 Diletta Fontana
Diletta Fontana Carlo Gambacorti-Passerini
Carlo Gambacorti-Passerini Rocco Piazza
Rocco Piazza
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 11 November 2021
Sec. Hematologic Malignancies
Volume 11 - 2021 | https://doi.org/10.3389/fonc.2021.756348
This article is part of the Research Topic Hybrid or Mixed Myelodyplastic / Myeloproliferative Disorders: Current Trends in Diagnosis and Treatment View all 10 articles
Atypical chronic myeloid leukemia is a rare disease whose pathogenesis has long been debated. It currently belongs to the group of myelodysplastic/myeloproliferative disorders. In this review, an overview on the current knowledge about diagnosis, prognosis, and genetics is presented, with a major focus on the recent molecular findings. We describe here the molecular pathogenesis of the disease, focusing on the mechanisms of action of the main mutations as well as on gene expression profiling. We also present the treatment options focusing on emerging targeted therapies.
Atypical chronic myeloid leukemia (aCML), BCR-ABL1-negative, is a rare hematological malignancy belonging to the overlap category of myelodysplastic/myeloproliferative neoplasms (MDS/MPN) of the WHO classification of myeloid neoplasms (1). The MDS/MPN category was introduced in the third edition of the WHO Classification of Tumors (2) and includes myeloid neoplasms that exhibit, at presentation, laboratory, clinical, and morphologic features that overlap between myelodysplastic syndromes (MDS) and myeloproliferative neoplasms (MPN) (3).
Initially, aCML was described as an atypical form of chronic myeloid leukemia (CML) BCR-ABL1-positive, since at the onset it presents with many features of classical CML (4–6). In 2008, in order to incorporate new laboratorial as well as clinical information to refine diagnostic criteria for previously described neoplasms and to introduce newly recognized disease entities, the WHO classification was updated and published as part of the fourth edition of the WHO monograph series (7, 8). In this classification, aCML has been modified to aCML BCR-ABL1-negative to highlight that it was not merely a variant of classical CML (9). Finally, in 2016, after 8 years since the previous revision and owing to the massive amount of information generated by the new sequencing technologies, a revision of the 2008 WHO classification of hematopoietic neoplasms (1) was deemed necessary.
aCML is a disorder of the elderly, since it typically affects patients with an age ranging between 60 and 76 years (10). Even though the first reports about aCML suggested a female predominance (11) or no sex predominance (12), in more recent years, reports analyzing larger cohorts of patients suggest a slight male predominance (13–15), though the biological reasons underlying aCML gender preference are poorly understood. Its estimated incidence is 1 out of 100 cases of t(9;22), BCR-ABL1-positive CML, meaning approximately 1 case per 1,000,000 persons per year (4, 11, 16, 17).
From a diagnostic point of view, aCML is a challenging myeloid malignancy characterized by features belonging to both myeloproliferative and myelodysplastic syndromes. As its clinical features may overlap with other myeloid malignancies, its diagnosis still relies primarily on morphological criteria, e.g., the evidence of dysgranulopoiesis in bone marrow or peripheral blood. Typically, aCML patients present clinical features similar to BCR-ABL1-positive CML including splenomegaly and a neutrophilic leukocytosis; they also show prominent granulocytic dysplasia (e.g., hypogranular and hypolobated neutrophils, abnormal chromatin clumping, and pseudo-Pelger–Huet neutrophils) (9). The white blood cell (WBC) count is >13 × 109/L, with ≥10% of immature granulocytes and <20% blasts in the peripheral blood (PB) and in the bone marrow (BM) (1, 9, 18). Due to the high WBC count, it is not uncommon to observe a monocyte count >1 × 109/L, but the percentage of monocytes at onset is always lower than 10% of the total leukocytes, which is critical to discriminate between aCML and a closely related MDS/MPN disorder, known as chronic myelomonocytic leukemia (CMML). In contrast to CML, basophilia is not prominent as basophils represent <2% of all PB white cells (9, 19). A hypercellular BM with myeloid hyperplasia and prominent granulocytic dysplasia is a consistent feature; however, trilineage dysplasia may be present (3, 20, 21). The leukocyte alkaline phosphatase level may be low, normal, or increased, therefore lacking diagnostic utility (9, 19). The recent discovery of somatic mutations occurring in SETBP1 (22) and ETNK1 (23) in up to a third of aCML cases (22–24) led to the insertion of these mutations in the 2016 WHO revision as supporting criteria for the diagnosis of aCML (1). However, the presence of these mutated genes does not ensure a certain diagnosis (see below and Table 1).
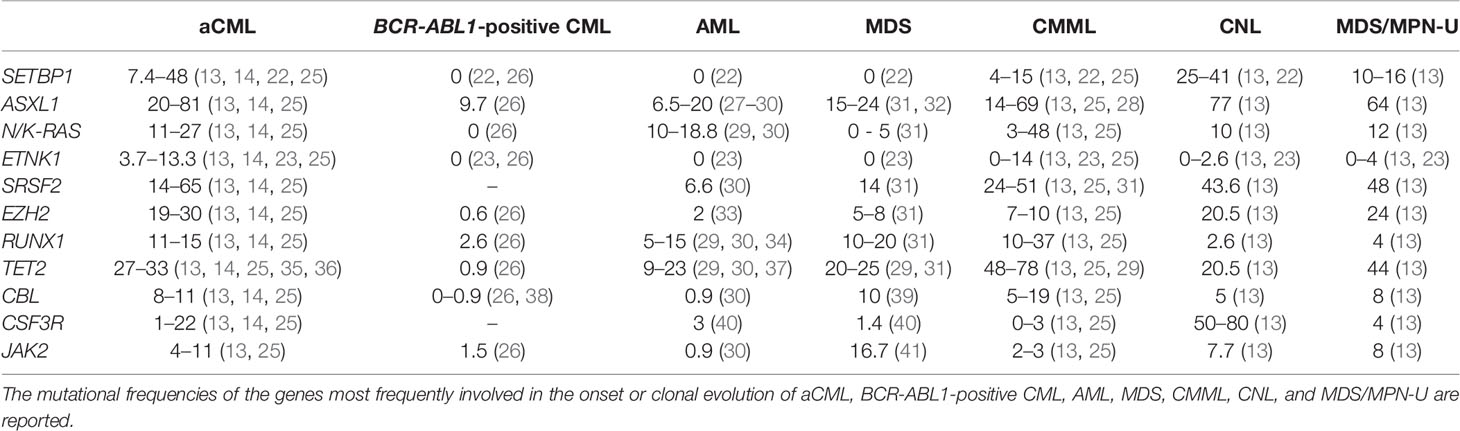
Table 1 Mutational frequencies.
The differential diagnosis of aCML includes BCR-ABL1-positive CML, CMML, chronic neutrophilic leukemia (CNL), and prefibrotic primary myelofibrosis (pre-PMF) (Table 2). Besides the lack of the Philadelphia chromosome and the BCR-ABL1 translocation, the major criteria that distinguish aCML from BCR-ABL1-positive CML are the dysgranulopoiesis, which is common in aCML but only occasionally reported in classical CML, and the normal basophil counts of the former (<2% of leukocytes) (1).

Table 2 Diagnostic criteria for BCR-ABL1-positive CML, CMML, CNL, and pre-PMF as defined by the WHO in the 2016 revision are listed.
The differential diagnosis between aCML and CNL was traditionally difficult since the proportion of immature myeloid cells (promyelocytes, myelocytes, and metamyelocytes) in PB (≥10% in aCML and <10% in CNL) and the presence of dysplasia were the only distinctive features (1, 17). Currently, the distinction between the two disorders is also supported by the high frequency of CSF3R mutations (42) in the latter and by the higher frequency of SETBP1 and ETNK1 mutations in the former. However, it is important to note that none of these variants are fully restricted to one of the two disorders (43), which suggests that they represent a continuum of related diseases rather than truly distinct entities.
On the other hand, CMML can be ruled out by the lack of monocytosis (1, 3), since in CMML, monocyte count must exceed 10% of the total leukocytes. Although effective, the not uncommon detection of borderline monocyte fractions at the onset occasionally renders the application of this hard threshold troublesome.
Finally, aCML diagnosis must not meet the WHO criteria for primary myelofibrosis (PMF), polycythemia vera (PV), or essential thrombocythemia (ET). In this context, the most challenging differential diagnosis is between aCML and pre-PMF, where the lack of an overt fibrosis and the common presence of myeloid leukocytosis with immature myeloid cells in PB of pre-PMF patients may render the differential diagnosis challenging. Luckily, the availability of the three myeloproliferative gene markers, JAK2, CALR, and MPL, allows to quickly discriminate between the two conditions. Conversely, the distinction between aCML and those rare myeloproliferative cases lacking all the three markers (i.e., the so-called triple-negative myeloproliferative disorders) is currently much more blurred.
aCML is an aggressive disease generally associated with poor outcome, with a median overall survival (OS) of 11–25 months and a 5-year OS of 25% (4, 11, 18, 19, 43–47). Fifteen percent to 40% of aCML patients progress to secondary acute myeloid leukemia (sAML), with a median time to leukemic evolution of 11.2 months (11); the remaining patients usually develop complications related to BM failure. Unfavorable prognostic factors for OS are an increased WBC count (>50 × 109/L) at presentation, increased immature precursors in the PB, age greater than 65 years at onset, hemoglobin (Hb) <10 g/dl, leukocyte count ≥50 × 109/L, and immature circulating precursors (10, 11, 15, 43, 47). In addition, mutations in ASXL1 and SETBP1 have been associated with a more aggressive disease (10, 48), although their prognostic impact is still unclear (14). The risk of progression to sAML seems to be higher in case of palpable hepato- or splenomegaly, monocytosis, BM blastosis >5%, marked dyserythropoiesis, and transfusional requirement (11). In a recent work, stratification based on RNA-sequencing data identified two populations in terms of overall survival, and the overexpression of DNPH1, GFI1B, and PARP1 genes has been correlated with poor prognosis (14).
The molecular features of aCML include an increased frequency of karyotypic abnormalities. In up to 80% of patients with aCML, additional chromosomal abnormalities (ACA) such as trisomy 8 or 9, del(20q), and −7/7q or isochromosomes 17q are the most common ones (4, 10, 11, 24, 49), but also aberrations involving chromosomes 12, 13, 14, 19, and 21 are reported (19, 44, 46, 50). Interestingly, trisomy 8, isochromosome 17q, and trisomy 19 are the most frequent anomalies observed in BCR-ABL1-positive CML, which are associated with blast crisis transformation (51–53).
The most frequent somatic mutations involve SETBP1, ASXL1, NRAS, KRAS, ETNK1, SRSF2, EZH2, RUNX1, and TET2, while mutations in CBL, CSF3R, and JAK2 are less frequent (10, 18, 20, 22–24, 43, 54–60). Even though mutations occurring in SETBP1 and ETNK1 are not univocally disease-specific, they represent the alterations most closely associated with aCML (1, 22–24, 58). SETBP1 and ASXL1 are considered as high-risk mutations (43, 54), while TET2 (61), CBL (62), and EZH2 (63) mutations may suggest a possible overlap between aCML and CMML at the molecular level. On the other hand, the absence of rearrangements involving PDGFRA or PDGFRB (3) and FGFR1 (1) and the negativity for JAK2 V617F mutation (64) all support a diagnosis of aCML. The mutation frequency of the main aCML oncogenes, compared with BCR-ABL1-positive CML, AML, MDS, CMML, CNL, and MDS/MPN-U, is reported in Table 1.
