S, Zhang W, Guo H, Zhao X. Hypoxia Influences Vasculogenic Mimicry Channel Formation and Tumor Invasion-Related Protein Expression in Melanoma. Cancer Lett (2007) 249:188–97. doi: 10.1016/j.canlet.2006.08.016
56. Zhang S, Guo H, Zhang D, Zhang W, Zhao X, Ren Z, et al. Microcirculation Patterns in Different Stages of Melanoma Growth. Oncol Rep (2006) 15:15–20. doi: 10.3892/or.15.1.15
57. Wechman SL, Emdad L, Sarkar D, Das SK, Fisher PB. Vascular Mimicry: Triggers, Molecular Interactions and In Vivo Models. Adv Cancer Res (2020) 148:27–67. doi: 10.1016/bs.acr.2020.06.001
58. Liu R, Yang K, Meng C, Zhang Z, Xu Y. Vasculogenic Mimicry is a Marker of Poor Prognosis in Prostate Cancer. Cancer Biol Ther (2012) 13:527–33. doi: 10.4161/cbt.19602
59. Brasch R, Pham C, Shames D, Roberts T, Van Dijke K, Van Bruggen N, et al. Assessing Tumor Angiogenesis Using Macromolecular MR Imaging Contrast Media. J Magn Reson Imaging (1997) 7:68–74. doi: 10.1002/jmri.1880070110
60. Pham CD, Roberts TP, Van Bruggen N, Melnyk O, Mann J, Ferrara N, et al. Magnetic Resonance Imaging Detects Suppression of Tumor Vascular Permeability After Administration of Antibody to Vascular Endothelial Growth Factor. Cancer Invest (1998) 16:225–30. doi: 10.3109/07357909809039771
61. Sweeney CJ, Miller KD, Sissons SE, Nozaki S, Heilman DK, Shen J, et al. The Antiangiogenic Property of Docetaxel Is Synergistic With a Recombinant Humanized Monoclonal Antibody Against Vascular Endothelial Growth Factor or 2-Methoxyestradiol But Antagonized by Endothelial Growth Factors. Cancer Res (2001) 61:3369–72. https://cancerres.aacrjournals.org/content/61/8/3369.long.
62. Saltz LB, Clarke S, Diaz-Rubio E, Scheithauer W, Figer A, Wong R, et al. Bevacizumab in Combination With Oxaliplatin-Based Chemotherapy as First-Line Therapy in Metastatic Colorectal Cancer: A Randomized Phase III Study. J Clin Oncol (2008) 26:2013–9. doi: 10.1200/JCO.2007.14.9930
63. Allegra CJ, Yothers G, O’connell MJ, Sharif S, Petrelli NJ, Colangelo LH, et al. Phase III Trial Assessing Bevacizumab in Stages II and III Carcinoma of the Colon: Results of NSABP Protocol C-08. J Clin Oncol (2011) 29:11–6. doi: 10.1200/JCO.2010.30.0855
64. De Gramont A, Van Cutsem E, Tabernero J, Moore MJ, Cunningham D, Riveras. Im F, et al. AVANT: Results From a Randomized, Three-Arm Multinational Phase III Study to Investigate Bevacizumab With Either XELOX or FOLFOX4 Versus FOLFOX4 Alone as Adjuvant Treatment for Colon Cancer. J Clin Oncol (2011) 29:362. doi: 10.1200/jco.2011.29.4_suppl.362
65. Manso L, Moreno F, Marquez R, Castelo B, Arcediano A, Arroyo M, et al. Use of Bevacizumab as a First-Line Treatment for Metastatic Breast Cancer. Curr Oncol (2015) 22:e51–60. doi: 10.3747/co.22.2210
66. Gracian AC, Dean A, Muñoz A, Hidalgo M, Pazo-Cid R, Martin M, et al. YOSEMITE: A 3 Arm Double-Blind Randomized Phase 2 Study of Gemcitabine, Paclitaxel Protein-Bound Particles for Injectable Suspension, and Placebo (GAP) Versus Gemcitabine, Paclitaxel Protein-Bound Particles for Injectable Suspension and Either 1 or 2 Truncated Courses of Demcizumab (GAD). Ann Oncol (2017) 28:211. doi: 10.1093/annonc/mdx369.004
67. Locatelli MA, Aftimos P, Dees EC, Lorusso PM, Pegram MD, Awada A, et al. Phase I Study of the Gamma Secretase Inhibitor PF-03084014 in Combination With Docetaxel in Patients With Advanced Triple-Negative Breast Cancer. Oncotarget (2017) 8:2320–8. doi: 10.18632/oncotarget.13727
68. Moore G, Annett S, Mcclements L, Robson T. Top Notch Targeting Strategies in Cancer: A Detailed Overview of Recent Insights and Current Perspectives. Cells (2020) 9:1503. doi: 10.3390/cells9061503
69. Dieras V, Wildiers H, Jassem J, Dirix LY, Guastalla JP, Bono P, et al. Trebananib (AMG 386) Plus Weekly Paclitaxel With or Without Bevacizumab as First-Line Therapy for HER2-Negative Locally Recurrent or Metastatic Breast Cancer: A Phase 2 Randomized Study. Breast (2015) 24:182–90. doi: 10.1016/j.breast.2014.11.003
70. Kienast Y, Klein C, Scheuer W, Raemsch R, Lorenzon E, Bernicke D, et al. Ang-2-VEGF-A CrossMab, a Novel Bispecific Human IgG1 Antibody Blocking VEGF-A and Ang-2 Functions Simultaneously, Mediates Potent Antitumor, Antiangiogenic, and Antimetastatic Efficacy. Clin Cancer Res (2013) 19:6730–40. doi: 10.1158/1078-0432.CCR-13-0081
71. Bendell JC, Sauri T, Gracian AC, Alvarez R, Lopez-Lopez C, Garcia-Alfonso P, et al. The McCAVE Trial: Vanucizumab Plus mFOLFOX-6 Versus Bevacizumab Plus mFOLFOX-6 in Patients With Previously Untreated Metastatic Colorectal Carcinoma (mCRC). Oncologist (2020) 25:e451–9. doi: 10.1634/theoncologist.2019-0291
72. Holmes DI, Zachary I. The Vascular Endothelial Growth Factor (VEGF) Family: Angiogenic Factors in Health and Disease. Genome Biol (2005) 6:209. doi: 10.1186/gb-2005-6-2-209
73. Kapoor P, Deshmukh R. VEGF: A Critical Driver for Angiogenesis and Subsequent Tumor Growth: An IHC Study. J Oral Maxillofac Pathol (2012) 16:330–7. doi: 10.4103/0973-029X.102478
74. Shweiki D, Itin A, Soffer D, Keshet E. Vascular Endothelial Growth Factor Induced by Hypoxia may Mediate Hypoxia-Initiated Angiogenesis. Nature (1992) 359:843–5. doi: 10.1038/359843a0
75. Minchenko A, Bauer T, Salceda S, Caro J. Hypoxic Stimulation of Vascular Endothelial Growth Factor Expression In Vitro and In Vivo. Lab Invest (1994) 71:374–9. https://pubmed.ncbi.nlm.nih.gov/7933988/.
