 Wentao Jia
Wentao Jia Shufang Liang
Shufang Liang Binbin Cheng
Binbin Cheng Changquan Ling
Changquan Ling- 1School of Traditional Chinese Medicine, Naval Medical University, Shanghai, China
- 2Department of Traditional Chinese Medicine, Changhai Hospital, Navy Medical University, Shanghai, China
Invasion and metastasis are the main reasons for the high mortality of liver cancer, which involve the interaction of tumor stromal cells and malignant cells. Cancer-associated fibroblasts (CAFs) are one of the major constituents of tumor stromal cells affecting tumor growth, invasion, and metastasis. The heterogeneous properties and sources of CAFs make both tumor-supporting and tumor-suppression effects possible. The mechanisms for CAFs in supporting hepatocellular carcinoma (HCC) progression can be categorized into upregulated aggressiveness and stemness, transformed metabolism toward glycolysis and glutamine reductive carboxylation, polarized tumor immunity toward immune escape of HCC cells, and increased angiogenesis. The tumor-suppressive effect of fibroblasts highlights the functional heterogenicity of CAF populations and provides new insights into tumor–stromal interplay mechanisms. In this review, we introduced several key inflammatory signaling pathways in the transformation of CAFs from normal stromal cells and the heterogeneous biofunctions of activated CAFs. In view of the pleiotropic regulation properties of traditional Chinese medicine (TCM) and heterogeneous effects of CAFs, we also introduced the application and values of TCM in the treatment of HCC through targeting CAFs.
Introduction
Hepatocellular carcinoma (HCC), one of the deadliest malignant diseases, claims more than 2,814,000 lives a year in China (1). To date, the 5-year survival rate of HCC patients after hepatectomy is still below 60% (2, 3). The recurrence and metastasis of HCC after radical resection account for more than 90% cases of early postoperative death (4). The dismal curative ratio of HCC has yet to be significantly improved despite several emerging therapies, which poses a great challenge to clinicians and researchers.
Currently, the therapeutic targets have been transformed from the tumor itself to the stromal cells in the tumor microenvironment (TME), which includes fibroblasts, endothelial cells, mesenchymal cells, and immune cells. HCC progression does not completely depend on tumor autonomous signaling (5). The cross-talks between stromal cells and cancer cells, as well as the communication among different kinds of stromal cells, are of great significance in constructing tumorigenic environments (6), which account for the dismal prognosis of HCC. Such interactions have gained much popularity in the treatment of HCC. Fibroblasts are one of the major constituents of tumor stromal cells, which originate from a variety of cell types in the presence of complex signaling. Several studies have identified components including tissue residual fibroblasts, endothelial cells undergoing endothelial-to-mesenchymal transition (EndMT), pericytes, vascular smooth muscle cells, cancer cells that undergo the epithelial–mesenchymal transformation (EMT), and bone marrow-derived cells (BMDCs) as the origins of cancer-associated fibroblasts (CAFs) (6, 7).
The multiple origins and diverse functions of CAFs make the heterogeneity of cell populations and distinct effects on tumor growth. With insufficient biomarker distinguishing all subtypes of fibroblasts, the identification of CAFs with different functions requires further investigation (8). Overall, the main types of CAFs exert a tumor-supporting effect. Several clinical studies have confirmed the tumor-supporting effect of CAFs and demonstrated that abundant CAFs in HCC tissue always predict poor prognosis after radical operation (9–11). Oddly, in some studies, the upregulation of CAF level in HCC tissue does not necessarily indicate poor prognosis, which means that some subtypes of CAFs as well as their cytokines and metabolic products show little effect on the progression of HCC and may even suppress the malignance of the tumor (12–17). Also, the neoplasm and metastasis lesions are commonly encased by fibrotic tissue and collagen, suggesting that tumor progression can be restrained by desmoplasia and some CAF populations (13, 18). In a cohort study, the high expression of fibroblast activation protein (FAP) is not associated with the disease-free survival time (DFS) of HCC patients after hepatic resection (16). This contradictory phenomenon merits long-term clinical studies with larger samples and more high-quality fundamental studies.
In this review, we introduced several key inflammatory signaling pathways in the transformation of CAFs from normal stromal cells, the metabolism reprogramming of activated CAFs, and the heterogeneous biofunctions of activated CAFs on HCC progression, metastasis, and recurrence. Finally, we also introduced the application and values of traditional Chinese medicine (TCM) in the treatment of HCC through targeting CAFs.
The Activation of CAFs in the Premalignant Microenvironment of HCC
In fibrosis and cirrhosis, the premalignant microenvironments are considered especially significant under the situation of tissue injury, hypoxia, and chronic inflammation. As tumors are regarded as “wounds that never heal”, the activation of fibroblasts is a critical process in tumor progression (19). Generally, fibroblasts in the liver environment are mainly distributed around the fibrous septum, fibrous capsule, and hepatic blood sinusoids and activated in the existence of tumor-associated signals (8, 19). Quiescent hepatic stellate cells (HSCs) and other normal fibroblasts suppress malignant cell growth via contact inhibition (18). While under liver injury, the inflammatory factors including sulfatase (SULF)-2, transforming growth factor (TGF)-β, CXCL6, and CXCR4 educate normal fibroblasts and other relevant cells toward CAFs (20–22) and increase the expression of CAF markers such as α-smooth muscle actin (SMA), FAP, vimentin, collagen, and fibroblast specific protein (FSP)-1 (23). The mechanisms for CAF activation are summarized in Figure 1.
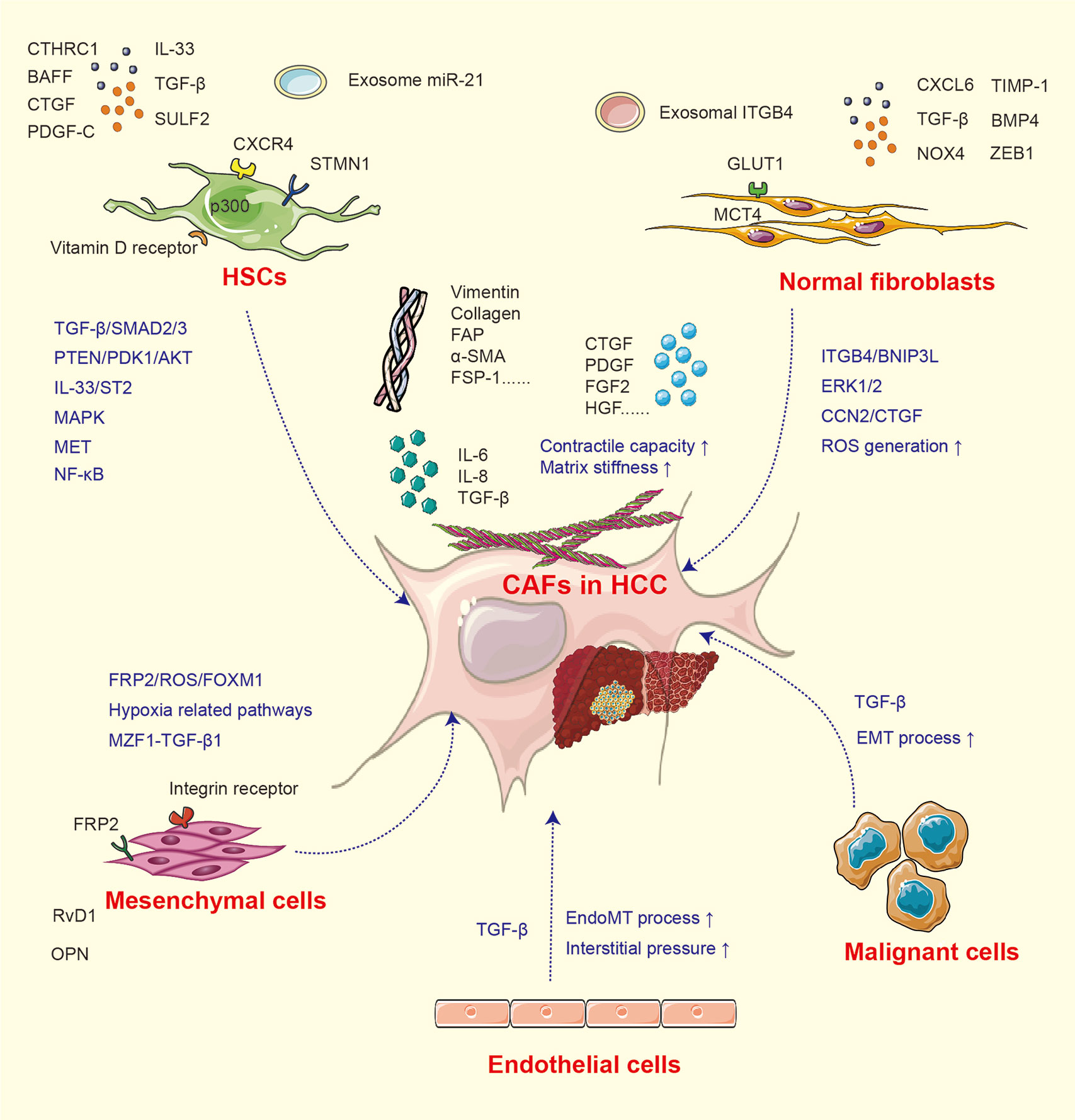
Figure 1 The activation of CAF from heterogeneous cells.
CAFs in HCC have heterogeneous cellular origins (7). As mentioned above, multiple populations undergoing mesenchymal transition and equipped with fibroblastic property can develop into CAFs under inflammatory signal. Quiescent HSCs are the major source of CAFs, and the activation of HSCs is established as a pivotal step of liver fibrosis and tumor initiation. Under normal situation, vitamin D receptor in HSCs is stimulated by p62/SQSTM1 (24), while under pathological state, this process is abolished due to the loss of p62, which induces HSCs into CAFs and further supports HCC initiation (24). In addition, numerous studies have investigated the HCC–HSC feedback loop. Co-culture with HCC cells enhances the expression of CAF biomarkers α-SMA, vimentin, FAP, and FSP-1 in HSCs (23). The conditioned medium collected from activated HSCs can in turn stimulate tumor cell proliferation, stemness, and invasion (18). TGF-β is considered as the most potent signal involved in HSC activation (21, 23, 25). The activation of the TGF-β/SMAD pathway induces type I and III collagen expression in human HSC line LX2. It has been confirmed that SULF2 expressed by HCC cells can induce HSCs into CAFs through the TGF-β/SMAD3 pathway, which may further inhibit apoptosis and promote EMT of HCC cells via the SDF-1/CXCR4-related signaling pathway (20). p300 acetyltransferase promotes TGF-β-stimulated HSC activation by both cytoplasm-to-nucleus shuttle for SMAD2/3-tafazzin (TAZ) and histone acetylation mechanisms (26). Recently, it was reported that HCC-derived SDF-1 upregulates the expression of TGF-β and activates LX2 into CAFs (27). Meanwhile, TGF-β can also activate mitogen-activated protein kinase (MAPK) signals, further endowing the tumor-supporting property of HSCs (6). Apart from TGF-β, factors including platelet-derived growth factor (PDGF)-C, microRNA (miRNA)-21, interleukin (IL)-33, CTHRC1, and bone morphogenetic protein (BMP)-4 can also activate HSCs and facilitate liver fibrosis (28–32).
In addition to HSCs, components including mesenchymal stem cells (MSCs), hepatic sinus endothelial cells (HSECs), and even tumor cells can be developed into CAFs. In a hypoxic microenvironment, MSCs are transformed toward CAFs (31, 33, 34), then enhance the expression of cyclooxygenase (COX)-2 and prostaglandin E2 (PGE2), and subsequently facilitate HCC progression (31). Another study found that after co-cultured with SK-Hep1 cells, the expression of CAF biomarkers tenascin-C and SDF-1 is significantly upregulated in human MSCs (34). The MSC-derived CAFs then facilitate the EMT process of SK-Hep1 cells through integrin-related signaling pathways. The high expression of osteopontin (OPN) in a cancer microenvironment can mediate the MSC–CAF transition via binding with the integrin receptor and stimulation of the downstream MZF1–TGF-β1 pathway (33). Additionally, cancer-derived exosomes can trigger the process of EndMT in HSECs. The transformed HSECs display CAF features and express increased CAF biomarkers (35). HCC cells located around the blood sinusoid and undergoing EMT commonly express the CAF biomarkers α-SMA and FAP, indicating that both TGF-β and hypoxia-inducible factor (HIF)-1α enriched under hypoxic condition can induce CAF features in HCC cells (22, 36).
Interestingly, the activation of CAFs is reversible. It has been proven that chronic hypoxia may deactivate CAFs, reduce contractile force and stiffness in the surrounding extracellular matrix, and thereby alleviate cell invasion (37). The reversible characteristics of CAFs may provide therapeutic targets and new mechanisms for HCC progression. The molecular factors and signaling pathways for CAF activation are listed in Table 1.
