 Mahnaz Rezaei
Mahnaz Rezaei Jiaxiong Tan
Jiaxiong Tan Chengwu Zeng
Chengwu Zeng Yangqiu Li
Yangqiu Li Mazdak Ganjalikhani-Hakemi
Mazdak Ganjalikhani-Hakemi
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol., 30 September 2021
Sec. Hematologic Malignancies
Volume 11 - 2021 | https://doi.org/10.3389/fonc.2021.753677
This article is part of the Research TopicCellular and Molecular Mechanisms of Immune Checkpoint Blockers in Anti-Leukemia and Lymphoma Immune TherapyView all 6 articles
T cell immunoglobulin and mucin domain 3 (TIM-3) expression on malignant cells has been reported in some leukemias. In myelodysplastic syndrome (MDS), increased TIM-3 expression on TH1 cells, regulatory T cells, CD8+ T cells, and hematopoietic stem cells (HSCs), which play a role in the proliferation of blasts and induction of immune escape, has been reported. In AML, several studies have reported overexpression of TIM-3 on leukemia stem cells (LSCs) but not on healthy HSCs. Overexpression of TIM-3 on exhausted CD4+ and CD8+ T cells and leukemic cells in CML, ALL, and CLL patients could be a prognostic risk factor for poor therapeutic response and relapse in patients. Currently, several TIM-3 inhibitors are used in clinical trials for leukemias, and some have shown encouraging response rates for MDS and AML treatment. For AML immunotherapy, blockade TIM-3 may have dual effects: directly inhibiting AML cell proliferation and restoring T cell function. However, blockade of PD-1 and TIM-3 fails to restore the function of exhausted CD8+ T cells in the early clinical stages of CLL, indicating that the effects of TIM-3 blockade may be different in AML and other leukemias. Thus, further studies are required to evaluate the efficacy of TIM-3 inhibitors in different types and stages of leukemia. In this review, we summarize the biological functions of TIM-3 and its contribution as it relates to leukemias. We also discuss the effects of TIM-3 blockade in hematological malignancies and clinical trials of TIM-3 for leukemia therapy.
T cell immunoglobulin and mucin domain 3 (TIM-3) is a cell surface molecule which was first identified approximately two decades ago on terminally differentiated CD4+ type 1 helper T cells (TH1 cells) and CD8+ cytotoxic T cells (CTLs) (1). Later, its expression was observed on other T cell subtypes, excluding TH2 cells, as well as some other immune cells including dendritic cells (DCs), natural killer (NK) cells, monocytes, macrophages, and mast cells (2–4). Also in some cancers, the malignant cells can express TIM-3 (5–8).
The gene that encodes TIM-3 is havcr2 (hepatitis A virus cellular receptor 2), which is located on chromosome 5q33.2. Due to the genes encoded in this region, including interleukin (IL)-4 and IL-5, this locus is known to be linked to allergies and autoimmune disease (9, 10).
TIM-3 is a single transmembrane (TM) molecule whose extracellular tail contains a N-terminal IgV domain. This domain is subsequently followed by a mucin domain with glycosylation sites, which explains where its name comes from. After the mucin domain, there is a link peptide with N-linked glycosylation sites and then the TM domain followed by the cytoplasmic tail in the C-terminus (2, 11).
TIM-3 does not contain classic inhibitory tyrosine-based motifs such as ITIM (immunoreceptor tyrosine-based inhibition) or ITSM (immunoreceptor tyrosine-based switch). However, there is a conserved region with five tyrosine residues, which two of them, Y265 and Y272 in humans (Y256 and Y263 in mice), are assumed to be phosphorylated after the interaction of TIM-3 with its ligands. Itk, a Tec family tyrosine kinase, and Fyn and Lck, two Src family kinase members, are known to be involved in the TIM-3 signaling pathway. A study of Jurkat T cells revealed that Tim-3 overexpression following T cell receptor and CD-28 induction promotes Lck or Fyn-dependent phosphorylation of Y256 and Y263. This event leads to accumulation of proteins with SH2 domains, such as the p85 subunit of phosphoinositide 3-kinase (PI3K) and phospholipase C-γ1 (PLC-γ1), to the cytoplasmic tail of TIM-3. Furthermore, TIM-3 activation enhances NFAT and NF-κB activation thorough interaction with ZAP-70 and SLP-76, components of the TCR signaling pathway. In contrast, another study of Jurkat cells demonstrated that TCR induction in TIM-3-expressing cells suppresses AP-1 and NFAT activation, resulting in impaired IL-2 production (12–14) (Figure 1). This discrepancy can likely be explained by differences in acute vs. chronic TCR induction and whether TIM-3 expression is secondary to TCR induction or vice versa. In addition, HLA-B associated transcript 3 (Bat3) can directly bind to the cytoplasmic tail of TIM-3 and prevent signal induction in the absence of TIM-3 ligands (15).
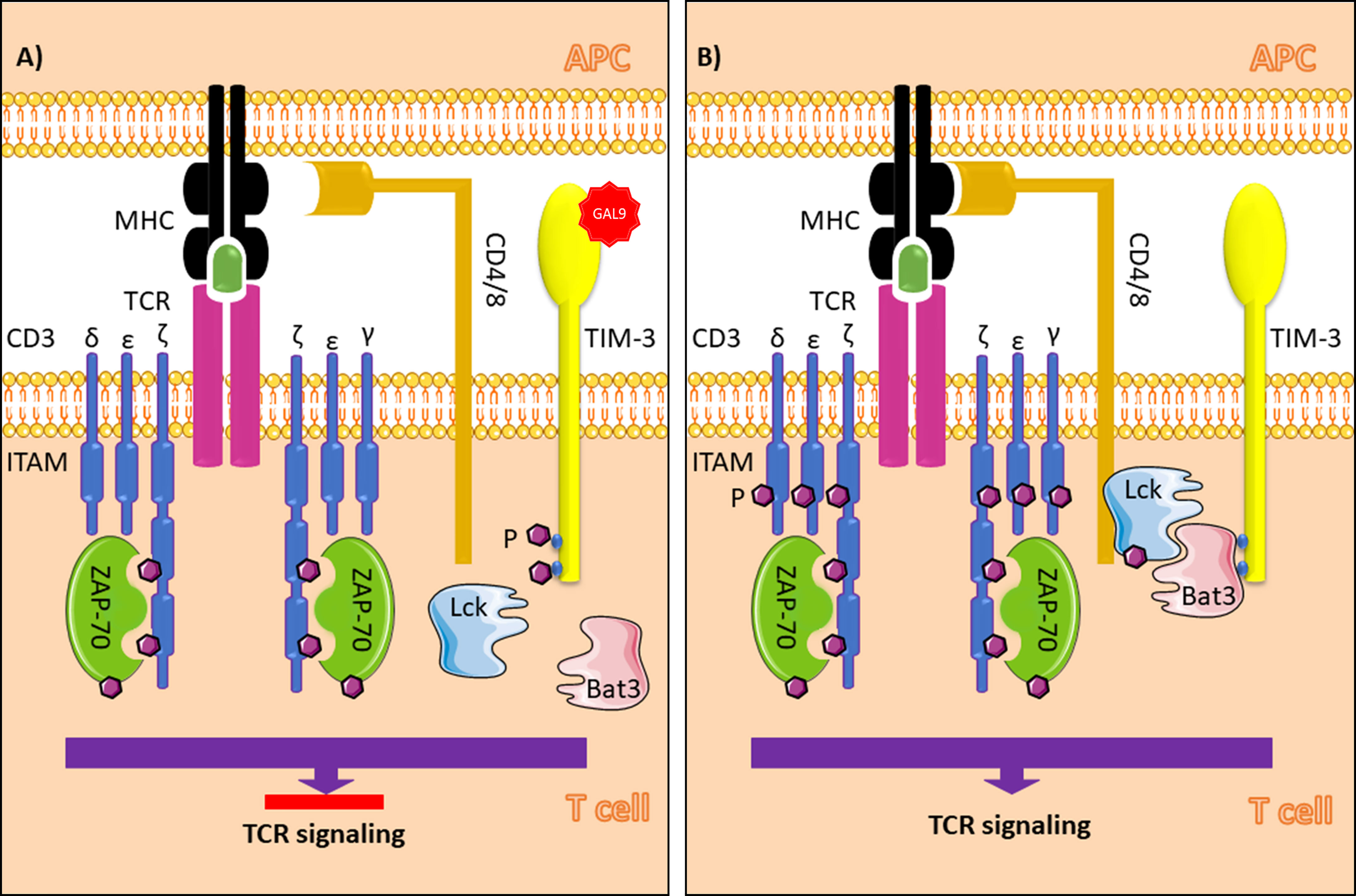
Figure 1 TIM-3 signaling in T cells in the presence (A) and absence (B) of galectin-9. In the absence of Gal-9, Bat3 binds to tyrosine residues in the cytoplasmic tail of TIM-3 (Y256 and Y263 in mice and Y26 and Y272 humans). This process leads to accumulation of the active form of Lck, which promotes phosphorylation of Zap70 and T cell signaling when the MHC peptide-TCR complex is formed. In the presence of Gal-9, as the ligand for TIM-3, phosphorylation of the tyrosine residues results in Bat3 release from the cytoplasmic tail. In this paradigm, Bat3 cannot form a complex with Lck; thus, TIM-3 induction inhibits T cell signaling. TIM-3, T cell immunoglobulin and mucin domain-3; Gal-9, galectin-9; Bat3, HLA-B associated transcript 3; TCR, T cell receptor; ITAM, immunoreceptor tyrosine-based activation motif; APC, antigen presenting cell; MHC, major histocompatibility complex; Ag, antigen. The figure was produced with the assistance of Servier Medical Art (https://smart.servier.com).