The molecular lesions responsible for the onset and progression of aCML were unknown until 2013, when, by applying NGS techniques, the presence of recurrent somatic mutations in SETBP1 was described (22, 24, 59, 65–70). SETBP1 mutations have been identified in about one-quarter of patients affected by aCML (22), but also in 10%–16% of MDS/MPN unclassifiable cases (24) and in 4%–15% of CMML patients (59). Moreover, SETBP1 mutations have been occasionally described in juvenile myelomonocytic leukemia (JMML) and in about 1.7%–7% of sAML arising from MPN or MDS (57). Several studies had shown that SETBP1 mutations are associated with an adverse clinical presentation, with a higher leukocyte count, a lower Hb level, and thrombocytopenia (22, 24, 71). These data suggest that this alteration is important not only for the dissection of the mechanisms of leukemogenesis, but also because it likely provides important prognostic value (72). SETBP1 maps on chromosome 18q21.1 and encodes for SET-binding protein 1a, a protein of 1,596 amino acids with a predicted molecular weight of 170 kDa. The protein contains a SET-binding region and a SKI homology region, in which the recurrent mutations are clustered. The latter is highly conserved among vertebrates, suggesting an important but still unknown biological function. Moreover, three AT hooks can be found in SETBP1 protein and they are likely responsible for the direct interaction occurring between SETBP1 and the genomic DNA. It is known that SETBP1 is a binding partner for the SET nuclear oncoprotein (73). In turn, SET binds and negatively regulates the phosphatase 2A (PP2A) (74) oncosuppressor, a major phosphatase implicated in many cellular processes, such as cellular proliferation (75–79). In particular, PP2A loss of function has been associated with cell transformation (80, 81). Indeed, PP2A is a tumor suppressor that acts by regulating several signaling pathways critical for malignant transformation, such as AKT and ERK1/2 (82–84). By directly interacting with SET, SETBP1 protects it from proteolytic cleavage, increasing the amount of full-length SET protein and leading to the formation of a SETBP1–SET–PP2A complex resulting in PP2A inhibition, ultimately causing increased proliferation and expansion of the leukemic clone (85). We originally demonstrated that the majority of SETBP1 somatic mutations cluster in a mutational hotspot within the SKI-homologous region of the protein, conferring a proliferative advantage to the mutated cells (22). This hotspot is part of a degron motif recognized by the F-box protein β-TrCP, one of the four subunits of the ubiquitin protein ligase complex known as SCF. Under physiological conditions, this interaction stimulates SETBP1 ubiquitination and degradation through the proteasome (Figure 1, upper panel). SETBP1 degron mutations severely decrease the affinity of β-TrCP to SETBP1, leading to the accumulation of SETBP1 protein, promoting its overexpression, and triggering the stabilization of SET at the protein level. The consequence of these events is the inhibition of PP2A (Figure 1, bottom panel). Besides the interaction with the SET–PP2A axis, SETBP1 is also able to directly interact with genomic DNA through its three conserved AT hooks (86), recruiting transcriptional modulators such as HCF1, KMT2A, PHF8, and PHF6, which belong to the SET/KMT2A (MLL1) COMPASS-like complex, forming a multiprotein complex that in turn causes the activation of gene expression. Notably, SETBP1 binds to the promoter of MECOM, which modulates the expression of several genes involved in the proliferation of hematopoietic stem cells and in the myeloid differentiation, upregulating it (86, 87). SETBP1 overexpression also confers self-renewal capability to myeloid progenitors in vitro by interacting with the homeobox A9 (HOXA9) and homeobox A10 (HOXA10) promoters (88). It is also reported to interact with the Runx1 promoter, resulting in Runx1 downregulation (89) and impairment of the Runx1-dependent program of myeloid differentiation (90, 91). Globally, these data suggest a complex role for SETBP1 as a transcriptional modulator, likely being able to activate or repress the expression of target genes depending on the coactivator/corepressor complexes co-recruited to the target locus.
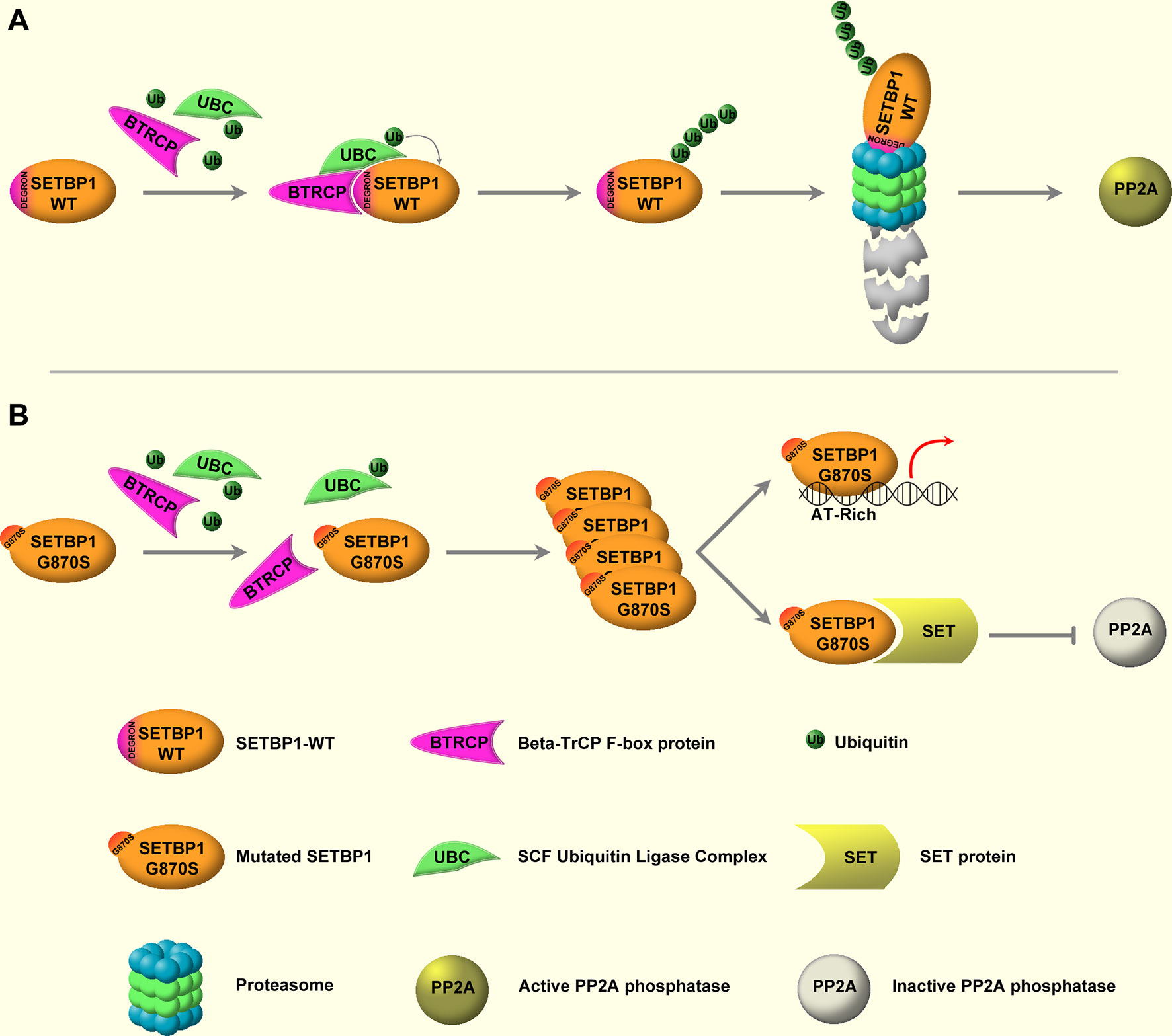
Figure 1 WT and mutated SETBP1 signaling. (A) SETBP1–WT is under the post-translational control of the ubiquitin protein ligase complex (UBC) known as SCF, which is mediated by the β-TrCP F-box protein. β-TrCP directly interacts with the SETBP1 degron motif, mediating the recruitment of SCF and the subsequent ubiquitination of SETBP1. Following ubiquitination, SETBP1 protein is degraded through the proteasome. (B) Degron mutations impair the interaction between SETBP1 and β-TrCP, protecting SETBP1 from proteasomal degradation and causing accumulation of the protein. This in turn leads to overactivation of the SETBP1 downstream signaling, specifically: 1) modulation of target genes through direct binding of SETBP1 to genomic DNA, mediated by its three AT-hook domains, and 2) activation of the SETBP1–SET axis, leading to SET accumulation and inhibition of PP2A phosphatase activity.
The ETNK1 gene (also known as EKI1) maps on chromosome 12p12.1. It spans 60.5 kb and consists of eight exons and seven introns (92). ETNK1 encodes a protein of 452 residues known as ethanolamine kinase, a cytoplasmic enzyme that catalyzes the first step of the de novo phosphatidylethanolamine (PE) biosynthesis through the Kennedy or cytidine diphosphate (CDP)-ethanolamine pathway (93). The Kennedy pathway is responsible for the de novo biosynthesis of the two major membrane phospholipids, phosphatidylcholine (PC) and PE. In particular, ETNK1 is responsible for the phosphorylation of ethanolamine to generate phosphoethanolamine (P-Et) (93). Somatic ETNK1 mutations were originally identified by our group in 13.3% of an aCML cohort (23). ETNK1 mutations were present as a heterozygous variant in the dominant clone and affected two contiguous residues: H243Y and N244S. Mutations clustering in the same hotspot of the kinase catalytic domain (N244S, N244T, N244K, G245A, G245V) were subsequently found also in 0%–14% of CMML cases (23, 94), in 20% of patients affected by aggressive systemic mastocytosis (SM) with eosinophilia (94), and in a single case of diffuse large B-cell lymphoma (DLBCL) (95). Recently, our group demonstrated that somatic ETNK1 mutations are responsible for a reduced activity of the enzyme, causing a decrease in P-Et synthesis (23, 96). Through the reduced competition of P-Et with succinate at mitochondrial complex II, an increased mitochondrial hyperactivation is triggered, which in turn is responsible for increased ROS production and subsequent DNA damage and accumulation of further mutations (96). Treatment with exogenous P-Et is able to restore a normal phenotype, protecting cells from ROS-mediated DNA damage (96, 97) (Figure 2). Notably, recent findings suggest that, whenever present, ETNK1 mutations occur at the initial stages of the clonal evolution of aCML (14), preceding other driver events, such as ASXL1 or SETBP1, which indirectly supports the role of ETNK1 as an inducer of a mutator phenotype.
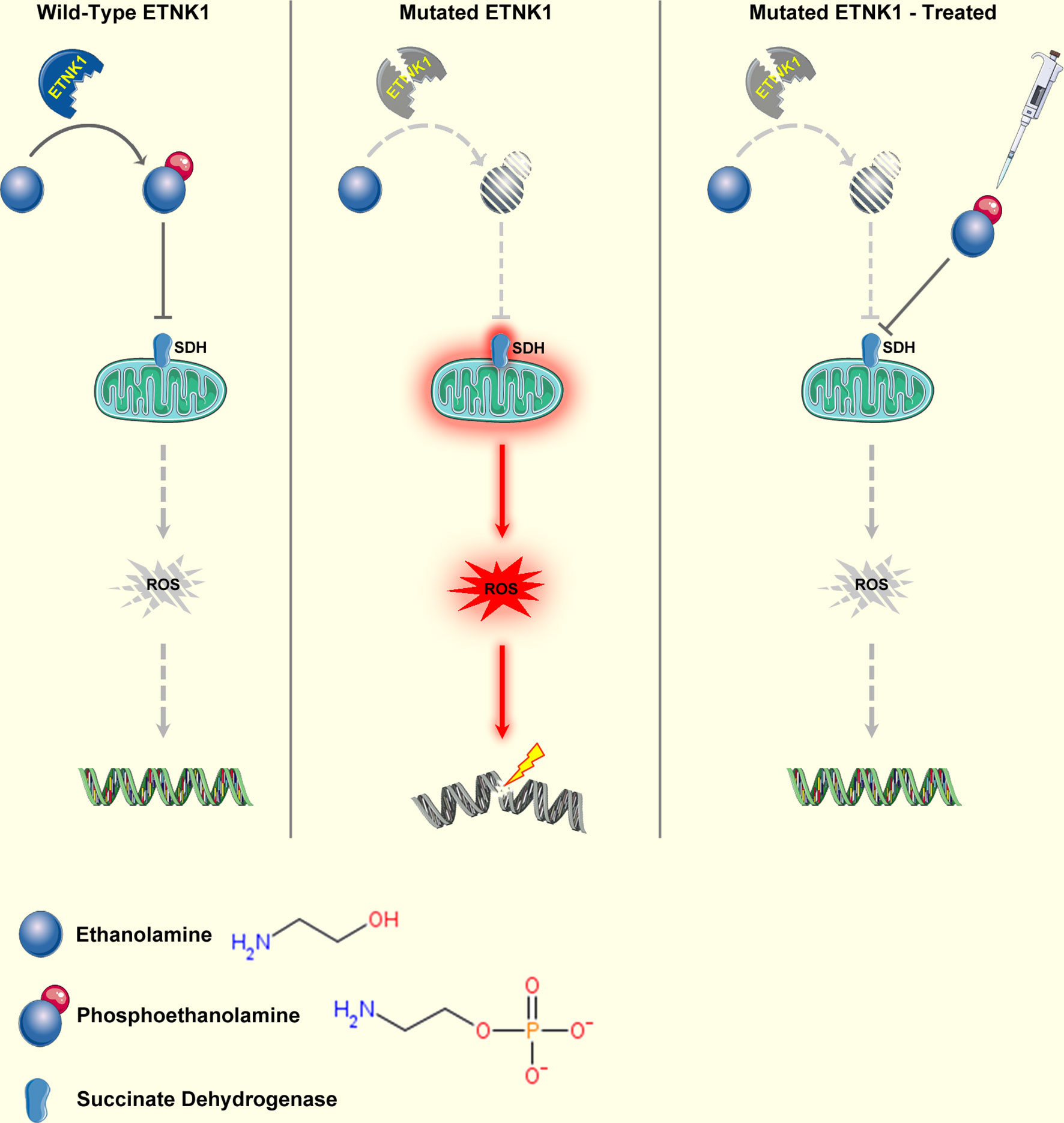
Figure 2 Mechanism of ETNK1 somatic mutations. Left panel: WT ETNK1 phosphorylates ethanolamine, causing the accumulation of phosphoethanolamine. Phosphoethanolamine directly inhibits mitochondrial complex II, also known as succinate dehydrogenase, therefore downmodulating mitochondrial activity and ROS production. Middle panel: mutated ETNK1 causes a decreased production of phosphoethanolamine, which in turn leads to an increased mitochondrial activation, increased ROS production, and DNA damage. Right panel: treatment of ETNK1-mutated cells with exogenous P-Et restores the normal mitochondrial activity through direct suppression of SDH activity, normalization of ROS production, and protection of DNA from ROS-mediated damage. The image was obtained from: Nat Commun. 2020 Nov 23;11(1):5938 under a Creative Commons Attribution 4.0 International License. Elements of the image were obtained from https://smart.servier.com/under a Creative Commons Attribution 3.0 License.
