 Juri Alessandro Giannotta
Juri Alessandro Giannotta Bruno Fattizzo
Bruno Fattizzo Wilma Barcellini
Wilma Barcellini- 1Hematology Unit, Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico, Milan, Italy
- 2Department of Oncology and Oncohematology, University of Milan, Milan, Italy
Paroxysmal nocturnal hemoglobinuria (PNH) is characterized by intravascular hemolytic anemia and thrombosis and is notoriously associated with aplastic anemia and myelodysplastic syndromes. Rarer associations include myeloproliferative neoplasms (MPNs), which are also burdened by increased thrombotic tendency. The therapeutic management of this rare combination has not been defined so far. Here, we describe a 62-year-old man who developed a highly hemolytic PNH more than 10 years after the diagnosis of MPN. The patient started eculizumab, obtaining good control of intravascular hemolysis but without amelioration of transfusion-dependent anemia. Moreover, we performed a review of the literature regarding the clinical and pathogenetic significance of the association of PNH and MPN. The prevalence of PNH clones in MPN patients is about 10%, mostly in association with JAK2V617F-positive myelofibrosis. Thrombotic events were a common clinical presentation (35% of subjects), sometimes refractory to combined treatment with cytoreductive agents, anticoagulants, and complement inhibitors. The latter showed only partial effectiveness in controlling hemolytic anemia and, due to the paucity of data, should be taken in consideration after a careful risk/benefit evaluation in this peculiar setting.
Introduction
Paroxysmal nocturnal hemoglobinuria (PNH) is a rare acquired disorder caused by the somatic mutations of phosphatidylinositol glycan A (PIGA). The consequent defect of glycosyl phosphatidylinositol (GPI)-anchored proteins on red blood cell (RBC) surface increases the susceptibility of PNH cells to complement-mediated destruction, leading to intravascular hemolytic anemia, which is the main clinical feature of the disease (1, 2). The natural history of PNH was burdened by high morbidity due to chronic anemia and considerably increased mortality, mainly related to fatal thrombotic events (3). With the advent of complement inhibitors, PNH patients significantly ameliorated their quality of life and survival (4). PNH has been described in the context of bone marrow failure (BMF) syndromes, namely, aplastic anemia (AA) and myelodysplastic syndrome (MDS) (4). However, with the development of more sensitive cytofluorimetric techniques (5), PNH clones of various sizes are increasingly being detected in various onco-hematologic and autoimmune disorders (6–9). The coexistence of PNH and myeloproliferative neoplasms (MPNs) has been reported, but its clinical/prognostic significance and therapeutic management are still poorly known. Moreover, these two conditions share an overlapping clinical presentation, represented by thrombotic events at usual and unusual sites (3, 10–13). Here, we provide the description of a patient with MPN who was subsequently diagnosed with PNH and required specific treatment for hemolytic anemia. In addition, we searched for the available evidence in literature about the association of PNH and MPN, collecting data over the last 40 years in MEDLINE via PubMed and the National Library of Medicine. In detail, we reviewed data about the coexistence of clinically overt PNH and MPN, the prevalence of PNH clones in MPN patients, and the prevalence of MPN driver mutations in PNH subjects.
Case Description
A 62-year-old Caucasian male was diagnosed with Janus kinase (JAK)2-negative essential thrombocythemia (ET) in May 2007 due to isolated asymptomatic thrombocytosis (Table 1). His medical history was unremarkable, except for moderate arterial hypertension on regular treatment; no previous thrombotic events were registered. Bone marrow (BM) evaluation showed normocellularity (40%), increased mature megakaryocytes, slight increase of reticulin fibers (MF-1), and normal karyotype. Once-daily acetylsalicylic acid (ASA) and low-dose hydroxyurea (HU) were started with adequate control of platelet count. From March 2016, a trend to increased lactate dehydrogenase (LDH) levels was noticed, and from February 2019, a mild macrocytic anemia [hemoglobin (Hb) 10.2 g/dL, mean corpuscular volume (MCV) 103 fL; normal iron and vitamin status] developed (Figure 1). Direct antiglobulin test (DAT) was negative, with mild elevation of unconjugated bilirubin (UB, 1.3 mg/dL), consumption of haptoglobin, and increased reticulocytes. HU dose was decreased, but anemia worsened (Hb nadir 7 g/dL), LDH rose to 4× upper limit of normal (ULN), and peripheral CD34-positive cells increased. The patient became strongly symptomatic for anemia, requiring about 1–2 RBC units/month. His physical examination was unremarkable. In October 2019, BM reevaluation showed increased cellularity (>95%) with dystrophic megakaryocytes and increased fibrosis (MF-2) and was therefore interpreted as fibrotic evolution of ET. Molecular tests on peripheral blood revealed the presence of a type-2 calreticulin (CALR) mutation at exon 9. Abdomen ultrasonography displayed normal spleen and liver size. HU and ASA were stopped, and an attempt with steroids (oral prednisone 50 mg/day) was made with a transient response and reappearance of transfusion dependency during tapering (2–3 packed RBC units/month). In February 2020, danazol was administered, again without anemia improvement. In November 2020, due to persistent intravascular hemolytic anemia and referred dark-colored urines, a flow cytometry for PNH was made and turned positive with a clone size of 95%/94% on neutrophils/monocytes. Low-molecular weight heparin (LMWH) prophylaxis (enoxaparin 4,000 units/day) was started, and the patient was referred to our center for treatment indications. No major PNH-related symptoms were registered. In December 2020, the patient started eculizumab after recommended vaccinations. In the subsequent months, Hb stabilized at 7.5–8 g/dL, LDH progressively lowered to 1.1×–1.2× ULN, and transfusion need returned to 1–2 units/month, with subjective amelioration of the quality of life and disappearance of hemoglobinuria. LMWH was stopped. In June 2021, due to a minor response to eculizumab, a re-evaluation was performed: DAT result turned positive for complement without evidence of cryoagglutinins (as frequently observed under eculizumab treatment), and UB levels slightly increased, indicative of extravascular hemolysis (EVH) during terminal complement inhibitor treatment. BM biopsy confirmed increased age-adjusted cellularity, granulocytic hyperplasia, and numerous megakaryocytes in loose and dense clusters, including both mature and atypical, dystrophic cells; fibrosis was stable with diffuse increase in reticulin and focal bundles of thick collagen fibers (MF-2); of note, hyperplasia of the erythroid lineage in the absence of dysplastic features was described. Karyotype analysis was normal. Analyzed by an expert hemopathologist, the BM trephine was consistent with MPN unclassifiable (MPN-U). A targeted next-generation sequencing (NGS) myeloid panel confirmed the presence of type-2 CALR mutation [variant allele frequency (VAF), 45%] and showed an additional somatic mutation in ten-eleven translocation 2 (TET2) gene (VAF, 9.7%). The patient is continuing regular fortnightly eculizumab infusions, with subjective benefit, although with persistent transfusion dependence.
