 Jing Li
Jing Li Nan Yu2
Nan Yu2 Qie Guo
Qie Guo
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 21 October 2021
Sec. Cancer Genetics
Volume 11 - 2021 | https://doi.org/10.3389/fonc.2021.759894
Tumorigenesis refers to the process of clonal dysplasia that occurs due to the collapse of normal growth regulation in cells caused by the action of various carcinogenic factors. These “successful” tumor cells pass on the genetic templates to their generations in evolutionary terms, but they also constantly adapt to ever-changing host environments. A unique peculiarity known as intratumor heterogeneity (ITH) is extensively involved in tumor development, metastasis, chemoresistance, and immune escape. An understanding of ITH is urgently required to identify the diversity and complexity of the tumor microenvironment (TME), but achieving this understanding has been a challenge. Single-cell sequencing (SCS) is a powerful tool that can gauge the distribution of genomic sequences in a single cell and the genetic variability among tumor cells, which can improve the understanding of ITH. SCS provides fundamental ideas about existing diversity in specific TMEs, thus improving cancer diagnosis and prognosis prediction, as well as improving the monitoring of therapeutic response. Herein, we will discuss advances in SCS and review SCS application in tumors based on current evidence.
It has been stated that inactivation of oncogenes boosts cancer remission, implying that oncogenes are the Achilles’ heel of cancers independent of the cellular heterogeneity remaining within the tumor. The existing model of cancer has kept oncogenes firmly in focus as therapeutic targets and is in agreement with the fact that all cancerous cells carry the same oncogenic genetic lesions (1). However, it is now recognized that tumor cells accumulate genetic alterations as a way of progressively optimizing themselves, resulting in highly diverse phenotypes of cancer cells. This diversity supports the complicated nature of the tumor microenvironment (TME), in which genetically distinct subclonal populations of cells coexist, resulting in intratumor heterogeneity (ITH) (2). Unfortunately, the understanding of ITH in tumor subsets is still poor. It is of utmost priority to reveal the underlying mechanisms promoting tumor development, metastasis, chemoresistance, and immune escape from the perspective of single cells (3, 4). Fortunately, genome, transcriptome, and epigenome profiling of single cells is well underway with the efforts of the Human Genome Project (HGP) and International HapMap Project, the obvious advances in high-throughput biochip technology have also aided these efforts (5). An understanding of ITH will not be delayed with the advent of single-cell sequencing (SCS) technology, which is expected to enable mapping of the “genetic story” of tumor development, metastasis, chemoresistance, and immune escape by providing centralized dynamic genomic DNA and transcriptome RNA data and epigenetic information scattered across single tumor cells (6). Here, we discuss the common themes emerging from initial studies of SCS and then highlight challenges in cancer biology for which single-cell genomics analysis may provide a compelling approach.
Benefiting from the advances in next-generation sequencing (NGS) and third-generation sequencing (TGS) technology, genome-wide scanning strategies have gradually evolved from analyses of mixed cell populations to single-cell targeted assessments (7). Accordingly, a novel method of bulk genomic analysis, advertised as SCS or single-cell RNA sequencing (scRNA-seq), by which the gene copy number in a single nucleus could be precisely measured was introduced by the Cold Spring Harbor laboratory and MD Anderson Cancer Center in 2011 (8).
SCS strategies can be divided into three categories: whole-genome sequencing (WGS), whole-transcriptome sequencing (WTS), and epigenetic sequencing (9). In particular, WGS provides a uniform amplification process for assessing genomic sequences in target cells using an exon-trapping method (10). WTS is aimed at all transcripts and is especially suitable for GeneScan assessment of stem cells and early embryonic cells with high heterogeneity (11). Alternatively, epigenetic sequencing can be used to elucidate the unfolding roles of DNA methylation, histone modification and structural and regulatory chromatin loops in transcriptional heterogeneity (12). Optimized SCS-based strategies such as transposase-accessible chromatin with sequencing (ATAC-seq), single-nucleus RNA sequencing (snRNA-seq), single-cell chromatin immunoprecipitation sequencing (scChIP-seq), topographic single-cell sequencing (TSCS), and clustered regularly interspaced short palindromic repeats (CRISPR) droplet sequencing (CROP-seq) have been rapidly developed (13).
Single-cell separation is the first step of SCS and can be accomplished by immunomagnetic separation (IMS), Raman optical tweezers (ROT), fluorescence-activated cell sorting (FACS), microfluidic, and laser-capture microdissection (LCM) sorting (14, 15). Genome-wide nucleotide amplification, including whole-genome amplification (WGA) and whole-transcriptome amplification (WTA), is a straightforward strategy for SCS. Polymerase chain reaction (PCR) derivatives, such as degenerate oligonucleotide-primed PCR (DOP-PCR), multiple displacement amplification (MDA), and multiple annealing- and looping-based amplification cycles (MLBACs), can be used to accomplish WGA (16). Successful WTA depends heavily on in vitro transcription (IVT) and RNA amplification mediated by Phi29 DNA polymerase because polyadenylated mRNA is reverse transcribed and then linked to a splice sequence with oligo-DT primers for template conversion and cDNA generation (17). The analysis of sequencing data is the last part of SCS. Uneven coverage in the data and chimeric reads with great regularity are surprisingly common (18). Standardizing the nucleic acid library and performing hybrid analysis for sequencing data with high genetic similarity using SmashCell, Velvet-SC, and SPAdes software are strategies to provide more reliable results (19).
SCS techniques have emerged as promising approaches to dissect human tumors at the resolution of individual cells and therefore represent useful tools for deciphering cancer biology. Here, we summarize current SCS data from various human cancers.
Lung cancer is a very heterogeneous disease composed of multiple unique histologic subtypes that harbor distinct molecular signatures (20). ITH in preneoplastic lesions is often addressed in the context of missed diagnosis, limited therapeutic success, high mortality, and poor prognosis of lung cancer (21, 22). Tracing a gene map of precursor lesions in lung nodules was crucial for gaining a better intelligence about the mechanisms underlying the occurrence and development of early lung cancer. A single-cell transcriptome atlas of lung adenocarcinoma featuring ground-glass nodule adenocarcinoma (GGN-ADC) was created using scRNA-seq, and in the atlas, eight cell types (cancer cells, endothelial cells, fibroblasts, T cells, B cells, natural killer cells, mast cells, and myeloid cells) were found in the TME (23). The cell cycle and the nuclear factor (NF)-κB, Toll-like receptor (TLR), and vascular endothelial growth factor (VEGF) signaling pathways, which are related to cell proliferation, were found to be downregulated in GGN-ADC compared with solid adenocarcinoma (SADC) cancer cells (24). Similarly, activation of the phosphoinositide 3-kinase/protein kinase B (PI3K/AKT), hypoxia-inducible factor-1 (HIF-1), and VEGF signalling pathways, which accelerates angiogenesis, was attenuated in GGN-ADC endothelial cells (25). However, immunosuppressive pathways were activated in T cells derived from GGN-ADC, and the cytotoxicity of NK cells was more robust. In addition, macrophages tended to become M1 polarized, and mast cells were more enriched in GGN-ADC (26). These findings suggest that imbalanced activation of innate and adaptive immune responses not only makes immune escape possible but also easily induces an inflammatory storm. Transcriptome data from GGN-ADC-derived cells were also updated with data from scRNA-seq using module G64. These cells can be clearly divided into two groups according to the upregulated or downregulated expression of cell cycle-related genes, which is considered a guideline to identify surgical patients with lung adenocarcinoma in The Cancer Genome Atlas (TCGA) database (27). In summary, it has been well documented using scRNA-seq that cancer cells and the TME together preferentially contribute to the growth of GGN-ADC over SADC. Encouragingly, the dissimilarities within the TME between GGN-ADC and SADC provide clear ideas for elucidating why GGN-ADC remains stable and has better survival.
ScRNA-seq has also revealed heterogeneous tumor and immune cell populations in the TME of early-stage lung adenocarcinomas harboring EGFR mutations, among which myeloid cells, T cells, tumor-associated macrophages (TAMs), and dendritic cells (DCs) are prominent. In particular, DCs in lung adenocarcinomas are mainly CD1C+, implying their function in the inhibition of effector T cells and the promotion of regulatory T cells (Tregs). These tumor-infiltrating T cells demonstrate exhausted and Treg features. Furthermore, TAMs display protumoral functions without M1 or M2 polarization (28). By assessing tumor-infiltrating myeloid cells (TIMs) in patients with non-small-cell lung cancer (NSCLC) using scRNA-seq, 25 TIM-specific genes, such as TREM2, CD81, MARCO, and APOE, were consistently identified, and their expression in bone marrow cells from different species was extremely heterogeneous (29, 30). Dimensionality cluster analysis of scRNA-seq data from NSCLC patients identified undetectable expression of TNFRSF9, which suggested that the T cells were suffering from exhaustion. A low abundance of TNFRSF9 in these “pre-exhausted” cells was positively associated with a worse prognosis of NSCLC (31). These findings highlight the role of differences among tumor-infiltrating lymphocytes (TILs), the dysfunction of which happens to be a biomarker of immunotherapy response in lung cancer.
