 Antonella Nicolò
Antonella Nicolò Alexandra Theresa Linder†
Alexandra Theresa Linder† Hassan Jumaa
Hassan Jumaa Palash Chandra Maity
Palash Chandra Maity
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
PERSPECTIVE article
Front. Oncol. , 21 December 2021
Sec. Hematologic Malignancies
Volume 11 - 2021 | https://doi.org/10.3389/fonc.2021.771669
This article is part of the Research Topic Biology and Treatment of High-Risk CLL View all 16 articles
Advanced genome-wide association studies (GWAS) identified several transforming mutations in susceptible loci which are recognized as valuable prognostic markers in chronic lymphocytic leukemia (CLL) and B cell lymphoma (BCL). Alongside, robust genetic manipulations facilitated the generation of preclinical mouse models to validate mutations associated with poor prognosis and refractory B cell malignancies. Taken together, these studies identified new prognostic markers that could achieve characteristics of precision biomarkers for molecular diagnosis. On the contrary, the idea of augmented B cell antigen receptor (BCR) signaling as a transforming cue has somewhat receded despite the efficacy of Btk and Syk inhibitors. Recent studies from several research groups pointed out that acquired mutations in BCR components serve as faithful biomarkers, which become important for precision diagnostics and therapy, due to their relevant role in augmented BCR signaling and CLL pathogenesis. For example, we showed that expression of a single point mutated immunoglobulin light chain (LC) recombined through the variable gene segment IGLV3-21, named IGLV3-21R110, marks severe CLL cases. In this perspective, we summarize the molecular mechanisms fine-tuning B cell transformation, focusing on immunoglobulin point mutations and recurrent mutations in tumor suppressors. We present a stochastic model for gain-of-autonomous BCR signaling and subsequent neoplastic transformation. Of note, additional mutational analyses on immunoglobulin heavy chain (HC) derived from non-subset #2 CLL IGLV3-21R110 cases endorses our perspective. Altogether, we propose a model of malignant transformation in which the augmented BCR signaling creates a conducive platform for the appearance of transforming mutations.
The B cell antigen receptor (BCR) signaling is the key survival and growth promoter for both normal and malignant B cells, controlling important cell fate decisions including proliferation and differentiation (1). Depending on quality, capacity and relevance, three different types of BCR signaling were described: cell-autonomous, tonic and standard ligand-dependent signaling (Figure 1A). B cells are engineered to control the BCR signaling strength and type during their development step-by-step (1–3). First, the autoreactive clones are self-eliminated by altered BCR activation threshold in the bone marrow compartment. Thereafter, tonic BCR signaling controls cell survival and proliferation, relying on the crosstalks integrating microenvironmental signals and cytoskeletal remodeling (1, 4). Finally, the antigen stimulated BCR signal promotes clonal expansion and differentiation during the germinal center (GC) reaction. A dysregulation of BCR signaling at any of the aforesaid levels, such as a gain-of-function mutation in signaling components, results in altered B cell survival, most often allowing escape from self-elimination process and resulting in primary immunodeficiencies, autoimmune diseases and B cell malignancies (Figure 1B) (5, 6). In particular, mutations or differential membrane organization of receptors which lead to the constitutive activation of the BCR are mainly associated with B cell malignancies, such as CLL and activated B cell-like diffuse large BCL (ABC DLBCL) (1, 2, 7, 8). Although sporadic and non-familial pathogenesis predominates in B cell neoplastic diseases, recent sequencing approaches and GWAS studies provided evidence of an inherited predisposition by identifying risk-associated loci in CLL and DLBCL (9–13). Recurrent mutations are mostly located in the non-coding regions of the genome, affecting active promoters or enhancers (12, 14). Very often, these recurrent genomic aberrations are functionally associated to genomic-instability, anti-apoptosis, and abnormal lymphoproliferation signaling networks (Figure 1B) (9, 11, 15, 16). Notably, the dysregulation of the Bcl-2 family of anti-apoptotic proteins, including Bcl-2, Bcl-xL and Mcl-1, permits escape from intrinsic and extrinsic apoptosis (17). Targeting this anti-apoptosis pathway using Bcl-2 inhibitors such as venetoclax, is a faithful alternative for treatment of resistant and refractory CLL as well as other B cell malignancies (18, 19). Along with identifying reliable signature mutations, continuous efforts have been made to analyze the leukemogenic potential of these sporadic mutations in genetically modified animals (20–23). For example, the TP53 and ATM mutations worsen the CLL prognosis and B lineage specific deletions of these genes induce early disease onset and aggressive leukemic features in Eµ-TCL1 mice model (22, 23). Similarly, gain-of-function mutation in the splicing factor SF3B, when combined with ATM deletion, presents CLL-like B cells in mouse models (23). These results strongly imply the role of prognostically adverse mutations and chromosomal anomalies in CLL pathogenesis but are unable to define the role of BCR signaling in the transforming process.