There are four known ligands for TIM-3. The first and most famous ligand is galectin-9 (Gal-9), which has been reported to induce apoptosis in TH1 cells. Gal-9 can also bind TIM-3 on other cells under other circumstances and result in activation of different pathways (16, 17). High-mobility group protein B1 (HMGB1) is a type of damage associated molecular pattern (DAMP), known as “alarmin”, which is released from damaged cells and induces the activation of phagocytes. HMGB-1 can bind TIM-3 in different contexts and does not always lead to one outcome (18). Another TIM-3 ligand is a well-known “eat-me” signal induction molecule phosphatidylserine (PtdSer). Appearance of PtdSer on the outer surface of cell membranes leads to phagocytosis. The same process is promoted when PtdSer interacts with TIM-3 on myeloid cells, but it does not always induce a same inflammatory response (19). The last ligand is carcinoembryonic antigen cell adhesion molecule 1 (CEACAM-1), which can have both cis and trans interactions with TIM-3 (20).
Myelodysplastic syndrome (MDS) is a heterogenous hematologic disorder, and its clinical manifestations include a wide spectrum ranging from mild and single lineage cytopenia to high-risk MDS, which rapidly progresses to acute myeloid leukemia (AML) (21–24).
Several studies of T cell populations in MDS patients have been performed so far. Ozkazanc et al. observed a subpopulation of CD4+ T cells in the bone marrow aspirate of MDS patients expressing the exhaustion markers programmed cell death 1 (PD-1), TIM-3, and lymphocyte activation gene 3 (LAG3) (25). Fu et al. classified CD4+ T cells in MDS patients and reported increased TIM-3 expression on type 1 helper T cells and regulatory T cells (26). In another study, Tao et al. reported that in addition to a decrease in the population of CD8+ T cells in MDS patients, these cells express high levels of TIM-3, and as expected, TIM-3+ CD8+ T cells express higher perforin and granzyme B and lower CD95 (also known as Fas) compared to TIM-3- CD8+ T cells in MDS patients (27). In a recent study, Tao et al. reported increased expression of Gal-9 on myeloid derived suppressor cells (MDSCs) in MDS patients and theorized that the interaction between the Gal-9 on MDSC cells and TIM-3 induces T cell exhaustion in MDS patients (28). All of these studies have suggested the use of immune checkpoint inhibition to restore immune surveillance in MDS patients.
Although different TIM-3 localization patterns (intracellular and extracellular expression) have been reported in different MDS cell lines, they all agree on increased overall expression of TIM-3 on hematopoietic stem cells (HSCs; CD34+ CD38- Lin-) in MDS patients compared to control groups (29, 30). Tao et al. reported that TIM-3 expression on HSCs in MDS patients is increased compared to healthy individuals and is close to the expression level in AML patients. Their study on TIM-3+ HSCs revealed decreased expression of differentiation related molecules, increased proliferation, and decreased apoptosis, compared to TIM-3- HSCs. This group also reported a correlation between a higher percentage of TIM-3 expression on HSCs in MDS patients and an increased WPSS score, greater than one cytopenia lineage, higher blast counts in bone marrow smears, and worse karyotype (21). Because these characteristics are known as indicators of disease progression, Tao et al. suggest that a higher percentage of TIM-3 reflects higher risk for MDS transformation to leukemia. Tcvetkov et al. have also reported increased levels of TIM-3 and its ligand, Gal-9 on bone marrow cells from MDS patients (31). In this regard, Asayama et al. claimed that the bone marrow microenvironment and Gal-9/TIM-3 axis in HSCs not only leads to MDS formation but is also related to disease progression to leukemia in MDS patients. This group also reported elevated levels of Gal-9 in MDS patients, highlighting the TIM-3/Gal-9 axis role in the proliferation of blasts and induction of immune escape, supporting disease progression (29) (Table 1). It is not clear that TIM-3 expression on HSCs is weather an early trigger for MDS formation and HSCs’ possible transformation to LSCs, or it is a secondary event to MDS formation and acts as an accelerator for disease progression. Hence, more studies are needed.

Table 1 TIM-3 alteration in leukemias and reported correlations.
Acute myeloid leukemia (AML) is a progressive myeloproliferative malignancy with low overall survival (32). AML has a heterogenous genetical nature; while about 45% of patients may display normal karyotype, others may show molecular mutations including internal tandem duplications in the FLT3 tyrosine kinase (FLT3-ITD). In some cases, cytogenetic abnormalities have also been reported. All of these abnormal alterations are defined with different prognostic values (33–37). Studies on the expression of TIM-3 in AML can be divided into two types: expression of TIM-3 on immune cells, particularly T cells and NK cells, and expression of TIM-3 on LSCs. Since genetic abnormalities in AML can be considered as prognostic risk factors, there may be correlations between them and the overexpression of TIM-3 in leukemia cells and/or immune cells in AML; hence more studies are required in this aspect.
When it comes to leukemia, there is a distinct group of stem cells (SCs) called leukemic stem cells (LSCs), which have different molecular patterns, sometimes including cytogenetic abnormalities and mutations, higher proliferation rates, and resistance to apoptosis, and they reproduce to form the malignant cell population (32, 38). One of the distinguishing characteristics of LSCs in AML is their upregulated TIM-3 expression. Many studies have reported overexpression of TIM-3 on LSCs but not on healthy hematopoietic stem cells (HSCs) (CD34+ CD38– Lin-) except in acute promyelocytic leukemia (M3) (8, 30, 39–41). Therefore, targeting TIM-3 with an antibody or microRNAs leads to LSC eradication with no effects on normal HSCs (8, 42–44). In addition, Jan et al. suggested that TIM-3 could be used to separate leukemic and healthy SCs (39).
Later, several studies have focused on the role of TIM-3 in leukemic cells in AML. It has been revealed that there is an autocrine stimulatory loop in AML cells that works through the interaction between TIM-3 with Gal-9, which leads to phosphorylation of ERK (extracellular signal-regulated kinase) and protein kinase B (PKB, also known as AKT). This process results in induction of the β-catenin pathway and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) activation, which is important for cell survival and disease progression, which explains why targeting TIM-3 on these cells promotes apoptosis (45, 46). Moreover, ligation of TIM-3 and Gal-9 in AML cell lines leads to phosphatidylinositol-3 kinase (PI-3K)/mammalian target of rapamycin (mTOR) pathway and ERK pathway activation, resulting in hypoxia-inducible factor 1-alpha (HIF-1α), vascular endothelial growth factor (VEGF), and TNF-α production (47).
TIM-3 is also involved in immune evasion in AML. Folgiero et al. ascertained that indoleamine 2,3-dioxygenase1 (IDO1), which is known as an anti-inflammatory enzyme (48), can be produced by AML blasts in response to TIM-3/Gal-9-dependent interferon gamma (IFN-γ) production from natural killer (NK) cells (49). Furthermore, Goncalves Silva et al. reported that increased amounts of TIM-3 in plasma, probably due to its secretion from AML blasts, and the soluble form of TIM-3 (sTIM-3), which is formed by its shedding from the surface of AML blasts, can inhibit the release of interleukin-2 (IL-2), a vital cytokine involved in the activation and function of T cells and NK cells, from immune cells (50).
Reports of the influence of TIM-3 on chemotherapeutic agents are controversial. Higher TIM-3 expression on AML blasts has been reported to be an enhancer for better response to chemotherapy by Xu et al. (51); however, Dama et al. noted that this overexpression is positively correlated with chemotherapy failure (52). It is evident that, more studies are required to address this controversy.
As mentioned above, upregulation of TIM-3 expression on immune cells leads to their dysfunctional activity in AML. Several studies have reported upregulation of TIM-3 on CD4+ and CD8+ T cells (25, 53–57). Ozkazanc et al. reported that the exhausted phenotype of CD4+ T cells, which is characterized by the expression of TIM-3, LAG-3, and PD-1, in AML patients is induced by the co-stimulatory signals of AML blasts (25).