Tumor invasion and metastasis are responsible for the majority of deaths in lung cancer (32). The subtypes of lung cancer cells deviating from the differentiation track were identified using scRNA-seq, and the dynamic changes in stromal and immune cells suggested tumor metastasis (33). Circulating tumor cells (CTCs) may be an initiator of metastasis and induce poor prognosis in lung cancer (34). Characteristic exon-related indel polymorphisms in CTCs have also been identified through scRNA-seq. The copy number variation (CNV) in each CTC is highly consistent regardless of the cancer subtype, thus indicating that CNV is a dominant factor influencing tumor metastasis (35). Overall, the characterization of CTCs has provided fascinating insights into the heterogeneity of lung cancer, which may raise new ideas for improving the diagnosis and treatment of lung cancer.
Chemotherapy has been approved as a standard treatment for lung cancer; unfortunately, tumor metastasis and local recurrence are becoming increasingly common due to chemoresistance (36). Emerging scRNA-seq data from biopsy specimens from lung cancer patients treated with or without chemotherapy indicate that the surviving tumor cells in the TME exist in an intricate and dynamic ecosystem and are typically activated in the alveolar pathway (37). Another study showed that patient-derived xenograft (PDX) cells with notable expression of KRASG12D were resistant to chemotherapy, thus confirming that KRAS mutation status and a risk score can be used to predict the outcome in lung cancer (38). Therefore, studies discerning the cell distribution in lung cancer via SCS have suggested that drug-resistant subpopulations are more resistant to the action of chemotherapeutic drugs, even influencing the TME to benefit themselves.
As the third leading cause of cancer-related deaths worldwide, hepatocellular carcinoma (HCC) is difficult to combat due to its high degree of malignancy and poor prognosis (39). Immunotherapy has brought hope for the treatment of HCC (40). To assess the immune microenvironment in HCC and identify innovative biomarkers for immunotherapy of HCC, T cells in HCC tissues were analyzed by scRNA-seq, and T-cell subpopulations were reviewed in a transcriptional map. A large number of CD8+ T cells and Tregs were found. Layilin inhibited the activation of CD8+ T cells but activated Tregs and thus might be a potential agent for immunotherapy in HCC (41). These findings demonstrated that CD8+ T-cell failure can be a hallmark of hepatocarcinogenesis, which may be instrumental for the development of novel approaches for effective immunotherapy for HCC.
Non-alcoholic steatohepatitis (NASH) represents one of the leading causes of HCC, but the underlying mechanisms associated with the carcinogenesis of NASH-related HCC have not been clarified (42). ATAC-seq identified chromatin-accessible regions in NASH-derived HCC tissue samples, and 139 upregulated and 60 downregulated genes were specifically found. Interestingly, 15 of the 139 upregulated genes had accessible chromatin sites within 5 kb of the transcription start site (TSS), including APOA4, SERPINE1, IGFBP1, ANXA2, and TUBB2a. The upregulation of these genes was found to be associated with the enrichment of transcription factors (TFs), especially NFATC2, and histone H3K4me1 and H3K27ac gene transcription-activating marks in chromatin-accessible regions (43). These data highlight the role of chromatin accessibility perturbations in reshaping the chromatin landscape in NASH-related HCC. Hepatitis B virus (HBV) represents one of the most well-known etiologic factors of HCC (44). The mechanism by which HBV regulates both host and viral gene transcription involves chromatin modification mediated by an RNA helicase, DEAD-box protein 5 (DDX5). ATAC-seq was performed in which the chromatin accessibility of wild-type DDX5 cells vs. DDX5KD cells was compared. ATAC-seq of DDX5KD and wild-type DDX5 cells further identified that DDX5 inhibits Wnt signaling by affecting chromatin accessibility of genes involved in Wnt signaling, and DDX5 may be a negative regulator of Wnt signalling and hepatocyte reprogramming in HCC (45).
CD24, CD133, and epithelial cell adhesion molecule (EpCAM) have been identified as markers of hepatocarcinoma stem cells (HSCs), which have been determined to have ITH (46). ScRNA-seq data have verified that CD24+/CD44+ cells are enriched in EpCAM+ HSC populations (47). Therefore, the differentially expressed genes between HSC subtypes affect the prognosis of HCC. In addition, HSC-induced transcriptome diversity is tightly related to ITH generation, thus providing new ideas for biomarkers for predicting prognosis and treatment response in HCC.
ScRNA-seq data from CTCs of HCC patients have attracted much attention. Overexpression of IGF2, a gene overexpressed in 20% of early HCCs and recently characterized as an HCC epidriver, was detected in CTCs (48, 49). Thus, scRNA-seq of CTCs introduced the application of liquid biopsy in the early screening and diagnosis of HCC based on the non-mutated druggable genomic aberrations such as IGF2 overexpression. CTC count ≥16 and mesenchymal CTC (M-CTC) percentage ≥2% prior to resection have been identified as significantly associated with early recurrence, multi-intrahepatic recurrence, and lung metastasis. Gene expression profiling has identified a scene in which a concomitant increase in mesenchymal marker expression (vimentin and Twist) and reduced epithelial marker expression (EpCAM and E-cadherin) are exclusively found in CTCs (50). Moreover, CTCs have been isolated from the peripheral vein, hepatic vein, inferior vena cava, and portal vein of HCC patients with early metastasis. Gene expression profiling of these CTCs suggests that they are undifferentiated compared with epithelial−mesenchymal transition (EMT)-like cells, thus indicating that CTCs were changed by the TME and participated in ITH induction in metastatic HCC (51). Collectively, systematic adhesion of CTCs shapes EMT-driven HCC metastasis, hence suggesting the utility of individualized treatments for HCC.
ScRNA-seq analysis of single colorectal cancer and adjacent normal intestinal gland cells has been carried out. The results confirmed that colorectal cancer cells possessed a wide range of genetic differences, and the levels of DNA methylation in tumor cells also varied (52). Another important finding from CROP-seq is that colorectal adenoma and colorectal cancer are of the same origin based on ITH and can be divided into different subclonal types based on the presence of non-random somatic mutations in the G protein-coupled receptor (GPCR), intracellular PI3K/AKT, and fibroblast growth factor receptor (FGFR) signaling pathways (53). Primary and metastatic tumor cells and CTCs in colorectal cancer have also been assessed using scRNA-seq. APC-, KRAS-, and PIK3CA driver mutations in the corresponding CTCs were revealed and were present in the premetastatic lesions (54). Alternatively, two vastly different cancer-associated fibroblast (CAF) profiles were identified in the TME of primary colorectal cancer, and mesenchymal markers such as vimentin and N-cadherin were upregulated only in the “CTC-CAF” subgroup, suggesting the utility of EMT marker-directed diagnostic methods and targeted therapies (55). Thus, the roles of ITH in tumor evolution and metastasis of colorectal cancer have been well validated.
SCS data in esophageal cancer are scarce, and studies have mainly focused on chemoresistance. The paclitaxel-resistant esophageal cell line KYSE-30 was successfully induced through exposure to low-dose paclitaxel. Results acquired from scRNA-seq advocated for the presence of differential expression of KRT19 between KYSE-30 and parental esophageal cancer cells, thus suggesting that there is inherent paclitaxel resistance in esophageal cancer (56). KYSE-30 cells are also characterized by noteworthy expression of genes related to the ubiquitin proteasome pathway, and downregulation of the expression of such genes provokes the HIF-1 signaling pathway, ultimately resulting in paclitaxel resistance in esophageal cancer (56). However, additional SCS data from esophageal cancer are needed.
