
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Oncol. , 06 December 2021
Sec. Hematologic Malignancies
Volume 11 - 2021 | https://doi.org/10.3389/fonc.2021.755188
This article is part of the Research Topic Insights in Hematologic Malignancies: 2021 View all 28 articles
 Lu-Lu Wang1†
Lu-Lu Wang1† Dehong Yan2†
Dehong Yan2† Xue Tang1
Xue Tang1 Mengqi Zhang2Shilin Liu1Ying Wang1Min Zhang1Guichi Zhou1Tonghui Li1Feifei Jiang1Xiaowen Chen1
Mengqi Zhang2Shilin Liu1Ying Wang1Min Zhang1Guichi Zhou1Tonghui Li1Feifei Jiang1Xiaowen Chen1 Feiqiu Wen1
Feiqiu Wen1 Sixi Liu1*
Sixi Liu1* Huirong Mai1*
Huirong Mai1*Background: Despite much improvement in the treatment for acute lymphoblastic leukemia (ALL), childhood ALLs with MLL-rearrangement (MLL-r) still have inferior dismal prognosis. Thus, defining mechanisms underlying MLL-r ALL maintenance is critical for developing effective therapy.
Methods: GSE13159 and GSE28497 were selected via the Oncomine website. Differentially expressed genes (DEGs) between MLL-r ALLs and normal samples were identified by R software. Next, functional enrichment analysis of these DEGs were carried out by Gene Ontology (GO), Kyoto Encyclopedia of Genes and Genomes (KEGG), Gene Set Enrichment Analysis (GSEA), and Search Tool for the Retrieval of Interacting Genes/Proteins (STRING). Then, the key hub genes and modules were identified by Weighted Gene Co-expression Network Analysis (WGCNA). Therapeutically Applicable Research to Generate Effective Treatments (TARGET) ALL (Phase I) of UCSC Xena analysis, qPCR, and Kaplan-Meier analysis were conducted for validating the expression of key hub genes from bone marrow cells of childhood ALL patients or ALL cell lines.
Results: A total of 1,045 DEGs were identified from GSE13159 and GSE28497. Through GO, KEGG, GSEA, and STRING analysis, we demonstrated that MLL-r ALLs were upregulating “nucleosome assembly” and “B cell receptor signal pathway” genes or proteins. WGCNA analysis found 18 gene modules using hierarchical clustering between MLL-r ALLs and normal. The Venn diagram was used to filter the 98 hub genes found in the key module with the 1,045 DEGs. We identified 18 hub genes from this process, 9 of which were found to be correlated with MLL-r status, using the UCSC Xena analysis. By using qPCR, we validated these 9 hub key genes to be upregulated in the MLL-r ALLs (RS4;11 and SEM) compared to the non-MLL-r ALL (RCH-ACV) cell lines. Three of these genes, BCL11A, GLT8D1 and NCBP2, were shown to be increased in MLL-r ALL patient bone marrows compared to the non-MLL-r ALL patient. Finally, Kaplan–Meier analysis indicated that childhood ALL patients with high BCL11A expression had significantly poor overall survival.
Conclusion: These findings suggest that upregulated BCL11A gene expression in childhood ALLs may lead to MLL-r ALL development and BCL11A represents a new potential therapeutic target for childhood MLL-r ALL.
Acute lymphoblastic leukemia (ALL) is the most common childhood malignancy, which is characterized by recurrent chromosomal and molecular abnormalities of immature lymphoid cells (1). About 9% of adult and 3%–5% of children ALL cases harbor rearrangements of the mixed-lineage leukemia (MLL) gene (2, 3). Despite the much improvement in treatment for ALL, childhood ALL with MLL-rearrangement (MLL-r) still displays inferior prognosis attributed to hyperleukocytosis, aggressive form with early relapse, and central nervous system involvement (4–6), distinguishing itself from other leukemia subgroups.
MLL-r genes form fusions with more than 80 partner genes, of which the most common ones are AF4, AF9, ELL, and ENL (7). MLL-AF4 fusion represents the largest subgroup and is associated with a particular poor prognosis (8). Recently, much progress has been made in understanding the MLL-r leukemia process by using array-based technologies. A subset of dysregulated genes were found in MLL-r ALL, especially posterior HOXA genes and MEIS1 genes (9, 10). Previous studies demonstrated that the MLL fusion proteins directly stimulated the histone methylation and target genes transcriptional elongation with the cooperation of DOT1L, PAFc, and pTEFb, leading to the persistent expression of genes that are important for the cell signaling, regulation of hematopoiesis, and transcription (11–13). Thus, exploring novel genes and pathways associated with MLL-r ALL may help to identify potential molecular mechanisms, diagnostic markers, and therapeutic targets in this special subgroup.
In this study, weighted gene co-expression network analysis (WGCNA) was first used to analyze the hub genes of MLL-r ALL samples mined from the Gene Expression Omnibus (GEO) database. The hub genes closely associated with MLL-r ALL were further confirmed in the data of Therapeutically Applicable Research to Generate Effective Treatments (TARGET) ALL (Phase I), ALL cell lines, and childhood ALL patients. We also evaluated the prognostic significance of hub gene expressions in the overall survival (OS) for childhood ALL patients. Our results provided the framework of co-expression gene modules of MLL-r ALL, which would be beneficial to the clinical diagnosis and treatment of childhood MLL-r ALL.
ONCOMINE database (https://www.oncomine.org) is an integrated data-mining platform that analyzes previously published or open-access cancer microarray data. Using the keywords “acute lymphoblastic leukemia” and “Cancer vs. Normal Analysis” for searching ALL databases, we identified two GEO Series (GSE, ID GSE13159 and GSE28497), which could be analyzed based on the same annotation platform. The GSE28497 was analyzed by the GPL96, while the GSE13159 was analyzed by GPL570. As described by the GEO website, all probe sets represented on the GPL96 are identically replicated on GPL570. Thus, we could analyze both GSEs by the same annotation platform GPL570.
GSE13159 contains 74 normal samples and 70 MLL-r ALL samples while GSE28497 includes 4 normal samples and 17 MLL-r ALL samples. Gene expression data from GSE13159 and GSE28497 were integrated manually to two parts, respectively, one containing 78 normal samples and another containing 87 leukemia samples for bioinformatics analysis. The combat function in the sva package was applied to remove the batch effects (Supplementary Figure 1) (14).
The robust multi-array average (RMA) in R software (version 3.6.5) was applied to explore the gene expression data (15). Differentially expressed genes (DEGs) between MLL-r ALL and normal cases were identified using the Bayesian method by the linear models for microarray expression data (LIMMA) package in R software (16). |Log2 fold change (FC)| > 1 and a p-value <0.01 were considered as threshold points. All the above operations were run with scripts in the R software Ggplot2 package was conducted to show the heatmap and volcano map.
DEGs were taken into Search Tool for the Retrieval of Interacting Genes/Proteins (STRING) with the maximum number of interactors = 0 and a confidence score ≥ 0.4 as the cutoff criterion. Then, the result was analyzed by Cytoscape (17). Top 40 genes were screened using the plug-in CytoHubba in Cytoscape (18). CytoHubba provides a simple interface to analyze the node essentiality on the selected network with 11 scoring methods. Then, the bio-functional modules in the top 40 genes were explored using a plug-in MCODE in Cytoscape with a Node Score Cutoff of 0.2, a degree cutoff of 2, and a k-Core of 2. Then, the genes in the three modules were taken into the DAVID website. Gene ontology (GO) and The Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses were carried out based on DAVID (http://david.ncifcrf.gov/summary.jsp). The significance threshold was p < 0.05.
