 Christian Münz
Christian Münz- Viral Immunobiology, Institute of Experimental Immunology, University of Zürich, Zürich, Switzerland
The oncogenic Epstein–Barr virus (EBV) persistently infects more than 95% of the human adult population. Even so it can readily transform human B cells after infection in vitro, it only rarely causes tumors in patients. A substantial proportion of the 1% of all human cancers that are associated with EBV occurs during coinfections, including those with the malaria parasite Plasmodium falciparum, the human immunodeficiency virus (HIV), and the also oncogenic and closely EBV-related Kaposi sarcoma-associated herpesvirus (KSHV). In this review, I will discuss how these infections interact with EBV, modify its immune control, and shape its tumorigenesis. The underlying mechanisms reveal new aspects of EBV-associated pathologies and point toward treatment possibilities for their prevention by the human immune system.
Introduction on EBV and Its Immune Control
The ubiquitous human γ-herpesvirus EBV persistently infects more than 95% of the human adult population (1, 2). While it is primarily acquired in early childhood, delayed primary infection during adolescence or young adulthood in one-third of the European and North American population can result in infectious mononucleosis (IM), an immunopathological CD8+ T cell lymphocytosis (3, 4). In some of these individuals, chronic active EBV (CAEBV) can develop which allows the virus to infect in addition to its main host cell, the human B cell, and other lymphocytes, including T and NK cells (5–7). From this EBV infection of T and NK cells, also virus-associated NK/T cell lymphomas are thought to develop (8). Most types of EBV-associated lymphomas, however, mirror the viral infection programs in B cells (8–10). Upon transmission via saliva, EBV crosses the mucosal epithelium to infect B cells in the underlying secondary lymphoid tissues, including tonsils (1, 11). As its default program, it expresses latent EBV gene products from circularized viral DNA episomes. In naïve B cells, all eight latent EBV proteins (six nuclear antigens or EBNAs, and two latent membrane proteins or LMPs) plus non-translated RNAs, including EBERs and miRNAs, can be detected (12, 13). This so-called latency III program drives B cells into proliferation and can also be found after in vitro immortalization of human B cells by EBV infection, as well as in lymphomas in immune compromised individuals (14). In germinal center B cells, only EBNA1 and the two LMPs are expressed at the protein level and thought to rescue infected cells from deletion without T cell help and B cell receptor engagement during this B cell differentiation stage (15). This latency IIa program is found in Hodgkin’s lymphoma (8). Via germinal center differentiation or directly from an early expressed latency IIb program (EBNA without LMP expression), EBV-infected B cells reach the memory B cell pool for persistence (16, 17). In these, EBV shuts down latent protein expression or transiently induces EBNA1 in homeostatically proliferating memory B cells during latency 0 or I, respectively (18). Latency I can also be found in Burkitt’s lymphoma (19). From latency 0 or I, EBV can reactivate upon plasma cell differentiation of its infected B cells into lytic replication and infectious viral particle production (20). Mucosal epithelial cell infection from the basolateral site might then allow for another round of lytic EBV replication prior to shedding into saliva (21). Such epithelial cell infection is thought to be the source for EBV-associated nasopharyngeal and gastric carcinomas, but it remains unclear under which circumstances EBV switches into the associated latency II and I programs in these cells (8). Nevertheless, all tumor-associated EBV infection programs are present in healthy EBV carriers and are thought to be kept in check by immune control.
This immune response is thought to be mainly dependent on cytotoxic lymphocytes, such as CD8+ T cells and NK cells, but not antibody responses or antiviral cytokine secretion (22–25). The strongest evidence for this notion comes from primary immunodeficiencies that predispose individuals to EBV-associated pathologies due to single-gene mutations. These identify the perforin/granzyme cytotoxic machinery, T cell receptor signaling, cytotoxic lymphocyte co-stimulation, development of cytotoxic lymphocytes, and their expansion as crucial elements of EBV-specific immune control (22–25). For example, patients with mutations in the co-stimulatory molecule CD27 or its ligand CD70 nearly uniformly suffer from EBV-associated pathologies (26). This co-stimulatory interaction is required for the expansion and cytotoxicity of a subset of EBV-specific CD8+ T cells recognizing lytic infection and EBV-specific immune control in mice with reconstituted human immune systems (humanized mice) (27). In the same preclinical model system of EBV infection, depletion of CD8+ T cells and NK cells or pharmacological inhibition of predominantly CD4+ T cell responses leads to higher viral loads and associated lymphomagenesis carrying primarily the latency III infection program (28–34). These findings suggest that primarily cytotoxic lymphocytes prevent transition of premalignant EBV infection to lymphomagenesis in the vast majority of asymptomatic EBV carriers.
Burkitt’s Lymphoma and Plasmodium falciparum
This balance between lymphomagenesis and its control by the immune system can be disrupted by coinfections. The most prominent case in point is the coinfection with the malaria parasite Plasmodium falciparum which primarily infects hepato- and erythrocytes (19, 35, 36). In Sub-Saharan Africa and Papua New Guinea, holoendemic exposure to this parasite is associated with Burkitt’s lymphoma (36, 37). In this endemic form, Burkitt’s lymphoma incidence is highest in the second half of the first decade of life and to 90% associated with EBV. In addition, it carries a characteristic somatic mutation in the form of c-myc translocation into the heavy- or light-chain immunoglobulin locus of the affected B cells (Figure 1A). This translocation is thought to occur downstream of the germinal center reaction and its expression of activation-induced deaminase (AID) (38). Germinal center induction and the associated AID expression are promoted by persistent Plasmodium infection (39, 40). However, EBV can also directly induce AID expression via its latent gene product EBNA3C (41). Thus, Burkitt’s lymphoma, the tumor entity in which EBV was originally discovered (42, 43), carries in its endemic form EBV latency I infection and a c-myc translocation into the immunoglobulin locus. P. falciparum, but not other malaria parasites, seems to promote Burkitt’s lymphoma development by driving more infected B cells into a differentiation stage in which c-myc translocation can occur (Figure 1A) and by weakening the immune control of the respective tumor cells.