An unbiased transcriptome-wide scRNA-seq analysis of gastric cancer (GC) cells from gastric adenocarcinoma patients identified five subgroups with distinct gene expression profiles, marking the beginning of a new era in delineating the cellular heterogeneity in GC. Among the subgroups, three subgroups exhibited different differentiation grades, which corresponded well to the histopathological features of Lauren’s subtypes. Interestingly, the other two subgroups displayed unique transcriptome features. One subgroup expressing chief-cell markers (e.g., LIPF and PGC) and RNF43 and with Wnt/β-catenin signaling pathway activation was consistent with the previously described entity fundic gland-type GA (chief cell-predominant, GA-FG-CCP). The other subgroup specifically expressed immune-related signature genes (e.g., LY6K and MHC II) and had Epstein−Barr virus infection. In addition, non-malignant epithelium analysis using scRNA-seq has offered molecular evidence for the potential transition of gastric chief cells into MUC6+/TFF2+ spasmolytic polypeptide-expressing metaplastic cells (57). Hereditary diffuse gastric cancer (HDGC) is a cancer syndrome caused by germline variants in CDH1, the gene encoding the EMT marker E-cadherin, but the mechanisms through which CDH1 loss initiates HDGC have not been determined. In an automated scRNA-seq analysis, gastric epithelium with CDH1 deletion was found to be enriched for stromal cells, macrophages, dendritic cells (DCs) and Tregs. These TME-exclusive cells with distinct expression of extracellular matrix components and cytokeratin 7 (CK7) decreased transcriptional heterogeneity. In particular, the macrophages did not conform to the binary M1/M2 paradigm, and gastric DCs had a gene expression program unique from that of peripheral blood mononuclear cell (PBMC)-derived DCs. Moreover, TME-specific helper T cells, cytotoxic T cells, Tregs, and NK cells were found to express multiple immune checkpoint and costimulatory molecules (58). In addition, CDH1 loss resulted in shifts along the squamous differentiation trajectory, as well as the aberrant expression of the KRT7 gene (encoding the CK7 protein), which was identified as the top candidate leading to gastrointestinal epithelial invasion and early metastasis in CDH1 carriers (59). Chromatin accessibility has been identified as one of the most relevant genomic characteristics associated with oncologic functions at a specific site, providing the structure for transcription TFs binding to regulate multiple genes, thus driving cancer progression and invasion (60). Dysregulated chromatin accessibility is critical for GC metastasis, but the mechanisms remain unclear. ATAC-seq suggested that bromodomain-containing protein 4 (BRD4) is a crucial regulator of chromatin remodeling that promotes GC progression by regulating multiple genes. JQ1, also known as a small-molecule inhibitor of BRD4, drives chromatin accessibility changes in GC cells, and differentially accessible regions are highly enriched for RUNX2-binding motif. JQ1 suppressed GC metastasis by downregulating chromatin accessibility, and JQ1-inducing inhibition of GC cell metastasis is dependent on EMT suppression caused by alleviated activation of the RUNX2/NID1 pathway (61). Therefore, GC cells trigger TME-exclusive intercellular communication (leading to EMT), thus affecting distant metastasis.
Pancreatic cancer is a lethal disease with extremely poor prognosis and is characterized by high heterogeneity of tumor cells and the TME, which lead existing therapies to offer only limited effectiveness (62). Cystomas are believed to be involved in inciting the development of pancreatic cancer. Of cystomas, intraductal papillary Mucinous neoplasms (IPMNs) are the most commonly identified, but there are no reliable markers to distinguish inert from invasive IPMNs (63). ScRNA-seq was used to trace precancerous lesions with 5,403 cells from low-grade IPMN (LGD), high-level IPMN (HGD), and pancreatic cancer specimens. Heterogeneity in epithelial cells of LGD specimens was found; this caused overexpression of the oncogene transcriptome in LGD specimens, and tumor growth-suppressing pathways were evidently activated at the same time. However, these tumor growth-suppressing pathways were repressed in HGD specimens and were inactivated in pancreatic cancer specimens (64). Acinar-to-ductal metaplasia is an early event in the initiation of pancreatic cancer but is generally poorly understood. SnRNA-seq indicates that the expression of the PDAC-associated oncogene GNASR201C more effectively produces cystic growth in ductal organoids than in acinar organoids, whereas KRASG12D more effectively models cancer in vivo when expressed in acinar organoids than when expressed in ductal organoids. KRASG12D, but not GNASR201C, induces acinar-to-ductal metaplasia-like changes in culture and in vivo (65). Furthermore, the transcription factor ONECUT2 was identified as a putative driver of early progression of pancreatic cancer whose role in the activation of the KRAS pathway was sufficient to induce acinar hyperplasia, although the neoplastic lesions developed focally (66). Regardless, SCS has identified a renewable source of ductal and acinar organoids for modeling exocrine development and diseases, and lineage tropism and plasticity in terms of oncogene action in the human pancreas have been well understood using SCS.
Most patients with pancreatic cancer will relapse within 4 years despite early diagnosis and surgical resection, suggesting that early micrometastasis develops silently (67). The response to adjuvant therapies is heterogeneous, with metastasis typically resulting due to mutations in the CDKN2A, SMAD4, TP53, and KRAS genes (68). A TSCS study described the spatial characteristics of metastasis of pancreatic cancer, in which a cutoff of at least 10 CTCs was established as a lower limit for reliable KRAS mutational analysis of CTCs (69). More recently, a variety of cell subsets in primary and metastatic tumor tissues were identified through scRNA-seq, among which CTCs with an EMT phenotype were located in only metastatic lesions and predicted a worse prognosis of pancreatic cancer (70). Another meaningful study using scRNA-seq identified heterogeneity between primary and metastatic lesions, confirming that BIRC5 expression was massively upregulated in metastatic CTCs. YM155 administration in a model with BIRC5 deletion shrank the metastatic foci in SW1990-bearing mice, thus indicating that BIRC5 in CTCs is a key coordinator of the metastasis of pancreatic cancer (71).
A gene map of fibroblasts in pancreatic cancer was drawn using scRNA-seq analysis, in which transforming growth factor (TGF)-β1 and LRRC15 expression were prominent. These fibroblasts appeared insensitive to anti-programmed death ligand 1 (PD-L1) therapy (72). Previous studies have shown that CD47 expression tempers the phagocytosis of macrophages (73), but little is known about CD47 expression in fibroblasts and its implication in pancreatic cancer. In fact, it has been found using scRNA-seq that anti-CD47 treatment leads to the chemotaxis of pro-inflammatory fibroblasts and the diminishment of immunosuppressive macrophages (74). More strikingly, the inflammatory fibroblasts overexpressed CXCL12, and these CXCL12-expressing fibroblasts were found to encourage the immune escape of pancreatic cancer cells (75). Therefore, these “unconventional” fibroblasts were reprogrammed to induce an immunotolerance framework in pancreatic cancer.
Breast cancer originates from ducts and epithelial cells and gradually develops from hyperplasia to atypical hyperplasia, in situ (adeno)carcinoma, and finally early and advanced invasive carcinoma (76). As early as 2011, the evolution of multicellularity in breast cancer was deduced, and three subpopulations in different clones were identified through scRNA-seq. An unexpectedly rich subgroup, namely, “pseudodiploid” cells, was reported. These “pseudodiploid” cells do not migrate to the site of disease progression and do not follow the path of tumor progression, which illustrates that breast cancer cells grow continuously with clonal expansion (8). The continuous growth of clonal subpopulations guides cell differentiation into five subtypes in accordance with the expression of estrogen receptor (ER), progesterone receptor (PR), and human epidermal growth factor receptor 2 (HER2) (77). Determining the changes in gene copy number with SCS is helpful for distinguishing cancer cells from non-cancer cells. Non-cancer cells include T cells, B cells, and macrophages. Both T cells and macrophages are regarded widely as immunosuppressive: Tregs can experience immune anergy and exhibit co-mutation of CTLA4, TIGHT, and GITR genes, and macrophages can take on an M2 phenotype (78, 79). Furthermore, aberrant expression of CDH1 and PVRL2, NK cell inhibitory receptor ligands, plays a very important role in the immune evasion of breast cancer cells. ATAC-seq data have demonstrated that CDH1 is positively regulated by TFs such as NFATC2, XBP1, YY1AR, GTF2I, IRF2, and NF1, and variations in CDH1 expression are mainly caused by CNV. However, CNV and the mRNA level of PVRL2 are very weakly correlated, and high expression of PVRL2 may also be induced by increases in TFs including FOXP3, GTF2I, YY1, NR3C1, SP1, TFAP2A, AR, ESR1, PAX5, and TP53 (80). This evidence indicates that ITH in breast cancer is boosted by the combined action of tumor cells and immune cells in the TME.
Somatic mutations provide a basis for the selection of clones with an advantage and imply that there is a causal relationship of oncogene dynamics with breast cancer (81). Somatic TP53 and PIK3CA driver mutations have been found to originate early in the progression of breast cancer (82). Changes in somatic copy number and the mutated genes encoding exogenous proteins in breast cancer have been identified, and strong correlations between mutations and histological grade have been corroborated using scRNA-seq (83, 84). In conclusion, SCS has contributed to a novel understanding of cell-level somatic mutation information that can facilitate investigation of cell heterogeneity.