76. Benjamin LE, Keshet E. Conditional Switching of Vascular Endothelial Growth Factor (VEGF) Expression in Tumors: Induction of Endothelial Cell Shedding and Regression of Hemangioblastoma-Like Vessels by VEGF Withdrawal. Proc Natl Acad Sci USA (1997) 94:8761–6. doi: 10.1073/pnas.94.16.8761
77. Shibuya M. Vascular Endothelial Growth Factor (VEGF) and Its Receptor (VEGFR) Signaling in Angiogenesis: A Crucial Target for Anti- and Pro-Angiogenic Therapies. Genes Cancer (2011) 2:1097–105. doi: 10.1177/1947601911423031
78. Presta LG, Chen H, O’connor SJ, Chisholm V, Meng YG, Krummen L, et al. Humanization of an Anti-Vascular Endothelial Growth Factor Monoclonal Antibody for the Therapy of Solid Tumors and Other Disorders. Cancer Res (1997) 57:4593–9. https://cancerres.aacrjournals.org/content/57/20/4593.long.
79. Miller K, Wang M, Gralow J, Dickler M, Cobleigh M, Perez EA, et al. Paclitaxel Plus Bevacizumab Versus Paclitaxel Alone for Metastatic Breast Cancer. N Engl J Med (2007) 357:2666–76. doi: 10.1056/NEJMoa072113
80. Gray R, Bhattacharya S, Bowden C, Miller K, Comis RL. Independent Review of E2100: A Phase III Trial of Bevacizumab Plus Paclitaxel Versus Paclitaxel in Women With Metastatic Breast Cancer. J Clin Oncol (2009) 27:4966–72. doi: 10.1200/JCO.2008.21.6630
81. Tolaney SM, Boucher Y, Duda DG, Martin JD, Seano G, Ancukiewicz M, et al. Role of Vascular Density and Normalization in Response to Neoadjuvant Bevacizumab and Chemotherapy in Breast Cancer Patients. Proc Natl Acad Sci USA (2015) 112:14325–30. doi: 10.1073/pnas.1518808112
82. Xu Y, Li Q, Li XY, Yang QY, Xu WW, Liu GL. Short-Term Anti-Vascular Endothelial Growth Factor Treatment Elicits Vasculogenic Mimicry Formation of Tumors to Accelerate Metastasis. J Exp Clin Cancer Res (2012) 31:16. doi: 10.1186/PREACCEPT-1800076486556870
83. Hendrix MJ, Seftor EA, Hess AR, Seftor RE. Vasculogenic Mimicry and Tumour-Cell Plasticity: Lessons From Melanoma. Nat Rev Cancer (2003) 3:411–21. doi: 10.1038/nrc1092
84. Graeber TG, Osmanian C, Jacks T, Housman DE, Koch CJ, Lowe SW, et al. Hypoxia-Mediated Selection of Cells With Diminished Apoptotic Potential in Solid Tumours. Nature (1996) 379:88–91. doi: 10.1038/379088a0
85. Ueda S, Saeki T, Osaki A, Yamane T, Kuji I. Bevacizumab Induces Acute Hypoxia and Cancer Progression in Patients With Refractory Breast Cancer: Multimodal Functional Imaging and Multiplex Cytokine Analysis. Clin Cancer Res (2017) 23:5769–78. doi: 10.1158/1078-0432.CCR-17-0874
86. Cacheux W, Boisserie T, Staudacher L, Vignaux O, Dousset B, Soubrane O, et al. Reversible Tumor Growth Acceleration Following Bevacizumab Interruption in Metastatic Colorectal Cancer Patients Scheduled for Surgery. Ann Oncol (2008) 19:1659–61. doi: 10.1093/annonc/mdn540
87. Stein WD, Yang J, Bates SE, Fojo T. Bevacizumab Reduces the Growth Rate Constants of Renal Carcinomas: A Novel Algorithm Suggests Early Discontinuation of Bevacizumab Resulted in a Lack of Survival Advantage. Oncologist (2008) 13:1055–62. doi: 10.1634/theoncologist.2008-0016
88. Alahmari AK, Almalki ZS, Alahmari AK, Guo JJ. Thromboembolic Events Associated With Bevacizumab Plus Chemotherapy for Patients With Colorectal Cancer: A Meta-Analysis of Randomized Controlled Trials. Am Health Drug Benefits (2016) 9:221–32. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5004819/.