In contrast, some studies have suggested meaningful associations between TIM-3 overexpression levels and prognosis. Li et al. revealed that TIM-3 overexpression on CD4+ T cells in AML patients that have an FLT3-ITD mutation, a poor prognostic factor, was higher than that in those who did not have this mutation (53). This group also reported higher levels of TIM-3 expression on CD8+ T cells in high-risk AML patients compared to low-risk patients. Kong et al. noted that for patients with elevated Tim3+ PD-1+ T cells after allogenic stem cell transplantation, leukemia relapse is more predictable (55). Later, Zahran et al. determined a positive correlation between TIM-3 expression and poor prognosis in AML patients with normal cytogenetics (56). Tan et al. demonstrated that higher TIM-3+ CD244+ CD8+ T cells are observed in M4 AML patients compared to M3 patients. This group also reported that AML patients have decreased TIM-3+ T cell portions by the time they are in the complete remission phase (57).
In a remarkable recent study, Rakova et al. observed NK cell activity in AML patients. This group revealed that TIM-3 upregulation on these cells was associated with increased activity of NK cells and resulted in better clinical outcome for AML patients (58) (Table 1). As noted, exceptional studies have been performed on TIM-3 in AML, yet so many aspects are still intact or not completely examined. For instance, how does TIM-3 affect the function of other immune cells, are there any other links between TIM-3 overexpression on different immune cells and different aspects of prognosis (i.e., resistance to different therapies, AML relapse, overall survival, refractory AML), are there mechanisms underlying upregulation of TIM-3 on LSCs, and how overexpression changes different aspects of function and stability of LSCs?
Reciprocal translocation between chromosome 9 and 22 [t(9,22)] creates the oncokinase BCR-ABL1, developing a myeloproliferative hematologic malignancy called chronic myeloid leukemia (CML). Tyrosine kinase inhibitors (TKIs) are considered first-line, approved treatments for CML patients that direct a deep molecular response hampering leukemia progression (59, 60).
Many studies reported impaired anti-tumor immunity in CML patients. Among them, Bruck et al. (61) observed TIM-3 overexpression on exhausted CD4+ and CD8+ T cells in untreated CML patients. This is not an unexpected report, since TIM-3 is known as an exhaustion marker in T cells. However, they also reported a correlation between PD-1+ TIM-3- CD8+ T cells and poor response to TKIs (Table 1). This may seem surprising, because we are more used to announce TIM-3 as a marker for disease progression, therapy resistance, probable relapse and more complicated situation for treatment. Unfortunately, there is not enough reports about TIM-3 in CML. TIM-3 and response to TKIs in CML is not well studied and more specified and detailed studies are required to clarify its possible role in better prognosis.
Dysfunctional immunity plays a major role in malignancy formation. Moreover, possibility of CML relapse due to the existence of undetectable residual leukemia stem cell, even after successful TKI therapy, magnifies the critical role of functional immune system. Therefore, much more clinical studies are required to examine the expression of TIM-3 in other immune cell types in CML as well and establish its role in formation, therapy resistance, relapse of CML, and immune scoring in this malignancy. Also, the possibility of the expression of TIM-3 on leukemic cells should not be ignored.
Acute lymphoblastic B-precursor leukemia (B-ALL) is the most common hematologic malignancy in children, which is formed by uncontrolled proliferation and a defect in lymphoid progenitors (62, 63). A few studies have been performed to determine the role of TIM-3 in B-ALL relapse, which is a challenging issue in this subtype. Although Liu et al. observed T cell exhaustion, characterized by TIM-3 and PD-1 expression, in post-allogenic-HSCT B-ALL relapse, they could not describe a more detailed relationship between increased immune checkpoint expression and relapse (64). Later in 2020 in a study on pediatric B-ALL, Blaeschke et al. reported overexpression of TIM-3 alone and in combination with PD-1 on CD4+ T cells in the bone marrow with no significant difference in T cell subpopulations, and defined TIM-3 as a substantial prognostic risk factor for relapse in B-ALL patients (65). This group also claimed that CD200 may be responsible for TIM-3 induction on T cells in ALL. These findings have not yet been shown in vivo.
While ALL is considered as the most common malignancy in children, about 15% of cases are T-cell acute lymphoblastic leukemia (known as T-ALL), in which almost 20% of them do not survive. According to records, this malignancy is not that common in adults (25% of all ALL cases), but mortality rate is higher than pediatric patients (about 50%) (66, 67).
Horlad et al. demonstrated that TIM-3 expression in leukemic cells is possibly related to resistance to chemotherapy in T-ALL patients (68), noting that more studies are needed. In a more recent study, Balajam et al. included both B-ALL and T-ALL cases and documented TIM-3 overexpression in bone marrow and peripheral blood mononuclear cells (PBMCs) in ALL patients compared to control individuals (69) (Table 1). Thus, more studies regarding the possible diagnostic and/or prognostic value of TIM-3 in ALL are recommended.
Chronic lymphocytic leukemia (CLL) is defined as a hematologic malignancy in which CD5+ B cells aggregate not only in peripheral blood but also in bone marrow and secondary lymphoid organs (70, 71). CLL has different contribution patterns all over the world (72, 73) and displays a wide spectrum of clinical manifestations ranging from asymptomatic individuals to fast progressing malignant cases (71, 74). Poor response to common treatments, refractory CLL, and leukemia relapse after a period of remission are other challenging issues in these patients (75).
Two major studies have been performed regarding the immunologic exhaustion profile in CLL patients. Allahmoradi et al. examined CD4+ T cells and Taghiloo et al. explored CD8+ T cells in CLL (76). Both studies reported that exhausted CD4+ and CD8+ T cells, characterized by PD-1 and TIM-3 expression, have a higher percentage and absolute count in CLL patients compared with healthy individuals. The same result was observed when only TIM-3+ T cells were considered. Moreover, lower proliferation and protective cytokine production by these TIM-3+ T cells has been reported. Furthermore, both Allahmoradi et al. and Taghiloo et al. claimed that the percentage and absolute count of TIM-3+ PD-1+ CD4+ T cells and TIM-3+ PD-1+ CD8+ T cells were positively correlated with advanced clinical stages for CLL patients. Later, Hadadi et al. reported TIM-3 overexpression in another immune cell type, called natural killer (NK) cells, which plays a critical role in eliminating malignant cells, in the peripheral blood of CLL patients. This study reported that an upregulated TIM-3 profile is a linked to poor prognostic factors for CLL patients (77) (Table 1). Together, these three studies have presented TIM-3 as a “promising biomarker and possible target for future immunotherapy” (76, 77). More detailed clinical studies are required to clarify the possible role of TIM-3 in CLL pathogenesis and its association with cases of leukemia relapse, response to standard therapeutic agents and molecular prognostic factors of CLL.
TIM-3 blockade alone was demonstrated to have anti-solid tumor effects in preclinical studies by improving the ex vivo proliferation of tumor-infiltrating T cells and increasing the secretion of the cytokines IFN-γ and TNF-α (78, 79). Other checkpoint inhibitors, such as the PD-1/PD-L1 axis and LAG-3, combined with TIM-3 blockade could further enhance the immune function of tumor-infiltrating T cells (78, 79). In clinical trials, TIM-3 blockade, especially in combination with PD-1 blockade, has demonstrated reliable preliminary results against solid tumors (79–81). Based on the expression characteristics of TIM-3, which is expressed both on AML cells and exhausted T cells (52, 57), TIM-3 blockade may have dual effects: directly inhibiting AML cell proliferation and revising the exhausted T cell phenotype and restoring T cell function. Moreover, overexpression of TIM-3/Gal-9 has been found in AML patients who failed chemotherapy, suggesting that targeting TIM-3/Gal-9 in combination with chemotherapy induction may be an alternative approach to increasing the complete remission rate of patients with AML (52, 82). The results of Kikushige Y et al., who found that injection of TIM-3+ AML cells in immune-deficient mice could establish an AML model and subsequent TIM-3 blockers could alleviate disease, supported the efficacy of this targeted approach (8). In AML allogeneic hematopoietic stem cell transplantation (allo-HSCT) models, TIM-3 blockade may lead to activation of macrophages to eradicate AML stem cells and ameliorate disease (39, 83). Recently, an in vitro study demonstrated that bispecific and split CAR T cells (BissCAR-T cells) that target CD13 and TIM-3 can specifically eliminate AML cells (84). Because there are no known life-essential cells that express both CD13 and TIM-3, BissCAR-T cells can selectively kill AML cells while reducing toxicity to human hematopoietic stem cells and other normal tissues. In addition, anti-human TIM-3 was identified as a potential strategy for curing AML by targeting LSCs (84).