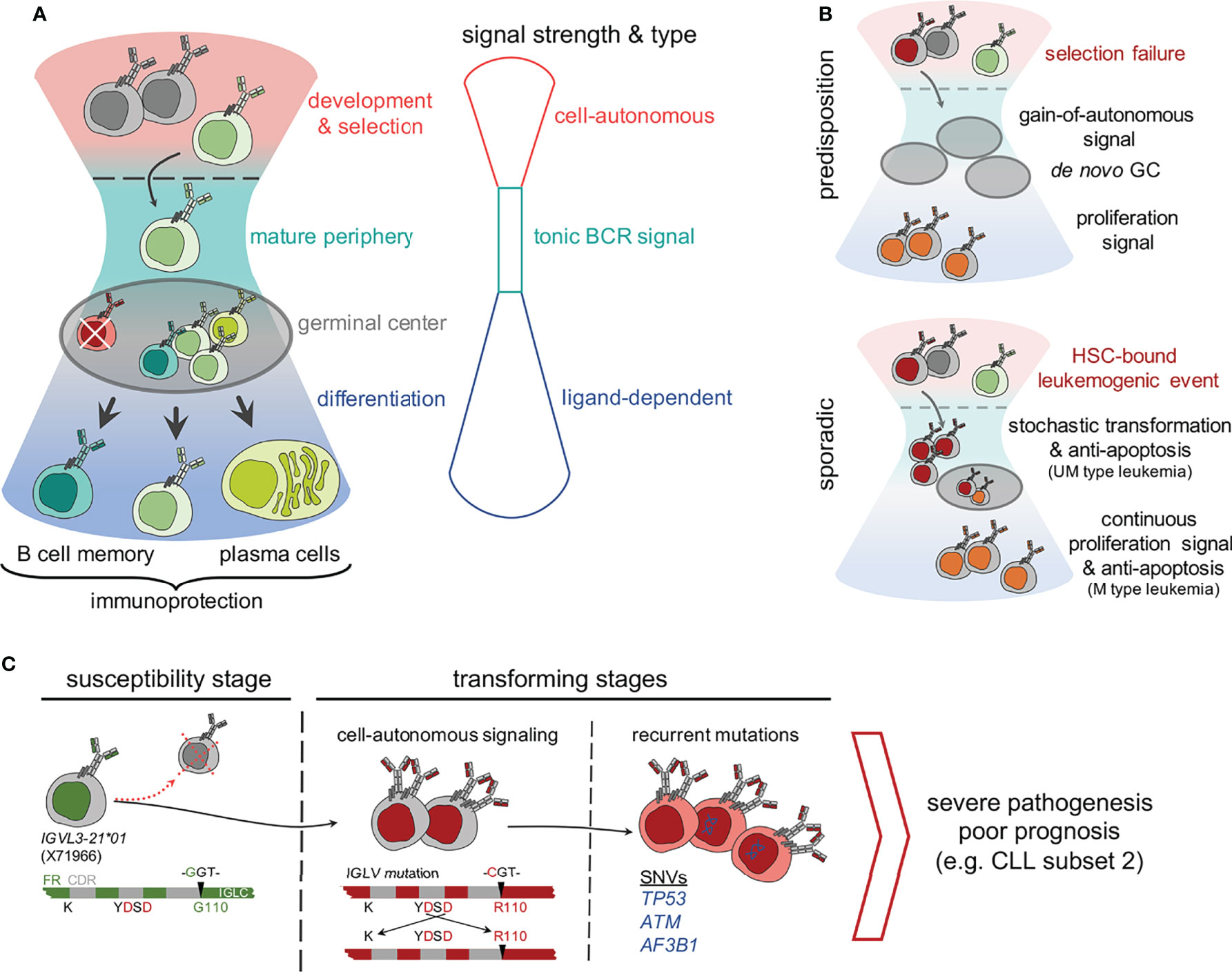
Figure 1 Compounding effect of molecular determinants of BCR signaling and recurrent mutations in genetically predisposed and sporadic origin CLL. (A) A schematic hourglass portrays the course of B cell development, selection, peripheral maintenance, activation and differentiation regulated through tight controls and varying strength of BCR signaling. As shown, each stage represents a predominant type of BCR (or pre-BCR) signaling, which controls the outcome as follows, 1) cell-autonomous signaling at bone marrow dwelled development and selection, 2) tonic signal at the mature peripheral compartments, and 3) antigen responsiveness or ligand-dependent activation signal for memory and plasma cell differentiation during the germinal center reaction. (B) Mutations that supersede the balance between BCR signal strength and developmental stages are a threat for genetic predisposition of leukemia. For example, a selection failure or gain-of-autonomous signaling mutation result in persisting autonomously active B clones in the peripheral compartment that promote proliferative boosts and prime de novo germinal center (GC) formations. In contrast, sporadic mutations have diverse origins. For example, a leukemogenic event occurring at haemopoietic stem cells (HSC) undergoing stochastic transformation might lead to anti-apoptosis and proliferation boost. (C) A stochastic model of neoplastic transformation through acquisition of biomarkers on susceptible genetic background exemplified by the acquired IGLV3-21R110 mutated CLL pathogenesis. As shown, individuals carrying the allele IGLV3-21*01 are predisposed to gain-of-autonomous BCR signaling through homotypic BCR : BCR interaction stabilized by IGLV3-21R110. Although mostly eliminated and undetected in the peripheral blood of a healthy IGLV3-21*01 carrier, a single point G→C mutation, possibly induced by activation-induced cytidine deaminase (AICDA) activity, converts a Glycine (G) to Arginine at 110th residue (R110) and initiates the neoplastic transformation. The gain-of-autonomous BCR signaling and enduring survival advocate the persistent single nucleotide variations (SNV’s) stochastically in TP53, ATM, splicing factor SF3B1 and in epigenetic modifiers to culminate the leukemogenesis and develop severe CLL.
Importantly, CLL prognosis is still strongly relying on the BCR and associated features, in particular on the mutational status, rearranged immunoglobulin genes and complementary determining region 3 (CDR3) (24, 25). These molecular determinants are potential functionally relevant biomarkers. Here, we review the current understanding of BCR associated biomarker exemplifying a novel IGL mutation, termed IGLV3-21R110 (where a glycine is replaced by arginine at position 110), in severe pathogenesis including stereotypic CLL subset #2 (26–28). Additionally, we show that the gain-of-autonomous BCR signaling requires point mutations in both IGLV and IGHV genes derived from a non-subset #2 CLL IGLV3-21R110 case. Thus, we postulate that the gain-of-autonomous BCR signaling and a convoluted signaling crosstalk might favor the recurrent mutations, thus contributing to genomic-instability and anti-apoptosis. Taken together, we endorse the use of BCR associated molecular biomarkers as a novel tool for an easy and comprehensive characterization of CLL.
The clinical course of CLL varies widely, ranging from patients with stable, asymptomatic disease without need of therapeutic intervention to patients suffering from a progressive disease requiring immediate treatment after diagnosis (15, 24, 25, 29). CLL diagnosis is established by routine laboratory tests such as blood counts, blood smear, immunophenotyping, and assessment of the IGHV mutational status or chromatin aberration by FISH (15, 24). Notably, the IGHV mutational status is a first-line molecular determinant or biomarker for prognosis and classification of CLL with highly significant differences in clinical behavior (30, 31). Notably, a biomarker indicates the disease states or medical signs of patients. The first working definition of biomarker from the U.S. National Institutes of Health (NIH) and the World Health Organization (WHO) delineated the objective characteristics, i.e., accurate and reproducible indicators of pathogenic processes or responses to a therapeutic intervention (32, 33). Moreover, there are at least two subclasses of biomarkers, namely prognostic and predictive, based on interaction and differential response to treatment (34, 35). In the last twenty years, this broad objective definition was extended, diversified, and subclassified by several authors (36). Nevertheless, the improvements in precision medicine compel the search for new biomarkers. More recently, we focus on the mechanism-centric approaches for functionally meaningful biomarker selection (36). Unfortunately, identification of a functionally relevant biomarker, specifically for B cell malignancies, requires multifaceted validations, including the generation of mouse models, which makes it meticulous and challenging (20, 21, 37).