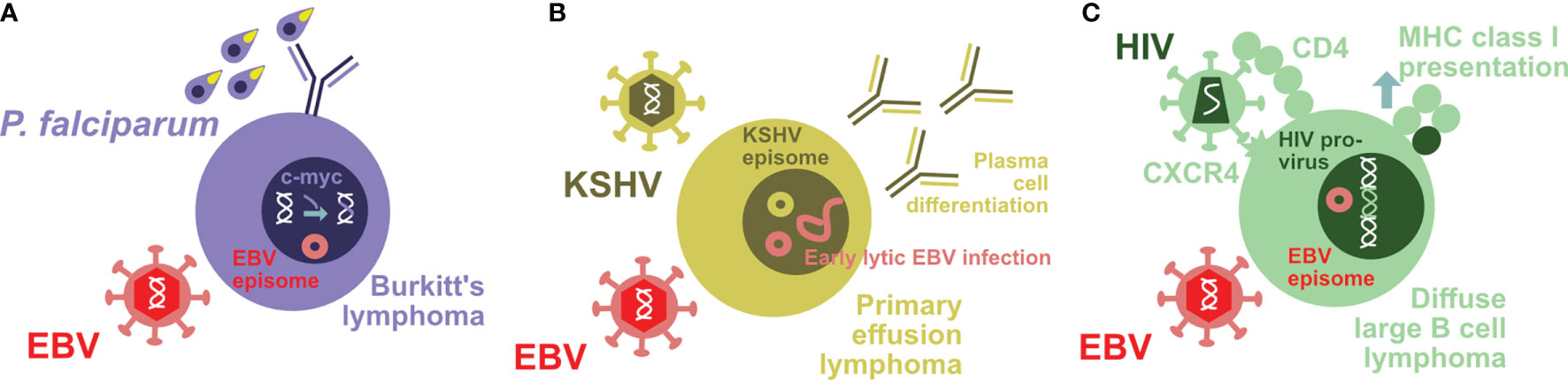
Figure 1 Coinfections modify EBV-associated lymphomagenesis. (A) Holoendemic Plasmodium falciparum (P. falciparum) exposure is associated with endemic Burkitt’s lymphoma. The characteristic c-myc translocation in these uniformly EBV-infected tumor cells might be driven by parasite-induced B cell differentiation. (B) Primary effusion lymphoma is to 100% KSHV and to 90% EBV infected. KSHV infection drives the characteristic plasmablastic differentiation of these tumor cells that in turn leads to increased early lytic EBV reactivation. (C) HIV is able to infect EBV-transformed B cells by virtue of their CD4 upregulation and maintained CXCR4 expression. The resulting double-infected B cells with integrated HIV provirus upregulate MHC class I antigen presentation and are therefore efficiently eliminated by CD8+ T cells.
Indeed, evidence for weakened immune control of EBV could be found in children with holoendemic P. falciparum exposure (44–49). This affects CD4+ T, CD8+ T, and NK cells. Loss of T cell responses against EBNA1, the sole viral protein that is expressed in latency I and Burkitt’s lymphoma cells, has been documented (47) (Figure 2A). But also NK cell differentiation to poorly functional CD56-CD16+ NK cells has been described in children with Burkitt’s lymphoma (48, 49) (Figure 2A) which is thought to compromise NK cell-mediated control of lytic EBV replication (33, 50, 51). NK cell differentiation to CD56-CD16+ cells could in part be driven by activating killer immunoglobulin-like receptors (KIRs) (52). Higher EBV reactivation and/or diminished immune control leads also to more than 100-fold elevated viral loads in children with holoendemic exposure to P. falciparum (53). The development and maintenance of optimal EBV-specific immune control could be compromised by the Th2 environment that is required for immune control of the P. falciparum blood stage (54). Indeed, Burkitt’s lymphoma patients carry malaria asymptomatically in Sub-Saharan Africa (55). Why, however, altered EBV-specific immune control and/or increased EBV-infected B cell activation that result in the elevated viral loads of children in holoendemic malaria regions are particularly associated with the P. falciparum parasite remains unclear. Unfortunately, an in vivo model to probe the interaction of these two important human pathogens is still lacking.
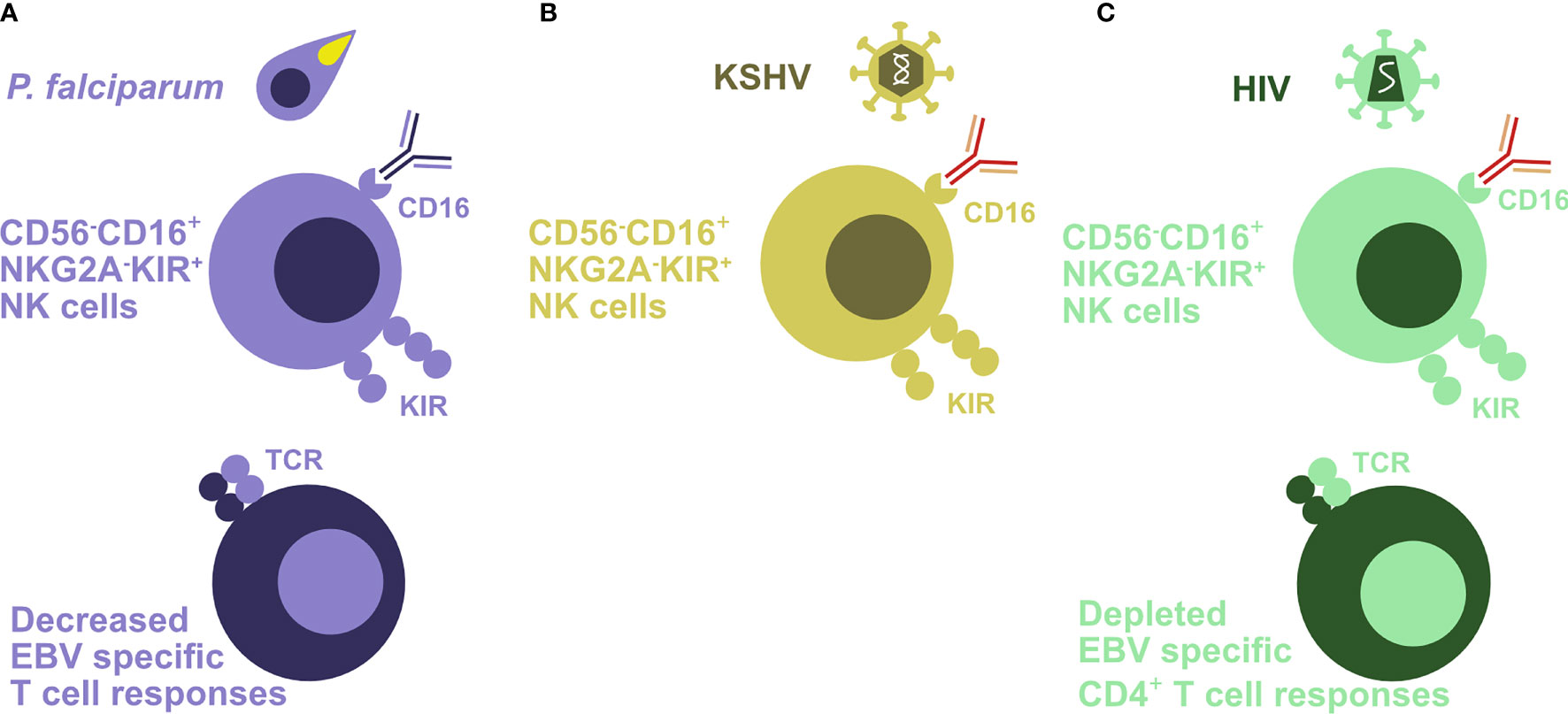
Figure 2 Coinfections attenuate EBV-specific immune control. (A) Holoendemic Plasmodium falciparum (P. falciparum) exposure and especially endemic Burkitt’s lymphoma are associated with an accumulation of terminally differentiated CD56-CD16+NKG2A-KIR+ NK cells. In addition, EBV-specific T cell responses are decreased in the affected patients. (B) KSHV coinfection drives CD56-CD16+NKG2A-KIR+ NK cell differentiation in the presence of EBV infection. (C) HIV-infected patients also demonstrate an accumulation of poorly functional CD56-CD16+NKG2A-KIR+ NK cells. In addition, this infection depletes EBV-specific CD4+ T cells.