Ductal carcinoma in situ (DCIS) constitutes the most common form of early breast cancer, but the metastasis of DCIS remains poorly understood (85). TSCS has been used to assess changes in gene copy number and to track the spatial information and clonal evolution of single tumor cells sorted from metastatic DCIS-IDC patients. A common genomic lineage was identified between in situ and invasive subpopulations, and most mutations and copy number aberrations evolved in the premetastatic lesion before tumor metastasis (86). The clones cooperated with each other to escape from the basement membrane and migrated to adjacent tissues to establish invasive carcinoma (87). Moreover, the heterogeneity between the primary tumor and metastases in lymph nodes was assessed using scRNA-seq, and a novel cell subpopulation called CXCL14 cancer cells was identified in the positive lymph nodes of breast cancer patients. ATAC-seq of the positive and negative lymph node samples further revealed the chromatin accessibility profile and identified potential TFs related to CXCL14 cancer cells, including ZNF467, bZIP, EBF1, and PIT1, in the lymph node metastases of breast cancer (88).
Triple-negative breast cancer (TNBC) is the most invasive and malignant type of breast cancer, and TNBC lacks ER, PR, and HER2 expression (89). Chemotherapy remains the standard therapeutic approach for TNBC, with neoadjuvant chemotherapy (NAC) being preferred (90). However, nearly half of the patients with TNBC have NAC resistance, leading to overall refractoriness of the disease. Two distinct clonal dynamic patterns were identified in NBC patients with NAC using scRNA-seq: “regressive” and “persistent.” NAC administration eliminated almost all the tumor cells in “regressive” patients, allowing only autosomal diploid cells such as fibroblasts and immune cells to thrive. In contrast, there were a large number of residual tumor cells in “persistent” patients whose genotypes were reprogrammed by NAC (91). Furthermore, the gene profile was selectively modulated, and the transcription profile in TNBC patients treated with docetaxel and epirubicin was modulated. However, a series of chemoresistance-driving genes had shown increased expression before NAC administration, implying that chemoresistance was preexisting (91). After employing scChIP-seq for breast cancer PDX samples, tumor heterogeneity at the chromatin level was characterized. Notably, loss of repressive chromatin marks such as H3K27me3 is associated with stable transcriptional repression of indicated genes that probably cause resistance to NAC (92). ScRNA-seq data have laid a foundation for clarifying the role of tissue-resident memory (TRM) cells differentiated from CD8+ T cells in breast cancer. These CD8+ TRM cells show remarkable expression of fatty acid-binding protein 4 (FABP4) and FABP5, which have received public attention for their role as immune checkpoint proteins that likely influence survival in TNBC patients (93). Overall, the development of SCS, by which genomic, transcriptomic, and epigenetic information from TNBC cells can be extracted, has been timely and has enabled the effective identification of unique mutations influencing ITH in rare subpopulations and an understanding of the evolutionary lineage.
The last decade has witnessed great advances in our understanding of the genetic basis of childhood T-cell acute lymphoblastic leukemia (T-ALL), but the effects of gene mutations in progenitor cells of T-ALL are ambiguous (94). In this regard, T-ALL mutations have been clarified using scRNA-seq. Specifically, loss of the chromosome 9p21 fragment was found to be an intermediate event, while Notch1 mutation was usually identified as a late event. Furthermore, STIL-TAL1 gene fusion and CDKN2A gene loss were identified as early and major events (95, 96).
Diffuse large B-cell lymphoma (DLBCL) is one of the most common subtypes of non-Hodgkin’s lymphoma (NHL). ScRNA-seq was used to reveal the phenotypic heterogeneity in DLBCL, revealing the close association of MHC II heterogeneity with DLBCL initiation (97). Vitreoretinal lymphoma (VRL) is a rare ocular malignant tumor (98). ScRNA-seq confirmed that the MyD88 L265P mutation accounts for more than 60% of all VRL mutations and can be used as a useful marker for the diagnosis of VRL (99–101). Mantle cell lymphoma (MCL) is an incurable form of NHL (102). MCL subsets with obvious TME heterogeneity have been identified through the implementation of scRNA-seq. Immune escape and drug resistance mechanisms in MCL are related to drug metabolism, DNA damage repair, apoptosis, and survival promotion (103).
Cutaneous T-cell lymphoma (CTCL) is a heterogeneous group of lymphoproliferative disorders, including Sézary syndrome, which has a poor prognosis and unclear pathogenesis (104). ScRNA-seq was performed and revealed that Foxp3+ malignant T cells induce the metastasis of GATA3+ and IKZF2+ clonal tumors, and Foxp3 might be a candidate for predicting early CTCL (105).
Langerhans cell histiocytosis (LCH), a spectrum of diseases formerly known as histiocytosis X, is characterized by diverse clinical manifestations involving bone, skin, lung, and pituitary tissue (106). Detailed maps of LCH lesions have been generated with scRNA-seq data, which confirmed the evolution of LCH and provided ground-breaking indicators for personalized treatment of LCH (107).
In conclusion, the potential roles of SCS in the diagnosis and treatment of hematological malignancies have been summarized, but more efforts should be made to highlight the advantages of SCS from a clinical standpoint.
SCS has obviously laid a foundation for a renewed understanding of tumor initiation and evolution. Here, the precise characterization of single-cell programs in lung cancer, HCC, colorectal cancer, esophageal cancer, GC, pancreatic cancer, breast cancer, and hematological malignancies was reviewed, highlighting the importance of cellular biology in cancer biology (Figure 1). However, there are many future challenges for this field that should not be ignored. In particular, the accuracy of targeted single-cell isolation, purification, and measurement is critical for ensuring the validity of SCS data. It is truly difficult to isolate the target cells and prevent them from being cross-contaminated with other cells. SCS technological developments are opening multiple new avenues, and cryopreservation has been shown to be suitable for cell capture and library preparation (108). Moreover, an automated single-cell analysis and isolation system was developed that showed superior noninvasive isolation of a single cell with the most favorable properties from arrays containing 105 cells comparing with a fluorescence-activated cell sorter. Such automated technology can be used for high-throughput screening for single-cell isolation with targeted labeling (109). In addition, amplification failure and allelic contamination with nonspecific products are frequent in SCS. Improvements in strategies such as template switching, second-strand synthesis for in vitro transcription mediated by RNaseH/DNA polymerase I, and poly(A) tagging have led to better quantitative performance of SCS (110). Additionally, SCS prevalence has suffered due to the substantial cost of bioinformatics analysis, and it is more difficult for patients who have already borne the heavy burden of cancer to agree to undergo SCS analysis. Fortunately, the development of alternate scRNA-seq platforms, in which thousands of cells are profiled, is enabling the exploration of tumor subpopulations, including those with adaptations underlying chemoresistance and metastatic advancement (111). Moreover, the Human Cell Atlas (HCA) project is attempting to conduct phenotyping at the single-cell level, hence generating cellular maps of cell lineages, organs, and organisms (112). Thus, deciphering the genome of a single cell will eventually be applicable in ordinary cancer patients, as many efforts have been made to continuously optimize single-cell separation and single-cell genome amplification technologies. The place of SCS in understanding ITH development and effects as they apply to tumor development, metastasis, chemoresistance, and immune escape cannot be discounted.
