 Yaolin Xu
Yaolin Xu Lijie He1*†
Lijie He1*†- 1Department of Oncology, The People’s Hospital of China Medical University/The People’s Hospital of LiaoNing Province, Shenyang, China
- 2Department of Cardiology, The People’s Hospital of China Medical University/The People’s Hospital of LiaoNing Province, Shenyang, China
- 3The Second Clinic Medical College, China Medical University, Shenyang, China
Immune checkpoint inhibitors (ICIs), Ipilimumab, Nivolumab, Pembrolizumab and Atezolizumab, have been applied in anti-tumor therapy and demonstrated exciting performance compared to conventional treatments. However, the unsatisfactory response rates, high recurrence and adaptive resistance limit their benefits. Metabolic reprogramming appears to be one of the crucial barriers to immunotherapy. The deprivation of required nutrients and altered metabolites not only promote tumor progression but also confer dysfunction on immune cells in the tumor microenvironment (TME). Glycolysis plays a central role in metabolic reprogramming and immunoregulation in the TME, and many therapies targeting glycolysis have been developed, and their combinations with ICIs are in preclinical and clinical trials. Additional attention has been paid to the role of amino acids, lipids, nucleotides and mitochondrial biogenesis in metabolic reprogramming and clinical anti-tumor therapy. This review attempts to describe reprogramming metabolisms within tumor cells and immune cells, from the aspects of glycolysis, amino acid metabolism, lipid metabolism, nucleotide metabolism and mitochondrial biogenesis and their impact on immunity in the TME, as well as the significance of targeting metabolism in anti-tumor therapy, especially in combination with ICIs. In particular, we highlight the expression mechanism of programmed cell death (ligand) 1 [PD-(L)1] in tumor cells and immune cells under reprogramming metabolism, and discuss in detail the potential of targeting key metabolic pathways to break resistance and improve the efficacy of ICIs based on results from current preclinical and clinical trials. Besides, we draw out biomarkers of potential predictive value in ICIs treatment from a metabolic perspective.
Introduction
ICIs have shown impressive clinical anti-tumor performance in various cancers. Ipilimumab [commercialized anti-cytotoxic T-lymphocyte antigen-4 (anti-CTLA-4)] was the first approved ICI for treating patients with advanced melanoma (1, 2). In 2012, Topalian SL et al. reported a rather unexpected result, detailing that anti-PD-1 antibody produced objective responses in approximately one in four to one in five patients with non-small-cell lung cancer (NSCLC), melanoma, or renal cell cancer, with tolerable adverse events (AEs) (3). Subsequently, commercialized PD-1, Nivolumab and Pembrolizumab have been gradually applied in clinical anti-tumor treatment. In 2018, a study of IMpower133 demonstrated significantly longer progression-free survival (PFS) and overall survival (OS) in patients with small cell lung cancer (SCLC), which is traditionally considered a “recalcitrant cancer” (4), with atezolizumab (commercialized anti-PD-L1) being used based on traditional chemotherapy (etoposide combined with carboplatin) (5). Currently, ICIs alone or in combination with chemotherapy have been included in the NCCN guidelines for first-line, second-line and salvage therapy in numerous cancers, including recurrent or metastatic head and neck squamous carcinoma, advanced esophageal cancer, advanced non-epidermal growth factor receptor (EGFR)/anaplastic lymphoma kinase (ALK) mutated NSCLC, extensive-stage SCLC, triple-negative breast cancer (TNBC), advanced gastric cancer, advanced hepatocellular carcinoma (HCC), advanced colorectal cancer, advanced renal cancer, advanced urothelium carcinoma and postoperative or advanced unresectable or metastatic melanoma. However, the unsatisfactory response rates, high recurrence and adaptive resistance limit their benefits. Despite superiority to chemotherapy, a favorable response to Nivolumab in second-line therapy is still absent in approximately 80% of patients with non-squamous NSCLC (6). In TNBC patients, the response to PD-1 inhibitors is also relatively moderate (19%) (7). Consequently, many researchers have sought to investigate biomarkers with more predictive value, as well as additional therapy that could overcome the current therapeutic bottleneck of ICIs.
The metabolic reprogramming of cancer cells contribute to their transformation, tumorigenesis, tumor progression and immune escape under genetic or environmental orchestrating. The primary energy supply changes from oxidative phosphorylation (OXPHOS) to glycolysis, enriched amino acid metabolism, lipid metabolism and nucleotide metabolism, to accommodate the rapid growth demands of tumor cells during tumorigenesis and tumor progression. Deprivation of essential nutrients, altered metabolic processes and changed metabolites of cancer cells promote their transformation while influencing the recognition, activation, expansion and cytotoxic functions of tumor-associated immune cells to facilitate escape from immune surveillance. Moreover, metabolic reprogramming is also involved in the expression and downstream signaling pathways of immune checkpoint PD-(L)1. Compounds targeting metabolic reprogramming have been developed in anti-tumor therapy and exciting advancements have been reported in overcoming the limitations of immunotherapy. In this review, we attempt to describe the reprogramming metabolism of glucose, amino acid, lipid, nucleotide and mitochondrial biogenesis in tumor cells and immune cells and the roles they play in immune compromise, as well as the progress on targeting metabolism in combination with ICIs for anti-tumor therapy. In particular, we highlight the expression mechanism of immune checkpoint PD-(L)1 in tumor cells and immune cells under reprogramming metabolism, and discuss in detail the potential of targeting vital metabolic pathways to break resistance and improve the efficacy of ICIs based on results from current preclinical and clinical trials. We also draw out biomarkers of potential predictive value in ICIs treatment from a metabolic perspective.
Immune Checkpoint With ICIs
Tumor cells, immune cells, metabolites, inflammatory factors, chemokines and other factors collectively construct the immunosuppressive TME. The immune checkpoint is one of the crucial factors participating in immune compromise, including co-inhibitory and co-stimulatory molecules. Tumor-associated or tumor-specific antigens are processed and expressed with major histocompatibility complex (MHC)-II by antigen-presenting cells (APCs). The processed antigens react with T cell receptor (TCR) expressed on T cells to provide the first stimulatory signal. There are also some ligands (such as CD28) expressed on T cells, reacting with their receptors, which act as co-stimulatory molecules to activate second stimulatory signals. After both signals are activated, T cells would proliferate, activate and serve cytotoxic functions. With a series of anti-tumor or anti-inflammatory cytokines [interferon-γ (IFN-γ), tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β)], or the inactivation of tumor suppressors (PTEN and RB1) (8, 9), the expression of co-inhibitory molecules up-regulates with the activation of toll like receptor (TLR)-/IFN-γ-mediated signaling pathways [nuclear factor κ-B (NF-κB), mitogen-activated protein kinase (MAPK), phosphatidylinositol 3 kinase (PI3K), mammalian target of rapamycin (mTOR) and janus kinase (JAK)] and downstream nuclear translocation of numerous transcription factors that transactivate expression of CD274 gene (10–13), which alleviate the immune reaction and avoid further lethal effects, resulting in immune compromise in the TME. In addition to competing with co-stimulatory molecules to exert immunosuppression, co-inhibitory checkpoints also exert intrinsic immunosuppressive effects. When stimulated, PD-1 becomes phosphorylated at its immune receptor tyrosine-based inhibitory motif (ITIM) and immune receptor tyrosine-based switch motif (ITSM), which then bind the Src homology 2 (SH2) domains of SH2-containing phosphatase 2 (SHP2), initiating T cell inactivation (14). Protein phosphatase-2A (PP2A) mediates CTLA-4 suppression of T-cell activation by interacting with the cytoplasmic tail of CTLA-4 with the lysine-rich motif and the tyrosine residue (15). To resolve the immune compromise of immune checkpoints on immunocytes, compounds targeting inhibitory immune checkpoints are being studied and applied in clinical anti-tumor therapy.
Ipilimumab, an anti-CTLA-4 antibody, has demonstrated improved anti-tumor efficacy with enhanced immunity (1). Ipilimumab is the pioneer ICI applied in the clinical anti-tumor treatment and has acquired responses of approximately 20% (16). In the early stage of the activation of T cells, usually within 48-72h, CTLA-4 will be over-expressed. Co-inhibitory CTLA-4 competes with co-stimulatory CD28 to bind to CD80 or CD86 on the surface of APCs to alleviate the anti-tumor immunity of tumor infiltrated T lymphocytes (TILs), such as CD4+T, CD8+T cells and regulatory T cells (Tregs). Anti-CTLA-4 could partially relieve the immunosuppressive TME with enhanced anti-tumor immunity of T lymphocytes. Nevertheless, it is associated with a wide range of autoimmune disorders, including thyroiditis, diabetes mellitus, coeliac disease, myasthenia gravis, Addison disease, systemic lupus erythematosus, and rheumatoid arthritis when blockading CTLA-4 (17). These may be some of the reasons for the early and widespread immune-related side effects of Ipilimumab, along with improved anti-tumor immunity.
Similarly, PD-1 inhibits the proliferation and cytotoxicity of lymphocytes when ligated with its ligands PD-L1 (encoded by CD274) or PD-L2 (encoded by PDCD1LG2). Compared with Ipilimumab, Nivolumab (anti-PD-1 antibody) has a better prognosis with less frequent and more manageable immune-related AEs (18, 19). Anti-PD-1 and anti-CTLA-4 have distinct cellular mechanisms in checkpoint blockade. Anti-PD-1 predominantly induces the expansion of exhausted CD8+ TILs, while anti-CTLA-4 can induce the expansion of ICOS+CD4+ T helper 1 (TH1) effector lymphocytes in addition to exhausted CD8+ TILs (20).
In addition, other immune checkpoints such as T cell immunoreceptor with immunoglobulin and ITIM domains (TIGIT)/CD155 and T cell immunoglobulin mucin-3 (TIM-3)/Galectin-9 are coming to the forefront of researcher attention for their crucial roles in immune compromise. Co-inhibitory TIGIT and CD96 compete with co-stimulatory CD226 for their ligands CD155 and CD112 on dendritic cells (DCs) or tumor cells to suppress the anti-tumor immunity of TILs (21, 22). And the interaction between TIGIT and CD155 could transfer DCs into a tolerant state with increasing immunosuppressive cytokines (23). It has been reported that CD155 and TIM-3 are over-expressed on many tumor cells and negatively correlated with the survival time of most cancer patients (24–29).
Intriguingly, metabolic reprogramming is involved in inhibitory or stimulatory immune checkpoint downstream signaling pathways in immune cells to compromise their immune function. Lymphocyte activation initiates a program of cell growth, proliferation, and differentiation that increases metabolic demand for glucose uptake, glycolysis and glutaminolysis (30). Lacking large internal glycogen storage, with the help of cell surface receptors, resting lymphocytes are highly dependent on extracellular glucose import to meet increased metabolic needs. CD28 co-stimulation, acting through PI3K and Akt, is required for T cells to increase their glycolytic rate in response to activation. Nevertheless, co-inhibitory CTLA-4 inhibits Akt activation and blocks T cell activation to prevent the upregulation of glucose transport and glycolysis that results from CD28 costimulation (30). In addition to inhibiting aerobic glycolysis and glutaminolysis as caused by CTLA-4 ligation, PD-1/PD-L1 downward signaling increases fatty acid β-oxidation (FAO) in immune cells (31–33). The suppressed glycolysis impairs the differentiation of T cells to effectors, and enhanced FAO sustains the longevity of the exhausted T cells (33), which is likely to be regulated by the PD-1-leptin-signal transducer and activator of transcription 3 (STAT3) signaling pathway (34). Moreover, isotopomer analysis in primary human TILs further showed that PD-1 signaling resulted in increased reductive carboxylation of pyruvate, anaplerosis of the cycle at acetyl coenzyme A (acetyl-CoA) and succinyl-CoA, and block in de novo nucleoside phosphate synthesis accompanied by decreased rapamycin complex 1 (mTORC1) signaling (35). Furthermore, other costimulatory molecules (such as CD137) and cytokines are crucial regulators of metabolic reprogramming in TILs (36). As reported, the co-stimulatory receptor GITR supports CD8+ T cell proliferation and effector cytokine production by upregulating nutrient uptake, lipid storage, glycolysis and oxygen consumption rate (37).
Furthermore, glucose consumption by tumors metabolically restricts T cells, leading to their dampened mTOR activity, glycolytic capacity, and IFN-γ production. And checkpoint blockade antibodies against CTLA-4, PD-1 and PD-L1 could restore immune metabolic dysfunction of TILs with restored glucose, permitting CD8+ T cell glycolysis and IFN-γ production (38). Besides, Strauss L et al. demonstrated that PD-1 ablation assists emergency myelopoiesis and differentiation of effector myeloid cells to strengthen anti-tumor immunity by increasing intermediates of glycolysis, tricarboxylic acid cycle (TCA cycle), pentose phosphate pathway (PPP) and elevated cholesterol (39). Chang CH et al. demonstrated that blocking PD-L1 on tumors could dampen their glycolysis with decreasing glycolysis enzymes by inhibiting the Akt-mTOR pathway (38). Alternatively, by [18F]FDG-positron emission tomography (PET) and flow cytometry, Tomita M et al. demonstrated that anti-PD-1 treatment raises glucose metabolism in cancer cells by increasing glucose transporter 1 (GLUT1) and hexokinase-II (HK-II) expression, which may be implicated with inflammatory factors (such as TNF-α) secreted by active immune cells (40).
Reprogrammed Metabolism of Glucose, Lipid, Amino Acid, Nucleotide and Mitochondrial Biogenesis in Cancer Cells With Immune Cells
With the orchestration of oncogenes and tumor suppressor genes, tumor cells not only re-edit their biological behaviors but also reprogram TME metabolism, which contributes to their invasion, migration, metastasis and even resistance to drugs (41, 42). Moreover, the reprogrammed metabolic processes of glucose, lipid, amino acid, nucleotide and mitochondrial biogenesis in tumor cells, and their altered metabolites, contribute to the evasion of immune surveillance and immune elimination (43–45). Metabolic competition in the TME is a driver of cancer progression (38) and immune compromise (46). The reprogrammed TME suppresses the immunity of anti-tumor TILs but supports the immunosuppressive TILs, thus sustaining tumor progression along with immune compromise. Besides, reprogramming metabolism in cancer cells and immune cells is one of the key obstacles in anti-tumor immunotherapy. There have been many attempts in targeting metabolic reprogramming to reinforce anti-tumor therapy, particularly in combination with ICIs.
Hypoxia in the Immunosuppresive TME
Hypoxia, one of the crucial stressors in the TME, accelerates tumor cell progression and suppresses the immunity of TILs. The mismatch of rapid growth and proliferation of tumor cells with the lack of blood vessels contribute to hypoxia in the TME. Hypoxia-inducible factors (HIFs) play a vital role in this process. Under normoxic conditions, HIF-1α can be downregulated through ubiquitination and von Hippel-Lindau protein-mediated proteasomal degradation, while stabilization of HIF-1α and/or HIF-2α under hypoxic stress leads to transcriptional upregulation of many hypoxia-responsive genes associated with the metabolism and immunity of tumor cells and immune cells (47). Long-lasting HIF signaling could stimulate oncogene driven tumor progress, invasion, metastasis, angiogenesis, metabolic reprogramming, cancer stem cell maintenance and even constructing an immunosuppressive TME in various cancers (48, 49). Nevertheless, it was also reported that HIF-1α suppresses tumor cell proliferation in VHL-deficient renal cell carcinoma through the repressed aspartate-producing enzymes GOT1 and GOT2 and thus impaired oxidative and reductive aspartate biogenesis (50). HIFs promote lipid peroxidation and endoplasmic reticulum (ER) stress, recruit the immunosuppressive myeloid-derived suppressor cells (MDSCs) (51, 52), M2 macrophages (53) and Tregs (54) to construct an immunosuppressive TME. Besides, in hypoxic conditions, T cell expansion and cytotoxicity are suppressed due to impaired TCR (55). Moreover, HIFs also upregulate the expression of the inhibitory checkpoints [PD-L1 (56, 57), cluster of differentiation 47 (CD47) (58) and human leukocyte antigen G (HLA-G) (59)] to assist immune compromise. Oxygen is a crucial metabolite required for the TILs to differentiate appropriately upon PD-1 blockade (60). Hypoxia appears to be a significant metabolic barrier to immunotherapy, and remodeling the hypoxic TME may allow patients to switch from resistance to immunotherapy to gaining clinical benefit (61, 62). Accordingly, investigations of combining hypoxia-targeting drugs with ICIs have been studied. Metformin, a classic hypoglycemic drug for type II diabetes, can inhibit oxygen consumption by tumor cells and alleviate intratumoral hypoxia. Although metformin demonstrated modest anti-tumor effects, the combination of metformin with anti-PD-1 has been reported to significantly improve intratumoral T-cell function and tumor clearance in mouse models, and even in individual tumor patients (62, 63). Although metformin demonstrates immunosuppressive effects in models of graft-versus-host disease, lupus and graft rejection, it is preferentially taken up by tumor cells rather than T cells in the TME with the help of the organic cation transporter (OCT), which enhances T cell depredation on oxygen from cancer cells thereby boosting the immunity (62).
Glucose Metabolism Reprogramming in Cancer Cells With Immune Cells and Their Effects on the Expression of Immune Checkpoints
Glucose metabolism is a significant supplier of energy and carbon sources in biological activities, including glycolysis (anaerobic oxidation), aerobic oxidation, PPP, glycogen metabolism, and gluconeogenesis. In the hypoxic TME, glycolysis is one of the critical metabolic reprogramming processes which could provide immediate and sufficient energy to satisfy the demand of tumor cells with rapid proliferation, adjusting to hypoxia environment and constructing an immunosuppressive TME. Tumor cell shifts metabolism to glycolysis, even in the presence of oxygen, which is called the Warburg effect (64). Hypoxia could activate multiple proglycolytic genes (SLC2A1, LDHA and PDK1) and glycolytic enzymes in favor of glucose uptake and glycolysis in both cancer cells and TILs, but depresses TCA cycle and aerobic oxidation (65–67). Lactate, the main metabolite of glycolysis involved in constructing an acidic TME, facilitates tumor progression and suppresses anti-tumor immunity through T cell suppression and PD-L1 upregulation (68).