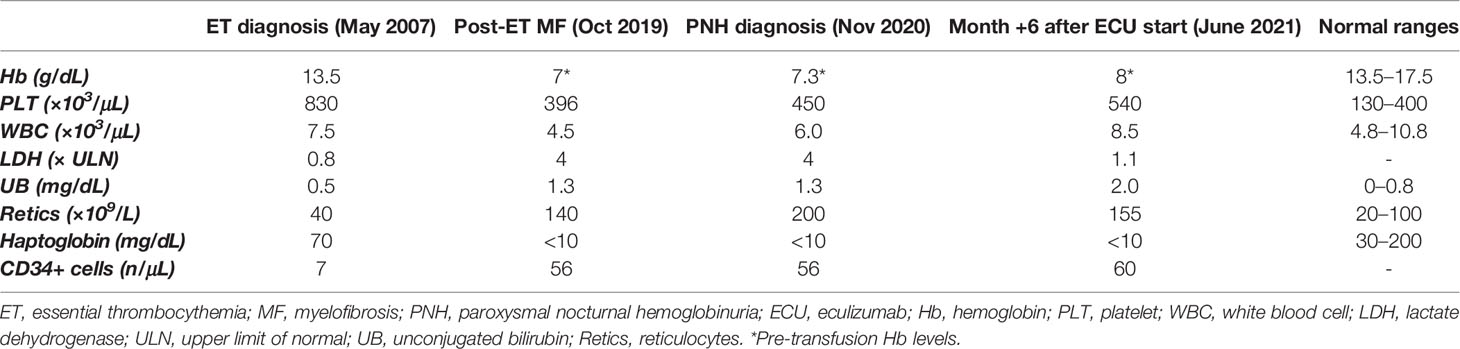
Table 1 Laboratory parameters at different time points of the clinical history of the patient.
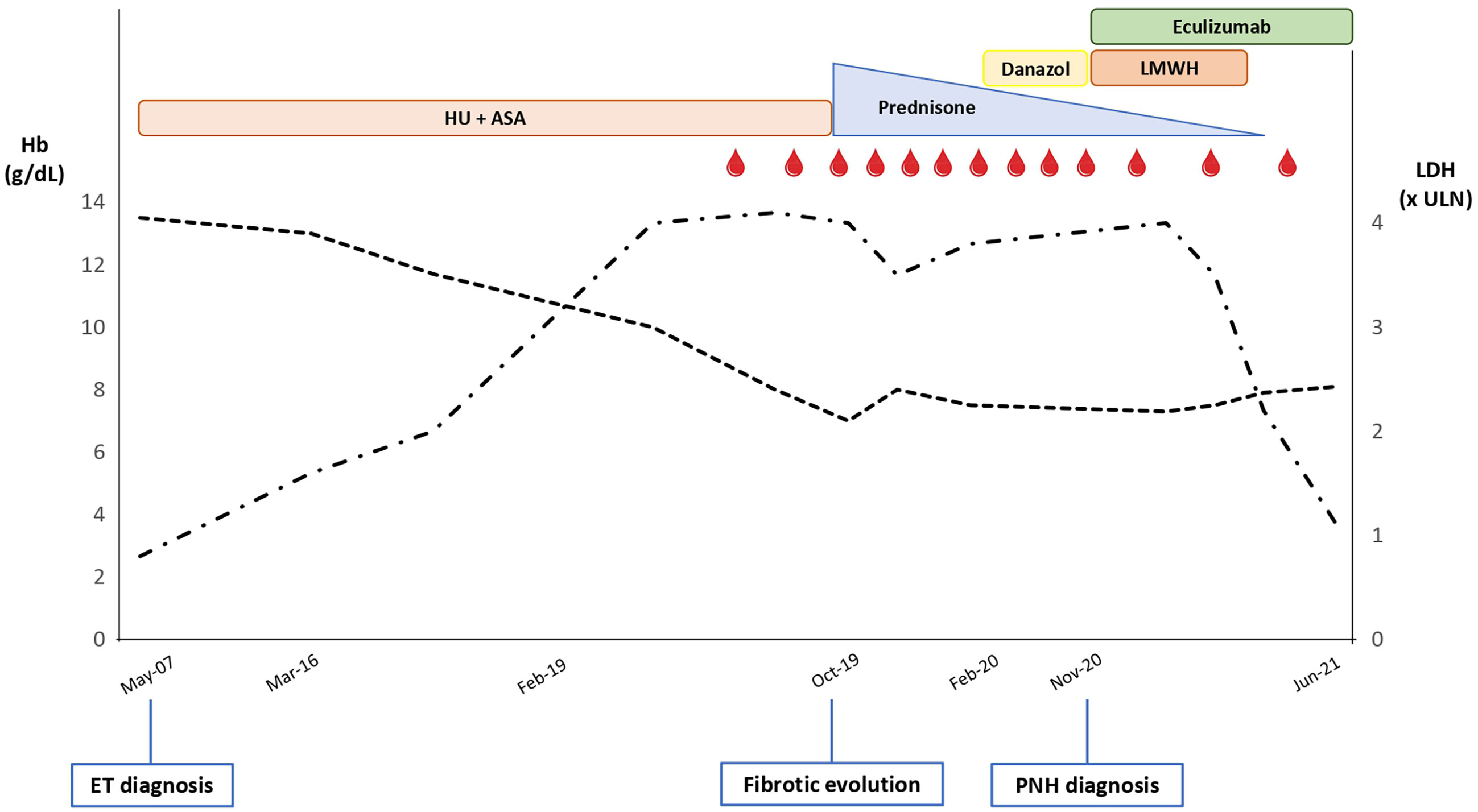
Figure 1 Trend of hemoglobin (Hb; dotted black line) and lactate dehydrogenase (LDH; dashed-dotted black line) levels along the patient’s clinical journey. ULN, upper limit of normal; HU, hydroxyurea; ASA, acetylsalicylic acid; LMWH, low-molecular weight heparin; ET, essential thrombocythemia; PNH, paroxysmal nocturnal hemoglobinuria. Red drops represent red blood cell transfusions.