Gene set enrichment analysis (GSEA) was conducted between MLL-r ALL and normal samples. Expression dataset from GSE13159 and GSE28497 was converted to the tab delimited gene cluster text (GCT) format following the operations according to the protocol (http://www.gsea-msigdb.org/gsea/). False discovery rate (FDR) < 0.05 and |normalized enrichment score (NES)| > 1 were regarded as the cutoff criteria.
WGCNA package of R software was applied to uncover the correlation among genes. Firstly, gene expression data from GSE13159 and GSE28497 were input into R software to inspect good genes and samples. Sample clustering was used to detect outliers and match the samples with their characteristics. The soft thresholding power of β was set at 6 to ensure a scale-free network. The minimum number of module genes was set at 30. The hierarchical clustering dendrogram summarized the gene modules with different colors. Heatmap and topological overlap matrix (TOM) plot were used to visualize the module structure. The clinical traits were classified into two subtypes, “Normal” and “Leukemia”, for constructing module–trait relationships. For each expression profile, the gene significance (GS) and module membership (MM) were defined as the correlation value for each trait and each module eigengene, respectively. Hub genes are a class of highly connected genes, which have high connectivity with other genes in the same gene module and clinical trait.
The gene expression data of TARGET ALL (Phase I) were analyzed by the University of California, Santa Cruz (UCSC) Xena (http://xena.ucsc.edu/). Hub gene expression levels were calculated between MLL-r and non-MLL-r ALL patients. The minimal residual disease (MRD) status on Day 8 and Day 29 of patients were also detected. The TARGET ALL (Phase I) project was obtained from patients enrolled on biology studies and clinical trials managed through the COG, POG 9906 (clinical trial for patients with newly diagnosed ALL between March 2000 and April 2003 that were defined as high risk for relapse). Patient samples for full characterization were chosen based on the following criteria: the disease onset at >9 years of age; did not have white blood cell count > 50,000/µl; did not express BCR/ABL fusion gene; were not known to be hypodiploid (DNA index > 0.95); and achieved remission (fewer than 5% blasts) following the standard two rounds of induction therapy. The primary patient samples were collected at diagnosis and gene expressions were analyzed following the protocol of Human Genome U133 Plus 2.0 Array (Affymetrix).
Five childhood MLL-r ALL and 30 childhood non-MLL-r ALL bone marrow (BM) samples were procured from the newly diagnosed patients being treated in Shenzhen Children’s Hospital from 2018 to 2020. Informed consent was obtained from all patients or their parents. Experiments involving human materials were approved by the ethics committee of Shenzhen Children’s Hospital (approval number 202105102) and carried out according to the Declaration of Helsinki. Primary mononuclear cells were collected from the bone marrow samples by Ficoll density gradient centrifugation.
RS4;11 and RCH-ACV cells (ATCC) were maintained in RPMI 1640 with 10% fetal bovine serum (FBS) supplemented with 1% penicillin and streptomycin. SEM cells (ATCC) were maintained in IMDM with 10% FBS, 1% penicillin, and streptomycin. Cells were cultured in a cell incubator and maintained at 5% CO2 and 37°C.
Total RNA of cells were reverse transcribed to cDNA with equal RNA volume (1 μg) using the PrimeScript™ RT Master Mix (Takara). Quantitative real-time PCR (qPCR) was performed using SYBR Green qPCR Master Mix (MedChem Express, USA) and detected on CFX96™ Real-Time System (BIO-RAD). Primer sequences for mRNA expression detection are shown in Supplementary Table 1. All experiments were repeated at least in triplicate. Gene expression levels were calculated relative to the housekeeping gene β-actin.
The gene expression data of TARGET ALL (Phase I) were obtained from USCS Xena, and the clinical information was downloaded from the official website of the TARGET program (https://ocg.cancer.gov/programs/target). Finally, a total of 207 patients with gene expression data were analyzed by Kaplan–Meier analysis. The ggplot2 of R software was used to plot the Kaplan–Meier survival curve. The population was stratified by the upper 25th percentile (high) versus the lower 25th percentile (low) expression for key hub genes mRNA. The correlations between hub gene expression and OS, the hazard ratio (HR) with 95% confidence intervals, and p-value were determined by the Log-rank test. Log-rank test p < 0.05 was considered a significant difference. The results published here are in whole or part based on data generated by the TARGET (https://ocg.cancer.gov/programs/target) initiative, phs000218.
Student’s t-test of variance was used for comparing the statistical differences of mRNA expression of BM samples and ALL cell lines. All the analyses were two‐sided and p < 0.05 was considered to be a significant difference.
Using the keywords “acute lymphoblastic leukemia” and “Cancer vs. Normal Analysis” for searching ALL databases in ONCOMINE website, we identified two GEO Series (GSE13159 and GSE28497) based on the same annotation platform. Gene expression data from GSE13159 and GSE28497 were integrated manually to two datasets (MLL-r ALL dataset containing 87 leukemia samples and normal samples dataset containing 78 normal samples, respectively). The gene expression data were analyzed using LIMMA package of R software and its extension packages. After removing the batch effect, we identified a total of 1,045 DEGs, 301 of which were upregulated genes while 744 were downregulated genes using adjusted |logFC| > 1 and p-value < 0.01 as cutoff threshold points. The most aberrantly expressed DEGs were marked in the volcano map, such as upregulated genes LAMP5, CSRP2, GPM6B, SOCS2, MEF2C, MEIS1, and FLT3 and downregulated genes NGFRAP1, PDXK, and DEF8 (Figure 1A). The heatmap of DEGs is shown in Figure 1B.
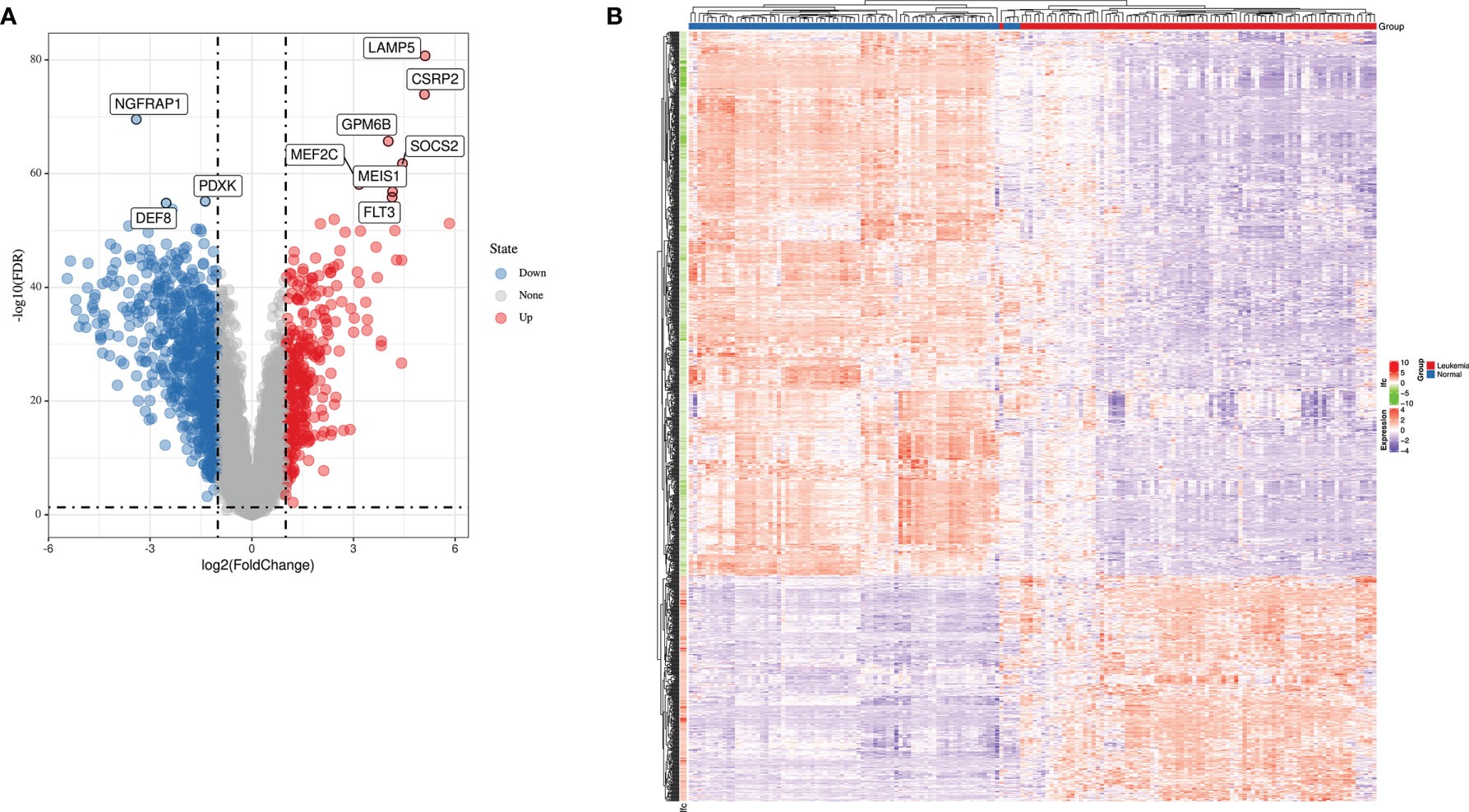
Figure 1 The expression profiles of MLL-r ALL. (A) Volcano map of differently expressed genes between MLL-r ALL and normal samples. (B) Heatmap of the DEGs according to the value of |logFC|.