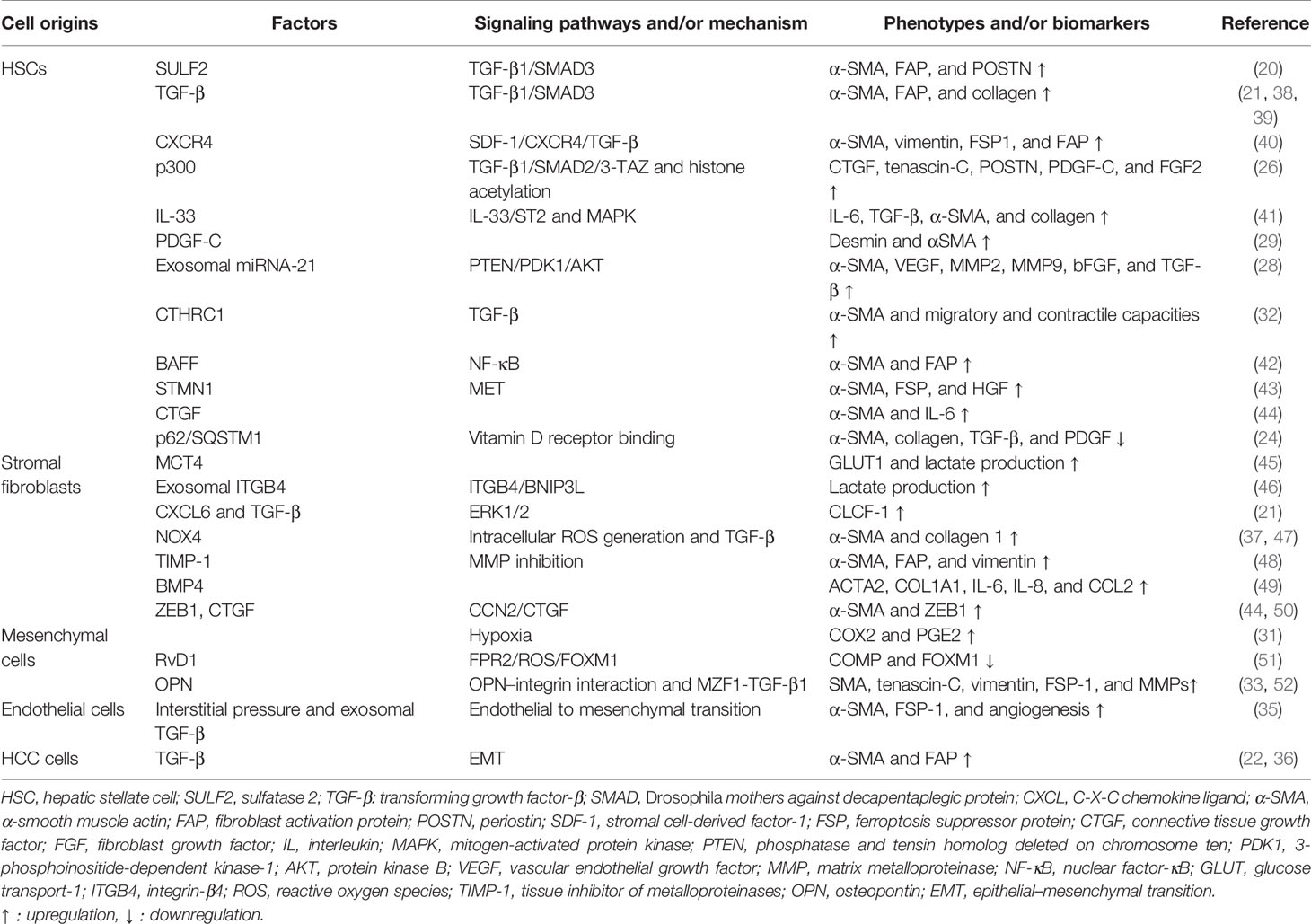
Table 1 The factors and pathways for CAF activation in the tumor/premalignant microenvironment.
The Mechanisms for CAFs in Supporting HCC Progression
Several studies have demonstrated the prognostic value of CAFs in predicting recurrence and metastasis of HCC after radical therapies. The high levels of CAFs as well as released proteins are commonly correlated with worse postoperative prognostic indices including early recurrence and metastasis; shortened DFS, OS, and PFS; and low survival rate (9–11), except for rare cases (12–17). In living donor liver transplantation, recipients with high levels of α-SMA+ CAFs in HCC display poorer prognosis (11). The high levels of CAF-related fibroblast growth factor (FGF)-2, endostatin, and VEGF may predict HCC recurrence (53). In HCC patients who underwent curative resection, factors including FGF3, FGFR2, Lysyl-oxidase-like (LOXL)-2, klotho-β, and FGF19 indicate a higher risk of tumor recurrence and worse survival rate. In patients who underwent HCC ablation, the level of serum FGF19 can serve as a potential prognostic biomarker (54–57).
The mechanisms for CAFs supporting tumor growth can be acknowledged as the direct influence on HCC cells and the reformation of the microenvironment. The role of CAFs in supporting HCC progression is concluded in Figure 2.
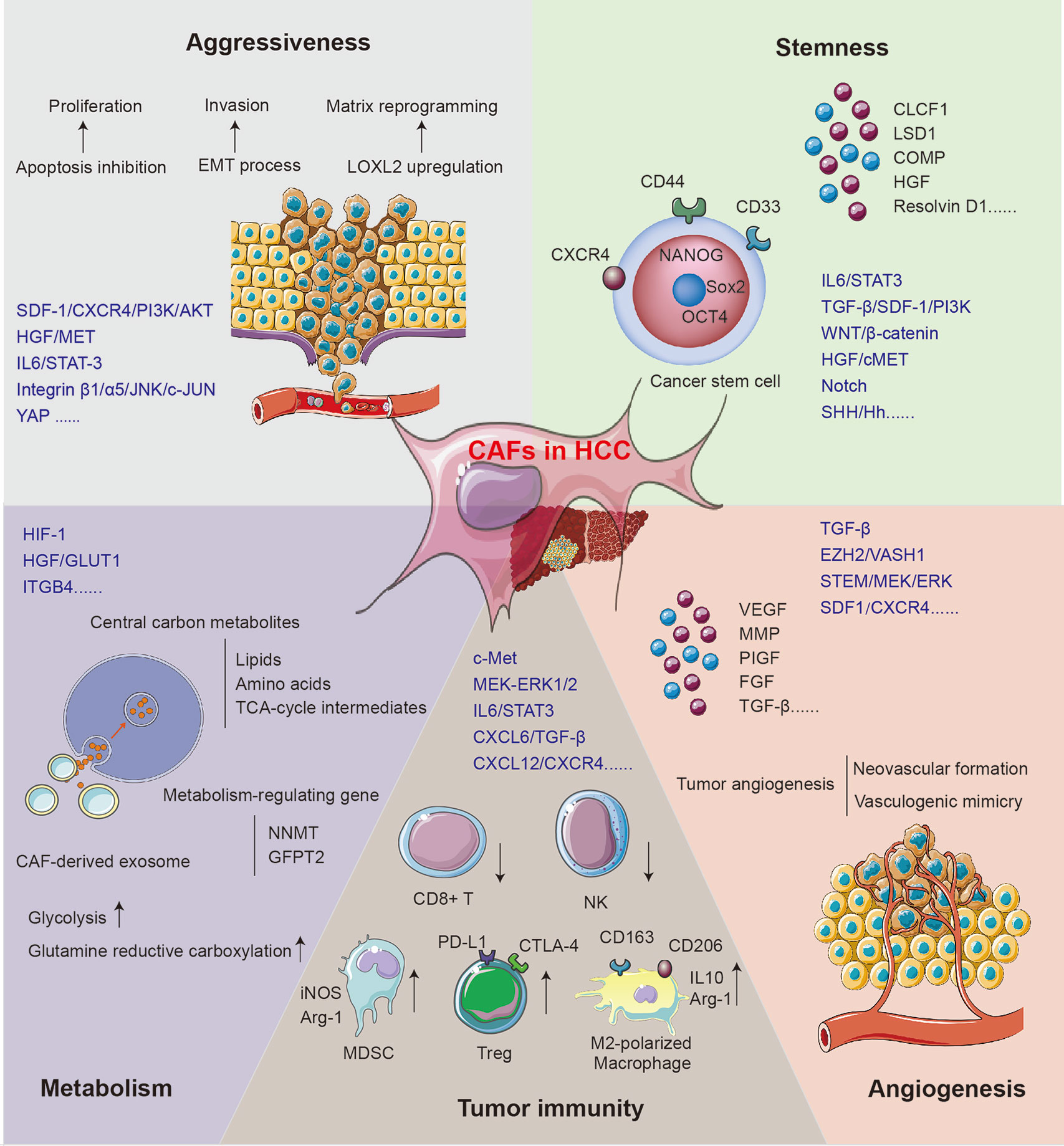
Figure 2 The mechanisms for CAFs in supporting HCC progression.
CAFs Promote the Aggressiveness of HCC Cells
The aggressiveness of HCC manifests in the upregulated proliferation and motility of cancer cells. SDF-1, one of the cytokines secreted by CAFs, may inhibit the apoptosis of HCC cells via the SDF-1/CXCR4/PI3K/AKT axis (48) and induce EMT through the SDF-1/CXCR4/OIP5-AS1/miR-153-3p/SNAI1 axis (20). Both classical and non-classical EMT processes play key roles in regulating tumor invasion and migration, which can lead to tumor recurrence and metastasis (58). Once treated with the conditioned medium of activated MRC5, the EMT phenotype was enhanced in human HCC cell lines Bel-7402 and LM3. Classical EMT markers including Snail, Twist, and integrin were regulated, while non-classical markers like E-cad/β-catenin complex, α/γ-catenin, and p120 catenin were redistributed in cells (58). Another study found that stathmin (STMN)-1 is highly expressed in HCC tissue, serving as a negative prognostic biomarker and mediating HCC recurrence and chemoresistance (43). STMN1 can facilitate the communication between CAFs and HCC via the HGF/MET pathway (43, 59). The IL-6/STAT3 signaling pathway is one of the most important mechanisms mediating HCC aggressiveness. Upregulated connective tissue growth factor (CTGF) in fibrosis environment can stimulate the IL-6 release from HSCs and accelerate HCC progression via the EMT process (44). Chemoresistance serves as another significant mechanism mediating HCC progression and metastasis after radical surgery. A recent study found that co-culture with CAFs maintains sorafenib resistance in HCC cells through B-cell-activating factor (BAFF)/NF-κB-dependent pathway (42); otherwise, overexpressed immunosuppressive cell differentiation (CD)-73 in cancer cells induced by CAFs can facilitate the resistance of sorafenib and cisplatin in HCC patients (60, 61).
CAFs can remodel ECM to gain cancer-supporting properties, and the function includes matrix degradation, deposition, and stiffening. Recently, tumor matrix stiffness is becoming an emerging target for tumor-aggressiveness study. Under high matrix stiffness condition, HCC releases a high level of LOXL2 through the integrin β1/α5/JNK/c-JUN signaling pathway. LOXL2 secreted by HCC can facilitate the formation of lung premetastatic niche via CD11b+ CD45+ BMDC recruitment, as well as fibronectin and matrix metalloproteinase (MMP)-9 upregulation (54, 62). A clinical study has confirmed the relationship between a high level of LOXL2 and worse prognosis in patients who underwent HCC radical surgery (54). Furthermore, overexpressed CXCR4 induced by high matrix stiffness can decrease the level of UBTD1 and promote proteasome-dependent degradation of YAP, contributing to HCC aggressiveness via the YAP signaling pathway (31). In addition, high matrix stiffness can promote the expression of the EMT transcription factor Snail through following three signaling pathways: integrin-mediated S100A11 membrane translocation, eIF4E phosphorylation, and TGF-β1 autocrine. The enhanced EMT process contributes to increased malignancy and progression of HCC (63).
CAFs Induce Stemness of HCC Cells
The residual cancer cells at the margins of the resection area are generally recognized as the main cause of HCC recurrence and metastasis. The liver cancer stem cells (LCSCs) are characterized by self-renewal and multilineage differentiation and play a key role in the chemoresistance, metastasis, and recurrence of HCC (64, 65). The stemness is stimulated by signals from TME including GSK-3β/β-catenin, Notch, Wnt, and IL-6/STAT3 (64, 66, 67). Recently, accumulating studies have evaluated the role of CAFs in HCC stemness induction. In the CAF–LCSC feedback loop, LCSCs can bolster the proliferation and transformation of CAFs. On the other hand, CAFs can in turn induce and maintain the stemness of HCC through the IL-6/STAT3, TGF-β/SDF-1/PI3K, Wnt/β-catenin, HGF/cMET, and SHH/Hh pathways (23, 68).
HGF, a pivotal factor in the regulation of liver fibrosis and hepatocyte growth, is extensively studied in CAF–HSC communication. Studies showed that the HGF expressed by CAFs can induce stemness through the MET-STEM and ERK1/2–FRA1–HEY1 signaling pathways (10, 69). Among other CAF-secreted factors, CLCF-1 increases the secretion of CXCL6 and TGF-β by HCC cells and promotes HCC stemness through autocrine effects (21). The nuclear Notch intracellular domain in HCC tissues, which symbolizes the activation of tumor stemness, is correlated with poor prognosis (70). The activated Notch signaling pathway can be attributed to the stimulation of high CAF-derived IL-6 and downstream STAT3 phosphorylation (70). Furthermore, Notch can also be stimulated by CAF-derived chromatin modification factor LSD1 (71). Another study showed that COMP secreted by CAFs can induce stemness and EMT in HCC cells, which can be impeded by an endogenous anti-inflammatory lipid mediator Resolvin D1 treatment via FPR2/ROS/FOXM1 (51). Changed matrix stiffness caused by CAFs can also promote the release of HGF via signals including ERK, PKB/Akt, and STAT3, which can further induce the stemness of HCC (72).
Not only CAFs but also peri-tumor fibroblasts can maintain the stemness of HCC cells. Studies showed that peri-tumor fibroblasts express more IL-6 than CAFs, which can promote stemness in an IL-6–STAT3-dependent manner (73). Interestingly, peri-tumor fibroblasts can also exert tumorigenesis effect and transform normal liver cells into malignant tumor cells (73). In addition to stemness induction effect, CAFs and peri-tumor fibroblasts can recruit cancer stem cells by releasing cytokines, including IL-6, CCL2, CXCL1, and CXCL8 (74). Peri-tumor fibroblasts can also facilitate intrahepatic metastasis via SCGF-β- and HGF-mediated signaling pathways (74).