Review of the Literature
Clinical Association of Paroxysmal Nocturnal Hemoglobinuria and Myeloproliferative Neoplasm
Since the 1970s, several case reports about the coexistence of PNH and MPN have been described (Table 2). A total of 23 cases have been reported so far, mainly in association with myelofibrosis (MF; n = 12), followed by polycythemia vera (PV; n = 3), MPN-U (n = 2), and only one case each of ET (20) and chronic myeloid leukemia (CML) (19). Nine cases were JAK2-positive, while only two CALR- and one MPL-mutated (24). In 12 cases, the diagnosis of MPN preceded that of PNH, and the clinical suspicion for the latter (when indicated) was the development of hemolytic or iron-deficient anemia (15, 17, 20). The diagnoses were concomitant in seven patients, whose main clinical presentation included atypical thromboses or the coexistence of hemolytic anemia and thrombocytosis (18, 21, 24, 25). In the remaining four subjects, the diagnosis of MPN followed that of PNH and derived from recurrent thrombosis or development of thrombocytosis/leukocytosis (14, 18, 19). Ten patients harbored PNH clones >10% (8/10 were >50%), while in four cases, it was <10%. PNH clone size showed a complessively wide distribution, as reported by Richards et al. (23) in a recent large retrospective study analyzing the clinical presentation of 1,081 PNH patients. The five patients with a previous MPN diagnosis had a PNH clone from 0.7% to 96.3%, while cytopenic/myelodysplastic patients generally harbored smaller clones, and hemolytic/thrombotic subjects harbored larger ones (23). With regard to treatments, eculizumab was administered in four patients (notably, all with JAK2V617F-positive MPN). In one patient, eculizumab monotherapy was able to resolve anemia (18); in another, the concomitant administration of eculizumab with acenocumarol and HU resolved a severe case of visceral thrombosis (25). In the remaining two cases, hemolytic anemia was refractory, and one experienced visceral thrombosis recurrence notwithstanding eculizumab plus HU and anticoagulation (18). A retrospective series of 55 PNH patients undergoing hematopoietic stem cell transplantation (HSCT) included two cases associated with MF. The reported overall survival at 5 years was 70%, although the outcome of these two patients is not specified (22). Regarding outcome, four out of the total 23 patients reported (17.4%) died due to acute lymphoblastic/myeloid leukemia progression (16, 17), infections, or liver failure secondary to refractory Budd–Chiari syndrome (18).
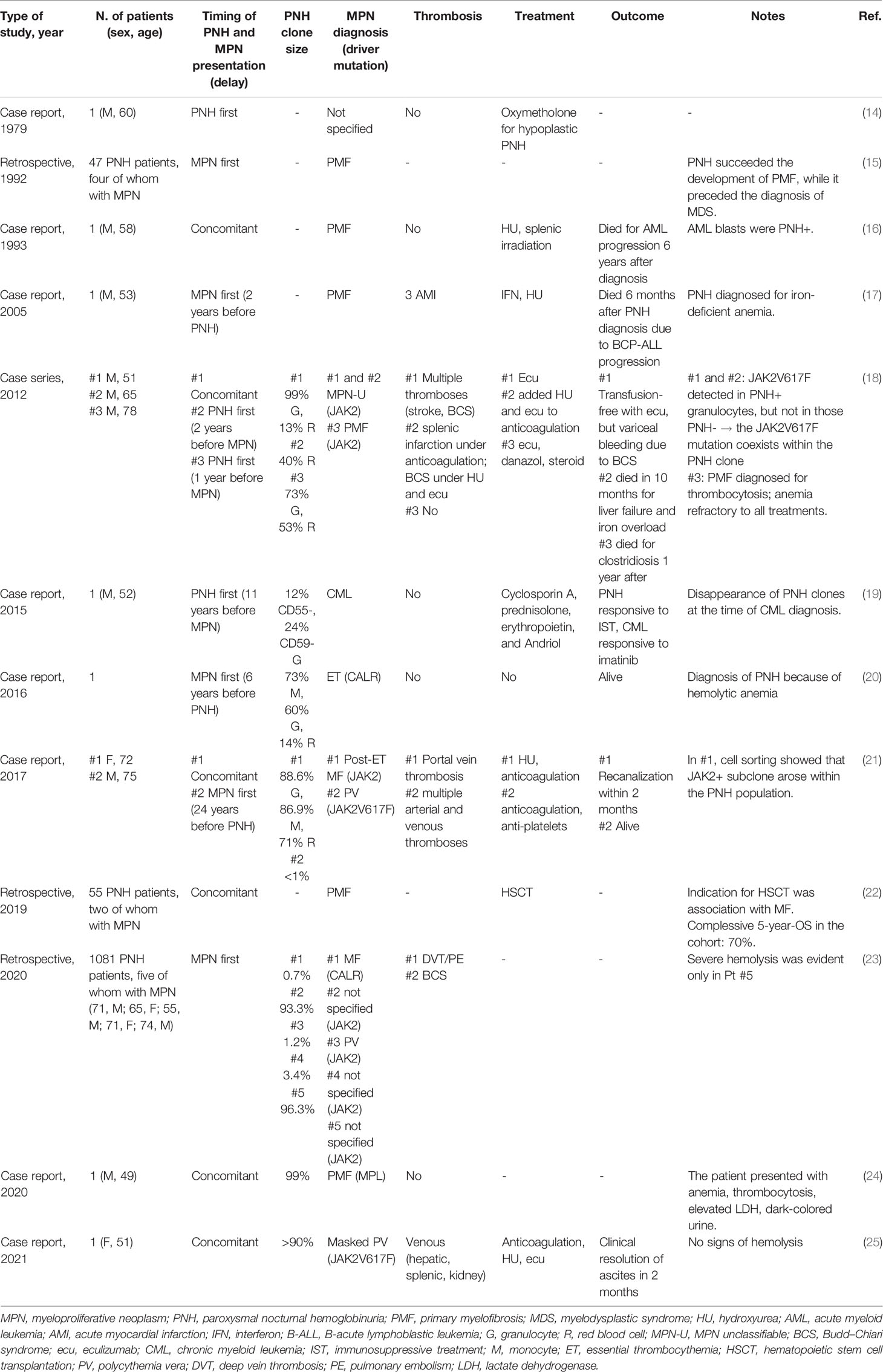
Table 2 Association of MPNs and PNH.