The ASXL1 gene is located on chromosome 20q11.1, spanning 81 kb. This gene belongs to the polycomb gene family and plays a role in the recruitment of the Polycomb Repressor Complex 2 (PRC2) to its target sequences. It is also a component of the H2AK119 complex, responsible for histone H2A deubiquitination (98). ASXL1 is mutated in more than 40% of aCML patients (14), and its mutations are associated with progression to acute phase and lower overall survival (22). It is currently known that ASXL1 contributes to the balance between the Polycomb Repressor Complex 1 (PRC1) and 2 (PRC2) in favor of the latter. Specifically, by interacting with the ubiquitin carboxy-terminal hydrolase BAP1, ASXL1 causes H2A Lysine 119 deubiquitination, therefore directly counteracting the activity of PRC1 (99). Instead, in combination with the PRC2, it promotes H3K27 trimethylation through the recruitment of the PRC2 effectors EZH1 and EZH2 at the target site, ultimately causing gene silencing.
ASXL1 mutations are typically frameshift or nonsense mutations causing a C-terminal truncation of the ASXL1 protein. Constitutive as well as hematopoietic-lineage-restricted, homozygous ASXL1 knockout causes impairment of the bone marrow self-renewal capacity, ultimately leading to an MDS-like disease in mice (100, 101). In line with its role in promoting PRC2 activity, ASXL1 knockout confers a pan-reduction of the H3K27 trimethylation mark (27), leading to derepression of posterior Hoxa genes and oncogenic miR125a microRNAs. Importantly, overexpression of a truncated form of ASXL1, in combination with overexpression of BAP1, caused an important reduction in the global level of H3K27me3, together with a depletion of the H2AK119ub mark, therefore suggesting that the C-terminal truncation mutations may impair the PRC2 activity of ASXL1 while preserving its interaction with BAP1. Globally, ASXL1 mutations act as loss-of-function events responsible for the promotion of myeloid transformation through loss of PRC2-mediated gene repression (27); however, their exact role is currently not entirely understood and likely multifaceted.
The TET2 gene maps on chromosome 4q24, spreading over 150 kb. TET2 is responsible for the modulation of DNA hydroxymethylation by converting 5-methylcytosine (5mC) to 5-hydroxymethylcytosine (5hmC) as the first step required to promote DNA demethylation (102, 103). Mutations occurring in TET2 are present in about 30% of aCML cases (14, 104–106); therefore, they are among the most frequent mutations occurring in this disorder, together with SETBP1 and ASXL1. TET2 mutations are invariably linked to a global decrease in the 5hmC mark, which suggests that they represent loss-of-function events. This evidence is also corroborated by the non-focal pattern of these variants, as TET2 mutations can be found throughout the entire coding region of the gene, and further supported by the frequent occurrence of nonsense and frameshift events.
TET2 plays a critical role in the bone marrow, mostly by promoting hematopoietic stem cell differentiation (107). In line with this role, it is expressed at high levels in progenitor cells and its deletion causes an increase in immature progenitor cells (35, 108), promoting myeloid as well as lymphoid malignancies in mice (109). From a prognostic point of view, the role of TET2 mutations is not univocal, but their presence probably does not negatively impact on the overall survival in most hematological malignancies (36, 110). In the context of MDS/MPN, individual reports suggest that TET2 mutations may be detrimental (111); however, its accurate prognostic role remains to be ascertained.
The RAS family of oncogenes comprises HRAS, NRAS, and KRAS genes. HRAS spans 3 kb and is located on chromosome 11p15.5, NRAS spans 7 kb and maps on chromosome 12p12.1, while KRAS spans more than 35 kb and is located at 1p13. These three genes share similar structures and sequences (112). The main product of the RAS genes is membrane-associated GTPases that control the MAP kinase cascade of serine/threonine kinases. Recurrent mutations in RAS genes occur in about 11%–27% of aCML patients (14, 22) and lead to a constitutively active expression of the protein. Usually aCML patients show NRAS or KRAS mutations (113, 114), and the most frequent mutations occur at codons 12, 31, and 61 (115–117).
The EZH2 gene is located at 7q36.1 and encodes the histone methyltransferase representing the catalytic subunit of the PRC2. In particular, EZH2 methylates histone H3 at lysine-27 (H3-K27), promoting the epigenetic silencing of genes involved in cell fate decisions (118, 119). The pattern of EZH2 mutations is particularly complex, as EZH2 variants can be both gain (GOF) as well as loss of function (LOF), with GOF mutations, such as EZH2-Y646X, frequently found in lymphoid malignancies and, in particular, in non-Hodgkin lymphoma (120) and solid, non-hematological tumors and LOF typically found in myeloid malignancies. In the context of aCML, LOF EZH2 mutations are seen in about 19%–30% of cases (14). The functional effect of EZH2 GOF mutations is to aberrantly increase H3K27me3, promoting transcriptional repression (121), which impairs B-cell differentiation and leads to an increased number and size of germinal centers (122). In contrast, LOF EZH2 mutations cause suppression of the H3K27me3 mark, causing overexpression of BCAT1 and leading to enhanced branched chain amino acid metabolism and activation of mTOR signaling (123). Association of EZH2 LOF mutations with poor prognosis was demonstrated in myelodysplastic syndromes (124), while their prognostic role in other disorders such as AML or aCML is less clear (125, 126).
The RUNX1 gene, also known as AML1, is located at chromosome band 21q22.12 and encodes the alpha subunit of the core-binding factor (CBF) complex (127). This complex is responsible for the transcriptional modulation of critical factors involved in growth, survival, and differentiation processes; ribosome biogenesis; cell cycle regulation; and p53 and transforming growth factor β signaling pathways (128). RUNX1 contains a runt-homology domain (RHD), which is responsible for DNA binding and interaction with the heterodimeric partner CBFβ and a TAD domain characterized by the presence of motifs binding to a large number of activating and repressor proteins. RUNX1 is known to be involved in more than 50 different chromosomal translocations. The t(8;21) involving RUNX1 and RUNXT1, the t(12;21) occurring in pediatric acute lymphoblastic leukemia and generating the ETV6–RUNX1 fusion, and the t(3;21) occurring in therapy-related AML and myelodysplastic syndrome and involving the MECOM oncogene are among those that are the most frequent. In the t(8;21), the persistence in the fusion of the RHD domain allows the binding of the protein to the normal RUNX1 gene targets. The presence of the RUNX1T1 fusion partner causes the recruitment of corepressors carrying deacetylase activity to the target promoters, therefore impairing the normal trans-activation and changing the function of the protein into a repressor, hence causing neomorphic activity (129).
Single-nucleotide somatic mutations are also commonly found in myeloid malignancies, such as AML and MDS. They typically occur in the RHD and, with a much lower frequency, in the TAD domain and can be mono- or biallelic. RUNX1 mutations include missense, nonsense, frameshift, deletions, and splicing mutations (130). Mechanistically, mutations occurring in the RHD domain usually inactivate the protein, while mutations occurring downstream of the RHD domain typically confer a weak dominant negative activity to the mutant (131). These mutations are functionally distinct from the chromosomal translocations and usually confer a worse prognosis. Mutations involving this gene are present in about 11%–15% of aCML cases (14), but are also found in 10%–37% of CMML patients (132–134).
The SRSF2 gene is located on chromosome 17q25.1 and encodes a protein that plays a role in the splicing of primary mRNA (135, 136). This protein contains an RNA recognition motif that promotes spliceosome assembly at adjacent splice sites allowing the removal of introns from the primary transcripts (137). Moreover, it plays an active role in transcription and elongation and in coupling transcription and splicing processes (138, 139). Mutations in key factors of the spliceosome, such as SRSF2, SF3B1, U2AF1, and ZRSR2, occur in a large fraction of myelodysplastic syndromes (140).
SRSF2 contains an RNA-binding domain (RBD) responsible for the interaction with exonic splicing enhancers and an SR (serine-arginine rich) domain directly interacting with the other splicing ribonucleoproteins.
p.P95H is by far the most common mutation occurring in the SRSF2 gene (141, 142). This mutation alters the RNA-binding affinity and specificity of the RBD domain, resulting in higher affinity for CCNG than to the standard GGNG motif, at least in vitro (143).
The frequency of SRSF2 mutations in aCML is 14%–65% of cases (10, 14, 58, 144, 145). Although SRSF2 mutations have been associated with worse survival outcomes in low-risk MDS patients (146), its prognostic role in aCML is currently unclear.
CBL is located on chromosome 11q23.3; it contains 16 exons and spans more than 110 kb (147). This gene encodes a protein that acts as an E3 ubiquitin ligase, being required for targeting substrates for degradation by the proteasome. CBL plays both positive and negative regulatory roles in tyrosine kinase signaling transduction pathways. CBL can bind to activated signaling complexes recruiting downstream signal transduction components or can target receptors that in turn trigger internalization of the receptor/ligand complex, promoting recycling or proteasomal degradation in endosomes (148–151). Besides aCML, CBL has also been found mutated in 5%–19% cases of CMML patients (152, 153). Moreover, mutations of CBL are frequently associated with uniparental disomy at 11q (14, 154).
The CSF3R gene is located at 1p34.3 and encodes the transmembrane receptor of the granulocyte colony-stimulating factor 3, which plays an essential role in the growth and differentiation of granulocytes (155, 156). Somatic CSF3R mutations are found in a large fraction (50%–80%) of patients affected by CNL (157, 158), and their presence is now one of the diagnostic criteria for CNL, according to the 2016 revision to the World Health Organization classification of myeloid neoplasms (1). In contrast, their association with aCML remains controversial. Although a single study reported CSF3R mutations to be frequent in aCML (157), several other works showed that CSF3R mutations are restricted to CNL and very rare in aCML (14, 22, 43, 58, 158). Currently, two types of CSF3R mutations are known: 1) extracellular domain/membrane proximal point mutations, such as the p.T618I variant, and 2) cytoplasmic truncation mutations. Mutations belonging to the first group result in a constitutive activation of the receptor, which occurs independently from the presence of the ligand. These mutations activate downstream JAK family tyrosine kinase pathways that drive the proliferation of neutrophil precursors, and are typically sensitive to JAK inhibitors. Truncation mutations instead interfere with receptor internalization and degradation, causing constitutive overexpression of CSF3R and ligand hypersensitivity, and show sensitivity to SRC kinase inhibitors (157).
To date, there are very few papers investigating the gene expression profile of aCML cases (13, 14, 25). Faisal and colleagues analyzed the mRNA expression of 26 aCML and 59 CMML cases, comparing them to a cohort of reference samples. Their analysis revealed a significant change in the expression levels of SETBP1, CDKN2A, GATA2, MPL, TMEM14C, CSF3R, and FLT3 genes. The strongest differential expression effect was detectable in FLT3 in CMML samples compared with aCML and references ones and in both SETBP1 and CSF3R in aCML and CMML cases. These findings are in line with the mutation frequency of these genes in aCML and CMML (25). In the work of Zhang, RNA-sequencing was performed on 76 samples of aCML, CNL, CMML, and MDS/MPN unclassifiable (MDS/MPN-U). Gene expression signatures identified three main sample clusters, with different proportions of all diagnoses in each group, associated with prognostic markers (13). On the other hand, our group performed RNA-sequencing on a cohort of 43 aCML patients, and stratification based on gene expression profile identified two different populations in terms of overall survival. In this context, overexpression of three genes (DNPH1, GFI1B, and PARP1) was predictive of poor prognosis (14). Contrary to these disorders characterized by high heterogeneity, several profiling studies in BCR-ABL1-positive CML have been reported. In particular, expression profiling analyses revealed a different signature associated with the classical t(9;22)(q34;q11) translocation or with variant t(9;22) rearrangements (159); similarly, signature analysis predicted imatinib response or resistance (160–166).