For many years, classical staging systems of Rai and Binet defined three major prognostic groups based on clinical parameters derived from physical examination and the aforementioned standard laboratory tests. However, with recent developments in CLL therapy and the sequencing techniques, a number of novel potential prognostic markers has been identified. Thus, the classical staging systems have become insufficient to distinguish prognostic groups and predict early disease progression (24, 38). To overcome these limits, a novel comprehensive prognostic score, the CLL international prognostic index (CLL-IPI) was created. The CLL-IPI combines biochemical and clinical parameters (age, clinical staging, serum ß-2 microglobulin) with cellular and genetic features such as TP53 and IGHV mutational status, to provide a more advanced risk stratification (24, 39). Of the five prognostic factors used by CLL-IPI, TP53 and IGHV mutation status have been described as particularly important in determining patient outcomes (15, 24, 39). In parallel, prognostic groups of stereotyped CLL subsets have also been defined according to the distinctive amino acid pattern within the IGHV CDR3 region (25, 40, 41). Nevertheless, not all CLL cases belong to stereotyped subsets, with roughly 60% of the CLL BCRs remaining unclassified (25, 41). Notably, neither the CLL-IPI grading system nor the subset classification considers the mutations in immunoglobulin light chain (LC), which are known to have direct impact on BCR signaling and prognostic relevance (26–28, 42). Intriguingly, most stereotypic BCR consist of an unique pair of HC and LC recombined through specific V(D)J segments (41, 43). Yet, the role of the LC is frequently underestimated. This is best exemplified by the light chain derived from variable gene segment IGLV3-21, associated with poor prognosis, independently of IGHV mutational status and stereotype (27, 28).
In particular, we identified the single point mutated IGLV3-21R110 as a novel molecular biomarker directly linked to BCR signaling and survival of CLL cells. Notably, crystallographic analyses identified the crucial residues of IGLV3-21R110 required for homotypic BCR-BCR interaction leading to autonomous signaling in CLL subset #2 (Figure 1C) (27, 44). Critical residues include a non-synonymous mutation in the IGLJ segment of the IGLV3-21 derived LC, introducing an indispensable arginin at the position 110 (R110). Of note, IGLV3-21 is the only LC variable gene segment which encodes the required D residues within the CDR2 region, for the homotypic interaction (Figure 1C). Among the three major IMGT annotated alleles of IGLV3-21 locus, only allele IGLV3-21*01 encodes the prerequisite K16 and YDSD motif in CDR2. Notably, a recent update in IMGT reference directory (release 202018-4) includes IGLV3-21*04, which also fulfills these requirements. Thus, the allelic variants IGLV3-21*01 or IGLV3-21*04 have intrinsic potential for the generation of autonomously active BCRs. Especially, B cells expressing the autonomously signaling IGLV3-21R110 variant are counter-selected in healthy donors (HDs) (27). Although IGLV3-21 is commonly found in association with its heavy chain counterpart IGHV3-21 in stereotyped subset #2 CLL, recent studies confirmed the prognostic value of IGLV3-21R110 beyond the IGHV identity and epigenetic classifications of CLL (26, 27, 42, 44). This prompted the idea of a novel subgroup of aggressive CLL expressing the mutated IGLV3-21R110, demonstrating the importance of the light chain as an inclusive criteria for CLL prognosis (26, 27). Notably, the recent finding of higher-order correlation between several major and minor CLL subsets are in line with the IGLV3-21R110 based broader classification which is best exemplified by CLL subset #2 and #169 (27, 41). Both stereotyped subsets employ the IGLV3-21R110 light chain and display structural and immunogenetic similarities with severe clinical courses suggesting a common antigen selection process in their pathogenesis (45). Alternatively, the homotypic BCR-BCR interaction caused by IGLV3-21R110 and subsequent autonomous signaling promote the clonal expansion (27). Given the important role of IGLV3-21R110, we developed two monoclonal antibodies, one to detect the expression of the functional biomarker IGLV3-21R110 and one to distinguish it from the unmutated founder LC. Indeed, the monoclonal anti-wild-type IGLV3-21 antibody can recognize the expression of the susceptible allele IGLV3-21*01 in HDs which is regarded as a risk index for CLL development. Altogether, the discovery of IGLV3-21R110 in severe CLL cases, the related mechanism of B cell activation and the development of antibody-based immunophenotyping tools established IGLV3-21R110 as functionally linked biomarker for CLL pathogenesis.
The process of neoplastic transformation in CLL and other B cell malignancies is controversial. Recent evidence proved the existence of susceptible alleles and mutations which might act as the main drivers of neoplastic transformation (46, 47). Conversely, clonal tracking of leukemic cells indicate that mutations arise at different stages prior and during the course of the disease leading to different cell fates depending on the affected genes (16, 48, 49). Moreover, clonal diversity analyses of IGHV-IGHD-IGHJ sequences revealed the continuous occurrence of mutations regardless of the initial mutation load in the parental (clinically dominant) leukemic clone (50). In short, the rate of intraclonal diversification is similar in UM- and M-CLL. Although this particular model suggests an AID-independent, non-canonical mode of somatic hypermutation, we and others found AID overexpression in M-CLL cases and in IGLV3-21R110 cases (27, 51, 52). The off-target effect of such non-canonical SHM results in non-IG genomic mutations (53, 54). When combined with the strength of BCR and other survival signals, the newly induced non-IG genomic mutations persist and clonally propagate. Alternatively, genomic mutations themselves deliver survival advantage in combination with structurally altered FRs of mutated BCRs allowing sustained signaling. In this context, we demonstrate that the survival signal is sustained by the gain-of-autonomous BCR signaling attained through point mutations in IGLV and IGHV genes (Figure 2) (27). Thus, we propose a stochastic transformation process, which continuously evokes new mutations that break-out the growth and survival dependencies and overrule the intrinsic DNA damage repair system (Figure 1C) (55, 56).
