
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol., 11 November 2021
Sec. Hematologic Malignancies
Volume 11 - 2021 | https://doi.org/10.3389/fonc.2021.769612
This article is part of the Research TopicRole of the Antigen Receptor in the Pathogenesis of B-Cell Lymphoid MalignanciesView all 10 articles
 Chrysi Galigalidou1,2
Chrysi Galigalidou1,2 Laura Zaragoza-Infante1,3
Laura Zaragoza-Infante1,3 Anastasia Iatrou1
Anastasia Iatrou1 Anastasia Chatzidimitriou1,4
Anastasia Chatzidimitriou1,4 Kostas Stamatopoulos1,4
Kostas Stamatopoulos1,4 Andreas Agathangelidis1,5*
Andreas Agathangelidis1,5*The term monoclonal B-cell lymphocytosis (MBL) describes the presence of a clonal B cell population with a count of less than 5 × 109/L and no symptoms or signs of disease. Based on the B cell count, MBL is further classified into 2 distinct subtypes: ‘low-count’ and ‘high-count’ MBL. High-count MBL shares a series of biological and clinical features with chronic lymphocytic leukemia (CLL), at least of the indolent type, and evolves to CLL requiring treatment at a rate of 1-2% per year, whereas ‘low-count’ MBL seems to be distinct, likely representing an immunological rather than a pre-malignant condition. That notwithstanding, both subtypes of MBL can carry ‘CLL-specific’ genomic aberrations such as cytogenetic abnormalities and gene mutations, yet to a much lesser extent compared to CLL. These findings suggest that such aberrations are mostly relevant for disease progression rather than disease onset, indirectly pointing to microenvironmental drive as a key contributor to the emergence of MBL. Understanding microenvironmental interactions is therefore anticipated to elucidate MBL ontogeny and, most importantly, the relationship between MBL and CLL.
Monoclonal B cell lymphocytosis (MBL) is an asymptomatic hematological condition characterized by the presence of clonal B cell expansion(s) in otherwise healthy individuals. The immunophenotype of the B cell clones in approximately 75% of all MBL cases is similar to that observed in chronic lymphocytic leukemia (CLL): CD5+, CD19+, CD20dim, CD23+ and low levels (dim) of surface immunoglobulin (sIgdim) that in most cases is IgM with or without IgD. Other immunophenotypic characteristics include the absence of FMC7 and the weak or no expression of CD79b and CD22. Based on this constellation of features, the most common subtype of MBL is defined as ‘CLL-like’. Another immunophenotype occasionally observed in MBL is characterized by strong expression of CD20 and sIg along with absence of CD23 (CD5+, CD19+, CD20high, CD23+, sIghigh); this MBL subtype is described as ‘atypical’ MBL. The last, most infrequent, MBL subtype is ‘CD5-negative’ MBL, in which the complete immunophenotypic characterization is CD5neg, CD19+, CD20+ with moderate to high sIg expression, with the ratio of κ: λ sIg being strongly skewed (either more than 3:1 or less than 1:3) (1–4).
Significant differences exist between the three MBL immunophenotypic subtypes regarding the expression of several cell surface markers, such as CD23, CD79b, and FMC7. Moreover, several established CLL risk factors are also differentially expressed between the immunophenotypic subtypes of MBL; for instance, markers associated with aggressive CLL, such as CD38, ZAP70, and CD49d are highly expressed in the majority of ‘atypical’ MBL cases yet are rarely expressed in ‘CLL-like’ MBL (1, 5). Moreover, the B cell memory marker CD27 shows higher expression in the ‘CLL-like’ type of MBL compared to ‘atypical’ and ‘CD5-negative’ MBL, while, in contrast, CD22, CD79a, CD79b, and CD1c (BDCA-1) are mainly expressed in the ‘non CLL-like’ types. Furthermore, FMC7 and CD43 are also characterized by differential expression among the three immunophenotypic types of MBL; more specifically, FMC7 exhibits low expression levels in ‘CLL-like’ MBL, intermediate levels in atypical, and high levels in the ‘CD5-negative’ type of MBL, while the exact opposite pattern is evident for CD43 expression (6). Differences are also observed regarding the expression of sIg, which is significantly lower in ‘CLL-like’ MBL compared to either the ‘atypical’ or the ‘CD5-negative’ types (1). Finally, expression of the ROR1 gene was statistically different among the three immunophenotypic subtypes of MBL being significantly higher in ‘CLL-like’ MBL versus the others (1).
Moving from the immunophenotype, the diagnostic criteria for distinguishing between MBL from CLL are primarily based on the number of circulating monoclonal B cells. Two distinct MBL subtypes are recognized: high-count MBL (HC-MBL), or ‘clinical’ MBL, with 0.5-4.99× 109 clonal B cells/L and low-count MBL (LC-MBL), or ‘general’ population MBL, characterized by <0.5 × 109 clonal B cells/L (4). It should be noted that most individuals diagnosed with MBL display a normal absolute B cell count, with the clonal B cell population accounting for less than 10% of the total number of B cells (3).
The incidence of MBL gradually increases with advancing age; in fact, MBL is very rarely detected in individuals under 40 years old, whereas it is present in more than 20% of individuals older than 70 years old and can reach even 75% among patients aged more than 90 years (3). Age is not the only factor strongly associated with MBL, as a familial history of hematological or solid malignancies has also been reported; in fact, 17% of first-degree relatives with no personal history of lymphoproliferative disorders from families with at least two cases of CLL were found to have MBL, in some cases at a young age (less than 40 years old) (7). Gender is another factor connected to the prevalence of MBL as, similar to CLL, males have a significantly higher risk for developing MBL compared to females (8).
Exposure to infections and immunodeficiency may also predispose to MBL development, at least in certain instances, especially when combined with advanced age and male gender. A characteristic example concerns the high frequency of MBL among patients with hepatitis C virus (HCV) infection (9). On the other hand, MBL was found to be significantly less common among individuals vaccinated against pneumococcal or influenza infections (3).
Unmutated somatic hypermutation (SHM) status of the rearranged immunoglobulin heavy variable (IGHV) gene and/or ‘CLL-related’ cytogenetic aberrations related to aggressive disease represent risk factors of progression from MBL to CLL and shorter overall survival (OS) (10). Furthermore, over 50% of HC-MBL cases carry at least one gene mutation in a CLL putative driver, evident up to 41 months before the progression to CLL. Thus, genomic characterization can be used to identify those MBL cases who will progress to CLL requiring treatment; this was more pronounced in cases with subclonal expansion of driver mutations (11, 12).
Along these lines, the genetic distance between LC-MBL and CLL is clearly greater than that of HC-MBL (13) and, thus, the question whether LC-MBL represents a very early stage in the natural history of CLL, remains elusive.
Monoclonal B cell populations can be detected long before CLL diagnosis, even up to 6.4 years (13). However, the majority of ‘CLL-like’ HC-MBL cases remain stable overtime; progression to CLL has been estimated at a rate of 1-4% per year (1). In more detail, a longitudinal analysis in 185 individuals with ‘CLL-like’ HC-MBL revealed a progressive lymphocytosis in 28% of the cases with a median follow-up of 6.7 years. However, only 7% of the cases progressed to CLL requiring treatment with the overall rate of progression to CLL being 1.1% per year (14). In a retrospective study by the Mayo Clinic, 302 ‘CLL-like’ HC-MBL cases were monitored for a median of 18 months with ~1.4% of cases per year requiring treatment due to disease progression. This study proposed a B cell count threshold of 10 x 109/L for discriminating between two categories of ‘CLL-like’ HC-MBL cases with higher and lower probability of treatment. This proposal was subsequently tested and validated in the GIMEMA database (15, 16). Finally, in an Italian study, 123 ‘CLL-like’ HC-MBL cases were monitored for a median of 43 months with 4% per year requiring treatment in the first 7 years (17). Irrespective of progression to CLL, periodic hematological follow-up was recommended after the detection of HC-MBL, since this group has shorter overall survival compared to the general population (18).
Many of the biological characteristics of HC-MBL are similar to CLL Rai stage 0 (19). Nowadays, the strongest predictive marker for MBL progressing to CLL is the B cell count. This has emerged after the establishment of a B cell count-based system transition for discriminating MBL from CLL. This has led to a sizeable fraction of patients, considered as Rai stage 0 CLL, ending up classified as HC-MBL (20, 21). Relevant findings suggested that the B cell count appears to be a better predictor of TFS and OS when a defined lymphocyte threshold is applied (10). In a relevant study, the authors proposed a distinction of 2 ‘CLL‐like’ cell count thresholds for the identification of a very low risk group (<1.2x109/L) and a high risk group (>3.7x109/L) within HC-MBL (10, 17).
Subsequent studies appraised the impact of the size of the MBL clone on several clinical metrics, such as progression-free-survival (PFS), TFS and OS. In regard to the former, it is rather difficult to draw robust conclusions, given that a slight increase in cell numbers in HC-MBL cases with clone sizes near the mathematical cutoff of 5.0x109/L could be classified as disease progression. On the other hand, the assessment of TFS through standard NCI/iwCLL criteria may be more informative (22).
In CLL, the study of prognostic markers for time-to-first-treatment (TTFT) and OS has been of great interest, especially towards the development of a multifactorial risk score. In this context, 28 individual prognostic variables were assessed in a series of 969 individuals with CLL Rai 0 or HC-MBL leading to the development of the CLL international Prognostic Index (CLL-IPI). Implementation of the CLL-IPI score led to the identification of 4 risk groups (low, intermediate, high, and very-high risk) with different 5-year OS (93%, 79%, 63%, and 23%, respectively). Multivariate analysis of absolute B-cell count with individual factors of the CLL-IPI showed that five parameters, namely age, Rai stage, serum beta-2 microglobulin, unmutated IGHV genes, and del(17p) or TP53 mutation were associated with shorter TTFT and OS. Overall, the CLL-IPI risk score, despite some limitations, is able to predict TTFT and OS in previously untreated CLL patients whose only disease symptom is the presence of a circulating clonal B cell population (23). Despite these advances, the relevance of CLL-IPI for the evolution of HC-MBL to CLL remains to be fully elucidated.
Several markers have been studied at MBL diagnosis for their capacity to predict progression to CLL, including B cell receptor immunoglobulin (BcR IG) stereotypy, ZAP70 expression, Hb, platelet count and LDH, however none of them was clearly associated with progression to CLL (p≥0.05 in all instances). In contrast, the SHM status of the clonotypic IGHV gene, high expression of CD38 (≥30%), high expression of CD49d (≥30%) and chromosomal abnormalities were found to be significant on univariate analysis regarding prediction of TTFT. However, on multivariate analysis trisomy 12 and del (16) (p13) were the sole independent predictors of treatment-free survival (TFS) in MBL after adjusting for IGHV SHM status and CD38 expression (5, 14).
HC-MBL represents the vast majority of MBL cases (approximately 85%) identified in the context of clinical practice following the investigation of lymphocytosis, with a median ‘CLL-like’ B cell count above 1.9x109/L. On the contrary, around 85% of MBL cases detected in population screening studies have a ‘CLL-like’ B cell count below 0.5x109/L, with 40% of them bearing fewer than 0.05x109 clonal B cells/L (24). Previous findings support a bimodal distribution with a lower peak below 0.05x109 clonal B cells/L, very few cases in the range of 0.05-0.5 x109 clonal B cells/L, and another peak accounting for cases with a clonal B cell count ranging between 0.5-5x109 clonal B cells/L. A possible explanation could be a bias towards the selection of cases with high lymphocyte counts against those with mild or borderline lymphocytosis in the hematology clinical routine (22). An alternative biological explanation is offered by the distinct immunogenetic profiles of HC-MBL and LC-MBL, whereby pronounced clonal expansions are seen only when cells with ‘CLL-like’ phenotype express BcR IG with certain distinctive features (19). Longitudinal studies support the latter explanation, since most of the LC-MBL cases persisted over large periods of time (up to 34 months), without any progression to clinically overt disease. In fact, LC-MBL seems to be stable over time, a characteristic that is more pronounced in ‘CLL-like’ LC-MBL (90%) compared to other immunophenotypic variants, such as atypical (44.4%) and CD5-negative MBL (66.7%) (2).