The reprogrammed glycolysis in cancer cells and immune cells contributes to immune evasion. In the glucose-deficient TME, its key participants involved in anti-tumor immune regulation, CD4+TILs, CD8+TILs, MDSCs, M1/M2 TAMs, Tregs and other immune cells (Table 1), are dysfunctional due to metabolic reprogramming (69, 70) (Figure 1). Activated T cells engage aerobic glycolysis and anabolic metabolism for growth, proliferation and effector functions. High glucose and oxygen consumption by rapidly proliferating tumor cells inhibit TILs’ glycolytic activity by depriving them of nutrients, leading to dampened cytotoxic and effector function. Meanwhile, the decreased downstream metabolite phosphoenolpyruvate (PEP) is necessary for sustaining Ca2+-NFAT signaling in T cells, mediating diminished anti-tumor responses in glucose-poor TME (69). Besides, reprogrammed low pH in the TME leads to anti-tumor effectors anergy followed by apoptosis and engages with immunosuppressive MDSCs and Tregs (71). Moreover, pro-inflammatory M1 macrophages rely mainly on glycolysis and present two breaks on the TCA cycle that result in the accumulation of itaconate and succinate. Excess of succinate leads to HIF-1α stabilization that, in turn, activates the transcription of glycolytic genes, thus sustaining the glycolytic metabolism of M1 macrophages (72). Alternatively, the immunosuppressive M2 macrophages, which are more dependent on OXPHOS, may be dampened in the hypoxic TME.
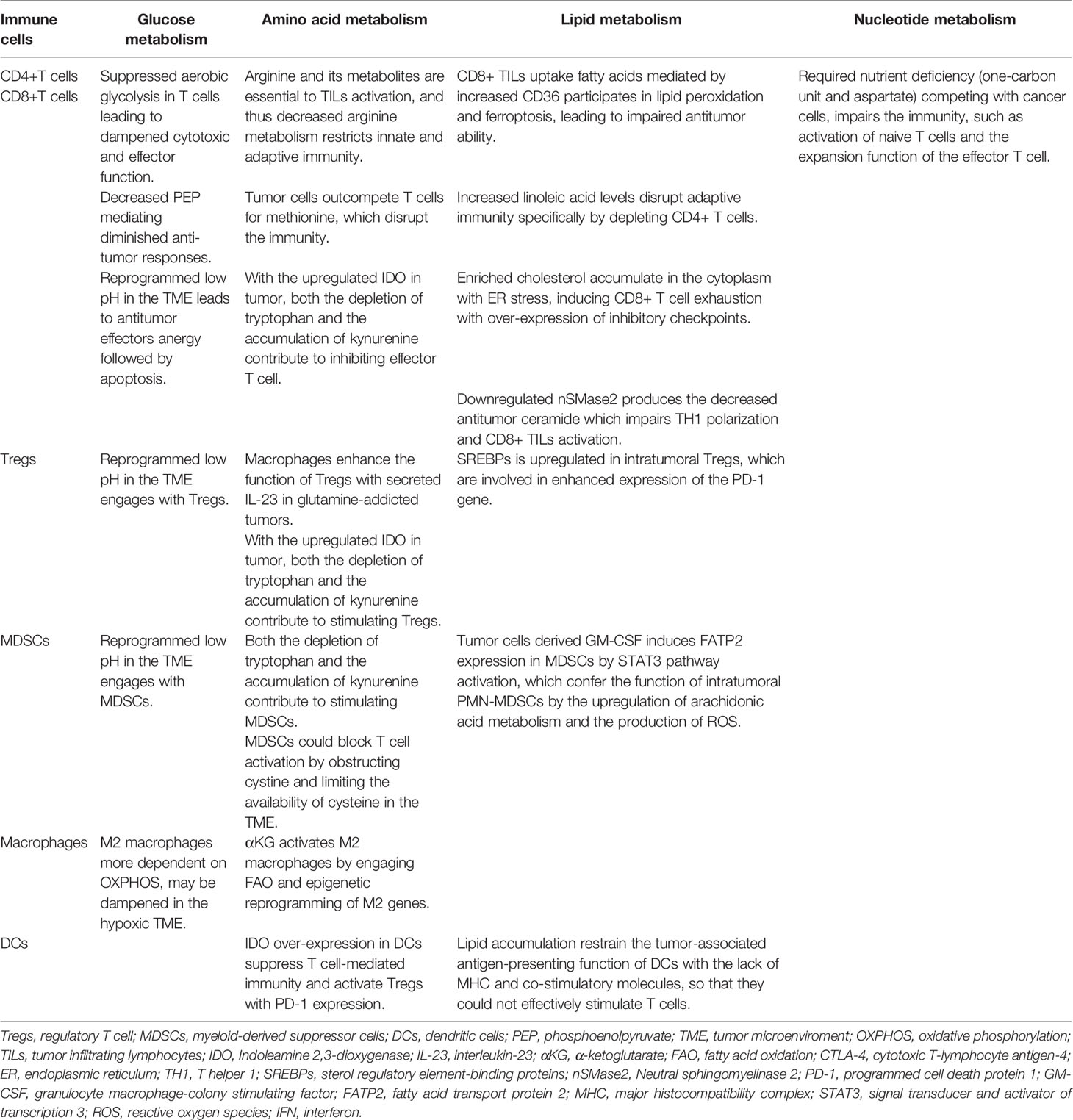
Table 1 The metabolic reprogramming with different types of metabolism in immune cells in the TME.
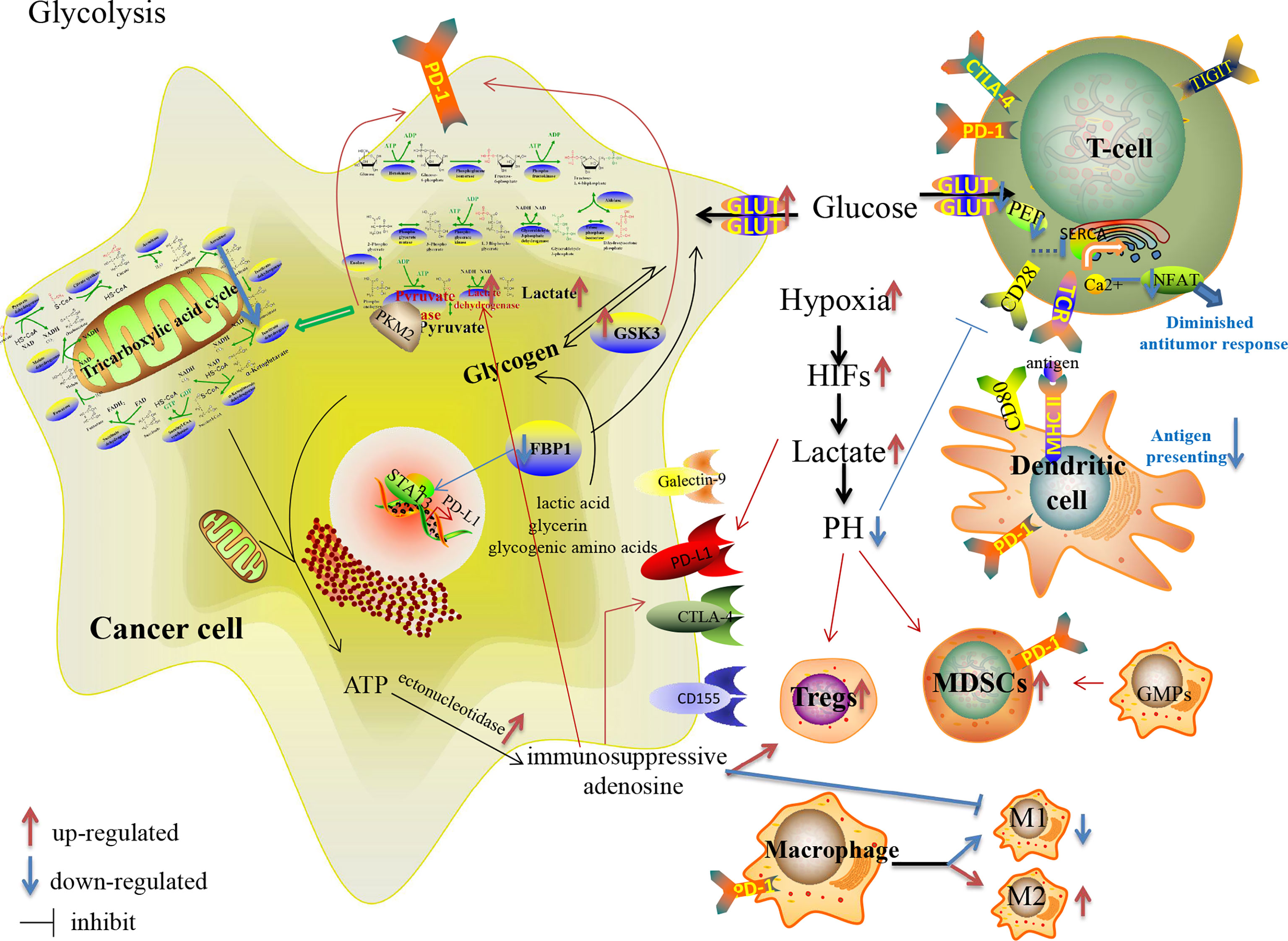
Figure 1 Glucose metabolism reprogramming in cancer cells and immune cells in the TME, and their effects on the expression of immune checkpoints.
Moreover, many glucose-metabolizing enzymes involved in glycolysis, such as HK, 6-phosphofructokinase-1 (PFK-1), pyruvate kinase and lactate dehydrogenase (LDH), mediate the escape of tumor cells from immune surveillance. In addition to HIF-1α, the activities of these enzymes are regulated by the oncogenes MYC and KRAS, the tumor suppressor TP53 and non-coding RNAs (73). The rate-limiting enzymes PFK-1, pyruvate kinase and HK play vital roles in glycolysis. Especially, pyruvate kinase isoform M2 (PKM2) is an essential participant in the processes of tumor progression and immune regulation, as well as glycolysis (74). Palsson-McDermott EM et al. reported that remodeled PKM2 up-regulated the expression of PD-L1 on tumor-associated macrophages (TAMs), DCs, T cells and tumor cells by regulating hypoxia response elements of Hif-1α target genes (75). Besides, in metabolism-reprogrammed TME, decreased expressions of GLUT and HK2 impair activated TILs, and these metabolic changes correlate with increased Tregs and expression of PD-L1 and Galectin-9 on cancer cells (76).
Increased expression of pH regulatory molecules, such as V-ATPase and carbonic anhydrase IX (CAIX)/CAXII, in the hypoxic TME, not only expel protons to sustain the intracellular homeostasis but also increase the immunosuppressive pressure mediated by the acidic metabolism (77). Neutralization of tumor acidity, such as bicarbonate, pH regulators and even RNAi nanoparticle, increases the infiltration with CD8+T and NK cells, decreases the number of immunosuppressive immune cells, and thus significantly inhibits the growth of tumors with potentiated anti-PD-1 therapy (78). Intratumoral acidosis and hypoxia may also dampen the bioactivity and distribution of ICI antibodies (79, 80). Zappasodi R et al. also suggested that the local glucose: lactate ratio may alter Tregs susceptibility to anti-CTLA-4, and decreasing tumor competition for glucose may facilitate the therapeutic activity of anti-CTLA-4 (81). Moreover, Shari Pilon-Thomas et al. reported that the combination of bicarbonate therapy with anti-CTLA-4 or anti-PD-1 improved anti-tumor responses in murine models (80). Besides, therapies targeting GLUTs (82) and LDH have been studied in anti-tumor therapy. Gong Y et al. reported that inhibition of LDH could enhance tumor response to anti-PD-1 immunotherapy in TNBC murine model (83).
In addition to glycolysis, glycogen metabolism and gluconeogenesis also reprogram the immunosuppressive TME. The process of glycogen synthesis is upregulated in many solid cancers, including renal, breast, bladder, uterine, ovarian, skin and brain cancers (84). Glycogen synthase kinase 3 (GSK3), encoded by GSK3A and GSK3B, is the crucial rate-limiting serine/threonine phosphatase in glycogen synthesis as well as PD-1 over-expression in cancer cells and CD8+T cells (85, 86). The downregulation or inhibition of GSK3 downregulates PD-1 expression in cancer (87). The use of GSK-3 inhibitors decreases PD-1 transcription by more than 80 percent, although the combination of anti-PD-1 with GSK-3 inhibitor could not further increase the killing of CD8+T cells (88). Gluconeogenesis converts non-glucose substances into glucose or glycogen. Fructose-1, 6-biphosphatase (FBP1), a negative regulator of glycolysis through inhibition of HIF-1α expression (89), is a critical enzyme in gluconeogenesis. It has been reported that FBP1 possesses a tumor suppressor function and it is often downregulated in many cancers, with the loss of FBP1 linked to tumor progression and poor prognosis in various cancer patients (89, 90). Bo Wang et al. further demonstrated that decreased FBP1 expression promotes tumor growth and resistance to ICIs in tumor-burdened mice with STAT3-PD-L1 over-expression, which provides a possible perspective for breaking therapeutic resistance to ICIs (91).
Amino Acid Metabolism Reprogramming in Cancer Cells With Immune Cells and Their Effects on the Expression of Immune Checkpoints
Apart from glycolysis and the Warburg effect, amino acids play critical roles in metabolic reprogramming to promote tumor progression in TME (Figure 2). Amino acids are avidly consumed to satisfy the demand of cancer cells proliferation with synthesizing proteins, nucleotides, hormones and neurotransmitters. Alternatively, the metabolism of amino acids is predominant in the activation and differentiation of T cells. When TCR signaling is activated, the expression of several amino acid transporters [solute carrier family (Slc7a5-Slc3a2), alanine, serine, and cysteine system amino acids transporter 2 (ASCT2)] are enhanced with T cell expansion and effector differentiation by activating mTOR (92). Nevertheless, the deprivation of essential nutrients from tumor cells dampens the function of immune cells (Table 1). Increasing attentions are focused on targeting reprogramming metabolism of amino acids, including glutamine, serine, glycine, arginine, tryptophan, methionine, cysteine and cystine, with increased clinical anti-tumor benefits.
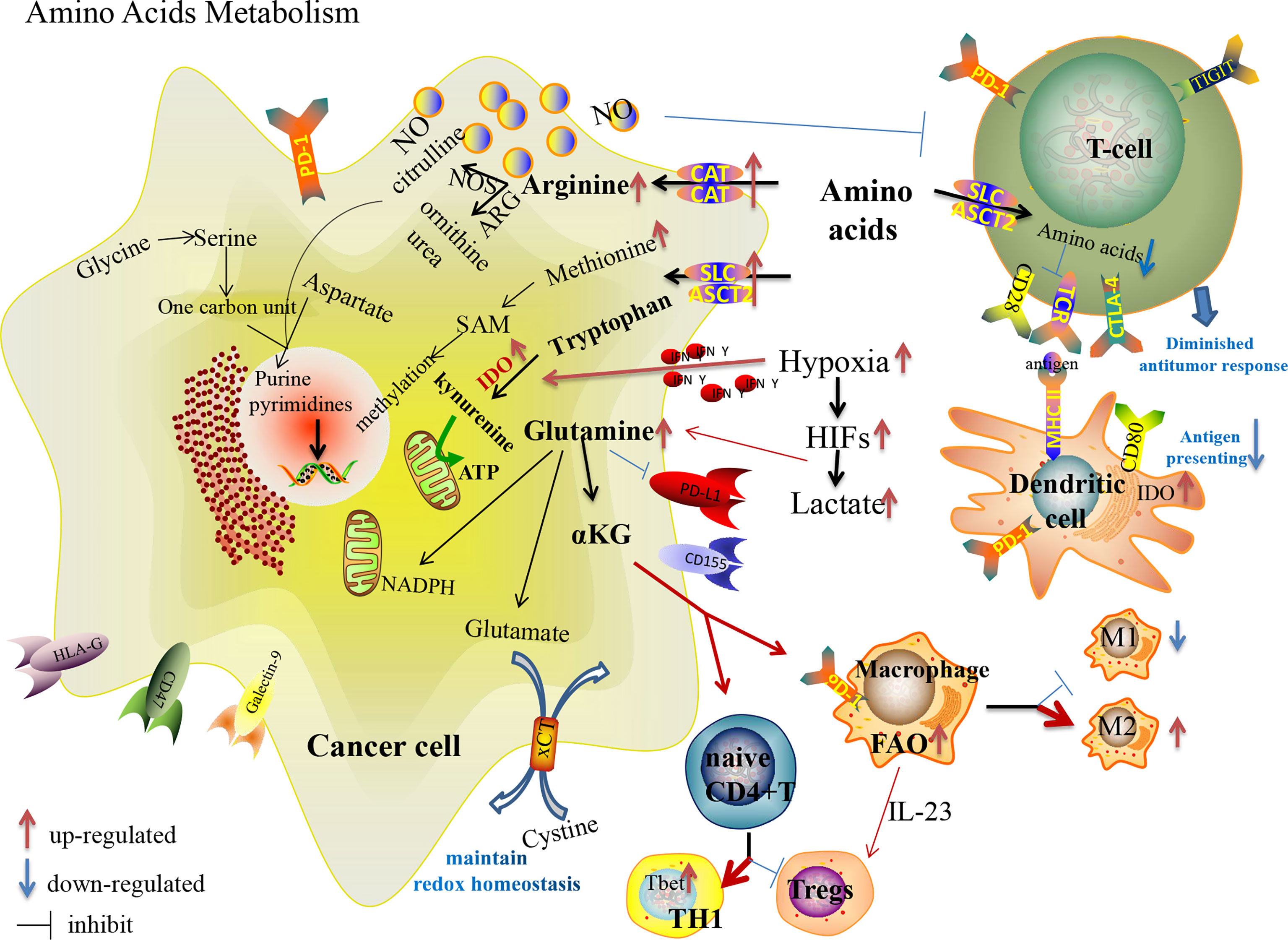
Figure 2 Amino acid metabolism reprogramming in cancer cells and immune cells in the TME, and their effects on the expression of immune checkpoints.