CAFs Regulate the Metabolism in Tumor Cells
The aberrant metabolism of glucose, lipid, protein, and oxygen is one of the hallmarks of HCC, which constitutes a selective advantage for tumor growth, invasion, and metastasis. It is well recognized that CAFs can reprogram energy metabolism in HCC and promote the malignant phenotype of HCC including EMT, LCSC-like property, and drug resistance (75). In HCC, metabolic coupling, which happens between catabolic CAFs and anabolic cancer cells, provides mitochondrial fuels for oxidative phosphorylation and drives higher disease stage and shorter DFS via glycolysis (76). Stromal–epithelial metabolic coupling can be initiated through downregulation of HGF, which was demonstrated by decreased glucose transporter (GLUT)-1 level and reduced glucose uptake in cancer cells (77). Furthermore, the regulation of GLUT level may influence the production of lactate and remodel the metabolic pattern (45, 77). This metabolic reprograming can be mediated by the HIF-1-dependent signaling pathway, leading to the energic metabolism dominated by lactic acid rather than glucose (76).
The exosome derived from CAFs provides central carbon metabolites including amino acids, lipids, and tricarboxylic acid (TCA)-cycle intermediates for HCC cells in a micropinocytosis manner, which can inhibit mitochondrial oxidative phosphorylation and promote glycolysis and glutamine-dependent reductive carboxylation in tumor cells. This mechanism makes it possible for tumor cells to proliferate and migrate in an energy-deprived environment (75). Furthermore, a study has confirmed that integrin subunit β (ITGB)-4 derived from tumor exosome can promote BNIP3L-dependent mitophagy and lactate production in CAFs, which in turn promotes the proliferation and EMT of cancer cells (46). Stromal nicotinamide N-methyltransferase (NNMT) expression is significant in functional aspects of the CAFs. It contributes to the depletion of S-adenosyl methionine and reduction in histone methylation, which regulate a series of gene expression associated with tumor progression (78). Glutamine-fructose-6-phosphate transaminase (GFPT)-2 is another prominent metabolism-regulating gene overexpressed in CAFs, which can drive the processes including glycolysis, pentose phosphate pathway, and TCA cycle process in cancer cells and promote tumor progression (79).
The mechanism of glucose metabolism is extensively studied in CAF–HCC cross-talk, while the processes like oxygen and lipid metabolism are rarely studied. The in-depth mechanisms underlying the metabolism regulation of CAF–HCC still warrant high-quality studies.
CAFs Promote Angiogenesis in HCC
Tumor angiogenesis is an important dimension of HCC progression, characterized by the degradation of blood vessel basement membrane; the activation, proliferation, and migration of vascular endothelial cell; and the reconstruction of a new blood vessel. Proangiogenic stimulators including VEGF, FGF, PIGF, and TGF, as well as signaling pathways like TGF-β, EZH2/VASH1, and STEM/MEK/ERK, are extensively investigated (80–82). Multiple clinical studies have indicated the positive correlation between αSMA+ CAFs and neovascular formation (9, 80). Activated CAFs can release multiple angiogenic cytokines, including VEGF, MMP2, MMP9, bFGF, and TGF-β, and further contribute to HCC progression. Also, CD90 high expressed in CAFs may promote the release of PIGF, which can facilitate the angiogenesis and predict worse prognosis (80).
Regarded as a supplement to traditional angiogenesis theory, tumor vasculogenic mimicry (VM) serves as an important pattern for a rich tumor blood supply within the microenvironment. In this process, aggressive tumor cells generate their own blood supply channels without the participation of endothelial cells. Clinical studies have found that the rate of VM in HCC tissue is about 35% (83). The levels of CAF biomarkers α-SMA and vimentin and functional molecules MMP2 and EphA2 are highly correlated with VM in HCC patients (83). Furthermore, CAF-induced activation of TGF-β and SDF-1–CXCR4 signaling pathways plays significant roles in educating HCC cells expressing VE-cadherin, MMP2, and laminin, which can induce VM formation (84).
CAFs Suppress Tumor Immunity in the HCC Microenvironment
As a critical frontline immune tissue, the liver is susceptible to the change of inflammation-related cells and cytokines in the microenvironment (85). The immunity components in HCC, including immunosuppressive regulatory T cells (Treg), tumor-associated macrophages (TAMs), tumor-associated neutrophils (TANs), pro-inflammatory CD4+/CD8+ T cells, natural killer (NK) cells, and dendritic cells (DCs), can be regulated by CAFs and their secreted factors (86). The tumor-expressed CD73 generates immunotolerant effect and promotes invasiveness via adenosine production from degradation of AMP in HCC (61, 87). In HCC, CAFs may activate c-Met and MEK–ERK1/2 signaling pathways, upregulate CD73 expression, and thereby promote the resistance to sorafenib and cisplatin (60). The CLCF-1–CXCL6/TGF-β axis plays an important role in the regulation of CAF-induced HCC stemness and immune evasion. HCC-derived CXCL6 and TGF-β stimulate ERK1/2 signal in CAFs, which upregulates the release of CLCF-1, forming a positive feedback loop between CAFs and HCC (21). It has been proven that activated HSCs can promote the apoptosis of effector T cells and NK cells and recruit Treg, causing harm to tumor immunity (88). Nicotinamide adenine dinucleotide phosphate oxidase (NOX)-4, a key regulator of CAF phenotype, can exclude CD8+ T-cell infiltration in HCC specifically (37). Inhibition of NOX4 can reverse CAFs to a quiescent state and restore CD8+ T-cell immunity, while blockade of TGF-β may prevent, rather than reverse, the activation of CAFs (37). NK cells can also be suppressed by the inhibitory factors PGE2 and IDO derived from CAFs (89).
In addition to the direct effects of CAFs on T cells and NK cells, they can also stimulate the IL-6/STAT3 pathway in neutrophils and further promote neutrophil proliferation, which impair T-cell function through the PD1/PD-L1 signaling pathway (90). Activated HSCs can enhance the activity of myeloid-derived suppressor cells (MDSCs) with increased iNOS and Arg-1 expression, which can inhibit the activation and function of effector T cells (88). Meanwhile, co-culture with activated HSCs can also stimulate the MDSCs through granulocyte–macrophage colony-stimulating factor (GM-CSF), M-CSF, COX-2, prostaglandin, and VEGF (91, 92).
M2-polarized macrophage is another prominent factor that impairs T-cell immunity through the deactivation of effector T cells and recruitment of Treg. High expression of CAF signature is associated with an immunosuppressive phenotype of infiltrating macrophage, suggesting the key role of CAF–macrophage interplay-mediated immunosuppression in the progression of HCC and fibrosis (27, 93). Studies have shown that TGF-β derived from macrophage can promote type I and III collagen expression in HSCs through the SMAD-dependent pathway (25, 94, 95). HSCs can also be activated by macrophage-derived cytokines including TGF-β, PDGF, oncostatin-M (OSM), tumor necrosis factor (TNF)-α, and IL-1β, as well as chemokines including CCL2 and CCL5 (94, 95). CAFs can in turn recruit monocytes via the SDF-1/CXCR4 pathway, which is associated with macrophage differentiation (96). CAFs can also inhibit pro-inflammatory features of M1-macrophages by reducing NO production and inhibiting pro-inflammatory cytokines (93). Meanwhile, CAFs promote M2 polarization of macrophage via release of SDF-1, IL-6, and GM-CSF and overexpression of PAI-1, producing an immunosuppressive tumor ecological niche (97, 98).
The Metabolic Reprogramming of CAFs in HCC Progression
Tumor microenvironments are characterized as hypoxia and energy deficiency, which render cancer cells and other stromal cells to go through metabolic reprogramming (99), so as to meet their needs for increasing biological process of bioenergy, biosynthesis, and redox balance (100). The activation of CAFs in HCC also requires a large quantity of energy to promote cell proliferation, ECM and cytokine release, and the migration toward certain pro-fibrotic and pro-inflammatory regions (101). As a consequence, metabolic reprogramming and energy expenditure regulation play key roles in the function change of CAFs during the HCC microenvironment and liver injury. It has been proven that the metabolic reprogramming of CAFs in HCC includes changes in carbohydrate and mitochondrial metabolism, protein glycosylation, vitamin and lipid metabolism, and redox metabolism. HSCs are the main source of CAFs in HCC, and the metabolic pathways of HSC activation are in pivotal status (102).
The glycolysis process is active in cancer cells even in a rich oxygen surrounding, which is known as the Warburg effect (103). Due to the cross-talk between stromal cells and cancer cells, the Warburg effect has also been observed in fibroblasts, which is identified as the “reverse Warburg effect” (76). Studies have revealed that similar to cancer cells, the increased glucose utilization rate as well as aerobic glycolytic activity (104) and glucose transport capacity (105) has been discovered in activated primary HSCs and LX2 cell lines. Upregulation of glucose transporter proteins including GLUT-1, 2, and 4 (106) and increased expression of hexokinase 2 (107), fructose-2,6-bisphosphatase-3 (108), and pyruvate kinase (109) in HSCs also indicate that the HCC environment may educate HSCs toward abnormal glucose metabolism. Lactate shuttle is also an important phenomenon observed between cancer cells and CAFs (45). In this way, lactate produced from cancer cells can be utilized by CAFs in MCT4-dependent manners, which in turn generate pyruvic acid for tumor metabolism (76, 110). Within the lactate shuttle, more glucose prioritizing ATP production by CAF metabolism can meet the large energy requirement of cancer cells, which also represents the coordination between CAFs and cancer cells.
The increase of mitochondrial number and activity is also observed in activated CAFs in HCC, indicating an enhanced energy supplement from oxidative phosphorylation besides glycolysis (105). According to some studies, the consequence of stimulated oxidative phosphorylation is not limited to ATP production, while ROS generation and oxidative stress can be also caused by oxidative phosphorylation change (111, 112).
Besides carbohydrate, the reprogramming of lipid metabolism and amino acid metabolism in CAFs also provides major energy for ECM remodeling and tumor support. The storage of vitamin A and the regulation of lipid homeostasis are the main functions of quiescent HSCs in the liver (102). During HSC activation, the loss of retinyl ester-containing cytoplasmic droplets can be identified as the consequence of aberrant lipid metabolism (113, 114). Meanwhile, upregulated activity of fatty acid β oxidation in activated HSCs is required to metabolize lipid droplets which are derived from autolysosomes in HSCs (115, 116). During this process, the autophagy of HSCs and retinyl ester hydrolase activity are also enhanced (117).
The amino acid metabolic pattern in CAFs is characterized by glutaminolysis, which is the transition of glutamine toward a-ketoglutarate (118). An increased expression of glutaminase is observed in activated CAFs in HCC, leading to the metabolism of glutamine and the ensuing decreased peroxisome proliferator-activated receptor γ (PPARγ) expression and increased collagen release (119, 120). Significantly, a study has revealed that aberrant glutaminolysis in activated HSCs is accompanied by upregulated fibrogenesis (121).
Some Types of CAFs May Exert a Tumor-Suppressive Effect
Although CAFs are mainly considered to support tumor growth, the antitumor functions of some subtypes of CAFs within a variety of cancers have been reported (13, 15, 17, 58, 122). The activated human fibroblast MRC5 is reported to induce apoptosis in the HCC LM3 cell line (58). Despite an EMT-enhancing effect, the apoptosis-inducing effect of fibroblasts provides new ideas and warrants further in-depth studies. Another recent study has shown the tumor-promoting effect of myofibroblastic CAF-secreted hyaluronan and inflammatory CAF-secreted HGF, which is responsible for the CAF–HCC interaction loop (17). On the other hand, not only cell contact and cell regulatory soluble factor but also the stiffness of the extracellular matrix remodeled by CAFs can also affect HCC development. Type I collagen expressed by myofibroblastic CAFs can inhibit HCC development by mechanically restraining tumor growth (17). This function may oppose the tumor-promoting effect of the CAF–HCC interaction loop. Another study indicated that exosome released from mesenchymal stem cell-derived CAFs contains miR-199a, which can sensitize HCC cells to the treatment of doxorubicin through the mTOR signaling pathway and reverse drug resistance in patients with advanced HCC (15).
Two CAF subtypes characterized by CD146 expression have distinct effects on tumor growth (122). The CD146− CAFs are reported to suppress estrogen receptor expression in breast cancer cells and increase the resistance to tamoxifen therapy, while CD146+ CAFs restore the sensitivity of cancer cells to tamoxifen treatment. The CD146− CAFs in tumor tissues predict worse patient outcomes, while CD146+ CAF is a positive prognostic index (122). Furthermore, in pancreatic ductal adenocarcinoma, the depletion of α-SMA+ myofibroblasts induced more aggressive and malignant tumor cells and enhanced the EMT process and cancer stemness (13). The tumor tissue-infiltrated CD4+ FOXP3+ Treg were also increased after CAF depletion, which reduced tumor response to gemcitabine treatment via cytotoxic T-lymphocyte-associated protein (CTLA)-4-induced immunosuppression (14). In another investigation related to CAFs in pancreatic ductal adenocarcinoma, sonic hedgehog is reported to form a fibroblast-rich desmoplastic stroma in a cancer environment. Once sonic hedgehog was depleted, unfavorable situations including undifferentiated histology, increased vascularity, and heightened proliferation were detected in the tumor (13). This suggested the antitumor function of sonic hedgehog and the ensuing fibroblast-rich stroma. This study also found that fibroblast can inhibit tumor angiogenesis through the hedgehog signaling pathway and prevent tumor progression.
The tumor-suppressive effect of some types of fibroblasts highlights the functional heterogenicity of CAF populations. When it comes to the development of CAF-targeting therapies, the positive role of this fibroblast subtype is supposed to be considered cautiously. Further studies are warranted to develop more reliable biomarkers in order to distinguish the distinct roles of CAFs.