To gain insight into a more comprehensive knowledge of DEGs of MLL-r ALL vs. normal samples, the GO and KEGG pathway analysis of DEGs was carried out via DAVID online analysis tool. GO analysis showed that MLL-r ALLs were mainly upregulating “transcription from RNA polymerase II promoter, cell proliferation, nucleosome assembly” genes and downregulating “immune response, inflammatory response, cell adhesion” genes (Figure 2A). KEGG analysis showed upregulation in “transcriptional misregulation in cancer, pathways in cancer, PI3K-Akt signaling pathway” and downregulation in “cytokine-cytokine receptor interaction, hematopoietic cell lineage, chemokine signaling pathways” (Figure 2B). Additionally, GSEA showed MLL-r ALLs to be enriched in “primary immunodeficiency, B cell receptor signaling pathway” pathways (Figure 3A) and negatively correlated with “complement and coagulation cascades, cytokine-cytokine receptor interaction, leukocyte transendothelial migration” pathways (Figure 3B).

Figure 2 PPI network by STRING. (A) GO enrichment analysis of up- and downregulated DEGs. (B) KEGG pathway analysis of up- and downregulated DEGs.
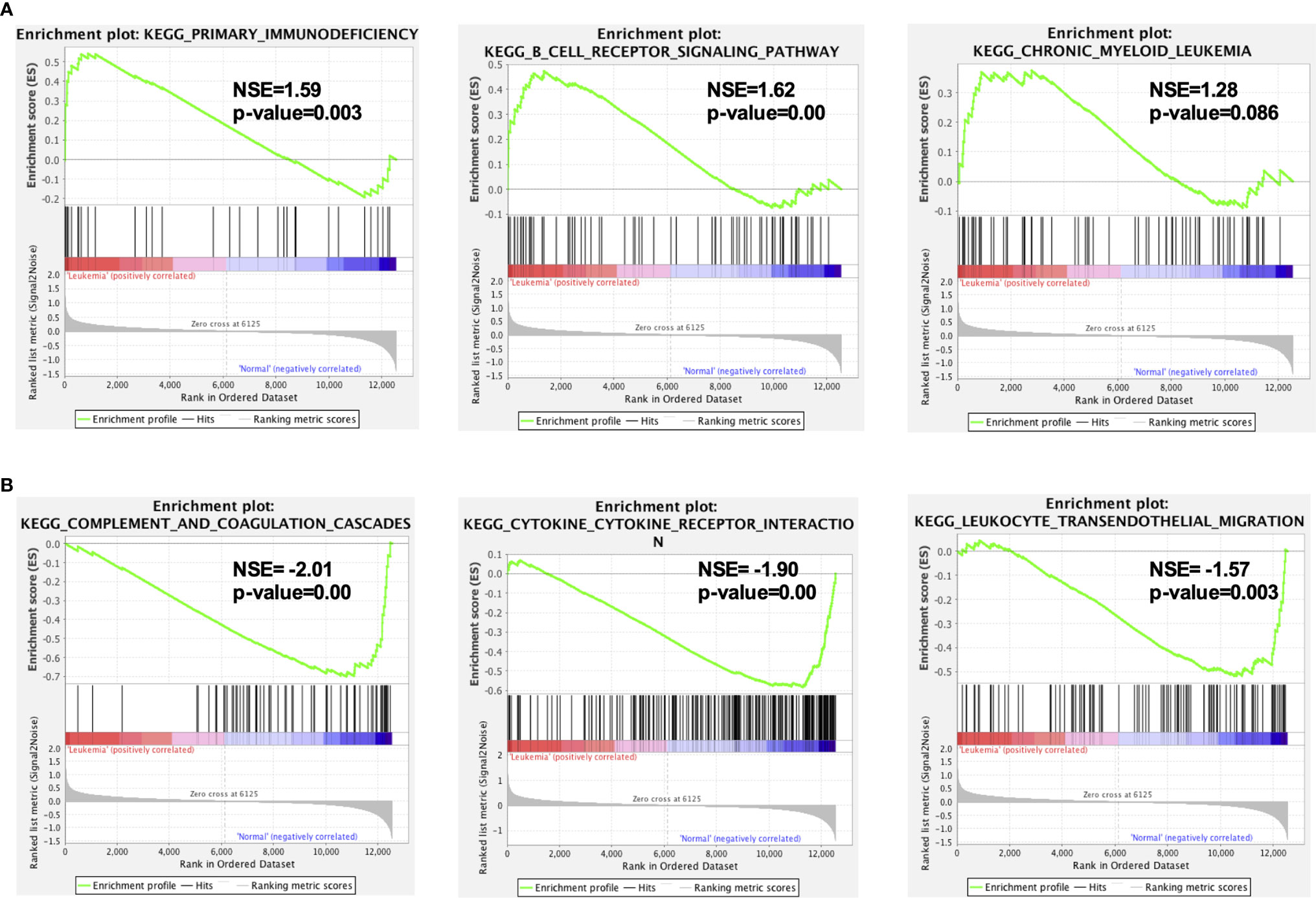
Figure 3 GSEA indicated the KEGG pathways in MLL-r ALL. (A) The gene sets enriched in MLL-r ALL samples were related to primary immunodeficiency and B-cell receptor signaling pathway (p < 0.05). (B) The gene sets enriched in normal samples were mainly related to complement and coagulation cascades, cytokine–cytokine receptor interaction, and leukocyte transendothelial migration (p < 0.05).
Based on the STRING website, we built a protein–protein interaction network of up- and downregulated DEGs from MLL-r ALL vs. normal samples (Figures 4A, 5A). In order to identify the key up- and downregulated DEGs between MLL-r ALL and normal samples, we adopted all the 11 methods in CytoHubba application, a plug-in of Cytoscape. Via calculation and analysis, the top 40 genes of up- and downregulated DEGs were identified separately (Figure 4B and Supplementary Figure 2A). Afterwards, the MCODE was employed to calculate the k-core of each gene and identify the critical networks in the top 40 genes. The upregulated gene with the highest k-core comprised three networks, which were involved in three important KEGG pathways: nucleosome assembly, B-cell proliferation, and transcriptional misregulation in cancer (Figure 4C). The top 40 downregulated DEGs with the highest k-score made up one important network, which was enriched in siderophore-dependent iron import into cell (Supplementary Figure 2B). Through GO, KEGG, GSEA, and STRING analysis, we demonstrated that MLL-r ALLs were upregulating “nucleosome assembly” and “B cell receptor signal pathway” genes or proteins and downregulating “complement and coagulation cascades” and “cytokine-cytokine receptor interaction” genes or proteins. These pathways were identified by at least two different biostatistics methods.