TIM-3 is highly expressed in peripheral blood and bone marrow exhausted T cells in a variety of hematological malignancies, including acute lymphoblastic leukemia (ALL), chronic lymphocytic leukemia (CLL), and multiple myeloma (MM) (57, 77, 85). However, few reports have demonstrated the significant effects of TIM-3 inhibitors alone in the above diseases, and the reason may be due to the fact that TIM-3 suppression can partially restore T cell activation but it is unable to overcome the T cell exhausted status. Moreover, there is high expression of a number of immune checkpoint proteins, such as PD-1 and TIGIT, in T cells from patients. For example, blockade of PD-1 and TIM-3 failed to restore the function of exhausted CD8+ T cells in the early clinical stages of CLL (85), indicating the effects of TIM-3 blockade may be different in AML and other leukemias, further studies are required to evaluate the efficacy of TIM-3 inhibitors in different types and stages of leukemia as well as in different leukemia bone marrow microenvironments.
Currently, the TIM-3 inhibitors used in clinical trials include MBG453 (also known as Sabatolimab), TSR-022, BMS-986258, LY3321367, SYM023, BGB-A425, and SHR-1702 (86, 87). However, MBG453 and SHR-1702 have begun to be used in clinical trials for leukemia immunotherapy only (86) (Table 2 and Figure 2).
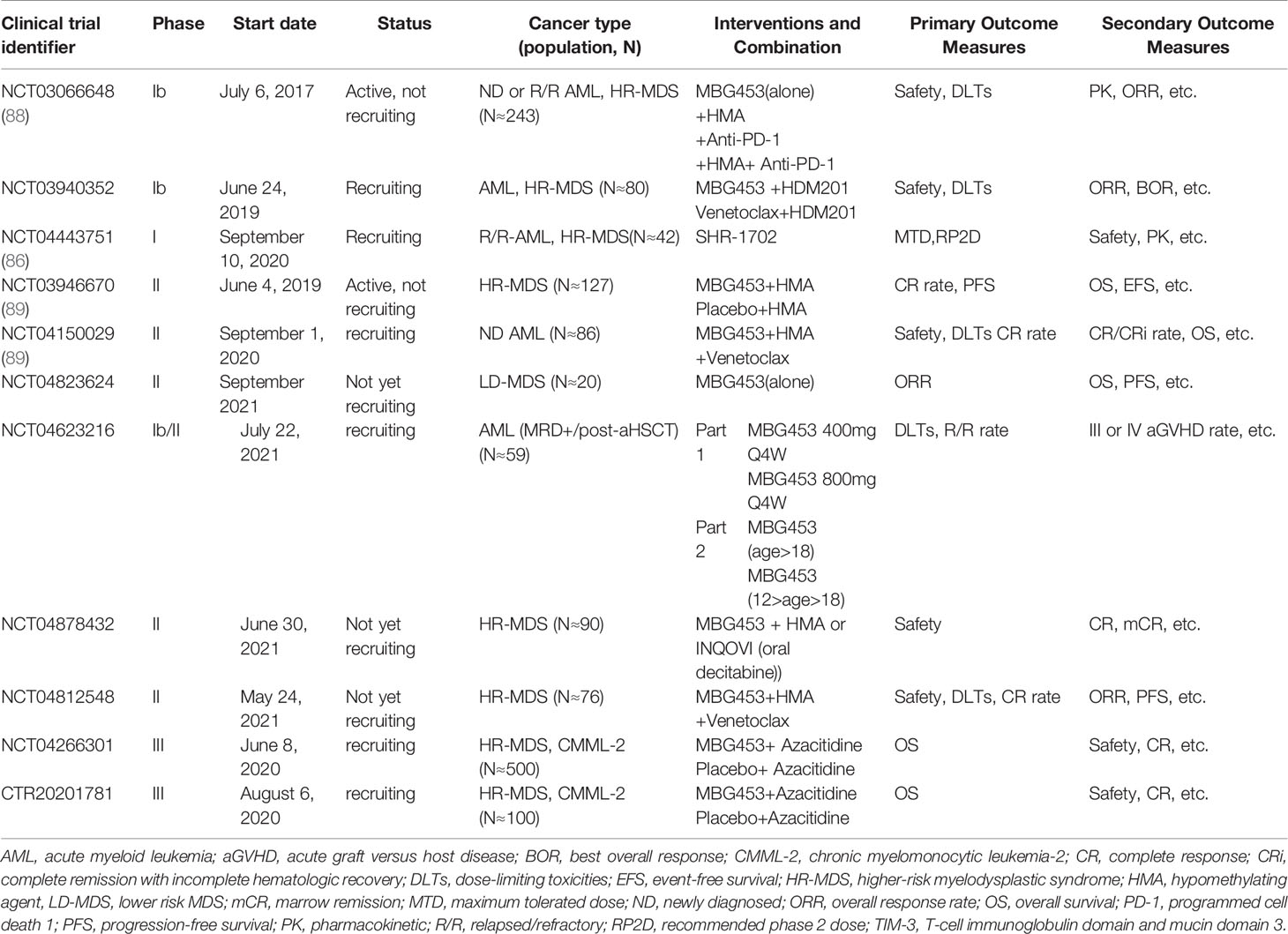
Table 2 Anti–TIM-3 agents and associated clinical trials in leukemia.
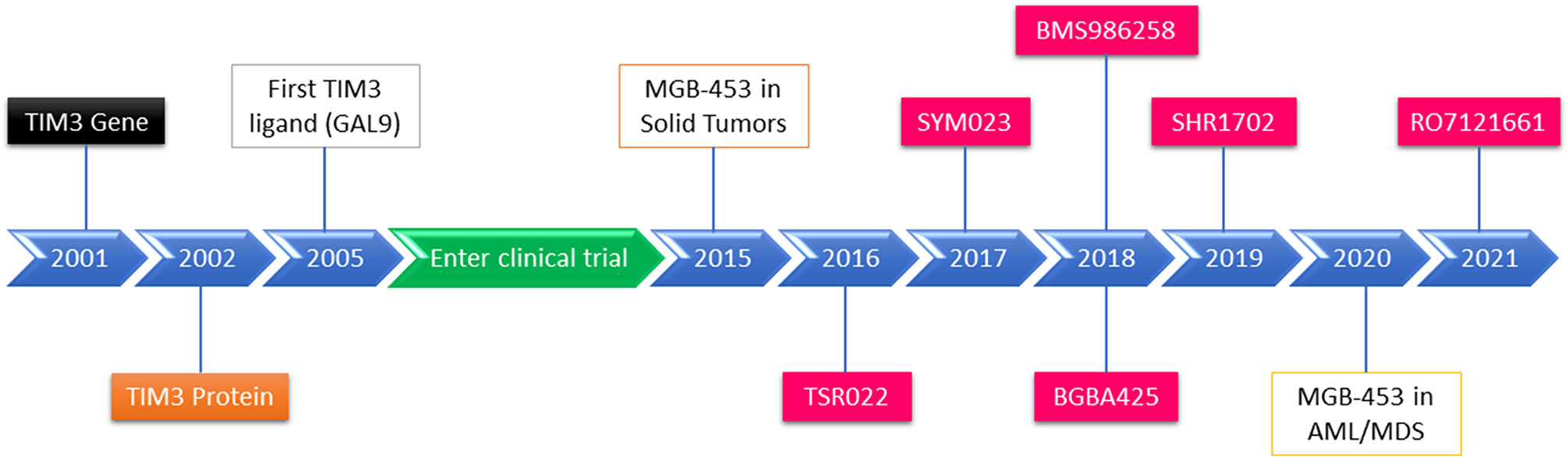
Figure 2 Schematic diagram of TIM-3 identification, TIM-3 inhibitor development, and TIM-3 blockade in cancer and leukemia therapies in clinical trials.
Although a number of TIM-3 blockade clinical trials for malignant tumor have been reported, MBG453 is the only inhibitor that has shown preliminary efficacy and safety in clinical studies for MDS and AML (86). Currently, eight phase I/II clinical trials are ongoing for AML and MDS with MBG453 monotherapy or the combination of different agents such as hypomethylating agents (HMAs), PD-1 inhibitors, HDM201 (an MDM2 inhibitor), and venetoclax. There is also just one phase I trial administering SHR-1702 for relapsed/refractory (R/R) AML and higher-risk myelodysplastic syndromes (HR-MDS). Moreover, there are two phase III trials of MBG453 + azacitidine for HR-MDS and chronic myelomonocytic leukemia-2 (CMML-2) (Table 2).