Even though progression to HC-MBL is very rare, LC-MBL cases may often display a small increase in the size of the B cell clone. Studies focusing on clonal dynamics revealed a correlation between clonal size and clonality; in specific, the majority of monoclonal cases increased in size over time, while a much smaller number of biclonal and multiclonal cases behaved accordingly (25). Mathematical modeling of these results led to the conclusion that, in most cases, the estimated time for the progression of LC-MBL to CLL far exceeds a normal life expectancy. However, assessment of the clinical impact of LC-MBL, if any, through the comparison against age- and sex-matched non-MBL subjects showed that the OS observed for individuals with LC-MBL was significantly shorter than that of the control group. Moreover, individuals with LC-MBL showed a significantly shortened survival compared to that of age-matched individuals of the general population from the same geographical region. Infections, cancer and cardiovascular diseases were the main causes of mortality among individuals with LC-MBL. On the other hand, non-infectious respiratory tract diseases or genitourinary diseases, diabetes, dementia or other nervous system disorders accounted for almost 30% of deaths in the age- and sex-matched general population cohort. In the entire cohort, advanced age, co-existing cardiovascular diseases, solid tumors and, to a lesser extent, the presence of LC-MBL clones, were independently associated with a shorter OS (25).
The cytogenetic profile of HC-MBL is highly similar to that of CLL, especially of the indolent type (Rai 0 stage), in that del(13q14) was found to be the most frequent aberration followed by trisomy 12 (1). On the other hand, compared to Rai 0 CLL, HC-MBL displayed a lower prevalence of cytogenetic aberrations related to aggressive disease, such as del(11q) and del(17p), that were often acquired as secondary abnormalities (17).
In contrast, LC-MBL showed a significantly lower frequency of ‘CLL-specific’ cytogenetic aberrations compared to HC-MBL and CLL (26). At the individual aberration level, a tendency was observed towards a greater frequency of del(13q) and trisomy 12 from LC-MBL to HC-MBL and CLL, yet the individual frequencies were not significantly different between the three. On these grounds, one could speculate that these lesions occur rather early during the development of MBL and may be associated with the acquisition of the ‘CLL-like’ immunophenotypic profile rather than overt disease (26). A much lower prevalence of del(11q) and del(17p) was observed in LC-MBL; even so, such cases were detected, with the aberration being present in the majority of clonal B cells (2). This finding indicates that the presence of del(17p) per se does not axiomatically correlate with aggressive disease, as also reported for a small group of patients with CLL carrying this abnormality yet remaining stable for prolonged periods, especially when expressing mutated IGHV genes (27, 28). The distribution of the most common “CLL-related” cytogenetic aberrations among LC-MBL, HC-MBL and CLL is depicted in Figure 1. Of note, although LC-MBL appears to display a relatively simple cytogenetic profile, longitudinal analysis disclosed a significant increase in the overall frequency of cytogenetic aberrations after seven years of follow-up (29% at baseline versus 62% at follow-up): importantly, all cytogenetic aberrations observed at baseline also persisted at follow-up (25).
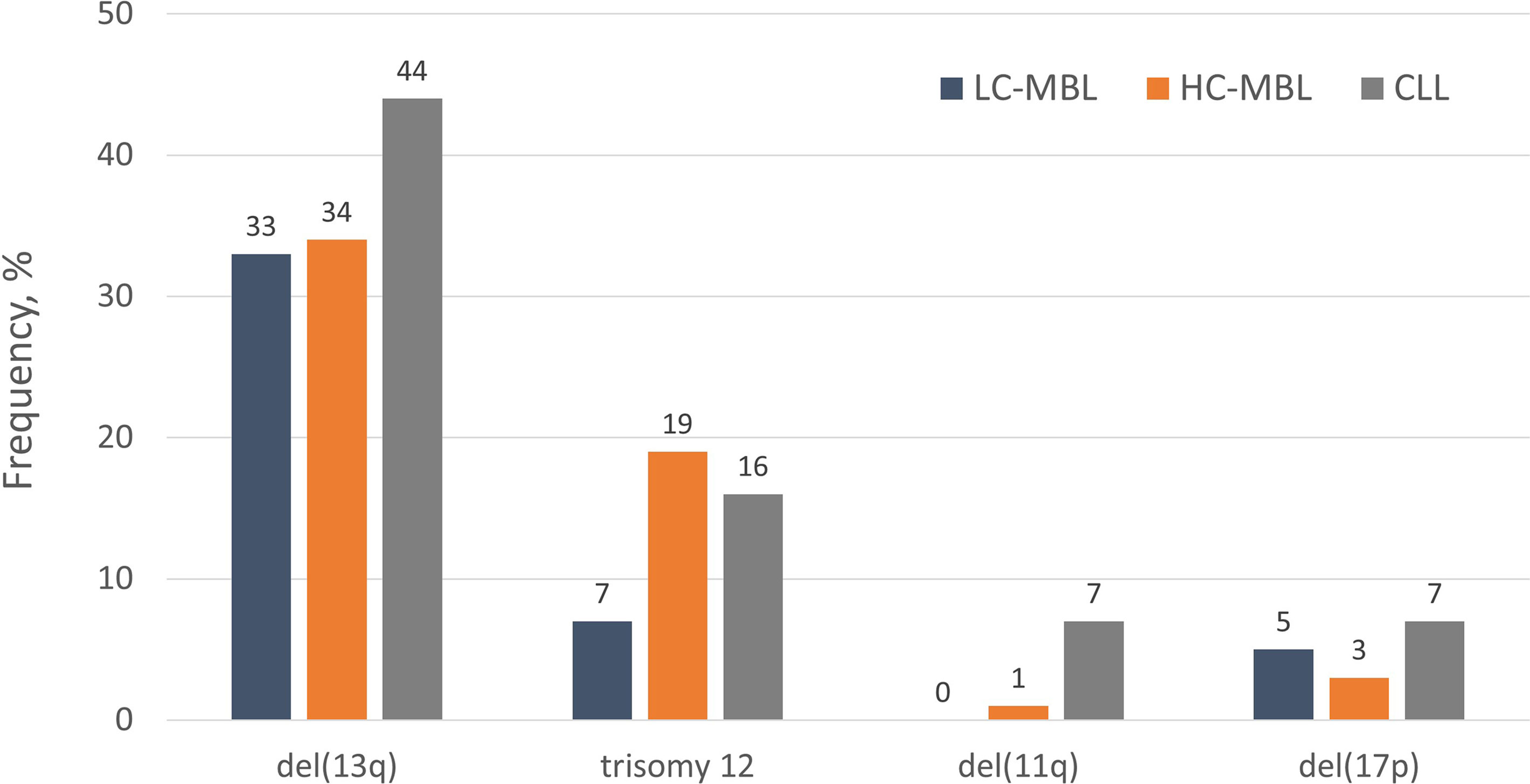
Figure 1 The distribution of “CLL-specific” cytogenetic aberrations in LC-MBL, HC-MBL and indolent CLL. Data was extracted from the studies by Rossi et al (17), Fazi et al. (2) and Henriques et al. (26).
Overall, these findings support that the progression from LC-MBL to HC-MBL and CLL is accompanied by the progressive acquisition of recurrent cytogenetic aberrations, each one of them enriched at specific stages along the natural history of the disease. In more detail, del(13q) and trisomy 12 occur rather early, at the LC-MBL stage, whereas del(17p) and del(11q) seem to emerge later, as secondary events at either the HC-MBL or the CLL stages. In line with this, the study of telomere length (TL) in both subtypes of MBL, indolent CLL and healthy subjects revealed the presence of significantly shorter telomeres already at the level of HC-MBL, suggesting that it may be part of the initial events in the process of CLL pathogenesis (26, 29).
Very few data is available for the profile of cytogenetic aberrations in ‘atypical’ and ‘CD5-negative’ subtypes of MBL; having said that, cases with del(13q), trisomy 12 and even del(17p) have been identified (1).
TP53 aberrations, including mutations, have an established role in shaping the outcome of CLL and, for this reason, their assessment is considered mandatory before treatment initiation (30). In recent years, the advent of NGS technologies led to the identification of additional gene mutations with putative clinical relevance in CLL. In particular, recurrent mutations in the NOTCH1, SF3B1, and BIRC3 genes were associated with distinct outcomes and were utilized in risk stratification schemes in the pivotal studies (31–33). Subsequent studies by several groups, including ours, have identified mutations in additional genes, e.g. RPS15, NFKBIE and EGR2, revealing a much more complicated genetic landscape for CLL (34–36). To complicate things even more, the detailed study of the clonal architecture in CLL in comparison to treatment revealed that the presence of mutations in putative CLL driver genes can adversely impact the clinical outcome of the disease, even when present in minor subclones, at least for certain mutations (37).
Some of the same recurrent gene mutations have been also found in HC-MBL. That said, at the individual gene level, NOTCH1 mutations exhibited a significantly lower prevalence in HC-MBL compared to CLL (3.2% versus 11.6%, respectively) (P=0.050) (38); moreover, SF3B1 mutations were even more scarce in HC-MBL (1.5%) versus CLL (~10%) (39). Interestingly, however, when larger cohorts were analyzed, the incidence of NOTCH1 and SF3B1 mutations was not significantly different between HC-MBL and Rai 0-CLL (NOTCH1: 8.2% vs. 13.1%; SF3B1: 4.7% vs. 3.8%) (Figure 2) (21). This finding supports the claim that MBL and early stage CLL are closely similar, differing only in clonal size, with MBL simply requiring more time to expand. This claim is further corroborated by the fact that low variant allele frequencies (VAF) were observed in almost all MBL cases but only in about half of CLL cases (41).
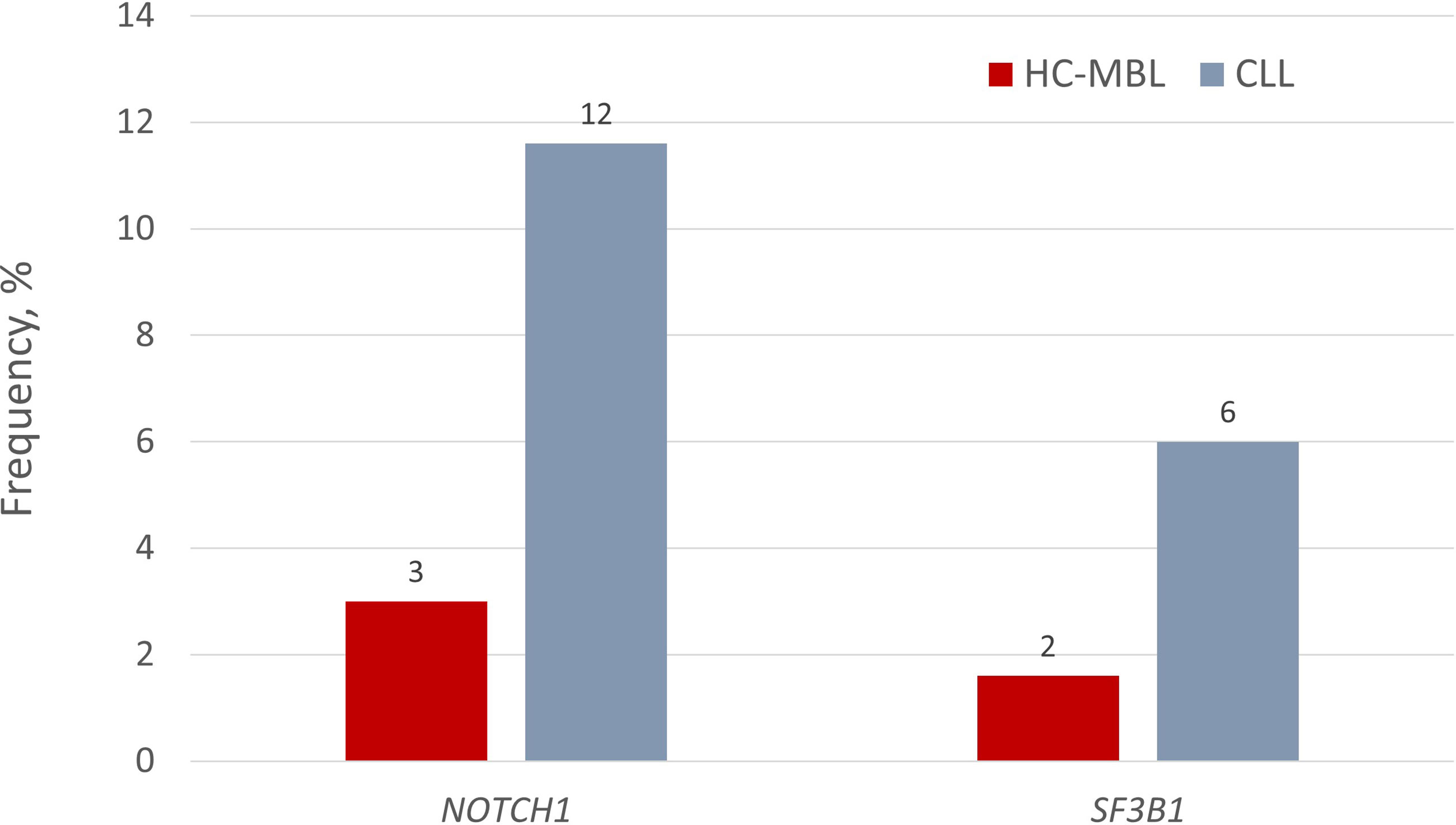
Figure 2 Incidence of NOTCH1 and SF3B1 gene mutations in HC-MBL and indolent CLL. Respective values for LC-MBL were 0% in both cases. Data regarding the presence of gene mutations in LC-MBL was extracted from Agathangelidis et al. (40), data regarding NOTCH1 mutations in HC-MBL and CLL from the study by Rasi et al. (38), and finally, the frequency of SF3B1 mutations in HC-MBL and CLL from the studies by Greco et al. (39) and Rossi et al. (33), respectively.