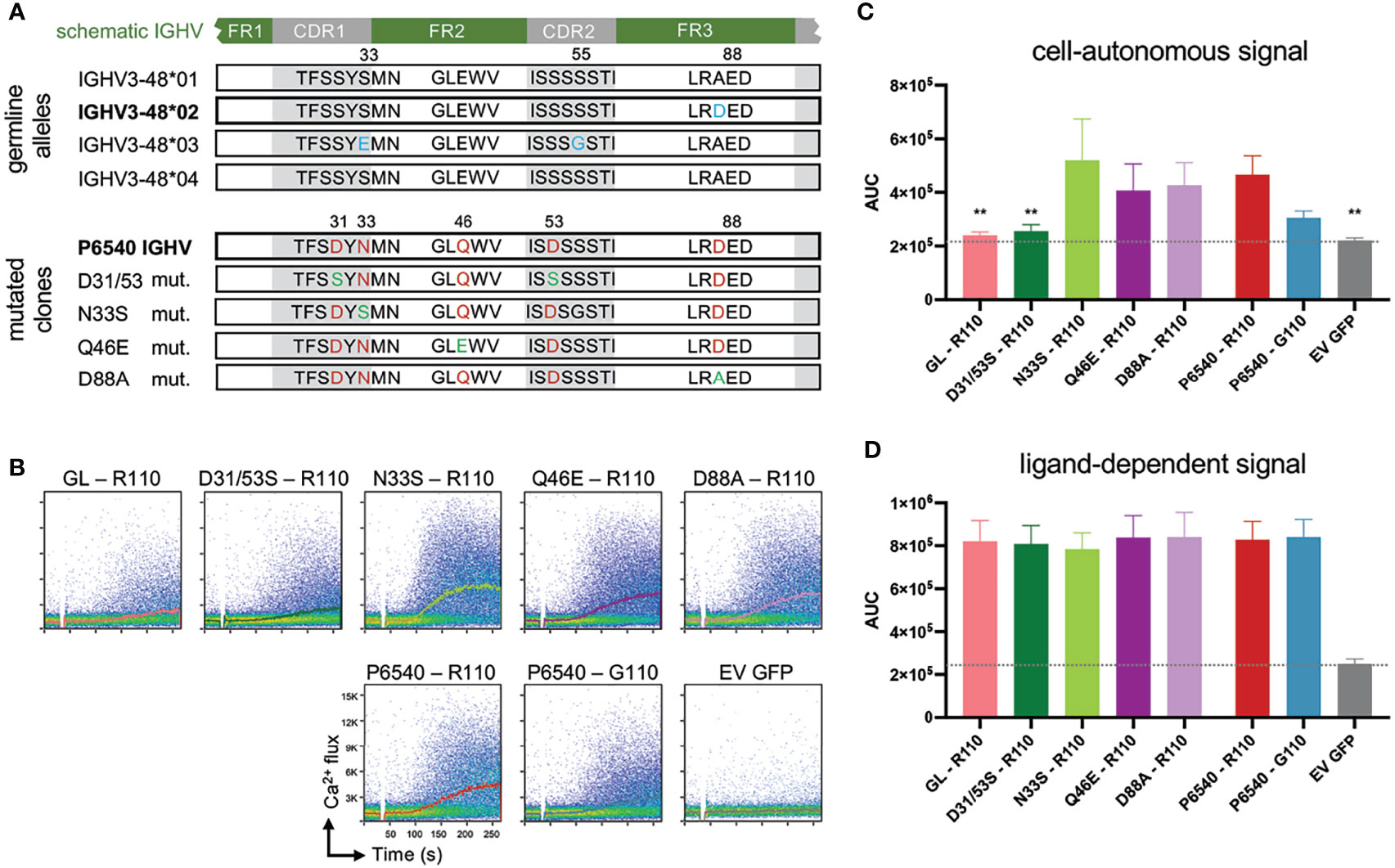
Figure 2 Specific IGHV associated point mutations of autonomous active BCR affect cell-autonomous signal. (A) In the upper part sequence alignment of 4 identified germline alleles of IGHV3-48 are shown, differences are marked in blue. Allele 01 and 04 are identical. In the lower part sequence alignment of CLL-patient P6540 derived IGHV is displayed, which is related to IGHV3-48*02 allele (Mutations marked in red). Several single point-mutations were reverted to their germline version, depicted in green. (B-D) IGHV and IGLV sequences obtained from the CLL patient P6540 were cloned into retroviral expression vectors for human μHC and λLC and expressed in the TKO system (see Supplementary Material and Method). Proper BCR expression on the TKO cells was assessed via flow cytometry (data not shown). Germline IGHV3-48*02 (GL) was included for comparison. IGHVs were co-expressed with autonomously active subset #2 IGLV3-21R110 LC variant. As control, P6540 IGHV was expressed with IGLV3-21G110. Indo-1 staining was performed to analyze intracellular calcium release. In particular, cell-autonomous signaling was assessed via calcium mobilization after 4-Hydroxytamoxifen administration, (B) shown in representative dot plots with kinetics and (C) statistically summarized as area under the curve of the kinetic (AUC). (D) As control B-cell receptors were additionally stimulated with anti-LC antibody to mimic ligand-dependent signal. One-Way ANOVA (Bonferroni Correction), significance of mean to mean of P6540 R110 is shown, (**p < 0,01) N = 5.
As described before, IGLV3-21R110 is originally identified in stereotyped CLL subset #2, which represents a BCR isotype IgM/λ encoded by the IGHV3-21/IGLV3-21 variable gene segments displaying distinctive SHMs (44). Nevertheless, we showed that IGLV3-21R110 LCs are compatible with a range of mutational status and different IGHV identities including IGHV3-48 derived IgM-BCRs (26, 27). Notably, stereotyped CLL subset #2 has eight related immunogenetic subsets, defined as satellite subsets, with the most frequent being CLL subset #169 (41). Intriguingly, multiple features of CLL subset #2, including IGHV mutational status, CDR3 length and mutated IGLV3-21R110 association, are adopted in the major satellite subset #169, e.g., case P6540 (45). Remarkably, P6540 derived IGHV sequence annotates to mutated IGHV3-48*02 among 4 major IMGT IGHV3-48 alleles (Figure 2A). Apart from the acquired LC mutation resulting in IGLV3-21R110, the IGHV sequence introduces three critical acidic D residues in each of CDR1, CDR2 and framework region 3 (FR3) regions. Considering P6540 as a model, we examined the combined effect of IGHV and IGLV mutations on autonomous BCR signaling. Using a bifluorescent expression system, the original P6540 derived IgM-BCR and its IGHV mutants were expressed and analyzed for Ca2+ influx in triple knockout (TKO) cell system, as described before (57). Interestingly, complete reversion of the IGHV3-48 segment to its germline version ceases the autonomous signaling, despite the use of the IGLV3-21R110 light chain (Figures 2B, C). Similar result is found in double mutant D35/53S replacing each of CDR1 and CDR2-bound aspartic acid (D) residues with serine (S). Therefore, the acquired point mutations in the CDR1 and CDR2 regions of the IGHV3-48 segment are crucial for the autonomous signaling of the P6540 BCR (Figures 2B, C), but irrelevant for ligand-dependent signaling (Figure 2D).
Interestingly, both subset #2 and its satellite subset #169, as IGLV3-21R110 CLL expressing cases, exhibit high frequency genomic aberrations in regulators of DNA damage response, including splicing factor SF3B1 (41, 49). Furthermore, we found that in non-subset #169 IGHV3-48 associated cases mutations also accumulate in TP53, ATM, chromatin modifying EP300, and PI3K signaling associated phosphatase PTEN (Figure 1C) (27). These recurrent mutations often arise in chemotherapy resistant patients (9, 16, 22, 58). Specifically, TP53 and ATM mutations are associated with poor outcome in CLL. Indeed, B lineage specific deletion of TP53 and ATM in Eµ-TCL-1 mice model results in earlier disease onset and more aggressive leukemic features compared to wild-type littermates (20–23). Therefore, recurrent mutations rejuvenate the malignant growth and contribute to the neoplastic transformation but may not resemble the primary transforming factor. On the contrary, B cell specific double transgenic mice overexpressing anti-apoptotic Bcl-2 and dominant negative form Traf2 adapter develop CLL/SLL-like phenotype with restricted IGHV usage mimicking CLL BCR stereotypes (59). This model suggests a conducive role of anti-apoptosis, allowing either selection of BCR clones from an indolent pool or escape of autoreactive clones from the self-elimination process. Thus, either aberrant BCR signaling or the compounding effect of ad hoc signaling crosstalk are the foremost transforming factor. On top, the appearance of further recurrent mutations in genes such as SF3B1 and TP53 accelerate and exacerbate the disease when combined with permissive BCR signaling or other conducive features - resembling a stochastic process.