Traditional Chinese Medicines as Resources of Treating Liver Fibrosis
The pathogenesis of HCC is a complex process, in which a chronic inflammatory environment and fibrotic deposition play a key role. Multiple strategies have been developed to attenuate liver fibrosis and exert antitumor effects (6, 7, 36). Traditional Chinese medicine has been proven effective in delaying tumor progression and preventing the recurrence and metastasis of HCC (123, 124). Recently, the antifibrotic effects of Chinese medicine decoction and natural product extracts have been extensively studied. Many studies have suggested that the antifibrotic and HCC-preventive effect of TCM components lies in the medicines with “qi-soothing in liver”, “blood stasis-removing”, and “heat-clearing and detoxifying” efficacy. Multiple medicines including Radix Salviae miltiorrhizae, Astragalus mongholicus, Radix bupleuri, Paeoniae radix rubra, Atractylodes macrocephala, Paeonia Lactiflora, Trionycis Carapax, and Angelica sinensis are commonly used clinically to achieve therapeutic goals (125). Several clinical studies have proven the effectiveness of TCM in preventing liver fibrosis and HCC recurrence after radical surgery. Huaier granule, an aqueous extract of Trametes robinophila Murr-based medicine approved by the Chinese State Food and Drug Administration, has been found to efficiently prolong recurrence-free survival and reduce extrahepatic recurrence in patients with HCC who underwent curative resection (126, 127). Jiedu granule, which is composed of four traditional Chinese herbal medicines including Actinidia valvata, Salvia chinensis, Cremastra appendiculata, and the gizzard membrane of Gallus gallus domesticus, has been proven effective in preventing recurrence as an adjuvant therapy with cinobufacini injection for HCC after curative resection (128, 129). Another randomized controlled trial revealed that Chunggan extract, an herbal formula, can ameliorated hepatic fibrosis and liver stiffness in patients with chronic liver disease (130). Liver fibrosis is a significant factor mediating recurrence after radical surgery, which can be attenuated by TCM administration in both single drug application and drug combination. The values of TCM in liver fibrosis treatment are supposed to lie in the prevention of cirrhosis–HCC transition and HCC progression; alleviation of symptoms in advanced HCC, which can be mediated by the regulation of CAFs/HSC activation; the suppression of hepatic inflammation; the improvement of antioxidant capacity; and the inhibition of vasculogenic mimicry (131).
The most prominent signaling pathway involved in the antifibrotic effect of TCM is TGF-β (25, 132). Total flavanones extracted from Sedum sarmentosum can suppress the activation of HSCs via the TGF-β1/SMAD pathway (133). Methyl ferulic acid, which is the primary monomer constituent of herbal medicines including Ferula assafoetida L., Ligusticum chuanxiong Hort., and Lycopodium selago L., can alleviate the progression of liver fibrosis through HSC-suppressing function via the TGF-β1/SMAD and NOX4/ROS signaling pathways (47). Salvianolic acid A, derived from Alvia miltiorrhiza Bge., can inhibit TGF-β and PI3K/AKT/mTOR signaling cascades in CCl-4-induced fibrosis mice, improve fibrotic degree, restore liver function, and reduce collagen deposition in the liver (134). The Xiaoyaosan and Yinchenhao decoction can inhibit the activation of HSCs via the TGF-β and Akt/FoxO3 pathways (135, 136). Yinchenhao decoction can also promote the apoptosis of parenchyma cells in a TNF-involved manner, which blocks the initiation of HCC in a cirrhosis environment (136). Besides medical treatment, Ammophila Arenaria umbilical moxibustion, a form of manual therapy, can promote the apoptosis of activated HSCs and prevent the progression of HCC (137). NF-κF and PPARγ are the other two key factors regulating TGF-β signaling and, thus, the activation of HSCs (138). In liver fibrosis, M2-polarized macrophage can bolster the transformation of HSCs to CAFs by extensively activating NF-κb signaling (139). Otherwise, the suppression of NF-κB and stimulation of PPARγ can inhibit the expression of TGF-β1. Ginkgo biloba extract (140, 141), 18α-glycyrrhizin (142), glycyrrhizic acid (143), and curcumin (144) can also suppress NF-κB signaling in the liver and alleviate liver fibrosis. Baicalin (145) and curcumin (146) can exert a protective effect on CCl4-induced liver injury by activating PPARγ and inhibiting TGF-β.
Ferroptosis is a novel pathway of cell death which is widely investigated in recent years. A study showed that Magnesium isoglycyrrhizinate attenuates fibrotic scar formation in CCl4-induced hepatic–fibrotic rats through ferroptosis-related cell death of HSCs (147). Artemether, extracted from Artemisia apiacea, promotes the accumulation of iron and lipid peroxides and, thus, induces ferroptosis in HSCs (148). This study also showed that tumor suppressor p53 plays a key role in artemether-induced HSC ferroptosis (148). In addition, iron regulatory protein 2 accumulation is another key mechanism of artemether-mediated antihepatic fibrosis process (149).
Activated CAFs can facilitate the development of HCC via stemness and autophagy induction, while chloroquine can downregulate stemness markers including Nanog, Sox2, and Oct4 in CAFs co-cultured with HCC cells, subsequently modulating the autophagy of cancer cells (150). GPR68 is a key protein regulating the proliferation and invasion of HCC cells. Conophylline can suppress the expression of GPR68 in CAFs and block the tumor-promoting effect of CAFs (52). A clinical study confirmed the therapeutic advantage of combined conophylline and sorafenib treatment in advance HCC (52). Exosomal circCCT3 derived from CAFs can stimulate the activity of glucose metabolism-related gene HK2 and further affect the glucose metabolism of HCC cells (151), which is blocked by coptisine, the main component of Chelidonium majus L., Corydalis uanhusuo W. T. Wang, and Coptis japonica Makino (152). Glycyrrhizin and its metabolites glycyrrhetinic acid can attenuate non-alcoholic steatohepatitis-induced liver fibrosis, which is mediated by suppression of NLR family pyrin domain-containing 3 (NLRP3) inflammasome and restoration of bile acid homeostasis (153). The CAF-induced immunosuppressive microenvironment is involved in HCC progression to a large extent, which can be improved by phillygenin derived from Forsythiae fructus (154). The expression of immunosuppressive IL-6, IL-1β, and TNF-α by CAFs can be inhibited by phillygenin treatment through the TLR4/MyD88/NF-κB signaling pathway (154).
The advantages of TCM treatment in liver fibrosis lie in the comprehensive treatment of multisite and multitarget conditions and overall regulation. Several extracts of Chinese medicine and their derivatives equipped with antifibrotic property are summarized in Table 2. The development of cirrhosis and the initiation of HCC are complex processes referring to numerous signaling pathway activation and multiple functional changes, making it possible for TCM to exert a multitarget–regulation effect.
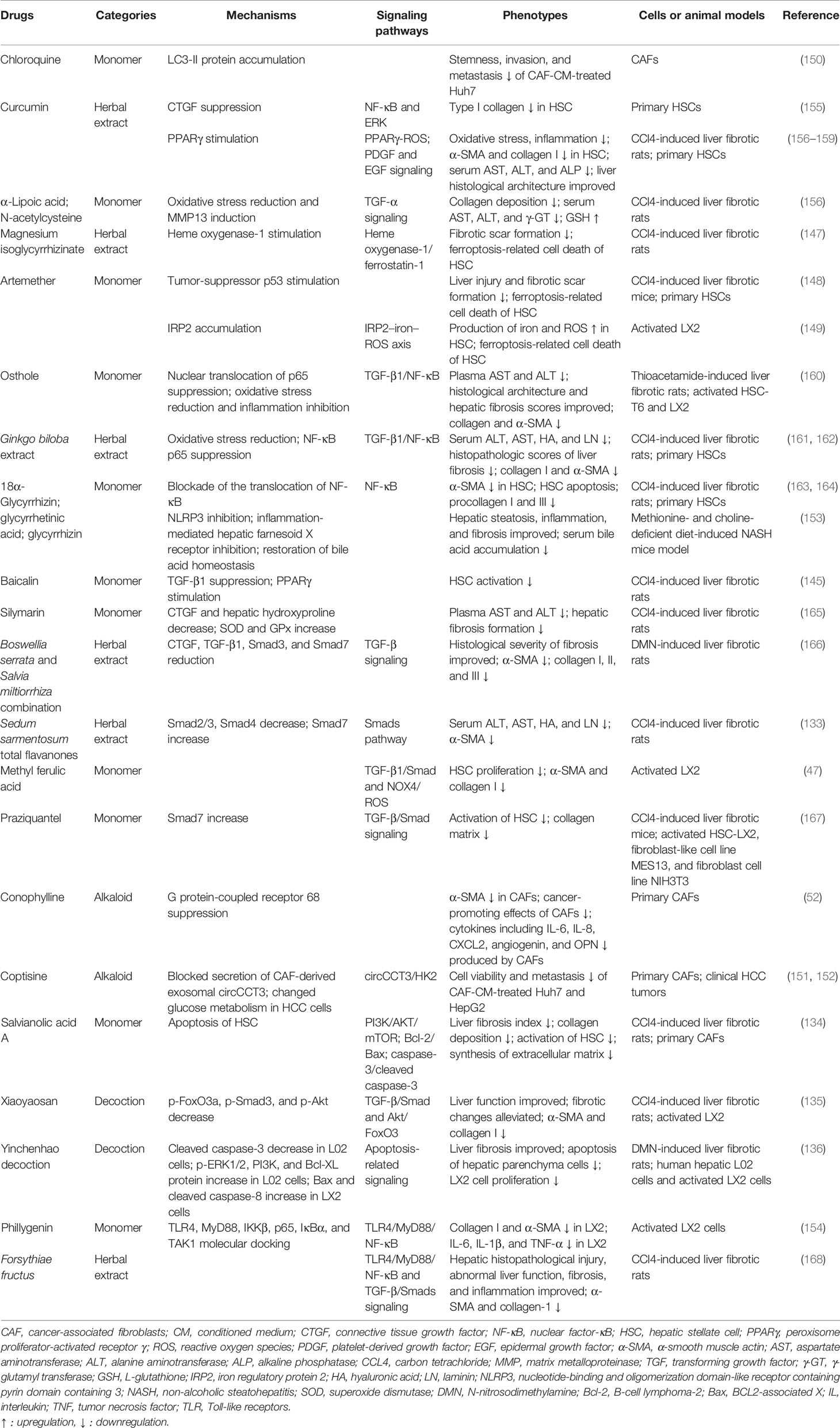
Table 2 Chinese medicine and their derivatives equipped with antifibrotic property.
Conclusion
The role of CAFs in HCC initiation and progression should not be ignored. The unique premalignant microenvironment in fibrosis tends to lead to the activation and tumor-supporting function of CAFs, which can be reversed or blocked by the application of TCM. HSCs are one of the dominant sources of CAFs in HCC. Generally, inactivated HSCs are quiescent in normal liver and equipped with the following functions significant for the maintenance of normal liver function: the metabolism and storage of vitamin A, the storage of lipid, and the production and secretion of ECM components including collagen and glycoprotein. Once stimulated by inflammatory factors and mechanical injuries in pathogenic conditions, the HSCs can be transformed to activated CAFs, which display myofibroblast-like phenotypes and highly express α-SMA, vimentin, desmin, etc. Meanwhile, the proliferation and mobility properties of HSCs can be enhanced to promote the migration toward the damage sites. Under precancerous conditions, the ECM can also be remodeled with increased release of ECM components and decreased decomposition capacity. Furthermore, the remodeled ECM can in turn facilitate the activation of CAFs and HCC progression by unfavorable matrix stiffness and contractile activity. The upregulated cytokines, chemokines, and receptor/ligand binding can also affect fibrosis and tumor progression.
The mechanisms for CAFs in supporting HCC progression can be categorized into upregulated aggressiveness and stemness, transformed metabolism toward glycolysis and glutamine reductive carboxylation, polarized tumor immunity toward immune escape of HCC cells, and increased angiogenesis. The TGF-β/SMAD signaling pathway plays a dominant role in all stages of liver fibrosis including the activation of HSCs, the deposition of ECM, the aggressiveness and stemness generation of CAF-educated HCC cells, and the process of CAF-induced angiogenesis. In the CAF activation process, TGF-β1 inhibits the apoptosis of normal fibroblasts and induces the excessive synthesis of collagen and fibronectin. The phosphorylation of SMAD2/3 is a canonical mechanism involved in the activation of CAFs. Besides the TGF-β1/SMAD pathways, TGF-β1 can also stimulate downstream MAPK, NF-κB, and PI3K to activate CAFs and generate fibrosis.
TCM plays a key role in fibrosis and cancer treatment. Both clinical and fundamental studies have verified the unique properties of TCM in its antifibrosis application. Based on the findings presented in this review, TCM can be regarded as a potential and potent strategy for the prevention and treatment of both liver fibrosis and liver cancer.