Prevalence of Paroxysmal Nocturnal Hemoglobinuria Clones in Myeloproliferative Neoplasm Patients
Some cross-sectional studies on MPN patients evaluated the association with PNH clones. Tanasi et al. (26) tested 32 patients with MPN and concomitant hemolysis or unexplained anemia and found a PNH clone in three (9.3%) with MF (two JAK2- and one CALR-mutated). Two of them harbored a PNH clone >90% but did not require specific PNH therapy (26). In a recent large monocentric study, more than 3,000 patients were tested for PNH because of unexplained cytopenia/thrombosis, including 92 patients diagnosed with MPN (27). A PNH clone was found in 16 patients (17.4%), mainly MF, and was generally smaller than 1% (except for one patient with a clone of 5%). It was associated with increased frequency of thrombosis without impact on overall survival (28). In a large cross-sectional study including 197 MPN, 14.2% of subjects had CD55/CD59-negative red cells; this prevalence rose to 21.3% in the subgroup of ET patients (29). At variance, in a series of 98 MPN subjects, PNH clones greater than 1% were detected in only two patients (2%) (30), one of whom with recurrent thrombotic complications. Finally, two studies failed to detect PNH clones in MPN patients. In detail, Nazha et al. (31) tested 62 MF patients with significant anemia (Hb <10 g/dL) and elevated LDH, but none of them harbored a PNH clone. Likewise, a study on 136 patients with myeloid disease, including five MF and 15 MDS/MPN overlap, found GPI-negative cells in 8% of low-risk MDS, but none in MPN subjects (32).
Prevalence of Myeloproliferative Neoplasm Driver Mutations in Paroxysmal Nocturnal Hemoglobinuria Patients
There are isolated reports of PNH patients harboring mutations in MPN-related driver genes, namely, JAK2. Shen et al. (33) performed targeted NGS in a cohort of 36 PNH patients and found JAK2V617F homozygous mutations in two patients (5.5%). Langabeer et al. (34) reported a case of AA-PNH with a concomitant JAK2V617F mutation at low allele ratio (1.8%) without any clinical feature of MPN; intriguingly, the JAK2-positive clone disappeared after cyclosporin therapy, while the PNH clone remained stable. More recently, Santagostino et al. (35) described a case of hemolytic PNH occurring 10 years after HSCT for acute myeloid leukemia (AML); concomitantly, somatic mutation analysis revealed the presence of JAK2 mutation with an allele ratio of 44% and TET2 with a VAF of 34%. Soon after, the patient relapsed for AML.
Discussion
Our patient, along with the others described, allows several clinical and pathogenic considerations about the rare coexistence of PNH and MPN. Besides AA and MDS, PNH clones have been detected also in the context of lymphoid disorders, such as acute lymphoblastic leukemia and lymphomas (9, 36), and in autoimmune/idiopathic cytopenias (6–8, 37). The review of the literature highlighted that about 10% of MPN patients harbor a PNH clone (26, 29, 30), and this frequency rises up to 17% if clones smaller than 1% are considered (27). More importantly, the disregarded association of these two conditions may cause a significant delay in PNH diagnosis, as observed in our case. Additionally, in the MPN setting, the differential diagnosis of hemolytic anemia may be hampered by several confounders: haptoglobin can be decreased in more than 30% of MF (38), LDH is often elevated as a consequence of disease burden (31), and reticulocytosis can be observed in case of myeloid metaplasia (39). Furthermore, the appearance of anemia in MPN should prompt the exclusion of fibrotic evolution, which was in fact observed in our patient, who met the diagnostic criteria for post-ET MF (40). However, at variance with post-ET MF, BM trephine revealed erythroid hyperplasia, which may be attributed to the concomitant peripheral hemolytic process and indicative of bone marrow compensation. Consultation with an expert hemopathologist may be thus advised when morphological findings are not fully consistent with a clear diagnosis and confounding factors coexist.