With the advent of GWAS and other NGS approaches, we witnessed the expansion of the previously used criteria for CLL prognosis and the introduction of new CLL-IPI classification (24, 25). Nevertheless, the CLL-IPI classification requires extensive genetic and molecular characterization (TP53 and IGHV) limiting a quick assessment. In contrast, novel diagnostic tools exploiting the existence of functionally relevant biomarkers delivers more quicker and easier assessment. To this aim, we developed monoclonal antibodies for specific detection of the point-mutated light chain IGLV3-21R110, which with appropriate and extensive validation could represent an easy and quick diagnostic tool for severe CLL cases. The presence of IGLV3-21R110 positive B cells or the expression of unmutated IGLV3-21 from susceptible allele IGLV3-21*01 in healthy donors (HD) is regarded as a risk index for CLL development (26–28). For early diagnosis and preemptive therapy, IGLV3-21R110 is a gem and we foresee future development of antibody-based direct cell-depletion or drug delivery interface. Additionally, quick immune-detection of unmutated IGLV3-21 LC expression combined with genetic analyses of IGLV3-21 allele in HD will predict the susceptibility to severe CLL.
Innovative genomic studies pointing out the existence of risk-associated loci and novel murine models of genomic alteration such as Sf3b mutation and ATM deletion, prompted the idea of a stochastic process of malignant transformation where the expression of a susceptible allele or mutations are regarded as the driver event for future development of neoplastic diseases. The existence of risk-associated loci also highlights the dynamics of familiar CLL, confirming that relatives of CLL patients have an increased risk of developing the disease (60, 61). In particular, CLL cases within the same family presented uniform BCR IG and similar genomic profiles, highlighting the correlation between germline predisposition and immunogenetic markers and pointing towards a shared mechanism of CLL development (62). Of note, recurrent unfavorable mutations are often associated with survival and anti-apoptotic pathways or with BCR signaling pathway. It is important to understand the role of these mutations as prognostic indicators, not only to improve diagnosis, but also to develop suitable preclinical models, which comprehend the heterogeneity of CLL.
So far, the majority of CLL mouse models mimic genetic aberrations or deregulated gene expression in CLL without taking into account the clonality of the BCR (63). Nevertheless, in a stochastic process of malignant transformation, cells accumulate numerous mutations which do not proliferate until the occurrence of additional events that overcome the checkpoints and drive the leukemogenesis. Thus, the generation of an inducible monoclonal CLL-mouse model to study CLL development in a stepwise manner would be of particular relevance to clarify the role of aberrant BCR signaling in the development of CLL. We propose that the use of a CLL-derived autonomous BCR, e.g. the aggressive subset #2 will help to elucidate the course of CLL. In fact, it is interesting to follow the growth of subset #2 BCR expressing B cells, preferably in inducible manner, in-vivo and to screen for acquired mutations. Moreover, subsequent changes in anti-apoptotic and pro-survival properties of these autonomously active B cells are of immense relevance for both indolent and aggressive outcome. Thus, an inclusion of functionally relevant prognostic parameters, even for indolent cases, can only be beneficial. Of course, further and larger independent studies must be carried out in order to validate these parameters.
Characterization of the molecular prognostic parameters and genomic landscape predict primary response but not relapse to treatment. Often, the underrated interactions between molecular cytogenetics features and the microenvironment specific survival signals mediated by direct contacts, adhesion molecules, chemokines and ligand-receptor interactions are of great prognostic value (15, 64–66). The kinases of BCR signaling pathway such as PI3K, BTK and SYK, are also involved in receptor mediated crosstalk (67). For example, ibrutinib, the BTK inhibitor, interferes with the homeostasis of the leukemic cells in the survival niches, thus demonstrating efficacy in high-risk CLL (22, 64, 68–70). Moreover, tissue infiltrating and colonizing ability of leukemic cells alters growth rate, metabolic dependency and proliferation of CLL cells (71). Recent studies have highlighted the importance of analyzing CLL cells in a physiologically relevant environment recreating the interactions between leukemic cells and their milieu (64). Such 3D-cell culture and bioreactor models analyze the role of the microenvironment comprehensively. This is of great resonance to characterize tumor cells in their milieu providing novel in vitro strategies to test new therapeutic agents and assess their effects under more in vivo-like conditions.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
PCM designed the study, led the experiments and interpreted the results together with HJ. AN and ATL performed the experiments, wrote the MS with PCM, and all authors reviewed, commented on, and approved the manuscript.
This work was supported by SFB 1074 (A10), SFB 1279 (B03) and the ERC 694992 to HJ; AN is PhD student through SFB1279; ATL is postdoctoral fellow through ERC 694992; PCM is research fellow at Institute of Immunology, Ulm University.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank Kostas Stamatopoulos and Paolo Ghia for the P6540 sequence and insightful discussions. We apologize to colleagues whose work could not be cited owing to space limitations.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2021.771669/full#supplementary-material