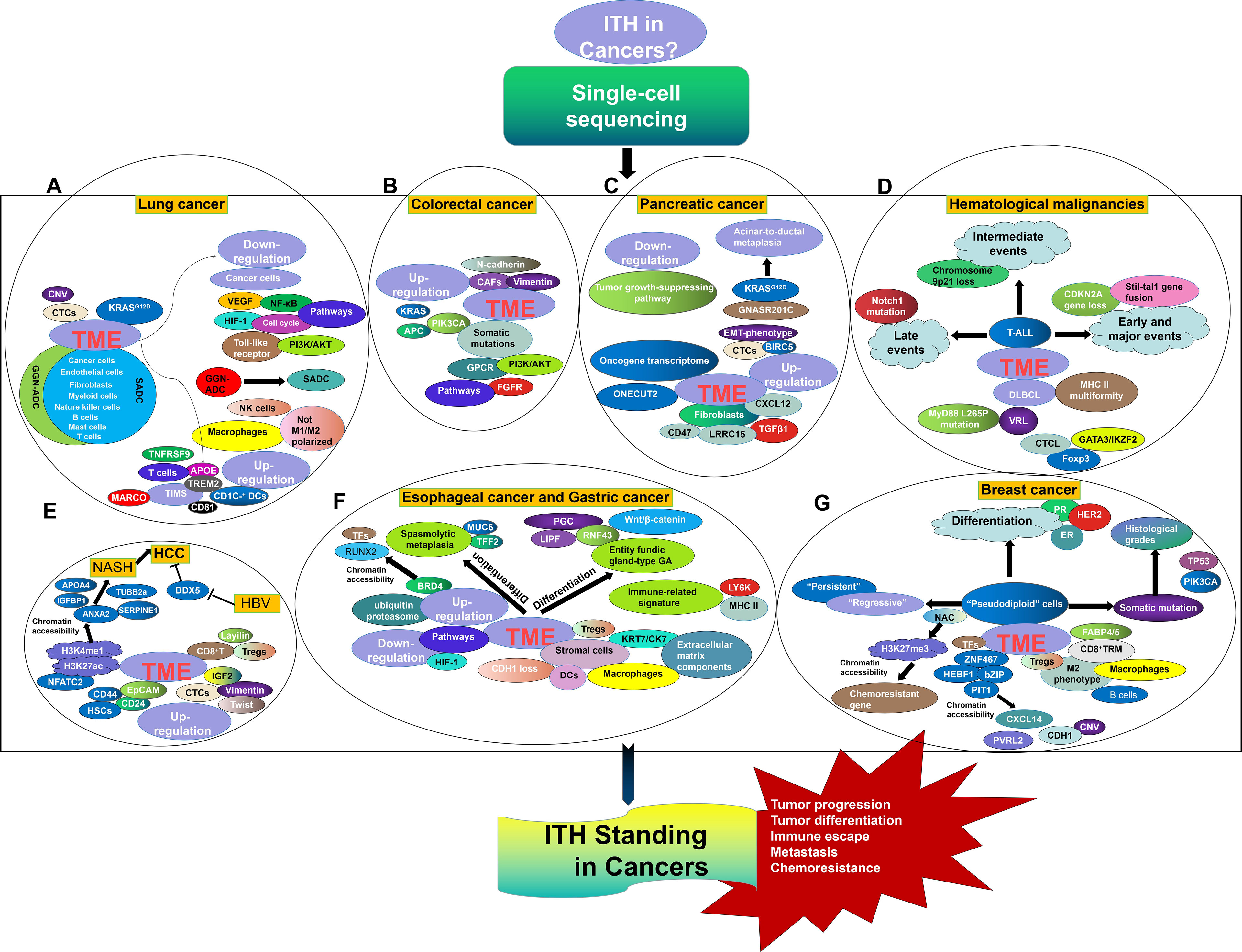
Figure 1 ITH Standing in Lung cancer (A), Colorectal cancer (B), Pancreatic cancer (C), Hematological malignancies (D), HCC (E), Esophageal cancer and gastric cancer (F), and Breast cancer (G) from the perspective of SCS.
JL contributed study concept design to this study. NY, XL, and MC consulted indicated references and refined interpreted information from acquired data. QG wrote the first draft of the article. All authors contributed to the article and approved the submitted version.
This work was supported by the Shandong Provincial Natural Science Foundation (ZR2020QH362).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.

1. Martín-Lorenzo A, Gonzalez-Herrero I, Rodríguez-Hernández G, García-Ramírez I, Vicente-Dueñas C, Sánchez-García I. Early Epigenetic Cancer Decisions. Biol Chem (2014) 395:1315–20. doi: 10.1515/hsz-2014-0185
2. Janiszewska M. The Microcosmos of Intratumor Heterogeneity: The Space-Time of Cancer Evolution. Oncogene (2020) 39:2031–9. doi: 10.1038/s41388-019-1127-5
3. Zhou H, Neelakantan D, Ford HL. Clonal Cooperativity in Heterogenous Cancers. Semin Cell Dev Biol (2017) 64:79–89. doi: 10.1016/j.semcdb.2016.08.028
4. Zhu X, Li S, Xu B, Luo H. Cancer Evolution: A Means by Which Tumors Evade Treatment. BioMed Pharmacother (2021) 133:111016. doi: 10.1016/j.biopha.2020.111016
5. Lindeboom RGH, Regev A, Teichmann SA. Towards a Human Cell Atlas: Taking Notes From the Past. Trends Genet (2021) 37:625–30. doi: 10.1016/j.tig.2021.03.007
6. Eberwine J, Sul JY, Bartfai T, Kim J. The Promise of Single-Cell Sequencing. Nat Methods (2014) 11:25–7. doi: 10.1038/nmeth.2769
7. Kraft F, Kurth I. Long-Read Sequencing to Understand Genome Biology and Cell Function. Int J Biochem Cell Biol (2020) 126:105799. doi: 10.1016/j.biocel.2020.105799
8. Navin N, Kendall J, Troge J, Andrews P, Rodgers L, McIndoo J, et al. Tumour Evolution Inferred by Single-Cell Sequencing. Nature (2011) 472:90–4. doi: 10.1038/nature09807
9. Lee J, Hyeon DY, Hwang D. Single-Cell Multiomics: Technologies and Data Analysis Methods. Exp Mol Med (2020) 52:1428–42. doi: 10.1038/s12276-020-0420-2
10. Zhao EY, Jones M, Jones SJM. Whole-Genome Sequencing in Cancer. old Spring Harb Perspect Med (2019) 9:a034579. doi: 10.1101/cshperspect.a034579
11. Shapira G, Shomron N. Single-Cell Transcriptome Profiling. Methods Mol Biol (2021) 2243:311–25. doi: 10.1007/978-1-0716-1103-6_16
12. Qi Z, Barrett T, Parikh AS, Tirosh I, Puram SV. Single-Cell Sequencing and its Applications in Head and Neck Cancer. Oral Oncol (2019) 99:104441. doi: 10.1016/j.oraloncology.2019.104441
13. Tang X, Huang Y, Lei J, Luo H, Zhu X. The Single-Cell Sequencing: New Developments and Medical Applications. Cell Biosci (2019) 9:53. doi: 10.1186/s13578-019-0314-y
14. Seth-Smith HM, Harris SR, Scott P, Parmar S, Marsh P, Unemo M, et al. Generating Whole Bacterial Genome Sequences of Low-Abundance Species From Complex Samples With IMS-MDA. Nat Protoc (2013) 8:2404–12. doi: 10.1038/nprot.2013.147
15. Bai X, Li Y, Zeng X, Zhao Q, Zhang Z. Single-Cell Sequencing Technology in Tumor Research. Clin Chim Acta (2021) 518:101–9. doi: 10.1016/j.cca.2021.03.013
16. Gawad C, Koh W, Quake SR. Single-Cell Genome Sequencing: Current State of the Science. Nat Rev Genet (2016) 17:175–88. doi: 10.1038/nrg.2015.16
17. Morin JA, Cao FJ, Lázaro JM, Arias-Gonzalez JR, Valpuesta JM, Carrascosa JL, et al. Active DNA Unwinding Dynamics During Processive DNA Replication. Proc Natl Acad Sci U S A (2012) 109:8115–20. doi: 10.1073/pnas.1204759109
18. Zheng Z, Chen E, Lu W, Mouradian G, Hodges M, Liang M, et al. Single-Cell Transcriptomic Analysis. Compr Physiol (2020) 10:767–83. doi: 10.1002/cphy.c190037
19. Olson ND, Lund SP, Colman RE, Foster JT, Sahl JW, Schupp JM, et al. Best Practices for Evaluating Single Nucleotide Variant Calling Methods for Microbial Genomics. Front Genet (2015) 6:235. doi: 10.3389/fgene.2015.00235
20. Wadowska K, Bil-Lula I, Trembecki Ł, Śliwińska-Mossoń M. Genetic Markers in Lung Cancer Diagnosis: A Review. Int J Mol Sci (2020) 21:4569. doi: 10.3390/ijms21134569
21. Hua X, Zhao W, Pesatori AC, Consonni D, Caporaso NE, Zhang T, et al. Genetic and Epigenetic Intratumor Heterogeneity Impacts Prognosis of Lung Adenocarcinoma. Nat Commun (2020) 11:2459. doi: 10.1038/s41467-020-16295-5
22. Zhao W, Zhu B, Hutchinson A, Pesatori AC, Consonni D, Caporaso NE, et al. Clinical Implications of Inter- and Intra-Tumor Heterogeneity of Immune Cell Markers in Lung Cancer. J Natl Cancer Inst (2021), djab157. doi: 10.1093/jnci/djab157
23. Lu T, Yang X, Shi Y, Zhao M, Bi G, Liang J, et al. Single-Cell Transcriptome Atlas of Lung Adenocarcinoma Featured With Ground Glass Nodules. Cell Discovery (2020) 6:69. doi: 10.1038/s41421-020-00200-x
24. Wang H, Meng D, Guo H, Sun C, Chen P, Jiang M, et al. Single-Cell Sequencing, an Advanced Technology in Lung Cancer Research. Onco Targets Ther (2021) 14:1895–909. doi: 10.2147/OTT.S295102
25. Chong ZX, Ho WY, Yeap SK, Wang ML, Chien Y, Verusingam ND, et al. Single-Cell RNA Sequencing in Human Lung Cancer: Applications, Challenges and Pathway Towards Personalized Therapy. J Chin Med Assoc (2021) 84:563–76. doi: 10.1097/JCMA.0000000000000535
26. Zhong R, Chen D, Cao S, Li J, Han B, Zhong H. Immune Cell Infiltration Features and Related Marker Genes in Lung Cancer Based on Single-Cell RNA-Seq. Clin Transl Oncol (2021) 23:405–17. doi: 10.1007/s12094-020-02435-2
27. Min JW, Kim WJ, Han JA, Jung YJ, Kim KT, Park WY, et al. Identification of Distinct Tumor Subpopulations in Lung Adenocarcinoma via Single-Cell RNA-Seq. PloS One (2015) 10:e0135817. doi: 10.1371/journal.pone.0135817
28. He D, Wang D, Lu P, Yang N, Xue Z, Zhu X, et al. Single-Cell RNA Sequencing Reveals Heterogeneous Tumor and Immune Cell Populations in Early-Stage Lung Adenocarcinomas Harboring EGFR Mutations. Oncogene (2021) 40:355–68. doi: 10.1038/s41388-020-01528-0
29. Lavin Y, Kobayashi S, Leader A, Amir ED, Elefant N, Bigenwald C, et al. Innate Immune Landscape in Early Lung Adenocarcinoma by Paired Single-Cell Analyses. Cell (2017) 169:750–65.e17. doi: 10.1016/j.cell.2017.04.014
