
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol., 18 November 2021
Sec. Molecular and Cellular Oncology
Volume 11 - 2021 | https://doi.org/10.3389/fonc.2021.762817
 Sikiru O. Imodoye1*
Sikiru O. Imodoye1* Kamoru A. Adedokun2
Kamoru A. Adedokun2 Abdurrasheed Ola Muhammed3Ibrahim O. Bello4Musa A. Muhibi5Taofeeq Oduola6Musiliu A. Oyenike7
Abdurrasheed Ola Muhammed3Ibrahim O. Bello4Musa A. Muhibi5Taofeeq Oduola6Musiliu A. Oyenike7Epithelial-mesenchymal transition (EMT) is a physiological program during which polarised, immobile epithelial cells lose connection with their neighbours and are converted to migratory mesenchymal phenotype. Mechanistically, EMT occurs via a series of genetic and cellular events leading to the repression of epithelial-associated markers and upregulation of mesenchymal-associated markers. EMT is very crucial for many biological processes such as embryogenesis and ontogenesis during human development, and again it plays a significant role in wound healing during a programmed replacement of the damaged tissues. However, this process is often hijacked in pathological conditions such as tumour metastasis, which constitutes the most significant drawback in the fight against cancer, accounting for about 90% of cancer-associated mortality globally. Worse still, metastatic tumours are not only challenging to treat with the available conventional radiotherapy and surgical interventions but also resistant to several cytotoxic agents during treatment, owing to their anatomically diffuse localisation in the body system. As the quest to find an effective method of addressing metastasis in cancer intervention heightens, understanding the molecular interplay involving the signalling pathways, downstream effectors, and their interactions with the EMT would be an important requisite while the challenges of metastasis continue to punctuate. Unfortunately, the molecular underpinnings that govern this process remain to be completely illuminated. However, it is becoming increasingly clear that EMT, which initiates every episode of metastasis, significantly requires some master regulators called EMT transcription factors (EMT-TFs). Thus, this review critically examines the roles of TFs as drivers of molecular rewiring that lead to tumour initiation, progression, EMT, metastasis, and colonisation. In addition, it discusses the interaction of various signalling molecules and effector proteins with these factors. It also provides insight into promising therapeutic targets that may inhibit the metastatic process to overcome the limitation of “undruggable” cancer targets in therapeutic design and upturn the current spate of drug resistance. More so, it extends the discussion from the basic understanding of the EMT binary switch model, and ultimately unveiling the E/M cellular plasticity along a phenotypic spectrum via multiple trans-differentiations. It wraps up on how this knowledge update shapes the diagnostic and clinical approaches that may demand a potential shift in investigative paradigm using novel technologies such as single-cell analyses to improve overall patient survival.
Embryological evidence of human development shows that the human body is derived from a single cell type, a totipotent cell, from which many other specialised cell types are generated in a bid to expand, differentiate, and grow to accommodate functional diversity. It is based on this understanding that the need for cell transition from one type to another is physiologically justified even though, understanding of similar cell transition in pathological situations, particularly in tumour metastasis, has recently emerged. Importantly, a phenomenon that facilitates such a cellular diversity during tissue/organ development and in adulthood is an epithelial-mesenchymal transition (EMT), characterised by reversible gradual loss of epithelial characteristics and the resultant development of mesenchymal features. As the knowledge of EMT roles in various tissue/organ development surges, three types of EMT have been recognised to occur in biological systems. Each type occurs in response to different biological signals and with different functional consequences (1).
Type 1 represents a physiological process that occurs in implantation, embryo formation, and organ development. Type 2 occurs in association with wound healing, tissue regeneration, and organ fibrosis. This type begins with the initial step of a repair-associated process that generates fibroblasts to regenerate tissue due to trauma and inflammatory damage. In contrast to type 1, type 2 does not continue indefinitely; it stops once the inflammatory process is attenuated (1, 2). In organ fibrosis, however, the EMT process has a propensity for persistent response to recurring inflammatory reactions with the possibility of leading to organ damage. Type 3 predominantly occurs in the neoplastic environment following initial genetic and epigenetic changes affecting oncogenes and tumour suppressor genes that conspire with normal EMT regulatory mechanisms to produce outcomes that are far distinct from the other two types of EMT. The cell phenotypes produced in this EMT type may invade and metastasise to distant organs via systemic circulation. In an attempt to decipher the molecular interplay of cell transition mechanisms—both in physiological and pathological states—a lot has been elucidated on the signalling pathways in type 1 and type 2 EMT (2); however, the specific cues that orchestrate type 3 EMT in epithelial carcinoma cells are yet to be completely understood.
In the past few decades, scientists have invested a lot of effort to unravel the pathogenesis of tumour metastasis. This has led to the conviction that a successful tumour spread requires several steps. These include the EMT process, tumour cell invasion, intravasation into the vascular system, transition through the circulatory system, extravasation out of the vasculature, seeding at the premetastatic niche, and, finally, survival and growth at the secondary metastatic site (3, 4). In addition, studies have established that tumour metastasis requires intimate interaction and collaboration between cancer cells and other stromal components of the tumour microenvironment (5), including the inflammatory signals (6), which significantly dictate several aspects of the metastatic cascade.
In this article, we review our current understanding of the multiplex interplays between the master regulators that orchestrate tumour progression in consideration of cellular plasticity along a phenotypic spectrum and beyond the concept of the binary switch model. In particular, we discuss the roles of TFs in EMT programming and potentially attractive therapeutic targets that could help mitigate tumour metastasis. We finally call into attention how this development could be better explored using the state-of-the-art therapeutic design during cancer control interventions.
The propensity of carcinoma to populate immediate tissues and migrate to remote organs has long been recognised as a dominant feature of tumour metastasis. This composite phenomenon results when tumour cells disconnect from the initial epithelial layer within the primary malignancies, invade the neighbouring microenvironment, intravasate into the vascular system, and ultimately permeate distant organs (7).
Scientists have proven beyond doubt that the local microenvironment provides necessary signals that determine the fate of the disseminated cells at the peripheral metastatic site, whether to proliferate and revert to a more epithelial phenotype or remain dormant for an extended period (8). In either case, metastasis results. Recent studies have signalled that these events may be supported by the acquisition of EMT features, which is characterised by the loss of apical-lateral polarity (9), even though the complex molecular interplay in this event remains a puzzle. Understanding the molecular mechanism in the EMT program for initiation of tumour invasion and metastasis is key to improved therapeutic interventions.
Despite the seemingly simple definition, EMT is an extremely complex process, activated by pleiotropic intrinsic and extrinsic factors and finely regulated temporally and spatially (10). It involves several molecular re-engineering that instigates polarised epithelial cells, which ordinarily interact with basement membrane through its basal surface to ultimately gain mesenchymal features. Consequently, loss of apical-basal polarity, enhanced motility, invasiveness, immunosuppression, and increased resistance to cell death and therapy result owing to the acquisition of stem cell-like properties (11). Meanwhile, the hallmark of EMT is the inhibition of E-cadherin. Suppression of E-cadherin results in reduced cell adhesion and imposition of migratory mesenchymal cells. This process is not a snap approach. Even though it was previously believed that the transition of tumour cells from pure epithelial to pure mesenchymal state followed a binary-switch model, the emerging knowledge shows that E→M transition is involved with a complex dynamic process through a phenotypic spectrum. Again, the EMT is a highly conserved phenomenon involving corporations among TFs, effector proteins, and signalling molecules that rely on the activity of TFs as effector molecules.
Generally, EMT is characterised by the suppression of epithelial markers particularly involving the intercellular junction protein complex made up of gap junction proteins (such as connexin) adherens junction proteins (particularly, E-cadherin), tight junction proteins (occludin, claudin, and junctional adhesion molecules), cytokeratins, and catenins, as shown in Figure 1. Primarily, E-cadherin is the most notable among all epithelial markers for its gatekeeping role to avert EMT by acting in concert with the intercellular adhesion system to maintain cellular polarity, differentiation, migration, and signalling in proliferation pathways. Consequently, loss of E-cadherin promotes the transformation of stationary epithelial cells to a migratory mesenchymal phenotype through a cascade process involving several intermediate E/M phenotypes. Apart from mutation, loss of E-cadherin may also be orchestrated by downregulation due to epigenetic or transcriptional silencing (12, 13). All the same, mesenchymal markers such as vimentin, N-cadherin, fibronectins, smooth muscle actin, and matrix metalloproteases (MMP) also become upregulated and function concurrently with the loss of E-cadherin during the EMT program (14) (Figure 1). Altogether, these effector proteins collaborate to induce rearrangement of cellular architecture leading to destabilisation in cellular adhesion, proteolytic degeneration of basement membrane, increased motility (15), and generation of circulating tumour cells (CTCs) (Figure 1).
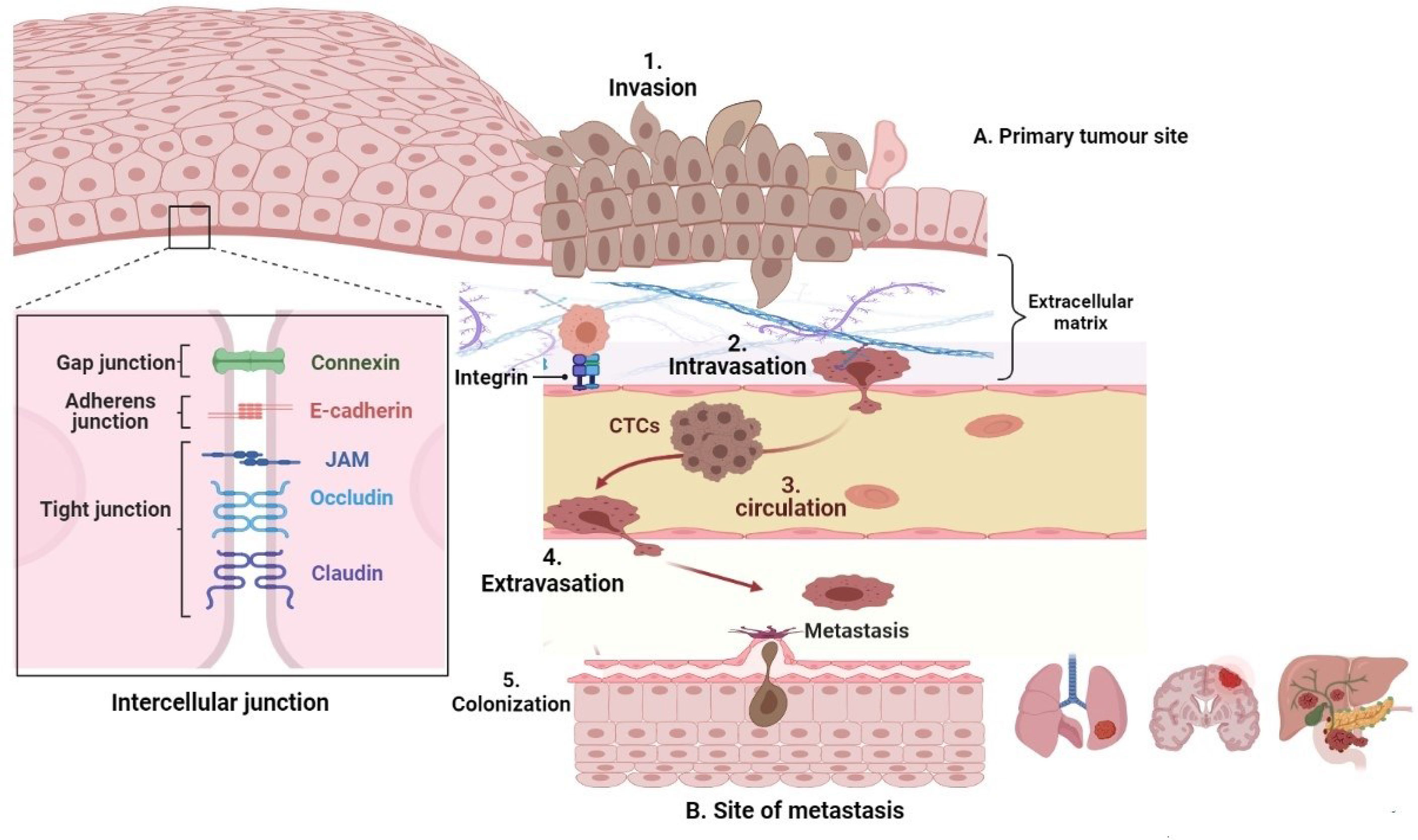
Figure 1 The metastatic cascade. Epithelial tumours may sometimes undergo EMT to generate mesenchymal cells with more motile and invasive properties that penetrate the basal lamina (invasion [1]) and enter the bloodstream or lymphatic system, becoming circulating tumour cells (intravasation [2]), which are transported via the systemic circulation (circulation [3]), migrate into distant tissues that have the favourable cellular cues (extravasation [4]). The microenvironmental signals then induce an EMT reversal (also called MET) to establish secondary micrometastases (colonization [5]).
Even though downregulation of E-cadherin has been shown to induce EMT independently in some cancer models (16, 17), a study carried out by Vafaizadeh and her colleagues (18) suggests that active β-catenin is essential for invasion in culture and experimental metastasis model attributed in part to detachment of adherens junctions during early EMT and subsequent degradation. Deducing in tandem with their structural arrangement, the implication of these findings is that β-catenin acts cooperatively with E-cadherin to initiate EMT (Figure 2), suggesting that β-catenin may be an important therapeutic target in certain types of cancer.