1. Corso J, Pan KT, Walter R, Doebele C, Mohr S, Bohnenberger H, et al. Elucidation of Tonic and Activated B-Cell Receptor Signaling in Burkitt's Lymphoma Provides Insights Into Regulation of Cell Survival. Proc Natl Acad Sci USA (2016) 113:5688–93. doi: 10.1073/pnas.1601053113
2. Gomes de Castro MA, Wildhagen H, Sograte-Idrissi S, Hitzing C, Binder M, Trepel M, et al. Differential Organization of Tonic and Chronic B Cell Antigen Receptors in the Plasma Membrane. Nat Commun (2019) 10:820. doi: 10.1038/s41467-019-08677-1
3. Yasuda S, Zhou Y, Wang Y, Yamamura M, Wang JY. A Model Integrating Tonic and Antigen-Triggered BCR Signals to Predict the Survival of Primary B Cells. Sci Rep (2017) 7:14888. doi: 10.1038/s41598-017-13993-x
4. Becker M, Hobeika E, Jumaa H, Reth M, Maity PC. CXCR4 Signaling and Function Require the Expression of the IgD-Class B-Cell Antigen Receptor. Proc Natl Acad Sci USA (2017) 114:5231–6. doi: 10.1073/pnas.1621512114
5. Shojaee S, Chan LN, Buchner M, Cazzaniga V, Cosgun KN, Geng H, et al. PTEN Opposes Negative Selection and Enables Oncogenic Transformation of Pre-B Cells. Nat Med (2016) 22:379–87. doi: 10.1038/nm.4062
6. Bach MP, Hug E, Werner M, Holch J, Sprissler C, Pechloff K, et al. Premature Terminal Differentiation Protects From Deregulated Lymphocyte Activation by ITK-Syk. J Immunol (2014) 192:1024–33. doi: 10.4049/jimmunol.1300420
7. Young RM, Wu T, Schmitz R, Dawood M, Xiao W, Phelan JD, et al. Survival of Human Lymphoma Cells Requires B-Cell Receptor Engagement by Self-Antigens. Proc Natl Acad Sci USA (2015) 112:13447–54. doi: 10.1073/pnas.1514944112
8. Ziegler CGK, Kim J, Piersanti K, Oyler-Yaniv A, Argyropoulos KV, van den Brink MRM, et al. Constitutive Activation of the B Cell Receptor Underlies Dysfunctional Signaling in Chronic Lymphocytic Leukemia. Cell Rep (2019) 28:923–37.e3. doi: 10.1016/j.celrep.2019.06.069
9. Schmitz R, Wright GW, Huang DW, Johnson CA, Phelan JD, Wang JQ, et al. Genetics and Pathogenesis of Diffuse Large B-Cell Lymphoma. N Engl J Med (2018) 378:1396–407. doi: 10.1056/NEJMoa1801445
10. Phelan JD, Young RM, Webster DE, Roulland S, Wright GW, Kasbekar M, et al. A Multiprotein Supercomplex Controlling Oncogenic Signalling in Lymphoma. Nature (2018) 560:387–91. doi: 10.1038/s41586-018-0290-0
11. Lin WY, Fordham SE, Sunter N, Elstob C, Rahman T, Willmore E, et al. Genome-Wide Association Study Identifies Risk Loci for Progressive Chronic Lymphocytic Leukemia. Nat Commun (2021) 12:665. doi: 10.1038/s41467-020-20822-9
12. Rani L, Mathur N, Gupta R, Gogia A, Kaur G, Dhanjal JK, et al. Genome-Wide DNA Methylation Profiling Integrated With Gene Expression Profiling Identifies PAX9 as a Novel Prognostic Marker in Chronic Lymphocytic Leukemia. Clin Epigenet (2017) 9:57. doi: 10.1186/s13148-017-0356-0
13. Berndt SI, Camp NJ, Skibola CF, Vijai J, Wang Z, Gu J, et al. Meta-Analysis of Genome-Wide Association Studies Discovers Multiple Loci for Chronic Lymphocytic Leukemia. Nat Commun (2016) 7:10933. doi: 10.1038/ncomms10933
14. Din L, Sheikh M, Kosaraju N, Smedby KE, Bernatsky S, Berndt SI, et al. Genetic Overlap Between Autoimmune Diseases and Non-Hodgkin Lymphoma Subtypes. Genet Epidemiol (2019) 43:844–63. doi: 10.1002/gepi.22242
15. Delgado J, Nadeu F, Colomer D, Campo E. Chronic Lymphocytic Leukemia: From Molecular Pathogenesis to Novel Therapeutic Strategies. Haematologica (2020) 105:2205–17. doi: 10.3324/haematol.2019.236000
16. Speedy HE, Beekman R, Chapaprieta V, Orlando G, Law PJ, Martin-Garcia D, et al. Insight Into Genetic Predisposition to Chronic Lymphocytic Leukemia From Integrative Epigenomics. Nat Commun (2019) 10:3615. doi: 10.1038/s41467-019-11582-2
17. Tzifi F, Economopoulou C, Gourgiotis D, Ardavanis A, Papageorgiou S, Scorilas A. The Role of BCL2 Family of Apoptosis Regulator Proteins in Acute and Chronic Leukemias. Adv Hematol (2012) 2012:524308. doi: 10.1155/2012/524308
18. Diepstraten ST, Anderson MA, Czabotar PE, Lessene G, Strasser A, Kelly GL. The Manipulation of Apoptosis for Cancer Therapy Using BH3-Mimetic Drugs. Nat Rev Cancer (2021). doi: 10.1038/s41568-021-00407-4
19. Cochrane T, Enrico A, Gomez-Almaguer D, Hadjiev E, Lech-Maranda E, Masszi T, et al. Impact of Venetoclax Monotherapy on the Quality of Life of Patients With Relapsed or Refractory Chronic Lymphocytic Leukemia: Results From the Phase 3b VENICE II Trial. Leuk Lymphoma (2021) 1–11. doi: 10.1080/10428194.2021.1986217
20. Knittel G, Rehkamper T, Korovkina D, Liedgens P, Fritz C, Torgovnick A, et al. Two Mouse Models Reveal an Actionable PARP1 Dependence in Aggressive Chronic Lymphocytic Leukemia. Nat Commun (2017) 8:153. doi: 10.1038/s41467-017-00210-6
21. Fabbri G, Holmes AB, Viganotti M, Scuoppo C, Belver L, Herranz D, et al. Common Nonmutational NOTCH1 Activation in Chronic Lymphocytic Leukemia. Proc Natl Acad Sci USA (2017) 114:E2911–9. doi: 10.1073/pnas.1702564114
22. Lee HJ, Gallardo M, Ma H, Zhang X, Larsson CA, Mejia A, et al. ). P53-Independent Ibrutinib Responses in an Emu-TCL1 Mouse Model Demonstrates Efficacy in High-Risk CLL. Blood Cancer J (2016) 6:e434. doi: 10.1038/bcj.2016.41
23. Yin S, Gambe RG, Sun J, Martinez AZ, Cartun ZJ, Regis FFD, et al. A Murine Model of Chronic Lymphocytic Leukemia Based on B Cell-Restricted Expression of Sf3b1 Mutation and Atm Deletion. Cancer Cell (2019) 35:283–96.e5. doi: 10.1016/j.ccell.2018.12.013