Although glutamine is one of the non-essential amino acids (NEAAs), many tumor cells exhibit “glutamine dependence” in the TME with increased glutamine uptake and catabolism regulated with oncogene KRAS (93) and by lactate in a HIF-2α/c-Myc/ASCT2/glutaminase manner (94). Maintaining a high glutamine level provides enough carbon and nitrogen sources to support biosynthesis, bioenergetics and cellular homeostasis to drive tumor growth. Glutamine participates in macromolecular synthesis (amino acids, lipids, nucleotides, hexosamines, polyamines), signaling in cancer cells (95), vital support of the glutathione and nicotinamide adenine dinucleotide phosphate (NADPH) production, contributing to intracellular OXPHOS (96). Glutaminolysis supports proliferation and the integrity of the TCA cycle in tumor cells (97). Dysregulation of glutaminase and glutamine synthetase are vital events that allow anabolic adaptation of tumors (98). The increased glutamine metabolism in T cell activation regulates the skewing of CD4+ T cells toward more inflammatory subtypes, while the activation status of glutaminolysis during TCR-stimulation determines the activation of CD8+ T cells (99, 100). Consequently, the competition of cancer cells towards glutamine appears to suppress the immunity of TILs. Besides, macrophages enhance the function of Tregs with secreted IL-23 in glutamine-addicted tumors (101). As a crucial metabolite of glutaminolysis, accumulated α-ketoglutarate (αKG) has controversial effects on immunoregulation. On the one hand, αKG could activate immunosuppressive M2 macrophages by engaging FAO and epigenetic reprogramming of M2 genes (99); on the other hand, the activation of glutamine-deprived naive CD4+ T cells in the presence of αKG could induce the differentiation of TH1 cells instead of immunosuppressive Tregs with the increased expression of the gene encoding the TH1 cell-associated transcription factor Tbet and stimulation of mTORC1 signaling (102). In addition, glutamine metabolism is involved in the expression of PD-(L)1 in cancer cells and immune cells. Nabe S et al. reported that glutamine restriction enhanced anti-tumor cytotoxicity of CD8+T lymphocytes with decreased PD-1 expression and prevented CD8+T-cell exhaustion (103). Ma G et al. reported that the expression of PD-L1 in cancer cells upregulated during glutamine deprivation, and the upregulated PD-L1 restored to the normal level after glutamine recovery (104). Many compounds targeting glutamine metabolism and transition of glutaminase have been developed from preclinical to clinical trials (105), though there is a lack of impressive achievement, even when utilized in combination with ICIs.
Arginine metabolism is one of the important mechanisms responsible for tumor progression and immune suppression. Increased arginine metabolism can sustain the growth of many cancers while causing immunosuppression (106, 107). Arginine metabolism depends on the activity of the nitric oxide synthases (NOS) and arginases (ARGs) enzyme families. NOS oxidizes arginine into citrulline and NO, and ARGs hydrolyze arginine into ornithine and urea. After arginine enters the cancer cells through the membrane-bound transporters [cationic amino acid transporters (CAT1 and CAT2B)], it is metabolized by NOS to produce NO, which is associated with immune suppression, neovascularization and tumor progression (106). Arginine and its downstream metabolites (ornithine and citrulline) are essential to TILs activation, and thus deprivation of arginine from cancer cells restricts innate and adaptive immunity to further promote tumor survival and growth. Due to the lack of key argininosuccinate synthetase 1 (ASS1), some tumor cells cannot synthesize arginine by themselves. In this way, suppression of external arginine supply is regarded as a promising anti-tumor therapy. PEGylated arginine deiminase (ADI-PEG 20) has been applied to clinical anti-tumor therapy of HCC, melanoma and other ASS1-deficient cancers by depleting the external supply of arginine (108), which has also been shown to affect the hypoxia-induced processes by inhibiting HIF signal (109). However, one of the challenges of ADI-PEG 20 is drug resistance with re-expression of ASS1 or inhibitory immune checkpoints. Elena Brin et al. reported that the combination of ADI-PEG 20, anti-PD-1 and anti-PD-L1 results in a more powerful anti-tumor therapy in mouse models compared to monotherapy (110). A phase 1b clinical trial of ADI-PEG 20 combined with pembrolizumab demonstrated well-tolerated drug AEs and a promising disease control rate in patients with advanced solid cancers (111). Further clinical trials are needed in patients enriched for defined arginine auxotrophic cancers. Besides, another preclinical trial showed that the combination of protein arginine methyltransferase inhibitor with ICIs also enhanced the anti-tumor immunity with increased CD8+TILs (112). These researches provide potential strategies to overcome anti-PD-L1 resistance.
Similarly, tryptophan, one of the essential amino acids (EAAs), is also deprived in TME with rapid growth and progression of tumor cells. Indoleamine 2,3-dioxygenase (IDO), a vital enzyme involved in tryptophan catabolism with kynurenine production, is upregulated in many malignant tumors (113). The end-products of kynurenine are involved in the metabolic pathway of NAD+ and adenosine triphosphate (ATP) (114). Both the depletion of tryptophan and the accumulation of kynurenine contribute to the formation of an immunosuppressive TME by inhibiting effector T cell and NK cell functions, stimulating Tregs and MDSCs, and inducing polarization of macrophages to turn into a tolerogenic phenotype (46). Moreover, IDO-expressing APCs might induce systemic tolerance to tumor antigens (115). Interestingly, hypoxia initially leads to a lower IDO1/kynurenine expression in “tumor elimination stage” and increases IDO1 mRNA expression with activation of immune cells and secretion of cytokines (especially IFN-γ), thereby increasing IDO1/kynurenine expression to participate in “tumor escape stage” (116). Besides IFN signals, the ligation of CTLA-4 with CD80/CD86 was found to induce IDO expression in DCs, resulting in the suppression of T cell-mediated immunity (115). And IDO-expressing DCs could activate Tregs with PD-1 expression (117). Highly expressed IDO in immunocytes seems to be an obstacle in ICI anti-tumor therapy (118, 119). Rikke B. Holmgaard et al. found that the anti-tumor effect of anti-CTLA-4 or anti-PD-L1/anti-PD-1 was significantly increased in IDO-deficient melanoma-bearing mice (113), suggesting that the combination of IDO inhibitors with ICIs may enhance the anti-tumor ability in clinical therapy. Although the combination of IDO1 inhibitor (INCB024360) and pembrolizumab had encouraging results in phase I/II clinical trials of melanoma patients, it unfortunately failed to meet the endpoints in phase III trials (120). More clinical trials of this combination therapy for multiple malignancies are under investigation (121).
Sulfur-containing amino acids consist of methionine, cysteine and cystine, which can be converted into each other. Methionine, one of the EAAs, is the precursor of S-adenosyl methionine (SAM) which directly supplies the methyl in methylation. It is also involved in lipid metabolism by activating endogenous antioxidant enzymes such as methionine sulfoxide reductase A, and by glutathione biosynthesis to counteract oxidative stress. Cancer cells cannot proliferate when methionine is replaced with its metabolic precursor, homocysteine, while the proliferation of non-tumor cells is unaffected by these conditions, known as methionine dependence or the Hoffman effect (122). Growing evidences indicate that methionine restriction inhibits the growth of cancer cells and enhances the efficacy of chemotherapy and radiotherapy in preclinical and clinical trials (123). In addition to serving as a universal methyl donor, SAM has been reported to be essential for T cell activation and proliferation and has significant anti-tumor effects in numerous cancers, though there is insufficient SAM available in the TME (124). Recently, a preclinical trial on mouse model demonstrated that the combination of SAM and ICI can effectively block melanoma by alteration of key genes and pathways implicated in cancer and immune responses compared to monotherapy, providing the rationale for initiating clinical trials with SAM and ICI (124). By over-expressing the methionine transporter SLC43A2, tumor cells avidly consume methionine and outcompete T cells for methionine, which disrupts the immunity (125). Bian Y et al. demonstrated that the inhibition of SLC43A2 boosts checkpoint-induced tumor immunity with increased T cell immunity in tumor-bearing mice and patients with colon cancer (125). Cysteine is an EAA for T-cell activation due to the lack of cystathionase which converts methionine to cysteine. T cells also cannot import cystine and reduce it intracellularly to cysteine with a lack of an intact transporter (126). In this way, MDSCs could block T cell activation by obstructing cystine and limiting the availability of cysteine in the TME. Through the cystine/glutamate transporter cystine-glutamate exchange (xCT), the uptake of extracellular cystine is orchestrated in exchange for intracellular glutamate to maintain the redox homeostasis and promote tumor progression (127). The xCT inhibitors (such as sulfasalazine) exert anti-tumor effects through the production of reactive oxygen species (ROS), which induces cell death by disrupting redox homeostasis. Liu N et al. demonstrated the impact of inhibition of xCT on anti-PD-1/PD-L1 therapy in tumor-bearing mice. They showed that inhibition of xCT blunted the efficacy of PD-1/PD-L1 blockade through upregulating PD-L1 expression via the transcription factors IRF4/EGR1, and thus exosomes carrying large amounts of PD-L1 secreted from melanoma cells induced macrophage M2 polarization and eventually induced anti-PD-1/PD-L1 therapy resistance (127).
As one of the crucial NEAAs, serine is involved in the anabolism of multiple macromolecular substances by forming one-carbon unit, participating in purine and pyrimidine nucleotide synthesis, NADH/NADPH production and methylation pathways. Cancer cells obtain serine through four main pathways: Acquisition from the TME, degradation of cellular proteins, transamination of glycine, and de novo synthesis from glucose and glutamate. Enhanced serine uptake and synthesis in cancer cells can satisfy the demand for the rapid growth of tumor cells. Serine/glycine/one-carbon unit also affect the growth, proliferation and differentiation of immune cells (128, 129). As the main product of the one-carbon unit, NADPH is also involved in B cell proliferation (130).
Lipid Metabolism Reprogramming in Cancer Cells With Immune Cells and Their Effects on the Expression of Immune Checkpoints
Lipids constitute the basic structure of membranes and function as signaling molecules and energy sources. Increased lipid uptake, storage and lipogenesis satisfy the demand of biological activities of rapidly proliferative cancer cells. Tumor cells could even synthesize new fatty acid by lipogenesis with activated fatty acids synthetase (FASN) which is partially controlled by mTORC2 (131), while normal cells can only uptake dietary fatty acid (132). Sterol regulatory element-binding proteins (SREBPs), a family of membrane-bound transcription factors in the ER, play a central role in regulating lipid metabolism and are linked to glucose metabolism by SREBPs cleavage-activating protein (133). Recent studies have shown that SREBPs are highly upregulated in various cancers and promote tumor growth (133).
Lipid metabolism in tumor-associated immune cells (Table 1) shapes an immunosuppressive TME favorable to tumor progression (Figure 3) (43, 134). A common metabolic alteration in the TME is lipid accumulation, a feature associated with immune dysfunction (135). Tumor cells derived granulocyte macrophage-colony stimulating factor (GM-CSF) induces fatty acid transport protein 2 (FATP2) expression in MDSCs by activation of STAT3 pathway, which confers the function of intratumoral PMN-MDSCs by the upregulation of arachidonic acid metabolism and the production of ROS (136). Adeshakin AO et al. reported that blockage of FATP2 expression in MDSCs by lipofermata decreased lipid accumulation, lower ROS, blocked immunosuppressive activity, down-regulated PD-L1 expression on CD8+ TILs, and even enhanced anti-PD-L1 anti-tumor efficacy in murine model (136). Fatty acid synthesis mediated by FASN contributes to the functional maturation of Tregs (137, 138). SREBPs are upregulated in intratumoral Tregs, which are involved in enhanced expression of the PD-1 gene (139). Inhibiting lipid synthesis and metabolic signaling dependent on SREBPs in Tregs unleash effective anti-tumor immune responses without autoimmune toxicity and even boost anti-PD-1 immunotherapy in murine model (139). Lipid accumulation restrain the tumor-associated antigen-presenting function of DCs with the lack of MHC and co-stimulatory molecules, so that they could not effectively stimulate T cells (43). Increased linoleic acid levels disrupt adaptive immunity specifically by depleting CD4+ T cells, which in turn promote carcinogenesis (140). CD8+ TILs uptake fatty acids mediated by increased CD36, which is induced by cholesterol in TME and participates in lipid peroxidation and ferroptosis, leading to decreased cytotoxic cytokine production and impaired anti-tumor ability in an oxidized lipid-CD36-p38 kinase manner (135, 141). Ma X et al. reported that blocking CD36 or inhibiting ferroptosis in CD8+ T cells effectively restored their anti-tumor activity and endowed greater anti-tumor efficacy in combination with anti-PD-1 antibodies in mice model (141). Rather than directly uptaking extracellular fatty acids in CD8+TILs, memory T cells rely on intrinsic mobilization fatty acids, engage FAO to a greater extent and support the metabolic programming necessary for development (142). Tissue-resident memory T cells (Trms), a subset of T-cells that produce higher amounts of cytokines than their circulating counterparts and provide enhanced local immunity, which is also associated with the success of ICIs therapy (143–146). Instead of utilizing glycolysis, Trms rely on FAO for cell survival, and deprivation of fatty acids results in Trms death (147). Targeting PD-L1 decreases fatty acid-binding protein (Fabp) 4 and Fabp5 expression in tumor cells, and the blockade of PD-L1 increases Fabp4/5 expression in Trms, promoting lipid uptake by Trm cells and resulting in better survival (147). However, a statistical study of large-scale datasets confirmed that tumors with activated lipid metabolism tend to have higher immune cells infiltration and better response to checkpoint immunotherapy (148). Harel M et al. also indicated that lipid metabolism is a regulatory mechanism that increases tumor immunogenicity by elevating antigen presentation and thus increasing sensitivity to T cell-mediated killing (149).
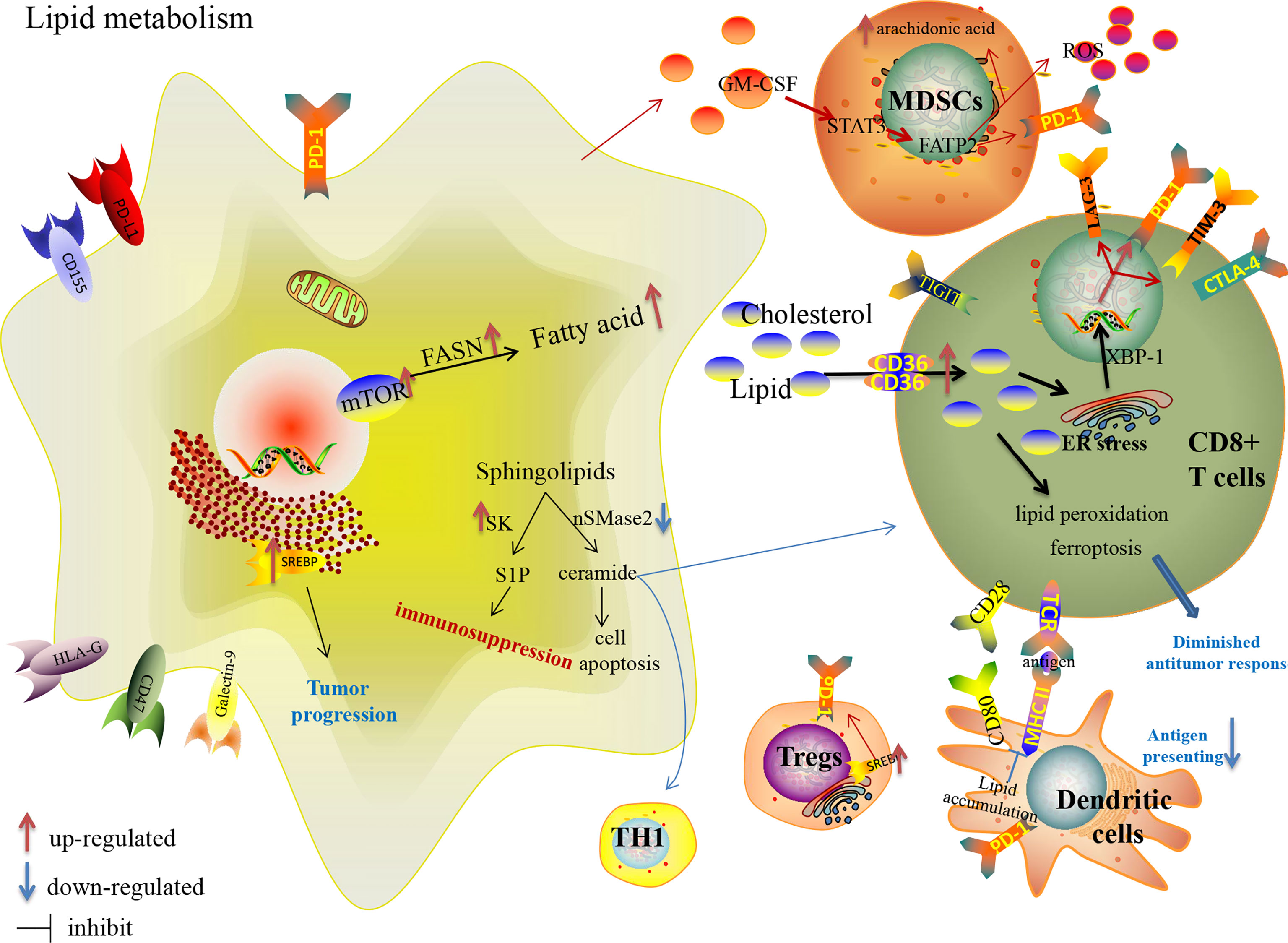
Figure 3 Lipid metabolism reprogramming in cancer cells and immune cells in the TME, and their effects on the expression of immune checkpoints.
Cholesterol, a crucial component of membrane lipids, is required for TCR clustering and T-cell immunological synapse to participate in the antigen-presenting, activation and differentiation function of macrophages and DCs (150–152). It has been reported that as enriched cholesterol accumulates in the cytoplasm, it induces the over-expression of inhibitory checkpoints [PD-1, TIM-3, and lymphocyte activation gene 3 (LAG-3)] in an ER-stress-X-box binding protein 1 (XBP1)-dependent manner leading to the functional depletion of CD8+ T cells. Ma X et al. showed that decreased cholesterol or ER stress could enhance CD8+ T cell anti-tumor function, highlighting a therapeutic avenue in improving T cell-based immunotherapy (153). Atorvastatin, one of the classic cholesterol-lowering drugs, down-regulates co-inhibitory receptors expression and increases IL-2 secretion by inhibiting the ras-activated MAPK and PI3K-Akt pathways and subsequent mTOR signaling to ameliorate activated T-cell function (154). In contrast, inhibiting cholesterol esterification with the increase in plasma membrane cholesterol level is a proven potentiated effector function and enhances proliferation of CD8+ TILs, but not CD4+ TILs, a result of the enhanced T-cell receptor clustering as well as the more efficient formation of the immunological synapse (155). Yang W et al. demonstrated that a combined therapy of avasimibe, the inhibitor of Acyl-coenzyme A:cholesterol acyltransferase1 (ACAT1) which is a crucial cholesterol esterification enzyme (156), with an anti-PD-1 antibody offers better efficacy than monotherapies in controlling tumor progression of melanoma-bearing mice (155). The combination therapy of targeting lipid metabolism with ICIs is under further study.