Most phase I/II trials of TIM-3 inhibitors for AML or MDS have been initiated in the past two years, and the trials are still ongoing; thus, the final results have yet to be released. Currently, positive results from several trials have indicated that the ORR of 69 patients with HR-MDS or AML who received MBG453 plus decitabine (Dec) (cutoff 27 Nov 2019, median exposure duration: 8.6 months) or that of 29 patients with HR-MDS or AML who received MBG453 plus azacitidine (Aza) (cutoff 14 Jan 2020, median exposure duration: 3.0 months) was 58% and 70% for HR-MDS, respectively, and 41% and 27%, respectively, for newly diagnosed (ND)-AML (NCT03066648) (88). Moreover, the ORR was only 24% for MBG453 plus Dec in patients with R/R AML (Supplementary Table 1). The most common grade 3/4 treatment-emergent (TE) adverse events (AEs) (in Dec and Aza group) were thrombocytopenia, febrile neutropenia, neutropenia, and anemia. For MBG453 plus Dec, only 4 patients experienced potential immune-related (IR) AEs that were reported as treatment-related (ALT increase, arthritis, hepatitis, hypothyroidism, rash). No treatment-related ≥ grade 3 potential IRAEs have been reported for MBG453+Aza (Supplementary Table 2). No treatment-related grade 4 IRAEs or deaths had been reported for either combination. Overall, MBG453 plus Dec or Aza is safe and well tolerated in HR-MDS and AML, which showed encouraging response rates and emerging durability as well (88, 90). However, whether TIM-3 inhibitors together with HMA treatment could improve clinical outcome requires more evidence from these trials. STIMULUS-MDS1 (N≈120; NCT03946670) is a double-blind, phase II clinical trial to evaluate whether MBG453 plus HMAs improves the CR rate and progression-free survival (PFS) vs. HMA alone in HR-MDS (89). STIMULUS-MDS2 (N≈500, NCT04266301) is a phase III clinical trial to further confirm whether MBG453 prolongs the OS of HR-MDS patients including CMML-2, and similar trials are ongoing in China and are recruiting (CTR20201781). STIMULUS-AML (N≈86; NCT04150029) is an open-label study evaluating the safety (DLTs) and efficacy (CR rate) of MBG453+HMA combined with venetoclax in ND-AML (89). In addition, ongoing clinical studies included the application of MBG453 in MRD-positive patients after allo-HSCT (NCT04623216) and in combination with a PD-1 inhibitor (NCT0306664), TP53-MDM2 inhibitor (NCT03940352), or Bcl-2 inhibitors plus HMAs (NCT04812548, NCT04150029) for efficacy and safety in MDS and AML. Similarly, a phase I clinical study of the efficacy and safety of another TIM-3 inhibitor, SHR-1702, for the treatment of R/R-AML and HR-MDS is ongoing as well (42 cases; NCT04443751) (86). Some studies have shown that TIM-3 inhibitors are well tolerated, and preliminary antitumor activity in advanced solid tumors has been demonstrated for this class of drugs (87, 91). Overall, TIM-3 blockade combined with other checkpoint inhibitors, targeted inhibitors, or HMAs may improve the clinical outcome of patients with leukemia; however, more clinical data are required to support the application of TIM-3 inhibitors in leukemia immunotherapy.
Numerous clinical studies have indicated that overexpression of TIM-3 is associated with poor prognosis in leukemia. As an immune checkpoint protein, TIM-3 is upregulated on T cells, resulting in increased T cell exhaustion in leukemia patients. TIM-3 is also expressed on leukemia cells and may serve as a biomarker and target for targeted therapy for different leukemias. However, blockade of TIM-3 may lead to different outcomes in AML in comparison with other leukemias. Therefore, further studies and more detailed clinical data are required to evaluate the efficacy of TIM-3 inhibitors in different types and stages of leukemia.
It is necessary to define how TIM-3 affects the function of other immune cells, and if there are other links between TIM-3 overexpression on different immune cells and resistance to different therapies, relapse, and overall survival. Some clinical trials have shown that blockade of TIM-3 alone fails to achieve clinical efficacy for most patients with AML or MDS. It is only when TIM-3 is combined with other checkpoint inhibitors, TKIs, or HMAs that improvements in clinical outcome are observed. Thus, the mechanism of TIM-3 blockade on leukemia cells, in the leukemia bone marrow microenvironment, and on T cells need to be characterized. This research may help to understand how to optimize TIM-3 blockade for leukemia immunotherapy.
MGH and MR conceptualized the manuscript. CZ and YL finalized the manuscript. JT and MR generated the figures. All authors contributed to the article and approved the submitted version.
This study was supported by grant from the National Natural Science Foundation of China (No. 82070152).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2021.753677/full#supplementary-material
1. Monney L, Sabatos CA, Gaglia JL, Ryu A, Waldner H, Chernova T, et al. Th1-Specific Cell Surface Protein Tim-3 Regulates Macrophage Activation and Severity of an Autoimmune Disease. Nature (2002) 415(6871):536–41. doi: 10.1038/415536a
2. Ferris RL, Lu B, Kane LP. Too Much of a Good Thing? Tim-3 and TCR Signaling in T Cell Exhaustion. J Immunol (2014) 193(4):1525–30. doi: 10.4049/jimmunol.1400557
3. Phong BL, Avery L, Sumpter TL, Gorman JV, Watkins SC, Colgan JD, et al. Tim-3 Enhances FcepsilonRI-Proximal Signaling to Modulate Mast Cell Activation. J Exp Med (2015) 212(13):2289–304. doi: 10.1084/jem.20150388
4. Gorman JV, Starbeck-Miller G, Pham NL, Traver GL, Rothman PB, Harty JT, et al. Tim-3 Directly Enhances CD8 T Cell Responses to Acute Listeria Monocytogenes Infection. J Immunol (2014) 192(7):3133–42. doi: 10.4049/jimmunol.1302290
5. Giraldo NA, Becht E, Vano Y, Petitprez F, Lacroix L, Validire P, et al. Tumor-Infiltrating and Peripheral Blood T-Cell Immunophenotypes Predict Early Relapse in Localized Clear Cell Renal Cell Carcinoma. Clin Cancer Res (2017) 23(15):4416–28. doi: 10.1158/1078-0432.CCR-16-2848
6. Solinas C, Garaud S, De Silva P, Boisson A, Van den Eynden G, de Wind A, et al. Immune Checkpoint Molecules on Tumor-Infiltrating Lymphocytes and Their Association With Tertiary Lymphoid Structures in Human Breast Cancer. Front Immunol (2017) 8:1412. doi: 10.3389/fimmu.2017.01412
7. Baitsch L, Baumgaertner P, Devevre E, Raghav SK, Legat A, Barba L, et al. Exhaustion of Tumor-Specific CD8(+) T Cells in Metastases From Melanoma Patients. J Clin Invest (2011) 121(6):2350–60. doi: 10.1172/JCI46102
8. Kikushige Y, Shima T, Takayanagi S, Urata S, Miyamoto T, Iwasaki H, et al. TIM-3 is a Promising Target to Selectively Kill Acute Myeloid Leukemia Stem Cells. Cell Stem Cell (2010) 7(6):708–17. doi: 10.1016/j.stem.2010.11.014
9. Meyers JH, Sabatos CA, Chakravarti S, Kuchroo VK. The TIM Gene Family Regulates Autoimmune and Allergic Diseases. Trends Mol Med (2005) 11(8):362–9. doi: 10.1016/j.molmed.2005.06.008
10. McIntire JJ, Umetsu SE, Akbari O, Potter M, Kuchroo VK, Barsh GS, et al. Identification of Tapr (an Airway Hyperreactivity Regulatory Locus) and the Linked Tim Gene Family. Nat Immunol (2001) 2(12):1109–16. doi: 10.1038/ni739
11. Anderson AC, Anderson DE, Bregoli L, Hastings WD, Kassam N, Lei C, et al. Promotion of Tissue Inflammation by the Immune Receptor Tim-3 Expressed on Innate Immune Cells. Science (2007) 318(5853):1141–3. doi: 10.1126/science.1148536
12. van de Weyer PS, Muehlfeit M, Klose C, Bonventre JV, Walz G, Kuehn EW. A Highly Conserved Tyrosine of Tim-3 is Phosphorylated Upon Stimulation by its Ligand Galectin-9. Biochem Biophys Res Commun (2006) 351(2):571–6. doi: 10.1016/j.bbrc.2006.10.079