Primary Effusion Lymphoma and KSHV
Another coinfection that modifies both EBV-associated tumor cells and EBV-specific immune control is the Kaposi sarcoma-associated herpesvirus (KSHV) (56). This human γ-herpesvirus that is closely related to EBV is associated with the tumors Kaposi sarcoma and primary effusion lymphoma (PEL) (57). PEL cells are 100% KSHV infected and carry simultaneously in 90% of cases EBV in the very same lymphoma cells (9, 58). In most cases, EBV infection displays latency I in these tumors. PEL are also the only tumor entity in which the KSHV genome is maintained after culturing in vitro (59). EBV enhances KSHV genome maintenance in these PEL cell lines (60). Accordingly, EBV supports KSHV persistence after human B cell infection in vivo in humanized mice (61, 62) and in vitro (63, 64) but needs to occur at nearly the same time to maintain KSHV. Probably as a result, KSHV infection is not found without EBV coinfection in African populations from Cameroon or Uganda (65, 66). In these studies, EBV infection was even identified as the most significant environmental factor for KSHV infection. Similarly, the orthologue viruses can be transmitted together in monkeys (67). Thus, EBV supports KSHV persistence.
Vice versa KSHV, however, also modifies EBV-associated lymphomagenesis and its immune control. PELs have a characteristic plasmablastic phenotype with a distinct gene expression pattern compared to solely EBV latency III carrying lymphomas (68). Accordingly, not only does KSHV coinfection increase lymphomagenesis in humanized mice, but also the resulting double-infected B and plasma cells show the characteristic gene expression pattern of PELs (61, 62) (Figure 1B). According to this plasmablastic differentiation, in vivo coinfected B cells also reactivate EBV at higher frequency into lytic infection (Figure 1B), and this can also be demonstrated in patient-derived PEL sections (61). This higher lytic EBV replication contributes to the increased lymphomagenesis of KSHV- and EBV-coinfected humanized mice, because coinfection with a lytic replication-deficient ΔBZLF1 EBV virus abolishes the increased tumor formation (61, 62). Indeed, the influence of lytic EBV replication on virus-associated tumorigenicity has also been observed in other settings with decreased lymphoma formation by ΔBZLF1 EBV infection alone or increased tumor frequency due to BZLF1 overexpression (69–72). In addition to regulating EBV-associated lymphomagenesis, KSHV coinfection also influences immune compartments (62). In both coinfected humanized mice and Kenyan children, CD56-CD16+CD38+CXCR6+ NK cells are enriched (Figure 2B). This terminal NK cell differentiation stage retains only limited abilities to expand, secrete cytokines, or kill infected cells (62), while early differentiated CD56+CD16+NKG2A+KIR- NK cells are the protective entity against lytic EBV replication and expand during infectious mononucleosis (33, 51, 73, 74). These studies suggest that KSHV influences EBV-associated lymphomagenesis by plasmablastic differentiation of coinfected B cells, associated induction of early lytic EBV replication, and compromising immune control of lytic EBV infection by causing NK cell differentiation to the less functional and weakly protective terminally differentiated CD56-negative subpopulation.
B Cell Lymphomas and HIV
PEL like most other EBV-associated lymphomas increase also during coinfection with the human immunodeficiency virus (HIV) (9, 14, 75). Primarily, loss of EBV-specific immune control upon HIV-mediated CD4+ T cell depletion and associated CD8+ T cell senescence are thought to contribute to this increase in EBV-associated malignancies. Accordingly, the frequency of EBV association is increased in Burkitt’s lymphoma, Hodgkin’s lymphoma, diffuse large B cell lymphoma, PEL, and primary CNS lymphoma in HIV-infected patients compared to patients without coinfections by HIV, KSHV, or P. falciparum (75). Accordingly, the selective loss of T cell responses to EBV antigens was found during progression to EBV-associated lymphomas in HIV-infected patients (76–78) (Figure 2C). The earlier occurrence of Burkitt’s and Hodgkin’s lymphoma and later emergence of EBV latency III lymphomas might indicate that progressive loss of T cell-mediated immune control against EBV allows more and more immunogenic virus-induced lymphomas to emerge. Only the frequencies of the latter have been significantly lowered by anti-retroviral therapies (ART) (14). Along these lines, T cell responses against the EBV latency I antigen, particularly in healthy virus carriers frequent EBNA1-specific CD4+ T cells (79, 80), seem to be lost prior to EBV-associated lymphoma development in HIV patients (76). Similarly, the protective function of CD8+ T cell-mediated immune control of EBV is lost during coinfection with HIV in humanized mice (31). While antibody-mediated CD8+ T cell depletion increased EBV viral loads in single infected humanized mice, the already increased EBV titers during HIV coinfection could not be further increased by CD8+ T cell depletion (31). Furthermore, HIV has also been described to differentiate NK cells to the less protective CD56-CD16+ phenotype (81–85) (Figure 2C). As discussed above, this terminal NK cell differentiation stage accumulates at the expense of early differentiated CD56+CD16+NKG2A+KIR- NK cells that target lytic EBV replication (51). Thus, HIV coinfection compromises EBV-specific immune control by T and NK cells, resulting in increased frequencies of EBV-associated lymphomas of all EBV latency patterns.
However, frequencies of some, mostly low immunogenic EBV-associated lymphomas remain high after CD4+ T cell count stabilization due to anti-retroviral treatment (ART) in HIV patients (14). Therefore, chronic inflammation due to HIV infection during ART or a more direct effect of HIV might continue to promote Burkitt’s and Hodgkin’s lymphoma despite restored general immune control of EBV against latency III-associated lymphomas. Along these lines, EBV-transformed B cells can be directly infected by HIV (31). During B cell infection, EBV upregulates CD4 on already CXCR4-expressing cells, and X4 tropic HIV strains can establish infection as well as reversely transcribed viral DNA integration in EBV-transformed B cells (31) (Figure 1C). Even under conditions of reduced CD8+ T cell function in HIV plus EBV-coinfected humanized mice, these double-infected B cells are efficiently cleared, presumably in part due to the upregulation of the antigen-processing machinery for MHC class I presentation in these cells (31) (Figure 1C). However, it is tempting to speculate that the HIV-induced expression of DNA-modifying enzymes, such as the APOBEC family of cytidine deaminases (86), might promote somatic mutations that are required for lymphomas with EBV latency I and II expression patterns to emerge. Thus, also HIV might contribute to EBV-associated lymphomagenesis by both immune suppression and modifying EBV-induced tumor cells directly.