Moreover, more attention has been focused on sphingolipid metabolism in tumor biological behavior, immune escape and anticancer therapy. Sphingolipids, including the two central bioactive lipids, ceramide and sphingosine-1-phosphate (S1P), have opposing roles in sustaining cancer cell survival (157). Neutral sphingomyelinase 2 (nSMase2) with SMPD3 encoding catalyzes the breakdown of sphingolipid to produce the anti-tumor ceramide linked to apoptosis, growth arrest and chemotherapeutic response (158), accumulates TH1 polarization and CD8+ TILs with increased IFN-γ and chemokine C-X-C motif ligand 9 (CXCL9), and even increases anti-PD-1 efficacy in murine models of melanoma and breast cancer (159). Nonetheless, nSMase2 is generally downregulated in many cancers (158). Sphingosine kinase (SK) catalyzes the phosphorylation of sphingosine to S1P. SK behaves as an immune escape lipid kinase with increased expression of immunosuppressive factors in many cancers. Increased expression of SK1 in tumor cells is significantly associated with shorter survival in metastatic melanoma patients treated with anti-PD-1, and targeting SK1 enhances the responses to ICIs in murine models of melanoma, breast and colon cancer with limited Tregs infiltration (160). These preclinical studies provide a solid foundation for further clinical trials to overcome ICI resistance by targeting sphingolipid metabolism.
Nucleotide Metabolism Reprogramming in Cancer Cells With Immune Cells and Their Effects on the Expression of Immune Checkpoints
The process of nucleotide synthesis includes de novo synthesis and salvage pathway. De novo synthesis, as the main synthesis pathway, refers to the process of forming ribonucleotides through a series of enzymatic reactions with phosphoribose, amino acids, one carbon unit and CO2. Then, the relevant deoxyribonucleotides diphosphate (dNDP), as the precursors of deoxyribonucleotide triphosphate (dNTP) participating in DNA synthesis, are produced by ribonucleotides catalyzed by ribonucleotide reductase (RNR). Acetylation and deacetylation of the ribonucleotide reductase small subunit M2 (RRM2) act as a crucial switch that impacts dNTP synthesis and DNA replication fork progression (161). Compared to normal cells, cancer cells have a greater demand for dNTP pool to satisfy the proliferative and invasive activities (162). And the increased nucleotide de novo synthesis drives the metastasis and stemness of cancer cells with more cyclic guanosine monophosphate (cGMP) (163). Required nutrient deficiency competing with cancer cells, such as one-carbon unit and aspartate, was also reported to impair the immunity of the TME, such as activation of naive T cells and the expansion function of the effector T cell (128, 164). Remarkably, enriched nucleosides or enhanced RRM2 expression promote tumorigenesis by suppressing oncogene-induced stable associated cell growth arrest, and dNTP repression with RRM2 knockdown could inhibit the growth of cancer cells (165). Additionally, statistical analysis demonstrated that high expression of RRM2 was relevant to immunosuppressive TME (166). Although several generations of RNR inhibitors have been developed in anti-tumor therapy (167), there has been a lack of exciting performance, especially in combination with ICIs. The dihydroorotate dehydrogenase (DHODH) is the enzyme responsible for the fourth step of de novo pyrimidine biosynthesis and coupled to the mitochondrial electron transport chain. Although DHODH is not known to be mutated or over-expressed in patients with cancer, DHODH inhibitors are able to shrink tumor burden and improve survival by inducting leukemic cells differentiation in preclinical AML models (168). Unexpectedly, a phase I/II clinical trial on the combination of leflunomide, a human DHODH inhibitor, and the BRAF inhibitor vemurafenib in metastatic melanoma patients with BRAFV600 mutations was terminated due to serious AEs (NCT01611675) (168).
Under endogenous replication stress, DNA breakage and DNA damage response and repair (DDR) play essential roles in tumorigenesis, tumor progression and immune regulation (169). DDR gene alternations are associated with increased TILs, higher genomic instability, increased tumor mutation load (TMB) and improved clinical outcomes in cancer (170). It was reported that DDR alterations are associated with higher ORR, longer PFS and OS in urothelial carcinoma and NSCLC patients treated with PD-1/PD-L1 blockade (170, 171). Besides, Tu X et al. demonstrated that intracellular PD-L1 regulates the DDR by binding and stabilizing the mRNAs of DNA damage genes, and a novel PD-L1 antibody, H1A, can sensitize cancer to DNA-damaging therapy, radiation or chemotherapy, by promoting PD-L1 degradation (172). Moreover, Sen T et al. demonstrated that targeting DDR proteins with poly ADP-ribose polymerase (PARP) or checkpoint kinase 1 significantly increases the expression of PD-L1, remarkably potentiated the anti-tumor effect of PD-L1 blockade and augmented cytotoxic T-cell infiltration in SCLC mouse models with the activation of STING/TBK1/IRF3 innate immune pathway (173). Although the ORR of ICIs in ovarian cancer is only 8-9%, preclinical studies have shown that combining ICIs augment the anti-tumor effects of DDR inhibitor olaparib (174). A phase 1a/b clinical trial showed that pamiparib, an oral PARP 1/2 inhibitor, with tislelizumab, a humanised anti-PD-1 monoclonal antibody, was generally well tolerated and had an ORR of 20% in patients with a variety of advanced solid tumors, including ovarian cancer (175). Furthermore, TOPACIO trial, a phase I/II clinical trial of the PARP inhibitor niraparib in combination with the anti-PD-1 antibody pembrolizumab in recurrent ovarian cancer frequently develop resistance to platinum-based chemotherapy, demonstrated an 18% ORR and a clinical benefit rate of 65%, clearly exceeding the expected activity of niraparib or pembrolizumab as monotherapies in recurrent platinum-resistant ovarian cancer (176).
Other than mutations of oncogenes and tumor suppressors, epigenetic reprogramming, such as histone modifications, DNA methylation, and noncoding RNAs, also drive the phenotypic changes of tumor cells to escape from immune surveillance and construct an immunosuppressive TME (177). In this way, a rational combination of PD-L1/PD-1 blockade and epigenetic agents may offer great potential in retraining the immune system and improving clinical outcomes of anti-tumor treatment. Some phase I/II clinical trials on the combination of ICIs and epigenetic agents among different cancer types are designed and ongoing (177).
Aberrant Mitochondrial Biogenesis in Cancer Cells With Immune Cells and Their Effects on the Expressions of Immune Checkpoints
ATP, as the dominant energy provider which plays a central role in energy generation, transformation, storage, utilization and other biological activities, is mainly produced by the catabolic metabolism of glucose, lipids and amino acids with TCA and OXPHOS. The protons and electrons removed from the metabolites are present as reducing equivalents (NADH+H+, FADH2) in mitochondria, which are pumped into the cytosol by an electron transport chain consisting of four complexes, forming an electrochemical gradient of H+ across the mitochondrial membrane, while the ATP synthase utilizes the potential energy released during its reflux down the gradient to drive ATP release. Although glycolysis predominates in the metabolic reprogramming of cancer cells, OXPHOS is also enhanced in tumor cells, and some compounds targeting OXPHOS to suppress tumors have been studied (178, 179). An example is metformin, which could decrease cancer cell proliferation by decreasing the NAD+/NADH ratio and inhibiting aspartate production (180), and stimulates the proliferation of CD8+ TILs with the production of mitochondrial ROS in an NF-E2-related factor 2 (Nrf2)/mTORC1/p62 pathway (181). It was also reported that metformin could further improve intratumoral T-cell function and tumor clearance when combined with ICIs (62, 181, 182). Phase II clinical trial showed metformin therapy was associated with better-than-expected OS on non-diabetic patients with advanced-stage ovarian cancer (183). The median PFS of 18.0 months and OS of 57.9 months compare favorably to other clinical trials with similar patient populations and historical expectations (183). The clinical trial focus on the combination therapy of Nivolumab with metformin for refractory or recurrent solid tumors is ongoing (184). Another example is IACS-010759, which has been reported to emphatically inhibit the proliferation of hypoxic tumor cells by interfering with the functions of mitochondrial NADH-ubiquinone oxidoreductase (complex I) without exhibiting cytotoxicity at tolerated doses in normal cells (185). And preclinical trial demonstrated that combined IACS-010759 and radiotherapy promoted anti-tumor effects in the PD-1-resistant model, though not in the sensitive model (186). Unfortunately, there has been no relevant clinical trial.
Different immunocyte populations have different metabolic educations in the metabolically reprogrammed TME. Quiescent T cells and Tregs depend on OXPHOS for energy production, while effector T cells mainly rely on glycolysis for proliferation (187). Besides, effector memory and central memory CD4+ T cells have elevated glycolysis and OXPHOS compared to naive T cells. When naive T cells respond to TCR stimulation, robust and rapid glycolysis and OXPHOS occur, accompanied by Akt or STAT5 signaling activation (188). Elevated OXPHOS also participates in immunoregulation with increased MHC presentation (189). Compared to pro-inflammatory M1 macrophages, immunosuppressive M2 macrophages are more dependent on OXPHOS (72). The immunosuppressive TME represses mitochondrial biogenesis (190), whereas 4-1BB costimulation increases mitochondria numbers in CD8+ T cells (191). Also, PD-1 engagement in CD8+ T cells would trigger a suppressed OXPHOS process with decreased mitochondrial cristae and unexpectedly greater assembly of respiratory supercomplexes different from resting cells and activated T cells (31). These results suggest that mitochondrion is the primary target of immune checkpoints engaged with the activated or inhibitory activity. Nevertheless, the role of OXPHOS in ICIs resistance is controversial. High-resolution mass spectrometry revealed higher OXPHOS in responders with TIL-based or anti-PD-1 immunotherapy than non-responders in melanoma patients (149). Alternatively, Chen D et al. considered the PD-1-resistant model, which seemed to utilize OXPHOS to a significantly greater extent than the PD-1-sensitive NSCLC murine model (186). They further demonstrated that the combination of OXPHOS inhibitor, IACS-010759 and radiotherapy promoted anti-tumor effects in the PD-1-resistant model, but not in the sensitive tumor-burdened animal model (186). Moreover, Jia L et al. tried to enhance the immunity of TILs by balancing the glycolysis and OXPHOS of tumor cells, and this balancing strategy provides a more reliable immune-boosting strategy to PD-L1 silencing than complete glycolysis inhibition (179).
Under the stress of hypoxia, inflammation and deficient nutrient, ATP is transferred to the extracellular space (192). Accumulated ATP in the TME is metabolized into immunosuppressive adenosine by upregulated ectonucleotidases in cancer. Increased adenosine binding to its receptors block proliferation of T cells, promote immunosuppressive Tregs, prevent the cytotoxic activity of NK cells and M1 macrophages, and induce the upregulation of co-inhibitory molecules (PD-1 and CTLA-4) and tolerogenic cytokines (193–195). It has been reported that the increased ectonucleotidase and adenosine are related to hypoxia exposure in many cancer cells (196, 197). Remarkably, adenosine drives the overexpression of LDH5, a crucial mediator promoting anaerobic glycolysis, creating a vicious cycle because the large amount of ATP produced by glycolysis feeds the production of adenosine in an ectonucleotidase-rich TME (198). Serra S et al. have shown that the A2A adenosine receptor blockade counteracts these effects, making leukemic cells more susceptible to pharmacological agents while restoring immune competence and T-cell proliferation in vitro studies (197). Moreover, dual blockade of PD-1 and A2A significantly enhances the cytotoxicity of CD8+ TILs, in turn inhibiting tumor progression and prolonging tumor-burden mice’s survival (199).
Biomarkers in ICIs Therapy From Reprogramming Metabolism
Although there have been exciting reports about the roles of ICIs in anti-tumor therapy, the response rate is not satisfactory. Low ORR, short survival and high recurrence have been reported in many cancer patients with ICIs treatment. Finding the appropriately predictive and prognostic biomarker is important in anti-tumor immunotherapy. Currently, the intensity and density of PD-1/PD-L1 expression in tumor tissue is commonly considered to be the standard biomarker for predicting the efficacy of immunotherapy. However, some cancer patients with high PD-(L)1 expression have been reported to have an inadequate response or even resistance to anti-PD-(L)1, whereas some patients with low PD-(L)1 expression have a strong response (200). It is necessary to search for more precise prognostic markers in the anti-tumor immunotherapy.
Based on the crucial role of the reprogramming metabolism in tumor progression and immunosuppression, many metabolism-related molecules have been investigated for their predictive and prognostic values in immunotherapy. TMB is reportedly an effective biomarker in many tumors with ICIs and have been applied in clinical studies (201, 202). Compared to TMB and aneuploidy, glycolytic activity is a stronger and more consistent predictor of immune signatures in a diverse range of cancers (203). Notably, Jiang Z et al. showed that highly glycolytic tumors exhibited a better immunotherapy response and favorable survival in the immunotherapy setting (203). LDH and tumor acidity are also reported as predictive and prognostic markers for immunotherapy (204). Besides, Xue G et al. even showed a better predictive and prognostic value of the CpG-based model than TMB in cancer patients (205). In addition, circulating tumor DNA seem to have predictive and prognostic values in anti-tumor immunotherapy (206).
Discussion
Immunometabolism has attracted increasing research interest on anti-tumor therapeutic applications recently (207). Metabolic reprogramming, hypoxia, genetic mutation and immune regulation control each other interdependently and collectively, contributing to tumor progression and immune compromise (208). Under the education of oncogenes and tumor suppressors, tumor cells and immune cells reprogram the metabolism with key molecular alterations to accommodate tumor progression and immune suppression (Table 2). Targeting altered metabolism within tumor cells provides the possibility of improving the efficacy of immunotherapy. In this article, we have demonstrated the reprogramming metabolism in cancer cells and immune cells, and described the regulatory mechanisms of immune checkpoints involved. More importantly, we tried to cite preclinical and clinical trial evidence to comprehensively demonstrate that combined targeting metabolic therapy with ICIs may enhance anti-tumor abilities of immune cells, overcome drug resistance, and even prolong the PFS/OS of cancer patients, which provide a new perspective in anti-tumor immunotherapy. In addition to this, we sought to find biomarkers with greater prognostic value for ICIs treatment from a metabolic perspective.
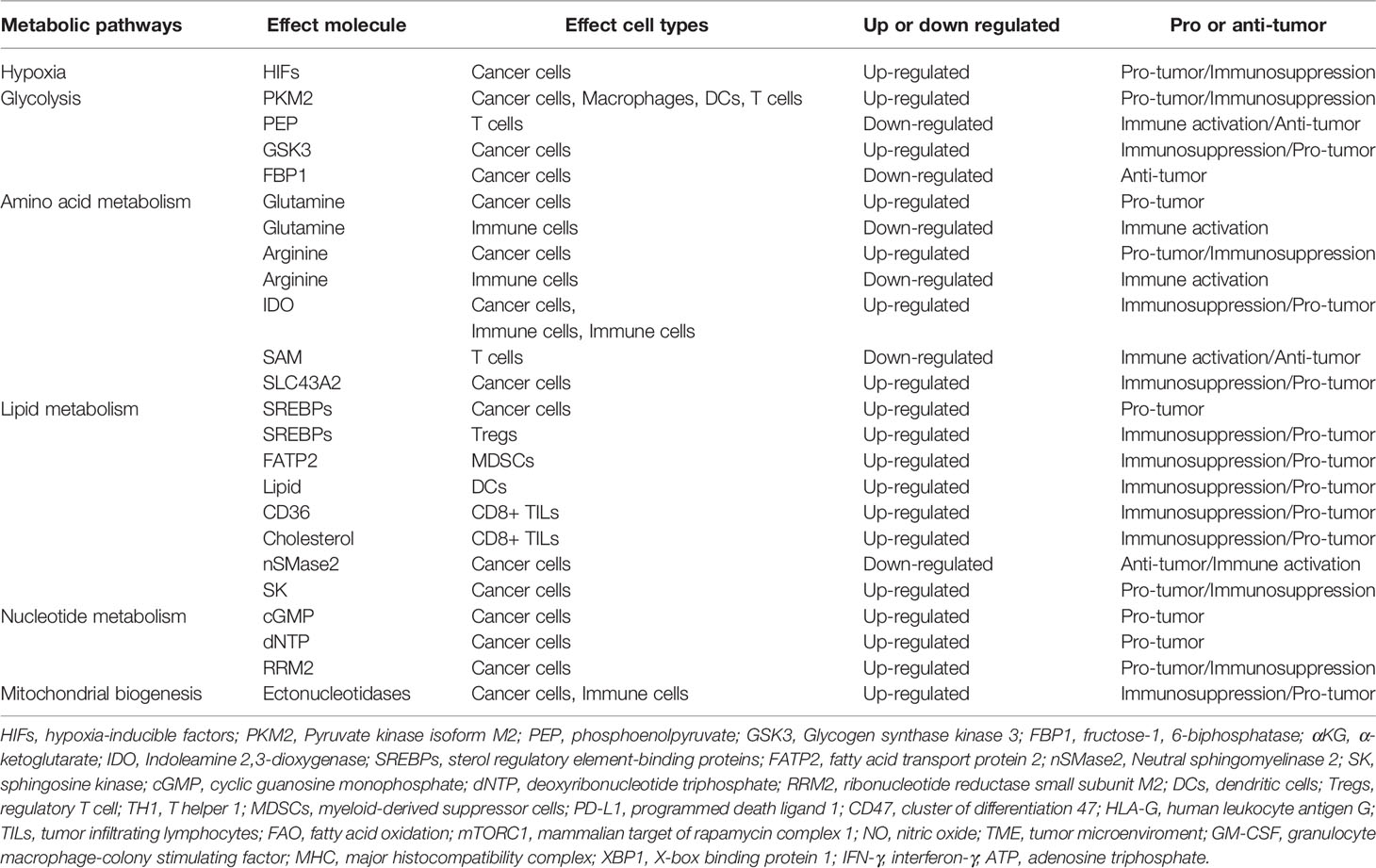
Table 2 The effect of key metabolic targets on cancer cells and immune cells in the TME.
Although ICIs have shown an impressive performance in the treatment of solid tumors, their ORR is around 20-40%. It is worth exploring how to improve its efficiency in anti-tumor treatment, overcome drug resistance and prolong the survival of cancer patients. A rising number of efforts have attempted to tip the balance of immune compromise to anti-tumor immunity by ICIs combined with chemotherapy, radiotherapy (209), targeted agents (210), innate immunity modulating drugs (211), even anti-platelet (212) and other therapies. Chemotherapy potentiates the immunogenicity of tumor cells to enhance immune recognition and immune elimination, and immunotherapy increases the sensitivity of tumor cells to chemotherapy (213). As shown in phase 3 clinical trial of locally advanced or metastatic non-squamous NSCLC patients without EGFR or ALK genomic aberration, NCT03607539, Sintilimab (anti-PD-1) plus pemetrexed and platinum had a longer median PFS (8.9 versus 5.0 months) and a better ORR (51.9% versus 29.8%) than that in the placebo plus pemetrexed and platinum group (214). In addition to destroying tumor cells to release antigens for enhanced antigen presentation, radiotherapy also enhances anti-tumor immunity by upregulating MHC-I expression and the cGAS-STING pathway (215), increasing TCR density (216) and the abscopal effect (209). Pacific trial, a phase III clinical trial on unresectable NSCLC patients, demonstrated longer PFS (17.2 vs 5.6 months), OS (28.3 vs 16.2 months) and a higher ORR (30% vs 17.8%) in patients treated with anti-PD-L1 Duvalumab within 1-14 days of completing chemoradiotherapy (217). CheckMate 067 further demonstrated a significantly enhanced ORR in melanoma patients treated with Nivolumab plus Ipilimumab (58%) with no apparent loss of quality of life, compared to Ipilimumab (19%), Nivolumab (45%) alone (218). NSCLC patients harboring EGFR mutations or ALK rearrangements are commonly thought to be associated with a low ORR to PD-1/PD-L1 inhibitors. However, a retrospective analysis revealed that the proportion of tumors with high-expressed PD-L1 was higher among T790M-negative patients than among T790M-positive patients after disease progression during EGFR-TKI treatment (219). This suggests the potential of combining ICIs with EGFR-TKIs to improve efficacy and overcome drug resistance.