13. Lee J, Su EW, Zhu C, Hainline S, Phuah J, Moroco JA, et al. Phosphotyrosine-Dependent Coupling of Tim-3 to T-Cell Receptor Signaling Pathways. Mol Cell Biol (2011) 31(19):3963–74. doi: 10.1128/MCB.05297-11
14. Ganjalikhani Hakemi M, Jafarinia M, Azizi M, Rezaeepoor M, Isayev O, Bazhin AV. The Role of TIM-3 in Hepatocellular Carcinoma: A Promising Target for Immunotherapy? Front Oncol (2020) 10:601661. doi: 10.3389/fonc.2020.601661
15. Rangachari M, Zhu C, Sakuishi K, Xiao S, Karman J, Chen A, et al. Bat3 Promotes T Cell Responses and Autoimmunity by Repressing Tim-3-Mediated Cell Death and Exhaustion. Nat Med (2012) 18(9):1394–400. doi: 10.1038/nm.2871
16. Su EW, Bi S, Kane LP. Galectin-9 Regulates T Helper Cell Function Independently of Tim-3. Glycobiology (2011) 21(10):1258–65. doi: 10.1093/glycob/cwq214
17. Zhu C, Anderson AC, Schubart A, Xiong H, Imitola J, Khoury SJ, et al. The Tim-3 Ligand Galectin-9 Negatively Regulates T Helper Type 1 Immunity. Nat Immunol (2005) 6(12):1245–52. doi: 10.1038/ni1271
18. Chiba S, Baghdadi M, Akiba H, Yoshiyama H, Kinoshita I, Dosaka-Akita H, et al. Tumor-Infiltrating DCs Suppress Nucleic Acid-Mediated Innate Immune Responses Through Interactions Between the Receptor TIM-3 and the Alarmin HMGB1. Nat Immunol (2012) 13(9):832–42. doi: 10.1038/ni.2376
19. DeKruyff RH, Bu X, Ballesteros A, Santiago C, Chim YL, Lee HH, et al. T Cell/Transmembrane, Ig, and Mucin-3 Allelic Variants Differentially Recognize Phosphatidylserine and Mediate Phagocytosis of Apoptotic Cells. J Immunol (2010) 184(4):1918–30. doi: 10.4049/jimmunol.0903059
20. Huang YH, Zhu C, Kondo Y, Anderson AC, Gandhi A, Russell A, et al. CEACAM1 Regulates TIM-3-Mediated Tolerance and Exhaustion. Nature (2015) 517(7534):386–90. doi: 10.1038/nature13848
21. Tao JL, Li LJ, Fu R, Wang HQ, Jiang HJ, Yue LZ, et al. Elevated TIM3+ Hematopoietic Stem Cells in Untreated Myelodysplastic Syndrome Displayed Aberrant Differentiation, Overproliferation and Decreased Apoptosis. Leuk Res (2014) 38(6):714–21. doi: 10.1016/j.leukres.2014.03.018
22. Zeidan AM, Shallis RM, Wang R, Davidoff A, Ma X. Epidemiology of Myelodysplastic Syndromes: Why Characterizing the Beast is a Prerequisite to Taming it. Blood Rev (2019) 34:1–15. doi: 10.1016/j.blre.2018.09.001
23. Ma X. Epidemiology of Myelodysplastic Syndromes. Am J Med (2012) 125(7 Suppl):S2–5. doi: 10.1016/j.amjmed.2012.04.014
24. Okuyama N, Sperr WR, Kadar K, Bakker S, Szombath G, Handa H, et al. Prognosis of Acute Myeloid Leukemia Transformed From Myelodysplastic Syndromes: A Multicenter Retrospective Study. Leuk Res (2013) 37(8):862–7. doi: 10.1016/j.leukres.2013.02.018
25. Ozkazanc D, Yoyen-Ermis D, Tavukcuoglu E, Buyukasik Y, Esendagli G. Functional Exhaustion of CD4(+) T Cells Induced by Co-Stimulatory Signals From Myeloid Leukaemia Cells. Immunology (2016) 149(4):460–71. doi: 10.1111/imm.12665
26. Fu R, Li L, Hu J, Wang Y, Tao J, Liu H, et al. Elevated TIM3 Expression of T Helper Cells Affects Immune System in Patients With Myelodysplastic Syndrome. J Investig Med (2019) 67(8):1125–30. doi: 10.1136/jim-2019-001059
27. Tao J, Li L, Wang Y, Fu R, Wang H, Shao Z. Increased Tim3+Cd8+T Cells in Myelodysplastic Syndrome Patients Displayed Less Perforin and Granzyme B Secretion and Higher CD95 Expression. Leuk Res (2016) 51:49–55. doi: 10.1016/j.leukres.2016.11.003
28. Tao J, Han D, Gao S, Zhang W, Yu H, Liu P, et al. CD8(+) T Cells Exhaustion Induced by Myeloid-Derived Suppressor Cells in Myelodysplastic Syndromes Patients Might be Through TIM3/Gal-9 Pathway. J Cell Mol Med (2020) 24(1):1046–58. doi: 10.1111/jcmm.14825
29. Asayama T, Tamura H, Ishibashi M, Kuribayashi-Hamada Y, Onodera-Kondo A, Okuyama N, et al. Functional Expression of Tim-3 on Blasts and Clinical Impact of its Ligand Galectin-9 in Myelodysplastic Syndromes. Oncotarget (2017) 8(51):88904–17. doi: 10.18632/oncotarget.21492
30. Goncalves Silva I, Gibbs BF, Bardelli M, Varani L, Sumbayev VV. Differential Expression and Biochemical Activity of the Immune Receptor Tim-3 in Healthy and Malignant Human Myeloid Cells. Oncotarget (2015) 6(32):33823–33. doi: 10.18632/oncotarget.5257
31. Tcvetkov N, Gusak A, Morozova E, Moiseev I, Baykov V, Barabanshikova M, et al. Immune Checkpoints Bone Marrow Expression as the Predictor of Clinical Outcome in Myelodysplastic Syndrome. Leuk Res Rep (2020) 14:100215. doi: 10.1016/j.lrr.2020.100215
32. Estey E, Dohner H. Acute Myeloid Leukaemia. Lancet (2006) 368(9550):1894–907. doi: 10.1016/S0140-6736(06)69780-8
33. Schlenk RF, Dohner K, Krauter J, Frohling S, Corbacioglu A, Bullinger L, et al. Mutations and Treatment Outcome in Cytogenetically Normal Acute Myeloid Leukemia. N Engl J Med (2008) 358(18):1909–18. doi: 10.1056/NEJMoa074306
34. Mrozek K, Marcucci G, Paschka P, Whitman SP, Bloomfield CD. Clinical Relevance of Mutations and Gene-Expression Changes in Adult Acute Myeloid Leukemia With Normal Cytogenetics: Are We Ready for a Prognostically Prioritized Molecular Classification? Blood (2007) 109(2):431–48. doi: 10.1182/blood-2006-06-001149
35. Grimwade D, Walker H, Oliver F, Wheatley K, Harrison C, Harrison G, et al. The Importance of Diagnostic Cytogenetics on Outcome in AML: Analysis of 1,612 Patients Entered Into the MRC AML 10 Trial. The Medical Research Council Adult and Children’s Leukaemia Working Parties. Blood (1998) 92(7):2322–33. doi: 10.1182/blood.V92.7.2322
36. Byrd JC, Mrozek K, Dodge RK, Carroll AJ, Edwards CG, Arthur DC, et al. Pretreatment Cytogenetic Abnormalities are Predictive of Induction Success, Cumulative Incidence of Relapse, and Overall Survival in Adult Patients With De Novo Acute Myeloid Leukemia: Results From Cancer and Leukemia Group B (CALGB 8461). Blood (2002) 100(13):4325–36. doi: 10.1182/blood-2002-03-0772
37. Nimer SD. Is it Important to Decipher the Heterogeneity of “Normal Karyotype AML”? Best Pract Res Clin Haematol (2008) 21(1):43–52. doi: 10.1016/j.beha.2007.11.010
38. Taussig DC, Miraki-Moud F, Anjos-Afonso F, Pearce DJ, Allen K, Ridler C, et al. Anti-CD38 Antibody-Mediated Clearance of Human Repopulating Cells Masks the Heterogeneity of Leukemia-Initiating Cells. Blood (2008) 112(3):568–75. doi: 10.1182/blood-2007-10-118331
39. Jan M, Chao MP, Cha AC, Alizadeh AA, Gentles AJ, Weissman IL, et al. Prospective Separation of Normal and Leukemic Stem Cells Based on Differential Expression of TIM3, a Human Acute Myeloid Leukemia Stem Cell Marker. Proc Natl Acad Sci U.S.A. (2011) 108(12):5009–14. doi: 10.1073/pnas.1100551108
40. Roth CG, Garner K, Eyck ST, Boyiadzis M, Kane LP, Craig FE. TIM3 Expression by Leukemic and non-Leukemic Myeloblasts. Cytometry B Clin Cytom (2013) 84(3):167–72. doi: 10.1002/cyto.b.21080
41. Darwish NH, Sudha T, Godugu K, Elbaz O, Abdelghaffar HA, Hassan EE, et al. Acute Myeloid Leukemia Stem Cell Markers in Prognosis and Targeted Therapy: Potential Impact of BMI-1, TIM-3 and CLL-1. Oncotarget (2016) 7(36):57811–20. doi: 10.18632/oncotarget.11063
42. Moghaddam Y, Andalib A, Mohammad-Ganji M, Homayouni V, Sharifi M, Ganjalikhani-Hakemi M. Evaluation of the Effect of TIM-3 Suppression by miR-498 and its Effect on Apoptosis and Proliferation Rate of HL-60 Cell Line. Pathol Res Pract (2018) 214(9):1482–8. doi: 10.1016/j.prp.2018.07.019
43. Hojati Z, Ganjalikhani-Hakemi M, Ameri M, Alimohammadi-Jelodar SF, Dehbashi M, Mohammad Ganji M, et al. Evaluation of Silencing Effect of miR-133a-5p Mimic on TIM-3 Expression in AML (HL-60) Cell Line. Indian J Clin Biochem (2020) 35(3):359–66. doi: 10.1007/s12291-019-00834-z