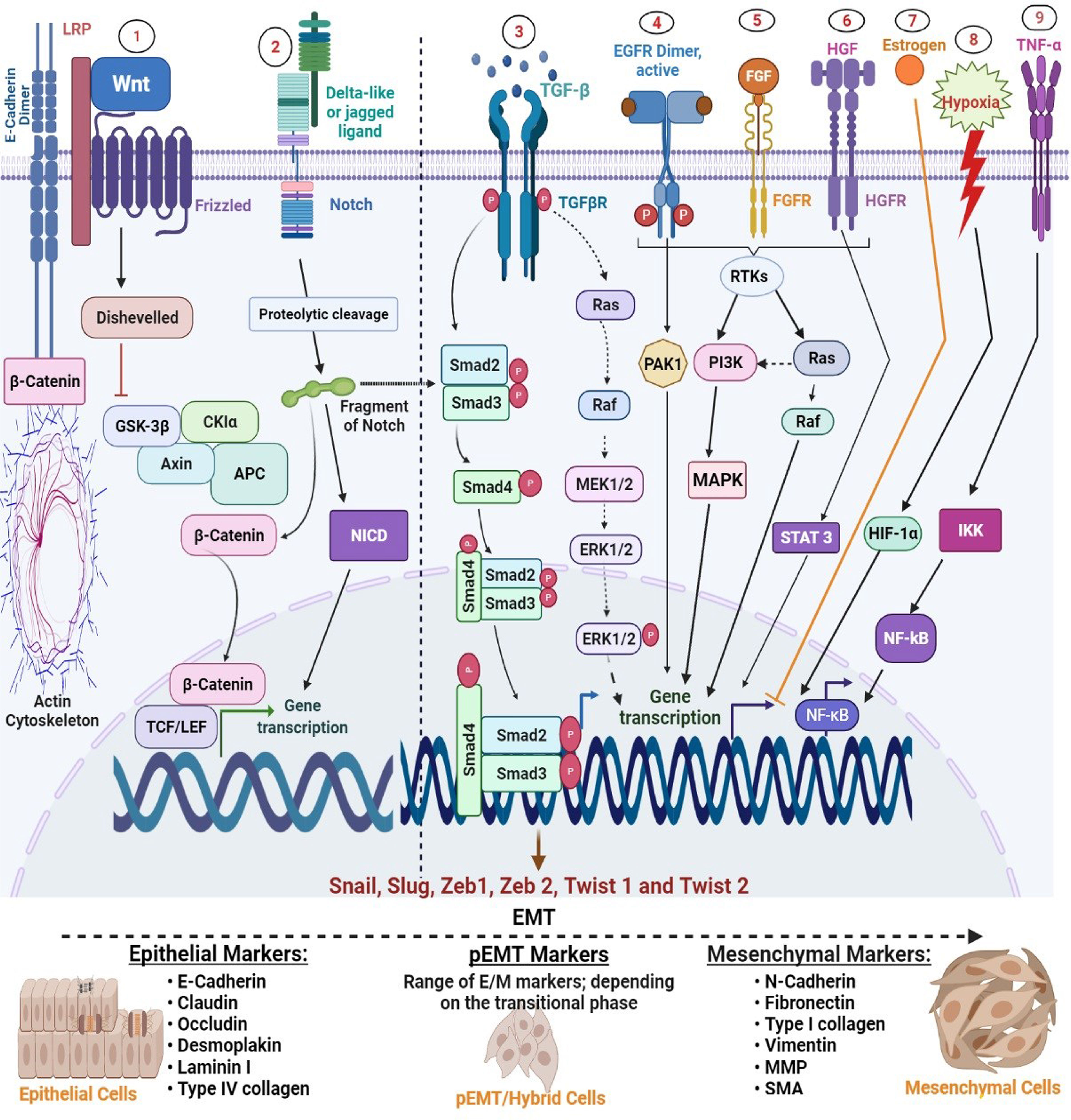
Figure 2 Regulation of major EMT transcription factors by signalling pathways. Epithelial-mesenchymal transition (EMT) is driven by SNAIL, zinc-finger E-box-binding (ZEB), and basic helix-loop-helix (bHLH) transcription factors that act as downstream effectors from several other signalling molecules. 1, Wnt signalling: WNT inhibits glycogen synthase kinase-3β (GSK3β) to stabilize β-catenin, which translocates to the nucleus to recruit the transcription factors; lymphoid enhancer-binding factor 1 (LEF) and T-cell factor (TCF) and promotes expression of SNAIL1 and SNAIL2. 2, Notch signalling: binding of a ligand (such as delta-like or jagged) with Notch receptors initiates the Notch signalling through the proteolytic cleavage by γ-secretase complex to release NICD. NICD then undergoes nuclear translocation to regulate the expression of target genes, including SNAIL, ZEB, and bHLH families. 3, Transforming growth factor-β (TGFβ) promotes EMT by acting at various strata; SMAD-mediated and non-SMAD signalling (i) SMAD-mediated signalling: Activation of the TβRI and TβRII turns on SMAD2 and SMAD3, which then integrate with SMAD4 to form a trimeric SMAD complex. This complex translocates into the nucleus to stimulate the expression of EMT transcription factors (ii) non-SMAD-mediated signalling: TGFβ also activates ERK through the RAS-MAKP pathway. Activated ERK can then stimulate the expression of SNAIL1 and SNAIL2 (unspecified in the image). 4–6, Growth factors (EGF, FGF, and HGF): various growth factors such as EGF, FGF, and PDGF activate receptor tyrosine kinases (RTKs), triggering dimerization and autophosphorylation of the intracellular domain of these receptors, which allows them to activate other downstream signalling molecules, including PI3K, PAK1, and STAT 3 and increase expression of SNAIL1 and SNAIL2. 7, Oestrogen; oestrogen is one of the few molecular candidates that negatively regulate EMT. It does this through direct inhibition of Slug transcription by forming a co-repressor complex consisting of ligand-activated ERα, HDAC1, and nuclear receptor co-repressor (NCoR) that binds to the oestrogen-response elements at the slug promoter sequence and inhibit its expression. 8, Hypoxia: in normoxic conditions, prolyl hydroxylases are activated which causes hydroxylation of HIF-1α and thereby hindering their activities, whereas, hypoxia stabilizes the enzymes, enabling their nuclear translocation and heterodimerization and binding to oversee stimulation of Twist and stabilization of SNAIL1/2. 9, Tumour necrotic factor-alpha (TNF-α): TNF-α signal activates IKB kinase complex (IKK), which in turn phosphorylates NF-kB inhibitor. Consequently, NF-kB becomes active and undergoes nuclear translocation to promote induction of Twist1 expression.
More so, various coordinated events that occur in EMT are controlled by a set of TFs in a cell-specific and context-dependent manner (19, 20). As illustrated in Figure 2, there is vast evidence that these factors are themselves controlled by various signalling pathways and microenvironment signals such as tumour growth factor-beta (TGF-β), epidermal growth factor (EGF), oestrogen, platelet-derived growth factor (PDGF), WNT, hedgehog (SHH), Notch, and integrins (21–26). In addition to induction of EMT by TFs and signalling molecules, certain types of cellular effectors, such as microRNAs, have also been implicated for their roles in stimulating EMT through TFs. In essence, epithelial-mesenchymal transition TFs (EMT-TFs) perform central roles during the EMT program, which is fundamental to various processes in tissue growth and organ development.
Before now, most experimental models have defined EMT as a binary transition of tumour cells between EMT and MET as mentioned above. However, this oversimplification has caused some confusion, as it fails to explain the real phenomena of the current knowledge, which may not address the limitations of modern clinical settings. Several attempts have been made to elucidate the mechanistic process of EMT through various studies and town hall discussions. Recently, during the 2017 and 2019 meetings of The EMT International Association (TEMTIA), the new concept of “epithelial-to-mesenchymal plasticity” (EMP) was introduced (27), thus broadening the traditional definition of EMT to accommodate the newly discovered features such as the partial activation of EMT, and the existence of a continuous spectrum of hybrid EMT/MET phenotype. Presently, the construction of mathematical models and EMT trajectory by single-cell transcriptomics has enhanced our understanding of the existence of multiple intermediate steps with various degrees of E or M states (28, 29) (Figure 3). This intermediate E/M phase is characterised by various degrees of molecular markers such as E-cadherin, vimentin, β-catenin, etc., depending on the level of EMT-TF expression or inhibition
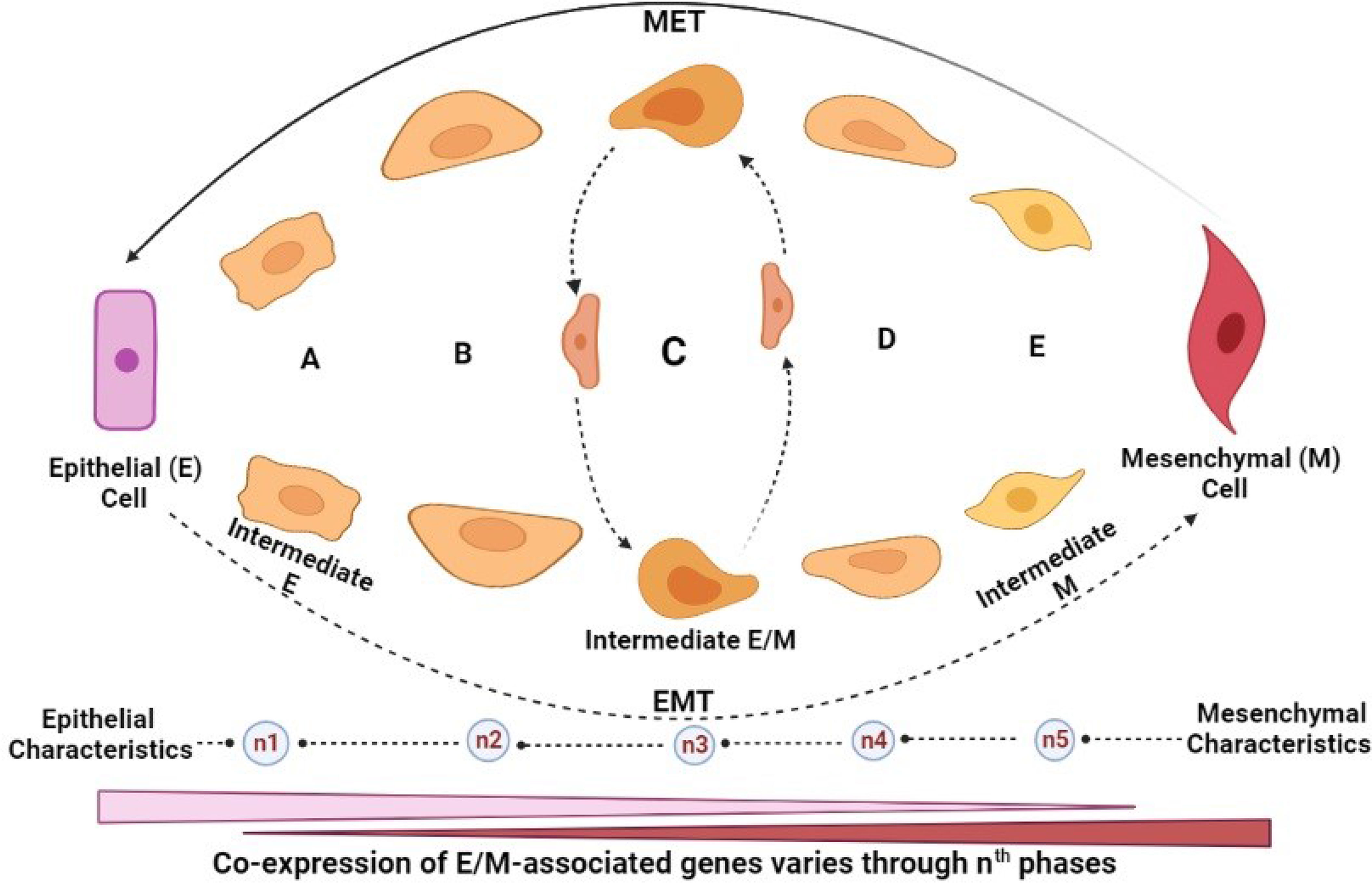
Figure 3 EMT/MET intermediates. (A–E) Intermediate cells: hybrid/partial EMT (pEMT) cells. nth: The transition phase indicates the number of transitions that may occur before M cells are formed, which is indeterminate but could be dictated by many factors including the tumour microenvironmental cues. Intermediate cells possess dynamic characteristics that are central to increased metastatic potential. Hybrid/pEMT “C”: indicates an unspecific hypothetical stage. The time taken (known as first arrival time (FAT) distribution) for E or M cells to transition to each other, or individual pMET/hybrid cell at the nth phase varies depending on (i) epigenetic changes (histone modification or methylation), (ii) presence of anti-EMT/pro-MET factors (such as OVOL 1/2, GRHL-2, ESRP-1/2, and various microRNAs (including miR20 family), among others. Generally, the stability of hybrid E/M is controlled by some factors known as phenotypic stability factors (PSFs) and their metabolic, genomic, and morphologic states. However, as these intermediate cells possess some similar characteristics, they also have different phenotypic behaviours. They possess identical properties such as tumour-initiating potential but are also significantly contrastive, based on (i) cellular plasticity, (ii) degree of invasiveness, and (iii) metastatic potential. Among all the intermediate phenotypes, however, intermediate M possesses the highest migratory potential, the highest degree of invasiveness, most primed to form spindle-shaped phenotype and the highest resistance to anoikis.
Remarkably, emerging studies have suggested that the hybrid E/M status can be further divided into several phases based on the combination of different markers they display, as shown in Figure 3. This proposal was supported by a genetically modified skin squamous cell carcinoma model, showing that neoplastic cells commit to spontaneous EMT and that the hybrid E/M status was divided into early and late hybrid E/M states according to the expression patterns of the surface markers CD106, CD61, and CD51 (30). The emerging concept of plasticity of EMT/MET through transitional states is further discussed extensively later in this report.
The cornerstone of every EMT and its reverse, i.e., mesenchymal-epithelial transition (MET) program, is the transcriptional regulators that control the gene expression required for the cellular transitions. The most widely studied among these factors are zinc-finger proteins (SNAIL), zinc-finger E-box-binding homeobox (ZEB), and Twist family of basic helix-loop-helix (bHLH) transcription factors (TWIST) (31, 32) which are altogether called EMT-TFs (Table 1).

Table 1 Signalling pathways regulate EMT-TFs.
EMT-TFs are gene expression regulators that direct selective gene expression according to the demand of a cell. Their activities are localized in the nucleus, where they have immediate accessibility to the DNA. A common feature of all EMT-TFs is their physiological roles in embryogenesis and organismal development. However, studies have confirmed their aberrant reappearance in cancer cells during tumour development and progression (40, 41). They are activated in the early events of EMT, and they could hence be said to play cardinal roles in development, fibrosis, and tumour aggressiveness. Sometimes, they control the expression of each other and function synergistically at target genes (42).
The conventional feature of all TFs is the direct/indirect inhibition of E-cadherin expression, resulting in the imposition of mesenchymal state and loss of epithelial cell surface biomarkers to gain effective change in phenotype (4). Barring genetic mutations, the overall effect of EMT-TFs on the degradation of E-cadherin to initiate EMT pathway characterizes a structural transformation in the biological properties of epithelial cells. While it is important to understand the mechanisms involved in phenotypic changes from one cellular architecture to another, the roles of individual EMT-inducing TFs cannot be overemphasised as a functional basis for loss of cellular configuration/polarity in epithelial cells, which in turn influences invasion, cell migration, and resistance to anoikis.
All snail protein members encode transcriptional repressors with a similar structural organisation. The C-terminal domain is exceptionally conserved, containing four- to five-type zinc fingers (C2H2) which oversee sequence-specific binding to the E-box element (5′-CAG GTG-3′) of the target genes (43). The N-terminal in humans contains the evolutionarily conserved SNAG domain, which is important for the binding of various transcriptional corepressor complexes to enforce repression of the target genes (44). The central region of Snail is characterised by a serine-rich domain (SRD) and a nuclear export sequence (NES) that regulate protein stability and subcellular localisation of Snail, respectively.
Snail member proteins are the most widely studied modulators of E-cadherin expression, including Snail, Slug, and Smuc. As opposed to Snail and Slug, the involvement of Smuc in the EMT process of human carcinomas is not yet established (45), according to the available data at present. However, as the paradigm of tumour metastasis keeps evolving, it may be worthwhile to investigate the possible involvement of Smuc in different cancer models.