89. An MM, Zou Z, Shen H, Liu P, Chen ML, Cao YB, et al. Incidence and Risk of Significantly Raised Blood Pressure in Cancer Patients Treated With Bevacizumab: An Updated Meta-Analysis. Eur J Clin Pharmacol (2010) 66:813–21. doi: 10.1007/s00228-010-0815-4
90. Allison M. Avastin’s Commercial March Suffers Setback. Nat Biotechnol (2010) 28:879–80. doi: 10.1038/nbt0910-879
91. Ferrara N, Adamis AP. Ten Years of Anti-Vascular Endothelial Growth Factor Therapy. Nat Rev Drug Discov (2016) 15:385–403. doi: 10.1038/nrd.2015.17
92. Takeshita K, Satoh M, Ii M, Silver M, Limbourg FP, Mukai Y, et al. Critical Role of Endothelial Notch1 Signaling in Postnatal Angiogenesis. Circ Res (2007) 100:70–8. doi: 10.1161/01.RES.0000254788.47304.6e
93. Andersson ER, Sandberg R, Lendahl U. Notch Signaling: Simplicity in Design, Versatility in Function. Development (2011) 138:3593–612. doi: 10.1242/dev.063610
94. Diez H, Fischer A, Winkler A, Hu CJ, Hatzopoulos AK, Breier G, et al. Hypoxia-Mediated Activation of Dll4-Notch-Hey2 Signaling in Endothelial Progenitor Cells and Adoption of Arterial Cell Fate. Exp Cell Res (2007) 313:1–9. doi: 10.1016/j.yexcr.2006.09.009
95. Leslie JD, Ariza-Mcnaughton L, Bermange AL, Mcadow R, Johnson SL, Lewis J. Endothelial Signalling by the Notch Ligand Delta-Like 4 Restricts Angiogenesis. Development (2007) 134:839–44. doi: 10.1242/dev.003244
96. Lobov IB, Renard RA, Papadopoulos N, Gale NW, Thurston G, Yancopoulos GD, et al. Delta-Like Ligand 4 (Dll4) is Induced by VEGF as a Negative Regulator of Angiogenic Sprouting. Proc Natl Acad Sci USA (2007) 104:3219–24. doi: 10.1073/pnas.0611206104
97. Boareto M, Jolly MK, Ben-Jacob E, Onuchic JN. Jagged Mediates Differences in Normal and Tumor Angiogenesis by Affecting Tip-Stalk Fate Decision. Proc Natl Acad Sci USA (2015) 112:E3836–44. doi: 10.1073/pnas.1511814112
98. Somanath PR, Razorenova OV, Chen J, Byzova TV. Akt1 in Endothelial Cell and Angiogenesis. Cell Cycle (2006) 5:512–8. doi: 10.4161/cc.5.5.2538
99. Deng Y, Zhang X, Simons M. Molecular Controls of Lymphatic VEGFR3 Signaling. Arterioscler Thromb Vasc Biol (2015) 35:421–9. doi: 10.1161/ATVBAHA.114.304881
100. Funahashi Y, Hernandez SL, Das I, Ahn A, Huang J, Vorontchikhina M, et al. A Notch1 Ectodomain Construct Inhibits Endothelial Notch Signaling, Tumor Growth, and Angiogenesis. Cancer Res (2008) 68:4727–35. doi: 10.1158/0008-5472.CAN-07-6499
101. Hoey T, Yen WC, Axelrod F, Basi J, Donigian L, Dylla S, et al. DLL4 Blockade Inhibits Tumor Growth and Reduces Tumor-Initiating Cell Frequency. Cell Stem Cell (2009) 5:168–77. doi: 10.1016/j.stem.2009.05.019
102. Haller BK, Brave A, Wallgard E, Roswall P, Sunkari VG, Mattson U, et al. Therapeutic Efficacy of a DNA Vaccine Targeting the Endothelial Tip Cell Antigen Delta-Like Ligand 4 in Mammary Carcinoma. Oncogene (2010) 29:4276–86. doi: 10.1038/onc.2010.176
103. Li JL, Sainson RC, Shi W, Leek R, Harrington LS, Preusser M, et al. Delta-Like 4 Notch Ligand Regulates Tumor Angiogenesis, Improves Tumor Vascular Function, and Promotes Tumor Growth In Vivo. Cancer Res (2007) 67:11244–53. doi: 10.1158/0008-5472.CAN-07-0969
104. Yan M, Callahan CA, Beyer JC, Allamneni KP, Zhang G, Ridgway JB, et al. Chronic DLL4 Blockade Induces Vascular Neoplasms. Nature (2010) 463:E6–7. doi: 10.1038/nature08751
105. Mukhopadhyay A. OncoMed Sinks as Cancer Drug Fails; Bayer Opts Against Licensing Drugs. HEALTHCARE PHARMA (2017).
106. Imbimbo BP. Therapeutic Potential of Gamma-Secretase Inhibitors and Modulators. Curr Top Med Chem (2008) 8:54–61. doi: 10.2174/156802608783334015
107. Aster JC, Blacklow SC, Pear WS. Notch Signalling in T-Cell Lymphoblastic Leukaemia/Lymphoma and Other Haematological Malignancies. J Pathol (2011) 223:262–73. doi: 10.1002/path.2789
108. Hemming ML, Elias JE, Gygi SP, Selkoe DJ. Proteomic Profiling of Gamma-Secretase Substrates and Mapping of Substrate Requirements. PloS Biol (2008) 6:e257. doi: 10.1371/journal.pbio.0060257
109. Patel NS, Li JL, Generali D, Poulsom R, Cranston DW, Harris AL. Up-Regulation of Delta-Like 4 Ligand in Human Tumor Vasculature and the Role of Basal Expression in Endothelial Cell Function. Cancer Res (2005) 65:8690–7. doi: 10.1158/0008-5472.CAN-05-1208
110. Trindade A, Djokovic D, Gigante J, Mendonca L, Duarte A. Endothelial Dll4 Overexpression Reduces Vascular Response and Inhibits Tumor Growth and Metastasization In Vivo. BMC Cancer (2017) 17:189. doi: 10.1186/s12885-017-3171-2
111. Vergote I, Scambia G, O’malley DM, Van Calster B, Park SY, Del Campo JM, et al. Trebananib or Placebo Plus Carboplatin and Paclitaxel as First-Line Treatment for Advanced Ovarian Cancer (TRINOVA-3/ENGOT-Ov2/GOG-3001): A Randomised, Double-Blind, Phase 3 Trial. Lancet Oncol (2019) 20:862–76. doi: 10.1016/S1470-2045(19)30178-0