An important clinical issue is the thrombotic risk in MPN-PNH patients and its management. In our literature review, 35% of MPN-PNH subjects had a severe thrombotic presentation, often refractory to combined anticoagulant, cytoreductive, and anti-complement treatments. This frequency appears higher than that reported for isolated untreated PNH (18.8%) (41) and for MPN (complessively 20%, higher in PV vs. ET/MF and JAK2-mutated patients) (12, 13, 42, 43), possibly due to the association of two thrombophilic conditions. With regard to therapy, it is well established that cytoreductive and anti-coagulant/platelet therapies are the cornerstones of thrombosis treatment in MPN, although recurrencies interest about 20% of treated patients (44, 45). In PNH, complement inhibition (Ci) has proven to significantly reduce the thrombotic risk, while anticoagulation alone is poorly effective (4, 46). Whether primary thromboprophylaxis is indicated in untreated PNH is still debated. Given the higher risk observed in patients with a larger clone size (i.e., >50%), they are generally candidates to primary prophylaxis if there are no contraindications. Prophylaxis may be then discontinued once complement inhibitors are started, as in the case described (47). With regard to secondary prophylaxis in patients on complement inhibitors, some experts discontinue anticoagulants when Intravascular hemolysis (IVH) is well controlled by anti-complement therapy (4, 48, 49). Finally, anti-platelet prophylaxis had been stopped in our patient when CALR-positive post-ET MF diagnosis was made. In fact, JAK2-negative ET is apparently associated with lower rates of thrombosis (50, 51); additionally, the indication to anti-thrombotic primary prophylaxis in MF is not clear-cut (52).
Another relevant clinical issue in MPN-PNH patients may be the infectious risk due to the treatment with anti-complement therapy and the known infectious diathesis observed in MPN (53). Despite the known risk of capsulated bacterial infections under Ci (54), no infectious complications occurred in our patient and in only one out of 23 patients (4%) in the literature (18).
With regard to therapy, in our patient, eculizumab showed effectiveness in controlling IVH but was not able to resolve transfusion-dependent anemia. Accordingly, the review of the literature showed that only a fraction of MPN-PNH patients responded to eculizumab (Table 2) (18, 25). In classic PNH, persistent anemia under Ci treatment can be caused by residual IVH, concomitant BMF, and EVH. The management of the latter still remains an unmet need, but promising results are coming from clinical trials with proximal complement inhibitors that avoid deposition of C3 on RBC surface (55).
Many speculations can be made regarding the pathogenic significance of the association of MPN and PNH clones. The evidence of MPN driver mutations, particularly JAK2, selectively in GPI-deficient cells (18, 21) has raised the hypothesis that they may confer an intrinsic growth advantage to PNH cells. This cooperative effect has also been proposed for other somatic mutations, such as TET2 (33, 35, 56, 57), also present in our patient. This view would provide a different explanation to the more common notion that PNH cells have an extrinsic growth advantage secondary to an autoimmune, GPI-selective process against bone marrow precursors (58, 59). Interestingly, MPN diagnosis preceded that of PNH in more than half of cases, and Shen et al. (33) demonstrated that PIGA mutation was subclonal to JAK2-mutated clone in their report. It is tempting to speculate that the MPN-associated inflammatory microenvironment (60–63) can impair normal hematopoiesis, exerting an “immune pressure” that favors PNH clone expansion, similarly to what happens in AA.
In conclusion, the association of MPN and PNH deserves attention because of the unpredictable clinical course often affected by dramatic thrombotic complications. Since PNH diagnosis is based on highly sensitive and cost-effective techniques, testing for PNH clones should be prompted in any MPN patient showing unexplained anemia with/without frank hemolysis, recurrent thrombosis, and/or atypical BM morphologic findings. Anti-complement treatment in the setting of MPN relies on a careful case-by-case evaluation, weighing the contribution of intravascular hemolysis to anemia and the thrombotic risk.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary files. Further inquiries can be directed to the corresponding author.
Ethics Statement
The studies involving human participants were reviewed and approved by Comitato Etico Milano Area 2. The patients/participants provided their written informed consent to participate in this study.
Author Contributions
JG, BF, and WB followed the patient, described his clinical history, and revised the literature and the paper for intellectual content. All authors contributed to the article and approved the submitted version.
Conflict of Interest
WB received consultancy honoraria from Agios, Alexion, Novartis, Annexon, Sanofi, and Sobi. BF received consultancy honoraria from Amgen, Alexion, Novartis, Momenta, Annexon, and Apellis.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Brodsky RA. Paroxysmal Nocturnal Hemoglobinuria. Blood (2014) 124:2804–11. doi: 10.1182/blood-2014-02-522128
2. Parker CJ. Update on the Diagnosis and Management of Paroxysmal Nocturnal Hemoglobinuria. Hematol Am Soc Hematol Educ Program (2016) 2016:208–16. doi: 10.1182/asheducation-2016.1.208
3. Hill A, Kelly RJ, Hillmen P. Thrombosis in Paroxysmal Nocturnal Hemoglobinuria. Blood (2013) 121:4985–96. doi: 10.1182/blood-2012-09-311381
4. Brodsky RA. How I Treat Paroxysmal Nocturnal Hemoglobinuria. Blood (2021) 137:1304–9. doi: 10.1182/blood.2019003812
5. Brando B, Gatti A, Preijers F. Flow Cytometric Diagnosis of Paroxysmal Nocturnal Hemoglobinuria: Pearls and Pitfalls - A Critical Review Article. EJIFCC (2019) 30:355–70.