No drug has so far proved to be effective and no established standards of care exist for the treatment of aCML (48, 54). Moreover, no consensus recommendations such as risk-based treatment algorithms exist to help clinicians in choosing between a watch-and-wait approach and initiation of therapy. During the last years, different therapeutic approaches have been proposed, but at present, allogeneic hematopoietic stem cell transplantation (allo-HSCT) remains the only potentially curative treatment option for aCML, even if only limited reports are available (167–171). However, allo-HSCT requires the presence of a suitable donor and is only available for young and middle-aged patients, since the toxicity of the transplant limits its use in the elderly (>70 years), where, especially with low-risk disease, monitoring or palliative chemotherapy may be more appropriate.
Regarding other medical therapies, different treatment strategies validated in other myeloid diseases have been evaluated in aCML. Hydroxyurea (HU) is used as a supportive care measure to control hyperleukocytosis and splenomegaly. Complete and partial hematological remissions have been reported in about 80% of patients, even if they are usually short-lived (10, 11, 19, 43, 44, 46, 47, 172–175). Moreover, complete and partial hematological remissions have also been reported after treatment with interferon alfa (IFN-alfa), even if many patients discontinued the treatment due to drug toxicity (19, 44, 172–174, 176, 177). However, both HU and IFN-alfa, despite being able to improve the WBC count, are unable to change the course of the disease and are typically used in a palliative setting where, due to the age of the patient or the presence of significant comorbidities, an allo-HSCT is not considered a valid option.
Among the other drugs that can be used in the treatment of aCML, especially in patients with high-risk disease, the hypomethylating agents (HMA), such as azacitidine or decitabine, are noteworthy (10, 47, 178–183). Indeed, based on their established activity in MDS and CMML, in which the overall response rates range from 25% to 70% and the overall survival from 12 to 37 months (184), these drugs could be used also in aCML. Nevertheless, up to now, the experience with HMA is still limited and the available data do not allow to accurately predict the efficacy of these compounds. Therefore, HMA cannot be considered as a standard of care for aCML yet and their use is off-label.
For selected patients with aggressive behavior, AML-like intensive chemotherapy is offered as a bridge to HSCT (185), even if this option has not been explored extensively.
Given the recent description of the mutational landscape of aCML, in the field of personalized therapies, different targeted drugs can be used for aCML treatment and are currently being investigated. Indeed, several actionable mutations have been identified in aCML which could be targeted, e.g., with tyrosine kinase inhibitors. Wang and colleagues administrated RAS, FLT3, MAPK, MYC, or AKT inhibitors to their patients (43). The MEK1/2 inhibitor trametinib, approved for malignant melanoma, has been used in RAS-mutated aCML (14, 186). Khanna and collaborators reported an aCML case with NRAS G12V mutation who experienced a notable response to trametinib with improvement in blood counts and 14 months of disease control (186), while another patient carrying NRAS G12D mutation treated with trametinib obtained a hematological response with blood cell count normalization and reduction of splenomegaly for 3 months (14).
For rare cases of aCML with JAK2 V617F mutation (62, 187, 188), ruxolitinib, a JAK2 inhibitor approved by the FDA in intermediate- to high-risk MF and PV intolerant or resistant to HU, can be used (189, 190). This drug is also effective in patients carrying CSF3R T618I mutation, since membrane proximal mutations result in JAK–STAT pathway activation (157), while truncation CSF3R mutations are reported to be sensitive to dasatinib (14, 157). An open-label, single-arm, phase II multicenter study evaluated the safety and efficacy of ruxolitinib in patients affected by CNL or aCML, regardless of their mutation status (191). Interestingly, 4/6 aCML patients carrying CSF3R mutations and 7/17 patients with CSF3R WT reached partial (PR) or complete responses (CR). PR was defined as >50% reduction of WBC, absolute neutrophil count (ANC), and granulocytic dysplasia and >25% reduction in spleen volume; CR was defined as normalization of WBC count and ANC, no evidence of granulocytic dysplasia, and normal spleen. A phase I, open-label study is currently ongoing to evaluate the safety and efficacy of TGR-1202, a PI3K-delta inhibitor, administered together with ruxolitinib in patients with MDS or MDS/MPN including aCML (ClinicalTrials.gov Identifier: NCT02493530).
Since the treatment of aCML remains a challenge, Gotlib proposed a new treatment algorithm (54), based on several decision nodes, including the potential candidacy for allogeneic hematopoietic stem cell transplantation, the results of myeloid mutation panel testing, the eligibility for enrollment in clinical trials, and the opportunity to adopt strategies used for MDS or MPN. Considering that two or more mutations in distinct genes often occur in aCML patients, combination therapies with different drugs could represent a promising approach. Since multiple actionable mutations are often present in various combinations in aCML patients, their enrollment in clinical trials should be considered whenever possible.
aCML is a BCR-ABL1-negative hematological disease characterized by poor survival. The challenges in the management of this leukemia comprise both the diagnosis, due to the overlap of several genetic mutations among different MDS/MPN disorders, and the treatment choices, since at present no standards of care are available, except for bone marrow transplantation that is the only curative option for younger patients. The application of NGS techniques led to the discovery of new genes involved in the onset of the disease, which is allowing the introduction of personalized therapies for aCML patients. Further studies will be required to thoroughly assess the efficacy of these new treatments. Importantly, transcriptomic studies done at single-cell resolution may also unravel new targetable pathways that could increase the therapeutic options available for the treatment of this aggressive disorder.
DF: writing—original draft preparation. CG-P: funding acquisition, writing—review and editing, and supervision. RP: funding acquisition, writing—review and editing, and supervision. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, et al. The 2016 Revision to the World Health Organization Classification of Myeloid Neoplasms and Acute Leukemia. Blood (2016) 127(20):2391–405. doi: 10.1182/blood-2016-03-643544
2. Jaffe ES, Harris NL, Stein H, Vardiman JW. Pathology and Genetics of Tumours of Haematopoietic and Lymphoid Tissues WHO Classification of Tumours. 3rd Edition Vol. 3. . Lyon: IARC Press (2001).
3. Orazi A, Germing U. The Myelodysplastic/Myeloproliferative Neoplasms: Myeloproliferative Diseases With Dysplastic Features. Leukemia (2008) 22(7):1308–19. doi: 10.1038/leu.2008.119
4. Vardiman JW, Bain B, Inbert M, Brunning RD, Pierre RV, Flandrin G. Atypical Chronic Myeloid Leukemia. In: Jaffe E, Harris NL, Stein H, Vardiman J, editors. WHO Classification of Tumours: Pathology and Genetics of Tumours of Haematopoietic and Lymphoid Tissues. Lyon: IARC Press (2001). p. 53–7. Lyon: IARC Press.
5. Oscier DG. Atypical Chronic Myeloid Leukaemia, a Distinct Clinical Entity Related to the Myelodysplastic Syndrome? Br J Haematol (1996) 92(3):582–6. doi: 10.1046/j.1365-2141.1996.396933.x
6. Bennett JM, Catovsky D, Daniel MT, Flandrin G, Galton DA, Gralnick H, et al. The Chronic Myeloid Leukaemias: Guidelines for Distinguishing Chronic Granulocytic, Atypical Chronic Myeloid, and Chronic Myelomonocytic Leukaemia. Proposals by the French-American-British Cooperative Leukaemia Group. Br J Haematol (1994) 87(4):746–54. doi: 10.1111/j.1365-2141.1994.tb06734.x
7. Sabattini E, Bacci F, Sagramoso C, Pileri SA. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues in 2008: An Overview. Pathologica (2010) 102(3):83–7.
8. Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, et al. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. In: WHO Classification of Tumours, Revised, 4th Edition, vol. 2. . Lyon: IARC Press (2008).
9. Vardiman JW, Thiele J, Arber DA, Brunning RD, Borowitz MJ, Porwit A, et al. The 2008 Revision of the World Health Organization (WHO) Classification of Myeloid Neoplasms and Acute Leukemia: Rationale and Important Changes. Blood (2009) 114(5):937–51. doi: 10.1182/blood-2009-03-209262
10. Patnaik MM, Barraco D, Lasho TL, Finke CM, Reichard K, Hoversten KP, et al. Targeted Next Generation Sequencing and Identification of Risk Factors in World Health Organization Defined Atypical Chronic Myeloid Leukemia. Am J Hematol (2017) 92(6):542–8. doi: 10.1002/ajh.24722
11. Breccia M, Biondo F, Latagliata R, Carmosino I, Mandelli F, Alimena G. Identification of Risk Factors in Atypical Chronic Myeloid Leukemia. Haematologica (2006) 91(11):1566–8. doi: 10.3324/%25x
12. Hernandez J, Villaescusa T, Arefi M, López L, Garcia J. Atypical Chronic Myeloid Leukemia (Acml). Atlas Genet Cytogenet Oncol Haematol (2009) 13(6):432–3. doi: 10.4267/2042/44494
13. Zhang H, Wilmot B, Bottomly D, Dao KT, Stevens E, Eide CA, et al. Genomic Landscape of Neutrophilic Leukemias of Ambiguous Diagnosis. Blood (2019) 134(11):867–79. doi: 10.1182/blood.2019000611
14. Fontana D, Ramazzotti D, Aroldi A, Redaelli S, Magistroni V, Pirola A, et al. Integrated Genomic, Functional, and Prognostic Characterization of Atypical Chronic Myeloid Leukemia. Hemasphere (2020) 4(6):e497. doi: 10.1097/HS9.0000000000000497
15. Giri S, Pathak R, Martin MG, Bhatt VR. Characteristics and Survival of BCR/ABL Negative Chronic Myeloid Leukemia: A Retrospective Analysis of the Surveillance, Epidemiology and End Results Database. Ther Adv Hematol (2015) 6(6):308–12. doi: 10.1177/2040620715607416
17. Cazzola M, Malcovati L, Invernizzi R. Myelodysplastic/Myeloproliferative Neoplasms. Hematol Am Soc Hematol Educ Program (2011) 1:264–72. doi: 10.1182/asheducation-2011.1.264
18. Gotlib J, Maxson JE, George TI, Tyner JW. The New Genetics of Chronic Neutrophilic Leukemia and Atypical CML: Implications for Diagnosis and Treatment. Blood (2013) 122(10):1707–11. doi: 10.1182/blood-2013-05-500959
19. Kurzrock R, Bueso-Ramos CE, Kantarjian H, Freireich E, Tucker SL, Siciliano M, et al. Bcr Rearrangement-Negative Chronic Myelogenous Leukemia Revisited. J Clin Oncol (2001) 19(11):2915–26. doi: 10.1200/JCO.2001.19.11.2915