At the clinical level, both CLL and MBL cases bearing a NOTCH1 mutation had a shorter progression free survival (PFS) compared to wildtype ones (41). Thus, arguably, gene mutations could be used as biomarkers for identifying those MBL cases that would eventually progress to CLL requiring treatment. Indeed, in a longitudinal analysis, 60% of MBL cases that progressed to require treatment exhibited a subclonal expansion bearing a driver gene mutation, compared to only 14% of untreated cases (11). On the other hand, a longitudinal analysis of eight MBL cases by whole exome sequencing (WES) at two time points of 65 months apart, revealed that the four cases who developed detectable lymphadenopathy by physical exam yet without need for treatment carried mutations in putative CLL driver genes, including ATM, DDX3X, EGR2, FBXW7, SAMHD1 and SF3B1 (42). Targeted re-sequencing not only validated the WES results, but also led to the identification of additional mutations in the BIRC3, POT1 and NOTCH1 genes. Interestingly, a damaging mutation affecting PRDM1 was identified in a fifth case, which is frequently inactivated in diffuse large B-cell lymphomas. Of all, FBXW7 was the only gene recurrently mutated and all mutations were found in highly evolutionarily conserved regions and considered damaging by PolyPhen and MutationTaster (42). This data supports the notion that ‘CLL-associated’ gene mutations are not necessarily clinically relevant when detected in MBL.
Along these lines, in our recent characterization of the genetic landscape in both subtypes of MBL as well as indolent CLL, we were able to detect gene mutations in CLL putative driver genes in all 3 entities, yet these were infrequent and did not have any obvious impact on disease progression after a prolonged follow up. On these grounds, we proposed that gene mutations may represent late events related mostly to disease progression, whereas interactions between the cell clone and its microenvironment, such as those mediated through the B cell receptor (BcR), could represent the major driver in the early stages of the natural history of CLL (40).
Findings from low-throughput studies using mostly subcloning techniques studies reported that oligoclonality is a common feature of ‘CLL-like’ MBL (43), present in both LC-MBL and HC-MBL cases. In 2011, similar findings were obtained in the first relevant NGS study of 9 individuals with HC-MBL in whom multiple, immunogenetically independent MBL clones were identified, clearly suggesting distinct origins (44). The coexistence of multiple B cell expansions was also observed in a subsequent study as well, where the authors concluded that different subsets of normal B cells in the same individual may be targets for oncogenetic events involved in lymphomagenesis, arguing that bi- and multi-clonal MBL cases may be associated with distinct chronic antigen-driven immune responses (44),. This could be particularly frequent at earlier stages (LC-MBL), further evidence for the potential reactive nature of MBL among individuals with normal lymphocyte counts, prior to the stepwise acquisition of genomic alterations and the (infrequent) progression to HC-MBL and CLL (45).
Of note, in the latter study, the median numbers of clonal B cells in both ‘CLL-like’ HC-MBL and CLL were significantly lower in multiclonal than in monoclonal cases (41). In contrast, the median number of clonal cells in ‘CLL-like’ LC-MBL was significantly higher in multiclonal than monoclonal cases. Yet, longitudinal analysis showed a significant increase in size in monoclonal compared to bi- and multi-clonal LC-MBL cases (86% versus 54%, respectively; p=0.004). In most studies, no statistically significant differences in IGHV gene usage were identified in monoclonal versus multiclonal cases. Interestingly, the number of multiclonal cases with stereotyped BcR IG was significantly higher than that of monoclonal cases. Finally, the frequency of cytogenetic alterations as well as genetic complexity in both ‘CLL-like’ MBL and CLL clones from multiclonal cases was significantly lower than that of monoclonal cases from these entities (45).
More recently, employing NGS, we systematically assessed the immunogenetic characteristics of LC-MBL. We found that 75% samples displayed a monoclonal profile characterized by the presence of a single clonotype dominating the repertoire (frequency range: 42.5-97.9%), whereas the remaining 25% of samples turned out to be oligoclonal (46). Clonality levels seemed to correlate with clone size, in line with previous studies reporting a lower frequency of oligoclonality along the transition from LC-MBL to HC-MBL and, eventually, CLL (47). Figure 3 summarizes graphically the distribution of monoclonal and oligoclonal cases among the 2 subtypes of MBL and CLL.
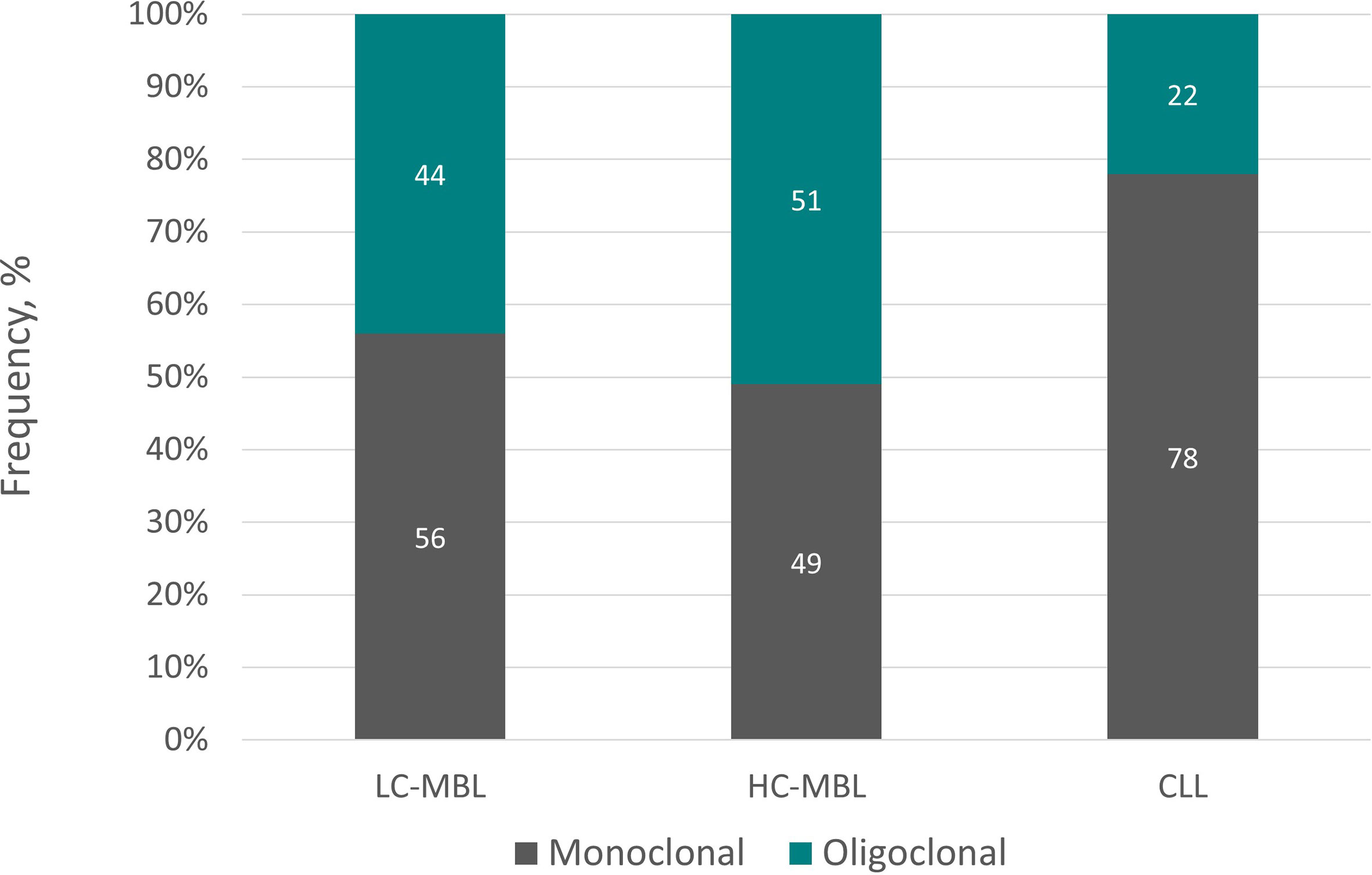
Figure 3 Monoclonal versus oligoclonal case distribution in LC-MBL, HC-MBL and CLL. Data regarding LC-MBL were extracted from Agathangelidis et al. (46) and Henriques et al. (26), whereas the data concerning HC-MBL and CLL were obtained from the latter.
Arguably, differences in clonality patterns among LC-MBL cases could indicate that these are captured at different stages along the ontogenetic trajectory. In this scenario, LC-MBL could initially involve a polyclonal B cell population that at some point acquired the CLL phenotype due to persistent antigenic stimulation. Microenvironmental interactions would induce further proliferation, eventually favoring the acquisition of genomic lesions, hence underlying the progressive transition to oligoclonal and, eventually, monoclonal LC-MBL cell populations. This evolutionary path could go in parallel with a continuous increase in clonal size, perhaps explaining the existence of multiclonal HC-MBL and CLL cases (25, 48).
The first studies reporting significant IGHV gene usage biases in the repertoire of CLL date from the 1990s (49). Subsequent studies in larger patient cohorts (50–52) cemented the notion that skewing of the IGHV repertoire in CLL results from strong selective pressures acting on the CLL progenitor cells by of a restricted group of antigens.
In order to obtain more insight into the reasons/ontogenetic timing repertoire skewing, several groups undertook comparative analyses of the IGHV gene repertoire in CLL and MBL. Early studies compared Rai 0 CLL with HC-MBL, yet the differences found were not statistically significant (17, 21). However, when comparisons extended to LC-MBL, that latter was found to display a distinct profile, including frequent expression of the IGHV4-59 and IGHV4-61 genes (rather infrequent in CLL), as well as pronounced scarcity of the IGHV4-34 and IGHV1-69 genes (26, 53). These findings suggested that HC-MBL and indolent CLL are closely related at the immunogenetic level, whereas LC-MBL is characterized by a rather unique repertoire, raising questions regarding its precise relationship to CLL (17, 43).