Nevertheless, the metabolic reprogramming of tumor cells and immune cells is one of the crucial obstacles to immunotherapy, and targeting reprogrammed metabolites provides a new perspective to improve the efficacy of ICIs in addition to chemotherapy, radiotherapy and targeted therapy. Many preclinical and phase I/II clinical trials have demonstrated promising performance of the combination of targeting metabolism with immunotherapy in overcoming drug resistance and minimizing tumor burden (Table 3). In particular, PARP inhibitors, ADI-PEG 20, IDO inhibitor and metformin have shown exciting performance in combination with ICIs in clinical trials. Furthermore, phase III clinical trials with extended samples on ORR, PFS and OS still need to be explored. Additionally, while pursuing an improved benefit rate, the combination therapy should pay attention to prevent the occurrence of serious AEs. The metabolic reprogramming process is integral and cannot be separated from each other completely. Although targeted tumor metabolic therapy provides access to more nutrients and oxygen to confers immune cell function, the metabolic pathways within tumor cells and immune cells are frequently similar, making tumor cells damaged while immune cells are injured by targeting metabolic signals. Such as rapamycin, which interferes with the proliferation, growth and survival of cancer cells as well as having immunosuppressive effects (220).
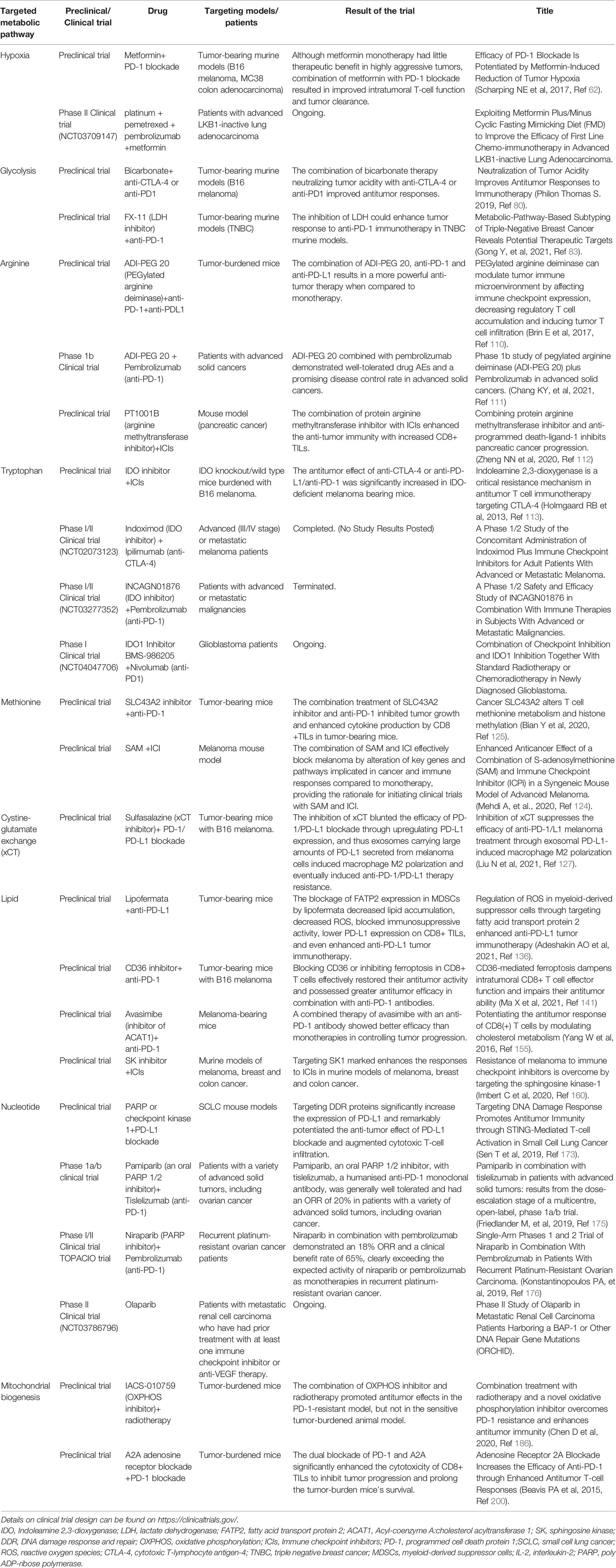
Table 3 Preclinical and clinical trials of drugs inhibiting metabolic targets with ICIs in anti-tumor therapy.
Among the reprogramming metabolic processes of cancer cells and immune cells, mTOR signaling seems to play a vital role in bridging metabolism and immunity. mTOR signaling controls cellular metabolism, immune cell differentiation, and effector function (131). The PI3K-mTOR-AKT signaling pathway not only promotes tumor cell growth and proliferation in the majority of cancers, but also controls the activation and polarization of macrophages, and decreases T-cell infiltration (131). Meanwhile, the mTOR signaling is also involved in immune checkpoint expression and downstream signaling in tumor cells and immune cells. It mediated the co-inhibitory checkpoints expression under the cytokines stimulation and participates in the PD-1/PD-L1 downstream signaling to reprogram cellular metabolism. mTOR signaling is also involved in the reprogramming lipid, amino acids and nucleotide metabolism in cancer cells and immune cells to promote tumor cell progression and escape immunological surveillance. Further studies on the role of the mTOR signaling in controlling the immunometabolism in the TME will better facilitate the efficacy and minimize the side effects of targeting metabolism in combination with ICIs.
In addition to the metabolic reprogramming of tumor cells and immune cells to manipulate the tumor-immune set point in TME, the metabolism of the gut microbiome has received increasing attention on the anti-tumor immune regulation. Microbiota-derived short-chain fatty acids, such as butyrate, promote the formation of long-term CD8+ memory T cells and modulate Tregs function (207, 220). Furthermore, in melanoma patients, a favorable gut microbiome combined with anti-PD-1 resulted in enhanced anti-tumor responses associated with improved Teff cell function (221). In this regard, supporting gut microbiota-related metabolism may be an effective adjuvant to anti-tumor immunotherapy.
Not all metabolic changes equally contribute to promoting the malignant transformation and progression of tumor cells. These metabolic changes can be divided into three categories: transforming activities, enabling activities and neutral activities (208). Only a tiny portion of metabolic activities are transforming activities that directly contribute to tumorigenesis and antagonize anti-tumor activities. Most metabolic reprogramming are enabling activities that change within tumor cells but are not involved in cell transformation, while neutral activities are dispensable to tumor cell progression. Nonetheless, neutral activities reportedly have predictive and prognostic values. Intriguingly, even the same kind of tumor cells may have different metabolic remodeling, thus even the same targeting metabolic therapy may have different effects. Gong Y et al. demonstrated that there are three heterogeneous metabolic-pathway-based subtypes (MPSs) with distinct metabolic features: MPS1, the lipogenic subtype with upregulated lipid metabolism; MPS2, the glycolytic subtype with upregulated carbohydrate and nucleotide metabolism; and MPS3, the mixed subtype with partial pathway dysregulation, which exists in TNBC patients with distinct prognoses, molecular subtype distributions and genomic alterations (83). Furthermore, even the same kind of metabolic reprogramming in the TME has different immune effects on different immune cells, and these differences manifest the charms of tumor heterogeneity. In an era of individualized precision treatment, providing the right personalized treatment to the right patients at the right time for optimizing benefits for each person requires close cooperation between various fields.
Author Contributions
YX and LH conceived this review. This review were performed by YX, QF, LH, and JH collectively. YX wrote the manuscript. LH polished this review. All authors contributed to the article and approved the submitted version.
Funding
This work was financially supported by the PhD Research Startup Foundation of Liaoning Province, China (No. 2020-BS-039).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Glossary

References
1. Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB. Et Al: Improved Survival With Ipilimumab in Patients With Metastatic Melanoma. N Engl J Med (2010) 363(8):711–23. doi: 10.1056/NEJMoa1003466
2. Robert C, Thomas L, Bondarenko I, O’Day S, Weber J, Garbe C. Et Al: Ipilimumab Plus Dacarbazine for Previously Untreated Metastatic Melanoma. N Engl J Med (2011) 364(26):2517–26. doi: 10.1056/NEJMoa1104621
3. Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF. Et Al: Safety, Activity, and Immune Correlates of Anti-PD-1 Antibody in Cancer. N Engl J Med (2012) 366(26):2443–54. doi: 10.1056/NEJMoa1200690
4. Gazdar AF, Bunn PA. And Minna JD: Small-Cell Lung Cancer: What We Know, What We Need to Know and the Path Forward. Nat Rev Cancer (2017) 17(12):725–37. doi: 10.1038/nrc.2017.87
5. Horn L, Mansfield AS, Szczęsna A, Havel L, Krzakowski M, Hochmair MJ. Et Al; IMpower133 Study Group: First-Line Atezolizumab Plus Chemotherapy in Extensive-Stage Small-Cell Lung Cancer. N Engl J Med (2018) 379(23):2220–9. doi: 10.1056/NEJMoa1809064
6. Borghaei H, Paz-Ares L, Horn L, Spigel DR, Steins M, Ready NE, et al. Nivolumab Versus Docetaxel in Advanced Nonsquamous Non-Small-Cell Lung Cancer. N Engl J Med (2015) 373(17):1627–39. doi: 10.1056/NEJMoa1507643
7. Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Rutkowski P, Lao CD, et al. Five-Year Survival With Combined Nivolumab and Ipilimumab in Advanced Melanoma. N Engl J Med (2019) 381(16):1535–46. doi: 10.1056/NEJMoa1910836
8. Parsa AT, Waldron JS, Panner A, Crane CA, Parney IF, Barry JJ, et al. Loss of Tumor Suppressor PTEN Function Increases B7-H1 Expression and Immunoresistance in Glioma. Nat Med (2007) 13(1):84–8. doi: 10.1038/nm1517
9. Jin X, Ding D, Yan Y, Li H, Wang B, Ma L, et al. Phosphorylated RB Promotes Cancer Immunity by Inhibiting NF-κb Activation and PD-L1 Expression. Mol Cell (2019) 73(1):22–35.e6. doi: 10.1016/j.molcel.2018.10.034
10. Liu J, Hamrouni A, Wolowiec D, Coiteux V, Kuliczkowski K, Hetuin D, et al. Plasma Cells From Multiple Myeloma Patients Express B7-H1 (PD-L1) and Increase Expression After Stimulation With IFN-{Gamma} and TLR Ligands via a MyD88-, TRAF6-, and MEK-Dependent Pathway. Blood (2007) 110(1):296–304. doi: 10.1182/blood-2006-10-051482
11. Kondo A, Yamashita T, Tamura H, Zhao W, Tsuji T, Shimizu M, et al. Interferon-Gamma and Tumor Necrosis Factor-Alpha Induce an Immunoinhibitory Molecule, B7-H1, via Nuclear factor-kappaB Activation in Blasts in Myelodysplastic Syndromes. Blood (2010) 116(7):1124–31. doi: 10.1182/blood-2009-12-255125
12. Qian Y, Deng J, Geng L, Xie H, Jiang G, Zhou L, et al. TLR4 Signaling Induces B7-H1 Expression Through MAPK Pathways in Bladder Cancer Cells. Cancer Invest (2008) 26(8):816–21. doi: 10.1080/07357900801941852
13. Kamran N, Takai Y, Miyoshi J, Biswas SK, Wong JS, Gasser S. Toll-Like Receptor Ligands Induce Expression of the Costimulatory Molecule CD155 on Antigen-Presenting Cells. PloS One (2013) 8(1):e54406. doi: 10.1371/journal.pone.0054406
14. Marasco M, Berteotti A, Weyershaeuser J, Thorausch N, Sikorska J, Krausze J, et al. Molecular Mechanism of SHP2 Activation by PD-1 Stimulation. Sci Adv (2020) 6(5):eaay4458. doi: 10.1126/sciadv.aay4458
15. Teft WA, Chau TA, Madrenas J. Structure-Function Analysis of the CTLA-4 Interaction With PP2A. BMC Immunol (2009) 10:23. doi: 10.1186/1471-2172-10-23
16. Sharma P, Allison JP. The Future of Immune Checkpoint Therapy. Sci (2015) 348(6230):56–61. doi: 10.1126/science.aaa8172
17. Hwang WL, Pike LRG, Royce TJ, Mahal BA, Loeffler JS. Safety of Combining Radiotherapy With Immune-Checkpoint Inhibition. Nat Rev Clin Oncol (2018) 15(8):477–94. doi: 10.1038/s41571-018-0046-7
18. Robert C, Schachter J, Long GV, Arance A, Grob JJ, Mortier L, et al. KEYNOTE-006 Investigators: Pembrolizumab Versus Ipilimumab in Advanced Melanoma. N Engl J Med (2015) 372(26):2521–32. doi: 10.1056/NEJMoa1503093
19. Gangadhar TC, Vonderheide RH. Mitigating the Toxic Effects of Anticancer Immunotherapy. Nat Rev Clin Oncol (2014) 11(2):91–9. doi: 10.1038/nrclinonc.2013.245
20. Wei SC, Levine JH, Cogdill AP, Zhao Y, Anang NAS, Andrews MC, et al. Distinct Cellular Mechanisms Underlie Anti-CTLA-4 and Anti-PD-1 Checkpoint Blockade. Cell (2017) 170(6):1120–1133.e17. doi: 10.1016/j.cell.2017.07.024
21. Mahnke K, Enk AH. TIGIT-CD155 Interactions in Melanoma: A Novel Co-Inhibitory Pathway With Potential for Clinical Intervention. J Invest Dermatol (2016) 136(1):9–11. doi: 10.1016/j.jid.2015.10.048
22. Nagumo Y, Iguchi-Manaka A, Yamashita-Kanemaru Y, Abe F, Bernhardt G, Shibuya A, et al. Increased CD112 Expression in Methylcholanthrene-Induced Tumors in CD155-Deficient Mice. PloS One (2014) 9(11):e112415. doi: 10.1371/journal.pone.0112415
23. Nakai R, Maniwa Y, Tanaka Y, Nishio W, Yoshimura M, Okita Y, et al. Overexpression of Necl-5 Correlates With Unfavorable Prognosis in Patients With Lung Adenocarcinoma. Cancer Sci (2010) 101(5):1326–30. doi: 10.1111/j.1349-7006.2010.01530.x
24. Atsumi S, Matsumine A, Toyoda H, Niimi R, Iino T, Sudo A. Prognostic Significance of CD155 mRNA Expression in Soft Tissue Sarcomas. Oncol Lett (2013) 5(6):1771–6. doi: 10.3892/ol.2013.1280
25. Nishiwada S, Sho M, Yasuda S, Shimada K, Yamato I, Akahori T, et al. Clinical Significance of CD155 Expression in Human Pancreatic Cancer. Anticancer Res (2015) 35(4):2287–97.
26. Yu X, Harden K, Gonzalez LC, Francesco M, Chiang E, Irving B, et al. The Surface Protein TIGIT Suppresses T Cell Activation by Promoting the Generation of Mature Immunoregulatory Dendritic Cells. Nat Immunol (2009) 10(1):48–57. doi: 10.1038/ni.1674
27. Wang Z, Yin N, Zhang Z, Zhang Y, Zhang G, Chen W. Upregulation of T-Cell Immunoglobulin and Mucin-Domain Containing-3 (Tim-3) in Monocytes/Macrophages Associates With Gastric Cancer Progression. Immunol Invest (2017) 46(2):134–48. doi: 10.1080/08820139.2016.1229790
28. Zhu S, Lin J, Qiao G, Wang X, Xu Y. Tim-3 Identifies Exhausted Follicular Helper T Cells in Breast Cancer Patients. Immunobiol (2016) 221(9):986–93. doi: 10.1016/j.imbio.2016.04.005
29. Li H, Wu K, Tao K, Chen L, Zheng Q, Lu X, et al. Tim-3/Galectin-9 Signaling Pathway Mediates T-Cell Dysfunction and Predicts Poor Prognosis in Patients With Hepatitis B Virus-Associated Hepatocellular Carcinoma. Hepatol (2012) 56(4):1342–51. doi: 10.1002/hep.25777
30. Frauwirth KA, Riley JL, Harris MH, Parry RV, Rathmell JC, Plas DR, et al. The CD28 Signaling Pathway Regulates Glucose Metabolism. Immun (2002) 16(6):769–77. doi: 10.1016/s1074-7613(02)00323-0
31. Ogando J, Sáez ME, Santos J, Nuevo-Tapioles C, Gut M, Esteve-Codina A, et al. PD-1 Signaling Affects Cristae Morphology and Leads to Mitochondrial Dysfunction in Human CD8+ T Lymphocytes. J Immunother Cancer (2019) 7(1):151. doi: 10.1186/s40425-019-0628-7
32. Qorraj M, Bruns H, Böttcher M, Weigand L, Saul D, Mackensen A, et al. The PD-1/PD-L1 Axis Contributes to Immune Metabolic Dysfunctions of Monocytes in Chronic Lymphocytic Leukemia. Leukemia (2017) 31(2):470–8. doi: 10.1038/leu.2016.214
33. Patsoukis N, Bardhan K, Chatterjee P, Sari D, Liu B, Bell LN, et al. PD-1 Alters T-Cell Metabolic Reprogramming by Inhibiting Glycolysis and Promoting Lipolysis and Fatty Acid Oxidation. Nat Commun (2015) 6:6692. doi: 10.1038/ncomms7692
34. Zhang C, Yue C, Herrmann A, Song J, Egelston C, Wang T, et al. STAT3 Activation-Induced Fatty Acid Oxidation in CD8+ T Effector Cells Is Critical for Obesity-Promoted Breast Tumor Growth. Cell Metab (2020) 31(1):148–161.e5. doi: 10.1016/j.cmet.2019.10.013
35. Palaskas NJ, Garcia JD, Shirazi R, Shin DS, Puig-Saus C, Braas D, et al. Global Alteration of T-Lymphocyte Metabolism by PD-L1 Checkpoint Involves a Block of De Novo Nucleoside Phosphate Synthesis. Cell Discov (2019) 5:62. doi: 10.1038/s41421-019-0130-x
36. Teijeira A, Garasa S, Etxeberria I, Gato-Cañas M, Melero I, Delgoffe GM. Metabolic Consequences of T-Cell Costimulation in Anticancer Immunity. Cancer Immunol Res (2019) 7(10):1564–9. doi: 10.1158/2326-6066.CIR-19-0115
37. Sabharwal SS, Rosen DB, Grein J, Tedesco D, Joyce-Shaikh B, Ueda R, et al. GITR Agonism Enhances Cellular Metabolism to Support CD8+ T-Cell Proliferation and Effector Cytokine Production in a Mouse Tumor Model. Cancer Immunol Res (2018) 6(10):1199–211. doi: 10.1158/2326-6066.CIR-17-0632
38. Chang CH, Qiu J, O’Sullivan D, Buck MD, Noguchi T, Curtis JD, et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell (2015) 162(6):1229–41. doi: 10.1016/j.cell.2015.08.016
39. Strauss L, Mahmoud MAA, Weaver JD, Tijaro-Ovalle NM, Christofides A, Wang Q, et al. Targeted Deletion of PD-1 in Myeloid Cells Induces Antitumor Immunity. Sci Immunol (2020) 5(43):eaay1863. doi: 10.1126/sciimmunol.aay1863
40. Tomita M, Yasui H, Higashikawa K, Nakajima K, Takakura H, Shiga T, et al. Anti PD-1 Treatment Increases [18F]FDG Uptake by Cancer Cells in a Mouse B16F10 Melanoma Model. EJNMMI Res (2018) 8(1):82. doi: 10.1186/s13550-018-0433-1
41. Jiang L, Xiao L, Sugiura H, Huang X, Ali A, Kuro-o M, et al. Metabolic Reprogramming During Tgfβ1-Induced Epithelial-to-Mesenchymal Transition. Oncogene (2015) 34(30):3908–16. doi: 10.1038/onc.2014.321
42. Viale A, Pettazzoni P, Lyssiotis CA, Ying H, Sánchez N, Marchesini M, et al. Oncogene Ablation-Resistant Pancreatic Cancer Cells Depend on Mitochondrial Function. Nat (2014) 514(7524):628–32. doi: 10.1038/nature13611
43. Herber DL, Cao W, Nefedova Y, Novitskiy SV, Nagaraj S, Tyurin VA, et al. Lipid Accumulation and Dendritic Cell Dysfunction in Cancer. Nat Med (2010) 16(8):880–6. doi: 10.1038/nm.2172
44. Patsoukis N, Bardhan K, Weaver J, Herbel C, Seth P, Li L, et al. The Role of Metabolic Reprogramming in T Cell Fate and Function. Curr Trends Immunol (2016) 17:1–12.