Figure 4 PPI networks of upregulated DEGs by STRING Search Tool. (A) The PPI network was constructed by STRING based on upregulated DEGs. (B) The 40 genes with highest CytoHubba scores were shown in a circle. The color scheme of each node from red to yellow represents the scores from high to low, respectively. (C) The genes with highest CytoHubba scores made up three critical subnetworks.
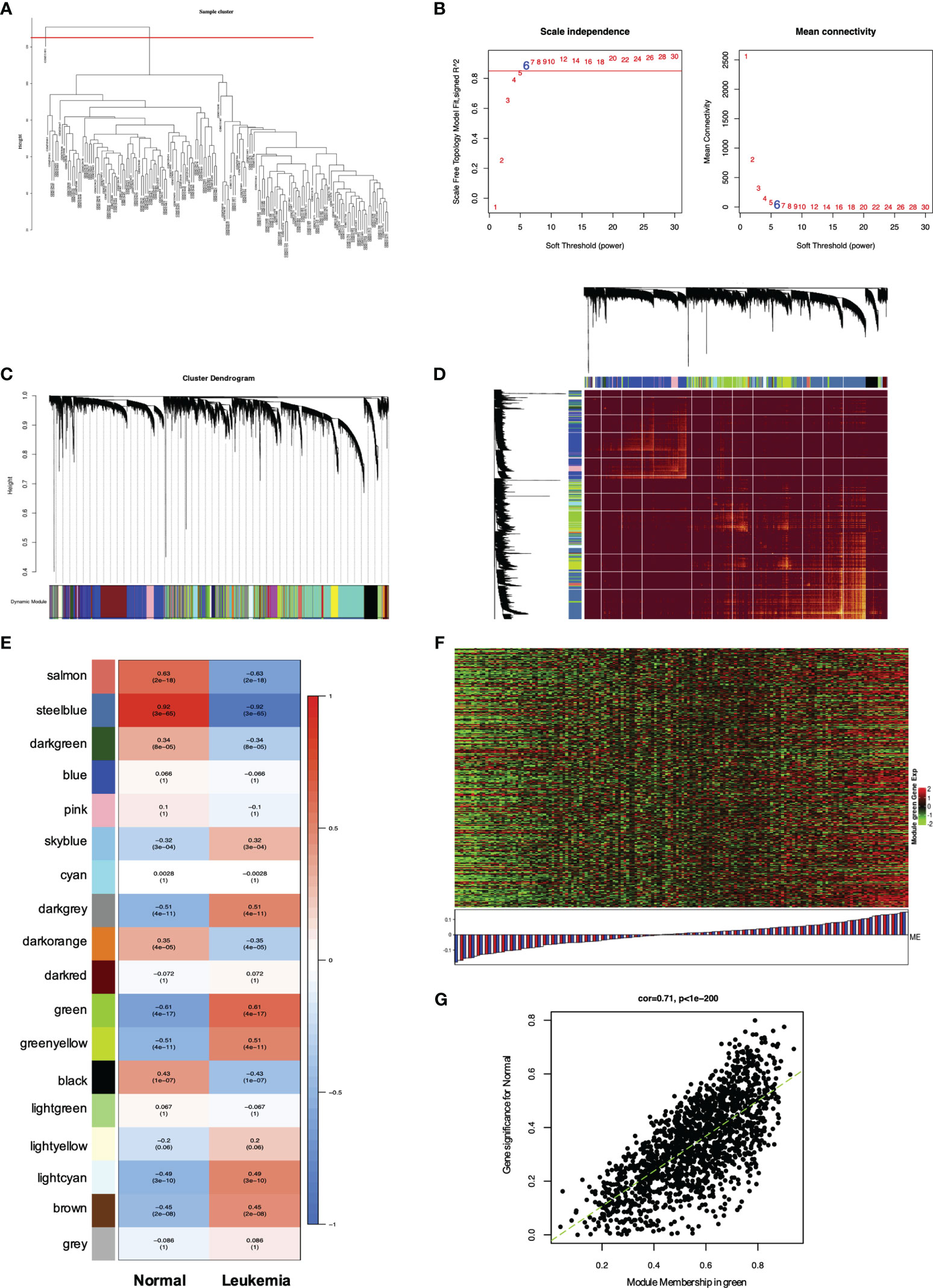
Figure 5 WGCNA of genes and modules in MLL-r ALL. (A) Cluster dendrogram of 165 samples. (B) Analysis of the scale-free fit index for various soft thresholding powers (β) and the mean connectivity for soft threshold power. (C) Hierarchical cluster tree showing adjacency modules based on topological overlap. (D) Heatmap of all DEGs formed a TOM plot to visualize the module structure. In the TOM plot, light yellow color represents low overlap and darker yellow color indicates higher overlap. Left side and the top side of the heatmap show the gene dendrogram and module assignment. (E) Correlation between each module and trait. Each row represents a module eigengene and each column represents a clinic trait. Each cell contains the correlation and p-value of each module. (F) Heatmap for the DEGs in the green module. (G) A scatter plot of eigengenes significance versus modular membership in the green module. Intramodular analysis of the genes found in the green module showed a high correlation with leukemia, with p < 1e-200 and correlation = 0.71.
WGCNA has been widely and successfully used to identify candidate biomarkers and therapeutic targets in various complicated diseases (19–21). To clarify the key modules and hub genes in MLL-r ALL, a total of 165 samples from GSE13159 and GSE28497 were input into R software for WGCNA, which was employed to uncover the highly correlated genes and the coexpression networks of MLL-r ALL. One GEO sample (GSM), GSM331691, was excluded from the analysis due to poor quality (Figure 5A). The power of β = 6 was set as the soft threshold for a scale-free network (Figure 5B). Subsequently, 18 gene modules were generated by the hierarchical clustering dendrogram, ranging in size from 2,947 (blue module) to 41 (gray module) (Figure 5C). After analyzing the interactions between the 18 modules, the TOM plot of a gene network was constructed with the corresponding hierarchical clustering dendrogram and the modules (Figure 5D). Based on the correlation between module eigengenes and clinical traits, the co-expressed genes were found primarily clustered in the green and steel blue modules. Among the modules, the green module was the most relevant with leukemia traits (Figure 5E). The heatmap of gene expression in the green module is shown in Figure 5F. We also explored the gene significance and module membership of the genes in the 18 modules. As shown in Figure 5G, the green module had significant correlations with leukemia trait. Eventually, 98 genes in the green module were selected for hub genes with a cutoff of intra-modular connectivity >0.85, demonstrating that these genes were not only the key components in module but also highly correlated with the leukemia trait. Taken together, we found 18 gene modules using hierarchical clustering between MLL-r ALLs and normal samples by WGCNA. The “Green” module was found to be most relevant with leukemia, 98 genes of which having the highest correlation with leukemia traits were selected for hub genes.