Also, E-cadherin is the hallmark of EMT, and its suppression is mainly attributed to the functions of Snail1 and Snail2 expressions by binding directly to the E-box of the promoter region and downregulate their expressions (8). EMT-TFs do not only inhibit E-cadherin expression but also downregulate the transcription of other genes encoding the epithelial junction proteins such as claudin (46), ZO-1 (47), both of which serve to stabilise the tight junctions. This implies that any biological process that tends to upregulate Snail expression will encourage EMT. In addition, Snail1 has also been shown to enhance the expressions of matrix degradation enzyme matrix metalloproteinases 9 (MMP9) (48), leading to EMT cascade, as a consequence of proteolytic action of MMP that reshapes the extracellular matrix and deconstructs the epithelial membrane (49).
Snail has long been known as an effector molecule for various signalling pathways that direct their functions. For instance, because Snail is a vital regulator of E-cadherin, scientists have asked whether the effects of the Notch on this process could be mediated through the Notch intracellular domain (NICD) via regulation of Snail1. The answer to this is affirmative since Notch has been shown to indirectly promote EMT through regulation of Snail (Figure 2). In further agreement with this, Kar et al. (50) reported that activation of Notch-1 promotes EMT via the repression of E-cadherin by Slug. Similarly, a study carried out by Niessen and his colleagues showed that activation of the Notch in the context of TGF-β stimulation results in synergistic upregulation of Snail in endothelial cells (51). From the above study, it is clearly understood that TGF-β signalling plays a key role in the pathogenesis of tumour metastasis and thus could function as a potential target in controlling tumour progression.
As indicated earlier, TFs regulate the expression of one another. For instance, when E-cadherin expression is suppressed, the expression of Snail proteins (SNAI1/SNAI2) is amplified by an autoactivation loop due to the inhibition of nuclear factor-κB (NF-κB) (52). Therefore, the self-stimulation loop of Snail is engendered by the downregulation of E-cadherin via SNAIL (Snail1/Snail2). This insinuates that Slug is not only a transcriptional repressor but also a transcriptional activator. Intriguingly, Twist1 also binds directly to E-box on the Slug promoter as a transcriptional activator to induce Slug transcription (53). Likewise, Slug is essential for Twist1 to induce EMT. Meanwhile, a knockdown of Slug completely blocks the ability of Twist1 to suppress E-cadherin transcription (54). By implication, Twist1 activity is insufficient to induce cancer cell invasion and distant metastasis in the absence of Slug, making it an attractive therapeutic target.
Apart from EMT modulation, Snail protein members also block the cell cycle, promote cell survival, and inhibit apoptosis, with additional roles in the induction of metastasis and acquisition of cancer stem cell-like characteristics (55, 56). These findings were further supported by separate reports establishing Slug to increase the amount of CD44, a marker of cancer stem cells (CSCs) (57, 58). By implication, the CSC membrane marker could be a potential target for tumour detection, metastatic prevention, selection of drug targets, and other possible intervention strategies.
Another important TF, known as zinc-finger E-box binding homeobox, or ZEB, also modulates the transition of epithelial cells to mesenchymal phenotype. Generally, homeobox genes are conserved in plants, fungi and animals. In humans, the ZEB family proteins are made up of two homologous proteins; ZEB1 (δEFI) and ZEB2 (SIP1) which constitute zinc-finger TFs (59). Structurally, they include two ZnF domains, N-terminal ZnF and C-terminal ZnF. The homeodomain consists of three-alpha helices. In this segment, the helix-loop-helix (or helix-turn-helix (HTH)) motif functions as the E-box-binding site and allows the protein to bind to the E-cadherin promoter region (42), and thus regulates the transcription of the adjacent DNA. Generally, the homeobox codes for a specific sequence that functions as part of evolutionarily conserved signalling pathways, which is particularly active in morphogenesis.
Currently, the roles of the proteins of ZEB have been further implicated in tumorigenesis and metastasis as a resultant disruption of morphogenesis. They function through transcriptional inhibition of the E-cadherin gene (CDH1) by binding the promoter site known as a consensus sequence in addition to corepressor recruitment, histone deacetylase process, and chromatin condensation ultimately initiating EMT (60).
In physiological states, however, the proteins of ZEB mainly occur in the heart, CNS, skeletal muscle, and hematopoietic cells to support organ development. Particularly, ZEB1 is commonly found in the thymus during T-lymphocyte development; whereas, ZEB2 is chiefly found in the spleen during B-lymphocyte development (61), suggesting specific functionality and expression. However, evidence of ZEB1 upregulation has been reported in different cancer types such as pancreatic, lung, liver, colon, and breast cancers (62–66). In the same vein, Yilmaz and Christofori (67) demonstrated that increased expression of ZEB1 decreased the response of cancer cells to therapy. These studies predict that ZEB1 could be an important target in anticancer drug resistance.
By interconnection, Snail1 and Twist1 mutually regulate the expression of ZEB1 (68), while the activated ZEB1, in turn, downregulates multiple genes that enforce epithelial characteristics (69), which is the main event in tumour metastasis. In other words, these proteins could be implicated as markers of poor clinical outcomes in patients with a solid tumour, most especially in the metastatic drive. Akin to the Snail family, various signalling molecules modulate ZEB1 and ZEB2 expression. For instance, TGF-β, Wnt/β-catenin, PI3K/Akt, and Ras/Erk signalling induce ZEB1 (70) (Figure 2), whereas micro-RNAs negatively regulate the expression.
Furthermore, posttranscriptional phosphorylation of ZEB1 also has the propensity to modulate their expression levels, the functional role that was not immediately understood. Although, recent findings have proposed an inhibitory effect on ZEB1 function. For instance, Llorens et al. (71) reported that phosphorylation of ZEB proteins within C-terminal ZnF inhibits the ZEB binding to DNA and its transcriptional activity. This study shows that ZEB1 phosphorylation could perform many functions particularly in crosslinking the TGF-β signalling with many other pathways involving signalling molecules (cytokines and growth factors) within the cellular microenvironment.
In mammals, two bHLH TFs have been identified, Twist1 and Twist2. These two factors share structural similarities and, as such, bind to the same E-box of DNA response elements of the target genes to regulate their expression (72). They are the most characterized indirect transcriptional repressors of the CDH1 promoter, and they play an essential role in embryo formation, wound healing, and tissue fibrosis (73, 74). In adulthood, they are either deficient or expressed at extremely low levels (75), making their molecular underpinning a striking phenomenon for further study, especially in pathogenesis.
Unsurprisingly, scientific studies have shown that Twist proteins are upregulated in carcinogenesis (76, 77) and metastasis (77). Apart from inhibiting the expression of the epithelial marker, Twist1 also increases the expression of mesenchymal markers, including vimentin, N-cadherin, and fibronectin, reducing cellular adhesion and promoting cellular motility (77, 78). In addition, Twist proteins generally promote cancer stem cell phenotype (79), which explains its association with poor prognosis.
By regulation, posttranslational modification of Twist proteins can impact their functions both positively and negatively; for instance, phosphorylation of Twist1 by MAP enhances its stability, promoting invasiveness and EMT in breast cancer cells (80). Contrastingly, IKKβ-mediated phosphorylation promotes the degradation of Twist proteins (81), providing an insight into possible regulation of EMT, which could further prevent tumour cell motility, invasion, and accordingly, cancer metastasis. Understanding the accumulation of Twist protein as a precursor for cancer metastasis, the “brake system” of this molecular purview renders IKKβ a potential novel target for future therapeutic design in the fight against cancer metastasis.
Apart from the TFs that have been studied extensively, previous studies have indicated other nonconventional TFs in the EMT process. For instance, goosecoid (GSC) represses E-cadherin expression indirectly (82) and has been found in various metastatic breast cancer (83). Also, Krüppel-like factor 8 (KLF8) enhances EMT induction through direct suppression of E-cadherin by regulating its promoter (84), resulting in improved motility and altered cell morphology, which has been implicated in breast, ovarian, and gastric cancer cell lines (84–87).
Additionally, another less-reported TF known as paired-related homeobox 1 (PRRX1) also plays important role in tumorigenesis and promotes tumour invasion. PRRX1 with two isoforms—PRRX1a and PRRX1b—exerts distinct functions in EMT. A report from recent studies shows crucial roles of PRRX1 via isoform switching during MET, tumour invasion, and metastasis of pancreatic malignancy (88). The study further suggests that PRRX1b isoform stimulates cellular de-differentiation, EMT initiation, and tumour invasiveness while the PRRX1a isoform could induce cell differentiation and the MET (88). Encoded in humans by the PRRX1 gene, the PRRX1 protein functions primarily act as a transcription coactivator, which enhances the DNA-binding activity of serum response factor that induces many genes by growth and differentiation factors.
Similarly, another superfamily of transcriptional regulators named forkhead box (FOX) proteins is emerging as important regulatory players in various gene expressions associated with cell growth, proliferation, and differentiation. Akin to the common TFs, some FOX proteins are also important drivers in embryonic development and biological shift in cellular phenotypes, which may be linked with maintenance of epithelial polarity via E-cadherin function.
In particular, a report in the literature shows that forkhead box C2 (FOXC2) induces EMT by indirect suppression of E-cadherin (89). In effect, FOXC2 plays a crucial role in embryogenesis and angiogenesis, among other physiological processes (90, 91). More importantly, FOXC2 contributes to the metastatic process via EMT activation in many cancer growths and developments involving cancers of the breast, prostate, and ovary (92–94). Furthermore, using human tumour cell lines, the investigation also reveals that suppression of FOXC2 by short hairpin RNA (shRNA) in an aggressive metastatic breast cancer model could halt the metastatic tendency (92) suggesting that FOCX2 is not only a promising molecular marker for cancer detection but also a potential therapeutic target. Currently, very few TFs remain “druggable.” Worse still, the existence of many TFs outside the nuclear receptor family poses difficulty in targeting those recognisable TFs with small molecule therapeutics, partly due to paucity of data. Therefore, it might be worthwhile to try out targeting the less-exploitable TFs such as GSC, KLF8, PRRX1, FOCX2, GRHL2, and other poorly investigated similar molecules considering their potential regulatory roles within various signalling pathways.
Despite being two clear-cut cellular states, epithelial and mesenchymal states can trans-differentiate into each other through EMT and its reverse, MET. This fluidity suggests that the regulatory loops among the transcription factors (TFs) involve complex interplays between EMT inducers and EMT suppressors. Several EMT promoters have been identified and elucidated earlier in this study; however, it is also important to discuss the roles of other factors with antimetastatic attributes.
In contrast to the EMT-inducing transcription factors, molecular mechanisms inhibiting EMT are inadequately characterised as of now. Meanwhile, as scientific efforts are intensifying to decipher the batteries of factors that regulate EMT, there seems to be some cheering news as emerging studies continue to provide evidence of possible spontaneous inhibition of EMT through some special transcription factors. These factors could be referred to as proepithelial factors or anti-EMT factors. Some of these factors include OVO-like transcriptional repressor (OVOL1, OVOL2), grainyhead-like transcription factor 2 (GRHL2), CCAAT-enhancer-binding protein alpha (C/EBPα), oestrogen receptor (ER), and p53. Their major role is to counterbalance the EMT-TFs thereby safeguarding the continued residence of cells in an epithelial state (93).
OVOL protein family, including OVOL1 and OVOL2, are the vertebrate homologs of Drosophila OVO and have been regarded as critical regulators of cellular transformation both in physiological and diseased states. Even though they are also members of the zinc-finger protein family, they are known as the anti-EMT transcription factors that promote the epithelial status. They are functionally important in the embryogenesis of vertebrates and for the maintenance of an epithelial state and terminal differentiation during tissue homeostasis (94). Mechanistically, OVOL1/2 can suppress EMT by direct inhibition of EMT-TFs, such as ZEB1, ZEB2, and TWIST. In addition, it promotes the reverse of EMT, that is, MET (95, 96), thereby inducing the expression of the cell-cell adhesion molecule called E-cadherin.
However, the extent of the reversal remains a conundrum and requires further investigations since the generation of hybrid EMT will amount to more aggressive malignancy and may portray OVOL proteins as spurious anti-EMT factors. Although the existence of varying hybrid EMT intermediates (as depicted in Figure 3) could explain different levels of invasiveness, plasticity, and migratory potential, a deeper understanding of how OVOL1 and OVOL2 promote epithelial differentiation and inhibit EMT in the context of different metastatic models, maybe rewarding to reliably label OVOL factors as useful therapeutic candidates. A report that seems to partially address this concern from Wu and colleagues revealed that OVOL2 thwarts TGF-β signalling and blocks EMT during breast tumour metastasis by repressing SMAD4 expression and interfering with SMAD4 and SMAD2/3 complex formation (97). More recently, Xu et al. proposed that OVOL1 coordinates the suppression of proliferation, invasion, and migration in oral squamous cell carcinoma cells by inhibiting ZEB1 expression via direct binding to its promoter (98). This further endorses the OVOL2/ZEB1 feedback loop as the controller of the epithelial-mesenchymal plasticity across several carcinomas.
Recently, mathematical modelling of the feedback circuit between ZEB1 and OVOL2 has revealed that in addition to epithelial and mesenchymal states, cells can acquire one or more hybrid epithelial/mesenchymal states (99). In other words, this has been considered the most plastic and aggressive state (99), hence reinforcing the concern about how beneficial OVOL could be as a novel therapeutic signature for overcoming tumour progression. While OVOL1 and OVOL2 have been described as gatekeepers that prevent mesenchymal trans-differentiation and maintain epithelial identity, their regulation is poorly understood.
OVOL1 or OVOL2 knockdown has been shown to significantly increase the mRNA levels of both vimentin and ZEB1. This led to an extensive functional study of OVOL proteins by Maho et al. (100). The study employed two precancerous conditions, actinic keratosis (AK) and cutaneous squamous cell carcinoma (cSCC) and showed that OVOL1 and OVOL2 were upregulated in AK and significantly downregulated in cSCC. While ZEB1 and vimentin were upregulated in cSCC, most AK cells were negative or faintly express the proteins, suggesting that downregulation of OVOL1/2 and upregulation of ZEB1 and vimentin may be associated with the progression of AK to cSCC. Taken together, this study provides abundant evidence to support the pro-MET features of OVOL factors and the importance of the OVOL2/ZEB1 axis in the maintenance of epithelial status. Thus, the OVOL1/2-ZEB1 axis can form an important axis of regulation of EMT in cancer progression.
The grainyhead-like (GRHL) family of transcription factors consist of three members, GRHL1, GRHL2, and GRHL3, which were first discovered in Drosophila melanogaster (101). Out of these, GRHL2 has been widely associated with neoplastic diseases. The roles of GRHL2 in tumour pathogenesis seem to be complex and controversial, varying with cancer type (102). Recently, the signalling pathways between EMT and GRHL2 have attracted considerable attention from researchers. In different cancer models, upregulation of GRHL2 expression has been directly correlated with lower EMT scores; whereas, cancers with mesenchymal features have reduced GRHL2 expression. In cultured human colorectal cancer cells, GRHL2 upregulation promotes epithelial states by reversing the epithelial-like shape from a spindle-like shape and increases E-cadherin, β-catenin, and ZO-1, while vimentin is significantly downregulated (103).