In the largest relevant study, our group applied Sanger sequencing in a cohort of 333 ‘CLL-like’ MBL cases and reported significant differences in the IGHV gene repertoire between HC-MBL versus LC-MBL; several genes were frequent only in the former (namely IGHV1-69, IGHV2-5, IGHV3-23, IGHV3-33, IGHV3-48 and IGHV4-34), whereas the IGHV4-59/61 genes was significantly more frequent in the latter. Comparison of both MBL subtypes against exclusively Rai-0 CLL cases or CLL cases from all stages disclosed several significant differences, again highlighting the distinct nature of the LC-MBL repertoire (Figure 4) (54). These findings strengthened the hypothesis that LC-MBL may not constitute a true pre-leukemic state but rather represent a manifestation of immune senescence.
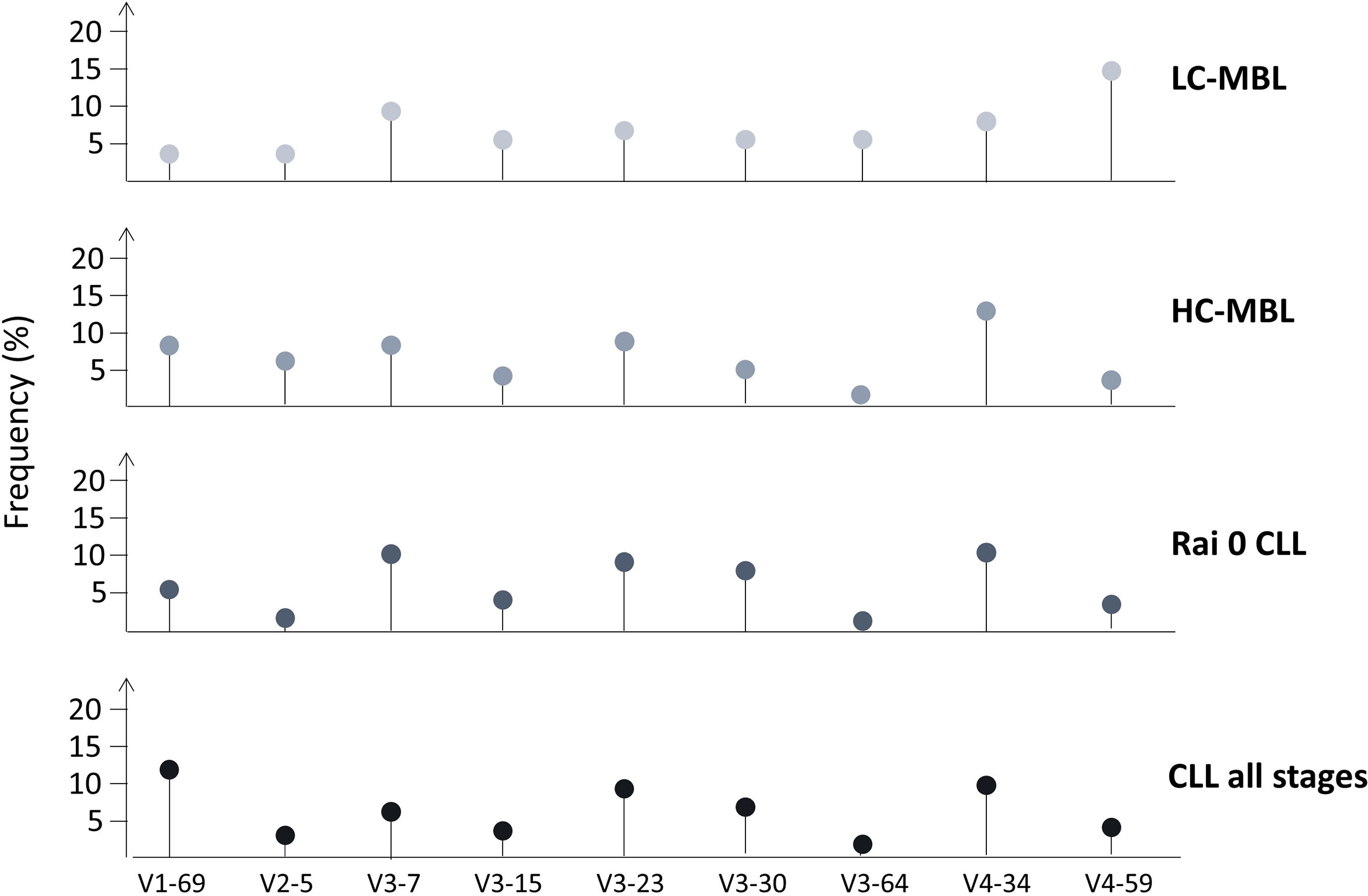
Figure 4 IGHV gene repertoire differences between LC-MBL, HC-MBL, Rai 0 CLL as well as in CLL from all stages. Data was obtained from Vardi et al. (54).
More recently, we re-appraised the immunogenetic profile of LC-MBL employing a high-throughput approach (46). More particularly, employing NGS, we studied the IGHV gene repertoire from clonal and normal B cell samples from 23 individuals with LC-MBL, which we compared against the corresponding repertoires of naïve and memory B cell samples from 6 healthy individuals. The most intriguing finding of our study concerned the stronger repertoire skewing in normal B cells from individuals with LC-MBL compared to healthy individuals. This argues for different selection processes and functions of these B cell subpopulations in LC-MBL compared to healthy individuals (55).
The SHM status of the clonotypic IGHV gene is now considered one of the most accurate biomarkers for clinical decision making in CLL (30). Based on the SHM status, CLL patients can be robustly classified CLL into 2 distinct groups: (i) CLL with unmutated IGHV genes (U-CLL), with few or no SHM [i.e. cases with a germline identity equal to or higher than 98%]; and, (ii) CLL with mutated IGHV genes (M-CLL), with a considerable SHM load and a germline identity below 98%. This classification scheme holds strong prognostic relevance, with U-CLL cases generally experiencing a worse prognosis and a more aggressive disease accompanied by early need for treatment compared to M-CLL (56, 57). It has been postulated that these two groups might also have distinct ontogeny, with M-CLL possibly deriving from B cells that have been activated by antigen(s) and have participated in the germinal center (GC) reaction, and U-CLL cases possibly originating from B cells following GC-independent maturation pathways (48, 58).
Following the same, CLL-relevant 98% identity cut-off value, several studies analysed the distribution of mutated and unmutated cases in MBL. In our series, out of the 355 IGH gene rearrangements analysed from 333 ‘CLL-like’ MBL cases, 267 (75.2%) were classified as mutated, while the rest (24.8%) were assigned to the unmutated subgroup. The distribution of unmutated and mutated IGH gene rearrangements in LC-MBL and HC-MBL was similar, and also comparable to Rai 0-CLL (26.4%, 24.4% and 25%, respectively). These 3 groups, however, differed significantly when compared against a CLL cohort containing 7,424 CLL patients from all stages, in which the frequency of unmutated cases was 45.1% (Figure 5) (54). In line with the above, both HC-MBL and Rai 0-CLL unmutated cases have been found to display distinct gene and miRNA expression profiles from the mutated MBL/CLL cases, independently of their classification (21).
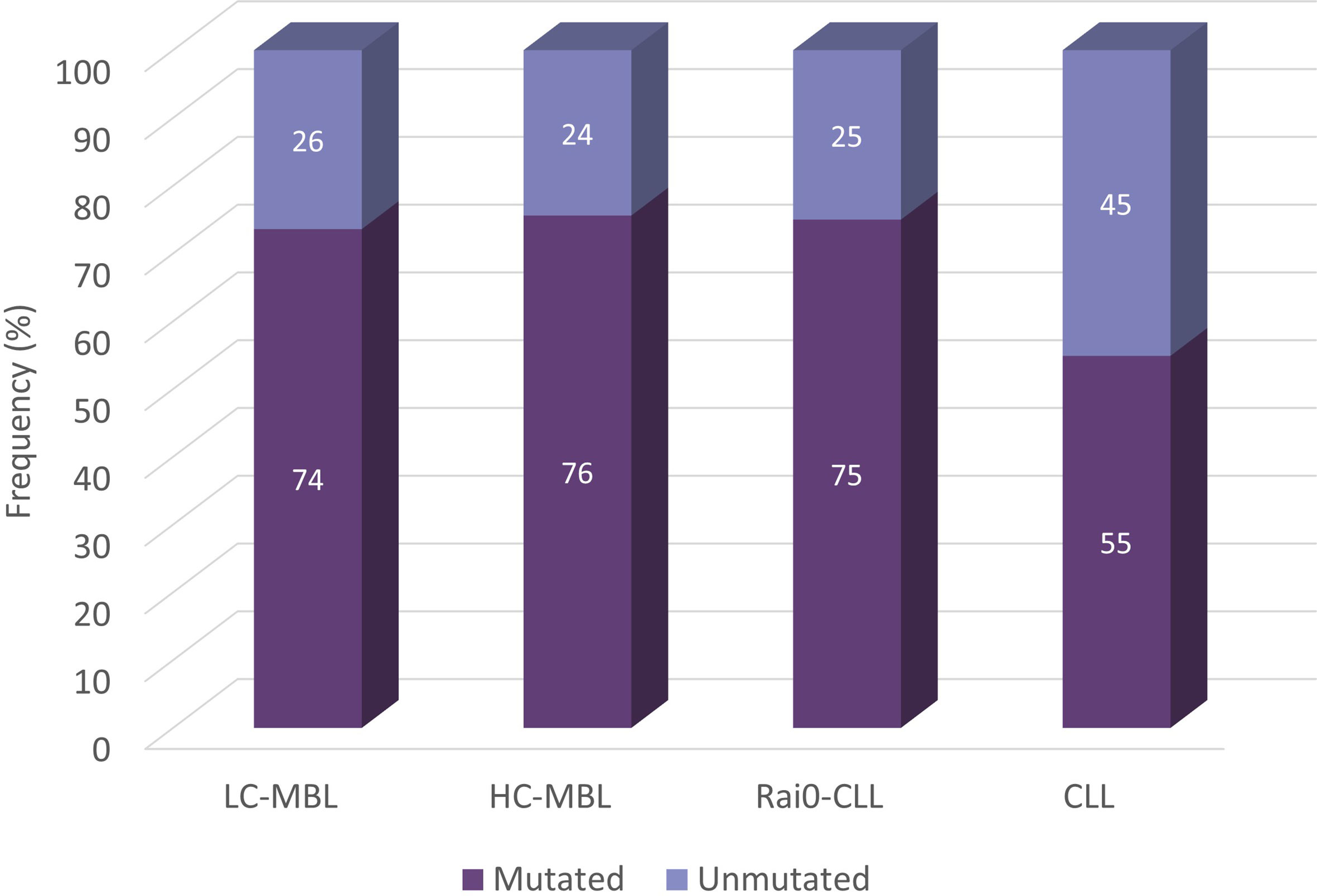
Figure 5 Somatic hypermutation status in cohorts from LC-MBL, HC-MBL and CLL. Data was obtained from Vardi et al. (54).
A distinctive feature of the IG gene repertoire of CLL concerns the presence of (quasi)identical BcR IG shared by different patients, a phenomenon termed BcR IG stereotypy. At odds with serendipity, whereby stereotyped BcR IG would be found with a negligible frequency (in the range of 10-12-10-16), stereotypy occurs in CLL at a remarkable frequency of 41% (59). Groups of patients expressing such restricted BCR IG are known as stereotyped subsets: they can be found in both U-CLL and M-CLL and vary in size from a handful to hundreds of cases, in which case they are deemed ‘major’ (52, 59). Notably, similarities between cases in stereotyped subsets extend from the primary IG sequences to biological and clinical profiles. Indeed, consistent clinical outcomes have been reported for patients assigned to the same subset (60–62), likely linked to consistent biological profiles: these pertain, amongst others, to distinctive gene expression, cytogenetic aberrations, BcR structure and signalling capacity, epigenetic modifications and antigenic reactivity profiles (63–71) that are also distinct between subsets but also from non-subset cases even of the same mutational category (i.e. U-CLL or M-CLL).
In this context, investigating whether ‘CLL-like’ stereotyped BcR IG could be detected also in MBL was a logical next step. When comparing HC-MBL with Rai 0-CLL, no significant differences were found, whereas, in sharp contrast, stereotyped BcR IG were exceedingly scarce in LC-MBL (17, 21). Interestingly, a correlation between the incidence of stereotypy and the absolute count of ‘CLL-like’ cells was identified: 5.5% in LC-MBL, 21.9% in HC-MBL, 20.2% in Rai 0-CLL, and 30.4% in CLL of all clinical stages (Figure 6) (54).