20. Dao KH, Tyner JW. What's Different About Atypical CML and Chronic Neutrophilic Leukemia? Hematol Am Soc Hematol Educ Program (2015) 2015:264–71. doi: 10.1182/asheducation-2015.1.264
21. Thakral B, Anastasi J, Sa AW. 17 - Myeloproliferative and “Overlap” Myelodysplastic/Myeloproliferative Neoplasms. Foundations Diagn Pathol (2018) 488–538.e484. doi: 10.1016/B978-0-323-47913-4.00017-3
22. Piazza R, Valletta S, Winkelmann N, Redaelli S, Spinelli R, Pirola A, et al. Recurrent SETBP1 Mutations in Atypical Chronic Myeloid Leukemia. Nat Genet (2013) 45(1):18–24. doi: 10.1038/ng.2495
23. Gambacorti-Passerini CB, Donadoni C, Parmiani A, Pirola A, Redaelli S, Signore G, et al. Recurrent ETNK1 Mutations in Atypical Chronic Myeloid Leukemia. Blood (2015) 125(3):499–503. doi: 10.1182/blood-2014-06-579466
24. Meggendorfer M, Bacher U, Alpermann T, Haferlach C, Kern W, Gambacorti-Passerini C, et al. SETBP1 Mutations Occur in 9% of MDS/MPN and in 4% of MPN Cases and are Strongly Associated With Atypical CML, Monosomy 7, Isochromosome I(17)(Q10), ASXL1 and CBL Mutations. Leukemia (2013) 27(9):1852–60. doi: 10.1038/leu.2013.133
25. Faisal M, Stark H, Busche G, Schlue J, Teiken K, Kreipe HH, et al. Comprehensive Mutation Profiling and Mrna Expression Analysis in Atypical Chronic Myeloid Leukemia in Comparison With Chronic Myelomonocytic Leukemia. Cancer Med (2019) 8(2):742–50. doi: 10.1002/cam4.1946
26. Branford S, Kim DDH, Apperley JF, Eide CA, Mustjoki S, Ong ST, et al. Laying the Foundation for Genomically-Based Risk Assessment in Chronic Myeloid Leukemia. Leukemia (2019) 33(8):1835–50. doi: 10.1038/s41375-019-0512-y
27. Abdel-Wahab O, Adli M, LaFave LM, Gao J, Hricik T, Shih AH, et al. ASXL1 Mutations Promote Myeloid Transformation Through Loss of PRC2-Mediated Gene Repression. Cancer Cell (2012) 22(2):180–93. doi: 10.1016/j.ccr.2012.06.032
28. Gelsi-Boyer V, Brecqueville M, Devillier R, Murati A, Mozziconacci MJ, Birnbaum D. Mutations in ASXL1 are Associated With Poor Prognosis Across the Spectrum of Malignant Myeloid Diseases. J Hematol Oncol (2012) 5:12. doi: 10.1186/1756-8722-5-12
29. DiNardo CD, Cortes JE. Mutations in AML: Prognostic and Therapeutic Implications. Hematol Am Soc Hematol Educ Program (2016) 2016(1):348–55. doi: 10.1182/asheducation-2016.1.348
30. Hou HA, Tien HF. Genomic Landscape in Acute Myeloid Leukemia and its Implications in Risk Classification and Targeted Therapies. J BioMed Sci (2020) 27(1):81. doi: 10.1186/s12929-020-00674-7
31. Awada H, Thapa B, Visconte V. The Genomics of Myelodysplastic Syndromes: Origins of Disease Evolution, Biological Pathways, and Prognostic Implications. Cells (2020) 9(11). doi: 10.3390/cells9112512
32. Paschka P, Schlenk RF, Gaidzik VI, Herzig JK, Aulitzky T, Bullinger L, et al. ASXL1 Mutations in Younger Adult Patients With Acute Myeloid Leukemia: A Study by the German-Austrian Acute Myeloid Leukemia Study Group. Haematologica (2015) 100(3):324–30. doi: 10.3324/haematol.2014.114157
33. Lund K, Adams PD, Copland M. EZH2 in Normal and Malignant Hematopoiesis. Leukemia (2014) 28(1):44–9. doi: 10.1038/leu.2013.288
34. Marcucci G, Haferlach T, Döhner H. Molecular Genetics of Adult Acute Myeloid Leukemia: Prognostic and Therapeutic Implications. J Clin Oncol (2011) 29(5):475–86. doi: 10.1200/JCO.2010.30.2554
35. Figueroa ME, Lugthart S, Li Y, Erpelinck-Verschueren C, Deng X, Christos PJ, et al. DNA Methylation Signatures Identify Biologically Distinct Subtypes in Acute Myeloid Leukemia. Cancer Cell (2010) 17(1):13–27. doi: 10.1016/j.ccr.2009.11.020
36. Nibourel O, Kosmider O, Cheok M, Boissel N, Renneville A, Philippe N, et al. Incidence and Prognostic Value of TET2 Alterations in De Novo Acute Myeloid Leukemia Achieving Complete Remission. Blood (2010) 116(7):1132–5. doi: 10.1182/blood-2009-07-234484
37. Metzeler KH, Becker H, Maharry K, Radmacher MD, Kohlschmidt J, Mrózek K, et al. ASXL1 Mutations Identify a High-Risk Subgroup of Older Patients With Primary Cytogenetically Normal AML Within the ELN Favorable Genetic Category. Blood (2011) 118(26):6920–9. doi: 10.1182/blood-2011-08-368225
38. Makishima H, Jankowska AM, McDevitt MA, O'Keefe C, Dujardin S, Cazzolli H, et al. CBL, CBLB, TET2, ASXL1, and IDH1/2 Mutations and Additional Chromosomal Aberrations Constitute Molecular Events in Chronic Myelogenous Leukemia. Blood (2011) 117(21):e198–206. doi: 10.1182/blood-2010-06-292433
39. Cuéllar C, Cánaves YP, de la Fuente EO, Sánchez JM, Rapado I, López JM, et al. Detection of CALR and CBL Mutations by Next Generation Sequencing (NGS) in Acute Myeloid Leukemia (AML) and Myelodisplasic Syndrome (MDS). EHA Library: Abstract: PB1662 (2018).
40. Liu H, Zhang Y, Wang F, Chen X, Teng W, Wang M, et al. CSF3R Mutation in Acute Leukemia and MDS Patients. Blood (2017) 130:2669. doi: 10.1182/blood.V130.Suppl_1.2669.2669
41. Jekarl DW, Han SB, Kim M, Lim J, Oh EJ, Kim Y, et al. JAK2 V617F Mutation in Myelodysplastic Syndrome, Myelodysplastic Syndrome/Myeloproliferative Neoplasm, Unclassifiable, Refractory Anemia With Ring Sideroblasts With Thrombocytosis, and Acute Myeloid Leukemia. Korean J Hematol (2010) 45(1):46–50. doi: 10.5045/kjh.2010.45.1.46
42. Maxson JE, Tyner JW. Genomics of Chronic Neutrophilic Leukemia. Blood (2017) 129(6):715–22. doi: 10.1182/blood-2016-10-695981
43. Wang SA, Hasserjian RP, Fox PS, Rogers HJ, Geyer JT, Chabot-Richards D, et al. Atypical Chronic Myeloid Leukemia is Clinically Distinct From Unclassifiable Myelodysplastic/Myeloproliferative Neoplasms. Blood (2014) 123(17):2645–51. doi: 10.1182/blood-2014-02-553800
44. Martiat P, Michaux JL, Rodhain J. Philadelphia-Negative (Ph-) Chronic Myeloid Leukemia (CML): Comparison With Ph+ CML and Chronic Myelomonocytic Leukemia. The Groupe Francais De Cytogenetique Hematologique. Blood (1991) 78(1):205–11. doi: 10.1182/blood.V78.1.205.205
45. Drozd-Sokołowska J, Mądry K, Waszczuk-Gajda A, Biecek P, Szwedyk P, Budziszewska K, et al. Atypical Chronic Myeloid Leukaemia: A Case of an Orphan Disease-a Multicenter Report by the Polish Adult Leukemia Group. Hematol Oncol (2018) 36(3):570–5. doi: 10.1002/hon.2501
46. Hernandez JM, del Canizo MC, Cuneo A, Garcia JL, Gutierrez NC, Gonzalez M, et al. Clinical, Hematological and Cytogenetic Characteristics of Atypical Chronic Myeloid Leukemia. Ann Oncol (2000) 11(4):441–4. doi: 10.1023/A:1008393002748
47. Onida F, Ball G, Kantarjian HM, Smith TL, Glassman A, Albitar M, et al. Characteristics and Outcome of Patients With Philadelphia Chromosome Negative, Bcr/Abl Negative Chronic Myelogenous Leukemia. Cancer (2002) 95(8):1673–84. doi: 10.1002/cncr.10832
48. Dhakal P, Gundabolu K, Amador C, Rayamajhi S, Bhatt VR. Atypical Chronic Myeloid Leukemia: A Rare Entity With Management Challenges. Future Oncol (2018) 14(2):177–85. doi: 10.2217/fon-2017-0334
49. Orazi A, O'Malley DP, Arber DA. Illustrated Pathology of the Bone Marrow. Cambridge Univ Press: Cambridge UK (2006) 2006:88–92. doi: 10.1017/CBO9780511543531
50. Tefferi A, Gilliland G. Classification of Chronic Myeloid Disorders: From Dameshek Towards a Semi-Molecular System. Best Pract Res Clin Haematol (2006) 19(3):365–85. doi: 10.1016/j.beha.2005.07.001
51. Krishna Chandran R, Geetha N, Sakthivel KM, Suresh Kumar R, Jagathnath Krishna KMN, Sreedharan H. Impact of Additional Chromosomal Aberrations on the Disease Progression of Chronic Myelogenous Leukemia. Front Oncol (2019) 9:88. doi: 10.3389/fonc.2019.00088
52. Clark RE, Apperley JF, Copland M, Cicconi S. Additional Chromosomal Abnormalities at Chronic Myeloid Leukemia Diagnosis Predict an Increased Risk of Progression. Blood Adv (2021) 5(4):1102–9. doi: 10.1182/bloodadvances.2020003570
53. Ochi Y, Yoshida K, Huang YJ, Kuo MC, Nannya Y, Sasaki K, et al. Clonal Evolution and Clinical Implications of Genetic Abnormalities in Blastic Transformation of Chronic Myeloid Leukaemia. Nat Commun (2021) 12(1):2833. doi: 10.1038/s41467-021-23097-w
54. Gotlib J. How I Treat Atypical Chronic Myeloid Leukemia. Blood (2017) 129(7):838–45. doi: 10.1182/blood-2016-08-693630
55. Mughal TI, Cross NC, Padron E, Tiu RV, Savona M, Malcovati L, et al. An International MDS/MPN Working Group's Perspective and Recommendations on Molecular Pathogenesis, Diagnosis and Clinical Characterization of Myelodysplastic/Myeloproliferative Neoplasms. Haematologica (2015) 100(9):1117–30. doi: 10.3324/haematol.2014.114660
56. Tiu RV, Sekeres MA. Making Sense of the Myelodysplastic/Myeloproliferative Neoplasms Overlap Syndromes. Curr Opin Hematol (2014) 21(2):131–40. doi: 10.1097/MOH.0000000000000021
57. Zoi K, Cross NC. Molecular Pathogenesis of Atypical CML, CMML and MDS/MPN-Unclassifiable. Int J Hematol (2015) 101(3):229–42. doi: 10.1007/s12185-014-1670-3
58. Meggendorfer M, Haferlach T, Alpermann T, Jeromin S, Haferlach C, Kern W, et al. Specific Molecular Mutation Patterns Delineate Chronic Neutrophilic Leukemia, Atypical Chronic Myeloid Leukemia, and Chronic Myelomonocytic Leukemia. Haematologica (2014) 99(12):e244–6. doi: 10.3324/haematol.2014.113159
59. Makishima H, Yoshida K, Nguyen N, Przychodzen B, Sanada M, Okuno Y, et al. Somatic SETBP1 Mutations in Myeloid Malignancies. Nat Genet (2013) 45(8):942–6. doi: 10.1038/ng.2696
60. Abdel-Wahab O, Mullally A, Hedvat C, Garcia-Manero G, Patel J, Wadleigh M, et al. Genetic Characterization of TET1, TET2, and TET3 Alterations in Myeloid Malignancies. Blood (2009) 114(1):144–7. doi: 10.1182/blood-2009-03-210039
61. Tefferi A, Elliot MA, Pardanani A. Atypical Myeloproliferative Disorders: Diagnosis and Management. Mayo Clinic Proc (2006) 81(4):553–63. doi: 10.4065/81.4.553
62. Jones AV, Kreil S, Zoi K, Waghorn K, Curtis C, Zhang LY, et al. Widespread Occurrence of the JAK2 V617F Mutation in Chronic Myeloproliferative Disorders. Blood (2005) 106(6):2162–8. doi: 10.1182/blood-2005-03-1320
63. Carulli G, Fabiani O, Azzara A. The Syndrome of Abnormal Chromatin Clumping in Leukocytes. Haematologica (1997) 82(5):635–6. doi: 10.3324/%25x
64. Fend F, Horn T, Koch I, Vela T, Orazi A. Atypical Chronic Myeloid Leukemia as Defined in the WHO Classification is a JAK2 V617F Negative Neoplasm. Leukemia Res (2008) 32(12):1931–5. doi: 10.1016/j.leukres.2008.04.024
65. Sakaguchi H, Okuno Y, Muramatsu H, Yoshida K, Shiraishi Y, Takahashi M, et al. Exome Sequencing Identifies Secondary Mutations of SETBP1 and JAK3 in Juvenile Myelomonocytic Leukemia. Nat Genet (2013) 45(8):937–41. doi: 10.1038/ng.2698
66. Inoue D, Kitaura J, Matsui H, Hou HA, Chou WC, Nagamachi A, et al. SETBP1 Mutations Drive Leukemic Transformation in ASXL1-Mutated MDS. Leukemia (2015) 29(4):847–57. doi: 10.1038/leu.2014.301