A recent study discovered that TGF-β-induced EMT is inhibited by GRHL2, preventing invasion and migration of gastric cancer. In turn, inhibition of TGF-β signalling pathways increased GRHL2 expression (104). Similarly, GRHL2 significantly inhibits TGF-β-induced, Twist-induced, and spontaneous EMT in breast cancer (105). Further mechanistic studies reveal that GRHL2 is directly inhibited by ZEB1, which in itself, a direct target for repression by GRHL2 (106), suggesting that GRHL2 and ZEB1 form a double-negative regulatory feedback loop in breast cancer cells.
The p53 tumour suppressor, also known as the “genome guardian,” plays a dominant role in maintaining genome stability and protects the DNA against mutagenesis (107). p53 plays diverse tissue regulatory roles through several mechanisms. It can be regarded as a transcription factor that acts in response to various stress cues, causing cell cycle arrest, cell ageing, and apoptosis. In addition, p53 plays a vital role in controlling the metabolism and antioxidant status of cells (108). Beyond this, p53 has been associated with anti-EMT roles, by restricting the plasticity of epithelial cells during EMT.
Essentially, p53 indirectly promotes M→E by attenuating EMT-TFs (109) via the upregulation of EMT-suppressing miRNAs. Thus, p53 has the potential to avert EMT and the associated stem cell-like phenotype across multiple cancers. Categorically, p53 induces the expression of miR-200c, miR-183, and miR-34, which target ZEB, SNAIL, and TWIST families of transcription factors (110). In other words, apart from maintaining genome integrity, a functioning p53 gene would also be crucial for the maintenance of epithelial integrity.
The transcription factor CCAAT enhancer-binding protein α (C/EBPa) is a widely expressed basic leucine zipper transcription factor that plays a critical role in cellular differentiation (111). It plays a pivotal role in the regulation of the cell cycle and the expression of several lineage-specific genes (112). Recent findings provided persuasive proof that C/EBPα is an important transcription factor required to sustain epithelial architecture of human mammary cells, preventing epithelial-to-mesenchymal transition and thereby acting as a repressor of breast cancer progression in vivo (113). C/EBPα factor can activate and represses several target genes, and its roles in the maintenance of epithelial traits have been attributed to its direct transcriptional activation of epithelial markers, such as CDH1 (E-cadherin) and suppression of EMT-TFs such as ZEB1 (114). This is consistent with previous data on hepatocellular carcinoma, where C/EBPα is shown to be a critical negative regulator of TGF-β-induced EMT, promoting inhibition of N-cadherin and maintenance of E-cadherin expression (114).
Since CEBPA mRNA levels were instantly suppressed upon TGF-β treatment, Ana et al. (113) proposed that SMAD3 may be responsible for the repression of CEBPA transcription. This sounds logical since SMAD3 was found to occupy the CEBPA locus upon TGF-β treatment. Depletion of SMAD3, owing to impaired TGF-β-mediated repression of CEBPA, supports a downstream regulatory role for SMAD3 as a transcriptional repressor of C/EBPα expression during TGF-β-induced EMT.
The oestrogen receptor, especially the alpha class, (ERα) plays a cardinal role in blocking the EMT process (Figure 2). For example, metastasis-associated protein 3 (MTA3), directly activated by ERα, downregulates Snail expression (115). Also, ERα directly inhibits Slug transcription by forming a corepressor complex consisting of ligand-activated ERα, histone deacetylase 1 (HDAC1), and nuclear receptor corepressor (NCoR) that binds to the oestrogen-response elements at the Slug promoter sequence (115, 116).
Therefore, the absence of ER or MTA3 results in abnormal expression of Snail and Slug diminishing the ER activity to promote metastasis, chemoresistance, and recurrence after treatment (117, 118). These findings justify why ER is regarded as a marker of poor clinical outcomes and an important therapeutic target in breast cancer (119). Thus, through many significant molecular mediations in connection with tumorigenesis, EMT, and metastatic cascade, it might be possible to develop a novel drug targeting the associated ER-signalling components in women breast cancer intervention.
As stated earlier, EMT-TFs are regulated by various intracellular signalling networks (Figure 2). It starts with the extracellular molecules binding to its particular membrane receptor to initiate intracellular signal transduction. The transforming growth factor, wnt, oestrogen, fibroblast growth factor (FGF), PDGF, and the Jagged family bind to TGF-β receptor (TGFβR), Frizzled, oestrogen receptor (ER), fibroblast growth factor receptor (FGFR), platelet-derived growth factor receptor (PDGF-R), and Notch, respectively (120–122). The ligand-bound receptors transduce intracellular signals via the pathways, such as MAPK, phosphatidylinositol 3-kinase (PI3K)/protein kinase B (Akt), nuclear factor-κB (NF-kB), β-catenin, or the Smad signalling, which regulate the expression and stability of EMT-TFs (121) (Figure 2).
Previous findings have shown that these networks of signalling interface at various strata and numerous feedback activation and inhibition mechanisms have been illustrated in different EMT contexts, with the possibility of showing overlapping and context-specific results (123, 124). This highlights the importance of delineating the induction of these pathways in various subpopulations of cancer cells following EMT activation to understand possible therapeutic targets in various human carcinomas.
Notably, the dominant roles of TGF-β in EMT must be acknowledged. It is a well-elucidated molecule that induces EMT in various cellular contexts, cancer inclusive. The functions of TGF-β in cancer vary from one biological setting to another, deciphering its dual relevance in tumour pathogenesis, which has been called the “TGF-β paradox” (125). In the early tumour stage, TGF-β supports apoptosis and inhibits the proliferation of tumour cells. Conversely, it plays a tumour-promoting role in the later stage by stimulating EMT, genomic instability, angiogenesis, cell motility, and metastasis.
Following successful binding to its receptor (TGF-βR), it activates several signalling cascades, including Ras-MAPK and SMAD-dependent signalling. Activation of the RAS-MAPK induces expression of Snail1 and Snail2, ultimately leading to the repression of E-cadherin (103). TGF-β can also initiate gene expression through activation of the Notch, and Wnt pathways, which then lead to activation of Snail1 and Snail2, expression of mesenchymal markers, and EMT induction via degradation of intracellular catenins (33) (Figure 2).
In addition to ligand-receptor interactions, environmental signals such as oxygen reduction in the cellular environment can as well directly activate TGF-β through hypoxia-inducible factor 1 alpha (HIF-1α) (126) which in turn leads to gene transcription. More so, HIF-1α can initiate epigenetic modulation of EMT by targeting the transcription of histone deacetylase 3 genes (HDAC3), cooperating with Snail and ZEB expressions to suppress epithelial expression (127), with the imposition of mesenchymal characteristics.
Furthermore, there is compelling evidence in recent years to support the significant role of Notch in the EMT process (128–130). This is not surprising because the pathway is the nexus of an adaptable signalling network that controls various cellular mechanisms in different biological contexts—determined by the tissue microenvironment (131, 132). Notch signalling explores two discrete channels to synergistically regulate the expression of EMT-TFs (133). Firstly, direct activation of TFs by mobilising the Notch intracellular domain (NICD) to their promoter and secondly, by potentiating HIF-1α recruitment to the lysyl oxidase (LOX) promoter to aid the hypoxia-induced upregulation of LOX, which stabilises Snail and safeguarding it from protein degradation (134). Importantly, the signals generated from this pathway usually stimulate transcriptional repressors (Snail1/2, ZEB1/2, and TWIST1/2) to promote and regulate EMT.
Fortunately, studies have proven that inhibition of Notch signalling by the small interfering RNA (siRNA) system promotes reversal of the EMT phenotype, resulting in the MET by inhibiting ZEB1, Slug, Snail, and NF-κB (135). These data present molecular evidence connecting Notch signalling with neoplastic drug resistance validated by EMT phenotype. This suggests that inactivation of Notch signalling with a novel targeted therapeutic approach could be conceived to subdue chemoresistance toward preventing tumour progression, for which present traditional therapeutic strategies are grossly dissatisfying.
Interestingly, Snail could also regulate the Notch expression. Kuphal et al. (136), for example, found downregulation of Notch-4 by antisense Snail cDNA transfection of melanoma cells, and the results are convincing in support of the complex crosstalk between Notch and Snail during the acquisition of EMT. These findings indicate the complex contribution of different TGF-β to EMT in carcinoma cells and, therefore, represent an intimidating obstacle in formulating a therapeutic model to control the EMT in human tumours.
MicroRNAs (miRNAs) are a small class of noncoding RNAs that modulate gene expression by RNA silencing, translational suppression, and mRNA degradation (137). They are involved in many biological processes involving cell differentiation, proliferation, cell death, and tumour invasion (138). Another report shows that miR-200 and miR-205 members can mediate EMT suppression by direct inhibition of ZEB1 and ZEB2, thus enhancing the tumour sensitivity to therapeutic interventions (139). In addition, the expression of miR-204/miR-29b demonstrates the blockage of metastasis and cancer invasion considerably in gastric cancer cell lines via Snail1-induced EMT suppression (140). Unsurprisingly, however, the low expression level of the miR-200 family in breast cancer enhances ZEB1/ZEB2 transcriptions, inducing TGF-β/BMP signalling to sustain EMT (141). Also, Kim et al. (110) reported that p53 negatively modulates EMT in cancer of the liver upregulating the miR-200 and miR-192 expression by targeting inhibition of ZEB1/ZEB2 expression.
Activation of p53 targets many microRNAs—miR-200c, miR-183, and miR-34—by inducing their expressions. This process, in turn, downregulates some EMT-TFs such as ZEB and Snail. Consequently, the chain reaction in the signalling cascade directly results in EMT suppression and thus MET activation. In essence, induction of TGF-β, mutations, or epigenetic silencing which may cause loss of p53 could ultimately stimulate the EMT process (142).
As mentioned earlier in this review, EMT was traditionally considered a binary model and that any intermediate state is just a transient snapshot acquired during the EMT process (143). However, our understanding has recently evolved through a plethora of scientific studies, revealing that cancer cells can acquire metastable intermediate transition, with a concoction of cells displaying the features of E or M phenotype or both (E/M) at the molecular and/or morphological level. While several authors have referred to these intermediary states as partial/hybrid EMT (pEMT) (144), some set of scholars (145) have consistently used “quasi-mesenchymal cells” because those cells express CDH1 gene (which encodes E-cadherin) at the transcript level without displaying E-cadherin at the cell surface (146), in addition to stem-like features and coexpression of certain epithelial and mesenchymal genes.
By studying the plasticity of EMT/MET through transitional states, investigations show a range of pEMT intermediates (30). Indeed, EMT is beyond the binary state; that EMT trans-differentiation phases exist as a spectrum of various intermediate hybrid states raise a big concern for cancer cell stemness and failure in many therapeutics. Although, there is a strong evidence to support that hybrid EMT/pEMT state is associated with increased metastatic tendency, hitherto, it is not understood whether each pEMT intermediate subpopulations have different cellular properties that may dictate their relative responses to treatment with immunotherapy, radiotherapy, and/or chemotherapy. Also, the precise mechanism that drives each transitional state through many intermediate phases is yet unknown. More so, the actual number of phases (here termed “n”, representing a definite number of transitional phases; Figure 3) each cancer phenotype undergoes from pure epithelial to mesenchymal state is still beyond the current understanding of EMT. By implication, tumours with pEMT may exhibit increased intratumour heterogeneity.
Although, all EMT-induced states possess aggressive features, those carcinoma cells in the pEMT state have demonstrated greater tumour-initiating and apoptotic potential as well as therapeutic resistance than purely epithelial or mesenchymal cells (147). Similar observations were made in a mouse model of prostate cancer, where both hybrid epithelial/mesenchymal and fully mesenchymal carcinoma cells were shown to initiate primary tumours, but the fully mesenchymal cells were unable to generate macroscopic metastases, while the hybrid cells were capable of doing so (148). Corroborating this finding, another study by Cornelia et al. (149) employed expression vectors and gene knockout models to establish that malignant cells that exist in the hybrid E/M state have highly tumorigenic features but lack plasticity. Instead, they were locked in the E/M state and thereby unable to transit into more E or more M states spontaneously, suggesting that residence in a hybrid E/M state is enough for the preservation of stem cell properties and the associated stemness is thus exhibited regardless of phenotypic plasticity.
More also, cells in pEMT state exhibit loss of apical-basal polarity and have better motility, while retaining adhesive features with the nearby cells and acquiring mesenchymal-like characteristics (144); hence, they can form cell clusters and move together. If these cell bundles are lucky to successfully emigrate to the bloodstream intact, they form clusters of CTCs that can migrate collectively.
Interestingly, hybrid cells coexpressing epithelial and mesenchymal markers have been detected among CTC clusters in the bloodstream of patients with different tumour types, including breast, lung, colon, and prostate cancer (150), thereby complicating our quest to understand the composite milieu of EMT spectrum. Tayoun and his colleagues reported that clustered CTCs have advanced plasticity, possess CSC characteristics and the ability to initiate tumours and form new lesions and are fatal in tumour patients (151). While a few factors that promote CTC cluster formation have been identified (152), the key mechanisms that allow CTC clusters to survive in the vascular system and allow them to metastasise effortlessly than freely circulating CTCs remain contestable.
However, it is convincing to predict that malignant cells that occupy pEMT locus along the EMT spectrum may be the most suitable for metastasis because CTCs that migrate collectively rather than singly may offer superior resistance against the onslaught of the protective machinery within the blood circulations, conforming with the axiom “united we stand, divided we fall!” Interestingly, an earlier study demonstrated that CTC clusters contribute to 50% of the total cells at the secondary metastatic niche despite only constituting 3% of the total CTCs (153). Reinforcing this discovery, Joosse et al. predicted that CTC aggregates have a high probability of being trapped in narrow blood vessels, hence facilitating extravasation (154).
In essence, the fact that neoplastic cells that occupy the pEMT state circulate in clusters, with a greater tendency to become CSC (155), make them behave more aggressively than their corresponding mesenchymal or epithelial counterparts. The mechanism by which pEMT regulates stemness in squamous cell carcinoma (SCC) is still largely unknown, although a study has proposed that clustered CTCs display binding sites for stemness-associated factors such as OCT4, NANOG, and SOX2 are more hypomethylated (155).
There is overwhelming evidence connecting EMT with tumour progression and metastasis and sometimes drug resistance, with transcription factors as the key players. Currently, emerging knowledge of cellular plasticity and the continuous spectrum of EMT transitional phases in metastasis establish the presence of multiple tumour cell subpopulations. Importantly, the translational significance of the interrelated events poses huge implications in the orbit of cancer treatment and other important intervention strategies. Here, we bring to the limelight some important approaches to circumvent the drawbacks facing the current cancer management, and further, provide how advancing the understanding of EMT transition beyond the binary model could shape the diagnostic and therapeutic strategies with a novel approach, as follows:
● Reversing the EMT to its opposite state, i.e., MET.