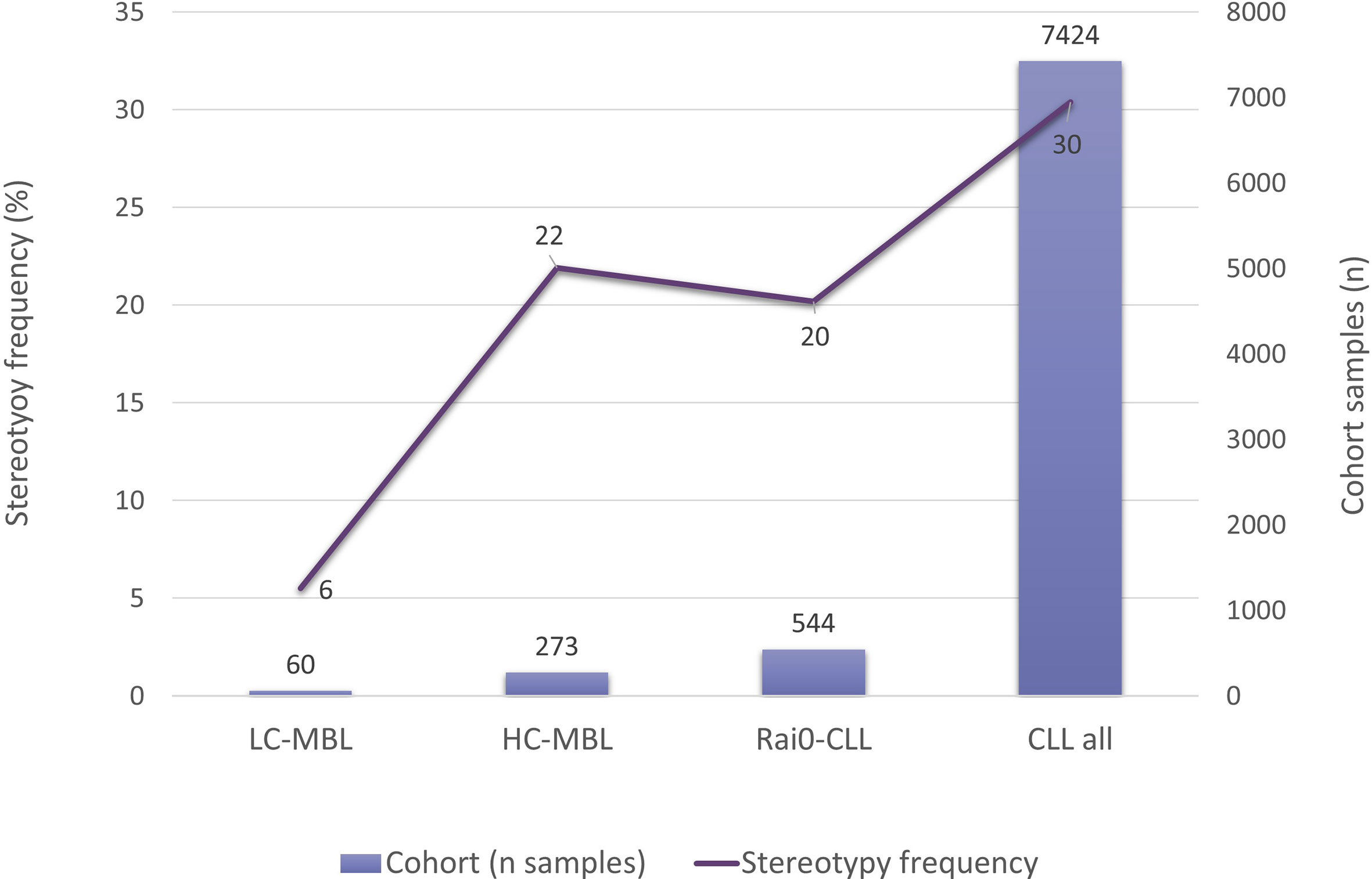
Figure 6 Frequency of BcR IG stereotypy in LC-MBL, HC-MBL, Rai 0 CLL and CLL from all clinical stages. Data was extracted from Vardi et al. (54).
The scarcity of BcR IG stereotypes in LC-MBL was also evident in our recent high-throughput study. Of the few stereotyped clonotypes, virtually all were classified to minor subsets with a small minority corresponding to stereotypes defining major CLL stereotyped subsets, particularly those associated with aggressive disease. On these grounds, we proposed that LC-MBL is immunogenetically distinct from CLL, at least for those cases with aggressive clinical behavior (46).
Multifarious abnormalities in the T cell compartment constitute a well characterized feature of the tumor microenvironment in CLL (72–75). However, although our understanding of the interactions between T cells and CLL leukemic cells is continuously growing, several important questions remain, not lest regarding the precise role of T cells i.e. whether they exert pro- or anti-neoplastic actions.
CLL is characterized by a relative loss of naïve CD4+ T cells accompanied by enrichment of antigen-experienced, memory and effector CD4+ T cells (73, 76). CD4+ T cells in CLL express higher levels of PD-1, human leukocyte antigen (HLA)-DR and Ki67, which are all activation markers (73, 77, 78). In vitro as well as in vivo studies in xenograft mouse models suggested that CD4+ T cells could enhance the survival and proliferation of clonal cells in CLL, a finding that was supported by correlations between CD4+ T cell counts and clinical outcome (79, 80).
CD8+ T cells from patients with CLL display high expression of PD-1 and other inhibitory receptors, accompanied by defective immune synapse formation (also occurring in CD4+ T cells as well) (78, 81). For these reasons, CD8+ T cells in CLL have been described as “pseudoexhausted” (78). Relevant experiments in mice showed that the exhaustion of T cells was anatomically restricted, being observed mainly in spleen samples and to a much lower extent in the blood (81). Impaired immune synapse formation was observed ex vivo when T cells from healthy donors were co-cultured with clonal CLL cells, suggesting that this defect was largely induced by the CLL cells themselves. Of interest, blocking inhibitory ligands, such as CD200, PD-L1, or CD276 with neutralizing antibodies led to restored immune synapse formation capacity (82). Regarding the biological function of CD8+ T cells in CLL, apparently they may recognize tumor specific antigens, however they ultimately fail to control the disease likely due to their functional exhaustion (76).
Characteristic T cell receptor (TR) gene repertoire restrictions have been established in CLL, mainly in the CD8+ but also in the CD4+ compartment (83). Even though clonal expansions occur frequently in the T cell repertoire of the elderly, restrictions were more pronounced in CLL, indicating the existence of stronger and perhaps different selective pressures. Whether these pressures are exerted by the same antigens interacting with the CLL clone or by CLL-derived antigens remains to be elucidated (83, 84).
Comparative analysis between HC-MBL, U-CLL and M-CLL cases revealed similar CD4+ T cell counts in all three conditions. In contrast, the CD8+ T cell count was higher in U-CLL compared to M-CLL patients and significantly higher than that of HC-MBL (85). Interestingly, both CD4+ and CD8+ clonal T cell expansions were identified in HC-MBL, similar to CLL. CD4+ T cell clonal expansions appeared to follow the numerical increase of clonal B cells, thus being more pronounced in CLL compared to HC-MBL (85). This finding suggests that CD4+ TR repertoire restrictions may be somehow influenced by the size of B cell clonal expansions, occurring early in clonal evolution and increasing concurrently with tumor progression. The fact that CLL displays higher T cell repertoire restrictions could also reflect the loss of effector T cell clones restraining CLL clonal expansions. In line with this, treatment with the Bruton’s tyrosine kinase inhibitor ibrutinib, led to an increase in the T cell repertoire diversity; this suggests that the eradication of the CLL malignant cells sets the stage for T cell reconstitution (86).
Concerning the clonality of the CD8+ T cell fraction, no significant differences were detected between HC-MBL and CLL (87). Furthermore, a longitudinal analysis showed that T cell clones, mainly from the CD8+ compartment, persisted over time in all MBL samples analyzed, similar to what was previously reported in CLL (83, 88), suggesting that interactions between CLL and T cells are evident prior to/at the early stages of CLL development (87). Moreover, decreased numbers of CD4+CD8+ double positive T cells were identified in HC-MBL compared to healthy control samples, alluding to impaired immunosurveillance which may actually favor the emergence of MBL clones. Furthermore, Tregs appeared increased in CLL, but not in MBL, compared to healthy controls (89).
Regarding LC-MBL, early studies reported an increase in the size of clonal T cell populations compared to the general population, especially for double positive CD4+CD8+ T cells, suggesting a general deregulation of the immune system, possibly related to chronic antigenic stimulation (90). Recently, a follow-up study of LC-MBL revealed statistically significant increase of total T cells as well as CD4+, CD8+ and the double negative cell subpopulations over time. Interestingly, LC-MBL with larger clone sizes over time also showed significantly higher (P<0.05) numbers of the distinct normal residual T cell subsets at follow up compared to the baseline, especially for CD4+ T cells (79, 91). This might imply that signals emanating from immune cells of the CLL microenvironment, potentially promoting activation, proliferation and/or survival of B cells, could contribute to the expansion of ‘CLL-like’ B cell clones at the earliest stages of CLL (25).
In our recent study of the TR beta chain gene repertoire in LC-MBL by NGS, we reported more pronounced T cell expansions in LC-MBL compared to aged-matched healthy individuals, yet lower than CLL. We attributed this finding to different antigenic pressures, leading to more pronounced T cell expansions in CLL versus LC-MBL versus healthy individuals (92). Moreover, a significant correlation between the level of T cell clonality and the size of the MBL clone was shown, in line with previous findings in CLL, further supporting the notion that T cell expansions could be driven by CLL-associated antigens (83, 88). Pairwise comparisons between all entities revealed distinct TRBV gene repertoire biases; this could perhaps imply that different antigen selection pressures operate in LC-MBL, CLL or healthy individuals. The latter assumption was further supported by the fact that shared clonotypes between different entities were scant. The nature of the implicated antigens and whether they are related to the expanded ‘CLL-like’ cell clones remains to be clarified (92).
Several defects in both the innate and the adaptive immune responses have been identified in CLL, including hypogammaglobulinemia (93). The exact mechanisms underlying these defects are poorly characterized, whereas even less information is available regarding the disease stage during which they take place. In this context, Criado and colleagues (55) studied the residual normal B cell compartment in the blood of individuals with LC-MBL and HC-MBL compared to Rai 0-CLL in order to get insight into the mechanisms that may contribute to the emergence of hypogammaglobulinemia in CLL. Both HC-MBL and Rai 0-CLL showed significantly reduced normal B cell counts, mostly at the expense of pre-GC B cells, both immature and naïve, suggesting diminished production and/or release of these cells in the circulation at the early stages of CLL. Considering that no such decrease has been identified in circulating immature and naïve B cell counts in relation to ageing (94), the decreased numbers of circulating pre-GC B cells in individuals with HC-MBL alludes to impaired production of newly generated B cells in the BM, even prior to the development of CLL (55).
When focusing on the memory B cells (MBC) and plasma cells (PC), there were no significant differences in either MBL (both subtypes) or CLL compared to healthy controls with the exception of PC counts between HC-MBL and the healthy controls. Of note, both MBC and PC showed significantly different distribution among B cell subsets, expressing distinct IG subclasses in both MBL and CLL. Of importance, the extent of these changes was greater from LC-MBL to HC-MBL and finally Rai0-CLL. Thus, the possibility of a progressive impairment of B cell responses driven by newly encountered antigens along the transition from LC-MBL to HC-MBL and finally Rai 0-CLL was proposed; impaired pre-GC B cell production has been considered as largely responsible, since it could lead to a progressive reduction of the diversity of the BcR IG repertoire from LC-MBL to HC-MBL and Rai 0-CLL (55). This would be in line with other features of CLL but also MBL, such as the launch of active, but silent, responses against common pathogens, particularly viruses such as cytomegalovirus (CMV) and the Epstein Barr virus (EBV), as well as bacteria such as S. pneumoniae (95). Additional, longitudinal studies in larger MBL and CLL cohorts would be necessary to support this hypothesis.