To gain insight into the valuable hub genes in the green module, we used a Venn diagram to filter the 98 hub genes found in the “Green” module with the 1,045 DEGs from Figure 1. As shown in the Venn diagram, we identified 18 hub genes from this process (Figure 6A). Since the higher expression of 18 hub genes in MLL-r ALL is compared to the normal samples, we wondered which hub genes were also highly expressed in MLL-r ALL compared to non-MLL-r ALL. Thus, we further selected the TARGET ALL (Phase I) dataset to verify the differential expression of 18 hub genes between MLL-r and non-MLL-r ALL patients (22). Based on the results from UCSC Xena browser, nine key hub genes (AKR1A1, BCL11A, DCLRE1C, DHTKD1, GLT8D1, NCBP2, PARP1, PTER, and STK39) were found highly expressed in the MLL-positive samples compared to the MLL-negative samples (Figure 6B). Accordingly, to further determine the potential biomarkers for MLL-r ALL, we assessed hub gene expression in ALL cell lines and childhood ALL bone marrow samples. We found that the higher expression level of nine key hub genes were also observed in MLL-AF4 ALL cell lines (RS4;11 and SEM) compared to the non-MLL-r ALL cell line (RCH-ACV) (Figure 7A). Moreover, in our single clinical center, MLL-r ALLs had significantly higher mRNA expressions of BCL11A, GLT8D1, and NCBP2 than non-MLL-r ALL patients (Figure 7B). However, there was no difference in AKR1A1, DCLRE1C, DHTKD1, PARP1, PTER, and STK39 expression levels between MLL-r ALL and non-MLL-r ALL patients (not shown). These data suggest that upregulated BCL11A, GLT8D1, and NCBP2 gene expression may be associated with the rearrangement of MLL in childhood ALL.

Figure 6 The expression level of key hub genes in childhood MLL-r ALL. (A) The Venn diagram showed hub genes identified from DEGs and WGCNA. (B) Expression of AKR1A1, BCL11A, DCLRE1C, DHTKD1, GLT8D1, NCBP2, PARP1, PTER, and STK39 in MLL-r ALL was highly correlated with the MLL status in the TARGET ALL (Phase I) dataset (p < 0.05). The red color represents positive status and the blue color represents negative status of MLL fusion and MRD.
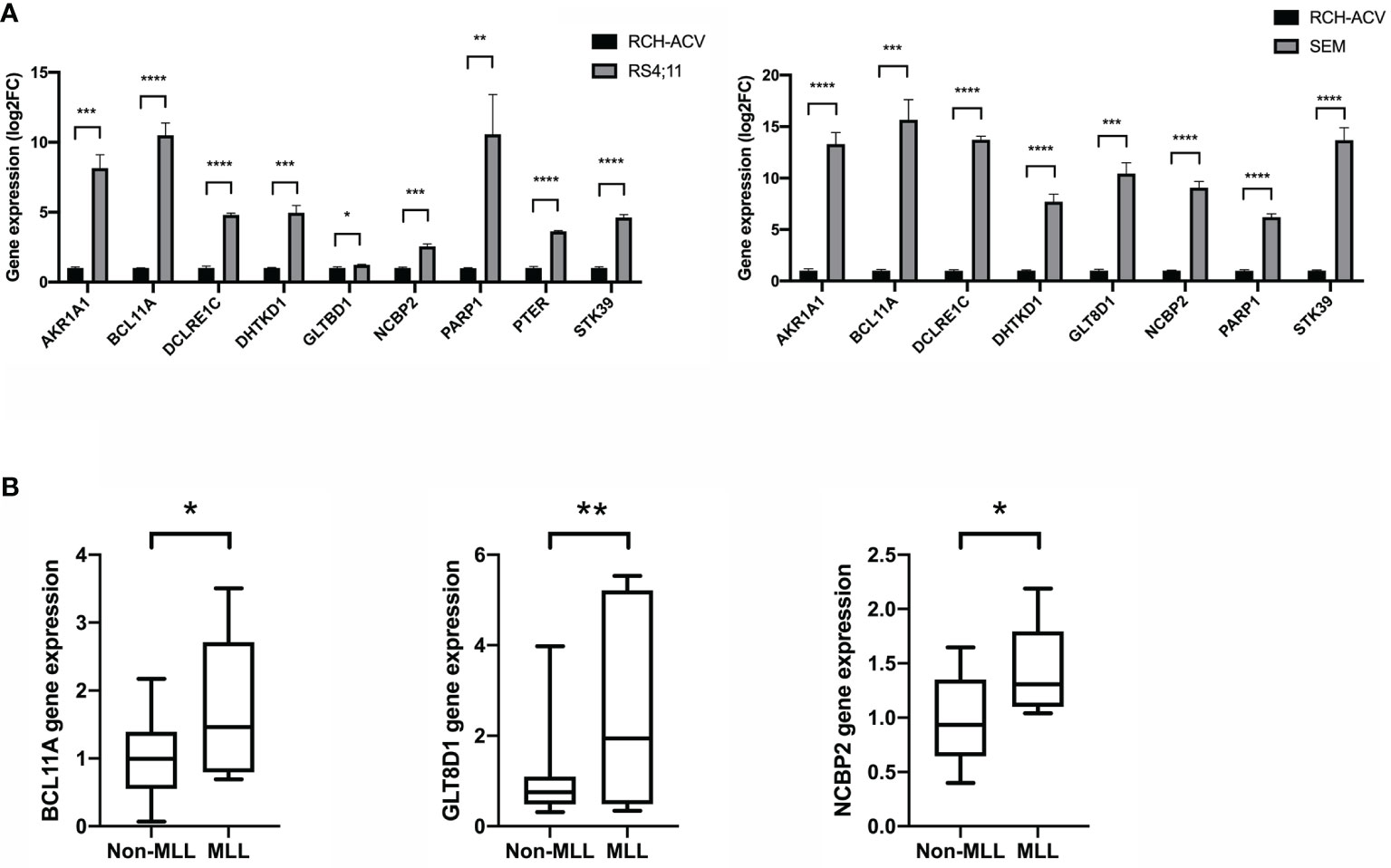
Figure 7 The gene expression level of key hub genes in MLL-r ALL and non-MLL-r ALL patients. (A) The mRNA expression of key hub genes were measured in RS4:11, SEM, and RCH-ACV cell lines. Data are normalized to β-actin and are representative of three independent experiments (*p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001). (B) The mRNA expression was detected in the BM of MLL-r ALL and non-MLL-r ALL patients. Results are expressed as a fold of the non-MLL-r ALL that is set as 1 (*p < 0.05; **p < 0.01).
Given the relationship between key hub genes and MLL status, we next examined the prognostic significance of hub genes in TARGET ALL (Phase I) datasets. We identified by Kaplan–Meier analysis that the high level of BCL11A mRNA expression was associated with inferior OS [HR = 0.35 (0.15–0.84), p = 0.014] (Figure 8). Nevertheless, there was no significant difference between the survival rate and the expression of AKR1A1, DCLRE1C, DHTKD1, GLT8D1, NCBP2, PARP1, PTER, and STK39 (Supplementary Figure 3). Together, these results demonstrated that childhood ALL patients with high BCL11A expression had significantly poor OS, and BCL11A represents a new potential therapeutic target for pediatric MLL-r ALL.