30. Zilionis R, Engblom C, Pfirschke C, Savova V, Zemmour D, Saatcioglu HD, et al. Single-Cell Transcriptomics of Human and Mouse Lung Cancers Reveals Conserved Myeloid Populations Across Individuals and Species. Immunity (2019) 50:1317–34.e10. doi: 10.1016/j.immuni.2019.03.009
31. Cho JW, Son J, Ha SJ, Lee I. Systems Biology Analysis Identifies TNFRSF9 as a Functional Marker of Tumor-Infiltrating Regulatory T-Cell Enabling Clinical Outcome Prediction in Lung Cancer. Comput Struct Biotechnol J (2021) 19:860–8. doi: 10.1016/j.csbj.2021.01.025
32. Talasaz A. Lung Cancer and a Bold New Vision. Future Oncol (2020) 16:701–3. doi: 10.2217/fon-2019-0323
33. Kim N, Kim HK, Lee K, Hong Y, Cho JH, Choi JW, et al. Single-Cell RNA Sequencing Demonstrates the Molecular and Cellular Reprogramming of Metastatic Lung Adenocarcinoma. Nat Commun (2020) 11:2285. doi: 10.1038/s41467-020-16164-1
34. Yousefi M, Ghaffari P, Nosrati R, Dehghani S, Salmaninejad A, Abarghan YJ, et al. Prognostic and Therapeutic Significance of Circulating Tumor Cells in Patients With Lung Cancer. Cell Oncol (Dordr) (2020) 43:31–49. doi: 10.1007/s13402-019-00470-y
35. Ni X, Zhuo M, Su Z, Duan J, Gao Y, Wang Z, et al. Reproducible Copy Number Variation Patterns Among Single Circulating Tumor Cells of Lung Cancer Patients. Proc Natl Acad Sci U S A (2013) 110:21083–8. doi: 10.1073/pnas.1320659110
36. Pirker R. Conquering Lung Cancer: Current Status and Prospects for the Future. Pulmonology (2020) 26:283–90. doi: 10.1016/j.pulmoe.2020.02.005
37. Maynard A, McCoach CE, Rotow JK, Harris L, Haderk F, Kerr DL, et al. Therapy-Induced Evolution of Human Lung Cancer Revealed by Single-Cell RNA Sequencing. Cell (2020) 182:1232–51.e22. doi: 10.1016/j.cell.2020.07.017
38. Kim KT, Lee HW, Lee HO, Kim SC, Seo YJ, Chung W, et al. Single-Cell mRNA Sequencing Identifies Subclonal Heterogeneity in Anti-Cancer Drug Responses of Lung Adenocarcinoma Cells. Genome Biol (2015) 16:127. doi: 10.1186/s13059-015-0692-3
39. Rubin JN, Gilman CAL, Gasmi B, Stern WR, Hernandez JM, Koh C. Hepatocellular Carcinoma Where You Least Expect it. Am J Gastroenterol (2020) 115:1134–6. doi: 10.14309/ajg.0000000000000616
40. Gryziak M, Woźniak K, Kraj L, Stec R. Milestones in the Treatment of Hepatocellular Carcinoma: A Systematic Review. Crit Rev Oncol Hematol (2021) 157:103179. doi: 10.1016/j.critrevonc.2020.103179
41. Zheng C, Zheng L, Yoo JK, Guo H, Zhang Y, Guo X, et al. Landscape of Infiltrating T Cells in Liver Cancer Revealed by Single-Cell Sequencing. Cell (2017) 169:1342–56.e16. doi: 10.1016/j.cell.2017.05.035
42. Lequoy M, Gigante E, Couty JP, Desbois-Mouthon C. Hepatocellular Carcinoma in the Context of Non-Alcoholic Steatohepatitis (NASH): Recent Advances in the Pathogenic Mechanisms. Horm Mol Biol Clin Investig (2020) 41:20190044. doi: 10.1515/hmbci-2019-0044/j/hmbci.2020.41.issue-1/hmbci-2019-0044/hmbci-2019-0044.xml
43. Dechassa ML, Tryndyak V, de Conti A, Xiao W, Beland FA, Pogribny IP. Identification of Chromatin-Accessible Domains in Non-Alcoholic Steatohepatitis-Derived Hepatocellular Carcinoma. Mol Carcinog (2018) 57:978–87. doi: 10.1002/mc.22818
44. McGlynn KA, Petrick JL, El-Serag HB. Epidemiology of Hepatocellular Carcinoma. Hepatology (2021) 73 Suppl 1:4–13. doi: 10.1002/hep.31288
45. Mani SKK, Yan B, Cui Z, Sun J, Utturkar S, Foca A, et al. Restoration of RNA Helicase DDX5 Suppresses Hepatitis B Virus (HBV) Biosynthesis and Wnt Signaling in HBV-Related Hepatocellular Carcinoma. Theranostics (2020) 10:10957–72. doi: 10.7150/thno.49629
46. Castelli G, Pelosi E, Testa U. Liver Cancer: Molecular Characterization, Clonal Evolution and Cancer Stem Cells. Cancers (Basel) (2017) 9:127. doi: 10.3390/cancers9090127
47. Ho DW, Tsui YM, Sze KM, Chan LK, Cheung TT, Lee E, et al. Single-Cell Transcriptomics Reveals the Landscape of Intra-Tumoral Heterogeneity and Stemness-Related Subpopulations in Liver Cancer. Cancer Lett (2019) 459:176–85. doi: 10.1016/j.canlet.2019.06.002
48. Martinez-Quetglas I, Pinyol R, Dauch D, Torrecilla S, Tovar V, Moeini A, et al. IGF2 Is Up-Regulated by Epigenetic Mechanisms in Hepatocellular Carcinomas and Is an Actionable Oncogene Product in Experimental Models. Gastroenterology (2016) 151:1192–205. doi: 10.1053/j.gastro.2016.09.001
49. D'Avola D, Villacorta-Martin C, Martins-Filho SN, Craig A, Labgaa I, von Felden J, et al. High-Density Single Cell mRNA Sequencing to Characterize Circulating Tumor Cells in Hepatocellular Carcinoma. Sci Rep (2018) 8:11570. doi: 10.1038/s41598-018-30047-y
50. Qi LN, Xiang BD, Wu FX, Ye JZ, Zhong JH, Wang YY, et al. Circulating Tumor Cells Undergoing EMT Provide a Metric for Diagnosis and Prognosis of Patients With Hepatocellular Carcinoma. Cancer Res (2018) 78:4731–44. doi: 10.1158/0008-5472.CAN-17-2459
51. Sun YF, Guo W, Xu Y, Shi YH, Gong ZJ, Ji Y, et al. Circulating Tumor Cells From Different Vascular Sites Exhibit Spatial Heterogeneity in Epithelial and Mesenchymal Composition and Distinct Clinical Significance in Hepatocellular Carcinoma. Clin Cancer Res (2018) 24:547–59. doi: 10.1158/1078-0432.CCR-17-1063
52. Roerink SF, Sasaki N, Lee-Six H, Young MD, Alexandrov LB, Behjati S, et al. Intra-Tumour Diversification in Colorectal Cancer at the Single-Cell Level. Nature (2018) 556:457–62. doi: 10.1038/s41586-018-0024-3
53. Wu H, Zhang XY, Hu Z, Hou Q, Zhang H, Li Y, et al. Evolution and Heterogeneity of Non-Hereditary Colorectal Cancer Revealed by Single-Cell Exome Sequencing. Oncogene (2017) 36:2857–67. doi: 10.1038/onc.2016.438
54. Leung ML, Davis A, Gao R, Casasent A, Wang Y, Sei E, et al. Single-Cell DNA Sequencing Reveals a Late-Dissemination Model in Metastatic Colorectal Cancer. Genome Res (2017) 27:1287–99. doi: 10.1101/gr.209973.116
55. Li H, Courtois ET, Sengupta D, Tan Y, Chen KH, Goh JJL, et al. Reference Component Analysis of Single-Cell Transcriptomes Elucidates Cellular Heterogeneity in Human Colorectal Tumors. Nat Genet (2017) 49:708–18. doi: 10.1038/ng.3818
56. Wu H, Chen S, Yu J, Li Y, Zhang XY, Yang L, et al. Single-Cell Transcriptome Analyses Reveal Molecular Signals to Intrinsic and Acquired Paclitaxel Resistance in Esophageal Squamous Cancer Cells. Cancer Lett (2018) 420:156–67. doi: 10.1016/j.canlet.2018.01.059