45. Sica A, Strauss L. Energy Metabolism Drives Myeloid-Derived Suppressor Cell Differentiation and Functions in Pathology. J Leukoc Biol (2017) 102(2):325–34. doi: 10.1189/jlb.4MR1116-476R
46. Kouidhi S, Ben Ayed F, Benammar Elgaaied A. Targeting Tumor Metabolism: A New Challenge to Improve Immunotherapy. Front Immunol (2018) 9:353. doi: 10.3389/fimmu.2018.00353
47. Li Y, Patel SP, Roszik J, Qin Y. Hypoxia-Driven Immunosuppressive Metabolites in the Tumor Microenvironment: New Approaches for Combinational Immunotherapy. Front Immunol (2018) 9:1591. doi: 10.3389/fimmu.2018.01591
48. Vadde R, Vemula S, Jinka R, Merchant N, Bramhachari PV, Nagaraju GP. Role of Hypoxia-Inducible Factors (HIF) in the Maintenance of Stemness and Malignancy of Colorectal Cancer. Crit Rev Oncol Hematol (2017) 113:22–7. doi: 10.1016/j.critrevonc.2017.02.025
49. Schito L, Semenza GL. Hypoxia-Inducible Factors: Master Regulators of Cancer Progression. Trends Cancer (2016) 2(12):758–70. doi: 10.1016/j.trecan.2016.10.016
50. Meléndez-Rodríguez F, Urrutia AA, Lorendeau D, Rinaldi G, Roche O, Böğürcü-Seidel N, et al. Hif1α Suppresses Tumor Cell Proliferation Through Inhibition of Aspartate Biosynthesis. Cell Rep (2019) 26(9):2257–2265.e4. doi: 10.1016/j.celrep.2019.01.106
51. Corzo CA, Condamine T, Lu L, Cotter MJ, Youn JI, Cheng P, et al. HIF-1α Regulates Function and Differentiation of Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. J Exp Med (2010) 207(11):2439–53. doi: 10.1084/jem.20100587
52. Hong J, Tobin NP, Rundqvist H, Li T, Lavergne M, García-Ibáñez Y, et al. Role of Tumor Pericytes in the Recruitment of Myeloid-Derived Suppressor Cells. J Natl Cancer Inst (2015) 107(10):djv209. doi: 10.1093/jnci/djv434
53. Ambade A, Satishchandran A, Saha B, Gyongyosi B, Lowe P, Kodys K, et al. Hepatocellular Carcinoma Is Accelerated by NASH Involving M2 Macrophage Polarization Mediated by Hif-1α Induced IL-10. Oncoimmunol (2016) 5(10):e1221557. doi: 10.1080/2162402X.2016.1221557
54. Ren L, Yu Y, Wang L, Zhu Z, Lu R, Yao Z. Hypoxia-Induced CCL28 Promotes Recruitment of Regulatory T Cells and Tumor Growth in Liver Cancer. Oncotarget (2016) 7(46):75763–73. doi: 10.18632/oncotarget.12409
55. Vuillefroy de Silly R, Dietrich PY, Walker PR. Hypoxia and Antitumor CD8+ T Cells: An Incompatible Alliance? Oncoimmunology (2016) 5(12):e1232236. doi: 10.1080/2162402X.2016.1232236
56. Noman MZ, Desantis G, Janji B, Hasmim M, Karray S, Dessen P, et al. PD-L1 Is a Novel Direct Target of HIF-1α, and Its Blockade Under Hypoxia Enhanced MDSC-Mediated T Cell Activation. J Exp Med (2014) 211(5):781–90. doi: 10.1084/jem.20131916
57. Barsoum IB, Smallwood CA, Siemens DR, Graham CH. A Mechanism of Hypoxia-Mediated Escape From Adaptive Immunity in Cancer Cells. Cancer Res (2014) 74(3):665–74. doi: 10.1158/0008-5472.CAN-13-0992
58. Zhang H, Lu H, Xiang L, Bullen JW, Zhang C, Samanta D, et al. HIF-1 Regulates CD47 Expression in Breast Cancer Cells to Promote Evasion of Phagocytosis and Maintenance of Cancer Stem Cells. Proc Natl Acad Sci USA (2015) 112(45):E6215–23. doi: 10.1073/pnas.1520032112
59. Yaghi L, Poras I, Simoes RT, Donadi EA, Tost J, Daunay A, et al. Hypoxia Inducible Factor-1 Mediates the Expression of the Immune Checkpoint HLA-G in Glioma Cells Through Hypoxia Response Element Located in Exon 2. Oncotarget (2016) 7(39):63690–707. doi: 10.18632/oncotarget.11628
60. Najjar YG, Menk AV, Sander C, Rao U, Karunamurthy A, Bhatia R, et al. Tumor Cell Oxidative Metabolism as a Barrier to PD-1 Blockade Immunotherapy in Melanoma. JCI Insight (2019) 4(5):e124989. doi: 10.1172/jci.insight.124989
61. Zandberg DP, Menk AV, Velez M, Normolle D, DePeaux K, Liu A, et al. Tumor Hypoxia Is Associated With Resistance to PD-1 Blockade in Squamous Cell Carcinoma of the Head and Neck. J Immunother Cancer (2021) 9(5):e002088. doi: 10.1136/jitc-2020-002088
62. Scharping NE, Menk AV, Whetstone RD, Zeng X, Delgoffe GM. Efficacy of PD-1 Blockade Is Potentiated by Metformin-Induced Reduction of Tumor Hypoxia. Cancer Immunol Res (2017) 5(1):9–16. doi: 10.1136/jitc-2020-002088
63. Kim Y, Vagia E, Viveiros P, Kang CY, Lee JY, Gim G, et al. Overcoming Acquired Resistance to PD-1 Inhibitor With the Addition of Metformin in Small Cell Lung Cancer (SCLC). Cancer Immunol Immunother (2021) 70(4):961–5. doi: 10.1007/s00262-020-02703-8
64. Warburg O. Über Den Stoffwechsel Der Carcinomzelle. Naturwissenschaften (1924) 12:1131–7. doi: 10.1007/BF01504608
65. Palsson-McDermott EM, Curtis AM, Goel G, Lauterbach MA, Sheedy FJ, Gleeson LE, et al. Pyruvate Kinase M2 Regulates Hif-1α Activity and IL-1β Induction and Is a Critical Determinant of the Warburg Effect in LPS-Activated Macrophages. Cell Metab (2015) 21(1):65–80. doi: 10.1016/j.cmet.2015.01.017
66. Huang D, Li C, Zhang H. Hypoxia and Cancer Cell Metabolism. Acta Biochim Biophys Sin (Shanghai) (2014) 46(3):214–9. doi: 10.1093/abbs/gmt148
67. Luo W, Hu H, Chang R, Zhong J, Knabel M, O’Meally R, et al. Pyruvate Kinase M2 Is a PHD3-Stimulated Coactivator for Hypoxia-Inducible Factor 1. Cell (2011) 145(5):732–44. doi: 10.1016/j.cell.2011.03.054
68. Feng J, Yang H, Zhang Y, Wei H, Zhu Z, Zhu B, et al. Tumor Cell-Derived Lactate Induces TAZ-Dependent Upregulation of PD-L1 Through GPR81 in Human Lung Cancer Cells. Oncogene (2017) 36(42):5829–39. doi: 10.1038/onc.2017.188
69. Ho PC, Bihuniak JD, Macintyre AN, Staron M, Liu X, Amezquita R, et al. Phosphoenolpyruvate Is a Metabolic Checkpoint of Anti-Tumor T Cell Responses. Cell (2015) 162(6):1217–28. doi: 10.1016/j.cell.2015.08.012
70. Siska PJ, Rathmell JC. T Cell Metabolic Fitness in Antitumor Immunity. Trends Immunol (2015) 36(4):257–64. doi: 10.1016/j.it.2015.02.007
71. Huber V, Camisaschi C, Berzi A, Ferro S, Lugini L, Triulzi T, et al. Cancer Acidity: An Ultimate Frontier of Tumor Immune Escape and a Novel Target of Immunomodulation. Semin Cancer Biol (2017) 43:74–89. doi: 10.1016/j.semcancer.2017.03.001
72. Viola A, Munari F, Sánchez-Rodríguez R, Scolaro T, Castegna A. The Metabolic Signature of Macrophage Responses. Front Immunol (2019) 10:1462. doi: 10.3389/fimmu.2019.01462
73. Li Z, Zhang H. Reprogramming of Glucose, Fatty Acid and Amino Acid Metabolism for Cancer Progression. Cell Mol Life Sci (2016) 73(2):377–92. doi: 10.1007/s00018-015-2070-4
74. Long L, Chen M, Yuan Y, Ming AL, Guo W, Wu K, et al. High Expression of PKM2 Synergizes With PD-L1 in Tumor Cells and Immune Cells to Predict Worse Survival in Human Lung Adenocarcinoma. J Cancer (2020) 11(15):4442–52. doi: 10.7150/jca.42610
75. Palsson-McDermott EM, Dyck L, Zasłona Z, Menon D, McGettrick AF, Mills KHG, et al. Pyruvate Kinase M2 Is Required for the Expression of the Immune Checkpoint PD-L1 in Immune Cells and Tumors. Front Immunol (2017) 8:1300. doi: 10.3389/fimmu.2017.01300
76. Siska PJ, van der Windt GJ, Kishton RJ, Cohen S, Eisner W, MacIver NJ, et al. Suppression of Glut1 and Glucose Metabolism by Decreased Akt/mTORC1 Signaling Drives T Cell Impairment in B Cell Leukemia. J Immunol (2016) 197(6):2532–40. doi: 10.4049/jimmunol.1502464
77. Kuchuk O, Tuccitto A, Citterio D, Huber V, Camisaschi C, Milione M, et al. pH Regulators to Target the Tumor Immune Microenvironment in Human Hepatocellular Carcinoma. Oncoimmunol (2018) 7(7):e1445452. doi: 10.1080/2162402X.2018.1445452
78. Zhang YX, Zhao YY, Shen J, Sun X, Liu Y, Liu H, et al. Nanoenabled Modulation of Acidic Tumor Microenvironment Reverses Anergy of Infiltrating T Cells and Potentiates Anti-PD-1 Therapy. Nano Lett (2019) 19(5):2774–83. doi: 10.1021/acs.nanolett.8b04296
79. Damaghi M, Wojtkowiak JW, Gillies RJ. pH Sensing and Regulation in Cancer. Front Physiol (2013) 4:370. doi: 10.3389/fphys.2013.00370
80. Pilon-Thomas S, Kodumudi KN, El-Kenawi AE, Russell S, Weber AM, Luddy K, et al. Neutralization of Tumor Acidity Improves Antitumor Responses to Immunotherapy. Cancer Res (2016) 76(6):1381–90. doi: 10.1158/0008-5472.CAN-15-1743
81. Zappasodi R, Serganova I, Cohen IJ, Maeda M, Shindo M, Senbabaoglu Y, et al. CTLA-4 Blockade Drives Loss of Treg Stability in Glycolysis-Low Tumours. Nat (2021) 591(7851):652–8. doi: 10.1038/s41586-021-03326-4
82. Chan DA, Sutphin PD, Nguyen P, Turcotte S, Lai EW, Banh A, et al. Targeting GLUT1 and the Warburg Effect in Renal Cell Carcinoma by Chemical Synthetic Lethality. Sci Transl Med (2011) 3(94):94ra70. doi: 10.1126/scitranslmed.3002394
83. Gong Y, Ji P, Yang YS, Xie S, Yu TJ, Xiao Y, et al. Metabolic-Pathway-Based Subtyping of Triple-Negative Breast Cancer Reveals Potential Therapeutic Targets. Cell Metab (2021) 33(1):51–64.e9. doi: 10.1016/j.cmet.2020.10.012
84. Bose S, Le A. Glucose Metabolism in Cancer. Adv Exp Med Biol (2018) 1063:3–12. doi: 10.1007/978-3-319-77736-8_1
85. Woodgett JR. Molecular Cloning and Expression of Glycogen Synthase Kinase-3/Factor A. EMBO J (1990) 9(8):2431–8. doi: 10.1002/j.1460-2075.1990.tb07419.x
86. Taylor A, Harker JA, Chanthong K, Stevenson PG, Zuniga EI, Rudd CE. Glycogen Synthase Kinase 3 Inactivation Drives T-Bet-Mediated Downregulation of Co-Receptor PD-1 to Enhance CD8(+) Cytolytic T Cell Responses. Immun (2016) 44(2):274–86. doi: 10.1016/j.immuni.2016.01.018
87. Taylor A, Rudd CE. Glycogen Synthase Kinase 3 Inactivation Compensates for the Lack of CD28 in the Priming of CD8+ Cytotoxic T-Cells: Implications for Anti-PD-1 Immunotherapy. Front Immunol (2017) 8:1653. doi: 10.3389/fimmu.2017.01653
88. Krueger J, Rudd CE, Taylor A. Glycogen Synthase 3 (GSK-3) Regulation of PD-1 Expression and and Its Therapeutic Implications. Semin Immunol (2019) 42:101295. doi: 10.1016/j.smim.2019.101295
89. Li B, Qiu B, Lee DS, Walton ZE, Ochocki JD, Mathew LK, et al. Fructose-1,6-Bisphosphatase Opposes Renal Carcinoma Progression. Nat (2014) 513(7517):251–5. doi: 10.1038/nature13557
90. Hirata H, Sugimachi K, Komatsu H, Ueda M, Masuda T, Uchi R, et al. Decreased Expression of Fructose-1,6-Bisphosphatase Associates With Glucose Metabolism and Tumor Progression in Hepatocellular Carcinoma. Cancer Res (2016) 76(11):3265–76. doi: 10.1158/0008-5472.CAN-15-2601
91. Wang B, Zhou Y, Zhang J, Jin X, Wu H, Huang H. Fructose-1,6-Bisphosphatase Loss Modulates STAT3-Dependent Expression of PD-L1 and Cancer Immunity. Theranostics (2020) 10(3):1033–45. doi: 10.7150/thno.38137
92. Le Bourgeois T, Strauss L, Aksoylar HI, Daneshmandi S, Seth P, Patsoukis N, et al. Targeting T Cell Metabolism for Improvement of Cancer Immunotherapy. Front Oncol (2018) 8:237. doi: 10.3389/fonc.2018.00237
93. Son J, Lyssiotis CA, Ying H, Wang X, Hua S, Ligorio M, et al. Glutamine Supports Pancreatic Cancer Growth Through a KRAS-Regulated Metabolic Pathway. Nat (2013) 496(7443):101–5. doi: 10.1038/nature12040
94. Pérez-Escuredo J, Dadhich RK, Dhup S, Cacace A, Van Hée VF, De Saedeleer CJ, et al. Lactate Promotes Glutamine Uptake and Metabolism in Oxidative Cancer Cells. Cell Cycle (2016) 15(1):72–83. doi: 10.1080/15384101.2015.1120930
95. Hensley CT, Wasti AT, DeBerardinis RJ. Glutamine and Cancer: Cell Biology, Physiology, and Clinical Opportunities. J Clin Invest (2013) 123(9):3678–84. doi: 10.1172/JCI69600
96. Matés JM, Campos-Sandoval JA, Márquez J. Glutaminase Isoenzymes in the Metabolic Therapy of Cancer. Biochim Biophys Acta Rev Cancer (2018) 1870(2):158–64. doi: 10.1016/j.bbcan.2018.07.007
97. Wise DR, Thompson CB. Glutamine Addiction: A New Therapeutic Target in Cancer. Trends Biochem Sci (2010) 35(8):427–33. doi: 10.1016/j.tibs.2010.05.003
98. Matés JM, Campos-Sandoval JA, Santos-Jiménez JL, Márquez J. Dysregulation of Glutaminase and Glutamine Synthetase in Cancer. Cancer Lett (2019) 467:29–39. doi: 10.1016/j.canlet.2019.09.011
99. Liu PS, Wang H, Li X, Chao T, Teav T, Christen S, et al. α-Ketoglutarate Orchestrates Macrophage Activation Through Metabolic and Epigenetic Reprogramming. Nat Immunol (2017) 18(9):985–94. doi: 10.1038/ni.3796
100. Altman BJ, Stine ZE, Dang CV. From Krebs to Clinic: Glutamine Metabolism to Cancer Therapy. Nat Rev Cancer (2016) 16(10):619–34. doi: 10.1038/nrc.2016.71
101. Fu Q, Xu L, Wang Y, Jiang Q, Liu Z, Zhang J, et al. Tumor-Associated Macrophage-Derived Interleukin-23 Interlinks Kidney Cancer Glutamine Addiction With Immune Evasion. Eur Urol (2019) 75(5):752–63. doi: 10.1016/j.eururo.2018.09.030
102. Klysz D, Tai X, Robert PA, Craveiro M, Cretenet G, Oburoglu L, et al. Glutamine-Dependent α-Ketoglutarate Production Regulates the Balance Between T Helper 1 Cell and Regulatory T Cell Generation. Sci Signal (2015) 8(396):ra97. doi: 10.1126/scisignal.aab2610
103. Nabe S, Yamada T, Suzuki J, Toriyama K, Yasuoka T, Kuwahara M, et al. Reinforce the Antitumor Activity of CD8+ T Cells via Glutamine Restriction. Cancer Sci (2018) 109(12):3737–50. doi: 10.1111/cas.13827
104. Ma G, Liang Y, Chen Y, Wang L, Li D, Liang Z, et al. : Glutamine Deprivation Induces PD-L1 Expression via Activation of EGFR/ERK/c-Jun Signaling in Renal Cancer. Mol Cancer Res (2020) 18(2):324–39. doi: 10.1158/1541-7786.MCR-19-0517
105. Li T, Le A. Glutamine Metabolism in Cancer. Adv Exp Med Biol (2018) 1063:13–32. doi: 10.1007/978-3-319-77736-8_2
106. Kim SH, Roszik J, Grimm EA, Ekmekcioglu S. Impact of L-Arginine Metabolism on Immune Response and Anticancer Immunotherapy. Front Oncol (2018) 8:67. doi: 10.3389/fonc.2018.00067