Author Contributions
WJ and SL drafted the manuscript with the support from CL and BC. BC revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by the grants from the National Natural Science Foundation of China (Nos. 82074203 and 82030117).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Wang H, Lu Z, Zhao X. Tumorigenesis, Diagnosis, and Therapeutic Potential of Exosomes in Liver Cancer. J Hematol Oncol (2019) 12(1):133. doi: 10.1186/s13045-019-0806-6
2. Nio K, Yamashita T, Kaneko S. The Evolving Concept of Liver Cancer Stem Cells. Mol Cancer (2017) 16(1):4. doi: 10.1186/s12943-016-0572-9
3. Ge Y, Mu W, Ba Q, Li J, Jiang Y, Xia Q, et al. Hepatocellular Carcinoma-Derived Exosomes in Organotropic Metastasis, Recurrence and Early Diagnosis Application. Cancer Lett (2020) 477:41–8. doi: 10.1016/j.canlet.2020.02.003
4. Sia D, Villanueva A, Friedman SL, Llovet JM. Liver Cancer Cell of Origin, Molecular Class, and Effects on Patient Prognosis. Gastroenterology (2017) 152(4):745–61. doi: 10.1053/j.gastro.2016.11.048
5. Anwanwan D, Singh SK, Singh S, Saikam V, Singh R. Challenges in Liver Cancer and Possible Treatment Approaches. Biochim Biophys Acta Rev Cancer (2020) 1873(1):188314. doi: 10.1016/j.bbcan.2019.188314
6. Yin Z, Dong C, Jiang K, Xu Z, Li R, Guo K, et al. Heterogeneity of Cancer-Associated Fibroblasts and Roles in the Progression, Prognosis, and Therapy of Hepatocellular Carcinoma. J Hematol Oncol (2019) 12(1):101. doi: 10.1186/s13045-019-0782-x
7. Zhang J, Gu C, Song Q, Zhu M, Xu Y, Xiao M, et al. Identifying Cancer-Associated Fibroblasts as Emerging Targets for Hepatocellular Carcinoma. Cell Biosci (2020) 10(1):127. doi: 10.1186/s13578-020-00488-y
8. Baglieri J, Brenner DA, Kisseleva T. The Role of Fibrosis and Liver-Associated Fibroblasts in the Pathogenesis of Hepatocellular Carcinoma. Int J Mol Sci (2019) 20(7):1723. doi: 10.3390/ijms20071723
9. Fang M, Yuan J, Chen M, Sun Z, Liu L, Cheng G, et al. The Heterogenic Tumor Microenvironment of Hepatocellular Carcinoma and Prognostic Analysis Based on Tumor Neo-Vessels, Macrophages and Alpha-SMA. Oncol Lett (2018) 15(4):4805–12. doi: 10.3892/ol.2018.7946
10. Lau EY, Lo J, Cheng BY, Ma MK, Lee JM, Ng JK, et al. Cancer-Associated Fibroblasts Regulate Tumor-Initiating Cell Plasticity in Hepatocellular Carcinoma Through C-Met/FRA1/HEY1 Signaling. Cell Rep (2016) 15(6):1175–89. doi: 10.1016/j.celrep.2016.04.019
11. Takamura H, Nakanuma S, Hayashi H, Tajima H, Kakinoki K, Sakai S, et al. Evaluation of Eligibility Criteria in Living Donor Liver Transplantation for Hepatocellular Carcinoma by Alpha-SMA-Positive Cancer-Associated Fibroblasts. Oncol Rep (2013) 30(4):1561–74. doi: 10.3892/or.2013.2616
12. Trimboli AJ, Cantemir-Stone CZ, Li F, Wallace JA, Merchant A, Creasap N, et al. Pten in Stromal Fibroblasts Suppresses Mammary Epithelial Tumours. Nature (2009) 461(7267):1084–91. doi: 10.1038/nature08486
13. Rhim AD, Oberstein PE, Thomas DH, Mirek ET, Palermo CF, Sastra SA, et al. Stromal Elements Act to Restrain, Rather Than Support, Pancreatic Ductal Adenocarcinoma. Cancer Cell (2014) 25(6):735–47. doi: 10.1016/j.ccr.2014.04.021
14. Ozdemir BC, Pentcheva-Hoang T, Carstens JL, Zheng X, Wu CC, Simpson TR, et al. Depletion of Carcinoma-Associated Fibroblasts and Fibrosis Induces Immunosuppression and Accelerates Pancreas Cancer With Reduced Survival. Cancer Cell (2014) 25(6):719–34. doi: 10.1016/j.ccr.2014.04.005
15. Lou G, Chen L, Xia C, Wang W, Qi J, Li A, et al. MiR-199a-Modified Exosomes From Adipose Tissue-Derived Mesenchymal Stem Cells Improve Hepatocellular Carcinoma Chemosensitivity Through mTOR Pathway. J Exp Clin Cancer Res (2020) 39(1):4. doi: 10.1186/s13046-019-1512-5
16. Kim GJ, Rhee H, Yoo JE, Ko JE, Lee JS, Kim H, et al. Increased Expression of CCN2, Epithelial Membrane Antigen, and Fibroblast Activation Protein in Hepatocellular Carcinoma With Fibrous Stroma Showing Aggressive Behavior. PLoS One (2014) 9(8):e105094. doi: 10.1371/journal.pone.0105094
17. Bhattacharjee S, Hamberger F, Ravichandra A, Miller M, Nair A, Affo S, et al. Tumor Restriction by Type I Collagen Opposes Tumor-Promoting Effects of Cancer-Associated Fibroblasts. J Clin Invest (2021) 131(11):e146987. doi: 10.1172/JCI146987
18. Piersma B, Hayward MK, Weaver VM. Fibrosis and Cancer: A Strained Relationship. Biochim Biophys Acta Rev Cancer (2020) 1873(2):188356. doi: 10.1016/j.bbcan.2020.188356
19. Li P, Gong Z, Shultz LD, Ren G. Mesenchymal Stem Cells: From Regeneration to Cancer. Pharmacol Ther (2019) 200:42–54. doi: 10.1016/j.pharmthera.2019.04.005
20. Wang C, Shang C, Gai X, Song T, Han S, Liu Q, et al. Sulfatase 2-Induced Cancer-Associated Fibroblasts Promote Hepatocellular Carcinoma Progression via Inhibition of Apoptosis and Induction of Epithelial-To-Mesenchymal Transition. Front Cell Dev Biol (2021) 9:631931. doi: 10.3389/fcell.2021.631931
21. Song M, He J, Pan QZ, Yang J, Zhao J, Zhang YJ, et al. Cancer-Associated Fibroblast-Mediated Cellular Crosstalk Supports Hepatocellular Carcinoma Progression. Hepatology (2021) 73(5):1717–35. doi: 10.1002/hep.31792
22. Jia XW, Li ZW, Dong LY, Sun GY, Wang X, Gao J, et al. Lack of Hepatic Stimulator Substance Expression Promotes Hepatocellular Carcinoma Metastasis Partly Through ERK-Activated Epithelial-Mesenchymal Transition. Lab Invest (2018) 98(7):871–82. doi: 10.1038/s41374-018-0039-2
23. Huang TX, Guan XY, Fu L. Therapeutic Targeting of the Crosstalk Between Cancer-Associated Fibroblasts and Cancer Stem Cells. Am J Cancer Res (2019) 9(9):1889–904.
24. Duran A, Hernandez ED, Reina-Campos M, Castilla EA, Subramaniam S, Raghunandan S, et al. P62/SQSTM1 by Binding to Vitamin D Receptor Inhibits Hepatic Stellate Cell Activity, Fibrosis, and Liver Cancer. Cancer Cell (2016) 30(4):595–609. doi: 10.1016/j.ccell.2016.09.004
25. Dooley S, ten Dijke P. TGF-Beta in Progression of Liver Disease. Cell Tissue Res (2012) 347(1):245–56. doi: 10.1007/s00441-011-1246-y
26. Wang Y, Tu K, Liu D, Guo L, Chen Y, Li Q, et al. P300 Acetyltransferase Is a Cytoplasm-To-Nucleus Shuttle for SMAD2/3 and TAZ Nuclear Transport in Transforming Growth Factor Beta-Stimulated Hepatic Stellate Cells. Hepatology (2019) 70(4):1409–23. doi: 10.1002/hep.30668
27. Chen S, Morine Y, Tokuda K, Yamada S, Saito Y, Nishi M, et al. Cancerassociated Fibroblastinduced M2polarized Macrophages Promote Hepatocellular Carcinoma Progression via the Plasminogen Activator Inhibitor1 Pathway. Int J Oncol (2021) 59(2):59. doi: 10.3892/ijo.2021.5239
28. Zhou Y, Ren H, Dai B, Li J, Shang L, Huang J, et al. Hepatocellular Carcinoma-Derived Exosomal miRNA-21 Contributes to Tumor Progression by Converting Hepatocyte Stellate Cells to Cancer-Associated Fibroblasts. J Exp Clin Cancer Res (2018) 37(1):324. doi: 10.1186/s13046-018-0965-2
29. Wright JH, Johnson MM, Shimizu-Albergine M, Bauer RL, Hayes BJ, Surapisitchat J, et al. Paracrine Activation of Hepatic Stellate Cells in Platelet-Derived Growth Factor C Transgenic Mice: Evidence for Stromal Induction of Hepatocellular Carcinoma. Int J Cancer (2014) 134(4):778–88. doi: 10.1002/ijc.28421
30. Tan Z, Liu Q, Jiang R, Lv L, Shoto SS, Maillet I, et al. Interleukin-33 Drives Hepatic Fibrosis Through Activation of Hepatic Stellate Cells. Cell Mol Immunol (2018) 15(4):388–98. doi: 10.1038/cmi.2016.63
31. Liu Y, Ren H, Zhou Y, Shang L, Zhang Y, Yang F, et al. The Hypoxia Conditioned Mesenchymal Stem Cells Promote Hepatocellular Carcinoma Progression Through YAP Mediated Lipogenesis Reprogramming. J Exp Clin Cancer Res (2019) 38(1):228. doi: 10.1186/s13046-019-1219-7
32. Li J, Wang Y, Ma M, Jiang S, Zhang X, Zhang Y, et al. Autocrine CTHRC1 Activates Hepatic Stellate Cells and Promotes Liver Fibrosis by Activating TGF-Beta Signaling. EBioMedicine (2019) 40:43–55. doi: 10.1016/j.ebiom.2019.01.009
33. Weber CE, Kothari AN, Wai PY, Li NY, Driver J, Zapf MA, et al. Osteopontin Mediates an MZF1-TGF-Beta1-Dependent Transformation of Mesenchymal Stem Cells Into Cancer-Associated Fibroblasts in Breast Cancer. Oncogene (2015) 34(37):4821–33. doi: 10.1038/onc.2014.410
34. Bhattacharya SD, Mi Z, Talbot LJ, Guo H, Kuo PC. Human Mesenchymal Stem Cell and Epithelial Hepatic Carcinoma Cell Lines in Admixture: Concurrent Stimulation of Cancer-Associated Fibroblasts and Epithelial-to-Mesenchymal Transition Markers. Surgery (2012) 152(3):449–54. doi: 10.1016/j.surg.2012.06.011
35. Yeon JH, Jeong HE, Seo H, Cho S, Kim K, Na D, et al. Cancer-Derived Exosomes Trigger Endothelial to Mesenchymal Transition Followed by the Induction of Cancer-Associated Fibroblasts. Acta Biomater (2018) 76:146–53. doi: 10.1016/j.actbio.2018.07.001
36. Affo S, Yu LX, Schwabe RF. The Role of Cancer-Associated Fibroblasts and Fibrosis in Liver Cancer. Annu Rev Pathol (2017) 12:153–86. doi: 10.1146/annurev-pathol-052016-100322
37. Ford K, Hanley CJ, Mellone M, Szyndralewiez C, Heitz F, Wiesel P, et al. NOX4 Inhibition Potentiates Immunotherapy by Overcoming Cancer-Associated Fibroblast-Mediated CD8 T-Cell Exclusion From Tumors. Cancer Res (2020) 80(9):1846–60. doi: 10.1158/0008-5472.CAN-19-3158
38. Friedman SL. Hepatic Stellate Cells: Protean, Multifunctional, and Enigmatic Cells of the Liver. Physiol Rev (2008) 88(1):125–72. doi: 10.1152/physrev.00013.2007
39. Breitkopf K, Godoy P, Ciuclan L, Singer MV, Dooley S. TGF-Beta/Smad Signaling in the Injured Liver. Z Gastroenterol (2006) 44(1):57–66. doi: 10.1055/s-2005-858989
40. Tan HX, Gong WZ, Zhou K, Xiao ZG, Hou FT, Huang T, et al. CXCR4/TGF-Beta1 Mediated Hepatic Stellate Cells Differentiation Into Carcinoma-Associated Fibroblasts and Promoted Liver Metastasis of Colon Cancer. Cancer Biol Ther (2020) 21(3):258–68. doi: 10.1080/15384047.2019.1685157
41. Andersson P, Yang Y, Hosaka K, Zhang Y, Fischer C, Braun H, et al. Molecular Mechanisms of IL-33-Mediated Stromal Interactions in Cancer Metastasis. JCI Insight (2018) 3(20):e122375. doi: 10.1172/jci.insight.122375
42. Gao L, Morine Y, Yamada S, Saito Y, Ikemoto T, Tokuda K, et al. The BAFF/NFkappaB Axis Is Crucial to Interactions Between Sorafenib-Resistant HCC Cells and Cancer-Associated Fibroblasts. Cancer Sci (2021) 112(9):3545–54. doi: 10.1111/cas.15041