Other Infections and EBV
Despite the near perfect immune control of EBV in the vast majority of asymptomatic virus carriers, it is associated with around 1% of all human tumors (8, 87). Most of these are, however, nasopharyngeal (NPC) or gastric carcinomas (GC), and not lymphomas (87). Unfortunately, it is quite unclear under which circumstances EBV establishes growth-transforming latent infection in epithelial cells, because this cannot yet be modeled in vitro and the only non-malignant EBV infection in the oropharyngeal epithelium, termed hairy leukoplakia, is caused by lytic EBV infection (88). However, it is assumed that premalignant genetic lesions that are introduced into the respective epithelial cells by carcinogenic substances in the diet and/or possible chronic inflammation in the oropharyngeal cavity or stomach provide fertile grounds for epithelial cell transformation by EBV, resulting in latency II NPC or latency I GC (89). One such coinfection that promotes premalignant genetic lesion development in the gastric epithelium could be Helicobacter pylori infection whose coinfection with EBV has been reported in GC patients (90, 91). The induced mutations are thought to allow EBV to establish latent transforming infection in epithelial cells. In addition to generating chronic inflammation, H. pylori might also directly promote EBV-driven proliferation in gastric epithelial cells by downregulating tumor suppressors (92). In contrast, coinfection of EBV with human papillomavirus (HPV) that is also associated with NPC is rare (93). Thus, in addition to P. falciparum, HIV, and KSHV, other coinfections might induce genetic lesions via chronic inflammation, particularly in epithelial cells, that are then explored by EBV during virus-associated carcinogenesis.
Conclusions and Outlook
More than 55 years after the discovery of EBV in Burkitt’s lymphoma (42, 43), it is clear that coinfections are some of the strongest environmental modulators of EBV-associated pathology. However, the mechanisms underlying these association remain largely unclear and require both clinical studies on affected patients as well as preclinical model systems to study coinfections. Along these lines, humanized mice offer the possibility to study lymphotropic coinfections such as KSHV or HIV together with EBV (31, 61, 62). In these instances, coinfections were able to regulate both lymphomagenesis in the EBV-infected B cells as well as their immune control. However, coinfections outside the hematopoietic lineage, such as for example in human hepatocytes, remain challenging (94–98). This includes P. falciparum for which both human hepatocytes and erythrocytes are required to complete its life cycle. Thus, endemic Burkitt’s lymphoma remains an enigma more than 60 years after its first description (19, 99–101). Nevertheless, the studies summarized in this review document that infections such as by the human tumor virus EBV never occur in isolation in humans. This suggests that also our preclinical animal models need to be adapted with coinfections in order to more faithfully recapitulate human physiology. Some previously established paradigms of microbiology and immunology might need to be revisited in such coinfection models.
Author Contributions
The author confirms being the sole contributor of this work and has approved it for publication.
Funding
Research in the author’s laboratory is funded by Cancer Research Switzerland (KFS-4962-02-2020), KFSP-Precision MS and HMZ ImmunoTargET of the University of Zurich, the Cancer Research Center Zurich, the Vontobel Foundation, the Sobek Foundation, the Swiss Vaccine Research Institute, Roche, Novartis, Innosuisse, and the Swiss National Science Foundation (310030_204470/1, 310030L_197952/1, and CRSII5_180323). The funders were not involved in the study design, collection, analysis, interpretation of data, the writing of this article, or the decision to submit it for publication.
Conflict of Interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Münz C. Latency and Lytic Replication in the Oncogenesis of the Epstein Barr Virus. Nat Rev Micobiol (2019) 17:691–700. doi: 10.1038/s41579-019-0249-7
2. Farrell PJ. Epstein-Barr Virus and Cancer. Annu Rev Pathol (2019) 14:29–53. doi: 10.1146/annurev-pathmechdis-012418-013023
3. Dunmire SK, Verghese PS, Balfour HH Jr. Primary Epstein-Barr Virus Infection. J Clin Virol (2018) 102:84–92. doi: 10.1016/j.jcv.2018.03.001
4. Luzuriaga K, Sullivan JL. Infectious Mononucleosis. N Engl J Med (2010) 362:1993–2000. doi: 10.1056/NEJMcp1001116
5. Kimura H, Hoshino Y, Kanegane H, Tsuge I, Okamura T, Kawa K, et al. Clinical and Virologic Characteristics of Chronic Active Epstein-Barr Virus Infection. Blood (2001) 98:280–6. doi: 10.1182/blood.V98.2.280
6. Kimura H, Okuno Y, Sato Y, Watanabe T, Murata T. Deletion of Viral microRNAs in the Oncogenesis of Epstein-Barr Virus-Associated Lymphoma. Front Microbiol (2021) 12:667968. doi: 10.3389/fmicb.2021.667968
7. Fournier B, Boutboul D, Bruneau J, Miot C, Boulanger C, Malphettes M, et al. Rapid Identification and Characterization of Infected Cells in Blood During Chronic Active Epstein-Barr Virus Infection. J Exp Med (2020) 217:e20192262. doi: 10.1084/jem.20192262
8. Shannon-Lowe C, Rickinson A. The Global Landscape of EBV-Associated Tumors. Front Oncol (2019) 9:713. doi: 10.3389/fonc.2019.00713
9. Cesarman E. Gammaherpesviruses and Lymphoproliferative Disorders. Annu Rev Pathol (2014) 9:349–72. doi: 10.1146/annurev-pathol-012513-104656