● Inhibiting the EMT initiation.
● Unveiling the prospect of single-cell analysis in EMT.
Reversing the EMT is a potential therapeutic approach involving a forced stimulation of MET, which in principle should be an exquisite way to prevent EMT [121]. However, the molecular basis of this process is only beginning to gain recognition in recent time compared with the EMT pathway, and therefore it may take a more extended period for a rational design of modulators of this direction to be explored. Mechanistically speaking, interfering EMT transition is crucial to the prevention of metastasis. Importantly, to achieve EMT→MET reversion, tumour staging may be a central point to consider. For instance, at early metastatic stages, Twist1 contributes to tumour invasion and vascular intravasation through repression of E-cadherin expression. Interestingly, however, epithelial cells that predominate metastatic lesions suggest that EMT→MET reversal is also fundamental to circulating tumour cells (CTCs) in colonising a new metastatic niche at distal organs, attributable to loss of Twist1 signal—the same signal that orchestrates tumour invasion and vascular intravasation at early metastatic stages. Without mincing words, the molecular mechanisms that dictate on-and-off switches of EMT-TFs as observable in Twist1 signal at different metastatic phases have received low attention. Thus, the connecting pathway involving Twist1 expression should be further explored, understanding that Twist1 and other associated downstream regulators could be exploitable in therapeutic designs for EMT→MET reversion, particularly at an early stage of tumour development.
Similar to the spatiotemporal signal programming in Twist1 expression in different metastatic phases, microRNAs equally regulate MET transition via down-regulation of many EMT-TFs such as ZEB1 and ZEB2 and Snail2 (Slug). In reverse, many EMT-TFs inhibit the transcription of microRNAs. In addition, microRNAs function in concert with the p53 to modulate the EMT pathway. Importantly, some molecular interactions involving the inactivation of p53 have connections with the downregulation of microRNAs. Inquisitively, the interconnections of p53, microRNA transcriptions, epigenetic mutation, and other associated factors remain an enticing area for further exploration as a possible target for induction of the EMT→MET pathway and potential therapeutic manipulations.
Inhibition of EMT in the early steps of invasion and metastasis, which perhaps promises to be the most cost-effective approach, has been the talking point in the scientific community of late. As earlier highlighted, the interplay and crosstalk between TFs and other signalling molecules orchestrate and mandate adjacent epithelial tumour cells to undergo EMT, leading to tumour progression, invasiveness, metastasis, and therapeutic resistance in some cancers. These TFs act as downstream effector molecules for the cascade. Thus, breaking the crosstalk between TFs and these pathways is a reasonable option for arresting tumour cells from metastasising and limiting drug resistance. Although these factors were for a long time considered “undruggable” targets (156), a deeper understanding of their specific expression pattern, degradation, mode of binding, protein/protein interaction, as well as interaction with other signalling pathways has changed this narrative. Additionally, it has also opened new possibilities to affect transcription factors as therapeutic targets for cancer treatment. Furthering the exploits through another perspective, signalling crosstalk, understanding that molecular modulators in one pathway are enzymatic (transcriptional) targets of another that sometimes compete with the original modulators, the undruggable bottleneck could end in a breakthrough.
Among the promising opportunities to indirectly or directly target a transcription factor are inhibition (or activation) at the expression level; inhibition through physical degradation; inhibition (or activation) at the protein interaction level; and inhibition (or activation) through the binding of a ligand-based molecule in an activation/inhibition pocket and inhibition (or activation) at the protein/DNA binding level. For example, when Snail was blocked in a high-grade serous ovarian cancer cell line (HGSOC), cancer stemness was reversed by decreasing CSC markers (CD117 and CD133) (157).
Similarly, Zhang et al. demonstrated that Snail knockout in MCF-7 breast cancer cells initially resistant to 5-FU resulted in reversal of EMT declined cell invasion and improved sensitivity to 5-FU (158). Consequent to the various studies about Snail and the correlation with chemoresistance and cancer stem cells, targeting Snail is a fascinating approach to overcome EMT and cancer resistance, which suggests that Snail inhibitors could prevent tumour recurrence. Inactivation of Twist1 expression by linolenic acid (ALA) treatment markedly reduces cell migration (159). Recently, a small molecule, CYD19, was identified to bind strongly with Snail and inhibited its interaction with CREB-binding protein (160). The altered interaction leads to impairment of CBP/p300-mediated Snail acetylation and rapid degeneration of Snail via the proteasomal pathway.
Furthermore, studies have shown that inhibition of γ-secretase would prevent the cleavage of the Notch receptor, thereby blocking Notch signal transduction (161, 162). In this way, the expression of Snail and ZEB proteins are kept in check so that E-cadherin retains its function to maintain epithelial characteristics. Thus, γ-secretase inhibitors can be clinically tried for treating human malignancies. Disappointingly, however, despite the repeated promising preclinical results on the control of tumour progression, gamma-secretase inhibitors (GSIs) have failed to demonstrate clinical benefit in most solid tumours. The poor clinical performance to date calls for important questions that are yet to be answered. Perhaps, refinement of tumour Notch expression profiles and further mechanistic understanding of GSIs will necessarily assist appropriate patient selection.
Also, more innovative approaches may include the rational design of combinatorial strategies to maximise the potential of these agents by sensitising tumours to traditional chemotherapeutics while also compromising tumour ability to engage treatment resistance programs. However, it is worthy of note here that caution should be applied while designing GSIs to eliminate undesired toxicity associated with the γ-secretase inhibitors, as they are involved in a wide array of cellular functions.
Even though, as earlier suggested, interfering EMT transition might be a step forward in the prevention of cancer metastasis, intratumour heterogeneity poses several hurdles against successful therapeutic design. As a result, understanding the E/M cellular transitional plasticity along a phenotypic spectrum would be central to more precise diagnoses and increasing the susceptibility of cancer cells to therapeutic manipulations, thus overcoming the current spate of drug resistance.
Several studies dispute the role of EMT in cancer metastasis as a result of their failure to detect EMT in the process, thus suggesting a striking controversy in cancer biology. Worse still, the use of individual EMT biomarkers from the bulk-tissue approach has repeatedly failed to answer the question of where gene upregulation or downregulation arises among the heterogeneous components involving malignant cells, the surrounding stroma and immune infiltrate. Thus, a method that can offer a direct and unbiased characterisation of individual cell’s (gene) expression to unveil intratumoural heterogeneity would therefore provide clarity over the age-old questions, and perhaps, paves way for targeted therapy. Therefore, the roles of novel technologies such as single-cell analysis would be invaluable to improve understanding of the underlying molecular mechanisms of tumour heterogeneity and investigation of diverse intermediates of E/M cells during EMT.
Intriguingly, it is now widely established that trans-differentiation of E/M cells and their intermediates are the main cause of intratumour heterogeneity and that the disseminated cancer cells need to revert towards a more epithelial phenotype to increase the sensitivity of neoplastic cells to therapeutic interventions. To therapeutically target EMT and indeed, the metastable pEMT state, it is essential to extensively evaluate the duplex modulations that shuttle cells towards or away from E and M states. Presumably, the bidirectional inhibitions that targets both EMT and MET might be an effective strategy to destabilise the M or hybrid states, while avoiding an escape route to a different resistant state. With this in mind, we anticipate the next few decades will represent a major acceleration in our knowledge of EMT and its entire spectrum of states. This is expected to aid in the stratification of patients into low- and high-risk cohorts while opening up entirely new avenues in the treatment of epithelial tumours.
Given the considerable heterogeneity of the tumour ecosystem, it is essential to analyse the molecular and genetic characteristics of each cell (163), rather than employing the bulk-tissue approaches. Fortunately, the emergence of single-cell sequencing technology has come to the rescue, enabling cells undergoing a transition to be inspected as single cell instead of collective cell populations. To simply put, with single-cell analysis, it would be possible to obtain complete genomic information of the entire cells concerning EMT gene regulatory circuits along the EMT continuum and compare them with the corresponding primary tumours. Through this, several clinically relevant genomic alterations could be discovered to provide valuable information for promising diagnostic and therapeutic opportunities. Specifically, single-cell RNA sequencing (ScRNA-seq), which provides high-throughput and high-resolution transcriptomic analyses of individual cells has been used to eliminate much of the background noise within a mixed population of cells. With a temporal profile, it will allow investigators to probe the genomic, transcriptomic, epigenomic, or other multiomics dynamics of heterogeneous cell populations because it can measure the distribution of genes and gene products (mRNA and proteins) from individual cells. With scRNA, one can encapsulate cell trajectories and developmental processes such as EMT by applying a scRNA-seq time course to assemble a cell trajectory map (164). Generating an EMT time-course to capture transient cell states at single-cell resolution informs the investigator with information on how this dynamic process occurs over time, thereby providing an elegant resource that is not available in any other known way. ScRNA-seq method is a recently developed technology with a promising prospect at providing a transcriptomic analysis of individual cells to address the inherent complexity of EMT milieu in tumour metastasis and its tumour environment. Despite the promising prospect of this novel method, some technical barriers have been identified.
Firstly, both cellular integrity and viability largely determines the success or otherwise of the subsequent single-cell analyses. It means that distinct cells need to be isolated from each other promptly while avoiding mechanical damage to the cells during the single-cell isolation process. However, this cellular segregation may impair cell integrity and enzymatic treatment using trypsin, collagenase, and/or papain to isolate single cells from tissues. In other words, it may affect cell viability or alter the transcriptional products. Hence, this necessitates the need for improve approaches to enable the efficient “gentle” extraction and capture of living cells and avoid the potential damage to single cells caused by enzymatic treatment.
Again, researchers have used scRNA-seq to analyse the association between therapeutic response and specific infiltrated immune cells in the tumour environment (165). At this point, another gap to be filled by ScRNA-seq is to enable rationale strategies that can identify drug-resistant cell populations associated with poor prognosis to achieve long-term treatment efficacy and combinatorial therapeutics that can aid in cotargeting multiple activated pathways.
There is overwhelming evidence that the dynamic EMT program, which oversees the transformation of polarised epithelial cells to a mesenchymal phenotype in embryogenesis, is also operational in the development of the secondary metastatic niche. Hitherto, the molecular event is understood as a consequent disruption of the tight regulation in morphogenesis, where transcription factors play significant roles. Unfortunately, the EMT program, primarily driven by transcriptional repressors, is not only involved in tumour metastasis but has now been discovered as a molecular driver in drug resistance. To date, the problems of drug resistance and pharmacological limitation of tumour targeted therapy remain a significant challenge in cancer control interventions. Meanwhile, in recent times, several EMT transcriptional repressors have been inculpated in the metastatic cascade. As research begins to gain momentum, it is becoming increasingly clear that these factors would play considerable roles in targeted therapeutic strategies in the fight against cancer metastasis as well as cancer resistance to therapies. Thus, while it is currently clear that the EMT program is beyond the binary switch concept, we believe that a deeper understanding of the emerging roles of cellular plasticity which adapts tumour cell phenotypes through a metastable spectrum would also require a paradigm shift in investigational approach to upscale the common bulk-tissue methods to novel techniques such as single-cell analysis to address the lingering challenges in cancer management.
SI conceptualised and designed the study. KA designed the images. Both SI and KA developed the first draft, refined, edited, and revised the manuscript. IB formatted the references and contributed to the final editing. AM, MM, TO, and MO contributed to the content materials and final editing. All the authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
bHLH, basic helix-loop-helix; CSC, cancer stem cell; CNS, central nervous system; CTC, circulating tumour cell; cDNA, complementary deoxyribonucleic acid; DNA, deoxyribonucleic acid; EGF, epidermal growth factor; EMT, epithelial-mesenchymal transition; ER, oestrogen receptor; FGF, fibroblast growth factor; FGFR, fibroblast growth factor receptor; FOX, Forkhead box; FOXC2, Forkhead box C2; GSI, gamma-secretase inhibitor; GSC, goosecoid; GRHL2, grainy head-like 2; HTH, helix-turn-helix; HDAC3, histone deacetylase 3; HIF-1α, hypoxia-inducible factor-1 alpha; KLF8, Krüppel-like factor 8; LOX, lysyl oxidase; MMP, matrix metalloproteases; MET, mesenchymal-epithelial transition; MTA3, metastasis-associated protein 3; MAPK, mitogen-activated protein kinase; NICD, Notch intracellular domain; NES, nuclear export sequence; NF-KB, nuclear factor-KB; NCoR, nuclear receptor corepressor; PRRX1, paired-related homeobox 1; PI3K, phosphatidylinositol 3-kinase; PDGF, platelet-derived growth factor; PDGFR, platelet-derived growth factor receptor; RNA, ribonucleic acid; SRD, serine-rich domain; shRNA, short hairpin ribonucleic acid; siRNA, small interfering ribonucleic acid; TF, transcription factor; TGF-β, tumour growth factor-β; TGF-βR, tumour growth factor-β receptor; ZEB, zinc-finger E-box binding; scRNA-seq, single-cell RNA sequencing; pEMT, partial epithelial-mesenchymal transition.