43. Zhang R, Gao X, Zuo J, Hu B, Yang J, Zhao J, et al. STMN1 Upregulation Mediates Hepatocellular Carcinoma and Hepatic Stellate Cell Crosstalk to Aggravate Cancer by Triggering the MET Pathway. Cancer Sci (2020) 111(2):406–17. doi: 10.1111/cas.14262
44. Makino Y, Hikita H, Kodama T, Shigekawa M, Yamada R, Sakamori R, et al. CTGF Mediates Tumor-Stroma Interactions Between Hepatoma Cells and Hepatic Stellate Cells to Accelerate HCC Progression. Cancer Res (2018) 78(17):4902–14. doi: 10.1158/0008-5472.CAN-17-3844
45. Fiaschi T, Marini A, Giannoni E, Taddei ML, Gandellini P, De Donatis A, et al. Reciprocal Metabolic Reprogramming Through Lactate Shuttle Coordinately Influences Tumor-Stroma Interplay. Cancer Res (2012) 72(19):5130–40. doi: 10.1158/0008-5472.CAN-12-1949
46. Sung JS, Kang CW, Kang S, Jang Y, Chae YC, Kim BG, et al. ITGB4-Mediated Metabolic Reprogramming of Cancer-Associated Fibroblasts. Oncogene (2020) 39(3):664–76. doi: 10.1038/s41388-019-1014-0
47. Cheng Q, Li C, Yang CF, Zhong YJ, Wu D, Shi L, et al. Methyl Ferulic Acid Attenuates Liver Fibrosis and Hepatic Stellate Cell Activation Through the TGF-Beta1/Smad and NOX4/ROS Pathways. Chem Biol Interact (2019) 299:131–9. doi: 10.1016/j.cbi.2018.12.006
48. Song T, Dou C, Jia Y, Tu K, Zheng X. TIMP-1 Activated Carcinoma-Associated Fibroblasts Inhibit Tumor Apoptosis by Activating SDF1/CXCR4 Signaling in Hepatocellular Carcinoma. Oncotarget (2015) 6(14):12061–79. doi: 10.18632/oncotarget.3616
49. Mano Y, Yoshio S, Shoji H, Tomonari S, Aoki Y, Aoyanagi N, et al. Bone Morphogenetic Protein 4 Provides Cancer-Supportive Phenotypes to Liver Fibroblasts in Patients With Hepatocellular Carcinoma. J Gastroenterol (2019) 54(11):1007–18. doi: 10.1007/s00535-019-01579-5
50. Lobe C, Vallette M, Arbelaiz A, Gonzalez-Sanchez E, Izquierdo L, Pellat A, et al. ZEB1 Promotes Cholangiocarcinoma Progression Through Tumor Dedifferentiation and Tumor-Stroma Paracrine Signaling. Hepatology (2021). doi: 10.1002/hep.32069
51. Sun L, Wang Y, Wang L, Yao B, Chen T, Li Q, et al. Resolvin D1 Prevents Epithelial-Mesenchymal Transition and Reduces the Stemness Features of Hepatocellular Carcinoma by Inhibiting Paracrine of Cancer-Associated Fibroblast-Derived COMP. J Exp Clin Cancer Res (2019) 38(1):170. doi: 10.1186/s13046-019-1163-6
52. Yamanaka T, Harimoto N, Yokobori T, Muranushi R, Hoshino K, Hagiwara K, et al. Conophylline Inhibits Hepatocellular Carcinoma by Inhibiting Activated Cancer-Associated Fibroblasts Through Suppression of G Protein-Coupled Receptor 68. Mol Cancer Ther (2021) 20(6):1019–28. doi: 10.1158/1535-7163.MCT-20-0150
53. Chiu KW, Nakano T, Chen KD, Hsu LW, Lai CY, Huang CY, et al. Repeated-Measures Implication of Hepatocellular Carcinoma Biomarkers in Living Donor Liver Transplantation. PLoS One (2015) 10(5):e0124943. doi: 10.1371/journal.pone.0124943
54. Wu S, Xing X, Wang Y, Zhang X, Li M, Wang M, et al. The Pathological Significance of LOXL2 in Pre-Metastatic Niche Formation of HCC and Its Related Molecular Mechanism. Eur J Cancer (2021) 147:63–73. doi: 10.1016/j.ejca.2021.01.011
55. Maeda T, Kanzaki H, Chiba T, Ao J, Kanayama K, Maruta S, et al. Serum Fibroblast Growth Factor 19 Serves as a Potential Novel Biomarker for Hepatocellular Carcinoma. BMC Cancer (2019) 19(1):1088. doi: 10.1186/s12885-019-6322-9
56. Lin ZZ, Hsu C, Jeng YM, Hu FC, Pan HW, Wu YM, et al. Klotho-Beta and Fibroblast Growth Factor 19 Expression Correlates With Early Recurrence of Resectable Hepatocellular Carcinoma. Liver Int (2019) 39(9):1682–91. doi: 10.1111/liv.14055
57. Hu L, Sham JS, Xie D, Wen JM, Wang WS, Wang Y, et al. Up-Regulation of Fibroblast Growth Factor 3 Is Associated With Tumor Metastasis and Recurrence in Human Hepatocellular Carcinoma. Cancer Lett (2007) 252(1):36–42. doi: 10.1016/j.canlet.2006.12.003
58. Ding S, Chen G, Zhang W, Xing C, Xu X, Xie H, et al. MRC-5 Fibroblast-Conditioned Medium Influences Multiple Pathways Regulating Invasion, Migration, Proliferation, and Apoptosis in Hepatocellular Carcinoma. J Transl Med (2015) 13:237. doi: 10.1186/s12967-015-0588-8
59. Yang T, Zhiheng H, Zhanhuai W, Qian X, Yue L, Xiaoxu G, et al. Increased RAB31 Expression in Cancer-Associated Fibroblasts Promotes Colon Cancer Progression Through HGF-MET Signaling. Front Oncol (2020) 10:1747. doi: 10.3389/fonc.2020.01747
60. Peng H, Xue R, Ju Z, Qiu J, Wang J, Yan W, et al. Cancer-Associated Fibroblasts Enhance the Chemoresistance of CD73(+) Hepatocellular Carcinoma Cancer Cells via HGF-Met-ERK1/2 Pathway. Ann Transl Med (2020) 8(14):856. doi: 10.21037/atm-20-1038
61. Ma XL, Hu B, Tang WG, Xie SH, Ren N, Guo L, et al. CD73 Sustained Cancer-Stem-Cell Traits by Promoting SOX9 Expression and Stability in Hepatocellular Carcinoma. J Hematol Oncol (2020) 13(1):11. doi: 10.1186/s13045-020-0845-z
62. Wu S, Zheng Q, Xing X, Dong Y, Wang Y, You Y, et al. Matrix Stiffness-Upregulated LOXL2 Promotes Fibronectin Production, MMP9 and CXCL12 Expression and BMDCs Recruitment to Assist Pre-Metastatic Niche Formation. J Exp Clin Cancer Res (2018) 37(1):99. doi: 10.1186/s13046-018-0761-z
63. Dong Y, Zheng Q, Wang Z, Lin X, You Y, Wu S, et al. Higher Matrix Stiffness as an Independent Initiator Triggers Epithelial-Mesenchymal Transition and Facilitates HCC Metastasis. J Hematol Oncol (2019) 12(1):112. doi: 10.1186/s13045-019-0795-5
64. Matthews VB, Yeoh GC. Liver Stem Cells. IUBMB Life (2005) 57(8):549–53. doi: 10.1080/15216540500215606
65. Liu YC, Yeh CT, Lin KH. Cancer Stem Cell Functions in Hepatocellular Carcinoma and Comprehensive Therapeutic Strategies. Cells (2020) 9(6):1331. doi: 10.3390/cells9061331
66. Wan S, Zhao E, Kryczek I, Vatan L, Sadovskaya A, Ludema G, et al. Tumor-Associated Macrophages Produce Interleukin 6 and Signal via STAT3 to Promote Expansion of Human Hepatocellular Carcinoma Stem Cells. Gastroenterology (2014) 147(6):1393–404. doi: 10.1053/j.gastro.2014.08.039
67. Craig AJ, von Felden J, Garcia-Lezana T, Sarcognato S, Villanueva A. Tumour Evolution in Hepatocellular Carcinoma. Nat Rev Gastroenterol Hepatol (2020) 17(3):139–52. doi: 10.1038/s41575-019-0229-4
68. Guo W, Sun YF, Shen MN, Ma XL, Wu J, Zhang CY, et al. Circulating Tumor Cells With Stem-Like Phenotypes for Diagnosis, Prognosis, and Therapeutic Response Evaluation in Hepatocellular Carcinoma. Clin Cancer Res (2018) 24(9):2203–13. doi: 10.1158/1078-0432.CCR-17-1753
69. Moritoki Y, Ueno Y, Kanno N, Yamagiwa Y, Fukushima K, Gershwin ME, et al. Lack of Evidence That Bone Marrow Cells Contribute to Cholangiocyte Repopulation During Experimental Cholestatic Ductal Hyperplasia. Liver Int (2006) 26(4):457–66. doi: 10.1111/j.1478-3231.2006.01250.x
70. Xiong S, Wang R, Chen Q, Luo J, Wang J, Zhao Z, et al. Cancer-Associated Fibroblasts Promote Stem Cell-Like Properties of Hepatocellular Carcinoma Cells Through IL-6/STAT3/Notch Signaling. Am J Cancer Res (2018) 8(2):302–16.
71. Liu C, Liu L, Chen X, Cheng J, Zhang H, Zhang C, et al. LSD1 Stimulates Cancer-Associated Fibroblasts to Drive Notch3-Dependent Self-Renewal of Liver Cancer Stem-Like Cells. Cancer Res (2018) 78(4):938–49. doi: 10.1158/0008-5472.CAN-17-1236
72. Schrader J, Gordon-Walker TT, Aucott RL, van Deemter M, Quaas A, Walsh S, et al. Matrix Stiffness Modulates Proliferation, Chemotherapeutic Response, and Dormancy in Hepatocellular Carcinoma Cells. Hepatology (2011) 53(4):1192–205. doi: 10.1002/hep.24108
73. Zhao Z, Xiong S, Wang R, Li Y, Wang X, Wang Y, et al. Peri-Tumor Fibroblasts Promote Tumorigenesis and Metastasis of Hepatocellular Carcinoma via Interleukin6/STAT3 Signaling Pathway. Cancer Manag Res (2019) 11:2889–901. doi: 10.2147/CMAR.S192263
74. Jiang J, Ye F, Yang X, Zong C, Gao L, Yang Y, et al. Peri-Tumor Associated Fibroblasts Promote Intrahepatic Metastasis of Hepatocellular Carcinoma by Recruiting Cancer Stem Cells. Cancer Lett (2017) 404:19–28. doi: 10.1016/j.canlet.2017.07.006
75. De Matteis S, Ragusa A, Marisi G, De Domenico S, Casadei Gardini A, Bonafe M, et al. Aberrant Metabolism in Hepatocellular Carcinoma Provides Diagnostic and Therapeutic Opportunities. Oxid Med Cell Longev (2018) 2018:7512159. doi: 10.1155/2018/7512159
76. Martinez-Outschoorn UE, Lisanti MP, Sotgia F. Catabolic Cancer-Associated Fibroblasts Transfer Energy and Biomass to Anabolic Cancer Cells, Fueling Tumor Growth. Semin Cancer Biol (2014) 25:47–60. doi: 10.1016/j.semcancer.2014.01.005
77. Brauer HA, Makowski L, Hoadley KA, Casbas-Hernandez P, Lang LJ, Roman-Perez E, et al. Impact of Tumor Microenvironment and Epithelial Phenotypes on Metabolism in Breast Cancer. Clin Cancer Res (2013) 19(3):571–85. doi: 10.1158/1078-0432.CCR-12-2123
78. Eckert MA, Coscia F, Chryplewicz A, Chang JW, Hernandez KM, Pan S, et al. Proteomics Reveals NNMT as a Master Metabolic Regulator of Cancer-Associated Fibroblasts. Nature (2019) 569(7758):723–8. doi: 10.1038/s41586-019-1173-8
79. Zhang W, Bouchard G, Yu A, Shafiq M, Jamali M, Shrager JB, et al. GFPT2-Expressing Cancer-Associated Fibroblasts Mediate Metabolic Reprogramming in Human Lung Adenocarcinoma. Cancer Res (2018) 78(13):3445–57. doi: 10.1158/0008-5472.CAN-17-2928
80. Liu Z, Chen M, Zhao R, Huang Y, Liu F, Li B, et al. CAF-Induced Placental Growth Factor Facilitates Neoangiogenesis in Hepatocellular Carcinoma. Acta Biochim Biophys Sin (Shanghai) (2020) 52(1):18–25. doi: 10.1093/abbs/gmz134
81. Jo M, Nishikawa T, Nakajima T, Okada Y, Yamaguchi K, Mitsuyoshi H, et al. Oxidative Stress Is Closely Associated With Tumor Angiogenesis of Hepatocellular Carcinoma. J Gastroenterol (2011) 46(6):809–21. doi: 10.1007/s00535-011-0392-z
82. Lin ZY, Chuang YH, Chuang WL. Cancer-Associated Fibroblasts Up-Regulate CCL2, CCL26, IL6 and LOXL2 Genes Related to Promotion of Cancer Progression in Hepatocellular Carcinoma Cells. BioMed Pharmacother (2012) 66(7):525–9. doi: 10.1016/j.biopha.2012.02.001
83. She Q, Hu S, Pu X, Guo Q, Mou C, Yang C. The Effect of Hepatocellular Carcinoma-Associated Fibroblasts on Hepatoma Vasculogenic Mimicry. Am J Cancer Res (2020) 10(12):4198–210.