112. Thurston G, Rudge JS, Ioffe E, Zhou H, Ross L, Croll SD, et al. Angiopoietin-1 Protects the Adult Vasculature Against Plasma Leakage. Nat Med (2000) 6:460–3. doi: 10.1038/74725
113. Scharpfenecker M, Fiedler U, Reiss Y, Augustin HG. The Tie-2 Ligand Angiopoietin-2 Destabilizes Quiescent Endothelium Through an Internal Autocrine Loop Mechanism. J Cell Sci (2005) 118:771–80. doi: 10.1242/jcs.01653
114. Holash J, Maisonpierre PC, Compton D, Boland P, Alexander CR, Zagzag D, et al. Vessel Cooption, Regression, and Growth in Tumors Mediated by Angiopoietins and VEGF. Science (1999) 284:1994–8. doi: 10.1126/science.284.5422.1994
115. Lobov IB, Brooks PC, Lang RA. Angiopoietin-2 Displays VEGF-Dependent Modulation of Capillary Structure and Endothelial Cell Survival In Vivo. Proc Natl Acad Sci USA (2002) 99:11205–10. doi: 10.1073/pnas.172161899
116. Imanishi Y, Hu B, Jarzynka MJ, Guo P, Elishaev E, Bar-Joseph I, et al. Angiopoietin-2 Stimulates Breast Cancer Metastasis Through the Alpha(5)Beta(1) Integrin-Mediated Pathway. Cancer Res (2007) 67:4254–63. doi: 10.1158/0008-5472.CAN-06-4100
117. Mita AC, Takimoto CH, Mita M, Tolcher A, Sankhala K, Sarantopoulos J, et al. Phase 1 Study of AMG 386, a Selective Angiopoietin 1/2-Neutralizing Peptibody, in Combination With Chemotherapy in Adults With Advanced Solid Tumors. Clin Cancer Res (2010) 16:3044–56. doi: 10.1158/1078-0432.CCR-09-3368
118. Winkler F, Kozin SV, Tong RT, Chae SS, Booth MF, Garkavtsev I, et al. Kinetics of Vascular Normalization by VEGFR2 Blockade Governs Brain Tumor Response to Radiation: Role of Oxygenation, Angiopoietin-1, and Matrix Metalloproteinases. Cancer Cell (2004) 6:553–63. doi: 10.1016/S1535-6108(04)00305-8
119. Porcu E, Maule F, Boso D, Rampazzo E, Barbieri V, Zuccolotto G, et al. BMP9 Counteracts the Tumorigenic and Pro-Angiogenic Potential of Glioblastoma. Cell Death Differ (2018) 25:1808–22. doi: 10.1038/s41418-018-0149-9
120. Hu-Lowe DD, Chen E, Zhang L, Watson KD, Mancuso P, Lappin P, et al. Targeting Activin Receptor-Like Kinase 1 Inhibits Angiogenesis and Tumorigenesis Through a Mechanism of Action Complementary to Anti-VEGF Therapies. Cancer Res (2011) 71:1362–73. doi: 10.1158/0008-5472.CAN-10-1451
121. Bendell JC, Gordon MS, Hurwitz HI, Jones SF, Mendelson DS, Blobe GC, et al. Safety, Pharmacokinetics, Pharmacodynamics, and Antitumor Activity of Dalantercept, an Activin Receptor-Like Kinase-1 Ligand Trap, in Patients With Advanced Cancer. Clin Cancer Res (2014) 20:480–9. doi: 10.1158/1078-0432.CCR-13-1840
122. Makker V, Filiaci VL, Chen LM, Darus CJ, Kendrick JE, Sutton G, et al. Phase II Evaluation of Dalantercept, a Soluble Recombinant Activin Receptor-Like Kinase 1 (ALK1) Receptor Fusion Protein, for the Treatment of Recurrent or Persistent Endometrial Cancer: An NRG Oncology/Gynecologic Oncology Group Study 0229n. Gynecol Oncol (2015) 138:24–9. doi: 10.1016/j.ygyno.2015.04.006
123. Voss MH, Bhatt RS, Vogelzang NJ, Fishman M, Alter RS, Rini BI, et al. A Phase 2, Randomized Trial Evaluating the Combination of Dalantercept Plus Axitinib in Patients With Advanced Clear Cell Renal Cell Carcinoma. Cancer (2019) 125:2400–8. doi: 10.1002/cncr.32061
124. Jimeno A, Posner MR, Wirth LJ, Saba NF, Cohen RB, Popa EC, et al. A Phase 2 Study of Dalantercept, an Activin Receptor-Like Kinase-1 Ligand Trap, in Patients With Recurrent or Metastatic Squamous Cell Carcinoma of the Head and Neck. Cancer (2016) 122:3641–9. doi: 10.1002/cncr.30317
125. Burger RA, Deng W, Makker V, Collins Y, Gray H, Debernardo R, et al. Phase II Evaluation of Dalantercept in the Treatment of Persistent or Recurrent Epithelial Ovarian Cancer: An NRG Oncology/Gynecologic Oncology Group Study. Gynecol Oncol (2018) 150:466–70. doi: 10.1016/j.ygyno.2018.06.017
126. Dirsch VM, Kiemer AK, Wagner H, Vollmar AM. The Triterpenoid Quinonemethide Pristimerin Inhibits Induction of Inducible Nitric Oxide Synthase in Murine Macrophages. Eur J Pharmacol (1997) 336:211–7. doi: 10.1016/S0014-2999(97)01245-4
127. Mu X, Shi W, Sun L, Li H, Jiang Z, Zhang L. Pristimerin, a Triterpenoid, Inhibits Tumor Angiogenesis by Targeting VEGFR2 Activation. Molecules (2012) 17:6854–68. doi: 10.3390/molecules17066854
128. Lei X, Zhong Y, Huang L, Li S, Fu J, Zhang L, et al. Identification of a Novel Tumor Angiogenesis Inhibitor Targeting Shh/Gli1 Signaling Pathway in Non-Small Cell Lung Cancer. Cell Death Dis (2020) 11:232. doi: 10.1038/s41419-020-2425-0
129. Howe LR, Subbaramaiah K, Patel J, Masferrer JL, Deora A, Hudis C, et al. Celecoxib, a Selective Cyclooxygenase 2 Inhibitor, Protects Against Human Epidermal Growth Factor Receptor 2 (HER-2)/Neu-Induced Breast Cancer. Cancer Res (2002) 62:5405–7. https://cancerres.aacrjournals.org/content/62/19/5405.long.