10. Kutok JL, Wang F. Spectrum of Epstein-Barr Virus-Associated Diseases. Annu Rev Pathol (2006) 1:375–404. doi: 10.1146/annurev.pathol.1.110304.100209
11. Tugizov SM, Herrera R, Palefsky JM. Epstein-Barr Virus Transcytosis Through Polarized Oral Epithelial Cells. J Virol (2013) 87:8179–94. doi: 10.1128/JVI.00443-13
12. Babcock JG, Hochberg D, Thorley-Lawson AD. The Expression Pattern of Epstein-Barr Virus Latent Genes In Vivo Is Dependent Upon the Differentiation Stage of the Infected B Cell. Immunity (2000) 13:497–506. doi: 10.1016/S1074-7613(00)00049-2
13. Hancock MH, Skalsky RL. Roles of Non-Coding RNAs During Herpesvirus Infection. Curr Top Microbiol Immunol (2018) 419:243–80. doi: 10.1007/82_2017_31
14. Totonchy J, Cesarman E. Does Persistent HIV Replication Explain Continued Lymphoma Incidence in the Era of Effective Antiretroviral Therapy? Curr Opin Virol (2016) 20:71–7. doi: 10.1016/j.coviro.2016.09.001
15. Thorley-Lawson DA. Epstein-Barr Virus: Exploiting the Immune System. Nat Rev Immunol (2001) 1:75–82. doi: 10.1038/35095584
16. Babcock GJ, Decker LL, Volk M, Thorley-Lawson DA. EBV Persistence in Memory B Cells In Vivo. Immunity (1998) 9:395–404. doi: 10.1016/S1074-7613(00)80622-6
17. Murer A, McHugh D, Caduff N, Kalchschmidt JS, Barros MH, Zbinden A, et al. EBV Persistence Without its EBNA3A and 3C Oncogenes In Vivo. PloS Pathog (2018) 14:e1007039. doi: 10.1371/journal.ppat.1007039
18. Hochberg D, Middeldorp JM, Catalina M, Sullivan JL, Luzuriaga K, Thorley-Lawson DA. Demonstration of the Burkitt’s Lymphoma Epstein-Barr Virus Phenotype in Dividing Latently Infected Memory Cells In Vivo. Proc Natl Acad Sci USA (2004) 101:239–44. doi: 10.1073/pnas.2237267100
19. Thorley-Lawson DA, Allday MJ. The Curious Case of the Tumour Virus: 50 Years of Burkitt’s Lymphoma. Nat Rev Microbiol (2008) 6:913–24. doi: 10.1038/nrmicro2015
20. Laichalk LL, Thorley-Lawson DA. Terminal Differentiation Into Plasma Cells Initiates the Replicative Cycle of Epstein-Barr Virus In Vivo. J Virol (2005) 79:1296–307. doi: 10.1128/JVI.79.2.1296-1307.2005
21. Tugizov SM, Berline JW, Palefsky JM. Epstein-Barr Virus Infection of Polarized Tongue and Nasopharyngeal Epithelial Cells. Nat Med (2003) 9:307–14. doi: 10.1038/nm830
22. Damania B, Münz C. Immunodeficiencies That Predispose to Pathologies by Human Oncogenic Gamma-Herpesviruses. FEMS Microbiol Rev (2019) 43:181–92. doi: 10.1093/femsre/fuy044
23. Latour S, Fischer A. Signaling Pathways Involved in the T-Cell-Mediated Immunity Against Epstein-Barr Virus: Lessons From Genetic Diseases. Immunol Rev (2019) 291:174–89. doi: 10.1111/imr.12791
24. Tangye SG, Latour S. Primary Immunodeficiencies Reveal the Molecular Requirements for Effective Host Defense Against EBV Infection. Blood (2020) 135:644–55. doi: 10.1182/blood.2019000928
25. Fournier B, Latour S. Immunity to EBV as Revealed by Immunedeficiencies. Curr Opin Immunol (2021) 72:107–15. doi: 10.1016/j.coi.2021.04.003
26. Ghosh S, Kostel Bal S, Edwards ESJ, Pillay B, Jimenez-Heredia R, Rao G, et al. Extended Clinical and Immunological Phenotype and Transplant Outcome in CD27 and CD70 Deficiency. Blood (2020) 136:2638–55. doi: 10.1182/blood.2020006738
27. Deng Y, Chatterjee B, Zens K, Zdimerova H, Müller A, Schuhmachers P, et al. CD27 Is Required for Protective Lytic EBV Antigen Specific CD8+ T Cell Expansion. Blood (2021) 137:3225–36. doi: 10.1182/blood.2020009482
28. Strowig T, Gurer C, Ploss A, Liu YF, Arrey F, Sashihara J, et al. Priming of Protective T Cell Responses Against Virus-Induced Tumors in Mice With Human Immune System Components. J Exp Med (2009) 206:1423–34. doi: 10.1084/jem.20081720
29. Chijioke O, Marcenaro E, Moretta A, Capaul R, Münz C. The SAP-Dependent 2B4 Receptor Mediates CD8+ T Cell Dependent Immune Control of Epstein Barr Virus Infection in Mice With Reconstituted Human Immune System Components. J Infect Dis (2015) 212:803–7. doi: 10.1093/infdis/jiv114
30. Murer A, Ruhl J, Zbinden A, Capaul R, Hammerschmidt W, Chijioke O, et al. MicroRNAs of Epstein-Barr Virus Attenuate T-Cell-Mediated Immune Control In Vivo. MBio (2019) 10:e01941–18. doi: 10.1128/mBio.01941-18
31. McHugh D, Myburgh R, Caduff N, Spohn M, Kok YL, Keller CW, et al. EBV Renders B Cells Susceptible to HIV-1 in Humanized Mice. Life Sci Alliance (2020) 3:e202000640. doi: 10.26508/lsa.202000640
32. Caduff N, McHugh D, Murer A, Ramer P, Raykova A, Landtwing V, et al. Immunosuppressive FK506 Treatment Leads to More Frequent EBV-Associated Lymphoproliferative Disease in Humanized Mice. PloS Pathog (2020) 16:e1008477. doi: 10.1371/journal.ppat.1008477
33. Chijioke O, Muller A, Feederle R, Barros MH, Krieg C, Emmel V, et al. Human Natural Killer Cells Prevent Infectious Mononucleosis Features by Targeting Lytic Epstein-Barr Virus Infection. Cell Rep (2013) 5:1489–98. doi: 10.1016/j.celrep.2013.11.041
34. Landtwing V, Raykova A, Pezzino G, Beziat V, Marcenaro E, Graf C, et al. Cognate HLA Absence in Trans Diminishes Human NK Cell Education. J Clin Invest (2016) 126:3772–82. doi: 10.1172/JCI86923
35. Rochford R, Cannon MJ, Moormann AM. Endemic Burkitt’s Lymphoma: A Polymicrobial Disease? Nat Rev Microbiol (2005) 3:182–7. doi: 10.1038/nrmicro1089
36. Quintana MDP, Smith-Togobo C, Moormann A, Hviid L. Endemic Burkitt Lymphoma - an Aggressive Childhood Cancer Linked to Plasmodium Falciparum Exposure, But Not to Exposure to Other Malaria Parasites. APMIS (2020) 128:129–35. doi: 10.1111/apm.13018
37. Morrow RH Jr. Epidemiological Evidence for the Role of Falciparum Malaria in the Pathogenesis of Burkitt’s Lymphoma. IARC Sci Publ (1985), 177–86.