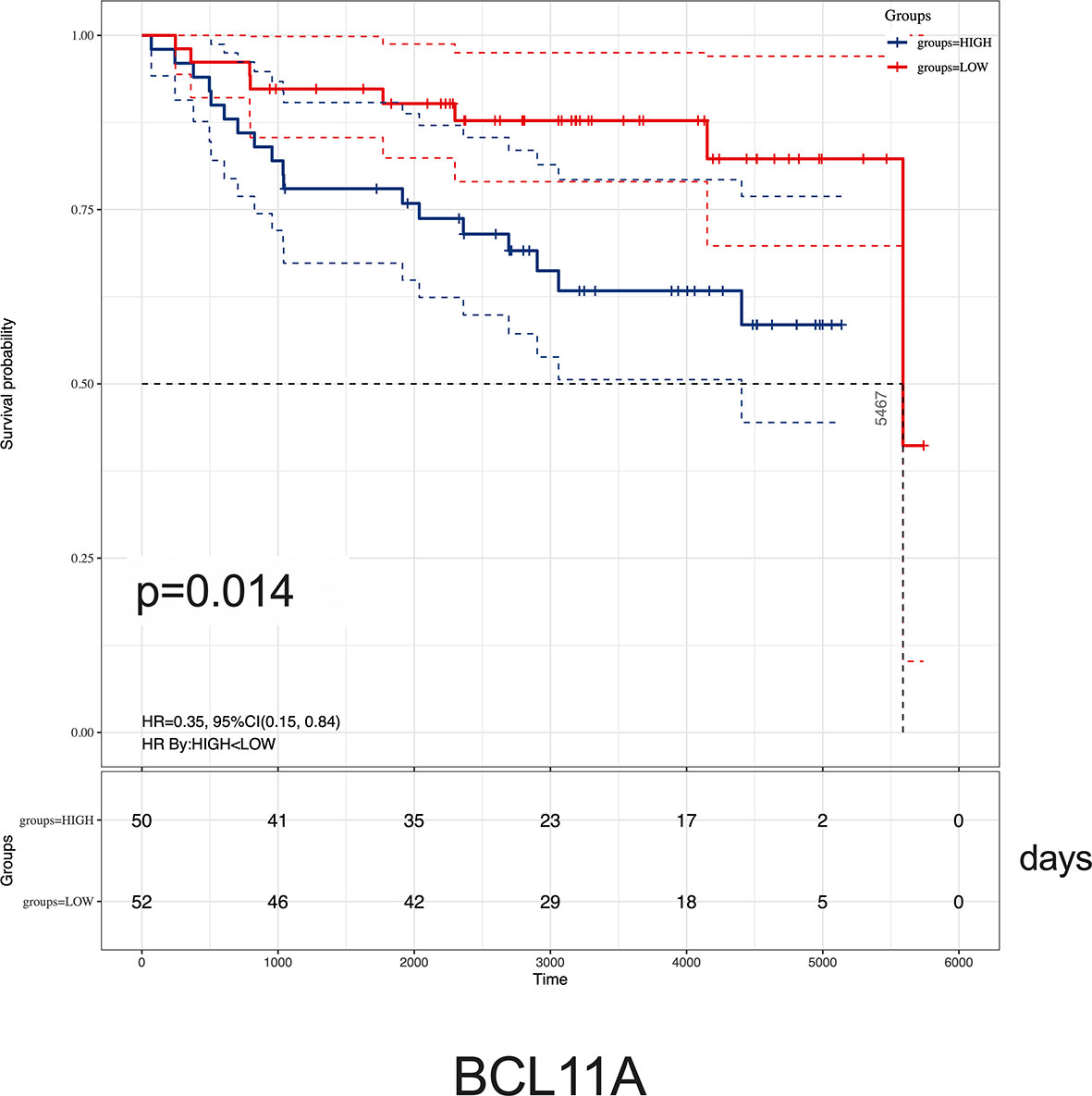
Figure 8 Kaplan–Meier analysis of BCL11A expression in childhood ALL. Survival cure comparing patients with high (blue) vs. low (red) BCL11A expression were plotted using a Log-rank test [HR = 0.35 (0.15–0.84), p = 0.014]. The unit of time is measured in days. The median survival time is 5,467 days.
Previous studies highlight the simple landscape of gene mutation, low DNA copy-number abnormalities, and limited window of genetic therapeutic targets for infant MLL-r ALLs (10, 23, 24). Transcriptional subversion and global gene expression alternation of MLL-r ALL play a greater role in this disease (25). Several microarray studies have already demonstrated the identification of DEGs related to distinct clinical and therapeutically relevant classes of leukemias (26, 27). However, the molecular alternation observed in MLL-r leukemia patients is rather complex and requires a more differentiated view. To gain a larger sample size, we combined two gene microarray expression datasets from GEO and conduct R software to analyze the DEGs in combined dataset.
As analyzed, in the upregulated gene or protein in MLL-r ALL samples, “B cell receptor signal pathway” was found to be significant in GO, KEGG pathway, and GSEA (Figures 2, 3A), while “nucleosome assembly” was significant in GO and PPI network (Figures 2, 4C). These results indicated that nucleosome assembly and B-cell receptor signal pathway were playing a core role in MLL-r ALLs. It is reported that MLL fusion genes enhance the process of transcriptional elongation by directly or indirectly binding to RNA polymerase II, leading to the alternation of whole genetic expression (28, 29). With the help of RNA polymerase II, MLL fusion genes drive the proliferation and self-renewal of immature hematopoietic cells by upregulating posterior HOXA genes and their cofactor MEIS1 (30, 31). Similarly, GO analysis in our study demonstrated that the upregulated DEGs were significantly related with RNA polymerase II pathway (Figure 2A). HOXA and MEIS1 were also identified in our PPI analysis (Figure 4C). Thus, the “transcription from RNA polymerase II” pathway may also be involved in the MLL-r ALL pathogenesis in this study.
WGCNA is widely used to explore the huge and complex relationships among genes across microarray or RNA sequence data. It provides a convenient and effective solution for screening core genes (32). Here, we first used WGCNA to explore the relationship of gene modules and clinical traits in MLL-r ALLs. As shown in the result, 18 upregulated genes were screened as hub genes by conducting WGCNA (Figure 7A), among which 9 key hub genes were confirmed to be highly associated with the MLL-r status in the TARGET ALL (Phase I) dataset (Figure 7B). In our childhood MLL-r ALL patients, BCL11A, GLT8D1, and NCBP2 had significantly higher expression in MLL-r ALL patients than that in the non-MLL-r ALL patients. One limitation of this study is that there were only five MLL-r ALL BM patients for qPCR in the data analysis of our single center. Thus, further investigation is required to demonstrate the relationship of MLL-r ALL patients and DEGs with high-throughput sequencing for multi-center data analysis.
It is noteworthy to mention that BCL11A, a C2H2 zinc finger transcription factor, was initially identified as a chromosomal translocation oncogene in B-cell malignancies (33, 34). It modulated hemoglobin switching by directing γ-globin gene promoter repression (35). Recent studies uncovered that BCL11A was a critical oncogene in many other cancers like triple-negative breast cancer, lung squamous carcinoma, colorectal cancer, and laryngeal squamous cell carcinoma (36–39). In acute myeloid leukemia, high expression of BCL11A and MDR1 was significantly associated with the poor prognosis (40). As analyzed using Kaplan–Meier, we reported that childhood ALL patients with high BCL11A expression showed inferior OS (Figure 8). Given the significant difference of BCL11A found by integrated analyses, we proposed that BCL11A could be considered as a potential target for ALL treatment and the molecular mechanisms study should be necessary to elucidate the role of BCL11A in MLL-r ALLs.
The high sensitivity of MRD has a profound influence on the treatment adjustment and the outcome of ALL patients during follow-up. MRD monitoring has been used to determine the ALL stratification in all childhood ALL. It was reported that MRD-positive status in Day 8 and Day 29 was associated with shorter event-free survival (EFS) in all risk groups (41). MLL-r ALL with positive MRD has significant worse EFS compared to those with negative MRD (42). To determine whether the poor prognosis that occurs with positive MRD is because of the hub gene expression, we examined the Day 8 and Day 29 MRD status of childhood ALL patients in the TARGET ALL (Phase I) dataset and their relationship with hub gene expression. However, there was no significant relationship between gene expression and MRD status (Figure 6B), neither in our single-center clinical data (not shown).