MBL detection is based on the identification of B cell expansions in the blood circulation with the characteristic “CLL-specific” phenotype, yet of a smaller size than the one required for CLL diagnosis. This criterion, based solely on a mathematical cutoff is devoid of any biological context, perhaps hampering our understanding of the mechanisms driving disease onset. Thus, the in-depth characterization of MBL holds great potential for understanding the mechanisms that represent major drivers in the process of its transformation to CLL.
Nowadays, it is well established that CLL is characterized by a highly complicated genomic landscape, including a series of cytogenetic aberrations as well as gene mutations. That said, the overall simple genetic background of MBL pointed towards the BcR IG as one of the major “players” prior to disease onset. The study of BcR IG clonality, especially through NGS-based methodologies, revealed much a much higher frequency of oligoclonality in MBL, especially LC-MBL, compared to CLL, most likely reflecting different staged along the ontogenetic trajectory. Relevant to mention, the BcR IG repertoire in HC-MBL was similar to CLL regarding both the IGHV gene repertoire as well as BcR IG stereotypy, whereas LC-MBL showed a highly distinct repertoire characterized by the dominance of different IGHV genes and a very low prevalence of stereotypes, alluding to different antigenic pressures shaping the BcR IG repertoire in LC-MBL versus HC-MBL and CLL. Further support for an early role of the microenvironment in CLL ontogenesis came from the study of the TR repertoire in both LC- and HC-MBL. Similar to CLL, T cell expansions were evident in both subtypes of MBL yet at a lower extent. As above, the TR repertoire was characterized by different biases in the repertoire of TRBV genes and the absence of shared clonotyped among MBL and CLL pointing towards different selection mechanisms. Last, the study of residual B cells in MBL and CLL revealed an impaired production of newly generated B cells in the BM, even prior to the development of CLL, as well as a progressive impairment of B cell responses driven by newly encountered antigens along the transition from LC-MBL to HC-MBL and finally Rai 0-CLL that could account for the clinical impact of MBL detection.
Overall, even if major questions still remain, parallel analysis of the B cell clone(s) and the tumor microenvironment in MBL holds strong potential for characterizing the mechanisms that are required for the emergence and malignant transformation of a monoclonal B cell population.
All authors contributed to the article and approved the submitted version. CG, LZ-I, and AI wrote the manuscript. AC, KS, and AA edited the text and gave final approval.
This work was supported in part by the Hellenic Foundation for Research and Innovation (HFRI) and the General Secretariat for Research and Technology (GSRT), under grant agreement No 336 (Project CLLon); COSMIC, a Marie Curie European Training Network funded from the European Union’s Horizon 2020 research and innovation programme under grant agreement no. 765158; the project ODYSSEAS (Intelligent and Automated Systems for enabling the Design, Simulation and Development of Integrated Processes and Products) implemented under the “Action for the Strategic Development on the Research and Technological Sector”, funded by the Operational Programme “Competitiveness, Entrepreneurship and Innovation” (NSRF 2014-2020) and co-financed by Greece and the European Union, with grant agreement no: MIS 5002462; NEoteRIC, funded from the European Union’s Horizon 2020 research and innovation programme under grant agreement No 871330; the Greek Precision Medicine Network (GPMN), a mission of the Research and Innovation sector of the Ministry of Education, Research and Religious Affairs of Greece.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Lanasa MC, Allgood SD, Slager SL, Dave SS, Love C, Marti GE, et al. Immunophenotypic and Gene Expression Analysis of Monoclonal B-Cell Lymphocytosis Shows Biologic Characteristics Associated With Good Prognosis CLL. Leukemia (2011) 25(9):1459–66. doi: 10.1038/leu.2011.117
2. Fazi C, Scarfò L, Pecciarini L, Cottini F, Dagklis A, Janus A, et al. General Population Low-Count CLL-Like MBL Persists Over Time Without Clinical Progression, Although Carrying the Same Cytogenetic Abnormalities of CLL. Blood (2011) 118(25):6618–25. doi: 10.1182/blood-2011-05-357251
3. Maitre E, Troussard X. Monoclonal B-Cell Lymphocytosis. Best Pract Res Clin Haematol (2019) 32(3):229–38. doi: 10.1016/j.beha.2019.06.002
4. Scarfò L, Ghia P. What Does It Mean I Have a Monoclonal B-Cell Lymphocytosis?: Recent Insights and New Challenges. Semin Oncol (2016) 43(2):201–8. doi: 10.1053/j.seminoncol.2016.02.013
5. Rawstron AC. Monoclonal B Cell Lymphocytosis - What Does It Really Mean? Curr Hematol Malig Rep (2013) 8(1):52–9. doi: 10.1007/s11899-012-0144-z
6. Kostopoulos IV, Paterakis G, Papadimitriou K, Pavlidis D, Tsitsilonis OE, Papadhimitriou SI. Immunophenotypic Analysis Reveals Heterogeneity and Common Biologic Aspects in Monoclonal B-Cell Lymphocytosis. Genes Chromosomes Cancer (2015) 54(4):210–21. doi: 10.1002/gcc.22234
7. de Tute R, Yuille M, Catovsky D, Houlston RS, Hillmen P, Rawstron AC. Monoclonal B-Cell Lymphocytosis (MBL) in CLL Families: Substantial Increase in Relative Risk for Young Adults [3]. Leukemia (2006) 20(4):728–9. doi: 10.1038/sj.leu.2404116
8. Goldin LR, Lanasa MC, Slager SL, Cerhan JR, Vachon CM, Strom SS, et al. Common Occurrence of Monoclonal B-Cell Lymphocytosis Among Members of High-Risk CLL Families. Br J Haematol (2010) 151(2):152–8. doi: 10.1111/j.1365-2141.2010.08339.x
9. Fazi C, Dagklis A, Cottini F, Scarfò L, Bertilaccio MTS, Finazzi R, et al. Monoclonal B Cell Lymphocytosis in Hepatitis C Virus Infected Individuals. Cytom Part B - Clin Cytom (2010) 78(SUPPL. 1):61–8. doi: 10.1002/cyto.b.20545
10. Shanafelt TD, Kay NE, Jenkins G, Call TG, Zent CS, Jelinek DF, et al. B-Cell Count and Survival: Differentiating Chronic Lymphocytic Leukemia From Monoclonal B-Cell Lymphocytosis Based on Clinical Outcome. Blood (2009) 113(18):4188–96. doi: 10.1182/blood-2008-09-176149
11. Barrio S, Shanafelt TD, Ojha J, Chaffee KG, Secreto C, Kortüm KM, et al. Genomic Characterization of High-Count MBL Cases Indicates That Early Detection of Driver Mutations and Subclonal Expansion Are Predictors of Adverse Clinical Outcome. Leukemia (2017) 31(1):170–6. doi: 10.1038/leu.2016.172
12. Angelillo P, Capasso A, Ghia P, Scarfò L. Monoclonal B-Cell Lymphocytosis: Does the Elderly Patient Need a Specialistic Approach? Eur J Intern Med (2018) 58:2–6. doi: 10.1016/j.ejim.2018.09.006
13. Landgren O, Albitar M, Ma W, Abbasi F, Hayes RB, Ghia P, et al. B-Cell Clones as Early Markers for Chronic Lymphocytic Leukemia. N Engl J Med (2009) 360(7):659–67. doi: 10.1056/NEJMoa0806122
14. Rawstron AC, Bennett FL, O’Connor SJM, Kwok M, Fenton JAL, Plummer M, et al. Monoclonal B-Cell Lymphocytosis and Chronic Lymphocytic Leukemia. N Engl J Med (2008) 359(6):575–83. doi: 10.1056/NEJMoa075290
15. Shanafelt TD, Kay NE, Rabe KG, Call TG, Zent CS, Maddocks K, et al. Brief Report: Natural History of Individuals With Clinically Recognized Monoclonal B-Cell Lymphocytosis Compared With Patients With Rai 0 Chronic Lymphocytic Leukemia. J Clin Oncol (2009) 27(24):3959–63. doi: 10.1200/JCO.2008.21.2704
16. Molica S, Mauro FR, Giannarelli D, Lauria F, Cortelezzi A, Brugiatelli M, et al. Differentiating Chronic Lymphocytic Leukemia From Monoclonal B-Lymphocytosis According to Clinical Outcome on Behalf of the GIMEMA Chronic Lymphoproliferative Diseases Working Group. Haematologica (2011) 96(2):277–83. doi: 10.3324/haematol.2010.030189
17. Rossi D, Sozzi E, Puma A, De Paoli L, Rasi S, Spina V, et al. The Prognosis of Clinical Monoclonal B Cell Lymphocytosis Differs From Prognosis of Rai 0 Chronic Lymphocytic Leukaemia and Is Recapitulated by Biological Risk Factors. Br J Haematol (2009) 146(1):64–75. doi: 10.1111/j.1365-2141.2009.07711.x
18. Parikh SA, Chaffee KG, Larson MC, Hampel PJ, Call TG, Ding W, et al. Outcomes of a Large Cohort of Individuals With Clinically Ascertained High-Count Monoclonal B-Cell Lymphocytosis. Haematologica (2018) 103(6):e237–40. doi: 10.3324/haematol.2017.183194
19. Strati P, Shanafelt TD. Monoclonal B-Cell Lymphocytosis and Early-Stage Chronic Lymphocytic Leukemia: Diagnosis, Natural History, and Risk Stratification. Blood (2015) 126(4):454–62. doi: 10.1182/blood-2015-02-585059
20. Mulligan CS, Thomas ME, Mulligan SP. Lymphocytes B Lymphocytes, and Clonal CLL Cells: Observations on the Impact of the New Diagnostic Criteria in the 2008 Guidelines for Chronic Lymphocytic Leukemia (CLL). Blood (2009) 113(25):6496–7. doi: 10.1182/blood-2008-07-166710
21. Morabito F, Mosca L, Cutrona G, Agnelli L, Tuana G, Ferracin M, et al. Clinical Monoclonal B Lymphocytosis Versus Rai 0 Chronic Lymphocytic Leukemia: A Comparison of Cellular, Cytogenetic, Molecular, and Clinical Features. Clin Cancer Res (2013) 19(21):5890–900. doi: 10.1158/1078-0432.CCR-13-0622
22. Rawstron AC, Shingles J, De Tute R, Bennett F, Jack AS, Hillmen P. Chronic Lymphocytic Leukaemia (CLL) and CLL-Type Monoclonal B-Cell Lymphocytosis (MBL) Show Differential Expression of Molecules Involved in Lymphoid Tissue Homing. Cytom Part B - Clin Cytom (2010) 78(SUPPL. 1):42–6. doi: 10.1002/cyto.b.20534
23. Parikh SA. Chronic Lymphocytic Leukemia Treatment Algorithm 2018. Blood Cancer J (2018) 8(10):93. doi: 10.1038/s41408-018-0131-2
24. Shanafelt TD, Ghia P, Lanasa MC, Landgren O, Rawstron AC. Monoclonal B-Cell Lymphocytosis (MBL): Biology, Natural History and Clinical Management. Leukemia (2010) 24(3):512–20. doi: 10.1038/leu.2009.287