38. Ramiro AR, Jankovic M, Eisenreich T, Difilippantonio S, Chen-Kiang S, Muramatsu M, et al. AID Is Required for C-Myc/IgH Chromosome Translocations In Vivo. Cell (2004) 118:431–8. doi: 10.1016/j.cell.2004.08.006
39. Robbiani DF, Deroubaix S, Feldhahn N, Oliveira TY, Callen E, Wang Q, et al. Plasmodium Infection Promotes Genomic Instability and AID-Dependent B Cell Lymphoma. Cell (2015) 162:727–37. doi: 10.1016/j.cell.2015.07.019
40. Torgbor C, Awuah P, Deitsch K, Kalantari P, Duca KA, Thorley-Lawson DA. A Multifactorial Role for P. Falciparum Malaria in Endemic Burkitt’s Lymphoma Pathogenesis. PloS Pathog (2014) 10:e1004170. doi: 10.1371/journal.ppat.1004170
41. Kalchschmidt JS, Bashford-Rogers R, Paschos K, Gillman AC, Styles CT, Kellam P, et al. Epstein-Barr Virus Nuclear Protein EBNA3C Directly Induces Expression of AID and Somatic Mutations in B Cells. J Exp Med (2016) 213:921–8. doi: 10.1084/jem.20160120
42. Epstein MA, Achong BG, Barr YM. Virus Particles in Cultured Lymphoblasts From Burkitt’s Lymphoma. Lancet (1964) 1:702–3. doi: 10.1016/S0140-6736(64)91524-7
43. Epstein MA, Henle G, Achong BG, Barr YM. Morphological and Biological Studies on a Virus in Cultured Lymphoblasts From Burkitt’s Lymphoma. J Exp Med (1964) 121:761–70. doi: 10.1084/jem.121.5.761
44. Moss DJ, Burrows SR, Castelino DJ, Kane RG, Pope JH, Rickinson AB, et al. A Comparison of Epstein-Barr Virus-Specific T-Cell Immunity in Malaria-Endemic and -Nonendemic Regions of Papua New Guinea. Int J Cancer (1983) 31:727–32. doi: 10.1002/ijc.2910310609
45. Whittle HC, Brown J, Marsh K, Greenwood BM, Seidelin P, Tighe H, et al. T-Cell Control of Epstein-Barr Virus-Infected B Cells Is Lost During P. Falciparum Malaria. Nature (1984) 312:449–50. doi: 10.1038/312449a0
46. Moormann AM, Chelimo K, Sumba PO, Tisch DJ, Rochford R, Kazura JW. Exposure to Holoendemic Malaria Results in Suppression of Epstein-Barr Virus-Specific T Cell Immunosurveillance in Kenyan Children. J Infect Dis (2007) 195:799–808. doi: 10.1086/511984
47. Moormann AM, Heller KN, Chelimo K, Embury P, Ploutz-Snyder R, Otieno JA, et al. Children With Endemic Burkitt Lymphoma Are Deficient in EBNA1-Specific IFN-Gamma T Cell Responses. Int J Cancer (2009) 124:1721–6. doi: 10.1002/ijc.24014
48. Forconi CS, Cosgrove CP, Saikumar-Lakshmi P, Nixon CE, Foley J, Ong’echa JM, et al. Poorly Cytotoxic Terminally Differentiated CD56negCD16pos NK Cells Accumulate in Kenyan Children With Burkitt Lymphomas. Blood Adv (2018) 2:1101–14. doi: 10.1182/bloodadvances.2017015404
49. Forconi CS, Oduor CI, Oluoch PO, Ong’echa JM, Munz C, Bailey JA, et al. A New Hope for CD56negCD16pos NK Cells as Unconventional Cytotoxic Mediators: An Adaptation to Chronic Diseases. Front Cell Infect Microbiol (2020) 10:162. doi: 10.3389/fcimb.2020.00162
50. Pappworth IY, Wang EC, Rowe M. The Switch From Latent to Productive Infection in Epstein-Barr Virus-Infected B Cells Is Associated With Sensitization to NK Cell Killing. J Virol (2007) 81:474–82. doi: 10.1128/JVI.01777-06
51. Azzi T, Lunemann A, Murer A, Ueda S, Beziat V, Malmberg KJ, et al. Role for Early-Differentiated Natural Killer Cells in Infectious Mononucleosis. Blood (2014) 124:2533–43. doi: 10.1182/blood-2014-01-553024
52. Muriuki BM, Forconi CS, Oluoch PO, Bailey JA, Ghansah A, Moormann AM, et al. Association of Killer Cell Immunoglobulin-Like Receptors With Endemic Burkitt Lymphoma in Kenyan Children. Sci Rep (2021) 11:11343. doi: 10.1038/s41598-021-90596-7
53. Moormann AM, Chelimo K, Sumba OP, Lutzke ML, Ploutz-Snyder R, Newton D, et al. Exposure to Holoendemic Malaria Results in Elevated Epstein-Barr Virus Loads in Children. J Infect Dis (2005) 191:1233–8. doi: 10.1086/428910
54. Langhorne J, Cross C, Seixas E, Li C, von der Weid T. A Role for B Cells in the Development of T Cell Helper Function in a Malaria Infection in Mice. Proc Natl Acad Sci USA (1998) 95:1730–4. doi: 10.1073/pnas.95.4.1730
55. Redmond LS, Ogwang MD, Kerchan P, Reynolds SJ, Tenge CN, Were PA, et al. Endemic Burkitt Lymphoma: A Complication of Asymptomatic Malaria in Sub-Saharan Africa Based on Published Literature and Primary Data From Uganda, Tanzania, and Kenya. Malar J (2020) 19:239. doi: 10.1186/s12936-020-03312-7
56. Cesarman E, Damania B, Krown SE, Martin J, Bower M, Whitby D. Kaposi Sarcoma. Nat Rev Dis Primers (2019) 5:9. doi: 10.1038/s41572-019-0060-9
57. Mariggio G, Koch S, Schulz TF. Kaposi Sarcoma Herpesvirus Pathogenesis. Philos Trans R Soc Lond B Biol Sci (2017) 372:20160275. doi: 10.1098/rstb.2016.0275
58. Cesarman E, Chang Y, Moore PS, Said JW, Knowles DM. Kaposi’s Sarcoma-Associated Herpesvirus-Like DNA Sequences in AIDS-Related Body-Cavity-Based Lymphomas. N Engl J Med (1995) 332:1186–91. doi: 10.1056/NEJM199505043321802
59. Cesarman E, Moore PS, Rao PH, Inghirami G, Knowles DM, Chang Y. In Vitro Establishment and Characterization of Two Acquired Immunodeficiency Syndrome-Related Lymphoma Cell Lines (BC-1 and BC-2) Containing Kaposi’s Sarcoma-Associated Herpesvirus-Like (KSHV) DNA Sequences. Blood (1995) 86:2708–14. doi: 10.1182/blood.V86.7.2708.2708
60. Bigi R, Landis JT, An H, Caro-Vegas C, Raab-Traub N, Dittmer DP. Epstein-Barr Virus Enhances Genome Maintenance of Kaposi Sarcoma-Associated Herpesvirus. Proc Natl Acad Sci USA (2018) 115:E11379–87. doi: 10.1073/pnas.1810128115