67. Fabiani E, Falconi G, Fianchi L, Criscuolo M, Leone G, Voso MT. SETBP1 Mutations in 106 Patients With Therapy-Related Myeloid Neoplasms. Haematologica (2014) 99(9):e152–3. doi: 10.3324/haematol.2014.108159
68. Patnaik MM, Itzykson R, Lasho T, Kosmider O, Finke C, Hanson C, et al. ASXL1 and SETBP1 Mutations and Their Prognostic Contribution in Chronic Myelomonocytic Leukemia: A Two-Center Study of 466 Patients. Leukemia (2014) 28(11):2206–12. doi: 10.1038/leu.2014.125
69. Thol F, Suchanek KJ, Koenecke C, Stadler M, Platzbecker U, Thiede C, et al. SETBP1 Mutation Analysis in 944 Patients With MDS and AML. Leukemia (2013) 27(10):2072–5. doi: 10.1038/leu.2013.145
70. Wang XA, Muramatsu H, Okuno Y, Sakaguchi H, Yoshida K, Kawashima N, et al. GATA2 and Secondary Mutations in Familial Myelodysplastic Syndromes and Pediatric Myeloid Malignancies. Haematologica (2015) 100(10):E398–401. doi: 10.3324/haematol.2015.127092
71. Linder K, Iragavarapu C, Liu D. Mutations as a Biomarker for Myelodysplasia /Myeloproliferative Neoplasm Overlap Syndrome. biomark Res (2017) 5:33. doi: 10.1186/s40364-017-0113-8
72. Lasho T. Atypical CML- the Role of Morphology and Precision Genomics. Best Pract Res Clin Haematol (2020) 33(2):101133. doi: 10.1016/j.beha.2019.101133
73. Chen M, Yao H, Chen S, Wang Q, Wen L, Xie J, et al. Rare Occurrence of SET Binding Protein 1 Mutation in Patients With Acute Lymphoblastic Leukemia, Mixed Phenotype Acute Leukemia and Chronic Myeloid Leukemia in Blast Crisis. Leuk Lymphoma (2014) 55(9):2209–10. doi: 10.3109/10428194.2013.873536
74. Minakuchi M, Kakazu N, Gorrin-Rivas MJ, Abe T, Copeland TD, Ueda K, et al. Identification and Characterization of SEB, a Novel Protein That Binds to the Acute Undifferentiated Leukemia-Associated Protein SET. Eur J Biochem (2001) 268(5):1340–51. doi: 10.1046/j.1432-1327.2001.02000.x
75. Janssens V, Goris J. Protein Phosphatase 2A: A Highly Regulated Family of Serine/Threonine Phosphatases Implicated in Cell Growth and Signalling. Biochem J (2001) 353:417–39. doi: 10.1042/bj3530417
76. Wang GL, Iakova P, Wilde M, Awad S, Timchenko NA. Liver Tumors Escape Negative Control of Proliferation via PI3K/Akt-Mediated Block of C/EBP Alpha Growth Inhibitory Activity. Genes Dev (2004) 18(8):912–25. doi: 10.1101/gad.1183304
77. Kawabe T, Muslin AJ, Korsmeyer SJ. HOX11 Interacts With Protein Phosphatases PP2A and PP1 and Disrupts a G2/M Cell-Cycle Checkpoint. Nature (1997) 385(6615):454–8. doi: 10.1038/385454a0
78. Mumby M. PP2A: Unveiling a Reluctant Tumor Suppressor. Cell (2007) 130(1):21–4. doi: 10.1016/j.cell.2007.06.034
79. Westermarck J, Hahn WC. Multiple Pathways Regulated by the Tumor Suppressor PP2A in Transformation. Trends Mol Med (2008) 14(4):152–60. doi: 10.1016/j.molmed.2008.02.001
80. Janssens V, Goris J, Van Hoof C. Pp2a: The Expected Tumor Suppressor. Curr Opin Genet Dev (2005) 15(1):34–41. doi: 10.1016/j.gde.2004.12.004
81. Schönthal AH. Role of Serine/Threonine Protein Phosphatase 2A in Cancer. Cancer Lett (2001) 170(1):1–13. doi: 10.1016/S0304-3835(01)00561-4
82. Cristobal I, Garcia-Orti L, Cirauqui C, Alonso MM, Calasanz MJ, Odero MD. PP2A Impaired Activity is a Common Event in Acute Myeloid Leukemia and its Activation by Forskolin has a Potent Anti-Leukemic Effect. Leukemia (2011) 25(4):606–14. doi: 10.1038/leu.2010.294
83. Arnold HK, Sears RC. A Tumor Suppressor Role for PP2A-B56 Alpha Through Negative Regulation of C-Myc and Other Key Oncoproteins. Cancer Metastasis Rev (2008) 27(2):147–58. doi: 10.1007/s10555-008-9128-9
84. Sablina AA, Hahn WC. The Role of PP2A a Subunits in Tumor Suppression. Cell Adhesion Migration (2007) 1(3):140–1. doi: 10.4161/cam.1.3.4986
85. Cristobal I, Blanco FJ, Garcia-Orti L, Marcotegui N, Vicente C, Rifon J, et al. SETBP1 Overexpression is a Novel Leukemogenic Mechanism That Predicts Adverse Outcome in Elderly Patients With Acute Myeloid Leukemia. Blood (2010) 115(3):615–25. doi: 10.1182/blood-2009-06-227363
86. Piazza R, Magistroni V, Redaelli S, Mauri M, Massimino L, Sessa A, et al. SETBP1 Induces Transcription of a Network of Development Genes by Acting as an Epigenetic Hub. Nat Commun (2018) 9(1):2192. doi: 10.1038/s41467-018-04462-8
87. Kataoka K, Kurokawa M. Ecotropic Viral Integration Site 1, Stem Cell Self-Renewal and Leukemogenesis. Cancer Sci (2012) 103(8):1371–7. doi: 10.1111/j.1349-7006.2012.02303.x
88. Oakley K, Han YF, Vishwakarma BA, Chu S, Bhatia R, Gudmundsson KO, et al. Setbp1 Promotes the Self-Renewal of Murine Myeloid Progenitors via Activation of Hoxa9 and Hoxa10. Blood (2012) 119(25):6099–108. doi: 10.1182/blood-2011-10-388710
89. Vishwakarma BA, Nguyen N, Makishima H, Hosono N, Gudmundsson KO, Negi V, et al. Runx1 Repression by Histone Deacetylation is Critical for Setbp1-Induced Mouse Myeloid Leukemia Development. Leukemia (2016) 30(1):200–8. doi: 10.1038/leu.2015.200
90. Ichikawa M, Asai T, Saito T, Seo S, Yamazaki I, Yamagata T, et al. AML-1 is Required for Megakaryocytic Maturation and Lymphocytic Differentiation, But Not for Maintenance of Hematopoietic Stem Cells in Adult Hematopoiesis. Nat Med (2004) 10(3):299–304. doi: 10.1038/nm997
91. Growney JD, Shigematsu H, Li Z, Lee BH, Adeisperger J, Rowan R, et al. Loss of Runx1 Perturbs Adult Hematopoiesis and is Associated With a Myeloproliferative Phenotype. Blood (2005) 106(2):494–504. doi: 10.1182/blood-2004-08-3280
92. Lykidis A, Wang J, Karim MA, Jackowski S. Overexpression of a Mammalian Ethanolamine-Specific Kinase Accelerates the CDP-Ethanolamine Pathway. J Biol Chem (2001) 276(3):2174–9. doi: 10.1074/jbc.M008794200
93. Kennedy EP, Weiss SB. The Function of Cytidine Coenzymes in the Biosynthesis of Phospholipides. J Biol Chem (1956) 222(1):193–214. doi: 10.1016/S0021-9258(19)50785-2
94. Lasho TL, Finke CM, Zblewski D, Patnaik M, Ketterling RP, Chen D, et al. Novel Recurrent Mutations in Ethanolamine Kinase 1 (ETNK1) Gene in Systemic Mastocytosis With Eosinophilia and Chronic Myelomonocytic Leukemia. Blood Cancer J (2015) 5:e275. doi: 10.1038/bcj.2014.94
95. Zhou XA, Louissaint A, Wenzel A, Yang J, Martinez-Escala ME, Moy AP, et al. Genomic Analyses Identify Recurrent Alterations in Immune Evasion Genes in Diffuse Large B-Cell Lymphoma, Leg Type. J Invest Dermatol (2018) 138(11):2365–76. doi: 10.1016/j.jid.2018.04.038
96. Fontana D, Mauri M, Renso R, Docci M, Crespiatico I, Røst LM, et al. ETNK1 Mutations Induce a Mutator Phenotype That can be Reverted With Phosphoethanolamine. Nat Commun (2020) 11(1):5938. doi: 10.1038/s41467-020-19721-w
97. Fontana D, Gambacorti-Passerini C, Piazza R. Impact of ETNK1 Somatic Mutations on Phosphoethanolamine Synthesis, ROS Production and DNA Damage. Mol Cell Oncol (2021) 8(2):1877598. doi: 10.1080/23723556.2021.1877598
98. Carbuccia N, Murati A, Trouplin V, Brecqueville M, Adélaïde J, Rey J, et al. Mutations of ASXL1 Gene in Myeloproliferative Neoplasms. Leukemia (2009) 23(11):2183–6. doi: 10.1038/leu.2009.141
99. Scheuermann JC, de Ayala Alonso AG, Oktaba K, Ly-Hartig N, McGinty RK, Fraterman S, et al. Histone H2A Deubiquitinase Activity of the Polycomb Repressive Complex PR-DUB. Nature (2010) 465(7295):243–7. doi: 10.1038/nature08966
100. Abdel-Wahab O, Gao J, Adli M, Dey A, Trimarchi T, Chung YR, et al. Deletion of Asxl1 Results in Myelodysplasia and Severe Developmental Defects In Vivo. J Exp Med (2013) 210(12):2641–59. doi: 10.1084/jem.20131141
101. Wang J, Li Z, He Y, Pan F, Chen S, Rhodes S, et al. Loss of Asxl1 Leads to Myelodysplastic Syndrome-Like Disease in Mice. Blood (2014) 123(4):541–53. doi: 10.1182/blood-2013-05-500272
102. Guallar D, Bi X, Pardavila JA, Huang X, Saenz C, Shi X, et al. RNA-Dependent Chromatin Targeting of TET2 for Endogenous Retrovirus Control in Pluripotent Stem Cells. Nat Genet (2018) 50(3):443–51. doi: 10.1038/s41588-018-0060-9
103. Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, et al. Leukemic IDH1 and IDH2 Mutations Result in a Hypermethylation Phenotype, Disrupt TET2 Function, and Impair Hematopoietic Differentiation. Cancer Cell (2010) 18(6):553–67. doi: 10.1016/j.ccr.2010.11.015
104. Haferlach T, Nagata Y, Grossmann V, Okuno Y, Bacher U, Nagae G, et al. Landscape of Genetic Lesions in 944 Patients With Myelodysplastic Syndromes. Leukemia (2014) 28(2):241–7. doi: 10.1038/leu.2013.336
105. Jankowska AM, Szpurka H, Tiu RV, Makishima H, Afable M, Huh J, et al. Loss of Heterozygosity 4q24 and TET2 Mutations Associated With Myelodysplastic/Myeloproliferative Neoplasms. Blood (2009) 113(25):6403–10. doi: 10.1182/blood-2009-02-205690
106. Reiter A, Invernizzi R, Cross NCP, Cazzola M. Molecular Basis of Myelodysplastic/Myeloproliferative Neoplasms. Haematologica-the Hematol J (2009) 94(12):1634–8. doi: 10.3324/haematol.2009.014001
107. Solary E, Bernard OA, Tefferi A, Fuks F, Vainchenker W. The Ten-Eleven Translocation-2 (TET2) Gene in Hematopoiesis and Hematopoietic Diseases. Leukemia (2014) 28(3):485–96. doi: 10.1038/leu.2013.337
108. Moran-Crusio K, Reavie L, Shih A, Abdel-Wahab O, Ndiaye-Lobry D, Lobry C, et al. Tet2 Loss Leads to Increased Hematopoietic Stem Cell Self-Renewal and Myeloid Transformation. Cancer Cell (2011) 20(1):11–24. doi: 10.1016/j.ccr.2011.06.001
109. Li Z, Cai X, Cai CL, Wang J, Zhang W, Petersen BE, et al. Deletion of Tet2 in Mice Leads to Dysregulated Hematopoietic Stem Cells and Subsequent Development of Myeloid Malignancies. Blood (2011) 118(17):4509–18. doi: 10.1182/blood-2010-12-325241
110. Tefferi A, Pardanani A, Lim KH, Abdel-Wahab O, Lasho TL, Patel J, et al. TET2 Mutations and Their Clinical Correlates in Polycythemia Vera, Essential Thrombocythemia and Myelofibrosis. Leukemia (2009) 23(5):905–11. doi: 10.1038/leu.2009.47
111. Kosmider O, Gelsi-Boyer V, Ciudad M, Racoeur C, Jooste V, Vey N, et al. TET2 Gene Mutation is a Frequent and Adverse Event in Chronic Myelomonocytic Leukemia. Haematologica (2009) 94(12):1676–81. doi: 10.3324/haematol.2009.011205
112. Lowy DR, Willumsen BM. Function and Regulation of Ras. Annu Rev Biochem (1993) 62:851–91. doi: 10.1146/annurev.bi.62.070193.004223
113. Hirai H, Tanaka S, Azuma M, Anraku Y, Kobayashi Y, Fujisawa M, et al. Transforming Genes in Human Leukemia Cells. Blood (1985) 66(6):1371–8. doi: 10.1182/blood.V66.6.1371.1371
114. Hirsch-Ginsberg C, LeMaistre AC, Kantarjian H, Talpaz M, Cork A, Freireich EJ, et al. RAS Mutations are Rare Events in Philadelphia Chromosome-Negative/Bcr Gene Rearrangement-Negative Chronic Myelogenous Leukemia, But are Prevalent in Chronic Myelomonocytic Leukemia. Blood (1990) 76(6):1214–9. doi: 10.1182/blood.V76.6.1214.1214
115. Braun BS, Tuveson DA, Kong N, Le DT, Kogan SC, Rozmus J, et al. Somatic Activation of Oncogenic Kras in Hematopoietic Cells Initiates a Rapidly Fatal Myeloproliferative Disorder. Proc Natl Acad Sci USA (2004) 101(2):597–602. doi: 10.1073/pnas.0307203101