84. Yang J, Lu Y, Lin YY, Zheng ZY, Fang JH, He S, et al. Vascular Mimicry Formation Is Promoted by Paracrine TGF-Beta and SDF1 of Cancer-Associated Fibroblasts and Inhibited by miR-101 in Hepatocellular Carcinoma. Cancer Lett (2016) 383(1):18–27. doi: 10.1016/j.canlet.2016.09.012
85. Pinter M, Scheiner B, Peck-Radosavljevic M. Immunotherapy for Advanced Hepatocellular Carcinoma: A Focus on Special Subgroups. Gut (2021) 70(1):204–14. doi: 10.1136/gutjnl-2020-321702
86. Ruf B, Heinrich B, Greten TF. Immunobiology and Immunotherapy of HCC: Spotlight on Innate and Innate-Like Immune Cells. Cell Mol Immunol (2021) 18(1):112–27. doi: 10.1038/s41423-020-00572-w
87. Sciarra A, Monteiro I, Menetrier-Caux C, Caux C, Gilbert B, Halkic N, et al. CD73 Expression in Normal and Pathological Human Hepatobiliopancreatic Tissues. Cancer Immunol Immunother (2019) 68(3):467–78. doi: 10.1007/s00262-018-2290-1
88. Hsieh CC, Hung CH, Chiang M, Tsai YC, He JT. Hepatic Stellate Cells Enhance Liver Cancer Progression by Inducing Myeloid-Derived Suppressor Cells Through Interleukin-6 Signaling. Int J Mol Sci (2019) 20(20):5079. doi: 10.3390/ijms20205079
89. Li T, Yang Y, Hua X, Wang G, Liu W, Jia C, et al. Hepatocellular Carcinoma-Associated Fibroblasts Trigger NK Cell Dysfunction via PGE2 and IDO. Cancer Lett (2012) 318(2):154–61. doi: 10.1016/j.canlet.2011.12.020
90. Cheng Y, Li H, Deng Y, Tai Y, Zeng K, Zhang Y, et al. Cancer-Associated Fibroblasts Induce PDL1+ Neutrophils Through the IL6-STAT3 Pathway That Foster Immune Suppression in Hepatocellular Carcinoma. Cell Death Dis (2018) 9(4):422. doi: 10.1038/s41419-018-0458-4
91. Yu MC, Chen CH, Liang X, Wang L, Gandhi CR, Fung JJ, et al. Inhibition of T-Cell Responses by Hepatic Stellate Cells via B7-H1-Mediated T-Cell Apoptosis in Mice. Hepatology (2004) 40(6):1312–21. doi: 10.1002/hep.20488
92. Chou HS, Hsieh CC, Charles R, Wang L, Wagner T, Fung JJ, et al. Myeloid-Derived Suppressor Cells Protect Islet Transplants by B7-H1 Mediated Enhancement of T Regulatory Cells. Transplantation (2012) 93(3):272–82. doi: 10.1097/TP.0b013e31823ffd39
93. Berzaghi R, Ahktar MA, Islam A, Pedersen BD, Hellevik T, Martinez-Zubiaurre I. Fibroblast-Mediated Immunoregulation of Macrophage Function Is Maintained After Irradiation. Cancers (Basel) (2019) 11(5):689. doi: 10.3390/cancers11050689
94. Breitkopf K, Roeyen C, Sawitza I, Wickert L, Floege J, Gressner AM. Expression Patterns of PDGF-A, -B, -C and -D and the PDGF-Receptors Alpha and Beta in Activated Rat Hepatic Stellate Cells (HSC). Cytokine (2005) 31(5):349–57. doi: 10.1016/j.cyto.2005.06.005
95. Bonner JC. Regulation of PDGF and Its Receptors in Fibrotic Diseases. Cytokine Growth Factor Rev (2004) 15(4):255–73. doi: 10.1016/j.cytogfr.2004.03.006
96. Li X, Bu W, Meng L, Liu X, Wang S, Jiang L, et al. CXCL12/CXCR4 Pathway Orchestrates CSC-Like Properties by CAF Recruited Tumor Associated Macrophage in OSCC. Exp Cell Res (2019) 378(2):131–8. doi: 10.1016/j.yexcr.2019.03.013
97. Cho H, Seo Y, Loke KM, Kim SW, Oh SM, Kim JH, et al. Cancer-Stimulated CAFs Enhance Monocyte Differentiation and Protumoral TAM Activation via IL6 and GM-CSF Secretion. Clin Cancer Res (2018) 24(21):5407–21. doi: 10.1158/1078-0432.CCR-18-0125
98. Iorio V, De Marco M, Basile A, Eletto D, Capunzo M, Remondelli P, et al. CAF-Derived IL6 and GM-CSF Cooperate to Induce M2-Like TAMs-Letter. Clin Cancer Res (2019) 25(2):892–3. doi: 10.1158/1078-0432.CCR-18-2455
99. Pavlova NN, Thompson CB. The Emerging Hallmarks of Cancer Metabolism. Cell Metab (2016) 23(1):27–47. doi: 10.1016/j.cmet.2015.12.006
100. Boroughs LK, DeBerardinis RJ. Metabolic Pathways Promoting Cancer Cell Survival and Growth. Nat Cell Biol (2015) 17(4):351–9. doi: 10.1038/ncb3124
101. Vandierendonck A, Degroote H, Vanderborght B, Verhelst X, Geerts A, Devisscher L, et al. NOX1 Inhibition Attenuates the Development of a Pro-Tumorigenic Environment in Experimental Hepatocellular Carcinoma. J Exp Clin Cancer Res (2021) 40(1):40. doi: 10.1186/s13046-021-01837-6
102. Trivedi P, Wang S, Friedman SL. The Power of Plasticity-Metabolic Regulation of Hepatic Stellate Cells. Cell Metab (2021) 33(2):242–57. doi: 10.1016/j.cmet.2020.10.026
103. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science (2009) 324(5930):1029–33. doi: 10.1126/science.1160809
104. Ban D, Hua S, Zhang W, Shen C, Miao X, Liu W. Costunolide Reduces Glycolysis-Associated Activation of Hepatic Stellate Cells via Inhibition of Hexokinase-2. Cell Mol Biol Lett (2019) 24:52. doi: 10.1186/s11658-019-0179-4
105. Gajendiran P, Vega LI, Itoh K, Sesaki H, Vakili MR, Lavasanifar A, et al. Elevated Mitochondrial Activity Distinguishes Fibrogenic Hepatic Stellate Cells and Sensitizes for Selective Inhibition by Mitotropic Doxorubicin. J Cell Mol Med (2018) 22(4):2210–9. doi: 10.1111/jcmm.13501
106. Pasanen I, Lehtonen S, Sormunen R, Skarp S, Lehtilahti E, Pietila M, et al. Breast Cancer Carcinoma-Associated Fibroblasts Differ From Breast Fibroblasts in Immunological and Extracellular Matrix Regulating Pathways. Exp Cell Res (2016) 344(1):53–66. doi: 10.1016/j.yexcr.2016.04.016
107. Hu JW, Sun P, Zhang DX, Xiong WJ, Mi J. Hexokinase 2 Regulates G1/S Checkpoint Through CDK2 in Cancer-Associated Fibroblasts. Cell Signal (2014) 26(10):2210–6. doi: 10.1016/j.cellsig.2014.04.015
108. Mejias M, Gallego J, Naranjo-Suarez S, Ramirez M, Pell N, Manzano A, et al. CPEB4 Increases Expression of PFKFB3 to Induce Glycolysis and Activate Mouse and Human Hepatic Stellate Cells, Promoting Liver Fibrosis. Gastroenterology (2020) 159(1):273–88. doi: 10.1053/j.gastro.2020.03.008
109. Zheng D, Jiang Y, Qu C, Yuan H, Hu K, He L, et al. Pyruvate Kinase M2 Tetramerization Protects Against Hepatic Stellate Cell Activation and Liver Fibrosis. Am J Pathol (2020) 190(11):2267–81. doi: 10.1016/j.ajpath.2020.08.002
110. Wilde L, Roche M, Domingo-Vidal M, Tanson K, Philp N, Curry J, et al. Metabolic Coupling and the Reverse Warburg Effect in Cancer: Implications for Novel Biomarker and Anticancer Agent Development. Semin Oncol (2017) 44(3):198–203. doi: 10.1053/j.seminoncol.2017.10.004
111. Nolfi-Donegan D, Braganza A, Shiva S. Mitochondrial Electron Transport Chain: Oxidative Phosphorylation, Oxidant Production, and Methods of Measurement. Redox Biol (2020) 37:101674. doi: 10.1016/j.redox.2020.101674
112. Cadenas S. Mitochondrial Uncoupling, ROS Generation and Cardioprotection. Biochim Biophys Acta Bioenerg (2018) 1859(9):940–50. doi: 10.1016/j.bbabio.2018.05.019
113. Khomich O, Ivanov AV, Bartosch B. Metabolic Hallmarks of Hepatic Stellate Cells in Liver Fibrosis. Cells (2019) 9(1):24. doi: 10.3390/cells9010024
114. Ajat M, Molenaar M, Brouwers J, Vaandrager AB, Houweling M, Helms JB. Hepatic Stellate Cells Retain the Capacity to Synthesize Retinyl Esters and to Store Neutral Lipids in Small Lipid Droplets in the Absence of LRAT. Biochim Biophys Acta Mol Cell Biol Lipids (2017) 1862(2):176–87. doi: 10.1016/j.bbalip.2016.10.013
115. Bates J, Vijayakumar A, Ghoshal S, Marchand B, Yi S, Kornyeyev D, et al. Acetyl-CoA Carboxylase Inhibition Disrupts Metabolic Reprogramming During Hepatic Stellate Cell Activation. J Hepatol (2020) 73(4):896–905. doi: 10.1016/j.jhep.2020.04.037
116. Tuohetahuntila M, Molenaar MR, Spee B, Brouwers JF, Wubbolts R, Houweling M, et al. Lysosome-Mediated Degradation of a Distinct Pool of Lipid Droplets During Hepatic Stellate Cell Activation. J Biol Chem (2017) 292(30):12436–48. doi: 10.1074/jbc.M117.778472
117. Shajari S, Saeed A, Smith-Cortinez NF, Heegsma J, Sydor S, Faber KN. Hormone-Sensitive Lipase Is a Retinyl Ester Hydrolase in Human and Rat Quiescent Hepatic Stellate Cells. Biochim Biophys Acta Mol Cell Biol Lipids (2019) 1864(9):1258–67. doi: 10.1016/j.bbalip.2019.05.012
118. Akins NS, Nielson TC, Le HV. Inhibition of Glycolysis and Glutaminolysis: An Emerging Drug Discovery Approach to Combat Cancer. Curr Top Med Chem (2018) 18(6):494–504. doi: 10.2174/1568026618666180523111351
119. Mestre-Farrera A, Bruch-Oms M, Pena R, Rodriguez-Morato J, Alba-Castellon L, Comerma L, et al. Glutamine-Directed Migration of Cancer-Activated Fibroblasts Facilitates Epithelial Tumor Invasion. Cancer Res (2021) 81(2):438–51. doi: 10.1158/0008-5472.CAN-20-0622
120. Du K, Chitneni SK, Suzuki A, Wang Y, Henao R, Hyun J, et al. Increased Glutaminolysis Marks Active Scarring in Nonalcoholic Steatohepatitis Progression. Cell Mol Gastroenterol Hepatol (2020) 10(1):1–21. doi: 10.1016/j.jcmgh.2019.12.006
121. Du K, Hyun J, Premont RT, Choi SS, Michelotti GA, Swiderska-Syn M, et al. Hedgehog-YAP Signaling Pathway Regulates Glutaminolysis to Control Activation of Hepatic Stellate Cells. Gastroenterology (2018) 154(5):1465–79.e13. doi: 10.1053/j.gastro.2017.12.022
122. Brechbuhl HM, Finlay-Schultz J, Yamamoto TM, Gillen AE, Cittelly DM, Tan AC, et al. Fibroblast Subtypes Regulate Responsiveness of Luminal Breast Cancer to Estrogen. Clin Cancer Res (2017) 23(7):1710–21. doi: 10.1158/1078-0432.CCR-15-2851
123. Ling CQ, Fan J, Lin HS, Shen F, Xu ZY, Lin LZ, et al. Clinical Practice Guidelines for the Treatment of Primary Liver Cancer With Integrative Traditional Chinese and Western Medicine. J Integr Med (2018) 16(4):236–48. doi: 10.1016/j.joim.2018.05.002
124. Tang KY, Du SL, Wang QL, Zhang YF, Song HY. Traditional Chinese Medicine Targeting Cancer Stem Cells as an Alternative Treatment for Hepatocellular Carcinoma. J Integr Med (2020) 18(3):196–202. doi: 10.1016/j.joim.2020.02.002
125. Xie Z, Qiang J, Pi X, Wang J, Chen Y, Yu Q, et al. Favorable Outcome of Adjunctive Traditional Chinese Medicine Therapy in Liver Cirrhosis: A Large Cohort Study in Southwest China. Complement Ther Med (2020) 51:102446. doi: 10.1016/j.ctim.2020.102446
126. Wang Z, Yu XL, Zhang J, Cheng ZG, Han ZY, Liu FY, et al. Huaier Granule Prevents the Recurrence of Early-Stage Hepatocellular Carcinoma After Thermal Ablation: A Cohort Study. J Ethnopharmacol (2021) 281:114539. doi: 10.1016/j.jep.2021.114539
127. Chen Q, Shu C, Laurence AD, Chen Y, Peng BG, Zhen ZJ, et al. Effect of Huaier Granule on Recurrence After Curative Resection of HCC: A Multicentre, Randomised Clinical Trial. Gut (2018) 67(11):2006–16. doi: 10.1136/gutjnl-2018-315983
128. Zhao HT, Meng YB, Zhai XF, Cheng BB, Yu SS, Yao M, et al. Comparable Effects of Jiedu Granule, a Compound Chinese Herbal Medicine, and Sorafenib for Advanced Hepatocellular Carcinoma: A Prospective Multicenter Cohort Study. J Integr Med (2020) 18(4):319–25. doi: 10.1016/j.joim.2020.05.003