130. Yoshinaka R, Shibata MA, Morimoto J, Tanigawa N, Otsuki Y. COX-2 Inhibitor Celecoxib Suppresses Tumor Growth and Lung Metastasis of a Murine Mammary Cancer. Anticancer Res (2006) 26:4245–54 https://ar.iiarjournals.org/content/26/6B/4245.
131. Solomon SD, Mcmurray JJ, Pfeffer MA, Wittes J, Fowler R, Finn P, et al. Cardiovascular Risk Associated With Celecoxib in a Clinical Trial for Colorectal Adenoma Prevention. N Engl J Med (2005) 352:1071–80. doi: 10.1056/NEJMoa050405
132. Limoge M, Safina A, Beattie A, Kapus L, Truskinovsky AM, Bakin AV. Tumor-Fibroblast Interactions Stimulate Tumor Vascularization by Enhancing Cytokine-Driven Production of MMP9 by Tumor Cells. Oncotarget (2017) 8:35592–608. doi: 10.18632/oncotarget.16022
133. Patnaik A, Haluska P, Tolcher AW, Erlichman C, Papadopoulos KP, Lensing JL, et al. A First-In-Human Phase I Study of the Oral P38 MAPK Inhibitor, Ralimetinib (LY2228820 Dimesylate), in Patients With Advanced Cancer. Clin Cancer Res (2016) 22:1095–102. doi: 10.1158/1078-0432.CCR-15-1718
134. Vergote I, Heitz F, Buderath P, Powell M, Sehouli J, Lee CM, et al. A Randomized, Double-Blind, Placebo-Controlled Phase 1b/2 Study of Ralimetinib, a P38 MAPK Inhibitor, Plus Gemcitabine and Carboplatin Versus Gemcitabine and Carboplatin for Women With Recurrent Platinum-Sensitive Ovarian Cancer. Gynecol Oncol (2020) 156:23–31. doi: 10.1016/j.ygyno.2019.11.006
135. Liu D, Wang J, Kinzel B, Mueller M, Mao X, Valdez R, et al. Dosage-Dependent Requirement of BMP Type II Receptor for Maintenance of Vascular Integrity. Blood (2007) 110:1502–10. doi: 10.1182/blood-2006-11-058594
136. David L, Mallet C, Keramidas M, Lamande N, Gasc JM, Dupuis-Girod S, et al. Bone Morphogenetic Protein-9 is a Circulating Vascular Quiescence Factor. Circ Res (2008) 102:914–22. doi: 10.1161/CIRCRESAHA.107.165530
137. Vattulainen-Collanus S, Southwood M, Yang XD, Moore S, Ghatpande P, Morrell NW, et al. Bone Morphogenetic Protein Signaling is Required for RAD51-Mediated Maintenance of Genome Integrity in Vascular Endothelial Cells. Commun Biol (2018) 1:149. doi: 10.1038/s42003-018-0152-1
138. Upton PD, Davies RJ, Trembath RC, Morrell NW. Bone Morphogenetic Protein (BMP) and Activin Type II Receptors Balance BMP9 Signals Mediated by Activin Receptor-Like Kinase-1 in Human Pulmonary Artery Endothelial Cells. J Biol Chem (2009) 284:15794–804. doi: 10.1074/jbc.M109.002881
139. Tillet E, Ouarne M, Desroches-Castan A, Mallet C, Subileau M, Didier R, et al. A Heterodimer Formed by Bone Morphogenetic Protein 9 (BMP9) and BMP10 Provides Most BMP Biological Activity in Plasma. J Biol Chem (2018) 293:10963–74. doi: 10.1074/jbc.RA118.002968
140. Sapkota G, Alarcon C, Spagnoli FM, Brivanlou AH, Massague J. Balancing BMP Signaling Through Integrated Inputs Into the Smad1 Linker. Mol Cell (2007) 25:441–54. doi: 10.1016/j.molcel.2007.01.006
141. Eivers E, Demagny H, De Robertis EM. Integration of BMP and Wnt Signaling via Vertebrate Smad1/5/8 and Drosophila Mad. Cytokine Growth Factor Rev (2009) 20:357–65. doi: 10.1016/j.cytogfr.2009.10.017
142. Larrivee B, Prahst C, Gordon E, Del Toro R, Mathivet T, Duarte A, et al. ALK1 Signaling Inhibits Angiogenesis by Cooperating With the Notch Pathway. Dev Cell (2012) 22:489–500. doi: 10.1016/j.devcel.2012.02.005
143. Hurst LA, Dunmore BJ, Long L, Crosby A, Al-Lamki R, Deighton J, et al. Tnfα Drives Pulmonary Arterial Hypertension by Suppressing the BMP Type-II Receptor and Altering NOTCH Signalling. Nat Commun (2017) 8. doi: 10.1038/ncomms14079
144. Scharpfenecker M, Van Dinther M, Liu Z, Van Bezooijen RL, Zhao Q, Pukac L, et al. BMP-9 Signals via ALK1 and Inhibits bFGF-Induced Endothelial Cell Proliferation and VEGF-Stimulated Angiogenesis. J Cell Sci (2007) 120:964–72. doi: 10.1242/jcs.002949
145. Long L, Ormiston ML, Yang X, Southwood M, Graf S, Machado RD, et al. Selective Enhancement of Endothelial BMPR-II With BMP9 Reverses Pulmonary Arterial Hypertension. Nat Med (2015) 21:777–85. doi: 10.1038/nm.3877
146. Lamouille S, Mallet C, Feige JJ, Bailly S. Activin Receptor-Like Kinase 1 is Implicated in the Maturation Phase of Angiogenesis. Blood (2002) 100:4495–501. doi: 10.1182/blood.V100.13.4495
147. Baeyens N, Larrivee B, Ola R, Hayward-Piatkowskyi B, Dubrac A, Huang B, et al. Defective Fluid Shear Stress Mechanotransduction Mediates Hereditary Hemorrhagic Telangiectasia. J Cell Biol (2016) 214:807–16. doi: 10.1083/jcb.201603106
148. Ntumba K, Akla N, Oh SP, Eichmann A, Larrivee B. BMP9/ALK1 Inhibits Neovascularization in Mouse Models of Age-Related Macular Degeneration. Oncotarget (2016) 7:55957–69. doi: 10.18632/oncotarget.11182