116. Chan IT, Kutok JL, Williams IR, Cohen S, Kelly L, Shigematsu H, et al. Conditional Expression of Oncogenic K-Ras From its Endogenous Promoter Induces a Myeloproliferative Disease. J Clin Invest (2004) 113(4):528–38. doi: 10.1172/JCI20476
117. Van Meter ME, Díaz-Flores E, Archard JA, Passegué E, Irish JM, Kotecha N, et al. K-Rasg12d Expression Induces Hyperproliferation and Aberrant Signaling in Primary Hematopoietic Stem/Progenitor Cells. Blood (2007) 109(9):3945–52. doi: 10.1182/blood-2006-09-047530
118. Cao R, Wang L, Wang H, Xia L, Erdjument-Bromage H, Tempst P, et al. Role of Histone H3 Lysine 27 Methylation in Polycomb-Group Silencing. Science (2002) 298(5595):1039–43. doi: 10.1126/science.1076997
119. Ernst T, Chase AJ, Score J, Hidalgo-Curtis CE, Bryant C, Jones AV, et al. Inactivating Mutations of the Histone Methyltransferase Gene EZH2 in Myeloid Disorders. Nat Genet (2010) 42(8):722–6. doi: 10.1038/ng.621
120. Morin RD, Johnson NA, Severson TM, Mungall AJ, An J, Goya R, et al. Somatic Mutations Altering EZH2 (Tyr641) in Follicular and Diffuse Large B-Cell Lymphomas of Germinal-Center Origin. Nat Genet (2010) 42(2):181–5. doi: 10.1038/ng.518
121. Yap DB, Chu J, Berg T, Schapira M, Cheng SW, Moradian A, et al. Somatic Mutations at EZH2 Y641 Act Dominantly Through a Mechanism of Selectively Altered PRC2 Catalytic Activity, to Increase H3K27 Trimethylation. Blood (2011) 117(8):2451–9. doi: 10.1182/blood-2010-11-321208
122. Béguelin W, Popovic R, Teater M, Jiang Y, Bunting KL, Rosen M, et al. EZH2 is Required for Germinal Center Formation and Somatic EZH2 Mutations Promote Lymphoid Transformation. Cancer Cell (2013) 23(5):677–92. doi: 10.1016/j.ccr.2013.04.011
123. Gu Z, Liu Y, Cai F, Patrick M, Zmajkovic J, Cao H, et al. Loss of EZH2 Reprograms BCAA Metabolism to Drive Leukemic Transformation. Cancer Discovery (2019) 9(9):1228–47. doi: 10.1158/2159-8290.CD-19-0152
124. Nakamura S, Takayama N, Hirata S, Seo H, Endo H, Ochi K, et al. Expandable Megakaryocyte Cell Lines Enable Clinically Applicable Generation of Platelets From Human Induced Pluripotent Stem Cells. Cell Stem Cell (2014) 14(4):535–48. doi: 10.1016/j.stem.2014.01.011
125. Mechaal A, Menif S, Abbes S, Safra I. EZH2, New Diagnosis and Prognosis Marker in Acute Myeloid Leukemia Patients. Adv Med Sci (2019) 64(2):395–401. doi: 10.1016/j.advms.2019.07.002
126. Stasik S, Middeke JM, Kramer M, Röllig C, Krämer A, Scholl S, et al. EZH2 Mutations and Impact on Clinical Outcome: An Analysis in 1,604 Patients With Newly Diagnosed Acute Myeloid Leukemia. Haematologica (2020) 105(5):e228–31. doi: 10.3324/haematol.2019.222323
127. Blyth K, Cameron ER, Neil JC. The RUNX Genes: Gain or Loss of Function in Cancer. Nat Rev Cancer (2005) 5(5):376–87. doi: 10.1038/nrc1607
128. Chuang LS, Ito K, Ito Y. RUNX Family: Regulation and Diversification of Roles Through Interacting Proteins. Int J Cancer (2013) 132(6):1260–71. doi: 10.1002/ijc.27964
129. Schindler JW, Van Buren D, Foudi A, Krejci O, Qin J, Orkin SH, et al. TEL-AML1 Corrupts Hematopoietic Stem Cells to Persist in the Bone Marrow and Initiate Leukemia. Cell Stem Cell (2009) 5(1):43–53. doi: 10.1016/j.stem.2009.04.019
130. Gaidzik VI, Teleanu V, Papaemmanuil E, Weber D, Paschka P, Hahn J, et al. RUNX1 Mutations in Acute Myeloid Leukemia are Associated With Distinct Clinico-Pathologic and Genetic Features. Leukemia (2016) 30(11):2282. doi: 10.1038/leu.2016.126
131. Matheny CJ, Speck ME, Cushing PR, Zhou Y, Corpora T, Regan M, et al. Disease Mutations in RUNX1 and RUNX2 Create Nonfunctional, Dominant-Negative, or Hypomorphic Alleles. EMBO J (2007) 26(4):1163–75. doi: 10.1038/sj.emboj.7601568
132. Kuo MC, Liang DC, Huang CF, Shih YS, Wu JH, Lin TL, et al. RUNX1 Mutations are Frequent in Chronic Myelomonocytic Leukemia and Mutations at the C-Terminal Region Might Predict Acute Myeloid Leukemia Transformation. Leukemia (2009) 23(8):1426–31. doi: 10.1038/leu.2009.48
133. Gelsi-Boyer V, Trouplin V, Adélaïde J, Aceto N, Remy V, Pinson S, et al. Genome Profiling of Chronic Myelomonocytic Leukemia: Frequent Alterations of RAS and RUNX1 Genes. BMC Cancer (2008) 8:299. doi: 10.1186/1471-2407-8-299
134. Ernst T, Chase A, Zoi K, Waghorn K, Hidalgo-Curtis C, Score J, et al. Transcription Factor Mutations in Myelodysplastic/Myeloproliferative Neoplasms. Haematologica (2010) 95(9):1473–80. doi: 10.3324/haematol.2010.021808
135. Fu XD, Maniatis T. The 35-Kda Mammalian Splicing Factor SC35 Mediates Specific Interactions Between U1 and U2 Small Nuclear Ribonucleoprotein Particles at the 3' Splice Site. Proc Natl Acad Sci USA (1992) 89(5):1725–9. doi: 10.1073/pnas.89.5.1725
136. Wahl MC, Will CL, Lührmann R. The Spliceosome: Design Principles of a Dynamic RNP Machine. Cell (2009) 136(4):701–18. doi: 10.1016/j.cell.2009.02.009
137. Liu HX, Chew SL, Cartegni L, Zhang MQ, Krainer AR. Exonic Splicing Enhancer Motif Recognized by Human SC35 Under Splicing Conditions. Mol Cell Biol (2000) 20(3):1063–71. doi: 10.1128/MCB.20.3.1063-1071.2000
138. Lin S, Coutinho-Mansfield G, Wang D, Pandit S, Fu XD. The Splicing Factor SC35 has an Active Role in Transcriptional Elongation. Nat Struct Mol Biol (2008) 15(8):819–26. doi: 10.1038/nsmb.1461
139. Das R, Yu J, Zhang Z, Gygi MP, Krainer AR, Gygi SP, et al. SR Proteins Function in Coupling RNAP II Transcription to Pre-Mrna Splicing. Mol Cell (2007) 26(6):867–81. doi: 10.1016/j.molcel.2007.05.036
140. Yoshida K, Sanada M, Shiraishi Y, Nowak D, Nagata Y, Yamamoto R, et al. Frequent Pathway Mutations of Splicing Machinery in Myelodysplasia. Nature (2011) 478(7367):64–9. doi: 10.1038/nature10496
141. Patnaik MM, Lasho TL, Finke CM, Hanson CA, Hodnefield JM, Knudson RA, et al. Spliceosome Mutations Involving SRSF2, SF3B1, and U2AF35 in Chronic Myelomonocytic Leukemia: Prevalence, Clinical Correlates, and Prognostic Relevance. Am J Hematol (2013) 88(3):201–6. doi: 10.1002/ajh.23373
142. Meggendorfer M, Roller A, Haferlach T, Eder C, Dicker F, Grossmann V, et al. SRSF2 Mutations in 275 Cases With Chronic Myelomonocytic Leukemia (CMML). Blood (2012) 120(15):3080–8. doi: 10.1182/blood-2012-01-404863
143. Kim E, Ilagan JO, Liang Y, Daubner GM, Lee SC, Ramakrishnan A, et al. SRSF2 Mutations Contribute to Myelodysplasia by Mutant-Specific Effects on Exon Recognition. Cancer Cell (2015) 27(5):617–30. doi: 10.1016/j.ccell.2015.04.006
144. Meggendorfer M, Jeromin S, Haferlach C, Kern W, Haferlach T. The Mutational Landscape of 18 Investigated Genes Clearly Separates Four Subtypes of Myelodysplastic/Myeloproliferative Neoplasms. Haematologica (2018) 103(5):e192–5. doi: 10.3324/haematol.2017.183160
145. Senín A, Arenillas L, Martínez-Avilés L, Fernández-Rodríguez C, Bellosillo B, Florensa L, et al. [Molecular Characterization of Atypical Chronic Myeloid Leukemia and Chronic Neutrophilic Leukemia]. Med Clin (Barc) (2015) 144(11):487–90. doi: 10.1016/j.medcle.2014.03.005
146. Makishima H, Visconte V, Sakaguchi H, Jankowska AM, Abu Kar S, Jerez A, et al. Mutations in the Spliceosome Machinery, a Novel and Ubiquitous Pathway in Leukemogenesis. Blood (2012) 119(14):3203–10. doi: 10.1182/blood-2011-12-399774
147. Nau MM, Lipkowitz S. Comparative Genomic Organization of the Cbl Genes. Gene (2003) 308:103–13. doi: 10.1016/S0378-1119(03)00471-2
148. Thien CB, Walker F, Langdon WY. RING Finger Mutations That Abolish C-Cbl-Directed Polyubiquitination and Downregulation of the EGF Receptor are Insufficient for Cell Transformation. Mol Cell (2001) 7(2):355–65. doi: 10.1016/S1097-2765(01)00183-6
149. Fu JF, Hsu JJ, Tang TC, Shih LY. Identification of CBL, a Proto-Oncogene at 11q23.3, as a Novel MLL Fusion Partner in a Patient With De Novo Acute Myeloid Leukemia. Genes Chromosomes Cancer (2003) 37(2):214–9. doi: 10.1002/gcc.10204
150. Sanada M, Suzuki T, Shih LY, Otsu M, Kato M, Yamazaki S, et al. Gain-of-Function of Mutated C-CBL Tumour Suppressor in Myeloid Neoplasms. Nature (2009) 460(7257):904–8. doi: 10.1038/nature08240
151. Makishima H, Cazzolli H, Szpurka H, Dunbar A, Tiu R, Huh J, et al. Mutations of E3 Ubiquitin Ligase Cbl Family Members Constitute a Novel Common Pathogenic Lesion in Myeloid Malignancies. J Clin Oncol (2009) 27(36):6109–16. doi: 10.1200/JCO.2009.23.7503
152. Jankowska AM, Makishima H, Tiu RV, Szpurka H, Huang Y, Traina F, et al. Mutational Spectrum Analysis of Chronic Myelomonocytic Leukemia Includes Genes Associated With Epigenetic Regulation: UTX, EZH2, and DNMT3A. Blood (2011) 118(14):3932–41. doi: 10.1182/blood-2010-10-311019
153. Kohlmann A, Grossmann V, Klein HU, Schindela S, Weiss T, Kazak B, et al. Next-Generation Sequencing Technology Reveals a Characteristic Pattern of Molecular Mutations in 72.8% of Chronic Myelomonocytic Leukemia by Detecting Frequent Alterations in TET2, CBL, RAS, and RUNX1. J Clin Oncol (2010) 28(24):3858–65. doi: 10.1200/JCO.2009.27.1361
154. Grand FH, Hidalgo-Curtis CE, Ernst T, Zoi K, Zoi C, McGuire C, et al. Frequent CBL Mutations Associated With 11q Acquired Uniparental Disomy in Myeloproliferative Neoplasms. Blood (2009) 113(24):6182–92. doi: 10.1182/blood-2008-12-194548
155. Beekman R, Touw IP. G-CSF and its Receptor in Myeloid Malignancy. Blood (2010) 115(25):5131–6. doi: 10.1182/blood-2010-01-234120
156. Liu F, Wu HY, Wesselschmidt R, Kornaga T, Link DC. Impaired Production and Increased Apoptosis of Neutrophils in Granulocyte Colony-Stimulating Factor Receptor-Deficient Mice. Immunity (1996) 5(5):491–501. doi: 10.1016/S1074-7613(00)80504-X
157. Maxson JE, Gotlib J, Pollyea DA, Fleischman AG, Agarwal A, Eide CA, et al. Oncogenic CSF3R Mutations in Chronic Neutrophilic Leukemia and Atypical CML. N Engl J Med (2013) 368(19):1781–90. doi: 10.1056/NEJMoa1214514
158. Pardanani A, Lasho TL, Laborde RR, Elliott M, Hanson CA, Knudson RA, et al. CSF3R T618I is a Highly Prevalent and Specific Mutation in Chronic Neutrophilic Leukemia. Leukemia (2013) 27(9):1870–3. doi: 10.1038/leu.2013.122
159. Albano F, Zagaria A, Anelli L, Coccaro N, Impera L, Minervini CF, et al. Gene Expression Profiling of Chronic Myeloid Leukemia With Variant T(9;22) Reveals a Different Signature From Cases With Classic Translocation. Mol Cancer (2013) 12:36. doi: 10.1186/1476-4598-12-36
160. Cammarata G, Augugliaro L, La Rosa M, Turri D, Rizzo V, Marfia A, et al. Gene Expression Profile of Chronic Myeloid Leukemia Innately Resistant to Imatinib. Clin Leukemia (2007) 1(4):234–42. doi: 10.3816/CLK.2007.n.014
161. Crossman LC, Mori M, Hsieh YC, Lange T, Paschka P, Harrington CA, et al. In Chronic Myeloid Leukemia White Cells From Cytogenetic Responders and non-Responders to Imatinib Have Very Similar Gene Expression Signatures. Haematologica (2005) 90(4):459–64.