44. Emamdoost F, Khanahmad H, Ganjalikhani-Hakemi M, Doosti A. The miR-125a-3p Inhibits TIM-3 Expression in AML Cell Line HL-60 In Vitro. Indian J Hematol Blood Transfus (2017) 33(3):342–7. doi: 10.1007/s12288-016-0733-4
45. Kikushige Y. [A TIM-3/Galectin-9 Autocrine Stimulatory Loop Drives Self-Renewal of Human Myeloid Leukemia Stem Cells and Leukemia Progression]. Rinsho Ketsueki (2016) 57(4):412–6. doi: 10.1016/j.stem.2015.07.011
46. Goncalves Silva I, Ruegg L, Gibbs BF, Bardelli M, Fruehwirth A, Varani L, et al. The Immune Receptor Tim-3 Acts as a Trafficker in a Tim-3/Galectin-9 Autocrine Loop in Human Myeloid Leukemia Cells. Oncoimmunology (2016) 5(7):e1195535. doi: 10.1080/2162402X.2016.1195535
47. Prokhorov A, Gibbs BF, Bardelli M, Ruegg L, Fasler-Kan E, Varani L, et al. The Immune Receptor Tim-3 Mediates Activation of PI3 Kinase/mTOR and HIF-1 Pathways in Human Myeloid Leukaemia Cells. Int J Biochem Cell Biol (2015) 59:11–20. doi: 10.1016/j.biocel.2014.11.017
48. Prendergast GC, Metz R, Muller AJ, Merlo LM, Mandik-Nayak L. IDO2 in Immunomodulation and Autoimmune Disease. Front Immunol (2014) 5:585. doi: 10.3389/fimmu.2014.00585
49. Folgiero V, Cifaldi L, Li Pira G, Goffredo BM, Vinti L, Locatelli F. TIM-3/Gal-9 Interaction Induces IFNgamma-Dependent IDO1 Expression in Acute Myeloid Leukemia Blast Cells. J Hematol Oncol (2015) 8:36. doi: 10.1186/s13045-015-0134-4
50. Goncalves Silva I, Yasinska IM, Sakhnevych SS, Fiedler W, Wellbrock J, Bardelli M, et al. The Tim-3-Galectin-9 Secretory Pathway is Involved in the Immune Escape of Human Acute Myeloid Leukemia Cells. EBioMedicine (2017) 22:44–57. doi: 10.1016/j.ebiom.2017.07.018
51. Xu L, Xu J, Ma S, Li X, Zhu M, Chen S, et al. High Tim-3 Expression on AML Blasts Could Enhance Chemotherapy Sensitivity. Oncotarget (2017) 8(60):102088–96. doi: 10.18632/oncotarget.22141
52. Dama P, Tang M, Fulton N, Kline J, Liu H. Gal9/Tim-3 Expression Level is Higher in AML Patients Who Fail Chemotherapy. J Immunother Cancer (2019) 7(1):175. doi: 10.1186/s40425-019-0611-3
53. Li C, Chen X, Yu X, Zhu Y, Ma C, Xia R, et al. Tim-3 is Highly Expressed in T Cells in Acute Myeloid Leukemia and Associated With Clinicopathological Prognostic Stratification. Int J Clin Exp Pathol (2014) 7(10):6880–8.
54. Sakuishi K, Apetoh L, Sullivan JM, Blazar BR, Kuchroo VK, Anderson AC. Targeting Tim-3 and PD-1 Pathways to Reverse T Cell Exhaustion and Restore Anti-Tumor Immunity. J Exp Med (2010) 207(10):2187–94. doi: 10.1084/jem.20100643
55. Kong Y, Zhang J, Claxton DF, Ehmann WC, Rybka WB, Zhu L, et al. PD-1(Hi)TIM-3(+) T Cells Associate With and Predict Leukemia Relapse in AML Patients Post Allogeneic Stem Cell Transplantation. Blood Cancer J (2015) 5:e330. doi: 10.1038/bcj.2015.58
56. Zahran AM, Mohammed Saleh MF, Sayed MM, Rayan A, Ali AM, Hetta HF. Up-Regulation of Regulatory T Cells, CD200 and TIM3 Expression in Cytogenetically Normal Acute Myeloid Leukemia. Cancer biomark (2018) 22(3):587–95. doi: 10.3233/CBM-181368
57. Tan J, Huang S, Huang J, Yu Z, Chen Y, Lu Y, et al. Increasing Tim-3+CD244+, Tim-3+CD57+, and Tim-3+PD-1+ T Cells in Patients With Acute Myeloid Leukemia. Asia Pac J Clin Oncol (2020) 16(3):137–41. doi: 10.1111/ajco.13304
58. Rakova J, Truxova I, Holicek P, Salek C, Hensler M, Kasikova L, et al. TIM-3 Levels Correlate With Enhanced NK Cell Cytotoxicity and Improved Clinical Outcome in AML Patients. Oncoimmunology (2021) 10(1):1889822. doi: 10.1080/2162402X.2021.1889822
59. Hochhaus A, Baccarani M, Silver RT, Schiffer C, Apperley JF, Cervantes F, et al. European LeukemiaNet 2020 Recommendations for Treating Chronic Myeloid Leukemia. Leukemia (2020) 34(4):966–84. doi: 10.1038/s41375-020-0776-2
60. Saussele S, Richter J, Guilhot J, Gruber FX, Hjorth-Hansen H, Almeida A, et al. Discontinuation of Tyrosine Kinase Inhibitor Therapy in Chronic Myeloid Leukaemia (EURO-SKI): A Prespecified Interim Analysis of a Prospective, Multicentre, non-Randomised, Trial. Lancet Oncol (2018) 19(6):747–57. doi: 10.1016/S1470-2045(18)30192-X
61. Bruck O, Blom S, Dufva O, Turkki R, Chheda H, Ribeiro A, et al. Immune Cell Contexture in the Bone Marrow Tumor Microenvironment Impacts Therapy Response in CML. Leukemia (2018) 32(7):1643–56. doi: 10.1038/s41375-018-0175-0
62. Moricke A, Zimmermann M, Reiter A, Henze G, Schrauder A, Gadner H, et al. Long-Term Results of Five Consecutive Trials in Childhood Acute Lymphoblastic Leukemia Performed by the ALL-BFM Study Group From 1981 to 2000. Leukemia (2010) 24(2):265–84. doi: 10.1038/leu.2009.257
63. Paul S, Kantarjian H, Jabbour EJ. Adult Acute Lymphoblastic Leukemia. Mayo Clin Proc (2016) 91(11):1645–66. doi: 10.1016/j.mayocp.2016.09.010
64. Liu L, Chang YJ, Xu LP, Zhang XH, Wang Y, Liu KY, et al. T Cell Exhaustion Characterized by Compromised MHC Class I and II Restricted Cytotoxic Activity Associates With Acute B Lymphoblastic Leukemia Relapse After Allogeneic Hematopoietic Stem Cell Transplantation. Clin Immunol (2018) 190:32–40. doi: 10.1016/j.clim.2018.02.009
65. Blaeschke F, Willier S, Stenger D, Lepenies M, Horstmann MA, Escherich G, et al. Leukemia-Induced Dysfunctional TIM-3(+)CD4(+) Bone Marrow T Cells Increase Risk of Relapse in Pediatric B-Precursor ALL Patients. Leukemia (2020) 34(10):2607–20. doi: 10.1038/s41375-020-0793-1
66. Karrman K, Johansson B. Pediatric T-Cell Acute Lymphoblastic Leukemia. Genes Chromosomes Cancer (2017) 56(2):89–116. doi: 10.1002/gcc.22416
67. Marks DI, Rowntree C. Management of Adults With T-Cell Lymphoblastic Leukemia. Blood (2017) 129(9):1134–42. doi: 10.1182/blood-2016-07-692608
68. Horlad H, Ohnishi K, Ma C, Fujiwara Y, Niino D, Ohshima K, et al. TIM-3 Expression in Lymphoma Cells Predicts Chemoresistance in Patients With Adult T-Cell Leukemia/Lymphoma. Oncol Lett (2016) 12(2):1519–24. doi: 10.3892/ol.2016.4774
69. Balajam NZ, Shabani M, Aghaei M, Haghighi M, Kompani F. Study of T-Cell Immunoglobulin and Mucin Domain-3 Expression Profile in Peripheral Blood and Bone Marrow of Human Acute Lymphoblastic Leukemia Patients. J Res Med Sci (2020) 25:69. doi: 10.4103/jrms.JRMS_759_19
70. Jadidi-Niaragh F, Ghalamfarsa G, Yousefi M, Tabrizi MH, Shokri F. Regulatory T Cells in Chronic Lymphocytic Leukemia: Implication for Immunotherapeutic Interventions. Tumour Biol (2013) 34(4):2031–9. doi: 10.1007/s13277-013-0832-x
71. Ghia P, Ferreri AM, Caligaris-Cappio F. Chronic Lymphocytic Leukemia. Crit Rev Oncol Hematol (2007) 64(3):234–46. doi: 10.1016/j.critrevonc.2007.04.008
72. Cha Z, Zang Y, Guo H, Rechlic JR, Olasnova LM, Gu H, et al. Association of Peripheral CD4+ CXCR5+ T Cells With Chronic Lymphocytic Leukemia. Tumour Biol (2013) 34(6):3579–85. doi: 10.1007/s13277-013-0937-2
73. Shanshal M, Haddad RY. Chronic Lymphocytic Leukemia. Dis Mon (2012) 58(4):153–67. doi: 10.1016/j.disamonth.2012.01.009
74. Ciszak L, Frydecka I, Wolowiec D, Szteblich A, Kosmaczewska A. Patients With Chronic Lymphocytic Leukaemia (CLL) Differ in the Pattern of CTLA-4 Expression on CLL Cells: The Possible Implications for Immunotherapy With CTLA-4 Blocking Antibody. Tumour Biol (2016) 37(3):4143–57. doi: 10.1007/s13277-015-4217-1
75. Jain N, O’Brien S. Targeted Therapies for CLL: Practical Issues With the Changing Treatment Paradigm. Blood Rev (2016) 30(3):233–44. doi: 10.1016/j.blre.2015.12.002
76. Allahmoradi E, Taghiloo S, Tehrani M, Hossein-Nattaj H, Janbabaei G, Shekarriz R, et al. CD4+ T Cells are Exhausted and Show Functional Defects in Chronic Lymphocytic Leukemia. Iran J Immunol (2017) 14(4):257–69.