6. Fattizzo B, Giannotta J, Zaninoni A, Kulasekararaj A, Cro L, Barcellini W. Small Paroxysmal Nocturnal Hemoglobinuria Clones in Autoimmune Hemolytic Anemia: Clinical Implications and Different Cytokine Patterns in Positive and Negative Patients. Front Immunol (2020) 11:1006. doi: 10.3389/fimmu.2020.01006
7. Damianaki A, Stagakis E, Mavroudi I, Spanoudakis M, Koutala H, Papadogiannis F, et al. Minor Populations of Paroxysmal Nocturnal Hemoglobinuria-Type Cells in Patients With Chronic Idiopathic Neutropenia. Eur J Haematol (2016) 97:538–46. doi: 10.1111/ejh.12766
8. Saito C, Ishiyama K, Yamazaki H, Zaimoku Y, Nakao S. Hypomegakaryocytic Thrombocytopenia (HMT): An Immune-Mediated Bone Marrow Failure Characterized by an Increased Number of PNH-Phenotype Cells and High Plasma Thrombopoietin Levels. Br J Haematol (2016) 175:246–51. doi: 10.1111/bjh.14210
9. Meletis J, Terpos E, Samarkos M, Meletis C, Apostolidou E, Komninaka V, et al. Detection of CD55- and/or CD59-Deficient Red Cell Populations in Patients With Lymphoproliferative Syndromes. Hematol J (2001) 2:33–7. doi: 10.1038/sj.thj.6200077
10. Van Bijnen ST, Van Heerde WL, Muus P. Mechanisms and Clinical Implications of Thrombosis in Paroxysmal Nocturnal Hemoglobinuria. J Thromb Haemost (2012) 10:1–10. doi: 10.1111/j.1538-7836.2011.04562.x
11. Barbui T, Finazzi G, Falanga A. Myeloproliferative Neoplasms and Thrombosis. Blood (2013) . 122:2176–84. doi: 10.1182/blood-2013-03-460154
12. Sant'Antonio E, Guglielmelli P, Pieri L, Primignani M, Randi ML, Santarossa C, et al. Splanchnic Vein Thromboses Associated With Myeloproliferative Neoplasms: An International, Retrospective Study on 518 Cases. Am J Hematol (2020) 95:156–66. doi: 10.1002/ajh.25677
13. Martinelli I, De Stefano V, Carobbio A, Randi ML, Santarossa C, Rambaldi A, et al. Cerebral Vein Thrombosis in Patients With Philadelphia-Negative Myeloproliferative Neoplasms. An European Leukemia Net Study. Am J Hematol (2014) 89:E200–5. doi: 10.1002/ajh.23809
14. Boyd AW, Parkin JD, Castaldi PA. A Case of Paroxysmal Nocturnal Haemoglobinuria Terminating in a Myeloproliferative Syndrome. Aust N Z J Med (1979) 9:181–3. doi: 10.1111/j.1445-5994.1979.tb04325.x
15. Graham DL, Gastineau DA. Paroxysmal Nocturnal Hemoglobinuria as a Marker for Clonal Myelopathy. Am J Med (1992) 93:671–4. doi: 10.1016/0002-9343(92)90201-l
16. Nakahata J, Takahashi M, Fuse I, Nakamori Y, Nomoto N, Saitoh H, et al. Paroxysmal Nocturnal Hemoglobinuria With Myelofibrosis: Progression to Acute Myeloblastic Leukemia. Leuk Lymphoma (1993) 12:137–42. doi: 10.3109/10428199309059582
17. Shaheen SP 2nd, Talwalkar SS, Simons R, Yam L. Acute Lymphoblastic Leukemic Transformation in a Patient With Chronic Idiopathic Myelofibrosis and Paroxysmal Nocturnal Hemoglobinuria: A Case Report and Review of the Literature. Arch Pathol Lab Med (2005) 129:96–9. doi: 10.5858/2005-129-96-ALLTIA
18. Sugimori C, Padron E, Caceres G, Shain K, Sokol L, Zhang L, et al. Paroxysmal Nocturnal Hemoglobinuria and Concurrent JAK2(V617F) Mutation. Blood Cancer J (2012) 2:e63. doi: 10.1038/bcj.2012.7
19. Chen Y, Tao S, Deng Y, Song L, Yu L. Chronic Myeloid Leukemia Transformation in a Patient With Paroxysmal Nocturnal Hemoglobinuria: A Rare Case Report With Literature Review. Int J Clin Exp Med (2015) 8:8226–9.
20. Fraiman YS, Cuka N, Batista D, Vuica-Ross M, Moliterno AR. Development of Paroxysmal Nocturnal Hemoglobinuria in CALR-Positive Myeloproliferative Neoplasm. J Blood Med (2016) 7:107–10. doi: 10.2147/JBM.S103473
21. Gaidano V, Geuna M, Cignetti A, Carnuccio F, Bernabei P, Santoro N, et al. Myeloproliferative Neoplasms, Thrombosis and Paroxysmal Nocturnal Hemoglobinuria: Is This Triad More Frequent Than We Thought? Hematol Med Oncol (2017). doi: 10.15761/HMO.1000132
22. Cooper JP, Farah RJ, Stevenson PA, Gooley TA, Storb R, Scott BL. Hematopoietic Cell Transplantation for Paroxysmal Nocturnal Hemoglobinuria in the Age of Eculizumab. Biol Blood Marrow Transplant (2019) 25:1331–9. doi: 10.1016/j.bbmt.2019.01.033
23. Richards SJ, Dickinson AJ, Cullen MJ, Griffin M, Munir T, McKinley C, et al. Presentation Clinical, Haematological and Immunophenotypic Features of 1081 Patients With GPI-Deficient (Paroxysmal Nocturnal Haemoglobinuria) Cells Detected by Flow Cytometry. Br J Haematol (2020) 189:954–66. doi: 10.1111/bjh.16427
24. Kirito K. Expansion of Paroxysmal Nocturnal Hemoglobinuria Clones in MPLW515L Mutation Harboring Primary Myelofibrosis. Ann Hematol (2020) 99:2707–9. doi: 10.1007/s00277-020-04088-1
25. Chatzidavid S, Giannakopoulou N, Diamantopoulos PT, Gavriilaki E, Katsiampoura P, Lakiotaki E, et al. JAK2V617F Positive Polycythemia Vera With Paroxysmal Nocturnal Hemoglobinuria and Visceral Thromboses: A Case Report and Review of the Literature. Thromb J (2021) 19:16. doi: 10.1186/s12959-021-00269-8
26. Tanasi I, Polino A, Bega G, Bonalumi A, Facchinelli D, Greco C, et al. Evidence of Paroxysmal Nocturnal Hemoglobinuria Clones in Some Patients With Myeloproliferative Neoplasms and Signs of Haemolysis or Unexplained Anemia. (2018). doi: 10.13140/RG.2.2.27063.16803. Abstract retrieved from SIES 2018.