25. Criado I, Rodríguez-Caballero A, Gutiérrez ML, Pedreira CE, Alcoceba M, Nieto W, et al. Low-Count Monoclonal B-Cell Lymphocytosis Persists After Seven Years of Follow Up and Is Associated With a Poorer Outcome. Haematologica (2018) 103(7):1198–208. doi: 10.3324/haematol.2017.183954
26. Henriques A, Rodríguez-Caballero A, Nieto WG, Langerak AW, Criado I, Lécrevisse Q, et al. Combined Patterns of IGHV Repertoire and Cytogenetic/Molecular Alterations in Monoclonal B Lymphocytosis Versus Chronic Lymphocytic Leukemia. PloS One (2013) 8(7):1–10. doi: 10.1371/journal.pone.0067751
27. Yu L, Kim HT, Kasar S, Benien P, Du W, Hoang K, et al. Survival of Del17p CLL Depends on Genomic Complexity and Somatic Mutation. Clin Cancer Res (2017) 23(3):735–45. doi: 10.1158/1078-0432.CCR-16-0594
28. Yi S, Li Z, Zou D, Xiong W, Li H, Cui R, et al. Del17p Does Not Always Significantly Influence the Survival of B-Cell Chronic Lymphoproliferative Disorders. Oncotarget (2018) 9(3):3353–64. doi: 10.18632/oncotarget.23261
29. Furtado FM, Scheucher PS, Santana BA, Scatena NF, Calado RT, Rego EM, et al. Telomere Length Analysis in Monoclonal B-Cell Lymphocytosis and Chronic Lymphocytic Leukemia Binet a. Braz J Med Biol Res (2017) 50(5):1–4. doi: 10.1590/1414-431x20176019
30. Hallek M, Cheson BD, Catovsky D, Caligaris-Cappio F, Dighiero G, Döhner H, et al. iwCLL Guidelines for Diagnosis, Indications for Treatment, Response Assessment, and Supportive Management of CLL. Blood (2018) 131(25):2745–60. doi: 10.1182/blood-2017-09-806398
31. Nadeu F, Delgado J, Royo C, Baumann T, Stankovic T, Pinyol M, et al. Clinical Impact of Clonal and Subclonal TP53, SF3B1, BIRC3, NOTCH1, and ATM Mutations in Chronic Lymphocytic Leukemia. Blood (2016) 127(17):2122–30. doi: 10.1182/blood-2015-07-659144
32. Cortese D, Sutton LA, Cahill N, Smedby KE, Geisler C, Gunnarsson R, et al. On the Way Towards a “CLL Prognostic Index”: Focus on TP53, BIRC3, SF3B1, NOTCH1 and MYD88 in a Population-Based Cohort. Leukemia (2014) 28(3):710–3. doi: 10.1038/leu.2013.333
33. Rossi D, Bruscaggin A, Spina V, Rasi S, Khiabanian H, Messina M, et al. Mutations of the SF3B1 Splicing Factor in Chronic Lymphocytic Leukemia: Association With Progression and Fludarabine-Refractoriness. Blood J Am Soc Hematol (2011) 118(26):6904–8. doi: 10.1182/blood-2011-08-373159
34. Ljungström V, Cortese D, Young E, Pandzic T, Mansouri L, Plevova K, et al. Whole-Exome Sequencing in Relapsing Chronic Lymphocytic Leukemia: Clinical Impact of Recurrent RPS15 Mutations. Blood (2016) 127(8):1007–16. doi: 10.1182/blood-2015-10-674572
35. Mansouri L, Sutton LA, Ljungström V, Bondza S, Arngården L, Bhoi S, et al. Functional Loss of Iκbϵ Leads to NF-κb Deregulation in Aggressive Chronic Lymphocytic Leukemia. J Exp Med (2015) 212(6):833–43. doi: 10.1084/jem.20142009
36. Young E, Noerenberg D, Mansouri L, Ljungström V, Frick M, Sutton LA, et al. EGR2 Mutations Define a New Clinically Aggressive Subgroup of Chronic Lymphocytic Leukemia. Leukemia (2017) 31(7):1547–54. doi: 10.1038/leu.2016.359
37. Rossi D, Khiabanian H, Spina V, Ciardullo C, Bruscaggin A, Famà R, et al. Clinical Impact of Small TP53 Mutated Subclones in Chronic Lymphocytic Leukemia. Blood (2014) 123(14):2139–47. doi: 10.1182/blood-2013-11-539726
38. Rasi S, Monti S, Spina V, Foà R, Gaidano G, Rossi D. Analysis of NOTCH1 Mutations in Monoclonal B-Cell Lymphocytosis. Haematologica (2012) 97(1):153–4. doi: 10.3324/haematol.2011.053090
39. Greco M, Capello D, Bruscaggin A, Spina V, Rasi S, Monti S, et al. Analysis of SF3B1 Mutations in Monoclonal B-Cell Lymphocytosis. Hematol Oncol (2013) 31(1):54–5. doi: 10.1002/hon.2013
40. Agathangelidis A, Ljungström V, Scarfò L, Fazi C, Gounari M, Pandzic T, et al. Highly Similar Genomic Landscapes in Monoclonal B-Cell Lymphocytosis and Ultra-stable Chronic Lymphocytic Leukemia With Low Frequency of Driver Mutations Haematologica (2018) 103(5):865–73. doi: 10.3324/haematol.2017.177212
41. Lionetti M, Fabris S, Cutrona G, Agnelli L, Ciardullo C, Matis S, et al. High-Throughput Sequencing for the Identification of NOTCH1 Mutations in Early Stage Chronic Lymphocytic Leukaemia: Biological and Clinical Implications. Br J Haematol (2014) 165(5):629–39. doi: 10.1111/bjh.12800
42. Ojha J, Secreto C, Rabe K, Ayres-Silva J, Tschumper R, Dyke DV, et al. Monoclonal B-Cell Lymphocytosis Is Characterized by Mutations in CLL Putative Driver Genes and Clonal Heterogeneity Many Years Before Disease Progression. Leukemia (2014) 28(12):2395–8. doi: 10.1038/leu.2014.226
43. Dagklis A, Fazi C, Sala C, Cantarelli V, Scielzo C, Massacane R, et al. The Immunoglobulin Gene Repertoire of Low-Count Chronic Lymphocytic Leukemia (CLL)-Like Monoclonal B Lymphocytosis Is Different From CLL: Diagnostic Implications for Clinical Monitoring. Blood (2009) 114(1):26–32. doi: 10.1182/blood-2008-09-176933
44. Lanasa MC, Allgood SD, Volkheimer AD, Gockerman JP, Whitesides JF, Goodman BK, et al. Single-Cell Analysis Reveals Oligoclonality Among Low-Count Monoclonal B-Cell Lymphocytosis. Leukemia (2010) 24(1):133–40. doi: 10.1038/leu.2009.192
45. Henriques A, Rodríguez-Caballero A, Criado I, Langerak AW, Nieto WG, Lécrevisse Q, et al. Molecular and Cytogenetic Characterization of Expanded B-Cell Clones From Multiclonal Versus Monoclonal B-Cell Chronic Lymphoproliferative Disorders. Haematologica (2014) 99(5):897–907. doi: 10.3324/haematol.2013.098913
46. Agathangelidis A, Galigalidou C, Scarfò L, Moysiadis T, Rovida A, Gounari M, et al. Infrequent “Chronic Lymphocytic Leukemia-Specific” Immunoglobulin Stereotypes in Aged Individuals With or Without Low-Count Monoclonal B-Cell Lymphocytosis. Haematologica (2021) 106(4):1178–81. doi: 10.3324/haematol.2020.247908
47. de Faria-Moss M, Yamamoto M, Arrais-Rodrigues C, Criado I, Gomes CP, de Lourdes Chauffaille M, et al. High Frequency of Chronic Lymphocytic Leukemia-Like Low-Count Monoclonal B-Cell Lymphocytosis in Japanese Descendants Living in Brazil. Haematologica (2020) 105(6):e298–301. doi: 10.3324/haematol.2019.230813
48. Gemenetzi K, Agathangelidis A, Zaragoza-Infante L, Sofou E, Papaioannou M, Chatzidimitriou A, et al. B Cell Receptor Immunogenetics in B Cell Lymphomas: Immunoglobulin Genes as Key to Ontogeny and Clinical Decision Making. Front Oncol (2020) 10:67. doi: 10.3389/fonc.2020.00067
49. Fais F, Ghiotto F, Hashimoto S, Sellars B, Valetto A, Allen SL, et al. Chronic Lymphocytic Leukemia B Cells Express Restricted Sets of Mutated and Unmutated Antigen Receptors. J Clin Invest (1998) 102(8):1515–25. doi: 10.1172/JCI3009
50. Murray F, Darzentas N, Hadzidimitriou A, Tobin G, Boudjogra M, Scielzo C, et al. Stereotyped Patterns of Somatic Hypermutation in Subsets of Patients With Chronic Lymphocytic Leukemia: Implications for the Role of Antigen Selection in Leukemogenesis. Blood (2008) 111(3):1524–33. doi: 10.1182/blood-2007-07-099564
51. Stamatopoulos K, Belessi C, Moreno C, Boudjograh M, Guida G, Smilevska T, et al. Over 20% of Patients With Chronic Lymphocytic Leukemia Carry Stereotyped Receptors: Pathogenetic Implications and Clinical Correlations. Blood (2007) 109(1):259–70. doi: 10.1182/blood-2006-03-012948
52. Agathangelidis A, Darzentas N, Hadzidimitriou A, Brochet X, Murray F, Yan XJ, et al. Stereotyped B-Cell Receptors in One-Third of Chronic Lymphocytic Leukemia: A Molecular Classification With Implications for Targeted Therapies. Blood (2012) 119(19):4467–75. doi: 10.1182/blood-2011-11-393694
53. Scarfò L, Zibellini S, Tedeschi A, Maura F, Neri A, Bertazzoni P, et al. Impact of B-Cell Count and Imaging Screening in cMBL: Any Need to Revise the Current Guidelines. Leukemia (2012) 26(7):1703–7. doi: 10.1038/leu.2012.20
54. Vardi A, Dagklis A, Scarfo L, Jelinek D, Newton D, Bennett F, et al. Immunogenetics Shows That Not All MBL Are Equal: The Larger the Clone, the More Similar to CLL. Blood (2013) 121(22):4521–8. doi: 10.1182/blood-2012-12-471698
55. Criado I, Blanco E, Rodríguez-Caballero A, Alcoceba M, Contreras T, Gutiérrez ML, et al. Residual Normal B-Cell Profiles in Monoclonal B-Cell Lymphocytosis Versus Chronic Lymphocytic Leukemia. Leukemia (2018) 32(12):2701–5. doi: 10.1038/s41375-018-0164-3
56. Damle RN, Wasil T, Fais F, Ghiotto F, Valetto A, Allen SL, et al. Ig V Gene Mutation Status and CD38 Expression as Novel Prognostic Indicators in Chronic Lymphocytic Leukemia. Blood (1999) 94(6):1840–7. doi: 10.1182/blood.V94.6.1840
57. Hamblin TJ, Davis Z, Gardiner A, Oscier DG, Stevenson FK. Unmutated Ig V(H) Genes Are Associated With a More Aggressive Form of Chronic Lymphocytic Leukemia. Blood (1999) 94(6):1848–54. doi: 10.1182/blood.V94.6.1848
58. Chiorazzi N, Ferrarini M. Cellular Origin(s) of Chronic Lymphocytic Leukemia: Cautionary Notes and Additional Considerations and Possibilities. Blood (2011) 117(6):1781–91. doi: 10.1182/blood-2010-07-155663
59. Agathangelidis A, Chatzidimitriou A, Gemenetzi K, Giudicelli V, Karypidou M, Plevova K, et al. Higher-Order Connections Between Stereotyped Subsets: Implications for Improved Patient Classification in CLL. Blood (2021) 137(10):1365–76. doi: 10.1182/blood.2020007039
60. Baliakas P, Hadzidimitriou A, Sutton LA, Minga E, Agathangelidis A, Nichelatti M, et al. Clinical Effect of Stereotyped B-Cell Receptor Immunoglobulins in Chronic Lymphocytic Leukaemia: A Retrospective Multicentre Study. Lancet Haematol (2014) 1(2):e74–84. doi: 10.1016/S2352-3026(14)00005-2
61. Baliakas P, Agathangelidis A, Hadzidimitriou A, Sutton LA, Minga E, Tsanousa A, et al. Not All IGHV3-21 Chronic Lymphocytic Leukemias Are Equal: Prognostic Considerations. Blood (2015) 125(5):856–9. doi: 10.1182/blood-2014-09-600874