107. Norian LA, Rodriguez PC, O’Mara LA, Zabaleta J, Ochoa AC, Cella M, et al. Tumor-Infiltrating Regulatory Dendritic Cells Inhibit CD8+ T Cell Function via L-Arginine Metabolism. Cancer Res (2009) 69(7):3086–94. doi: 10.1158/0008-5472.CAN-08-2826
108. Phillips MM, Sheaff MT, Szlosarek PW. Targeting Arginine-Dependent Cancers With Arginine-Degrading Enzymes: Opportunities and Challenges. Cancer Res Treat (2013) 45(4):251–62. doi: 10.4143/crt.2013.45.4.251
109. Burrows N, Cane G, Robson M, Gaude E, Howat WJ, Szlosarek PW, et al. Hypoxia-Induced Nitric Oxide Production and Tumour Perfusion Is Inhibited by Pegylated Arginine Deiminase (ADI-Peg20). Sci Rep (2016) 6:22950. doi: 10.1038/srep22950
110. Brin E, Wu K, Lu HT, He Y, Dai Z, He W. PEGylated Arginine Deiminase can Modulate Tumor Immune Microenvironment by Affecting Immune Checkpoint Expression, Decreasing Regulatory T Cell Accumulation and Inducing Tumor T Cell Infiltration. Oncotarget (2017) 8(35):58948–63. doi: 10.18632/oncotarget.19564
111. Chang KY, Chiang NJ, Wu SY, Yen CJ, Chen SH, Yeh YM, et al. Phase 1b Study of Pegylated Arginine Deiminase (ADI-PEG 20) Plus Pembrolizumab in Advanced Solid Cancers. Oncoimmunol (2021) 10(1):1943253. doi: 10.1080/2162402X.2021.1943253
112. Zheng NN, Zhou M, Sun F, Huai MX, Zhang Y, Qu CY, et al. Combining Protein Arginine Methyltransferase Inhibitor and Anti-Programmed Death-Ligand-1 Inhibits Pancreatic Cancer Progression. World J Gastroenterol (2020) 26(26):3737–49. doi: 10.3748/wjg.v26.i26.3737
113. Holmgaard RB, Zamarin D, Munn DH, Wolchok JD, Allison JP. Indoleamine 2,3-Dioxygenase Is a Critical Resistance Mechanism in Antitumor T Cell Immunotherapy Targeting CTLA-4. J Exp Med (2013) 210(7):1389–402. doi: 10.1084/jem.20130066
114. Bilir C, Sarisozen C. Indoleamine 2,3-Dioxygenase (IDO): Only an Enzyme or a Checkpoint Controller? J Oncol Sci (2017) 3(2):52–6. doi: 10.1016/j.jons.2017.04.001
115. Mellor AL, Munn DH. IDO Expression by Dendritic Cells: Tolerance and Tryptophan Catabolism. Nat Rev Immunol (2004) 4(10):762–74. doi: 10.1038/nri1457
116. Hornyák L, Dobos N, Koncz G, Karányi Z, Páll D, Szabó Z, et al. The Role of Indoleamine-2,3-Dioxygenase in Cancer Development, Diagnostics, and Therapy. Front Immunol (2018) 9:151. doi: 10.3389/fimmu.2018.00151
117. Sharma MD, Shinde R, McGaha TL, Huang L, Holmgaard RB, Wolchok JD, et al. The PTEN Pathway in Tregs Is a Critical Driver of the Suppressive Tumor Microenvironment. Sci Adv (2015) 1(10):e1500845. doi: 10.1126/sciadv.1500845
118. Toulmonde M, Penel N, Adam J, Chevreau C, Blay JY, Le Cesne A, et al. Use of PD-1 Targeting, Macrophage Infiltration, and IDO Pathway Activation in Sarcomas: A Phase 2 Clinical Trial. JAMA Oncol (2018) 4(1):93–7. doi: 10.1001/jamaoncol.2017.1617
119. Moon PK, Tran S, Minhas PS. Revisiting IDO and Its Value as a Predictive Marker for Anti-PD-1 Resistance. J Transl Med (2019) 17(1):31. doi: 10.1186/s12967-019-1784-8
120. Platten M, Nollen EAA, Röhrig UF, Fallarino F, Opitz CA. Tryptophan Metabolism as a Common Therapeutic Target in Cancer, Neurodegeneration and Beyond. Nat Rev Drug Discov (2019) 18(5):379–401. doi: 10.1038/s41573-019-0016-5
121. Davar D, Bahary N. Modulating Tumor Immunology by Inhibiting Indoleamine 2,3-Dioxygenase (IDO): Recent Developments and First Clinical Experiences. Target Oncol (2018) 13(2):125–40. doi: 10.1007/s11523-017-0547-9
122. Kaiser P. Methionine Dependence of Cancer. Biomolecules (2020) 10(4):568. doi: 10.3390/biom10040568
123. Wanders D, Hobson K, Ji X. Methionine Restriction and Cancer Biology. Nutrients (2020) 12(3):684. doi: 10.3390/nu12030684
124. Mehdi A, Attias M, Mahmood N, Arakelian A, Mihalcioiu C, Piccirillo CA, et al. Enhanced Anticancer Effect of a Combination of S-Adenosylmethionine (SAM) and Immune Checkpoint Inhibitor (ICPi) in a Syngeneic Mouse Model of Advanced Melanoma. Front Oncol (2020) 10:1361. doi: 10.3389/fonc.2020.01361
125. Bian Y, Li W, Kremer DM, Sajjakulnukit P, Li S, Crespo J, et al. Cancer SLC43A2 Alters T Cell Methionine Metabolism and Histone Methylation. Nat (2020) 585(7824):277–82. doi: 10.1038/s41586-020-2682-1
126. Srivastava MK, Sinha P, Clements VK, Rodriguez P, Ostrand-Rosenberg S. Myeloid-Derived Suppressor Cells Inhibit T-Cell Activation by Depleting Cystine and Cysteine. Cancer Res (2010) 70(1):68–77. doi: 10.1158/0008-5472.CAN-09-2587
127. Liu N, Zhang J, Yin M, Liu H, Zhang X, Li J, et al. Inhibition of xCT Suppresses the Efficacy of Anti-PD-1/L1 Melanoma Treatment Through Exosomal PD-L1-Induced Macrophage M2 Polarization. Mol Ther (2021) 29(7):2321–34. doi: 10.1016/j.ymthe.2021.03.013
128. Ma EH, Bantug G, Griss T, Condotta S, Johnson RM, Samborska B, et al. Serine Is an Essential Metabolite for Effector T Cell Expansion. Cell Metab (2017) 25(2):345–57. doi: 10.1016/j.cmet.2016.12.011
129. Wu Q, Chen X, Li J, Sun S. Serine and Metabolism Regulation: A Novel Mechanism in Antitumor Immunity and Senescence. Aging Dis (2020) 11(6):1640–53. doi: 10.14336/AD.2020.0314
130. Wang LW, Shen H, Nobre L, Ersing I, Paulo JA, Trudeau S, et al. Epstein-Barr-Virus-Induced One-Carbon Metabolism Drives B Cell Transformation. Cell Metab (2019) 30(3):539–555.e11. doi: 10.1016/j.cmet.2019.06.003
131. Guri Y, Nordmann TM, Roszik J. mTOR at the Transmitting and Receiving Ends in Tumor Immunity. Front Immunol (2018) 9:578. doi: 10.3389/fimmu.2018.00578
132. Menendez JA, Lupu R. Fatty Acid Synthase and the Lipogenic Phenotype in Cancer Pathogenesis. Nat Rev Cancer (2007) 7(10):763–77. doi: 10.1016/j.ymthe.2021.03.013
133. Cheng C, Geng F, Cheng X, Guo D. Lipid Metabolism Reprogramming and Its Potential Targets in Cancer. Cancer Commun (Lond) (2018) 38(1):27. doi: 10.1186/s40880-018-0301-4
134. Bensinger SJ, Bradley MN, Joseph SB, Zelcer N, Janssen EM, Hausner MA, et al. LXR Signaling Couples Sterol Metabolism to Proliferation in the Acquired Immune Response. Cell (2008) 134(1):97–111. doi: 10.1016/j.cell.2008.04.052
135. Xu S, Chaudhary O, Rodríguez-Morales P, Sun X, Chen D, Zappasodi R, et al. Uptake of Oxidized Lipids by the Scavenger Receptor CD36 Promotes Lipid Peroxidation and Dysfunction in CD8+ T Cells in Tumors. Immun (2021) 54(7):1561–1577.e7. doi: 10.1016/j.immuni.2021.05.003
136. Adeshakin AO, Liu W, Adeshakin FO, Afolabi LO, Zhang M, Zhang G, et al. Regulation of ROS in Myeloid-Derived Suppressor Cells Through Targeting Fatty Acid Transport Protein 2 Enhanced Anti-PD-L1 Tumor Immunotherapy. Cell Immunol (2021) 362:104286. doi: 10.1016/j.cellimm.2021.104286
137. Savage PA, Klawon DEJ, Miller CH. Regulatory T Cell Development. Annu Rev Immunol (2020) 38:421–53. doi: 10.1146/annurev-immunol-100219-020937
138. Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell (2017) 168(4):707–23. doi: 10.1016/j.cell.2017.01.017
139. Lim SA, Wei J, Nguyen TM, Shi H, Su W, Palacios G, et al. Lipid Signalling Enforces Functional Specialization of Treg Cells in Tumours. Nat (2021) 591(7849):306–11. doi: 10.1038/s41586-021-03235-6
140. Ma C, Kesarwala AH, Eggert T, Medina-Echeverz J, Kleiner DE, Jin P, et al. NAFLD Causes Selective CD4(+) T Lymphocyte Loss and Promotes Hepatocarcinogenesis. Nat (2016) 531(7593):253–7. doi: 10.1038/nature16969
141. Ma X, Xiao L, Liu L, Ye L, Su P, Bi E, et al. CD36-Mediated Ferroptosis Dampens Intratumoral CD8+ T Cell Effector Function and Impairs Their Antitumor Ability. Cell Metab (2021) 33(5):1001–1012.e5. doi: 10.1016/j.cmet.2021.02.015
142. O’Sullivan D, van der Windt GJ, Huang SC, Curtis JD, Chang CH, Buck MD, et al. Memory CD8(+) T Cells Use Cell-Intrinsic Lipolysis to Support the Metabolic Programming Necessary for Development. Immun (2014) 41(1):75–88. doi: 10.1016/j.immuni.2014.06.005
143. Mueller SN, Mackay LK. Tissue-Resident Memory T Cells: Local Specialists in Immune Defence. Nat Rev Immunol (2016) 16(2):79–89. doi: 10.1038/nri.2015.3
144. Schenkel JM, Masopust D. Tissue-Resident Memory T Cells. Immun (2014) 41(6):886–97. doi: 10.1016/j.immuni.2014.12.007
145. Watanabe R, Gehad A, Yang C, Scott LL, Teague JE, Schlapbach C, et al. Human Skin Is Protected by Four Functionally and Phenotypically Discrete Populations of Resident and Recirculating Memory T Cells. Sci Transl Med (2015) 7(279):279ra39. doi: 10.1126/scitranslmed.3010302
146. Gebhardt T, Wakim LM, Eidsmo L, Reading PC, Heath WR, Carbone FR. Memory T Cells in Nonlymphoid Tissue That Provide Enhanced Local Immunity During Infection With Herpes Simplex Virus. Nat Immunol (2009) 10(5):524–30. doi: 10.1038/ni.1718
147. Lin R, Zhang H, Yuan Y, He Q, Zhou J, Li S, et al. Fatty Acid Oxidation Controls CD8+ Tissue-Resident Memory T-Cell Survival in Gastric Adenocarcinoma. Cancer Immunol Res (2020) 8(4):479–92. doi: 10.1158/2326-6066.CIR-19-0702
148. Wu XN, Su D, Mei YD, Xu MQ, Zhang H, Wang ZY, et al. Identified Lung Adenocarcinoma Metabolic Phenotypes and Their Association With Tumor Immune Microenvironment. Cancer Immunol Immunother (2021) 70(10):2835–50. doi: 10.1007/s00262-021-02896-6
149. Harel M, Ortenberg R, Varanasi SK, Mangalhara KC, Mardamshina M, Markovits E, et al. Proteomics of Melanoma Response to Immunotherapy Reveals Mitochondrial Dependence. Cell (2019) 179(1):236–250.e18. doi: 10.1016/j.cell.2019.08.012
150. Westerterp M, Gautier EL, Ganda A, Molusky MM, Wang W, Fotakis P, et al. Cholesterol Accumulation in Dendritic Cells Links the Inflammasome to Acquired Immunity. Cell Metab (2017) 25(6):1294–304.e6. doi: 10.1016/j.cmet.2017.04.005
151. Spann NJ, Garmire LX, McDonald JG, Myers DS, Milne SB, Shibata N, et al. Regulated Accumulation of Desmosterol Integrates Macrophage Lipid Metabolism and Inflammatory Responses. Cell (2012) 151(1):138–52. doi: 10.1016/j.cell.2012.06.054
152. Anderson HA, Roche PA. MHC Class II Association With Lipid Rafts on the Antigen Presenting Cell Surface. Biochim Biophys Acta (2015) 1853(4):775–80. doi: 10.1016/j.bbamcr.2014.09.019
153. Ma X, Bi E, Lu Y, Su P, Huang C, Liu L, et al. Cholesterol Induces CD8+ T Cell Exhaustion in the Tumor Microenvironment. Cell Metab (2019) 30(1):143–156.e5. doi: 10.1016/j.cmet.2019.04.002
154. Okoye I, Namdar A, Xu L, Crux N, Elahi S. : Atorvastatin Downregulates Co-Inhibitory Receptor Expression by Targeting Ras-Activated mTOR Signalling. Oncotarget (2017) 8(58):98215–32. doi: 10.18632/oncotarget.21003
155. Yang W, Bai Y, Xiong Y, Zhang J, Chen S, Zheng X, et al. Potentiating the Antitumour Response of CD8(+) T Cells by Modulating Cholesterol Metabolism. Nat (2016) 531(7596):651–5. doi: 10.1038/nature17412
156. Chang TY, Chang CC, Ohgami N, Yamauchi Y. Cholesterol Sensing, Trafficking, and Esterification. Annu Rev Cell Dev Biol (2006) 22:129–57. doi: 10.1146/annurev.cellbio.22.010305.104656
157. Ogretmen B. Sphingolipid Metabolism in Cancer Signalling and Therapy. Nat Rev Cancer (2018) 18(1):33–50. doi: 10.1038/nrc.2017.96
158. Clarke CJ. Neutral Sphingomyelinases in Cancer: Friend or Foe? Adv Cancer Res (2018) 140:97–119. doi: 10.1016/bs.acr.2018.04.010
159. Montfort A, Bertrand F, Rochotte J, Gilhodes J, Filleron T, Milhès J, et al. Neutral Sphingomyelinase 2 Heightens Anti-Melanoma Immune Responses and Anti-PD-1 Therapy Efficacy. Cancer Immunol Res (2021) 9(5):568–82. doi: 10.1158/2326-6066.CIR-20-0342
160. Imbert C, Montfort A, Fraisse M, Marcheteau E, Gilhodes J, Martin E, et al. Resistance of Melanoma to Immune Checkpoint Inhibitors Is Overcome by Targeting the Sphingosine Kinase-1. Nat Commun (2020) 11(1):437. doi: 10.1038/s41467-019-14218-7
161. Chen G, Luo Y, Warncke K, Sun Y, Yu DS, Fu H, et al. Acetylation Regulates Ribonucleotide Reductase Activity and Cancer Cell Growth. Nat Commun (2019) 10(1):3213. doi: 10.1038/s41467-019-11214-9
162. Kohnken R, Kodigepalli KM, Wu L. Regulation of Deoxynucleotide Metabolism in Cancer: Novel Mechanisms and Therapeutic Implications. Mol Cancer (2015) 14:176. doi: 10.1186/s12943-015-0446-6
163. Lv Y, Wang X, Li X, Xu G, Bai Y, Wu J, et al. Nucleotide De Novo Synthesis Increases Breast Cancer Stemness and Metastasis via cGMP-PKG-MAPK Signaling Pathway. PloS Biol (2020) 18(11):e3000872. doi: 10.1371/journal.pbio.3000872
164. Ron-Harel N, Notarangelo G, Ghergurovich JM, Paulo JA, Sage PT, Santos D, et al. Defective Respiration and One-Carbon Metabolism Contribute to Impaired Naïve T Cell Activation in Aged Mice. Proc Natl Acad Sci USA (2018) 115(52):13347–52. doi: 10.1073/pnas.1804149115
165. Aird KM, Zhang G, Li H, Tu Z, Bitler BG, Garipov A, et al. Suppression of Nucleotide Metabolism Underlies the Establishment and Maintenance of Oncogene-Induced Senescence. Cell Rep (2013) 3(4):1252–65. doi: 10.1016/j.celrep.2013.03.004
166. Mazzu YZ, Armenia J, Nandakumar S, Chakraborty G, Yoshikawa Y, Jehane LE, et al. Ribonucleotide Reductase Small Subunit M2 Is a Master Driver of Aggressive Prostate Cancer. Mol Oncol (2020) 14(8):1881–97. doi: 10.1002/1878-0261.12706
167. Knighton LE, Delgado LE, Truman AW. Novel Insights Into Molecular Chaperone Regulation of Ribonucleotide Reductase. Curr Genet (2019) 65(2):477–82. doi: 10.1007/s00294-018-0916-7
168. Sykes DB, Kfoury YS, Mercier FE, Wawer MJ, Law JM, Haynes MK, et al. Inhibition of Dihydroorotate Dehydrogenase Overcomes Differentiation Blockade in Acute Myeloid Leukemia. Cell (2016) 167(1):171–186.e15. doi: 10.1016/j.cell.2016.08.057