27. Fattizzo B, Ireland R, Dunlop A, Yallop D, Kassam S, Large J, et al. Clinical and Prognostic Significance of Small Paroxysmal Nocturnal Hemoglobinuria Clones in Myelodysplastic Syndrome and Aplastic Anemia. Leukemia (2021). doi: 10.1038/s41375-021-01190-9
28. Fattizzo B, Dunlop A, Ireland R, Kassam S, Yallop D, Mufti G, et al. Prevalence of Small PNH Clones and Their Prognostic Significance in Patients Tested for Unusual Indications: A Single Center Experience. Br J Haematol (2019) 185:1–3. doi: 10.1111/bjh.15854. Special Issue: Abstracts of the 59th Annual Scientific Meeting of the British Society for HematologyApril 2019, Glasgow, UK.
29. Meletis J, Terpos E, Samarkos M, Meletis C, Konstantopoulos K, Komninaka V, et al. Detection of CD55 and/or CD59 Deficient Red Cell Populations in Patients With Aplastic Anaemia, Myelodysplastic Syndromes and Myeloproliferative Disorders. Haematologia (Budap) (2001) 31:7–16. doi: 10.1163/15685590151092643
30. Gutwein O, Englander Y, Herzog-Tzarfati K, Filipovich-Rimon T, Apel A, Marcus R, et al. Prevalence of Paroxysmal Nocturnal Hemoglobinuria Clones in Myeloproliferative Neoplasm Patients: A Cross-Sectional Study. Clin Lymphoma Myeloma Leuk. (2019) 19:812–4. doi: 10.1016/j.clml.2019.07.441
31. Nazha A, Jorgensen JL, Verstovsek S. Paroxysmal Nocturnal Hemoglobinuria is Not a Cause of Anemia in Patients With Myelofibrosis. Leuk Lymphoma (2014) 55:2215–6. doi: 10.3109/10428194.2013.876628
32. Wang SA, Pozdnyakova O, Jorgensen JL, Medeiros LJ, Stachurski D, Anderson M, et al. Detection of Paroxysmal Nocturnal Hemoglobinuria Clones in Patients With Myelodysplastic Syndromes and Related Bone Marrow Diseases, With Emphasis on Diagnostic Pitfalls and Caveats. Haematologica (2009) 94:29–37. doi: 10.3324/haematol.13601
33. Shen W, Clemente MJ, Hosono N, Yoshida K, Przychodzen B, Yoshizato T, et al. Deep Sequencing Reveals Stepwise Mutation Acquisition in Paroxysmal Nocturnal Hemoglobinuria. J Clin Invest (2014) 124:4529–38. doi: 10.1172/JCI74747
34. Langabeer SE, Haslam K, O'Brien D, Enright H, Leahy M. Transient JAK2 V617F Mutation in an Aplastic Anaemia Patient With a Paroxysmal Nocturnal Haemoglobinuria Clone. Br J Haematol (2013) 161:297–8. doi: 10.1111/bjh.12224
35. Santagostino A, Lombardi L, Dine G, Hirsch P, Misra SC. Paroxysmal Nocturnal Hemoglobinuria With a Distinct Molecular Signature Diagnosed Ten Years After Allogenic Bone Marrow Transplantation for Acute Myeloid Leukemia. Case Rep Hematol (2019) 2019:8928623. doi: 10.1155/2019/8928623
36. Isoda A, Ogawa Y, Matsumoto M, Sawamura M. Coexistence of Paroxysmal Nocturnal Hemoglobinuria (PNH) and Acute Lymphoblastic Leukemia (ALL): Is PNH a Prodrome of ALL? Leuk Res (2009) 33:e3–5. doi: 10.1016/j.leukres.2008.05.016
37. Kalam S, Beale R, Hughes D, Kulasekararaj A, Srirangalingam U. Coombs-Positive Paroxysmal Nocturnal Haemoglobinuria. Oxf Med Case Rep (2020) 2020):omz125. doi: 10.1093/omcr/omz125
38. Strati P, Masarova L, Bose P, Daver N, Pemmaraju N, Verstovsek S. Haptoglobin is Frequently Low in Patients With Myelofibrosis: Clinical Relevance. Leuk Res (2017) 57:85–8. doi: 10.1016/j.leukres.2017.03.006
39. Barosi G. Myelofibrosis With Myeloid Metaplasia: Diagnostic Definition and Prognostic Classification for Clinical Studies and Treatment Guidelines. J Clin Oncol (1999) 17:2954–70. doi: 10.1200/JCO.1999.17.9.2954
40. Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, et al. The 2016 Revision to the World Health Organization Classification of Myeloid Neoplasms and Acute Leukemia. Blood (2016) 127:2391–405. doi: 10.1182/blood-2016-03-643544
41. Schrezenmeier H, Röth A, Araten DJ, Kanakura Y, Larratt L, Shammo JM, et al. Baseline Clinical Characteristics and Disease Burden in Patients With Paroxysmal Nocturnal Hemoglobinuria (PNH): Updated Analysis From the International PNH Registry. Ann Hematol (2020) 99:1505–14. doi: 10.1007/s00277-020-04052-z
42. Sekhar M, McVinnie K, Burroughs AK. Splanchnic Vein Thrombosis in Myeloproliferative Neoplasms. Br J Haematol (2013) 162:730–47. doi: 10.1111/bjh.12461
43. Rungjirajittranon T, Owattanapanich W, Ungprasert P, Siritanaratkul N, Ruchutrakool T. A Systematic Review and Meta-Analysis of the Prevalence of Thrombosis and Bleeding at Diagnosis of Philadelphia-Negative Myeloproliferative Neoplasms. BMC Cancer (2019) 19:184. doi: 10.1186/s12885-019-5387-9
44. Finazzi G, De Stefano V, Barbui T. Splanchnic Vein Thrombosis in Myeloproliferative Neoplasms: Treatment Algorithm 2018. Blood Cancer J (2018) 8:64. doi: 10.1038/s41408-018-0100-9
45. De Stefano V, Za T, Rossi E, Vannucchi AM, Ruggeri M, Elli E, et al. GIMEMA CMD-Working Party. Recurrent Thrombosis in Patients With Polycythemia Vera and Essential Thrombocythemia: Incidence, Risk Factors, and Effect of Treatments. Haematologica (2008) 93:372–80. doi: 10.3324/haematol.12053