57. Zhang M, Hu S, Min M, Ni Y, Lu Z, Sun X, et al. Dissecting Transcriptional Heterogeneity in Primary Gastric Adenocarcinoma by Single Cell RNA Sequencing. Gut (2021) 70:464–75. doi: 10.1136/gutjnl-2019-320368
58. Sathe A, Grimes SM, Lau BT, Chen J, Suarez C, Huang RJ, et al. Single-Cell Genomic Characterization Reveals the Cellular Reprogramming of the Gastric Tumor Microenvironment. Clin Cancer Res (2020) 26:2640–53. doi: 10.1158/1078-0432.CCR-19-3231
59. Dixon K, Brew T, Farnell D, Godwin TD, Cheung S, Chow C, et al. Modelling Hereditary Diffuse Gastric Cancer Initiation Using Transgenic Mouse-Derived Gastric Organoids and Single-Cell Sequencing. J Pathol (2021) 254:254–64. doi: 10.1002/path.5675
60. Stadhouders R, Vidal E, Serra F, Di Stefano B, Le Dily F, Quilez J, et al. Transcription Factors Orchestrate Dynamic Interplay Between Genome Topology and Gene Regulation During Cell Reprogramming. Nat Genet (2018) 50:238–49. doi: 10.1038/s41588-017-0030-7
61. Zhou S, Zhang S, Wang L, Huang S, Yuan Y, Yang J, et al. BET Protein Inhibitor JQ1 Downregulates Chromatin Accessibility and Suppresses Metastasis of Gastric Cancer via Inactivating RUNX2/NID1 Signaling. Oncogenesis (2020) 9:33. doi: 10.1038/s41389-020-0218-z
62. Jain T, Dudeja V. The War Against Pancreatic Cancer in 2020-Advances on All Fronts. Nat Rev Gastroenterol Hepatol (2021) 18:99–100. doi: 10.1038/s41575-020-00410-4
63. Rezaee N, Barbon C, Zaki A, He J, Salman B, Hruban RH, et al. Intraductal Papillary Mucinous Neoplasm (IPMN) With High-Grade Dysplasia Is a Risk Factor for the Subsequent Development of Pancreatic Ductal Adenocarcinoma. HPB (Oxford) (2016) 18:236–46. doi: 10.1016/j.hpb.2015.10.010
64. Bernard V, Semaan A, Huang J, San Lucas FA, Mulu FC, Stephens BM, et al. Single-Cell Transcriptomics of Pancreatic Cancer Precursors Demonstrates Epithelial and Microenvironmental Heterogeneity as an Early Event in Neoplastic Progression. Clin Cancer Res (2019) 25:2194–205. doi: 10.1158/1078-0432.CCR-18-1955
65. Huang L, Desai R, Conrad DN, Leite NC, Akshinthala D, Lim CM, et al. Commitment and Oncogene-Induced Plasticity of Human Stem Cell-Derived Pancreatic Acinar and Ductal Organoids. Cell Stem Cell (2021) 28:1090–104.e6. doi: 10.1016/j.stem.2021.03.022
66. Schlesinger Y, Yosefov-Levi O, Kolodkin-Gal D, Granit RZ, Peters L, Kalifa R, et al. Single-Cell Transcriptomes of Pancreatic Preinvasive Lesions and Cancer Reveal Acinar Metaplastic Cells' Heterogeneity. Nat Commun (2020) 11:4516. doi: 10.1038/s41467-020-18207-z
68. Wood LD, Hruban RH. Pathology and Molecular Genetics of Pancreatic Neoplasms. Cancer J (2012) 18:492–501. doi: 10.1097/PPO.0b013e31827459b6
69. Court CM, Ankeny JS, Sho S, Hou S, Li Q, Hsieh C, et al. Reality of Single Circulating Tumor Cell Sequencing for Molecular Diagnostics in Pancreatic Cancer. J Mol Diagn (2016) 18:688–96. doi: 10.1016/j.jmoldx.2016.03.006
70. Lin W, Noel P, Borazanci EH, Lee J, Amini A, Han IW, et al. Single-Cell Transcriptome Analysis of Tumor and Stromal Compartments of Pancreatic Ductal Adenocarcinoma Primary Tumors and Metastatic Lesions. Genome Med (2020) 12:80. doi: 10.1186/s13073-020-00776-9
71. Dimitrov-Markov S, Perales-Patón J, Bockorny B, Dopazo A, Muñoz M, Baños N, et al. Discovery of New Targets to Control Metastasis in Pancreatic Cancer by Single-Cell Transcriptomics Analysis of Circulating Tumor Cells. Mol Cancer Ther (2020) 19:1751–60. doi: 10.1158/1535-7163.MCT-19-1166
72. Dominguez CX, Müller S, Keerthivasan S, Koeppen H, Hung J, Gierke S, et al. Single-Cell RNA Sequencing Reveals Stromal Evolution Into LRRC15+ Myofibroblasts as a Determinant of Patient Response to Cancer Immunotherapy. Cancer Discovery (2020) 10:232–53. doi: 10.1158/2159-8290.CD-19-0644
73. McCracken MN, Cha AC, Weissman IL. Molecular Pathways: Activating T Cells After Cancer Cell Phagocytosis From Blockade of CD47 "Don't Eat Me" Signals. Clin Cancer Res (2015) 21:3597–601. doi: 10.1158/1078-0432.CCR-14-2520
74. Pan Y, Lu F, Fei Q, Yu X, Xiong P, Yu X, et al. Single-Cell RNA Sequencing Reveals Compartmental Remodeling of Tumor-Infiltrating Immune Cells Induced by Anti-CD47 Targeting in Pancreatic Cancer. J Hematol Oncol (2019) 12:124. doi: 10.1186/s13045-019-0822-6
75. Mannarapu M, Dariya B, Bandapalli OR. Application of Single-Cell Sequencing Technologies in Pancreatic Cancer. Mol Cell Biochem (2021) 476:2429–37. doi: 10.1007/s11010-021-04095-4
76. Hwang KT. Clinical Databases for Breast Cancer Research. Adv Exp Med Biol (2021) 1187:493–509. doi: 10.1007/978-981-32-9620-6_26
77. Narod SA. Which Genes for Hereditary Breast Cancer? N Engl J Med (2021) 384:471–3. doi: 10.1056/NEJMe2035083
78. Chung W, Eum HH, Lee HO, Lee KM, Lee HB, Kim KT, et al. Single-Cell RNA-Seq Enables Comprehensive Tumour and Immune Cell Profiling in Primary Breast Cancer. Nat Commun (2017) 8:15081. doi: 10.1038/ncomms15081
79. Azizi E, Carr AJ, Plitas G, Cornish AE, Konopacki C, Prabhakaran S, et al. Single-Cell Map of Diverse Immune Phenotypes in the Breast Tumor Microenvironment. Cell (2018) 174:1293–308.e36. doi: 10.1016/j.cell.2018.05.060
80. Chen X, Lin Y, Qu Q, Ning B, Chen H, Cai L. A Multi-Source Data Fusion Framework for Revealing the Regulatory Mechanism of Breast Cancer Immune Evasion. Front Genet (2020) 11:595324. doi: 10.3389/fgene.2020.595324
81. Low SK, Zembutsu H, Nakamura Y. Breast Cancer: The Translation of Big Genomic Data to Cancer Precision Medicine. Cancer Sci (2018) 109:497–506. doi: 10.1111/cas.13463
82. Yates LR, Gerstung M, Knappskog S, Desmedt C, Gundem G, Van Loo P, et al. Subclonal Diversification of Primary Breast Cancer Revealed by Multiregion Sequencing. Nat Med (2015) 21:751–9. doi: 10.1038/nm.3886
83. Stephens PJ, Tarpey PS, Davies H, Van Loo P, Greenman C, Wedge DC, et al. The Landscape of Cancer Genes and Mutational Processes in Breast Cancer. Nature (2012) 486:400–4. doi: 10.1038/nature11017
84. Baslan T, Kendall J, Volyanskyy K, McNamara K, Cox H, D'Italia S, et al. Novel Insights Into Breast Cancer Copy Number Genetic Heterogeneity Revealed by Single-Cell Genome Sequencing. Elife (2020) 9:e51480. doi: 10.7554/eLife.51480