169. Ragu S, Matos-Rodrigues G, Lopez BS. Replication Stress, DNA Damage, Inflammatory Cytokines and Innate Immune Response. Genes (Basel) (2020) 11(4):409. doi: 10.3390/genes11040409
170. Ricciuti B, Recondo G, Spurr LF, Li YY, Lamberti G, Venkatraman D, et al. Impact of DNA Damage Response and Repair (DDR) Gene Mutations on Efficacy of PD-(L)1 Immune Checkpoint Inhibition in Non-Small Cell Lung Cancer. Clin Cancer Res (2020) 26(15):4135–42. doi: 10.1158/1078-0432.CCR-19-3529
171. Teo MY, Seier K, Ostrovnaya I, Regazzi AM, Kania BE, Moran MM, et al. Alterations in DNA Damage Response and Repair Genes as Potential Marker of Clinical Benefit From PD-1/PD-L1 Blockade in Advanced Urothelial Cancers. J Clin Oncol (2018) 36(17):1685–94. doi: 10.1200/JCO.2017.75.7740
172. Tu X, Qin B, Zhang Y, Zhang C, Kahila M, Nowsheen S, et al. PD-L1 (B7-H1) Competes With the RNA Exosome to Regulate the DNA Damage Response and Can Be Targeted to Sensitize to Radiation or Chemotherapy. Mol Cell (2019) 74(6):1215–1226.e4. doi: 10.1016/j.molcel.2019.04.005
173. Sen T, Rodriguez BL, Chen L, Corte CMD, Morikawa N, Fujimoto J, et al. Targeting DNA Damage Response Promotes Antitumor Immunity Through STING-Mediated T-Cell Activation in Small Cell Lung Cancer. Cancer Discov (2019) 9(5):646–61. doi: 10.1158/2159-8290.CD-18-1020
174. Färkkilä A, Gulhan DC, Casado J, Jacobson CA, Nguyen H, Kochupurakkal B, et al. Immunogenomic Profiling Determines Responses to Combined PARP and PD-1 Inhibition in Ovarian Cancer. Nat Commun (2020) 11(1):1459. doi: 10.1038/s41467-020-15315-8
175. Friedlander M, Meniawy T, Markman B, Mileshkin L, Harnett P, Millward M, et al. Pamiparib in Combination With Tislelizumab in Patients With Advanced Solid Tumours: Results From the Dose-Escalation Stage of a Multicentre, Open-Label, Phase 1a/B Trial. Lancet Oncol (2019) 20(9):1306–15. doi: 10.1016/S1470-2045(19)30396-1
176. Konstantinopoulos PA, Waggoner S, Vidal GA, Mita M, Moroney JW, Holloway R, et al. Single-Arm Phases 1 and 2 Trial of Niraparib in Combination With Pembrolizumab in Patients With Recurrent Platinum-Resistant Ovarian Carcinoma. JAMA Oncol (2019) 5(8):1141–9. doi: 10.1001/jamaoncol.2019.1048
177. Chen X, Pan X, Zhang W, Guo H, Cheng S, He Q, et al. Epigenetic Strategies Synergize With PD-L1/PD-1 Targeted Cancer Immunotherapies to Enhance Antitumor Responses. Acta Pharm Sin B (2020) 10(5):723–33. doi: 10.1016/j.apsb.2019.09.006
178. Moreno-Sánchez R, Rodríguez-Enríquez S, Marín-Hernández A, Saavedra E. Energy Metabolism in Tumor Cells. FEBS J (2007) 274(6):1393–418. doi: 10.1111/j.1742-4658.2007.05686.x
179. Jia L, Gao Y, Zhou T, Zhao XL, Hu HY, Chen DW, et al. Enhanced Response to PD-L1 Silencing by Modulation of TME via Balancing Glucose Metabolism and Robust Co-Delivery of siRNA/Resveratrol With Dual-Responsive Polyplexes. Biomaterials (2021) 271:120711. doi: 10.1016/j.biomaterials.2021.120711
180. Gui DY, Sullivan LB, Luengo A, Hosios AM, Bush LN, Gitego N, et al. Environment Dictates Dependence on Mitochondrial Complex I for NAD+ and Aspartate Production and Determines Cancer Cell Sensitivity to Metformin. Cell Metab (2016) 24(5):716–27. doi: 10.1016/j.cmet.2016.09.006
181. Nishida M, Yamashita N, Ogawa T, Koseki K, Warabi E, Ohue T, et al. Mitochondrial Reactive Oxygen Species Trigger Metformin-Dependent Antitumor Immunity via Activation of Nrf2/mTORC1/p62 Axis in Tumor-Infiltrating CD8T Lymphocytes. J Immunother Cancer (2021) 9(9):e002954. doi: 10.1136/jitc-2021-002954
182. Afzal MZ, Mercado RR, Shirai K. Efficacy of Metformin in Combination With Immune Checkpoint Inhibitors (Anti-PD-1/Anti-CTLA-4) in Metastatic Malignant Melanoma. J Immunother Cancer (2018) 6(1):64. doi: 10.1186/s40425-018-0375-1
183. Brown JR, Chan DK, Shank JJ, Griffith KA, Fan H, Szulawski R, et al. Phase II Clinical Trial of Metformin as a Cancer Stem Cell-Targeting Agent in Ovarian Cancer. JCI Insight (2020) 5(11):e133247. doi: 10.1172/jci.insight.133247
184. Kubo T, Ninomiya T, Hotta K, Kozuki T, Toyooka S, Okada H, et al. Study Protocol: Phase-Ib Trial of Nivolumab Combined With Metformin for Refractory/Recurrent Solid Tumors. Clin Lung Cancer (2018) 19(6):e861–4. doi: 10.1016/j.cllc.2018.07.010
185. Tsuji A, Akao T, Masuya T, Murai M, Miyoshi H. IACS-010759, a Potent Inhibitor of Glycolysis-Deficient Hypoxic Tumor Cells, Inhibits Mitochondrial Respiratory Complex I Through a Unique Mechanism. J Biol Chem (2020) 295(21):7481–91. doi: 10.1074/jbc.RA120.013366
186. Chen D, Barsoumian HB, Fischer G, Yang L, Verma V, Younes AI, et al. Combination Treatment With Radiotherapy and a Novel Oxidative Phosphorylation Inhibitor Overcomes PD-1 Resistance and Enhances Antitumor Immunity. J Immunother Cancer (2020) 8(1):e000289. doi: 10.1136/jitc-2019-000289
187. He N, Fan W, Henriquez B, Yu RT, Atkins AR, Liddle C, et al. Metabolic Control of Regulatory T Cell (Treg) Survival and Function by Lkb1. Proc Natl Acad Sci U S A (2017) 114(47):12542–7. doi: 10.1073/pnas.1715363114
188. Jones N, Vincent EE, Cronin JG, Panetti S, Chambers M, Holm SR, et al. Akt and STAT5 Mediate Naïve Human CD4+ T-Cell Early Metabolic Response to TCR Stimulation. Nat Commun (2019) 10(1):2042. doi: 10.1038/s41467-019-10023-4
189. Charni S, de Bettignies G, Rathore MG, Aguiló JI, van den Elsen PJ, Haouzi D, et al. Oxidative Phosphorylation Induces De Novo Expression of the MHC Class I in Tumor Cells Through the ERK5 Pathway. J Immunol (2010) 185(6):3498–503. doi: 10.4049/jimmunol.1001250
190. Scharping NE, Menk AV, Moreci RS, Whetstone RD, Dadey RE, Watkins SC, et al. The Tumor Microenvironment Represses T Cell Mitochondrial Biogenesis to Drive Intratumoral T Cell Metabolic Insufficiency and Dysfunction. Immun (2016) 45(2):374–88. doi: 10.1016/j.immuni.2016.07.009
191. Teijeira A, Labiano S, Garasa S, Etxeberria I, Santamaría E, Rouzaut A, et al. Mitochondrial Morphological and Functional Reprogramming Following CD137 (4-1bb) Costimulation. Cancer Immunol Res (2018) 6(7):798–811. doi: 10.1158/2326-6066.CIR-17-0767
192. Eltzschig HK, Sitkovsky MV, Robson SC. Purinergic Signaling During Inflammation. N Engl J Med (2012) 367(24):2322–33. doi: 10.1056/NEJMra1205750
193. Allard B, Longhi MS, Robson SC, Stagg J. The Ectonucleotidases CD39 and CD73: Novel Checkpoint Inhibitor Targets. Immunol Rev (2017) 276(1):121–44. doi: 10.1111/imr.12528
194. Blay J, White TD, Hoskin DW. The Extracellular Fluid of Solid Carcinomas Contains Immunosuppressive Concentrations of Adenosine. Cancer Res (1997) 57(13):2602–5.
195. Arab S, Hadjati J. Adenosine Blockage in Tumor Microenvironment and Improvement of Cancer Immunotherapy. Immune Netw (2019) 19(4):e23. doi: 10.4110/in.2019.19.e23
196. Li J, Wang L, Chen X, Li L, Li Y, Ping Y, et al. CD39/CD73 Upregulation on Myeloid-Derived Suppressor Cells via TGF-β-mTOR-HIF-1 Signaling in Patients With Non-Small Cell Lung Cancer. Oncoimmunol (2017) 6(6):e1320011. doi: 10.1080/2162402X.2017.1320011
197. Serra S, Vaisitti T, Audrito V, Bologna C, Buonincontri R, Chen SS, et al. Adenosine Signaling Mediates Hypoxic Responses in the Chronic Lymphocytic Leukemia Microenvironment. Blood Adv (2016) 1(1):47–61. doi: 10.1182/bloodadvances.2016000984
198. Giatromanolaki A, Kouroupi M, Pouliliou S, Mitrakas A, Hasan F, Pappa A, et al. Ectonucleotidase CD73 and CD39 Expression in Non-Small Cell Lung Cancer Relates to Hypoxia and Immunosuppressive Pathways. Life Sci (2020) 259:118389. doi: 10.1016/j.lfs.2020.118389
199. Beavis PA, Milenkovski N, Henderson MA, John LB, Allard B, Loi S, et al. Adenosine Receptor 2a Blockade Increases the Efficacy of Anti-PD-1 Through Enhanced Antitumor T-Cell Responses. Cancer Immunol Res (2015) 3(5):506–17. doi: 10.1158/2326-6066.CIR-14-0211
200. Patel SP, Kurzrock R. PD-L1 Expression as a Predictive Biomarker in Cancer Immunotherapy. Mol Cancer Ther (2015) 14(4):847–56. doi: 10.1158/1535-7163.MCT-14-0983
201. Hellmann MD, Callahan MK, Awad MM, Calvo E, Ascierto PA, Atmaca A, et al. Tumor Mutational Burden and Efficacy of Nivolumab Monotherapy and in Combination With Ipilimumab in Small-Cell Lung Cancer. Cancer Cell (2018) 33(5):853–861.e4. doi: 10.1016/j.ccell.2018.04.001
202. Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Cancer Immunology. Mutational Landscape Determines Sensitivity to PD-1 Blockade in Non-Small Cell Lung Cancer. Sci (2015) 348(6230):124–8. doi: 10.1126/science.aaa1348
203. Jiang Z, Liu Z, Li M, Chen C, Wang X. Increased Glycolysis Correlates With Elevated Immune Activity in Tumor Immune Microenvironment. EBioMed (2019) 42:431–42. doi: 10.1016/j.ebiom.2019.03.068
204. Lacroix R, Rozeman EA, Kreutz M, Renner K, Blank CU. Targeting Tumor-Associated Acidity in Cancer Immunotherapy. Cancer Immunol Immunother (2018) 67(9):1331–48. doi: 10.1007/s00262-018-2195-z
205. Xue G, Cui ZJ, Zhou XH, Zhu YX, Chen Y, Liang FJ, et al. DNA Methylation Biomarkers Predict Objective Responses to PD-1/PD-L1 Inhibition Blockade. Front Genet (2019) 10:724. doi: 10.3389/fgene.2019.00724
206. Forschner A, Battke F, Hadaschik D, Schulze M, Weißgraeber S, Han CT, et al. Tumor Mutation Burden and Circulating Tumor DNA in Combined CTLA-4 and PD-1 Antibody Therapy in Metastatic Melanoma- Results of a Prospective Biomarker Study. J Immunother Cancer (2019) 7(1):180. doi: 10.1186/s40425-019-0659-0
207. DePeaux K, Delgoffe GM. Metabolic Barriers to Cancer Immunotherapy. Nat Rev Immunol (2021). doi: 10.1038/s41577-021-00541-y
208. Vander Heiden MG, DeBerardinis RJ. Understanding the Intersections Between Metabolism and Cancer Biology. Cell (2017) 168(4):657–69. doi: 10.1016/j.cell.2016.12.039
209. Koo T, Kim IA. Radiotherapy and Immune Checkpoint Blockades: A Snapshot in 2016. Radiat Oncol J (2016) 34(4):250–9. doi: 10.3857/roj.2016.02033
210. Liu L, Mayes PA, Eastman S, Shi H, Yadavilli S, Zhang T, et al. The BRAF and MEK Inhibitors Dabrafenib and Trametinib: Effects on Immune Function and in Combination With Immunomodulatory Antibodies Targeting PD-1, PD-L1, and CTLA-4. Clin Cancer Res (2015) 21(7):1639–51. doi: 10.1158/1078-0432.CCR-14-2339
211. Moore E, Clavijo PE, Davis R, Cash H, Van Waes C, Kim Y, et al. Established T Cell-Inflamed Tumors Rejected After Adaptive Resistance Was Reversed by Combination STING Activation and PD-1 Pathway Blockade. Cancer Immunol Res (2016) 4(12):1061–71. doi: 10.1158/2326-6066.CIR-16-0104
212. Kanikarla-Marie P, Lam M, Sorokin AV, Overman MJ, Kopetz S, Menter DG. Platelet Metabolism and Other Targeted Drugs; Potential Impact on Immunotherapy. Front Oncol (2018) 8:107. doi: 10.3389/fonc.2018.00107
213. Wangpaichitr M, Kandemir H, Li YY, Wu C, Nguyen D, Feun LG, et al. Relationship of Metabolic Alterations and PD-L1 Expression in Cisplatin Resistant Lung Cancer. Cell Dev Biol (2017) 6(2):183. doi: 10.4172/2168-9296.1000183
214. Yang Y, Wang Z, Fang J, Yu Q, Han B, Cang S, et al. Efficacy and Safety of Sintilimab Plus Pemetrexed and Platinum as First-Line Treatment for Locally Advanced or Metastatic Nonsquamous NSCLC: A Randomized, Double-Blind, Phase 3 Study (Oncology Program by InnovENT Anti-PD-1-11). J Thorac Oncol (2020) 15(10):1636–46. doi: 10.1016/j.jtho.2020.07.014
215. Sato H, Okonogi N, Nakano T. Rationale of Combination of Anti-PD-1/PD-L1 Antibody Therapy and Radiotherapy for Cancer Treatment. Int J Clin Oncol (2020) 25(5):801–9. doi: 10.1007/s10147-020-01666-1
216. Gupta A, Probst HC, Vuong V, Landshammer A, Muth S, Yagita H, et al. Radiotherapy Promotes Tumor-Specific Effector CD8+ T Cells via Dendritic Cell Activation. J Immunol (2012) 189(2):558–66. doi: 10.4049/jimmunol.1200563
217. Antonia SJ, Villegas A, Daniel D, Vicente D, Murakami S, Hui R, et al. PACIFIC Investigators. Overall Survival With Durvalumab After Chemoradiotherapy in Stage III NSCLC. N Engl J Med (2018) 379(24):2342–50. doi: 10.1056/NEJMoa1809697
218. Polk A, Svane IM, Andersson M, Nielsen D. Checkpoint Inhibitors in Breast Cancer -Current Status. Cancer Treat Rev (2018) 63:122–34. doi: 10.1016/j.ctrv.2017.12.008
219. Haratani K, Hayashi H, Tanaka T, Kaneda H, Togashi Y, Sakai K, et al. Tumor Immune Microenvironment and Nivolumab Efficacy in EGFR Mutation-Positive Non-Small-Cell Lung Cancer Based on T790M Status After Disease Progression During EGFR-TKI Treatment. Ann Oncol (2017) 28(7):1532–9. doi: 10.1093/annonc/mdx183
220. Guerra L, Bonetti L, Brenner D. Metabolic Modulation of Immunity: A New Concept in Cancer Immunotherapy. Cell Rep (2020) 32(1):107848. doi: 10.1016/j.celrep.2020.107848
Keywords: glycolysis, amino acid metabolism, lipid metabolism, nucleotide metabolism, mitochondrial biogenesis, the tumor microenvironment, immune checkpoints, PD-1
Citation: Xu Y, He L, Fu Q and Hu J (2021) Metabolic Reprogramming in the Tumor Microenvironment With Immunocytes and Immune Checkpoints. Front. Oncol. 11:759015. doi: 10.3389/fonc.2021.759015
Received: 16 August 2021; Accepted: 27 October 2021;
Published: 11 November 2021.
Edited by:
Nelida Ines Noguera, University of Rome Tor Vergata, ItalyReviewed by:
Guangyong Peng, Saint Louis University, United StatesStephen John Ralph, Griffith University, Australia
Copyright © 2021 Xu, He, Fu and Hu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lijie He, MTc3MDI0ODg4OTZAMTYzLmNvbQ==; Yaolin Xu, eHlsOTEzNkAxNjMuY29t
†These authors have contributed equally to this work