149. Ouarne M, Bouvard C, Boneva G, Mallet C, Ribeiro J, Desroches-Castan A, et al. BMP9, But Not BMP10, Acts as a Quiescence Factor on Tumor Growth, Vessel Normalization and Metastasis in a Mouse Model of Breast Cancer. J Exp Clin Cancer Res (2018) 37:209. doi: 10.1186/s13046-018-0885-1
150. Ye L, Kynaston H, Jiang WG. Bone Morphogenetic Protein-9 Induces Apoptosis in Prostate Cancer Cells, the Role of Prostate Apoptosis Response-4. Mol Cancer Res (2008) 6:1594–606. doi: 10.1158/1541-7786.MCR-08-0171
151. Herrera B, Van Dinther M, Ten Dijke P, Inman GJ. Autocrine Bone Morphogenetic Protein-9 Signals Through Activin Receptor-Like Kinase-2/Smad1/Smad4 to Promote Ovarian Cancer Cell Proliferation. Cancer Res (2009) 69:9254–62. doi: 10.1158/0008-5472.CAN-09-2912
152. Ye L, Kynaston H, Jiang WG. Bone Morphogenetic Protein-10 Suppresses the Growth and Aggressiveness of Prostate Cancer Cells Through a Smad Independent Pathway. J Urol (2009) 181:2749–59. doi: 10.1016/j.juro.2009.01.098
153. Herrera B, Garcia-Alvaro M, Cruz S, Walsh P, Fernandez M, Roncero C, et al. BMP9 is a Proliferative and Survival Factor for Human Hepatocellular Carcinoma Cells. PloS One (2013) 8:e69535. doi: 10.1371/journal.pone.0069535
154. Eleftheriou NM, Sjolund J, Bocci M, Cortez E, Lee SJ, Cunha SI, et al. Compound Genetically Engineered Mouse Models of Cancer Reveal Dual Targeting of ALK1 and Endoglin as a Synergistic Opportunity to Impinge on Angiogenic TGF-Beta Signaling. Oncotarget (2016) 7:84314–25. doi: 10.18632/oncotarget.12604
155. Gou L, Liu M, Xia J, Wan Q, Jiang Y, Sun S, et al. BMP9 Promotes the Proliferation and Migration of Bladder Cancer Cells Through Up-Regulating lncRNA Uca1. Int J Mol Sci (2018) 19:1116. doi: 10.3390/ijms19041116
156. Cunha SI, Pardali E, Thorikay M, Anderberg C, Hawinkels L, Goumans MJ, et al. Genetic and Pharmacological Targeting of Activin Receptor-Like Kinase 1 Impairs Tumor Growth and Angiogenesis. J Exp Med (2010) 207:85–100. doi: 10.1084/jem.20091309
157. Suzuki Y, Ohga N, Morishita Y, Hida K, Miyazono K, Watabe T. BMP-9 Induces Proliferation of Multiple Types of Endothelial Cells In Vitro and In Vivo. J Cell Sci (2010) 123:1684–92. doi: 10.1242/jcs.061556
158. Goumans MJ, Valdimarsdottir G, Itoh S, Lebrin F, Larsson J, Mummery C, et al. Activin Receptor-Like Kinase (ALK)1 is an Antagonistic Mediator of Lateral TGFbeta/ALK5 Signaling. Mol Cell (2003) 12:817–28. doi: 10.1016/S1097-2765(03)00386-1
159. Van Meeteren LA, Thorikay M, Bergqvist S, Pardali E, Stampino CG, Hu-Lowe D, et al. Anti-Human Activin Receptor-Like Kinase 1 (ALK1) Antibody Attenuates Bone Morphogenetic Protein 9 (BMP9)-Induced ALK1 Signaling and Interferes With Endothelial Cell Sprouting. J Biol Chem (2012) 287:18551–61. doi: 10.1074/jbc.M111.338103
160. Voss MH, Bhatt RS, Plimack ER, Rini BI, Alter RS, Beck JT, et al. The DART Study: Results From the Dose-Escalation and Expansion Cohorts Evaluating the Combination of Dalantercept Plus Axitinib in Advanced Renal Cell Carcinoma. Clin Cancer Res (2017) 23:3557–65. doi: 10.1158/1078-0432.CCR-16-2395
161. David L, Feige JJ, Bailly S. Emerging Role of Bone Morphogenetic Proteins in Angiogenesis. Cytokine Growth Factor Rev (2009) 20:203–12. doi: 10.1016/j.cytogfr.2009.05.001
162. Ye L, Mason MD, Jiang WG. Bone Morphogenetic Protein and Bone Metastasis, Implication and Therapeutic Potential. Front Biosci (Landmark Ed) (2011) 16:865–97. doi: 10.2741/3725
163. Ormiston ML, Upton PD, Li W, Morrell NW. The Promise of Recombinant BMP Ligands and Other Approaches Targeting BMPR-II in the Treatment of Pulmonary Arterial Hypertension. Glob Cardiol Sci Pract (2015) 2015:47. doi: 10.5339/gcsp.2015.47
164. Theilmann AL, Hawke LG, Hilton LR, Whitford MKM, Cole DV, Mackeil JL, et al. Endothelial BMPR2 Loss Drives a Proliferative Response to BMP (Bone Morphogenetic Protein) 9 via Prolonged Canonical Signaling. Arterioscler Thromb Vasc Biol (2020) 40:2605–18. doi: 10.1161/ATVBAHA.119.313357
165. Di Mauro C, Rosa R, D’amato V, Ciciola P, Servetto A, Marciano R, et al. Hedgehog Signalling Pathway Orchestrates Angiogenesis in Triple-Negative Breast Cancers. Br J Cancer (2017) 116:1425–35. doi: 10.1038/bjc.2017.116
166. Carballo GB, Honorato JR, De Lopes GPF, Spohr T. A Highlight on Sonic Hedgehog Pathway. Cell Commun Signal (2018) 16:11. doi: 10.1186/s12964-018-0220-7
167. Blotta S, Jakubikova J, Calimeri T, Roccaro AM, Amodio N, Azab AK, et al. Canonical and Noncanonical Hedgehog Pathway in the Pathogenesis of Multiple Myeloma. Blood (2012) 120:5002–13. doi: 10.1182/blood-2011-07-368142