162. Frank O, Brors B, Fabarius A, Li L, Haak M, Merk S, et al. Gene Expression Signature of Primary Imatinib-Resistant Chronic Myeloid Leukemia Patients. Leukemia (2006) 20(8):1400–7. doi: 10.1038/sj.leu.2404270
163. McLean LA, Gathmann I, Capdeville R, Polymeropoulos MH, Dressman M. Pharmacogenomic Analysis of Cytogenetic Response in Chronic Myeloid Leukemia Patients Treated With Imatinib. Clin Cancer Res (2004) 10(1 Pt 1):155–65. doi: 10.1158/1078-0432.CCR-0784-3
164. Ohno R, Nakamura Y. Prediction of Response to Imatinib by Cdna Microarray Analysis. Semin Hematol (2003) 40(2 Suppl 2):42–9. doi: 10.1053/shem.2003.50041
165. Tipping AJ, Deininger MW, Goldman JM, Melo JV. Comparative Gene Expression Profile of Chronic Myeloid Leukemia Cells Innately Resistant to Imatinib Mesylate. Exp Hematol (2003) 31(11):1073–80. doi: 10.1016/j.exphem.2003.08.006
166. Villuendas R, Steegmann JL, Pollán M, Tracey L, Granda A, Fernández-Ruiz E, et al. Identification of Genes Involved in Imatinib Resistance in CML: A Gene-Expression Profiling Approach. Leukemia (2006) 20(6):1047–54. doi: 10.1038/sj.leu.2404197
167. Mittal P, Saliba RM, Giralt SA, Shahjahan M, Cohen AI, Karandish S, et al. Allogeneic Transplantation: A Therapeutic Option for Myelofibrosis, Chronic Myelomonocytic Leukemia and Philadelphia-Negative/BCR-ABL-Negative Chronic Myelogenous Leukemia. Bone Marrow Transplant (2004) 33(10):1005–9. doi: 10.1038/sj.bmt.1704472
168. Koldehoff M, Beelen DW, Trenschel R, Steckel NK, Peceny R, Ditschkowski M, et al. Outcome of Hematopoietic Stem Cell Transplantation in Patients With Atypical Chronic Myeloid Leukemia. Bone Marrow Transplant (2004) 34(12):1047–50. doi: 10.1038/sj.bmt.1704686
169. Koldehoff M, Steckel NK, Hegerfeldt Y, Ditschkowski M, Beelen DW, Elmaagacli AH. Clinical Course and Molecular Features in 21 Patients With Atypical Chronic Myeloid Leukemia. Int J Lab Hematol (2012) 34(1):e3–5. doi: 10.1111/j.1751-553X.2011.01351.x
170. Onida F, de Wreede LC, van Biezen A, Eikema DJ, Byrne JL, Iori AP, et al. Allogeneic Stem Cell Transplantation in Patients With Atypical Chronic Myeloid Leukaemia: A Retrospective Study From the Chronic Malignancies Working Party of the European Society for Blood and Marrow Transplantation. Br J Haematol (2017) 177(5):759–65. doi: 10.1111/bjh.14619
171. Lim SN, Lee JH, Kim DY, Kim SD, Kang YA, Lee YS, et al. Allogeneic Hematopoietic Cell Transplantation in Adult Patients With Myelodysplastic/Myeloproliferative Neoplasms. Blood Res (2013) 48(3):178–84. doi: 10.5045/br.2013.48.3.178
172. Kurzrock R, Kantarjian HM, Shtalrid M, Gutterman JU, Talpaz M. Philadelphia Chromosome-Negative Chronic Myelogenous Leukemia Without Breakpoint Cluster Region Rearrangement: A Chronic Myeloid Leukemia With a Distinct Clinical Course. Blood (1990) 75(2):445–52. doi: 10.1182/blood.V75.2.445.445
173. Montefusco E, Alimena G, Lo Coco F, De Cuia MR, Wang YZ, Aloe Spiriti MA, et al. Ph-Negative and Bcr-Negative Atypical Chronic Myelogenous Leukemia: Biological Features and Clinical Outcome. Ann Hematol (1992) 65(1):17–21. doi: 10.1007/BF01715120
174. Costello R, Lafage M, Toiron Y, Brunel V, Sainty D, Arnoulet C, et al. Philadelphia Chromosome-Negative Chronic Myeloid Leukaemia: A Report of 14 New Cases. Br J Haematol (1995) 90(2):346–52. doi: 10.1111/j.1365-2141.1995.tb05157.x
175. Costello R, Sainty D, LafagePochitaloff M, Gabert J. Clinical and Biological Aspects of Philadelphia-Negative/BCR-Negative Chronic Myeloid Leukemia. Leuk Lymphoma (1997) 25(3-4):225–32. doi: 10.3109/10428199709114162
176. Jabbour E, Kantarjian H, Cortes J, Thomas D, Garcia-Manero G, Ferrajoli A, et al. PEG-IFN-Alpha-2b Therapy in BCR-ABL-Negative Myeloproliferative Disorders - Final Result of a Phase 2 Study. Cancer (2007) 110(9):2012–8. doi: 10.1002/cncr.23018
177. Gerds AT. I Walk the Other Line: Myelodysplastic/Myeloproliferative Neoplasm Overlap Syndromes. Curr Hematol Malignancy Rep (2014) 9(4):400–6. doi: 10.1007/s11899-014-0233-2
178. Hausmann H, Bhatt VR, Yuan J, Maness LJ, Ganti AK. Activity of Single-Agent Decitabine in Atypical Chronic Myeloid Leukemia. J Oncol Pharm Pract (2016) 22(6):790–4. doi: 10.1177/1078155215605662
179. Ades L, Sekeres MA, Wolfromm A, Teichman ML, Tiu RV, Itzykson R, et al. Predictive Factors of Response and Survival Among Chronic Myelomonocytic Leukemia Patients Treated With Azacitidine. Leuk Res (2013) 37(6):609–13. doi: 10.1016/j.leukres.2013.01.004
180. Kantarjian HM, O'Brien S, Cortes J, Giles FJ, Faderl S, Issa JP, et al. Results of Decitabine (5-Aza-2 ' Deoxycytidine) Therapy in 130 Patients With Chronic Myelogenous Leukemia. Cancer (2003) 98(3):522–8. doi: 10.1002/cncr.11543
181. Tong XZ, Li J, Zhou ZH, Zheng D, Liu JR, Su C. Efficacy and Side-Effects of Decitabine in Treatment of Atypical Chronic Myeloid Leukemia. Leuk Lymphoma (2015) 56(6):1911–3. doi: 10.3109/10428194.2014.986477
182. Mao L, You L, Yang M, Li Y, Ye X, Tong H. The First Case of Decitabine Successfully in Treatment of Atypical Chronic Myeloid Leukemia With CEBPA Double Mutation. Chemotherapy (2013) 2:114. doi: 10.4172/2167-7700.1000114
183. Jiang HF, Wu ZL, Ren L, Tao DH, Tong HY. Decitabine for the Treatment of Atypical Chronic Myeloid Leukemia: A Report of Two Cases. Oncol Lett (2016) 11(1):689–92. doi: 10.3892/ol.2015.3977
184. Patnaik MM, Tefferi A. Chronic Myelomonocytic Leukemia: 2016 Update on Diagnosis, Risk Stratification, and Management. Am J Hematol (2016) 91(6):632–42. doi: 10.1002/ajh.24396
185. Talati C, Padron E. An Exercise in Extrapolation: Clinical Management of Atypical CML, MDS/MPN-Unclassifiable, and MDS/MPN-RS-T. Curr Hematol Malig Rep (2016) 11(6):425–33. doi: 10.1007/s11899-016-0350-1
186. Khanna V, Pierce ST, Dao KH, Tognon CE, Hunt DE, Junio B, et al. Durable Disease Control With MEK Inhibition in a Patient With NRAS-Mutated Atypical Chronic Myeloid Leukemia. Cureus (2015) 7(12):e414. doi: 10.7759/cureus.414
187. Levine RL, Loriaux M, Huntly BJ, Loh ML, Beran M, Stoffregen E, et al. The JAK2V617F Activating Mutation Occurs in Chronic Myelomonocytic Leukemia and Acute Myeloid Leukemia, But Not in Acute Lymphoblastic Leukemia or Chronic Lymphocytic Leukemia. Blood (2005) 106(10):3377–9. doi: 10.1182/blood-2005-05-1898
188. Steensma DP, Dewald GW, Lasho TL, Powell HL, McClure RF, Levine RL, et al. The JAK2 V617F Activating Tyrosine Kinase Mutation is an Infrequent Event in Both "Atypical" Myeloproliferative Disorders and Myelodysplastic Syndromes. Blood (2005) 106(4):1207–9. doi: 10.1182/blood-2005-03-1183
189. Gao T, Yu C, Xia S, Liang T, Gu X, Liu Z. A Rare Atypical Chronic Myeloid Leukemia Bcr-Abl1 Negative With Concomitant. Ther Adv Hematol (2020) 11:2040620720927105. doi: 10.1177/2040620720927105
190. Pardanani A, Tefferi A. Is There a Role for JAK Inhibitors in BCR-ABL1-Negative Myeloproliferative Neoplasms Other Than Myelofibrosis? Leuk Lymphoma (2014) 55(12):2706–11. doi: 10.3109/10428194.2014.985159
Keywords: aCML, SETBP1, ETNK1, BCR-ABL1-negative, molecular pathogenesis, MDS/MPN
Citation: Fontana D, Gambacorti-Passerini C and Piazza R (2021) Molecular Pathogenesis of BCR-ABL-Negative Atypical Chronic Myeloid Leukemia. Front. Oncol. 11:756348. doi: 10.3389/fonc.2021.756348
Received: 10 August 2021; Accepted: 22 October 2021;
Published: 11 November 2021.
Edited by:
Argiris Symeonidis, University of Patras, GreeceReviewed by:
Katerina Machova Polakova, Institute of Haematology and Blood Transfusion, CzechiaCopyright © 2021 Fontana, Gambacorti-Passerini and Piazza. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rocco Piazza, cm9jY28ucGlhenphQHVuaW1pYi5pdA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.