1. Marconi GD, Fonticoli L, Rajan TS, Pierdomenico SD, Trubiani O, Pizzicannella J, et al. Epithelial-Mesenchymal Transition (EMT): The Type-2 EMT in Wound Healing, Tissue Regeneration and Organ Fibrosis. Cells (2021) 10(7):1587. doi: 10.3390/cells10071587
2. Weiskirchen R, Weiskirchen S, Tacke F. Organ and Tissue Fibrosis: Molecular Signals, Cellular Mechanisms and Translational Implications. Mol Aspects Med (2019) 65:2–15. doi: 10.1016/j.mam.2018.06.003
3. Krebs AM, Mitschke J J, Lasierra Losada M, Schmalhofer O, Boerries M, Busch H, et al. The EMT-Activator Zeb1 Is a Key Factor for Cell Plasticity and Promotes Metastasis in Pancreatic Cancer. Nat Cell Biol (2017) 19:518–29. doi: 10.1038/ncb3513
4. Arvelo F, Sojo F, Cotte C. Tumour Progression and Metastasis. Ecancermedicalscience (2016) 10:617. doi: 10.3332/ecancer.2016.617
5. Motohara T, Masuda K, Morotti M, Zheng Y, El-Sahhar S, Yi Cho C, et al. An Evolving Story of the Metastatic Voyage of Ovarian Cancer Cells: Cellular and Molecular Orchestration of the Adipose-Rich Metastatic Microenvironment. Oncogene (2019) 38(16):2885–98. doi: 10.1038/s41388-018-0637-x
6. Seager RJ, Hajal C, Spill F, Kamm RD, Zaman MH. Dynamic Interplay Between Tumour, Stroma and Immune System can Drive or Prevent Tumour Progression. Converg Sci Phys Oncol (2017) 3:034002. doi: 10.1088/2057-1739/aa7e86
7. Mrozik KM, Blaschuk OW, Cheong CM, Zannettino AC, Vandyke K. N-Cadherin in Cancer Metastasis, Its Emerging Role in Haematological Malignancies and Potential as a Therapeutic Target in Cancer. BMC Cancer (2018) 18(1):939. doi: 10.1186/s12885-018-4845-0
8. Deshmukh AP, Vasaikar SV, Tomczak K, Tripathi S, den Hollander P, Arslan E, et al. Identification of EMT Signaling Cross-Talk and Gene Regulatory Networks by Single-Cell RNA Sequencing. Proc Natl Acad Sci USA (2021) 118:e2102050118. doi: 10.1073/pnas.2102050118
9. Jung HY, Fattet L, Tsai JH, Kajimoto T, Chang Q, Newton AC, et al. Apical-Basal Polarity Inhibits Epithelial-Mesenchymal Transition and Tumour Metastasis by PAR-Complex-Mediated SNAI1 Degradation. Nat Cell Biol (2019) 21(3):359–71. doi: 10.1038/s41556-019-0291-8
10. Lu W, Kang Y. Epithelial-Mesenchymal Plasticity in Cancer Progression and Metastasis. Dev Cell (2019) 49(3):361–74. doi: 10.1016/j.devcel.2019.04.010
11. Zhang N, Ng AS, Cai S, Li Q, Yang L, Kerr D. Novel Therapeutic Strategies: Targeting Epithelial-Mesenchymal Transition in Colorectal Cancer. Lancet Oncol (2021) 22(8):358–68. doi: 10.1016/S1470-2045(21)00343-0
12. Fan X, Jin S, Li Y, Khadaroo PA, Dai Y, He L, et al. Genetic And Epigenetic Regulation Of E-Cadherin Signaling In Human Hepatocellular Carcinoma. Cancer Manag Res (2019) 11:8947–63. doi: 10.2147/CMAR.S225606
13. Otake S, Itoh Y, Omata C, Saitoh M, Miyazawa K. ZEB1 and Oncogenic Ras Constitute a Regulatory Switch for Stimulus-Dependent E-Cadherin Downregulation. Cancer Sci (2021) 112(1):205–16. doi: 10.1111/cas.14701
14. Wang Y, Zhou BP. Epithelial-Mesenchymal Transition-A Hallmark of Breast Cancer Metastasis. Cancer Hallm (2013) 1:38–49. doi: 10.1166/ch.2013.1004
15. Jayachandran J, Srinivasan H, Mani KP. Molecular Mechanism Involved in Epithelial to Mesenchymal Transition. Arch Biochem Biophys (2021) 710:108984. doi: 10.1016/j.abb.2021.108984
16. Aban CE, Lombardi A, Neiman G, Biani MC, La Greca A, Waisman A, et al. Downregulation of E-Cadherin in Pluripotent Stem Cells Triggers Partial EMT. Sci Rep (2021) 11(1):2048. doi: 10.1038/s41598-021-81735-1
17. Na TY, Schecterson L, Mendonsa AM, Gumbiner BM. The Functional Activity of E-Cadherin Controls Tumor Cell Metastasis at Multiple Steps. Proc Natl Acad Sci USA (2020) 117(11):5931–7. doi: 10.1073/pnas.1918167117
18. Vafaizadeh V, Buechel D, Rubinstein N, Kalathur KR, Bazzani L, Saxena M, et al. The Interactions of Bcl9/Bcl9L With β-Catenin and Pygopus Promote Breast Cancer Growth, Invasion, and Metastasis. Oncogene (2021) 40:6195–209. doi: 10.1038/s41388-021-02016-9
19. Roche J. The Epithelial-To-Mesenchymal Transition in Cancer. Cancers (Basel) (2018) 10:79. doi: 10.3390/cancers10030079
20. Vasaikar SV, Deshmukh AP, den Hollander P, Addanki S, Kuburich NA, Kudaravalli S, et al. EMTome: A Resource for Pan-Cancer Analysis of Epithelial-Mesenchymal Transition Genes and Signatures. Br J Cancer (2021) 124:259–69. doi: 10.1038/s41416-020-01178-9
21. Imani S, Hosseinifard H, Cheng J, Wei C, Fu J. Prognostic Value of EMT-Inducing Transcription Factors (EMT-TFs) in Metastatic Breast Cancer: A Systematic Review and Meta-Analysis. Sci Rep (2016) 6:28587. doi: 10.1038/srep28587
22. Li L, Qi L, Liang Z, Song W, Liu Y, Wang Y, Sun B, et al. Transforming Growth Factor-β1 Induces EMT by the Transactivation of Epidermal Growth Factor Signaling Through HA/CD44 in Lung and Breast Cancer Cells. Int J Mol Med (2015) 36:113–22. doi: 10.3892/ijmm.2015.2222
23. Nwabo Kamdje AH, Takam Kamga P, Tagne Simo R, Vecchio L, Seke Etet PF, Muller JM, et al. Developmental Pathways Associated With Cancer Metastasis: Notch, Wnt, and Hedgehog. Cancer Biol Med (2017) 14(2):109–20. doi: 10.20892/j.issn.2095-3941.2016.0032
24. Padilla-Rodriguez M, Parker SS, Adams DG, Westerling T, Puleo JI, Watson AW, et al. The Actin Cytoskeletal Architecture of Estrogen Receptor Positive Breast Cancer Cells Suppresses Invasion. Nat Commun (2018) 9(1):2980. doi: 10.1038/s41467-018-05367-2
25. Hamidi H, Ivaska J. Every Step of the Way: Integrins in Cancer Progression and Metastasis. Nat Rev Cancer (2018) 18(9):533–48. doi: 10.1038/s41568-018-0038-z. Erratum in: Nat Rev Cancer (2019) 19(3):179.
26. Schlesinger M. Role of Platelets and Platelet Receptors in Cancer Metastasis. J Hematol Oncol (2018) 11(1):125. doi: 10.1186/s13045-018-0669-2
27. Yang J, Antin P, Berx G, Blanpain C, Brabletz T, Bronner M, et al. Guidelines and Definitions for Research on Epithelial-Mesenchymal Transition. Nat. Rev. Mol Cel Biol (2020) 21:341–52. doi: 10.1038/s41580-020-0237-9
28. Tripathi S, Xing J, Levine H, Jolly MK. Mathematical Modeling of Plasticity and Heterogeneity in EMT. Methods Mol Biol (2021) 2179:385–413. doi: 10.1007/978-1-0716-0779-4_28
29. Carstens JL, Yang S, Correa de Sampaio P, Zheng X, Barua S, McAndrews KM, et al. Stabilized Epithelial Phenotype of Cancer Cells in Primary Tumors Leads to Increased Colonization of Liver Metastasis in Pancreatic Cancer. Cell Rep (2021) 35:108990. doi: 10.1016/j.celrep.2021.108990
30. Pastushenko I, Brisebarre A, Sifrim A, Fioramonti M, Revenco T, Boumahdi S, et al. Identification of the Tumour Transition States Occurring During EMT. Nature (2018) 556:463–8. doi: 10.1038/s41586-018-0040-3
31. Diepenbruck M, Christofori G. Epithelial-Mesenchymal Transition (EMT) and Metastasis: Yes, No, Maybe? Curr Opin Cell Biol (2016) 43:7–13. doi: 10.1016/j.ceb.2016.06.002
32. Mittal V. Epithelial Mesenchymal Transition in Tumor Metastasis. Annu Rev Pathol (2018) 13:395–412. doi: 10.1146/annurev-pathol-020117-043854
33. Chen HT, Liu H, Mao MJ, Tan Y, Mo XQ, Men XJ, et al. Crosstalk Between Autophagy and Epithelial-Mesenchymal Transition and Its Application in Cancer Therapy. Mol Cancer (2019) 18:101. doi: 10.1186/s12943-019-1030-2
34. Xie F, Ling L, van Dam H, Zhou F, Zhang L. TGF-β Signaling in Cancer Metastasis. Acta Biochim Biophys Sin (2018) 50:121–32. doi: 10.1093/abbs/gmx123
35. Gudey SK, Sundar R, Heldin CH, Bergh A, Landstrom M. Pro-Invasive Properties of Snail1 Are Regulated by Sumoylation in Response to TGFbeta Stimulation in Cancer. Oncotariget (2017) 8:97703–26. doi: 10.18632/oncotarget.20097
36. Rohan K, Niraj KJ, Saurabh KJ, Ankur S, Sunny D, Asthana N, et al. A “Notch” Deeper Into the Epithelial-Mesenchymal Transition in Breast Cancer. Genes (2019) 10(12):961. doi: 10.3390/genes10120961
37. Li X, Deng W, Nail CD, Bailey SK, Kraus MH, Ruppert JM, Lobo-Ruppert SM. Snail Induction Is an Early Response to Gli1 That Determines the Efficiency of Epithelial Transformation. Oncogene (2006) 25:609–21. doi: 10.1038/sj.onc.1209077
38. Yoo YA, Kang MH, Kim JS, Oh SC. Sonic Hedgehog Signaling Promotes Motility and Invasiveness of Gastric Cancer Cells Through TGF-β-Mediated Activation of the ALK5–Smad 3 Pathway. Carcinogenesis (2008) 29:480–90. doi: 10.1093/carcin/bgm281
39. Cruceriu D, Baldasici O, Balacescu O, Berindan-Neagoe I. The Dual Role of Tumor Necrosis Factor-Alpha (TNF-α) in Breast Cancer: Molecular Insights and Therapeutic Approaches. Cell Oncol (2019) 43:1–18. doi: 10.1007/s13402-019-00489-1
40. Johnston SJ, Carroll JS. Transcription Factors and Chromatin Proteins as Therapeutic Targets in Cancer. Biochim Biophys Acta (2015) 185:183–92. doi: 10.1016/j.bbcan.2015.02.002
41. Tania M, Khan MA, Fu J. Epithelial to Mesenchymal Transition Inducing Transcription Factors and Metastatic Cancer. Tumour Biol (2014) 35:7335–42. doi: 10.1007/s13277-014-2163-y
42. Papanikolaou S, Vourda A, Syggelos S, Gyftopoulos K. Cell Plasticity and Prostate Cancer: The Role of Epithelial-Mesenchymal Transition in Tumor Progression, Invasion, Metastasis and Cancer Therapy Resistance. Cancers (Basel) (2021) 13(11):2795. doi: 10.3390/cancers13112795
43. Yastrebova MA, Khamidullina AI, Tatarskiy VV, Scherbakov AM. Snail-Family Proteins: Role in Carcinogenesis and Prospects for Antitumor Therapy. Acta Naturae (2021) 13(1):76–90. doi: 10.32607/actanaturae.11062
44. Wang Y, Shi J, Chai K, Ying X, Zhou B. The Role of Snail in EMT and Tumorigenesis. Curr Cancer Drug Targets (2013) 13(9):963–72. doi: 10.2174/15680096113136660102
45. Kang E, Seo J, Yoon H, Cho S. The Post-Translational Regulation of Epithelial–Mesenchymal Transition-Inducing Transcription Factors in Cancer Metastasis. Int J Mol Sci (2021) 22:3591. doi: 10.3390/ijms22073591
46. Stemmler MP, Eccles RL, Brabletz S, Brabletz T. Non Redundant Functions of EMT Transcription Factors. Nat Cell Biol (2019) 21:102–12. doi: 10.1038/s41556-018-0196-y
47. Bolós V, Peinado H, Pérez-Moreno MA, Fraga MF, Esteller M, Cano A. The Transcription Factor Slug Represses E-Cadherin Expression and Induces Epithelial to Mesenchymal Transitions: A Comparison With Snail and E47 Repressors. Erratum in: J Cell Sci (2016) 129(6):1283. doi: 10.1242/jcs.188243
48. Wu W, You R, Cheng C, Lee M, Lin T, Hu C. Snail Collaborates With EGR-1 and SP-1 to Directly Activate Transcription of MMP 9 and ZEB1. Sci Rep (2017) 7:17753. doi: 10.1038/s41598-017-18101-7
49. Saray Q, Rodrigo A, Enrique B, Julio CT, Victor A, Julio L. Role of Matrix Metalloproteinases in Angiogenesis and Cancer. Front Oncol (2019) 9:1370. doi: 10.3389/fonc.2019.01370
50. Kar R, Jha NK, Jha SK, Sharma A, Dholpuria S, Asthana N, et al. Deeper Into the Epithelial-To-Mesenchymal Transition (EMT) Program in Breast Cancer. Genes (2019) 10(12):961. doi: 10.3390/genes10120961