61. McHugh D, Caduff N, Barros MHM, Rämer P, Raykova A, Murer A, et al. Persistent KSHV Infection Increases EBV-Associated Tumor Formation In Vivo via Enhanced EBV Lytic Gene Expression. Cell Host Microbe (2017) 22:61–73. doi: 10.1016/j.chom.2017.06.009
62. Caduff N, McHugh D, Rieble L, Forconi CS, Ong’echa JM, Oluoch PO, et al. KSHV Infection Drives Poorly Cytotoxic CD56 Negative Natural Killer Cell Differentiation In Vivo Upon KSHV/EBV Dual Infection. Cell Rep (2021) 35:109056. doi: 10.1016/j.celrep.2021.109056
63. Faure A, Hayes M, Sugden B. How Kaposi’s Sarcoma-Associated Herpesvirus Stably Transforms Peripheral B Cells Towards Lymphomagenesis. Proc Natl Acad Sci USA (2019) 116:16519–28. doi: 10.1073/pnas.1905025116
64. Blackbourn DJ, Lennette E, Klencke B, Moses A, Chandran B, Weinstein M, et al. The Restricted Cellular Host Range of Human Herpesvirus 8. Aids (2000) 14:1123–33. doi: 10.1097/00002030-200006160-00009
65. Labo N, Marshall V, Miley W, Davis E, McCann B, Stolka KB, et al. Mutual Detection of Kaposi’s Sarcoma-Associated Herpesvirus and Epstein-Barr Virus in Blood and Saliva of Cameroonians With and Without Kaposi’s Sarcoma. Int J Cancer (2019) 145:2468–77. doi: 10.1002/ijc.32546
66. Sallah N, Miley W, Labo N, Carstensen T, Fatumo S, Gurdasani D, et al. Distinct Genetic Architectures and Environmental Factors Associate With Host Response to the Gamma2-Herpesvirus Infections. Nat Commun (2020) 11:3849. doi: 10.1038/s41467-020-17696-2
67. Bruce AG, Barcy S, Staheli J, Bielefeldt-Ohmann H, Ikoma M, Howard K, et al. Experimental Co-Transmission of Simian Immunodeficiency Virus (SIV) and the Macaque Homologs of the Kaposi Sarcoma-Associated Herpesvirus (KSHV) and Epstein-Barr Virus (EBV). PloS One (2018) 13:e0205632. doi: 10.1371/journal.pone.0205632
68. Klein U, Gloghini A, Gaidano G, Chadburn A, Cesarman E, Dalla-Favera R, et al. Gene Expression Profile Analysis of AIDS-Related Primary Effusion Lymphoma (PEL) Suggests a Plasmablastic Derivation and Identifies PEL-Specific Transcripts. Blood (2003) 101:4115–21. doi: 10.1182/blood-2002-10-3090
69. Ma SD, Hegde S, Young KH, Sullivan R, Rajesh D, Zhou Y, et al. A New Model of Epstein-Barr Virus Infection Reveals an Important Role for Early Lytic Viral Protein Expression in the Development of Lymphomas. J Virol (2011) 85:165–77. doi: 10.1128/JVI.01512-10
70. Antsiferova O, Müller A, Rämer P, Chijioke O, Chatterjee B, Raykova A, et al. Adoptive Transfer of EBV Specific CD8+ T Cell Clones can Transiently Control EBV Infection in Humanized Mice. PloS Pathog (2014) 10:e1004333. doi: 10.1371/journal.ppat.1004333
71. Ma SD, Yu X, Mertz JE, Gumperz JE, Reinheim E, Zhou Y, et al. An Epstein-Barr Virus (EBV) Mutant With Enhanced BZLF1 Expression Causes Lymphomas With Abortive Lytic EBV Infection in a Humanized Mouse Model. J Virol (2012) 86:7976–87. doi: 10.1128/JVI.00770-12
72. Bristol JA, Djavadian R, Albright ER, Coleman CB, Ohashi M, Hayes M, et al. A Cancer-Associated Epstein-Barr Virus BZLF1 Promoter Variant Enhances Lytic Infection. PloS Pathog (2018) 14:e1007179. doi: 10.1371/journal.ppat.1007179
73. Lam JKP, Azzi T, Hui KF, Wong AMG, McHugh D, Caduff N, et al. Co-Infection of Cytomegalovirus and Epstein-Barr Virus Diminishes the Frequency of CD56dimNKG2A+KIR- NK Cells and Contributes to Suboptimal Control of EBV in Immunosuppressed Children With Post-Transplant Lymphoproliferative Disorder. Front Immunol (2020) 11:1231. doi: 10.3389/fimmu.2020.01231
74. Panisova E, Lunemann A, Burgler S, Kotur M, Lazarovici J, Danu A, et al. Reduced Frequency of Cytotoxic CD56dim CD16+ NK Cells Leads to Impaired Antibody-Dependent Degranulation in EBV-Positive Classical Hodgkin Lymphoma. Cancer Immunol Immunother (2021). doi: 10.1007/s00262-021-02956-x
75. Shindiapina P, Ahmed EH, Mozhenkova A, Abebe T, Baiocchi RA. Immunology of EBV-Related Lymphoproliferative Disease in HIV-Positive Individuals. Front Oncol (2020) 10:1723. doi: 10.3389/fonc.2020.01723
76. Piriou E, van Dort K, Nanlohy NM, van Oers MH, Miedema F, van Baarle D. Loss of EBNA1-Specific Memory CD4+ and CD8+ T Cells in HIV-Infected Patients Progressing to AIDS-Related non-Hodgkin Lymphoma. Blood (2005) 106:3166–74. doi: 10.1182/blood-2005-01-0432
77. Hernandez DM, Valderrama S, Gualtero S, Hernandez C, Lopez M, Herrera MV, et al. Loss of T-Cell Multifunctionality and TCR-Vbeta Repertoire Against Epstein-Barr Virus Is Associated With Worse Prognosis and Clinical Parameters in HIV+ Patients. Front Immunol (2018) 9:2291. doi: 10.3389/fimmu.2018.02291
78. Gasser O, Bihl FK, Wolbers M, Loggi E, Steffen I, Hirsch HH, et al. HIV Patients Developing Primary CNS Lymphoma Lack EBV-Specific CD4+ T Cell Function Irrespective of Absolute CD4+ T Cell Counts. PloS Med (2007) 4:e96. doi: 10.1371/journal.pmed.0040096
79. Münz C, Bickham KL, Subklewe M, Tsang ML, Chahroudi A, Kurilla MG, et al. Human CD4+ T Lymphocytes Consistently Respond to the Latent Epstein-Barr Virus Nuclear Antigen EBNA1. J Exp Med (2000) 191:1649–60. doi: 10.1084/jem.191.10.1649
80. Leen A, Meij P, Redchenko I, Middeldorp J, Bloemena E, Rickinson A, et al. Differential Immunogenicity of Epstein-Barr Virus Latent-Cycle Proteins for Human CD4+ T-Helper 1 Responses. J Virol (2001) 75:8649–59. doi: 10.1128/JVI.75.18.8649-8659.2001