77. Hadadi L, Hafezi M, Amirzargar AA, Sharifian RA, Abediankenari S, Asgarian-Omran H. Dysregulated Expression of Tim-3 and NKp30 Receptors on NK Cells of Patients With Chronic Lymphocytic Leukemia. Oncol Res Treat (2019) 42(4):202–8. doi: 10.1159/000497208
78. Zhou G, Sprengers D, Boor PPC, Doukas M, Schutz H, Mancham S, et al. Antibodies Against Immune Checkpoint Molecules Restore Functions of Tumor-Infiltrating T Cells in Hepatocellular Carcinomas. Gastroenterology (2017) 153(4):1107–19 e10. doi: 10.1053/j.gastro.2017.06.017
79. Liu F, Zeng G, Zhou S, He X, Sun N, Zhu X, et al. Blocking Tim-3 or/and PD-1 Reverses Dysfunction of Tumor-Infiltrating Lymphocytes in HBV-Related Hepatocellular Carcinoma. Bull Cancer (2018) 105(5):493–501. doi: 10.1016/j.bulcan.2018.01.018
80. Schnell A, Bod L, Madi A, Kuchroo VK. The Yin and Yang of Co-Inhibitory Receptors: Toward Anti-Tumor Immunity Without Autoimmunity. Cell Res (2020) 30(4):285–99. doi: 10.1038/s41422-020-0277-x
81. Liu F, Liu Y, Chen Z. Tim-3 Expression and its Role in Hepatocellular Carcinoma. J Hematol Oncol (2018) 11(1):126. doi: 10.1186/s13045-018-0667-4
82. Wang Z, Chen J, Wang M, Zhang L, Yu L. One Stone, Two Birds: The Roles of Tim-3 in Acute Myeloid Leukemia. Front Immunol (2021) 12:618710. doi: 10.3389/fimmu.2021.618710
83. Akashi K. TIM-3 Is a Novel Therapeutic Target for Eradicating Acute Myelogenous Leukemia Stem Cells. In: Nakao K, Minato N, Uemoto S, editors. Innovative Medicine: Basic Research and Development. Japan: Springer (2015). p. 307–15.
84. Tan J, Yu Z, Huang J, Chen Y, Huang S, Yao D, et al. Increased PD-1+Tim-3+ Exhausted T Cells in Bone Marrow may Influence the Clinical Outcome of Patients With AML. biomark Res (2020) 8:6. doi: 10.1186/s40364-020-0185-8
85. Rezazadeh H, Astaneh M, Tehrani M, Hossein-Nataj H, Zaboli E, Shekarriz R, et al. Blockade of PD-1 and TIM-3 Immune Checkpoints Fails to Restore the Function of Exhausted CD8(+) T Cells in Early Clinical Stages of Chronic Lymphocytic Leukemia. Immunologic Res (2020) 68(5):269–79. doi: 10.1007/s12026-020-09146-4
86. Zeidan AM, Komrokji RS, Brunner AM. TIM-3 Pathway Dysregulation and Targeting in Cancer. Expert Rev Anticancer Ther (2021) 21(5):523–34. doi: 10.1080/14737140.2021.1865814
87. Harding JJ, Moreno V, Bang YJ, Hong MH, Patnaik A, Trigo J, et al. Blocking TIM-3 in Treatment-Refractory Advanced Solid Tumors: A Phase Ia/b Study of LY3321367 With or Without an Anti-PD-L1 Antibody. Clin Cancer Res (2021) 27(8):2168–78. doi: 10.1158/1078-0432.CCR-20-4405
88. Brunner A, Borate U, Esteve J, Porkka K, Knapper S, Vey N, et al. AML-190: Anti-TIM-3 Antibody MBG453 in Combination With Hypomethylating Agents (HMAs) in Patients With High-Risk Myelodysplastic Syndrome (HR-MDS) and Acute Myeloid Leukemia: A Phase 1 Study. Clin Lymphoma Myeloma Leukemia (2020) 20:S188–9.
89. Zeidan A, Esteve J, Kim HJ, Miyazaki Y, Platzbecker U, Schuh A, et al. AML-187: The STIMULUS Clinical Trial Program: Evaluating Combination Therapy With MBG453 in Patients With Higher-Risk Myelodysplastic Syndrome (HR-MDS) or Acute Myeloid Leukemia. Clin Lymphoma Myeloma Leukemia (2020) 2020(20):S188. doi: 10.1016/S2152-2650(20)30727-8
90. Borate U, Esteve J, Porkka K, Knapper S, Wei AH. Phase Ib Study of the Anti-TIM-3 Antibody MBG453 in Combination With Decitabine in Patients With High-Risk Myelodysplastic Syndrome (MDS) and Acute Myeloid Leukemia (AML). Blood (2019) 134(Supplement_1):570. doi: 10.1182/blood-2019-128178
Keywords: acute lymphoblastic leukemia, acute myeloid leukemia, chronic lymphoblastic leukemia, chronic myeloid leukemia, myelodysplastic syndrome, TIM-3
Citation: Rezaei M, Tan J, Zeng C, Li Y and Ganjalikhani-Hakemi M (2021) TIM-3 in Leukemia; Immune Response and Beyond. Front. Oncol. 11:753677. doi: 10.3389/fonc.2021.753677
Received: 05 August 2021; Accepted: 10 September 2021;
Published: 30 September 2021.
Edited by:
Naval Daver, University of Texas MD Anderson Cancer Center, United StatesReviewed by:
Premal Lulla, Baylor College of Medicine, United StatesCopyright © 2021 Rezaei, Tan, Zeng, Li and Ganjalikhani-Hakemi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yangqiu Li, eWFuZ3FpdWxpQGhvdG1haWwuY29t; Chengwu Zeng, YmlvLXpjd0AxNjMuY29t; Mazdak Ganjalikhani-Hakemi, bWdoYWtlbWlAbWVkLm11aS5hYy5pcg==
†ORCID: Jiaxiong Tan, orcid.org/0000-0002-5606-3962
Chengwu Zeng, orcid.org/0000-0002-1333-3918
Yangqiu Li, orcid.org/0000-0002-0974-4036
Mazdak Ganjalikhani-Hakemi, orcid.org/0000-0002-4764-7616
‡These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.