51. Niessen K, Fu Y, Chang L, Hoodless PA, McFadden D, Karsan A. Slug Is a Direct Notch Target Required for Initiation of Cardiac Cushion Cellularization. J Cell Biol (2008) 182:315–25. doi: 10.1083/jcb.200710067
52. Assani G, Zhou Y. Effect of Modulation of Epithelial-Mesenchymal Transition Regulators Snail1 and Snail2 on Cancer Cell Radiosensitivity by Targeting of the Cell Cycle, Cell Apoptosis and Cell Migration/Invasion (Review). Onc Lett (2019) 17(1):23–30. doi: 10.3892/ol.2018.9636
53. Casas E, Kim J, Bendesky A, Ohno-Machado L, Wolfe CJ, Yang J. Snail2 Is an Essential Mediator of Twist1-Induced Epithelial Mesenchymal Transition and Metastasis. Cancer Res (2011) 71:245–54. doi: 10.1158/0008-5472.CAN-10-2330
54. Shih JY, Yang PC. The EMT Regulator Slug and Lung Carcinogenesis. Carcinogenesis (2011) 32:1299–304. doi: 10.1093/carcin/bgr110
55. Georgakopoulos-Soares I, Chartoumpekis DV, Kyriazopoulou V, Zaravinos A. EMT Factors and Metabolic Pathways in Cancer. Front Oncol (2020) 10:499. doi: 10.3389/fonc.2020.00499
56. Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, et al. The Epithelial-Mesenchymal Transition Generates Cells With Properties of Stem Cells. Cell (2008) 133:704–15. doi: 10.1016/j.cell.2008.03.027
57. Nassour M, Idoux-Gillet Y, Selmi A, Côme C, Faraldo ML, Deugnier MA, et al. Slug Controls Stem/Progenitor Cell Growth Dynamics During Mammary Gland Morphogenesis. PloS One (2012) 7:53498. doi: 10.1371/journal.pone.0053498
58. Ye X, Tam WL, Shibue T, Kaygusuz Y, Reinhardt F, Ng Eaton E, et al. Distinct EMT Programs Control Normal Mammary Stem Cells and Tumour-Initiating Cells. Nature (2015) 525:256–60. doi: 10.1038/nature14897
59. Gheldof A, Hulpiau P, Van Roy F, De Craene B, Berx G. Evolutionary Functional Analysis and Molecular Regulation of the ZEB Transcription Factors. Cell Mol Life Sci (2012) 69:2527–41. doi: 10.1007/s00018-012-0935-3
60. Singh A, Settleman J. EMT, Cancer Stem Cells and Drug Resistance: An Emerging Axis of Evil in the War on Cancer. Oncogene (2010) 29:4741–51. doi: 10.1038/onc.2010.215
61. Postigo AA, Dean DC. Differential Expression and Function of Members of the Zfh-1 Family of Zinc Finger/Homeodomain Repressors. Proc Natl Acad Sci USA (2000) 97:6391–96. doi: 10.1073/pnas.97.12.6391
62. De Craene B, Berx G. Regulatory Networks Defining EMT During Cancer Initiation and Progression. Nat Rev Cancer (2013) 13:97–110. doi: 10.1038/nrc3447
63. St Johnston D, Ahringer J. Cell Polarity in Eggs and Epithelia: Parallels and Diversity. Cell (2010) 141:757–74. doi: 10.1016/j.cell.2010.05.011
64. Navarro C, Nola S, Audebert S, Santoni MJ, Arsanto JP, Ginestier C, et al. Junctional Recruitment of Mammalian Scribble Relies on E-Cadherin Engagement. Oncogene (2005) 24:4330–39. doi: 10.1038/sj.onc.1208632
65. Qin Y, Capaldo C, Gumbiner BM, Macara IG. The Mammalian Scribble Polarity Protein Regulates Epithelial Cell Adhesion and Migration Through E-Cadherin. J Cell Biol (2005) 171:1061–71. doi: 10.1083/jcb.200506094
66. Moreno-Bueno G, Portillo F, Cano A. Transcriptional Regulation of Cell Polarity in EMT and Cancer. Oncogene (2008) 27:6958–69. doi: 10.1038/onc.2008.346
67. Yilmaz M, Christofori G. Mechanisms of Motility in Metastasizing Cells. Mol Cancer Res (2010) 8:629–42. doi: 10.1158/1541-7786.MCR-10-0139
68. Dave N, Guaita-Esteruelas S, Gutarra S, Frias À, Beltran M, Peiró S, et al. Functional Cooperation Between Snail1 and Twist in the Regulation of ZEB1 Expression During Epithelial to Mesenchymal Transition. J Biol Chem (2011) 286:12024–32. doi: 10.1074/jbc.M110.168625
69. Spaderna S, Schmalhofer O, Wahlbuhl M, Dimmler A, Bauer K, Sultan A, et al. The Transcriptional Repressor ZEB1 Promotes Metastasis and Loss of Cell Polarity in Cancer. Cancer Res (2008) 68:537–44. doi: 10.1158/0008-5472.CAN-07-5682
70. Zhang Y, Xu L, Li A, Han X. The Roles of ZEB1 in Tumorigenic Progression and Epigenetic Modifications. Biomed Pharmacother (2019) 110:400–8. doi: 10.1016/j.biopha.2018.11.112
71. Llorens MC, Lorenzatti G, Cavallo NL, Vaglienti MV, Perrone AP, Carenbauer AL, et al. Phosphorylation Regulates Functions of ZEB1 Transcription Factor. J Cell Physiol (2016) 231:2205–17. doi: 10.1002/jcp.25338
72. Franco HL, Casasnovas J, Rodríguez-Medina JR, Cadilla CL. Redundant or Separate Entities?–Roles of Twist1 and Twist2 as Molecular Switches During Gene Transcription. Nucleic Acids Res (2011) 39:1177–86. doi: 10.1093/nar/gkq890
73. Soldatov R, Kaucka M, Kastriti ME, Petersen J, Chontorotzea T, Englmaier L, et al. Spatiotemporal Structure of Cell Fate Decisions in Murine Neural Crest. Science (2019) 364:eaas9536. doi: 10.1126/science.aas9536
74. Yeo SY, Lee KW, Shin D, An S, Cho KH, Kim SH. A Positive Feedback Loop Bi-Stably Activates Fibroblasts. Nat Commun (2018) 9:3016. doi: 10.1038/s41467-018-05274-6
75. Qin Q, Xu Y, He T, Qin C, Xu J. Normal and Disease-Related Biological Functions of Twist1 and Underlying Molecular Mechanisms. Cell Res (2012) 22:90–106. doi: 10.1038/cr.2011.144
76. Norozi F, Ahmadzadeh A, Shahjahani M, Shahrabi S, Saki N. Twist as a New Prognostic Marker in Hematological Malignancies. Clin Transl Oncol (2016) 18:113–24. doi: 10.1007/s12094-015-1357-0
77. Yang J, Mani SA, Weinberg RA. Exploring a New Twist on Tumor Metastasis. Cancer Res (2006) 66:4549–52. doi: 10.1158/0008-5472.CAN-05-3850
78. Meng J, Chen S, Han JX, Qian B, Wang XR, Zhong WL, et al. Twist1 Regulates Vimentin Through Cul2 Circular RNA to Promote EMT in Hepatocellular Carcinoma. Cancer Res (2018) 78:4150–62. doi: 10.1158/0008-5472.CAN-17-3009
79. Wang SS, Jiang J, Liang XH, Tang YL. Links Between Cancer Stem Cells and Epithelial-Mesenchymal Transition. Onco Targets Ther (2015) 8:2973–80. doi: 10.2147/OTT.S91863
80. Hong J, Zhou J, Fu J, He T, Qin J, Wang L, et al. Phosphorylation of Serine 68 of Twist1 by MAPKs Stabilizes Twist1 Protein and Promotes Breast Cancer Cell Invasiveness. Cancer Res (2011) 71:3980–90. doi: 10.1158/0008-5472.CAN-10-2914
81. Zhong J, Ogura K, Wang Z, Inuzuka H. Degradation of the Transcription Factor Twist, an Oncoprotein That Promotes Cancer Metastasis. Discov Med (2013) 15:7–15. doi: pmc5522964/
82. Shimoda Y, Ubukata Y, Handa T, Yokobori T, Watanabe T, Dolgormaa G, et al. High Expression of Forkhead Box Protein C2 Is Associated With Aggressive Phenotypes and Poor Prognosis in Clinical Hepatocellular Carcinoma. BMC Cancer (2018) 18:597. doi: 10.1186/s12885-018-4503-6
83. Hartwell KA, Muir B, Reinhardt F, Carpenter AE, Sgroi DC, Weinberg RA. The Spemann Organizer Gene, Goosecoid, Promotes Tumor Metastasis. Proc Natl Acad Sci USA (2006) 103:18969–74. doi: 10.1073/pnas.0608636103
84. Wang X, Zheng M, Liu G, Xia W, McKeown-Longo PJ, Hung MC, et al. Krüppel-Like Factor 8 Induces Epithelial to Mesenchymal Transition and Epithelial Cell Invasion. Cancer Res (2007) 67:7184–93. doi: 10.1158/0008-5472.CAN-06-4729
85. Wang X, Lu H, Urvalek AM, Li T, Yu L, Lamar J, et al. KLF8 Promotes Human Breast Cancer Cell Invasion and Metastasis by Transcriptional Activation of MMP9. Oncogene (2011) 30:1901–11. doi: 10.1038/onc.2010.563
86. Lu H, Wang X, Urvalek AM, Li T, Xie H, Yu L, et al. Transformation of Human Ovarian Surface Epithelial Cells by Krüppel-Like Factor 8. Oncogene (2014) 33:10–8. doi: 10.1038/onc.2012.545
87. Zhang H, Liu L, Wang Y, Zhao G, Xie R, Liu C, et al. KLF8 Involves in TGF-Beta-Induced EMT and Promotes Invasion and Migration in Gastric Cancer Cells. J Cancer Res Clin Oncol (2013) 139:1033–42. doi: 10.1007/s00432-012-1363-3
88. Takano S, Reichert M, Bakir B, Das KK, Nishida T, Miyazaki M, et al. Prrx1 Isoform Switching Regulates Pancreatic Cancer Invasion and Metastatic Colonization. Genes Dev (2016) 30:233–47. doi: 10.1101/gad.263327.115
89. Yang J, Weinberg RA. Epithelial-Mesenchymal Transition: At the Crossroads of Development and Tumor Metastasis. Dev Cell (2008) 14:818–29. doi: 10.1016/j.devcel.2008.05.009
90. Kume T, Deng K, Hogan BL. Minimal Phenotype of Mice Homozygous for a Null Mutation in the Forkhead/Winged Helix Gene, Mf2. Mol Cell Biol (2000) 20:1419–25. doi: 10.1128/MCB.20.4.1419-1425.2000
91. Cederberg A, Grønning LM, Ahrén B, Taskén K, Carlsson P, Enerbäck S. FOXC2 Is a Winged Helix Gene That Counteracts Obesity, Hypertriglyceridemia, and Diet-Induced Insulin Resistance. Cell (2001) 106:563–73. doi: 10.1016/s0092-8674(01)00474-3
92. Mani SA, Yang J, Brooks M, Schwaninger G, Zhou A, Miura N, et al. Mesenchyme Forkhead 1 (FOXC2) Plays a Key Role in Metastasis and Is Associated With Aggressive Basal-Like Breast Cancers. Proc Natl Acad Sci USA (2007) 104:10069–74. doi: 10.1073/pnas.0703900104
93. Chung VY, Tan TZ, Tan M, Wong MK, Kuay KT, Yang Z, et al. GRHL2-miR-200-ZEB1 Maintains the Epithelial Status of Ovarian Cancer Through Transcriptional Regulation and Histone Modification. Sci Rep (2016) 6:19943. doi: 10.1038/srep19943
94. Sun X, Li Z, Niu Y, Zhao L, Huang Y, Li Q, et al. Jarid1b Promotes Epidermal Differentiation by Mediating the Repression of Ship1 and Activation of the AKT/Ovol1 Pathway. Cell Prolif (2019) 52(5):12638. doi: 10.1111/cpr.12638
95. Kitazawa K, Hikichi T, Nakamura T, Nakamura M, Sotozono C, Masui S, et al. Direct Reprogramming Into Corneal Epithelial Cells Using a Transcriptional Network Comprising PAX6, OVOL2, and KLF4. Cornea (2019) 38(1):34–41. doi: 10.1097/ICO.0000000000002074
96. Watanabe K, Liu Y, Noguchi S, Murray M, Chang JC, Kishima M, et al. OVOL2 Induces Mesenchymal-to-Epithelial Transition in Fibroblasts and Enhances Cell-State Reprogramming Towards Epithelial Lineages. Sci Rep (2019) 9(1):6490. doi: 10.1038/s41598-019-43021-z
97. Wu RS, Hong JJ, Wu JF, Yan S, Wu D, Liu N, et al. OVOL2 Antagonizes TGF-β Signaling to Regulate Epithelial to Mesenchymal Transition During Mammary Tumor Metastasis. Oncotarget (2017) 8(24):39401–16. doi: 10.18632/oncotarget.17031
98. Xu C, Yan T, Yang J. OVOL1 Inhibits Oral Squamous Cell Carcinoma Growth and Metastasis by Suppressing Zinc Finger E-Box Binding Homeobox 1. Int J Clin Exp Pathol (2019) 12(7):2801–8.
99. Jolly MK, Mani SA, Levine H. Hybrid Epithelial/ Mesenchymal Phenotype(s): The ‘Fittest’ for Metastasis? Biochim Biophys Acta Rev Cancer (2018) 1870:151–7. doi: 10.1016/j.bbcan.2018.07.001
100. Murata M, Ito T, Tanaka Y, Yamamura K, Furue K, Furue M. OVOL2-Mediated ZEB1 Downregulation May Prevent Promotion of Actinic Keratosis to Cutaneous Squamous Cell Carcinoma. J Clin Med (2020) 3:618. doi: 10.3390/jcm9030618
101. Mlacki M. Recent Discoveries Concerning the Involvement of Transcription Factors From the Grainyhead-Like Family in Cancer. Exp Biol Med (2015) 240:1396–401. doi: 10.1177/1535370215588924
102. He J, Feng C, Zhu H, Wu S, Jin P, Xu T. Grainyhead-Like 2 as a Double-Edged Sword in Development and Cancer. Am J Transl Res (2020) 12(2):310–31.