62. Jaramillo S, Agathangelidis A, Schneider C, Bahlo J, Robrecht S, Tausch E, et al. Prognostic Impact of Prevalent Chronic Lymphocytic Leukemia Stereotyped Subsets: Analysis Within Prospective Clinical Trials of the German CLL Study Group. Haematologica (2020) 105(11):2598–607. doi: 10.3324/haematol.2019.231027
63. Marincevic M, Cahill N, Gunnarsson R, Isaksson A, Mansouri M, Göransson H, et al. High-Density Screening Reveals a Different Spectrum of Genomic Aberrations in Chronic Lymphocytic Leukemia Patients With “Stereotyped” IGHV3-21 and IGHV4-34 B-Cell Receptors. Haematologica (2010) 95(9):1519–25. doi: 10.3324/haematol.2009.021014
64. Kanduri M, Marincevic M, Halldórsdóttir AM, Mansouri L, Junevik K, Ntoufa S, et al. Distinct Transcriptional Control in Major Immunogenetic Subsets of Chronic Lymphocytic Leukemia Exhibiting Subset-Biased Global DNA Methylation Profiles. Epigenetics (2012) 7(12):1435–42. doi: 10.4161/epi.22901
65. Marincevic M, Mansouri M, Kanduri M, Isaksson A, Göransson H, Smedby KE, et al. Distinct Gene Expression Profiles in Subsets of Chronic Lymphocytic Leukemia Expressing Stereotyped IGHV4-34 B Cell Receptors. Haematologica (2010) 95(12):2072–9. doi: 10.3324/haematol.2010.028639
66. Papakonstantinou N, Ntoufa S, Chartomatsidou E, Papadopoulos G, Hatzigeorgiou A, Anagnostopoulos A, et al. Differential microRNA Profiles and Their Functional Implications in Different Immunogenetic Subsets of Chronic Lymphocytic Leukemia. Mol Med (2013) 19(1):115–23. doi: 10.2119/molmed.2013.00005
67. Ntoufa S, Vardi A, Papakonstantinou N, Anagnostopoulos A, Aleporou-Marinou V, Belessi C, et al. Distinct Innate Immunity Pathways to Activation and Tolerance in Subgroups of Chronic Lymphocytic Leukemia With Distinct Immunoglobulin Receptors. Mol Med (2012) 18(9):1281–91. doi: 10.2119/molmed.2011.00480
68. Gounari M, Ntoufa S, Apollonio B, Papakonstantinou N, Ponzoni M, Chu CC, et al. Excessive Antigen Reactivity may Underlie the Clinical Aggressiveness of Chronic Lymphocytic Leukemia Stereotyped Subset 8. Blood (2015) 125(23):3580–7. doi: 10.1182/blood-2014-09-603217
69. Ntoufa S, Papakonstantinou N, Apollonio B, Gounari M, Galigalidou C, Fonte E, et al. B Cell Anergy Modulated by TLR1/2 and the miR-17∼92 Cluster Underlies the Indolent Clinical Course of Chronic Lymphocytic Leukemia Stereotyped Subset 4. J Immunol (2016) 196(10):4410–7. doi: 10.4049/jimmunol.1502297
70. Minici C, Gounari M, Übelhart R, Scarfò L, Dühren-von Minden M, Schneider D, et al. Distinct Homotypic B-Cell Receptor Interactions Shape the Outcome of Chronic Lymphocytic Leukaemia. Nat Commun (2017) 8:15746. doi: 10.1038/ncomms15746
71. Gemenetzi K, Psomopoulos F, Carriles AA, Gounari M, Minici C, Plevova K, et al. Higher-Order Immunoglobulin Repertoire Restrictions in CLL: The Illustrative Case of Stereotyped Subsets 2 and 169. Blood (2021) 137(14):1895–904. doi: 10.1182/blood.2020005216
72. Vlachonikola E, Stamatopoulos K, Chatzidimitriou A. T Cells in Chronic Lymphocytic Leukemia: A Two-Edged Sword. Front Immunol (2021) 11:612244. doi: 10.3389/fimmu.2020.612244
73. Palma M, Gentilcore G, Heimersson K, Mozaffari F, Näsman-Glaser B, Young E, et al. T Cells in Chronic Lymphocytic Leukemia Display Dysregulated Expression of Immune Checkpoints and Activation Markers. Haematologica (2017) 102(3):562–72. doi: 10.3324/haematol.2016.151100
74. Scrivener S, Goddard RV, Kaminski ER, Prentice AG. Abnormal T-Cell Function in B-Cell Chronic Lymphocytic Leukaemia. Leuk Lymphoma (2003) 44(3):383–9. doi: 10.1080/1042819021000029993
75. Brusa D, Serra S, Coscia M, Rossi D, D’Arena G, Laurenti L, et al. The PD-1/PD-L1 Axis Contributes to T-Cell Dysfunction in Chronic Lymphocytic Leukemia. Haematologica (2013) 98(6):953. doi: 10.3324/haematol.2012.077537
76. Roessner PM, Seiffert M. T-Cells in Chronic Lymphocytic Leukemia: Guardians or Drivers of Disease? Leukemia (2020) 34(8):2012–24. doi: 10.1038/s41375-020-0873-2
77. Elston L, Fegan C, Hills R, Hashimdeen SS, Walsby E, Henley P, et al. Increased Frequency of CD4+ PD-1+ HLA-DR+ T Cells Is Associated With Disease Progression in CLL. Br J Haematol (2020) 188(6):872–80. doi: 10.1111/bjh.16260
78. Riches JC, Davies JK, McClanahan F, Fatah R, Iqbal S, Agrawal S, et al. T Cells From CLLpatients Exhibit Features of T-Cell Exhaustion But Retain Capacity for Cytokine Production. Blood (2013) 121(9):1612–21. doi: 10.1182/blood-2012-09-457531
79. Os A, Bürgler S, Ribes AP, Funderud A, Wang D, Thompson KM, et al. Chronic Lymphocytic Leukemia Cells Are Activated and Proliferate in Response to Specific T Helper Cells. Cell Rep (2013) 4(3):566–77. doi: 10.1016/j.celrep.2013.07.011
80. Bagnara D, Kaufman MS, Calissano C, Marsilio S, Patten PEM, Simone R, et al. A Novel Adoptive Transfer Model of Chronic Lymphocytic Leukemia Suggests a Key Role for T Lymphocytes in the Disease. Blood J Am Soc Hematol (2011) 117(20):5463–72. doi: 10.1182/blood-2010-12-324210
81. Hanna BS, Roessner PM, Scheffold A, Jebaraj BMC, Demerdash Y, Öztürk S, et al. Pi3kδ Inhibition Modulates Regulatory and Effector T-Cell Differentiation and Function in Chronic Lymphocytic Leukemia. Leukemia (2019) 33(6):1427–38. doi: 10.1038/s41375-018-0318-3
82. Ramsay AG, Clear AJ, Fatah R, Gribben JG. Multiple Inhibitory Ligands Induce Impaired T-Cell Immunologic Synapse Function in Chronic Lymphocytic Leukemia That Can Be Blocked With Lenalidomide: Establishing a Reversible Immune Evasion Mechanism in Human Cancer. Blood (2012) 120(7):1412–21. doi: 10.1182/blood-2012-02-411678
83. Vardi A, Vlachonikola E, Karypidou M, Stalika E, Bikos V, Gemenetzi K, et al. Restrictions in the T-Cell Repertoire of Chronic Lymphocytic Leukemia: High-Throughput Immunoprofiling Supports Selection by Shared Antigenic Elements. Leukemia (2017) 31(7):1555–61. doi: 10.1038/leu.2016.362
84. Rawstron AC. Monoclonal B-Cell Lymphocytosis. Hematol Am Soc Hematol Educ Progr (2009) 430–9. doi: 10.1182/asheducation-2009.1.430
85. Lanasa MC, Weinberg JB. Immunologic Aspects of Monoclonal B-Cell Lymphocytosis. Immunol Res (2011) 49(1):269–80. doi: 10.1007/s12026-010-8188-4
86. Yin Q, Sivina M, Robins H, Yusko E, Vignali M, O’Brien S, et al. Ibrutinib Therapy Increases T Cell Repertoire Diversity in Patients With Chronic Lymphocytic Leukemia. J Immunol (2017) 198(4):1740–7. doi: 10.4049/jimmunol.1601190
87. Blanco G, Vardi A, Puiggros A, Gómez-Llonín A, Muro M, Rodríguez-Rivera M, et al. Restricted T Cell Receptor Repertoire in CLL-Like Monoclonal B Cell Lymphocytosis and Early Stage CLL. Oncoimmunology (2018) 7(6):e1432328. doi: 10.1080/2162402X.2018.1432328
88. Vardi A, Agathangelidis A, Stalika E, Karypidou M, Siorenta A, Anagnostopoulos A, et al. Antigen Selection Shapes the T-Cell Repertoire in Chronic Lymphocytic Leukemia. Clin Cancer Res (2016) 22(1):167–74. doi: 10.1158/1078-0432.CCR-14-3017
89. Te Raa GD, Tonino SH, Remmerswaal EBM, Van Houte AJ, Koene HR, Van Oers MH, et al. Chronic Lymphocytic Leukemia Specific T-Cell Subset Alterations Are Clone-Size Dependent and Not Present in Monoclonal B Lymphocytosis. Leuk Lymphoma (2012) 53(11):2321–5. doi: 10.3109/10428194.2012.698277
90. Ghia P, Prato G, Stella S, Scielzo C, Geuna M, Caligaris-Cappio F. Age-Dependent Accumulation of Monoclonal CD4+ CD8+ Double Positive T Lymphocytes in the Peripheral Blood of the Elderly. Br J Haematol (2007) 139(5):780–90. doi: 10.1111/j.1365-2141.2007.06867.x
91. Purroy N, Wu CJ. Coevolution of Leukemia and Host Immune Cells in Chronic Lymphocytic Leukemia. Cold Spring Harb Perspect Med (2017) 7(4):a026740. doi: 10.1101/cshperspect.a026740
92. Agathangelidis A, Galigalidou C, Scarfò L, Moysiadis T, Rovida A, Vlachonikola E, et al. High-Throughput Analysis of the T-Cell Receptor Gene Repertoire in Low-Count Monoclonal B-Cell Lymphocytosis Reveals a Distinct Profile From Chronic Lymphocytic Leukemia. Haematologica (2020) 105(10):515–8. doi: 10.3324/haematol.2019.221275
93. Parikh SA, Leis JF, Chaffee KG, Call TG, Hanson CA, Ding W, et al. Hypogammaglobulinemia in Newly Diagnosed Chronic Lymphocytic Leukemia: Natural History, Clinical Correlates, and Outcomes. Cancer (2015) 121(17):2883–91. doi: 10.1002/cncr.29438
94. Blanco E, Pérez-Andrés M, Arriba-Méndez S, Contreras-Sanfeliciano T, Criado I, Pelak O, et al. Age-Associated Distribution of Normal B-Cell and Plasma Cell Subsets in Peripheral Blood. J Allergy Clin Immunol (2018) 141(6):2208–19. doi: 10.1016/j.jaci.2018.02.017
95. Criado I, Muñoz-Criado S, Rodríguez-Caballero A, Nieto WG, Romero A, Fernández-Navarro P, et al. Host Virus and Pneumococcus-Specific Immune Responses in High-Count Monoclonal B-Cell Lymphocytosis and Chronic Lymphocytic Leukemia: Implications for Disease Progression. Haematologica (2017) 102(7):1238–46. doi: 10.3324/haematol.2016.159012
Keywords: monoclonal B cell lymphocytosis (MBL), chronic Lymphocutic Leukemia (CLL), genetics, immunogenetics, tumor microenvironment, ontogenesis, B cell receptor, immunoglobulin
Citation: Galigalidou C, Zaragoza-Infante L, Iatrou A, Chatzidimitriou A, Stamatopoulos K and Agathangelidis A (2021) Understanding Monoclonal B Cell Lymphocytosis: An Interplay of Genetic and Microenvironmental Factors. Front. Oncol. 11:769612. doi: 10.3389/fonc.2021.769612
Received: 02 September 2021; Accepted: 26 October 2021;
Published: 11 November 2021.
Edited by:
Dimitar G. Efremov, International Centre for Genetic Engineering and Biotechnology, ItalyReviewed by:
Paolo Strati, University of Texas MD Anderson Cancer Center, United StatesCopyright © 2021 Galigalidou, Zaragoza-Infante, Iatrou, Chatzidimitriou, Stamatopoulos and Agathangelidis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Andreas Agathangelidis, YWdhdGhhbkBiaW9sLnVvYS5ncg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.