In the current study, we first integrated the information on DEGs through different bioinformatics analysis and identified that the nuclear assembly and B-cell receptor signaling pathway were the key pathways in MLL-r leukemia. Furthermore, we combined the results of WGCNA and DEGs, validated by clinical samples, and found that BCL11A, GLT8D1, and NCBP2 were the key hub genes highly associated with the MLL status in childhood MLL-r ALL patients. Childhood ALL patients with high BCL11A expression had significantly poor OS. These findings suggest that upregulated BCL11A hub gene expression in childhood ALLs may lead to MLL-r ALL development and BCL11A represents a new potential therapeutic target for childhood MLL-r ALL.
Publicly available datasets were analyzed in this study. This data can be found here: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE13159; https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE28497; https://ocg.cancer.gov/programs/target/resources.
The studies involving human participants were reviewed and approved by the ethics committee of Shenzhen Children’s Hospital. Written informed consent was obtained from all patients or their parents.
L-LW and DY conceived the project, analyzed the datasets, and wrote the manuscript. XT, MQZ, and MZ performed the experiments. SLL, YW, GZ, TL, and FJ contributed analysis tools. XC, FW, SXL, and HM revised the manuscript and discussed the data. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We appreciate the GEO datasets (GSE13159 and GSE28497) and TARGET ALL project for free using. This work was supported by Sanming Project of Medicine in Shenzhen (SZSM201512033), Guangdong Medical Science and Technology Research Project (A2020101), Shenzhen Fund for Guangdong Provincial High-level Clinical Key Specialties (SZGSP012), Shenzhen Key Medical Discipline Construction Fund (SZXK034), Shenzhen Healthcare Research Project (SZLY2018015), National Key R&D Program of China (2019YFA0906100), National Natural Science Foundation of China (82071772), and Shenzhen Basic Science Research Project (JCYJ20190807161419228).
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2021.755188/full#supplementary-material
1. Dores GM, Devesa SS, Curtis RE, Linet MS, Morton LM. Acute Leukemia Incidence and Patient Survival Among Children and Adults in the United States, 2001-2007. Blood (2012) 119(1):34–43. doi: 10.1182/blood-2011-04-347872
2. Moorman AV, Ensor HM, Richards SM, Chilton L, Schwab C, Kinsey SE, et al. Prognostic Effect of Chromosomal Abnormalities in Childhood B-Cell Precursor Acute Lymphoblastic Leukaemia: Results From the UK Medical Research Council ALL97/99 Randomised Trial. Lancet Oncol (2010) 11(5):429–38. doi: 10.1016/S1470-2045(10)70066-8
3. Armstrong SA, Look AT. Molecular Genetics of Acute Lymphoblastic Leukemia. J Clin Oncol Off J Am Soc Clin Oncol (2005) 23(26):6306–15. doi: 10.1200/JCO.2005.05.047
4. Stam RW, den Boer ML, Schneider P, Nollau P, Horstmann M, Beverloo HB, et al. Targeting FLT3 in Primary MLL-Gene-Rearranged Infant Acute Lymphoblastic Leukemia. Blood (2005) 106(7):2484–90. doi: 10.1182/blood-2004-09-3667
5. Pieters R, Schrappe M, De Lorenzo P, Hann I, De Rossi G, Felice M, et al. A Treatment Protocol for Infants Younger Than 1 Year With Acute Lymphoblastic Leukaemia (Interfant-99): An Observational Study and a Multicentre Randomised Trial. Lancet (London England) (2007) 370(9583):240–50. doi: 10.1016/S0140-6736(07)61126-X
6. Tomizawa D, Koh K, Hirayama M, Miyamura T, Hatanaka M, Saikawa Y, et al. Outcome of Recurrent or Refractory Acute Lymphoblastic Leukemia in Infants With MLL Gene Rearrangements: A Report From the Japan Infant Leukemia Study Group. Pediatr Blood Cancer (2009) 52(7):808–13. doi: 10.1002/pbc.21975
7. Meyer C, Burmeister T, Gröger D, Tsaur G, Fechina L, Renneville A, et al. The MLL Recombinome of Acute Leukemias in 2017. Leukemia (2018) 32(2):273–84. doi: 10.1038/leu.2017.213
8. Thomas M, Gessner A, Vornlocher H-P, Hadwiger P, Greil J, Heidenreich O. Targeting MLL-AF4 With Short Interfering RNAs Inhibits Clonogenicity and Engraftment Of T (-Positive Human Leukemic Cells. Blood (2005) 106(10):3559–66. doi: 10.1182/blood-2005-03-1283
9. Krivtsov AV, Armstrong SA. MLL Translocations, Histone Modifications and Leukaemia Stem-Cell Development. Nat Rev Cancer (2007) 7(11):823–33. doi: 10.1038/nrc2253
10. Andersson AK, Ma J, Wang J, Chen X, Gedman AL, Dang J, et al. The Landscape of Somatic Mutations in Infant MLL-Rearranged Acute Lymphoblastic Leukemias. Nat Genet (2015) 47(4):330–7. doi: 10.1038/ng.3230
11. Muntean AG, Tan J, Sitwala K, Huang Y, Bronstein J, Connelly JA, et al. The PAF Complex Synergizes With MLL Fusion Proteins at HOX Loci to Promote Leukemogenesis. Cancer Cell (2010) 17(6):609–21. doi: 10.1016/j.ccr.2010.04.012
12. Lin C, Smith ER, Takahashi H, Lai KC, Martin-Brown S, Florens L, et al. AFF4, a Component of the ELL/P-TEFb Elongation Complex and a Shared Subunit of MLL Chimeras, can Link Transcription Elongation to Leukemia. Mol Cell (2010) 37(3):429–37. doi: 10.1016/j.molcel.2010.01.026
13. Yokoyama A, Lin M, Naresh A, Kitabayashi I, Cleary ML. A Higher-Order Complex Containing AF4 and ENL Family Proteins With P-TEFb Facilitates Oncogenic and Physiologic MLL-Dependent Transcription. Cancer Cell (2010) 17(2):198–212. doi: 10.1016/j.ccr.2009.12.040
14. Leek JT, Johnson WE, Parker HS, Jaffe AE, Storey JD. The Sva Package for Removing Batch Effects and Other Unwanted Variation in High-Throughput Experiments. Bioinformatics (2012) 28(6):882–3. doi: 10.1093/bioinformatics/bts034
15. Gautier L, Cope L, Bolstad BM, Irizarry RA. Affy–Analysis of Affymetrix GeneChip Data at the Probe Level. Bioinformatics (2004) 20(3):307–15. doi: 10.1093/bioinformatics/btg405
16. Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, et al. Limma Powers Differential Expression Analyses for RNA-Sequencing and Microarray Studies. Nucleic Acids Res (2015) 43(7):e47. doi: 10.1093/nar/gkv007
17. Franceschini A, Szklarczyk D, Frankild S, Kuhn M, Simonovic M, Roth A, et al. STRING V9.1: Protein-Protein Interaction Networks, With Increased Coverage and Integration. Nucleic Acids Res (2013) 41(Database issue):D808–15. doi: 10.1093/nar/gks1094