24. Hallek M. Chronic Lymphocytic Leukemia: 2020 Update on Diagnosis, Risk Stratification and Treatment. Am J Hematol (2019) 94:1266–87. doi: 10.1002/ajh.25595
25. Stamatopoulos K, Agathangelidis A, Rosenquist R, Ghia P. Antigen Receptor Stereotypy in Chronic Lymphocytic Leukemia. Leukemia (2017) 31:282–91. doi: 10.1038/leu.2016.322
26. Nadeu F, Royo R, Clot G, Duran-Ferrer M, Navarro A, Martin S, et al. IGLV3-21R110 Identifies an Aggressive Biological Subtype of Chronic Lymphocytic Leukemia With Intermediate Epigenetics. Blood (2021) 137(21):2935–46. doi: 10.1182/blood.2020008311
27. Maity PC, Bilal M, Koning MT, Young M, van Bergen CAM, Renna V, et al. IGLV3-21*01 Is an Inherited Risk Factor for CLL Through the Acquisition of a Single-Point Mutation Enabling Autonomous BCR Signaling. Proc Natl Acad Sci USA (2020) 117:4320–7. doi: 10.1073/pnas.1913810117
28. Stamatopoulos B, Smith T, Crompot E, Pieters K, Clifford R, Mraz M, et al. The Light Chain IgLV3-21 Defines a New Poor Prognostic Subgroup in Chronic Lymphocytic Leukemia: Results of a Multicenter Study. Clin Cancer Res (2018) 24:5048–57. doi: 10.1158/1078-0432.CCR-18-0133
29. Li AJ, Dhanraj JP, Lopes G, Parker JL. Clinical Trial Risk in Leukemia: Biomarkers and Trial Design. Hematol Oncol (2021) 39:105–13. doi: 10.1002/hon.2818
30. Chiorazzi N, Stevenson FK. Celebrating 20 Years of IGHV Mutation Analysis in CLL. Hemasphere (2020) 4:e334. doi: 10.1097/HS9.0000000000000334
31. Damle RN, Wasil T, Fais F, Ghiotto F, Valetto A, Allen SL, et al. Ig V Gene Mutation Status and CD38 Expression as Novel Prognostic Indicators in Chronic Lymphocytic Leukemia. Blood (1999) 94:1840–7. doi: 10.1182/blood.V94.6.1840
32. Strimbu K, Tavel JA. What Are Biomarkers? Curr Opin HIV AIDS (2010) 5:463–6. doi: 10.1097/COH.0b013e32833ed177
33. Biomarkers Definitions Working, G. Biomarkers and Surrogate Endpoints: Preferred Definitions and Conceptual Framework. Clin Pharmacol Ther (2001) 69:89–95. doi: 10.1067/mcp.2001.113989
34. Rossi D, Gaidano G. The Clinical Implications of Gene Mutations in Chronic Lymphocytic Leukaemia. Br J Cancer (2016) 114:849–54. doi: 10.1038/bjc.2016.78
35. Lee J, Wang YL. Prognostic and Predictive Molecular Biomarkers in Chronic Lymphocytic Leukemia. J Mol Diagn (2020) 22:1114–25. doi: 10.1016/j.jmoldx.2020.06.004
36. Yu CY, Mitrofanova A. Mechanism-Centric Approaches for Biomarker Detection and Precision Therapeutics in Cancer. Front Genet (2021) 12:687813. doi: 10.3389/fgene.2021.687813
37. Kohlhas V, Hallek M, Nguyen PH. Constitutive Activation of Lyn Kinase Enhances BCR Responsiveness, But Not the Development of CLL in Emicro-TCL1 Mice. Blood Adv (2020) 4:6106–16. doi: 10.1182/bloodadvances.2020002584
38. Fabbri G, Dalla-Favera R. The Molecular Pathogenesis of Chronic Lymphocytic Leukaemia. Nat Rev Cancer (2016) 16:145–62. doi: 10.1038/nrc.2016.8
39. Hallek M, Cheson BD, Catovsky D, Caligaris-Cappio F, Dighiero G, Dohner H, et al. iwCLL Guidelines for Diagnosis, Indications for Treatment, Response Assessment, and Supportive Management of CLL. Blood (2018) 131:2745–60. doi: 10.1182/blood-2017-09-806398
40. Agathangelidis A, Darzentas N, Hadzidimitriou A, Brochet X, Murray F, Yan XJ, et al. Stereotyped B-Cell Receptors in One-Third of Chronic Lymphocytic Leukemia: A Molecular Classification With Implications for Targeted Therapies. Blood (2012) 119:4467–75. doi: 10.1182/blood-2011-11-393694
41. Agathangelidis A, Chatzidimitriou A, Gemenetzi K, Giudicelli V, Karypidou M, Plevova K, et al. Higher-Order Connections Between Stereotyped Subsets: Implications for Improved Patient Classification in CLL. Blood (2021) 137:1365–76. doi: 10.1182/blood.2020007039
42. Baliakas P, Agathangelidis A, Hadzidimitriou A, Sutton LA, Minga E, Tsanousa A, et al. Not All IGHV3-21 Chronic Lymphocytic Leukemias Are Equal: Prognostic Considerations. Blood (2015) 125:856–9. doi: 10.1182/blood-2014-09-600874
43. Gerousi M, Laidou S, Gemenetzi K, Stamatopoulos K, Chatzidimitriou A. Distinctive Signaling Profiles With Distinct Biological and Clinical Implications in Aggressive CLL Subsets With Stereotyped B-Cell Receptor Immunoglobulin. Front Oncol (2021) 11:771454. doi: 10.3389/fonc.2021.771454
44. Minici C, Gounari M, Übelhart R, Scarfò L, Dühren-von Minden M, Schneider D, et al. Distinct Homotypic B-Cell Receptor Interactions Shape the Outcome of Chronic Lymphocytic Leukaemia. Nat Commun (2017) 8:15746. doi: 10.1038/ncomms15746
45. Gemenetzi K, Psomopoulos F, Carriles AA, Gounari M, Minici C, Plevova K, et al. Higher-Order Immunoglobulin Repertoire Restrictions in CLL: The Illustrative Case of Stereotyped Subsets 2 and 169. Blood (2021) 137:1895–904. doi: 10.1182/blood.2020005216
46. Locher M, Jukic E, Bohn JP, Untergasser G, Steurer M, Cramer CA, et al. Clonal Dynamics in a Composite Chronic Lymphocytic Leukemia and Hairy Cell Leukemia-Variant. Genes Chromosomes Cancer (2021) 60:287–92. doi: 10.1002/gcc.22925
47. Schuh A, Becq J, Humphray S, Alexa A, Burns A, Clifford R, et al. Monitoring Chronic Lymphocytic Leukemia Progression by Whole Genome Sequencing Reveals Heterogeneous Clonal Evolution Patterns. Blood (2012) 120:4191–6. doi: 10.1182/blood-2012-05-433540
48. Koczkodaj D, Popek-Marciniec S, Zmorzynski S, Wasik-Szczepanek E, Filip AA. Examination of Clonal Evolution in Chronic Lymphocytic Leukemia. Med Oncol (2019) 36:79. doi: 10.1007/s12032-019-1300-2
49. Hotinski AK, Best OG, Thurgood LA, Lower KM, Kuss BJ. A Biclonal Case of Chronic Lymphocytic Leukaemia With Discordant Mutational Status of the Immunoglobulin Heavy Chain Variable Region and Bimodal CD49d Expression. Br J Haematol (2021) 192:e77–81. doi: 10.1111/bjh.17257