46. Loschi M, Porcher R, Barraco F, Terriou L, Mohty M, de Guibert S, et al. Impact of Eculizumab Treatment on Paroxysmal Nocturnal Hemoglobinuria: A Treatment Versus No-Treatment Study. Am J Hematol (2016) 91:366–70. doi: 10.1002/ajh.24278
47. Griffin M, Munir T. Management of Thrombosis in Paroxysmal Nocturnal Hemoglobinuria: A Clinician's Guide. Ther Adv Hematol (2017) 8:119–26. doi: 10.1177/2040620716681748
48. Al-Jafar HA, AlDallal SM, Askar HA, Aljeraiwi AM, Al-Alansari A. Long Standing Eculizumab Treatment Without Anticoagulant Therapy in High-Risk Thrombogenic Paroxysmal Nocturnal Hemoglobinuria. Hematol Rep (2015) 7:5927. doi: 10.4081/hr.2015.5927
49. Emadi A, Brodsky RA. Successful Discontinuation of Anticoagulation Following Eculizumab Administration in Paroxysmal Nocturnal Hemoglobinuria. Am J Hematol (2009) 84:699–701. doi: 10.1002/ajh.21506
50. Finazzi G, Carobbio A, Guglielmelli P, Cavalloni C, Salmoiraghi S, Vannucchi AM, et al. Calreticulin Mutation Does Not Modify the IPSET Score for Predicting the Risk of Thrombosis Among 1150 Patients With Essential Thrombocythemia. Blood (2014) 124:2611–2. doi: 10.1182/blood-2014-08-596676
51. Gangat N, Wassie EA, Lasho TL, Finke C, Ketterling RP, Hanson CA, et al. Mutations and Thrombosis in Essential Thrombocythemia: Prognostic Interaction With Age and Thrombosis History. Eur J Haematol (2015) 94:31–6. doi: 10.1111/ejh.12389
52. Koschmieder S. How I Manage Thrombotic/Thromboembolic Complications in Myeloproliferative Neoplasms. Hamostaseologie (2020) 40:47–53. doi: 10.1055/s-0040-1701474
53. Landtblom AR, Andersson TM, Dickman PW, Smedby KE, Eloranta S, Batyrbekova N, et al. Risk of Infections in Patients With Myeloproliferative Neoplasms-A Population-Based Cohort Study of 8363 Patients. Leukemia (2021) 35:476–84. doi: 10.1038/s41375-020-0909-7
54. Socié G, Caby-Tosi MP, Marantz JL, Cole A, Bedrosian CL, Gasteyger C, et al. Eculizumab in Paroxysmal Nocturnal Haemoglobinuria and Atypical Haemolytic Uraemic Syndrome: 10-Year Pharmacovigilance Analysis. Br J Haematol (2019) 185:297–310. doi: 10.1111/bjh.15790
55. Risitano AM, Marotta S, Ricci P, Marano L, Frieri C, Cacace F, et al. Anti-Complement Treatment for Paroxysmal Nocturnal Hemoglobinuria: Time for Proximal Complement Inhibition? A Position Paper From the SAAWP of the EBMT. Front Immunol (2019) 10:1157. doi: 10.3389/fimmu.2019.01157
56. Lobry C, Bains A, Zamechek LB, Ibrahim S, Aifantis I, Araten DJ. Analysis of TET2 Mutations in Paroxysmal Nocturnal Hemoglobinuria (PNH). Exp Hematol Oncol (2019) 8:17. doi: 10.1186/s40164-019-0142-0
57. Sun L, Babushok DV. Secondary Myelodysplastic Syndrome and Leukemia in Acquired Aplastic Anemia and Paroxysmal Nocturnal Hemoglobinuria. Blood (2020) 136:36–49. doi: 10.1182/blood.2019000940
58. Gargiulo L, Papaioannou M, Sica M, Talini G, Chaidos A, Richichi B, et al. Glycosylphosphatidylinositol-Specific, CD1d-Restricted T Cells in Paroxysmal Nocturnal Hemoglobinuria. Blood (2013) 121:2753–61. doi: 10.1182/blood-2012-11-469353
59. Luzzatto L, Notaro R. The "Escape" Model: A Versatile Mechanism for Clonal Expansion. Br J Haematol (2019) 184:465–6. doi: 10.1111/bjh.15111
60. Hasselbalch HC. Chronic Inflammation as a Promotor of Mutagenesis in Essential Thrombocythemia, Polycythemia Vera and Myelofibrosis. A Human Inflammation Model for Cancer Development? Leuk Res (2013) 37:214–20. doi: 10.1016/j.leukres.2012.10.020
61. Hermouet S, Bigot-Corbel E, Gardie B. Pathogenesis of Myeloproliferative Neoplasms: Role and Mechanisms of Chronic Inflammation. Mediators Inflamm (2015) 2015:145293. doi: 10.1155/2015/145293
62. Lussana F, Rambaldi A. Inflammation and Myeloproliferative Neoplasms. J Autoimmun (2017) 85:58–63. doi: 10.1016/j.jaut.2017.06.010
Keywords: paroxysmal nocturnal hemoglobinuria, myeloproliferative neoplasm, complement inhibitors, thrombosis, bone marrow failure, case report
Citation: Giannotta JA, Fattizzo B and Barcellini W (2021) Paroxysmal Nocturnal Hemoglobinuria in the Context of a Myeloproliferative Neoplasm: A Case Report and Review of the Literature. Front. Oncol. 11:756589. doi: 10.3389/fonc.2021.756589
Received: 10 August 2021; Accepted: 15 October 2021;
Published: 11 November 2021.
Edited by:
Abbas Ghaderi, Shiraz University of Medical Sciences, IranReviewed by:
Francesca Schieppati, Papa Giovanni XXIII Hospital, ItalyMarco Santoro, Università degli Studi di Palermo, Italy
Copyright © 2021 Giannotta, Fattizzo and Barcellini. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Juri Alessandro Giannotta, anVyaWdpYW5uQGdtYWlsLmNvbQ==