18. Chin C-H, Chen S-H, Wu H-H, Ho C-W, Ko M-T, Lin C-Y. Cytohubba: Identifying Hub Objects and Sub-Networks From Complex Interactome. BMC Syst Biol (2014) 8 Suppl 4(Suppl 4):S11. doi: 10.1186/1752-0509-8-S4-S11
19. Liu H, Liu M, You H, Li X, Li X. Oncogenic Network and Hub Genes for Natural Killer/T-Cell Lymphoma Utilizing WGCNA. Front Oncol (2020) 10:223. doi: 10.3389/fonc.2020.00223
20. Li X, Liu Z, Mi M, Zhang C, Xiao Y, Liu X, et al. Identification of Hub Genes and Key Pathways Associated With Angioimmunoblastic T-Cell Lymphoma Using Weighted Gene Co-Expression Network Analysis. Cancer Manag Res (2019) 11:5209–20. doi: 10.2147/CMAR.S185030
21. Liu Z, Sun T, Zhang Z, Bi J, Kong C. An 18-Gene Signature Based on Glucose Metabolism and DNA Methylation Improves Prognostic Prediction for Urinary Bladder Cancer. Genomics (2021) 113(1 Pt 2):896–907. doi: 10.1016/j.ygeno.2020.10.022
22. Mullighan CG, Zhang J, Harvey RC, Collins-Underwood JR, Schulman BA, Phillips LA, et al. JAK Mutations in High-Risk Childhood Acute Lymphoblastic Leukemia. Proc Natl Acad Sci U.S.A. (2009) 106(23):9414–8. doi: 10.1073/pnas.0811761106
23. Dobbins SE, Sherborne AL, Ma YP, Bardini M, Biondi A, Cazzaniga G, et al. The Silent Mutational Landscape of Infant MLL-AF4 Pro-B Acute Lymphoblastic Leukemia. Genes Chromosomes Cancer (2013) 52(10):954–60. doi: 10.1002/gcc.22090
24. Bardini M, Spinelli R, Bungaro S, Mangano E, Corral L, Cifola I, et al. DNA Copy-Number Abnormalities do Not Occur in Infant ALL With T (/MLL-Af4. Leukemia (2010) 24(1):169–76. doi: 10.1038/leu.2009.203
25. Agraz-Doblas A, Bueno C, Bashford-Rogers R, Roy A, Schneider P, Bardini M, et al. Unraveling the Cellular Origin and Clinical Prognostic Markers of Infant B-Cell Acute Lymphoblastic Leukemia Using Genome-Wide Analysis. Haematologica (2019) 104(6):1176–88. doi: 10.3324/haematol.2018.206375
26. Krivtsov AV, Feng Z, Lemieux ME, Faber J, Vempati S, Sinha AU, et al. H3K79 Methylation Profiles Define Murine and Human MLL-AF4 Leukemias. Cancer Cell (2008) 14(5):355–68. doi: 10.1016/j.ccr.2008.10.001
27. Armstrong SA, Golub TR, Korsmeyer SJ. MLL-Rearranged Leukemias: Insights From Gene Expression Profiling. Semin Hematol (2003) 40(4):268–73. doi: 10.1016/S0037-1963(03)00196-3
28. Bitoun E, Oliver PL, Davies KE. The Mixed-Lineage Leukemia Fusion Partner AF4 Stimulates RNA Polymerase II Transcriptional Elongation and Mediates Coordinated Chromatin Remodeling. Hum Mol Genet (2007) 16(1):92–106. doi: 10.1093/hmg/ddl444
29. Basu S, Nandy A, Biswas D. Keeping RNA Polymerase II on the Run: Functions of MLL Fusion Partners in Transcriptional Regulation. Biochim Biophys Acta Gene Regul Mech (2020) 1863(8):194563. doi: 10.1016/j.bbagrm.2020.194563
30. Armstrong SA, Staunton JE, Silverman LB, Pieters R, den Boer ML, Minden MD, et al. MLL Translocations Specify a Distinct Gene Expression Profile That Distinguishes a Unique Leukemia. Nat Genet (2002) 30(1):41–7. doi: 10.1038/ng765
31. Rozovskaia T, Feinstein E, Mor O, Foa R, Blechman J, Nakamura T, et al. Upregulation of Meis1 and HoxA9 in Acute Lymphocytic Leukemias With the T(4 : 11) Abnormality. Oncogene (2001) 20(7):874–8. doi: 10.1038/sj.onc.1204174
32. Langfelder P, Horvath S. WGCNA: An R Package for Weighted Correlation Network Analysis. BMC Bioinf (2008) 9:559. doi: 10.1186/1471-2105-9-559
33. Satterwhite E, Sonoki T, Willis TG, Harder L, Nowak R, Arriola EL, et al. The BCL11 Gene Family: Involvement of BCL11A in Lymphoid Malignancies. Blood (2001) 98(12):3413–20. doi: 10.1182/blood.V98.12.3413
34. Yin B, Delwel R, Valk PJ, Wallace MR, Loh ML, Shannon KM, et al. A Retroviral Mutagenesis Screen Reveals Strong Cooperation Between Bcl11a Overexpression and Loss of the Nf1 Tumor Suppressor Gene. Blood (2009) 113(5):1075–85. doi: 10.1182/blood-2008-03-144436
35. Liu N, Hargreaves VV, Zhu Q, Kurland JV, Hong J, Kim W, et al. Direct Promoter Repression by BCL11A Controls the Fetal to Adult Hemoglobin Switch. Cell (2018) 173(2):430–442.e17. doi: 10.1016/j.cell.2018.03.016
36. Seachrist DD, Hannigan MM, Ingles NN, Webb BM, Weber-Bonk KL, Yu P, et al. The Transcriptional Repressor BCL11A Promotes Breast Cancer Metastasis. J Biol Chem (2020) 295(33):11707–19. doi: 10.1074/jbc.RA120.014018
37. Lazarus KA, Hadi F, Zambon E, Bach K, Santolla M-F, Watson JK, et al. BCL11A Interacts With SOX2 to Control the Expression of Epigenetic Regulators in Lung Squamous Carcinoma. Nat Commun (2018) 9(1):3327. doi: 10.1038/s41467-018-05790-5
38. Park SY, Lee S-J, Cho HJ, Kim J-T, Yoon HR, Lee KH, et al. Epsilon-Globin HBE1 Enhances Radiotherapy Resistance by Downregulating BCL11A in Colorectal Cancer Cells. Cancers (Basel) (2019) 11(4):498. doi: 10.3390/cancers11040498
39. Zhou J, Zhou L, Zhang D, Tang W-J, Tang D, Shi X-L, et al. BCL11A Promotes the Progression of Laryngeal Squamous Cell Carcinoma. Front Oncol (2020) 10:375. doi: 10.3389/fonc.2020.00375
40. Xutao G, PengCheng S, Yin L, Huijuan D, Yan W, Haiqing Z, et al. BCL11A and MDR1 Expressions Have Prognostic Impact in Patients With Acute Myeloid Leukemia Treated With Chemotherapy. Pharmacogenomics (2018) 19(4):343–8. doi: 10.2217/pgs-2017-0157
41. Borowitz MJ, Devidas M, Hunger SP, Bowman WP, Carroll AJ, Carroll WL, et al. Clinical Significance of Minimal Residual Disease in Childhood Acute Lymphoblastic Leukemia and its Relationship to Other Prognostic Factors: A Children’s Oncology Group Study. Blood (2008) 111(12):5477–85. doi: 10.1182/blood-2008-01-132837
Keywords: acute lymphoblastic leukemia, MLL rearrangement, BCL11A, differentially expressed gene, WGCNA
Citation: Wang LL, Yan D, Tang X, Zhang M, Liu S, Wang Y, Zhang M, Zhou G, Li T, Jiang F, Chen X, Wen F, Liu S and Mai H (2021) High Expression of BCL11A Predicts Poor Prognosis for Childhood MLL-r ALL. Front. Oncol. 11:755188. doi: 10.3389/fonc.2021.755188
Received: 08 August 2021; Accepted: 15 November 2021;
Published: 06 December 2021.
Edited by:
Shimin Hu, University of Texas MD Anderson Cancer Center, United StatesReviewed by:
Habibe Kurt, Warren Alpert Medical School of Brown University, United StatesCopyright © 2021 Wang, Yan, Tang, Zhang, Liu, Wang, Zhang, Zhou, Li, Jiang, Chen, Wen, Liu and Mai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sixi Liu, dGlnZXI2NDdAMTI2LmNvbQ==; Huirong Mai, bWFpaHVpcm9uZ0AxNjMuY29t
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.