129. Wang J, Luo J, Yin X, Huang W, Cao H, Wang G, et al. Jiedu Granule Combined With Transcatheter Arterial Chemoembolization and Gamma Knife Radiosurgery in Treating Hepatocellular Carcinoma With Portal Vein Tumor Thrombus. BioMed Res Int (2019) 2019:4696843. doi: 10.1155/2019/4696843
130. Kim HG, Kim JM, Han JM, Lee JS, Choi MK, Lee DS, et al. Chunggan Extract, a Traditional Herbal Formula, Ameliorated Alcohol-Induced Hepatic Injury in Rat Model. World J Gastroenterol (2014) 20(42):15703–14. doi: 10.3748/wjg.v20.i42.15703
131. Hou ZJ, Zhang JH, Zhang X, Ling QH, Zheng C, Zhu XJ, et al. Long-Term Traditional Chinese Medicine Combined With NA Antiviral Therapy on Cirrhosis Incidence in Chronic Hepatitis B Patients in the Real-World Setting: A Retrospective Study. Evid Based Complement Alternat Med (2020) 2020:3826857. doi: 10.1155/2020/3826857
132. Shan L, Liu Z, Ci L, Shuai C, Lv X, Li J. Research Progress on the Anti-Hepatic Fibrosis Action and Mechanism of Natural Products. Int Immunopharmacol (2019) 75:105765. doi: 10.1016/j.intimp.2019.105765
133. Lin YC, Luo HY, Liu HF, Du XH. Anti-Fibrotic Mechanism of Sedum Sarmentosum Total Flavanones in Inhibiting Activation of HSC by Regulating Smads. Zhongguo Zhong Yao Za Zhi (2020) 45(3):631–5. doi: 10.19540/j.cnki.cjcmm.20190829.401
134. Wang R, Song F, Li S, Wu B, Gu Y, Yuan Y. Salvianolic Acid A Attenuates CCl4-Induced Liver Fibrosis by Regulating the PI3K/AKT/mTOR, Bcl-2/Bax and Caspase-3/Cleaved Caspase-3 Signaling Pathways. Drug Des Devel Ther (2019) 13:1889–900. doi: 10.2147/DDDT.S194787
135. Zhou Y, Wu R, Cai FF, Zhou WJ, Lu YY, Zhang H, et al. Xiaoyaosan Decoction Alleviated Rat Liver Fibrosis via the TGFbeta/Smad and Akt/FoxO3 Signaling Pathways Based on Network Pharmacology Analysis. J Ethnopharmacol (2021) 264:113021. doi: 10.1016/j.jep.2020.113021
136. Cai FF, Bian YQ, Wu R, Sun Y, Chen XL, Yang MD, et al. Yinchenhao Decoction Suppresses Rat Liver Fibrosis Involved in an Apoptosis Regulation Mechanism Based on Network Pharmacology and Transcriptomic Analysis. BioMed Pharmacother (2019) 114:108863. doi: 10.1016/j.biopha.2019.108863
137. Zhang B, Chen YQ, Zhou CX, Chen RX. Technical Elements and Clinical Application of Umbilical Refining of Heat-Sensitive Moxibustion. Zhongguo Zhen Jiu (2020) 40(9):965–7. doi: 10.13703/j.0255-2930.20191214-k0006
138. Wu HM, Ni XX, Xu QY, Wang Q, Li XY, Hua J. Regulation of Lipid-Induced Macrophage Polarization Through Modulating Peroxisome Proliferator-Activated Receptor-Gamma Activity Affects Hepatic Lipid Metabolism via a Toll-Like Receptor 4/NF-kappaB Signaling Pathway. J Gastroenterol Hepatol (2020) 35(11):1998–2008. doi: 10.1111/jgh.15025
139. Sun YY, Li XF, Meng XM, Huang C, Zhang L, Li J. Macrophage Phenotype in Liver Injury and Repair. Scand J Immunol (2017) 85(3):166–74. doi: 10.1111/sji.12468
140. Wang Y, Wang R, Wang Y, Peng R, Wu Y, Yuan Y. Ginkgo Biloba Extract Mitigates Liver Fibrosis and Apoptosis by Regulating P38 MAPK, NF-Kappab/IkappaBalpha, and Bcl-2/Bax Signaling. Drug Des Devel Ther (2015) 9:6303–17. doi: 10.2147/DDDT.S93732
141. Ding J, Yu J, Wang C, Hu W, Li D, Luo Y, et al. Ginkgo Biloba Extract Alleviates Liver Fibrosis Induced by CCl in Rats. Liver Int (2005) 25(6):1224–32. doi: 10.1111/j.1478-3231.2005.01169.x
142. Qu Y, Zong L, Xu M, Dong Y, Lu L. Effects of 18alpha-Glycyrrhizin on TGF-Beta1/Smad Signaling Pathway in Rats With Carbon Tetrachloride-Induced Liver Fibrosis. Int J Clin Exp Pathol (2015) 8(2):1292–301.
143. Liang B, Guo XL, Jin J, Ma YC, Feng ZQ. Glycyrrhizic Acid Inhibits Apoptosis and Fibrosis in Carbon-Tetrachloride-Induced Rat Liver Injury. World J Gastroenterol (2015) 21(17):5271–80. doi: 10.3748/wjg.v21.i17.5271
144. Gowifel AMH, Khalil MG, Nada SA, Kenawy SA, Ahmed KA, Salama MM, et al. Combination of Pomegranate Extract and Curcumin Ameliorates Thioacetamide-Induced Liver Fibrosis in Rats: Impact on TGF-Beta/Smad3 and NF-kappaB Signaling Pathways. Toxicol Mech Methods (2020) 30(8):620–33. doi: 10.1080/15376516.2020.1801926
145. Qiao H, Han H, Hong D, Ren Z, Chen Y, Zhou C. Protective Effects of Baicalin on Carbon Tetrachloride Induced Liver Injury by Activating PPARgamma and Inhibiting Tgfbeta1. Pharm Biol (2011) 49(1):38–45. doi: 10.3109/13880209.2010.493179
146. Zhang F, Zhang Z, Chen L, Kong D, Zhang X, Lu C, et al. Curcumin Attenuates Angiogenesis in Liver Fibrosis and Inhibits Angiogenic Properties of Hepatic Stellate Cells. J Cell Mol Med (2014) 18(7):1392–406. doi: 10.1111/jcmm.12286
147. Sui M, Jiang X, Chen J, Yang H, Zhu Y. Magnesium Isoglycyrrhizinate Ameliorates Liver Fibrosis and Hepatic Stellate Cell Activation by Regulating Ferroptosis Signaling Pathway. BioMed Pharmacother (2018) 106:125–33. doi: 10.1016/j.biopha.2018.06.060
148. Wang L, Zhang Z, Li M, Wang F, Jia Y, Zhang F, et al. P53-Dependent Induction of Ferroptosis Is Required for Artemether to Alleviate Carbon Tetrachloride-Induced Liver Fibrosis and Hepatic Stellate Cell Activation. IUBMB Life (2019) 71(1):45–56. doi: 10.1002/iub.1895
149. Li Y, Jin C, Shen M, Wang Z, Tan S, Chen A, et al. Iron Regulatory Protein 2 Is Required for Artemether -Mediated Anti-Hepatic Fibrosis Through Ferroptosis Pathway. Free Radic Biol Med (2020) 160:845–59. doi: 10.1016/j.freeradbiomed.2020.09.008
150. Zhao Z, Bai S, Wang R, Xiong S, Li Y, Wang X, et al. Cancer-Associated Fibroblasts Endow Stem-Like Qualities to Liver Cancer Cells by Modulating Autophagy. Cancer Manag Res (2019) 11:5737–44. doi: 10.2147/CMAR.S197634
151. Li W, Xu Y, Wang X, Cao G, Bu W, Wang X, et al. Circcct3 Modulates Vascular Endothelial Growth Factor A and Wnt Signaling to Enhance Colorectal Cancer Metastasis Through Sponging miR-613. DNA Cell Biol (2020) 39(1):118–25. doi: 10.1089/dna.2019.5139
152. Lv B, Zhu W, Feng C. Coptisine Blocks Secretion of Exosomal Circcct3 From Cancer-Associated Fibroblasts to Reprogram Glucose Metabolism in Hepatocellular Carcinoma. DNA Cell Biol (2020) 39:12. doi: 10.1089/dna.2020.6058
153. Yan T, Wang H, Cao L, Wang Q, Takahashi S, Yagai T, et al. Glycyrrhizin Alleviates Nonalcoholic Steatohepatitis via Modulating Bile Acids and Meta-Inflammation. Drug Metab Dispos (2018) 46(9):1310–9. doi: 10.1124/dmd.118.082008
154. Hu N, Wang C, Dai X, Zhou M, Gong L, Yu L, et al. Phillygenin Inhibits LPS-Induced Activation and Inflammation of LX2 Cells by TLR4/MyD88/NF-kappaB Signaling Pathway. J Ethnopharmacol (2020) 248:112361. doi: 10.1016/j.jep.2019.112361
155. Chen A, Zheng S. Curcumin Inhibits Connective Tissue Growth Factor Gene Expression in Activated Hepatic Stellate Cells In Vitro by Blocking NF-kappaB and ERK Signalling. Br J Pharmacol (2008) 153(3):557–67. doi: 10.1038/sj.bjp.0707542
156. Morsy MA, Abdalla AM, Mahmoud AM, Abdelwahab SA, Mahmoud ME. Protective Effects of Curcumin, Alpha-Lipoic Acid, and N-Acetylcysteine Against Carbon Tetrachloride-Induced Liver Fibrosis in Rats. J Physiol Biochem (2012) 68(1):29–35. doi: 10.1007/s13105-011-0116-0
157. Fu Y, Zheng S, Lin J, Ryerse J, Chen A. Curcumin Protects the Rat Liver From CCl4-Caused Injury and Fibrogenesis by Attenuating Oxidative Stress and Suppressing Inflammation. Mol Pharmacol (2008) 73(2):399–409. doi: 10.1124/mol.107.039818
158. Cheng Y, Ping J, Xu LM. Effects of Curcumin on Peroxisome Proliferator-Activated Receptor Gamma Expression and Nuclear Translocation/Redistribution in Culture-Activated Rat Hepatic Stellate Cells. Chin Med J (Engl) (2007) 120(9):794–801. doi: 10.1097/00029330-200705010-00011
159. Zhou Y, Zheng S, Lin J, Zhang QJ, Chen A. The Interruption of the PDGF and EGF Signaling Pathways by Curcumin Stimulates Gene Expression of PPARgamma in Rat Activated Hepatic Stellate Cell In Vitro. Lab Invest (2007) 87(5):488–98. doi: 10.1038/labinvest.3700532
160. Liu YW, Chiu YT, Fu SL, Huang YT. Osthole Ameliorates Hepatic Fibrosis and Inhibits Hepatic Stellate Cell Activation. J BioMed Sci (2015) 22:63. doi: 10.1186/s12929-015-0168-5
161. Liu SQ, Yu JP, He L, Yu HG, Luo HS. Effects of Nuclear Factor kappaB and Transforming Growth Factor Beta1 in the Anti-Liver Fibrosis Process Using Ginkgo Biloba Extract. Zhonghua Gan Zang Bing Za Zhi (2005) 13(12):903–7.
162. Liu SQ, Yu JP, Chen HL, Luo HS, Chen SM, Yu HG. Therapeutic Effects and Molecular Mechanisms of Ginkgo Biloba Extract on Liver Fibrosis in Rats. Am J Chin Med (2006) 34(1):99–114. doi: 10.1142/S0192415X06003679
163. Wang JY, Zhang QS, Guo JS, Hu MY. Effects of Glycyrrhetinic Acid on Collagen Metabolism of Hepatic Stellate Cells at Different Stages of Liver Fibrosis in Rats. World J Gastroenterol (2001) 7(1):115–9. doi: 10.3748/wjg.v7.i1.115
164. Qu Y, Chen WH, Zong L, Xu MY, Lu LG. 18alpha-Glycyrrhizin Induces Apoptosis and Suppresses Activation of Rat Hepatic Stellate Cells. Med Sci Monit (2012) 18(1):BR24–32. doi: 10.12659/MSM.882196
165. Tzeng JI, Chen MF, Chung HH, Cheng JT. Silymarin Decreases Connective Tissue Growth Factor to Improve Liver Fibrosis in Rats Treated With Carbon Tetrachloride. Phytother Res (2013) 27(7):1023–8. doi: 10.1002/ptr.4829
166. Sferra R, Vetuschi A, Catitti V, Ammanniti S, Pompili S, Melideo D, et al. Boswellia Serrata and Salvia Miltiorrhiza Extracts Reduce DMN-Induced Hepatic Fibrosis in Mice by TGF-Beta1 Downregulation. Eur Rev Med Pharmacol Sci (2012) 16(11):1484–98.
167. Liu J, Kong D, Qiu J, Xie Y, Lu Z, Zhou C, et al. Praziquantel Ameliorates CCl4 -Induced Liver Fibrosis in Mice by Inhibiting TGF-Beta/Smad Signalling via Up-Regulating Smad7 in Hepatic Stellate Cells. Br J Pharmacol (2019) 176(24):4666–80. doi: 10.1111/bph.14831
Keywords: cancer-associated fibroblasts (CAFs), traditional Chinese medicine, hepatocellular carcinoma (HCC), stromal cell, tumor recurrence
Citation: Jia W, Liang S, Cheng B and Ling C (2021) The Role of Cancer-Associated Fibroblasts in Hepatocellular Carcinoma and the Value of Traditional Chinese Medicine Treatment. Front. Oncol. 11:763519. doi: 10.3389/fonc.2021.763519
Received: 24 August 2021; Accepted: 28 October 2021;
Published: 18 November 2021.
Edited by:
Bin-Yan Zhong, The First Affiliated Hospital of Soochow University, ChinaCopyright © 2021 Jia, Liang, Cheng and Ling. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Changquan Ling, bGluZ2NoYW5ncXVhbkBob3RtYWlsLmNvbQ==; Binbin Cheng, Y2JiODIwMkBzbW11LmVkdS5jbg==
†These authors have contributed equally to this work