85. Badve SS, Gökmen-Polar Y. Ductal Carcinoma in Situ of Breast: Update 2019. Pathology (2019) 51:563–69. doi: 10.1016/j.pathol.2019.07.005
86. Casasent AK, Schalck A, Gao R, Sei E, Long A, Pangburn W, et al. Multiclonal Invasion in Breast Tumors Identified by Topographic Single Cell Sequencing. Cell (2018) 172:205–17.e12. doi: 10.1016/j.cell.2017.12.007
87. Taurin S, Alkhalifa H. Breast Cancers, Mammary Stem Cells, and Cancer Stem Cells, Characteristics, and Hypotheses. Neoplasia (2020) 22:663–78. doi: 10.1016/j.neo.2020.09.009
88. Xu K, Zhang W, Wang C, Hu L, Wang R, Wang C, et al. Integrative Analyses of scRNA-Seq and scATAC-Seq Reveal CXCL14 as a Key Regulator of Lymph Node Metastasis in Breast Cancer. Hum Mol Genet (2021) 30:37/0–80. doi: 10.1093/hmg/ddab042
89. Yin L, Duan JJ, Bian XW, Yu SC. Triple-Negative Breast Cancer Molecular Subtyping and Treatment Progress. Breast Cancer Res (2020) 22:61. doi: 10.1186/s13058-020-01296-5
90. Mo H, Xu B. Progress in Systemic Therapy for Triple-Negative Breast Cancer. Front Med (2021) 15:1–10. doi: 10.1007/s11684-020-0741-5
91. Kim C, Gao R, Sei E, Brandt R, Hartman J, Hatschek T, et al. Chemoresistance Evolution in Triple-Negative Breast Cancer Delineated by Single-Cell Sequencing. Cell (2018) 173:879–93.e13. doi: 10.1016/j.cell.2018.03.041
92. Grosselin K, Durand A, Marsolier J, Poitou A, Marangoni E, Nemati F, et al. High-Throughput Single-Cell ChIP-Seq Identifies Heterogeneity of Chromatin States in Breast Cancer. Nat Genet (2019) 51:1060–6. doi: 10.1038/s41588-019-0424-9
93. Savas P, Virassamy B, Ye C, Salim A, Mintoff CP, Caramia F, et al. Single-Cell Profiling of Breast Cancer T Cells Reveals a Tissue-Resident Memory Subset Associated With Improved Prognosis. Nat Med (2018) 24:986–93. doi: 10.1038/s41591-018-0078-7
94. Inaba H, Mullighan CG. Pediatric Acute Lymphoblastic Leukemia. Haematologica (2020) 105:2524–39. doi: 10.3324/haematol.2020.247031
95. De Bie J, Demeyer S, Alberti-Servera L, Geerdens E, Segers H, Broux M, et al. Single-Cell Sequencing Reveals the Origin and the Order of Mutation Acquisition in T-Cell Acute Lymphoblastic Leukemia. Leukemia (2018) 32:1358–69. doi: 10.1038/s41375-018-0127-8
96. Furness CL, Mansur MB, Weston VJ, Ermini L, van Delft FW, Jenkinson S, et al. The Subclonal Complexity of STIL-TAL1+ T-Cell Acute Lymphoblastic Leukaemia. Leukemia (2018) 32:1984–93. doi: 10.1038/s41375-018-0046-8
97. Wright GW, Huang DW, Phelan JD, Coulibaly ZA, Roulland S, Young RM, et al. A Probabilistic Classification Tool for Genetic Subtypes of Diffuse Large B Cell Lymphoma With Therapeutic Implications. Cancer Cell (2020) 37:551–68.e14. doi: 10.1016/j.ccell.2020.03.015
98. Almas T, Stephenson KA, Ehtesham M, Murphy CC. Beware the Quiet Eye: Primary Vitreoretinal Lymphoma Masquerading as Posterior Uveitis. BMJ Case Rep (2020) 13:e240802. doi: 10.1136/bcr-2020-240802
99. Miserocchi E, Ferreri AJM, Giuffrè C, Cangi MG, Francaviglia I, Calimeri T. Et al.MYD88 L265P Mutation Detection In The Aqueous Humor Of Patients With Vitreoretinal Lymphoma. Retina J Ret Vit Dis (2019) 39:679–84. doi: 10.1097/IAE.0000000000002319
100. Carreno E, Clench T, Steeples LR, Salvatore S, Lee RWJ, Dick AD, et al. Clinical Spectrum of Vitreoretinal Lymphoma and Its Association With MyD88 L265P Mutation. Acta Ophthalmol (2019) 97:e138–9. doi: 10.1111/aos.13808
101. Tan WJ, Wang MM, Ricciardi-Castagnoli P, Tang T, Chee SP, Lim TS. Et al.Single-Cell MYD88 Sequencing of Isolated B Cells From Vitreous Biopsies Aids Vitreoretinal Lymphoma Diagnosis. Blood (2019) 134:709–12. doi: 10.1182/blood.2019000022
102. Silkenstedt E, Dreyling M. Mantle Cell Lymphoma-Advances in Molecular Biology, Prognostication and Treatment Approaches. Hematol Oncol (2021) 39 Suppl 1:31–8. doi: 10.1002/hon.2860
103. Wang L, Mo S, Li X, He Y, Yang J. Single-Cell RNA-Seq Reveals the Immune Escape and Drug Resistance Mechanisms of Mantle Cell Lymphoma. Cancer Biol Med (2020) 17:726–39. doi: 10.20892/j.issn.2095-3941.2020.0073
104. Phyo ZH, Shanbhag S, Rozati S. Update on Biology of Cutaneous T-Cell Lymphoma. Front Oncol (2020) 10:765. doi: 10.3389/fonc.2020.00765
105. Borcherding N, Voigt AP, Liu V, Link BK, Zhang W, Jabbari A. Single-Cell Profiling of Cutaneous T-Cell Lymphoma Reveals Underlying Heterogeneity Associated With Disease Progression. Clin Cancer Res (2019) 25:2996–3005. doi: 10.1158/1078-0432.CCR-18-3309
106. Rodriguez-Galindo C, Allen CE. Langerhans Cell Histiocytosis. Blood (2020) 135:1319–31. doi: 10.1182/blood.2019000934
107. Halbritter F, Farlik M, Schwentner R, Jug G, Fortelny N, Schnöller T. Et al. Epigenomics and Single-Cell Sequencing Define a Developmental Hierarchy in Langerhans Cell Histiocytosis. Cancer Discov (2019) 9:1406–21. doi: 10.1158/2159-8290.CD-19-0138
108. Guillaumet-Adkins A, Rodríguez-Esteban G, Mereu E, Mendez-Lago M, Jaitin DA, Villanueva A, et al. Single-Cell Transcriptome Conservation in Cryopreserved Cells and Tissues. Genome Biol (2017) 18:45. doi: 10.1186/s13059-017-1171-9
109. Tatematsu K, Kuroda S. Automated Single-Cell Analysis and Isolation System: A Paradigm Shift in Cell Screening Methods for Bio-Medicines. Adv Exp Med Biol (2018) 1068:7–17. doi: 10.1007/978-981-13-0502-3_2
110. Hrdlickova R, Toloue M, Tian B. RNA-Seq Methods for Transcriptome Analysis. Wiley Interdiscip Rev RNA (2017) 8:10.1002/wrna.1364. doi: 10.1002/wrna.1364. 10.1002/wrna.1364.
111. Gonzalez Castro LN, Tirosh I, Suvà ML. Decoding Cancer Biology One Cell at a Time. Cancer Discov (2021) 11:960–70. doi: 10.1158/2159-8290.CD-20-1376
Keywords: cancer, single-cell sequencing, intratumor heterogeneity, tumor microenvironment, single tumor cell
Citation: Li J, Yu N, Li X, Cui M and Guo Q (2021) The Single-Cell Sequencing: A Dazzling Light Shining on the Dark Corner of Cancer. Front. Oncol. 11:759894. doi: 10.3389/fonc.2021.759894
Received: 17 August 2021; Accepted: 30 September 2021;
Published: 21 October 2021.
Edited by:
Xiao Zhu, Guangdong Medical University, ChinaReviewed by:
Dacheng Fan, Emory University, United StatesCopyright © 2021 Li, Yu, Li, Cui and Guo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qie Guo, Z3VvcWllODIyQDE2My5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.