168. Lee H, Ko HW. Ciliary Smoothened-Mediated Noncanonical Hedgehog Signaling Promotes Tubulin Acetylation. Biochem Biophys Res Commun (2016) 480:574–9. doi: 10.1016/j.bbrc.2016.10.093
169. Taipale J, Cooper MK, Maiti T, Beachy PA. Patched Acts Catalytically to Suppress the Activity of Smoothened. Nature (2002) 418:892–7. doi: 10.1038/nature00989
170. Chen SC, Huang M, He QW, Zhang Y, Opoku EN, Yang H, et al. Administration of Sonic Hedgehog Protein Induces Angiogenesis and has Therapeutic Effects After Stroke in Rats. Neuroscience (2017) 352:285–95. doi: 10.1016/j.neuroscience.2017.03.054
171. Lin Y, Shao Y, Li J, Zhang W, Zheng K, Zheng X, et al. The Hierarchical Micro-/Nanotextured Topographies Promote the Proliferation and Angiogenesis-Related Genes Expression in Human Umbilical Vein Endothelial Cells by Initiation of Hedgehog-Gli1 Signaling. Artif Cells Nanomed Biotechnol (2018) 46:S1141–51. doi: 10.1080/21691401.2018.1533845
172. Cao X, Geradts J, Dewhirst MW, Lo HW. Upregulation of VEGF-A and CD24 Gene Expression by the Tgli1 Transcription Factor Contributes to the Aggressive Behavior of Breast Cancer Cells. Oncogene (2012) 31:104–15. doi: 10.1038/onc.2011.219
173. Cui D, Chen X, Yin J, Wang W, Lou M, Gu S. Aberrant Activation of Hedgehog/Gli1 Pathway on Angiogenesis in Gliomas. Neurol India (2012) 60:589–96. doi: 10.4103/0028-3886.105192
174. Harris LG, Pannell LK, Singh S, Samant RS, Shevde LA. Increased Vascularity and Spontaneous Metastasis of Breast Cancer by Hedgehog Signaling Mediated Upregulation of Cyr61. Oncogene (2012) 31:3370–80. doi: 10.1038/onc.2011.496
175. Goel HL, Pursell B, Chang C, Shaw LM, Mao J, Simin K, et al. GLI1 Regulates a Novel Neuropilin-2/Alpha6beta1 Integrin Based Autocrine Pathway That Contributes to Breast Cancer Initiation. EMBO Mol Med (2013) 5:488–508. doi: 10.1002/emmm.201202078
176. Duan ZH, Wang HC, Zhao DM, Ji XX, Song M, Yang XJ, et al. Cooperatively Transcriptional and Epigenetic Regulation of Sonic Hedgehog Overexpression Drives Malignant Potential of Breast Cancer. Cancer Sci (2015) 106:1084–91. doi: 10.1111/cas.12697
177. Riaz SK, Khan JS, Shah STA, Wang F, Ye L, Jiang WG, et al. Involvement of Hedgehog Pathway in Early Onset, Aggressive Molecular Subtypes and Metastatic Potential of Breast Cancer. Cell Commun Signal (2018) 16:3. doi: 10.1186/s12964-017-0213-y
178. Riobo-Del Galdo NA, Lara Montero A, Wertheimer EV. Role of Hedgehog Signaling in Breast Cancer: Pathogenesis and Therapeutics. Cells (2019) 8:375. doi: 10.3390/cells8040375
179. Cevatemre B, Erkisa M, Aztopal N, Karakas D, Alper P, Tsimplouli C, et al. And Ulukaya, EA Promising Natural Product, Pristimerin, Results in Cytotoxicity Against Breast Cancer Stem Cells In Vitro and Xenografts In Vivo Through Apoptosis and an Incomplete Autopaghy in Breast Cancer. Pharmacol Res (2018) 129:500–14. doi: 10.1016/j.phrs.2017.11.027
180. Huang S, He P, Peng X, Li J, Xu D, Tang Y. Pristimerin Inhibits Prostate Cancer Bone Metastasis by Targeting PC-3 Stem Cell Characteristics and VEGF-Induced Vasculogenesis of BM-EPCs. Cell Physiol Biochem (2015) 37:253–68. doi: 10.1159/000430350
181. Cheng S, Zhang Z, Hu C, Xing N, Xia Y, Pang B. Pristimerin Suppressed Breast Cancer Progression via miR-542-5p/DUB3 Axis. Onco Targets Ther (2020) 13:6651–60. doi: 10.2147/OTT.S257329
182. St-Louis I, Singh M, Brasseur K, Leblanc V, Parent S, Asselin E. Expression of COX-1 and COX-2 in the Endometrium of Cyclic, Pregnant and in a Model of Pseudopregnant Rats and Their Regulation by Sex Steroids. Reprod Biol Endocrinol (2010) 8:103. doi: 10.1186/1477-7827-8-103
183. Wu G, Luo J, Rana JS, Laham R, Sellke FW, Li J. Involvement of COX-2 in VEGF-Induced Angiogenesis via P38 and JNK Pathways in Vascular Endothelial Cells. Cardiovasc Res (2006) 69:512–9. doi: 10.1016/j.cardiores.2005.09.019
184. Xu K, Shu HK. EGFR Activation Results in Enhanced Cyclooxygenase-2 Expression Through P38 Mitogen-Activated Protein Kinase-Dependent Activation of the Sp1/Sp3 Transcription Factors in Human Gliomas. Cancer Res (2007) 67:6121–9. doi: 10.1158/0008-5472.CAN-07-0141
185. Xu L, Stevens J, Hilton MB, Seaman S, Conrads TP, Veenstra TD, et al. COX-2 Inhibition Potentiates Antiangiogenic Cancer Therapy and Prevents Metastasis in Preclinical Models. Sci Transl Med (2014) 6:242ra284. doi: 10.1126/scitranslmed.3008455
186. Ristimaki A, Sivula A, Lundin J, Lundin M, Salminen T, Haglund C, et al. Prognostic Significance of Elevated Cyclooxygenase-2 Expression in Breast Cancer. Cancer Res (2002) 62:632–5 https://cancerres.aacrjournals.org/content/62/3/632.long.