103. Yang Z, Wu D, Chen Y, Min Z, Quan Y. GRHL2 Inhibits Colorectal Cancer Progression and Metastasis via Oppressing Epithelial-Mesenchymal Transition. Cancer Biol Ther (2019) 20:1195–205. doi: 10.1080/15384047.2019.1599664
104. Xiang J, Fu X, Ran W, Wang Z. Grhl2 Reduces Invasion and Migration Through Inhibition of TGFbeta-Induced EMT in Gastric Cancer. Oncogenesis (2017) 6:284. doi: 10.1038/oncsis.2016.83
105. Werner S, Frey S, Riethdorf S, Schulze C, Alawi M, Kling L. Dual Roles of the Transcription Factor Grainyhead-Like 2 (GRHL2) in Breast Cancer. J Biol Chem (2013) 288:22993–3008. doi: 10.1074/jbc.M113.456293
106. Mooney SM, Talebian V, Jolly MK, Jia D, Gromala M, Levine, et al. The GRHL2/ZEB Feedback Loop-a Key Axis in the Regulation of EMT in Breast Cancer. J Cell Biochem (2017) 118:2559–70. doi: 10.1002/jcb.25974;;k
107. Jose M, Rupasinghe V, Dellaire G, Murphy K. Role of Dietary Antioxidants in P53-Mediated Cancer Chemoprevention and Tumor Suppression. Oxid Med Cell Longevity (2021) 2021:9924328. doi: 10.1155/2021/9924328
108. Bieging KT, Mello SS, Attardi LD. Unravelling Mechanisms of P53- Mediated Tumour Suppression. Nat Rev Cancer (2014) 14:359–70. doi: 10.1038/nrc3711
109. Yang-Hartwich Y, Roslyn T, Cai MR, Jamie G, Carlous C, Marta N, et al. P53–Pirh2 Complex Promotes Twist1 Degradation and Inhibits EMT. Mol Cancer Res (2019) 17:153–64. doi: 10.1158/1541-7786
110. Kim T, Veronese A, Pichiorri F, Lee TJ, Jeon YJ, Volinia S, et al. P53 Regulates Epithelial-Mesenchymal Transition Through microRNAs Targeting ZEB1 and ZEB2. J Exp Med (2011) 208:875–83. doi: 10.1084/jem.20110235
111. Jeffrey H, Albrecht MD. An Epigenetic Switch Between Differentiation and Hepatoblastoma. Cel Mol Gastr Hepatol (2021) 15:882. doi: 10.1016/j.jcmgh.2021.08.020
112. Johnson PF. Molecular Stop Signs: Regulation of Cell-Cycle Arrest by C/EBP Transcription Factors. J Cell Sci (2005) 118:2545–55. doi: 10.1242/jcs.02459
113. Lourenço AR, Roukens MG, Seinstra D, Frederiks CL, Pals CE, Vervoort SJ, et al. C/EBPα Is Crucial Determinant of Epithelial Maintenance by Preventing Epithelial-to-Mesenchymal Transition. Nat Commun (2020) 11:785. doi: 10.1038/s41467-020-14556-x
114. Lu J, Du C, Yao J, Wu B, Duan Y, Zhou L, et al. C/Ebpα Suppresses Lung Adenocarcinoma Cell Invasion and Migration by Inhibiting β-Catenin. Cell Physiol Biochem (2017) 42:1779–88. doi: 10.1159/000479457
115. Ye Y, Xiao Y, Wang W, Yearsley K, Gao JX, Barsky SH. ERalpha Suppresses Slug Expression Directly by Transcriptional Repression. Biochem J (2008) 416:179–87. doi: 10.1042/BJ20080328
116. Ye Y, Xiao Y, Wang W, Yearsley K, Gao JX, Shetuni B, et al. ERalpha Signaling Through Slug Regulates E-Cadherin and EMT. Oncogene (2010) 29:1451–62. doi: 10.1038/onc.2009.433
117. Dhasarathy A, Kajita M, Wade PA. The Transcription Factor Snail Mediates Epithelial to Mesenchymal Transitions by Repression of Estrogen Receptor-Alpha. Mol Endocrinol (2007) 21:2907–18. doi: 10.1210/me.2007-0293
118. Scherbakov AM, Andreeva OE, Shatskaya VA, Krasil'nikov MA. The Relationships Between Snail1 and Estrogen Receptor Signaling in Breast Cancer Cells. J Cell Biochem (2012) 113:2147–55. doi: 10.1002/jcb.24087
119. Fujita N, Jaye DL, Kajita M, Geigerman C, Moreno CS, Wade PA. MTA3, a Mi-2/NuRD Complex Subunit, Regulates an Invasive Growth Pathway in Breast Cancer. Cell (2003) 113:207–19. doi: 10.1016/s0092-8674(03)00234-4
120. Kim BN, Ahn DH, Kang N, Yeo CD, Kim YK, Lee Ky, et al. TGF-β Induced EMT and Stemness Characteristics Are Associated With Epigenetic Regulation in Lung Cancer. Sci Rep (2020) 10:10597. doi: 10.1038/s41598-020-67325-7
121. Eswarakumar VP, Lax I, Schlessinger J. Cellular Signaling by Fibroblast Growth Factor Receptors. Cytokine Growth Factor Rev (2005) 16:139–49. doi: 10.1016/j.cytogfr.2005.01.001
122. Espinoza I, Miele L. Deadly Crosstalk: Notch Signaling at the Intersection of EMT and Cancer Stem Cells. Cancer Lett (2013) 341:41–5. doi: 10.1016/j.canlet.2013.08.027
123. Bradley CK, Norton CR, Chen Y, Han X, Booth CJ, Yoon JK, et al. The Snail Family Gene Snai3 Is Not Essential for Embryogenesis in Mice. PLoS One (2013) 8:e65344. doi: 10.1371/journal.pone.0065344
124. Polyak K, Weinberg RA. Transitions Between Epithelial and Mesenchymal States: Acquisition of Malignant and Stem Cell Traits. Nat Rev Cancer (2009) 9:265–73. doi: 10.1038/nrc2620
125. Wu F, Weigel KJ, Zhou H, Wang X. Paradoxical Roles of TGF-β Signaling in Suppressing and Promoting Squamous Cell Carcinoma. Acta Biochim Biophys Sin (2018) 50:98–105. doi: 10.1093/abbs/gmx127
126. Moustakas A, Heldin CH. Induction of Epithelial-Mesenchymal Transition by Transforming Growth Factor β. Semin Cancer Biol (2012) 22:446–54. doi: 10.1016/j.semcancer.2012.04.002
127. Wu MZ, Tsai YP, Yang MH, Huang CH, Chang SY, Chang CC, et al. Interplay Between HDAC3 and WDR5 Is Essential for Hypoxia-Induced Epithelial-Mesenchymal Transition. Mol Cell (2011) 43:811–22. doi: 10.1016/j.molcel.2011.07.012
128. Chen J, Imanaka N, Chen J, Griffin JD. Hypoxia Potentiates Notch Signaling in Breast Cancer Leading to Decreased E-Cadherin Expression and Increased Cell Migration and Invasion. Br J Cancer (2010) 102:351–60. doi: 10.1038/sj.bjc.6605486
129. Noseda M, McLean G, Niessen K, Chang L, Pollet I, Montpetit R, et al. Notch Activation Results in Phenotypic and Functional Changes Consistent With Endothelial-to-Mesenchymal Transformation. Circ Res (2004) 94:910–17. doi: 10.1161/01.RES.0000124300.76171.C9
130. Zavadil J, Cermak L, Soto-Nieves N, Böttinger EP. Integration of TGF-Beta/Smad and Jagged1/Notch Signalling in Epithelial-to-Mesenchymal Transition. EMBO J (2004) 23:1155–65. doi: 10.1038/sj.emboj.7600069
131. Brzozowa M, Mielańczyk L, Michalski M, Malinowski L, Kowalczyk-Ziomek G, Helewski K, et al. Role of Notch Signaling Pathway in Gastric Cancer Pathogenesis. Contemp Oncol (Pozn) (2013) 17:1–5. doi: 10.5114/wo.2013.33765
132. Brzozowa M, Wojnicz R, Kowalczyk-Ziomek G, Helewski K. The Notch Ligand Delta-Like 4 (DLL4) as a Target in Angiogenesis-Based Cancer Therapy? Contemp Oncol (Pozn) (2013) 17:234–37. doi: 10.5114/wo.2013.35588
133. Sahlgren C, Gustafsson MV, Jin S, Poellinger L, Lendahl U. Notch Signaling Mediates Hypoxia-Induced Tumor Cell Migration and Invasion. Proc Natl Acad Sci USA (2008) 105:6392–97. doi: 10.1073/pnas.0802047105
134. Grego-Bessa J, Díez J, Timmerman L, de la Pompa JL. Notch and Epithelial-Mesenchyme Transition in Development and Tumor Progression: Another Turn of the Screw. Cell Cycle (2004) 3:718–21. doi: 10.4161/cc.3.6.949
135. Wang Z, Li Y, Kong D, Banerjee S, Ahmad A, Azmi AS, et al. Acquisition of Epithelial-Mesenchymal Transition Phenotype of Gemcitabine-Resistant Pancreatic Cancer Cells Is Linked With Activation of the Notch Signaling Pathway. Cancer Res (2009) 69:2400–07. doi: 10.1158/0008-5472.CAN-08-4312
136. Kuphal S, Palm HG, Poser I, Bosserhoff AK. Snail-Regulated Genes in Malignant Melanoma. Melanoma Res (2005) 15:305–13. doi: 10.1097/00008390-200508000-00012
137. Shyu AB, Wilkinson MF, van Hoof A. Messenger RNA Regulation: To Translate or to Degrade. EMBO J (2008) 27:471–81. doi: 10.1038/sj.emboj.7601977
138. Zhang J, Ma L. MicroRNA Control of Epithelial-Mesenchymal Transition and Metastasis. Cancer Metastasis Rev (2012) 31:653–62. doi: 10.1007/s10555-012-9368-6
139. Fischer KR, Durrans A, Lee S, Sheng J, Li F, Wong ST, et al. Epithelial-To-Mesenchymal Transition Is Not Required for Lung Metastasis But Contributes to Chemoresistance. Nature (2015) 527:472–76. doi: 10.1038/nature15748
140. Liu Z, Long J, Du R, Ge C, Guo K, Xu Y. miR-204 Regulates the EMT by Targeting Snai1 to Suppress the Invasion and Migration of Gastric Cancer. Tumour Biol (2016) 37:8327–35. doi: 10.1007/s13277-015-4627-0
141. Saydam O, Shen Y, Würdinger T, Senol O, Boke E, James MF, et al. Downregulated microRNA-200a in Meningiomas Promotes Tumor Growth by Reducing E-Cadherin and Activating the Wnt/beta-Catenin Signaling Pathway. Mol Cell Biol (2009) 29:5923–40. doi: 10.1128/MCB.00332-09
142. Yan C, Higgins PJ. Drugging the Undruggable: Transcription Therapy for Cancer. Biochim Biophys Acta (2013) 183:76–85. doi: 10.1016/j.bbcan.2012.11.002
143. Liao C, Wang Q, An J, Long Q, Wang H, Xiang M, et al. Partial EMT in Squamous Cell Carcinoma: A Snapshot. Int J Bio Sci (2021) 17(12):3036–47. doi: 10.7150/ijbs.61566
144. Pastushenko I, Blanpain C. EMT Transition States During Tumor Progression and Metastasis. Trends Cell Biol (2019) 29:212–26. doi: 10.1016/j.tcb.2018.12.001
145. Dongre A, Weinberg RA. New Insights Into the Mechanisms of Epithelial–Mesenchymal Transition and Implications for Cancer. Nat Rev Mol Cell Bio (2019) 20:69–84. doi: 10.1038/s41580-018-0080-4
146. Yu M, Bardia A, Wittner BA, Stott SL, Smas ME, Ting DT, et al. Circulating Breast Tumor Cells Exhibit Dynamic Changes in Epithelial and Mesenchymal Composition. Science (2013) 339:580–4. doi: 10.1126/science.1228522
147. Saxena K, Jolly MK, Balamurugan K. Hypoxia, Partial EMT and Collective Migration: Emerging Culprits in Metastasis. Transl Oncol (2020) 13:100845. doi: 10.1016/j.tranon.2020.100845
148. Ruscetti M, Quach B, Dadashian EL, Mulholland DT, Wu H. Tracking and Functional Characterization of Epithelial-Mesenchymal Transition and Mesenchymal Tumor Cells During Prostate Cancer Metastasis. Cancer Res (2015) 75:2749–59. doi: 10.1158/0008-5472.CAN-14-3476
149. Cornelia K, Alexander A, Jasmin M, Eaton EN, Thiru P, Khodor YK, et al. Acquisition of a Hybrid E/M State Is Essential for Tumorigenicity of Basal Breast Cancer Cells. PINAS (2019) 116(15):7353–62. doi: 10.1073/pnas.1812876116
150. Wu S, Liu S, Liu Z, Huang J, Pu X, Li J, et al. Classification of Circulating Tumor Cells by Epithelial-Mesenchymal Transition Markers. PLoS One (2015) 10:e0123976. doi: 10.1371/journal.pone.0123976
151. Tayoun T, Faugeroux V, Oulhen M, Aberlenc A, Pawlikowska P, Farace F. CTC-Derived Models: A Window Into the Seeding Capacity of Circulating Tumor Cells (CTCs). Cells (2019) 10:1145. doi: 10.3390/cells8101145
152. Szczerba BM, Castro-Giner F, Vetter M, Krol I, Gkountela S. Neutrophils Escort Circulating Tumour Cells to Enable Cell Cycle Progression. Nat (2019) 566:553–7. doi: 10.1038/s41586-019-0915-y
153. Aceto N, Bardia A, Miyamoto DT, Donaldson MC, Wittner BS, Spencer JA, et al. Circulating Tumor Cell Clusters Are Oligoclonal Precursors of Breast Cancer Metastasis. Cell (2014) 158:1110–22. doi: 10.1016/j.cell.2014.07.013
154. Joosse SA, Gorges TM, Pantel K. Biology, Detection, and Clinical Implications of Circulating Tumor Cells. EMBO Mol Med (2015) 7:1–11. doi: 10.15252/emmm.201303698
155. Pasani S, Sahoo S, Jolly MK. Hybrid E/M Phenotype(s) and Stemness: A Mechanistic Connection Embedded in Network Topology. J Clin Med (2021) 1:60. doi: 10.3390/jcm10010060
156. Haslehurst AM, Koti M, Dharsee M, Nuin P, Evans K, Geraci J, et al. EMT Transcription Factors Snail and Slug Directly Contribute to Cisplatin Resistance in Ovarian Cancer. BMC Cancer (2012) 12:91. doi: 10.1186/1471-2407-12-91
157. Hojo N, Huisken AL, Wang H, Chirshev E, Kim NS, Nguyen SM, et al. Snail Knockdown Reverses Stemness and Inhibits Tumour Growth in Ovarian Cancer. Sci Rep (2018) 8:8704. doi: 10.1038/s41598-018-27021-z
158. Zhu X, Shen H, Yin X, Long L, Xie C, Liu Y, et al. miR-186 Regulation of Twist1 and Ovarian Cancer Sensitivity to Cisplatin. Oncogene (2016) 35:323–32. doi: 10.1038/onc.2015.84
159. Wang SC, Sun HL, Hsu YH, Liu S, Lii C, Tsai C, et al. α-Linolenic Acid Inhibits the Migration of Human Triple-Negative Breast Cancer Cells by Attenuating Twist1 Expression and Suppressing Twist1-Mediated Epithelial-Mesenchymal Transition. Biochem Pharm (2020) 180:114152. doi: 10.1016/j.bcp.2020.114152
160. Li HM, Bi YR, Li Y, Fu R, Lv WC, Jiang N, et al. A Potent CBP/p300-Snail Interaction Inhibitor Suppresses Tumor Growth and Metastasis in Wild-Type P53-Expressing Cancer. Sci Adv (2020) 6(17):eaaw8500. doi: 10.1126/sciadv.aaw8500
161. Christopoulos PF, Gjølberg TT, Krüger S, Haraldsen G, Andersen JT, Sundlisaeter E. Targeting the Notch Signaling Pathway in Chronic Inflammatory Diseases. Front Immunol (2021) 12:668207. doi: 10.3389/fimmu.2021.668207
162. Irene K, Guttilla R. Mechanism and Regulation of Epithelial–Mesenchymal Transition in Cancer. Cell Health Cytoskeleton (2015) 7:155–66. doi: 10.2147/CHC.S73822
163. Keller L, Pantel K. Unravelling Tumour Heterogeneity by Single-Cell Profiling of Circulating Tumour Cells. Nat Rev Cancer (2019) 19:553–67. doi: 10.1038/s41568-019-0180-2
164. Griffiths JA, Scialdone A, Marioni JC. Using Single-Cell Genomics to Understand Developmental Processes and Cell Fate Decisions. Mol Syst Biol (2018) 14(4):8046. doi: 10.15252/msb.20178046
Keywords: EMT, embryogenesis, ontogenesis, tumour, metastasis, microenvironment, transcription factors
Citation: Imodoye SO, Adedokun KA, Muhammed AO, Bello IO, Muhibi MA, Oduola T and Oyenike MA (2021) Understanding the Complex Milieu of Epithelial-Mesenchymal Transition in Cancer Metastasis: New Insight Into the Roles of Transcription Factors. Front. Oncol. 11:762817. doi: 10.3389/fonc.2021.762817
Received: 22 August 2021; Accepted: 04 October 2021;
Published: 18 November 2021.
Edited by:
Leonardo Freire-de-Lima, Federal University of Rio de Janeiro, BrazilReviewed by:
Daniele Vergara, University of Salento, ItalyCopyright © 2021 Imodoye, Adedokun, Muhammed, Bello, Muhibi, Oduola and Oyenike. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sikiru O. Imodoye, T3BleWVtaS5pbW9kb3llQGdtYWlsLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.