50. Bagnara D, Tang C, Brown JR, Kasar S, Fernandes S, Colombo M, et al. Post-Transformation IGHV-IGHD-IGHJ Mutations in Chronic Lymphocytic Leukemia B Cells: Implications for Mutational Mechanisms and Impact on Clinical Course. Front Oncol (2021) 11:640731. doi: 10.3389/fonc.2021.640731
51. Patten PE, Chu CC, Albesiano E, Damle RN, Yan XJ, Kim D, et al. IGHV-Unmutated and IGHV-Mutated Chronic Lymphocytic Leukemia Cells Produce Activation-Induced Deaminase Protein With a Full Range of Biologic Functions. Blood (2012) 120:4802–11. doi: 10.1182/blood-2012-08-449744
52. Morande PE, Yan XJ, Sepulveda J, Seija N, Marquez ME, Sotelo N, et al. AID Overexpression Leads to Aggressive Murine CLL and Nonimmunoglobulin Mutations That Mirror Human Neoplasms. Blood (2021) 138:246–58. doi: 10.1182/blood.2020008654
53. Innes CL, Hesse JE, Palii SS, Helmink BA, Holub AJ, Sleckman BP, et al. DNA Damage Activates a Complex Transcriptional Response in Murine Lymphocytes That Includes Both Physiological and Cancer-Predisposition Programs. BMC Genomics (2013) 14:163. doi: 10.1186/1471-2164-14-163
54. Sherman MH, Kuraishy AI, Deshpande C, Hong JS, Cacalano NA, Gatti RA, et al. AID-Induced Genotoxic Stress Promotes B Cell Differentiation in the Germinal Center via ATM and LKB1 Signaling. Mol Cell (2010) 39:873–85. doi: 10.1016/j.molcel.2010.08.019
55. Ahn IE, Underbayev C, Albitar A, Herman SE, Tian X, Maric I, et al. Clonal Evolution Leading to Ibrutinib Resistance in Chronic Lymphocytic Leukemia. Blood (2017) 129:1469–79. doi: 10.1182/blood-2016-06-719294
56. Zapatka M, Tausch E, Ozturk S, Yosifov DY, Seiffert M, Zenz T, et al. Clonal Evolution in Chronic Lymphocytic Leukemia Is Scant in Relapsed But Accelerated in Refractory Cases After Chemo(Immune)Therapy. Haematologica (2021). doi: 10.3324/haematol.2020.265777
57. Meixlsperger S, Kohler F, Wossning T, Reppel M, Muschen M, Jumaa H. Conventional Light Chains Inhibit the Autonomous Signaling Capacity of the B Cell Receptor. Immunity (2007) 26:323–33. doi: 10.1016/j.immuni.2007.01.012
58. Ljungstrom V, Cortese D, Young E, Pandzic T, Mansouri L, Plevova K, et al. Whole-Exome Sequencing in Relapsing Chronic Lymphocytic Leukemia: Clinical Impact of Recurrent RPS15 Mutations. Blood (2016) 127:1007–16. doi: 10.1182/blood-2015-10-674572
59. Perez-Chacon G, Zapata JM. The Traf2DNxBCL2-Tg Mouse Model of Chronic Lymphocytic Leukemia/Small Lymphocytic Lymphoma Recapitulates the Biased IGHV Gene Usage, Stereotypy, and Antigen-Specific HCDR3 Selection of Its Human Counterpart. Front Immunol (2021) 12:627602. doi: 10.3389/fimmu.2021.627602
60. Brown JR. Inherited Predisposition to Chronic Lymphocytic Leukemia. Expert Rev Hematol (2008) 1:51–61. doi: 10.1586/17474086.1.1.51
61. Cerhan JR, Slager SL. Familial Predisposition and Genetic Risk Factors for Lymphoma. Blood (2015) 126:2265–73. doi: 10.1182/blood-2015-04-537498
62. Kolijn PM, Muggen AF, Ljungstrom V, Agathangelidis A, Wolvers-Tettero ILM, Beverloo HB, et al. Consistent B Cell Receptor Immunoglobulin Features Between Siblings in Familial Chronic Lymphocytic Leukemia. Front Oncol (2021) 11:740083. doi: 10.3389/fonc.2021.740083
63. Simonetti G, Bertilaccio MT, Ghia P, Klein U. Mouse Models in the Study of Chronic Lymphocytic Leukemia Pathogenesis and Therapy. Blood (2014) 124:1010–9. doi: 10.1182/blood-2014-05-577122
64. Barbaglio F, Belloni D, Scarfo L, Sbrana FV, Ponzoni M, Bongiovanni L, et al. 3D Co-Culture Model of Chronic Lymphocytic Leukemia Bone Marrow Microenvironment Predicts Patient-Specific Response to Mobilizing Agents. Haematologica (2021) 106(9):2334–44. doi: 10.3324/haematol.2020.248112
65. Auge H, Notarantonio AB, Morizot R, Quinquenel A, Fornecker LM, Hergalant S, et al. Microenvironment Remodeling and Subsequent Clinical Implications in Diffuse Large B-Cell Histologic Variant of Richter Syndrome. Front Immunol (2020) 11:594841. doi: 10.3389/fimmu.2020.594841
66. Sivina M, Xiao L, Kim E, Vaca A, Chen SS, Keating MJ, et al. CXCL13 Plasma Levels Function as a Biomarker for Disease Activity in Patients With Chronic Lymphocytic Leukemia. Leukemia (2021) 35:1610–20. doi: 10.1038/s41375-020-01063-7
67. Seda V, Mraz M. B-Cell Receptor Signalling and its Crosstalk With Other Pathways in Normal and Malignant Cells. Eur J Haematol (2015) 94:193–205. doi: 10.1111/ejh.12427
68. Ahn IE, Tian X, Wiestner A. Ibrutinib for Chronic Lymphocytic Leukemia With TP53 Alterations. N Engl J Med (2020) 383:498–500. doi: 10.1056/NEJMc2005943
69. Roy Chowdhury S, Bouchard EDJ, Saleh R, Nugent Z, Peltier C, Mejia E, et al. Mitochondrial Respiration Correlates With Prognostic Markers in Chronic Lymphocytic Leukemia and Is Normalized by Ibrutinib Treatment. Cancers (2020) 12:650. doi: 10.3390/cancers12030650
70. Chen SS, Chang BY, Chang S, Tong T, Ham S, Sherry B, et al. BTK Inhibition Results in Impaired CXCR4 Chemokine Receptor Surface Expression, Signaling and Function in Chronic Lymphocytic Leukemia. Leukemia (2016) 30:833–43. doi: 10.1038/leu.2015.316
Keywords: BCR signaling, CLL, biomarkers, transformation, immunoglobulin genes
Citation: Nicolò A, Linder AT, Jumaa H and Maity PC (2021) The Determinants of B Cell Receptor Signaling as Prototype Molecular Biomarkers of Leukemia. Front. Oncol. 11:771669. doi: 10.3389/fonc.2021.771669
Received: 06 September 2021; Accepted: 02 December 2021;
Published: 21 December 2021.
Edited by:
Jitka Malcikova, Masaryk University, CzechiaReviewed by:
Srimoyee Mukherjee, Tufts University School of Medicine, United StatesCopyright © 2021 Nicolò, Linder, Jumaa and Maity. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Palash Chandra Maity, cGFsYXNoLm1haXR5QHVuaS11bG0uZGU=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.