187. Bresalier RS, Sandler RS, Quan H, Bolognese JA, Oxenius B, Horgan K, et al. Cardiovascular Events Associated With Rofecoxib in a Colorectal Adenoma Chemoprevention Trial. N Engl J Med (2005) 352:1092–102. doi: 10.1056/NEJMoa050493
188. Baron JA, Sandler RS, Bresalier RS, Quan H, Riddell R, Lanas A, et al. A Randomized Trial of Rofecoxib for the Chemoprevention of Colorectal Adenomas. Gastroenterology (2006) 131:1674–82. doi: 10.1053/j.gastro.2006.08.079
189. Zarubin T, Han J. Activation and Signaling of the P38 MAP Kinase Pathway. Cell Res (2005) 15:11–8. doi: 10.1038/sj.cr.7290257
190. Limoge M, Safina A, Truskinovsky AM, Aljahdali I, Zonneville J, Gruevski A, et al. Tumor P38mapk Signaling Enhances Breast Carcinoma Vascularization and Growth by Promoting Expression and Deposition of Pro-Tumorigenic Factors. Oncotarget (2017) 8:61969–81. doi: 10.18632/oncotarget.18755
191. Hotchkiss KA, Ashton AW, Klein RS, Lenzi ML, Zhu GH, Schwartz EL. Mechanisms by Which Tumor Cells and Monocytes Expressing the Angiogenic Factor Thymidine Phosphorylase Mediate Human Endothelial Cell Migration. Cancer Res (2003) 63:527–33 https://cancerres.aacrjournals.org/content/63/2/527.long.
192. Fantin A, Vieira JM, Gestri G, Denti L, Schwarz Q, Prykhozhij S, et al. Tissue Macrophages Act as Cellular Chaperones for Vascular Anastomosis Downstream of VEGF-Mediated Endothelial Tip Cell Induction. Blood (2010) 116:829–40. doi: 10.1182/blood-2009-12-257832
193. Schmidt T, Carmeliet P. Blood-Vessel Formation: Bridges That Guide and Unite. Nature (2010) 465:697–9. doi: 10.1038/465697a
194. Campbell RM, Anderson BD, Brooks NA, Brooks HB, Chan EM, De Dios A, et al. Characterization of LY2228820 Dimesylate, a Potent and Selective Inhibitor of P38 MAPK With Antitumor Activity. Mol Cancer Ther (2014) 13:364–74. doi: 10.1158/1535-7163.MCT-13-0513
195. Tate CM, Blosser W, Wyss L, Evans G, Xue Q, Pan Y, et al. LY2228820 Dimesylate, a Selective Inhibitor of P38 Mitogen-Activated Protein Kinase, Reduces Angiogenic Endothelial Cord Formation In Vitro and In Vivo. J Biol Chem (2013) 288:6743–53. doi: 10.1074/jbc.M112.425553
196. Zonneville J, Colligan S, Grant S, Miller A, Wallace P, Abrams SI, et al. Blockade of P38 Kinase Impedes the Mobilization of Protumorigenic Myeloid Populations to Impact Breast Cancer Metastasis. Int J Cancer (2020) 147:2279–92. doi: 10.1002/ijc.33050
197. Rousseau S, Houle F, Landry J, Huot J. P38 MAP Kinase Activation by Vascular Endothelial Growth Factor Mediates Actin Reorganization and Cell Migration in Human Endothelial Cells. Oncogene (1997) 15:2169–77. doi: 10.1038/sj.onc.1201380
198. Goldsmith CS, Kim SM, Karunarathna N, Neuendorff N, Toussaint LG, Earnest DJ, et al. Inhibition of P38 MAPK Activity Leads to Cell Type-Specific Effects on the Molecular Circadian Clock and Time-Dependent Reduction of Glioma Cell Invasiveness. BMC Cancer (2018) 18:43. doi: 10.1186/s12885-017-3896-y
199. Aesoy R, Sanchez BC, Norum JH, Lewensohn R, Viktorsson K, Linderholm B. An Autocrine VEGF/VEGFR2 and P38 Signaling Loop Confers Resistance to 4-Hydroxytamoxifen in MCF-7 Breast Cancer Cells. Mol Cancer Res (2008) 6:1630–8. doi: 10.1158/1541-7786.MCR-07-2172
200. Lin L, Chen YS, Yao YD, Chen JQ, Chen JN, Huang SY, et al. CCL18 From Tumor-Associated Macrophages Promotes Angiogenesis in Breast Cancer. Oncotarget (2015) 6:34758–73. doi: 10.18632/oncotarget.5325
201. Chen J, Yao Y, Gong C, Yu F, Su S, Chen J, et al. CCL18 From Tumor-Associated Macrophages Promotes Breast Cancer Metastasis via PITPNM3. Cancer Cell (2011) 19:541–55. doi: 10.1016/j.ccr.2011.02.006
202. Liu Y, Zheng H, Li Q, Li S, Lai H, Song E, et al. Discovery of CCL18 Antagonist Blocking Breast Cancer Metastasis. Clin Exp Metastasis (2019) 36:243–55. doi: 10.1007/s10585-019-09965-2
Keywords: angiogenesis, breast cancer, vascular endothelial growth factor, bone morphogenetic protein 9, notch signaling
Citation: Harry JA and Ormiston ML (2021) Novel Pathways for Targeting Tumor Angiogenesis in Metastatic Breast Cancer. Front. Oncol. 11:772305. doi: 10.3389/fonc.2021.772305
Received: 07 September 2021; Accepted: 12 November 2021;
Published: 03 December 2021.
Edited by:
Qingxin Mu, University of Washington, United StatesReviewed by:
Luis E Arias-Romero, National Autonomous University of Mexico, MexicoVaishali Aggarwal, University of Pittsburgh, United States
Copyright © 2021 Harry and Ormiston. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mark L. Ormiston, bWFyay5vcm1pc3RvbkBxdWVlbnN1LmNh