81. Mavilio D, Lombardo G, Benjamin J, Kim D, Follman D, Marcenaro E, et al. Characterization of CD56-/CD16+ Natural Killer (NK) Cells: A Highly Dysfunctional NK Subset Expanded in HIV-Infected Viremic Individuals. Proc Natl Acad Sci USA (2005) 102:2886–91. doi: 10.1073/pnas.0409872102
82. Alter G, Teigen N, Davis BT, Addo MM, Suscovich TJ, Waring MT, et al. Sequential Deregulation of NK Cell Subset Distribution and Function Starting in Acute HIV-1 Infection. Blood (2005) 106:3366–9. doi: 10.1182/blood-2005-03-1100
83. Bjorkstrom NK, Ljunggren HG, Sandberg JK. CD56 Negative NK Cells: Origin, Function, and Role in Chronic Viral Disease. Trends Immunol (2010) 31:401–6. doi: 10.1016/j.it.2010.08.003
84. Milush JM, Lopez-Verges S, York VA, Deeks SG, Martin JN, Hecht FM, et al. CD56negCD16+ NK Cells Are Activated Mature NK Cells With Impaired Effector Function During HIV-1 Infection. Retrovirology (2013) 10:158. doi: 10.1186/1742-4690-10-158
85. Voigt J, Malone DFG, Dias J, Leeansyah E, Bjorkstrom NK, Ljunggren HG, et al. Proteome Analysis of Human CD56neg NK Cells Reveals a Homogeneous Phenotype Surprisingly Similar to CD56dim NK Cells. Eur J Immunol (2018) 48:1456–69. doi: 10.1002/eji.201747450
86. Venkatesan S, Rosenthal R, Kanu N, McGranahan N, Bartek J, Quezada SA, et al. Perspective: APOBEC Mutagenesis in Drug Resistance and Immune Escape in HIV and Cancer Evolution. Ann Oncol (2018) 29:563–72. doi: 10.1093/annonc/mdy003
87. Cohen JI, Fauci AS, Varmus H, Nabel GJ. Epstein-Barr Virus: An Important Vaccine Target for Cancer Prevention. Sci Transl Med (2011) 3:107fs7. doi: 10.1126/scitranslmed.3002878
88. Greenspan JS, Greenspan D, Webster-Cyriaque J. Hairy Leukoplakia; Lessons Learned: 30-Plus Years. Oral Dis (2016) 22(Suppl 1):120–7. doi: 10.1111/odi.12393
89. Wong KCW, Hui EP, Lo KW, Lam WKJ, Johnson D, Li L, et al. Nasopharyngeal Carcinoma: An Evolving Paradigm. Nat Rev Clin Oncol (2021) 18(11):679–95. doi: 10.1038/s41571-021-00524-x
90. Davila-Collado R, Jarquin-Duran O, Dong LT, Espinoza JL. Epstein-Barr Virus and Helicobacter Pylori Co-Infection in Non-Malignant Gastroduodenal Disorders. Pathogens (2020) 9:104. doi: 10.3390/pathogens9020104
91. de Souza CRT, Almeida MCA, Khayat AS, da Silva EL, Soares PC, Chaves LC, et al. Association Between Helicobacter Pylori, Epstein-Barr Virus, Human Papillomavirus and Gastric Adenocarcinomas. World J Gastroenterol (2018) 24:4928–38. doi: 10.3748/wjg.v24.i43.4928
92. Pandey S, Jha HC, Shukla SK, Shirley MK, Robertson ES. Epigenetic Regulation of Tumor Suppressors by Helicobacter Pylori Enhances EBV-Induced Proliferation of Gastric Epithelial Cells. mBio (2018) 9:e00649–18. doi: 10.1128/mBio.00649-18
93. Tham T, Machado R, Russo DP, Herman SW, Teegala S, Costantino P. Viral Markers in Nasopharyngeal Carcinoma: A Systematic Review and Meta-Analysis on the Detection of P16(INK4a), Human Papillomavirus (HPV), and Ebstein-Barr Virus (EBV). Am J Otolaryngol (2021) 42:102762. doi: 10.1016/j.amjoto.2020.102762
94. Washburn ML, Bility MT, Zhang L, Kovalev GI, Buntzman A, Frelinger JA, et al. A Humanized Mouse Model to Study Hepatitis C Virus Infection, Immune Response, and Liver Disease. Gastroenterology (2011) 140:1334–44. doi: 10.1053/j.gastro.2011.01.001
95. Kremsdorf D, Strick-Marchand H. Modeling Hepatitis Virus Infections and Treatment Strategies in Humanized Mice. Curr Opin Virol (2017) 25:119–25. doi: 10.1016/j.coviro.2017.07.029
96. Vaughan AM, Pinapati RS, Cheeseman IH, Camargo N, Fishbaugher M, Checkley LA, et al. Plasmodium Falciparum Genetic Crosses in a Humanized Mouse Model. Nat Methods (2015) 12:631–3. doi: 10.1038/nmeth.3432
97. Schafer C, Roobsoong W, Kangwanrangsan N, Bardelli M, Rawlinson TA, Dambrauskas N, et al. A Humanized Mouse Model for Plasmodium Vivax to Test Interventions That Block Liver Stage to Blood Stage Transition and Blood Stage Infection. iScience (2020) 23:101381. doi: 10.1016/j.isci.2020.101381
98. Minkah NK, Schafer C, Kappe SHI. Humanized Mouse Models for the Study of Human Malaria Parasite Biology, Pathogenesis, and Immunity. Front Immunol (2018) 9:807. doi: 10.3389/fimmu.2018.00807
99. Burkitt D. A Sarcoma Involving the Jaws in African Children. Br J Surg (1958) 46:218–23. doi: 10.1002/bjs.18004619704
100. Burkitt D. A Children’s Cancer Dependent on Climatic Factors. Nature (1962) 194:232–4. doi: 10.1038/194232a0
Keywords: Burkitt’s lymphoma, primary effusion lymphoma, cytotoxic lymphocytes, humanized mice, diffuse large B cell lymphoma, T cells, NK cells
Citation: Münz C (2021) Modification of EBV-Associated Pathologies and Immune Control by Coinfections. Front. Oncol. 11:756480. doi: 10.3389/fonc.2021.756480
Received: 10 August 2021; Accepted: 08 October 2021;
Published: 28 October 2021.
Edited by:
Paola Chabay, CONICET Instituto Multidisciplinario de Investigación en Patologías Pediátricas (IMIPP), ArgentinaReviewed by:
Elisabeth Littwitz-Salomon, University Hospital Essen, GermanyRosemary Rochford, University of Colorado, United States
Claire Shannon-Lowe, University of Birmingham, United Kingdom
Copyright © 2021 Münz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christian Münz, Y2hyaXN0aWFuLm11ZW56QHV6aC5jaA==