
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
SYSTEMATIC REVIEW article
Front. Oncol., 30 September 2021
Sec. Hematologic Malignancies
Volume 11 - 2021 | https://doi.org/10.3389/fonc.2021.751668
This article is part of the Research TopicHybrid or Mixed Myelodyplastic / Myeloproliferative Disorders: Current Trends in Diagnosis and TreatmentView all 10 articles
 Klaus Geissler1,2*
Klaus Geissler1,2*Numerous examples in oncology have shown that better understanding the pathophysiology of a malignancy may be followed by the development of targeted treatment concepts with higher efficacy and lower toxicity as compared to unspecific treatment. The pathophysiology of chronic myelomonocytic leukemia (CMML) is heterogenous and complex but applying different research technologies have yielded a better and more comprehensive understanding of this disease. At the moment treatment for CMML is largely restricted to the unspecific use of cytotoxic drugs and hypomethylating agents (HMA). Numerous potential molecular targets have been recently detected by preclinical research which may ultimately lead to treatment concepts that will provide meaningful benefits for certain subgroups of patients.
● CMML is a clinically, molecularly and biologically heterogenous disease
● The combination of molecular data, functional in vitro findings, and in vivo preclinical models provide a comprehensive view of CMML pathogenesis
● Mutations in TET2 are common initial clonal driver abnormalities in CMML
● ASXL1 mutations play a major role in the transformation process into AML
● There is a close correlation between growth factor-independent myeloid colony-formation and the presence of RAS-pathway mutations
● RAS-pathway activation is a crucial pathophysiologic process for GM-CSF hypersensitivity, myeloproliferation, progressive disease and transformation into AML
● Numerous molecular targets provide the rationale for individualized treatment concepts in patients with CMML
Although the term chronic myelomonocytic leukemia (CMML) has been used previously, CMML has been officially, based on morphological criteria/phenotype, acknowledged as a specific entity in the FAB classification 1976 (1, 2). It is characterized by leukocytosis with monocytes and granulocytic cells in all stages of development, marked dysmyelopoiesis, a variable course, unresponsiveness to aggressive chemotherapy and an inherent risk of transformation to acute myeloid leukemia (AML) (3). With regard to the presence of myeloproliferation CMML was originally subdivided into myeloproliferative disorder MP-CMML (WBC count >13 x 109/L) versus myelodysplastic syndrome MD-CMML (WBC count ≤13 x 109/L MD-CMML) by the FAB criteria (4). Since CMML is characterized by features of both a MDS and a MPN the World Health Organization (WHO) classification of 2002 assigned CMML to the mixed category MDS/MPN (5). CMML is further subclassified by WHO into three groups based on blast equivalent (blasts plus promonocytes) in peripheral blood (PB) and bone marrow (BM) as follows: CMML-0 if PB <2% and BM <5% blast equivalent, CMML-1 if PB 2-4% or BM 5-9% blast equivalent, and CMML-2 if PB 5-19% or BM 10-19% blast equivalent, and/or Auer rods are present (6). The median survival of reported series is highly variable indicating a significant clinical heterogeneity of the disease (7–12).
Cancer is a biologically complex disease with characteristics acquired during the course of a multistep development process. In the past many research tools have been applied to better characterize the phenotypic, genotypic and functional features of cancer and to deeper understand the pathophysiology of malignancy with the ultimate goal to identify prognostic and predictive biomarkers, to render diagnosis more precisely and to develop targeted therapeutics for personalized medicine. No single technology is sufficient to consider all aspects of tumor complexity and information from different technologies are required to provide a comprehensive picture of cancer.
In 1987 a mutation within codon 12 of the NRAS gene was reported for the first time by Janssen et al. in a patient with CMML in a study investigating molecular alterations of RAS genes in a variety of preleukemic disorders and leukemias of myeloid origin (13). Subsequently is has been shown that RAS mutations are rare events in BCR/ABL negative chronic myeloid leukemia (CML) but are prevalent in CMML (14). In this study mutations in the RAS oncogene were found in 17 of 30 (57%) CMML patients. In the last years the molecular landscape in patients with CMML has been described by several groups. Molecular abnormalities can be seen in >90% of patients with CMML (15) with a marked heterogeneity among CMML patients. A large number of gene mutations in genes encoding epigenetic regulators (TET2, ASXL1, EZH2, UTX, IDH1, IDH2, DNMT3A) (9, 16–22) splicing factors (SF3B1, SRSF2, ZRSF2, U2AF1) (23, 24), and cytokine signaling molecules (NRAS, KRAS, CBL, JAK2, FLT3) have been reported (9, 25–29). Mutations in the transcription regulators RUNX1, NPM1, and TP53 have also been found in CMML (9, 30, 31). Table 1 shows the frequencies of gene mutations in 3 different CMML cohorts in which comprehensive molecular analyses has been reported (9, 11, 32). Considering all molecular data reported mutations in TET2 (~60%), SRSF2 (~50%), ASXL1 (~40%) and RAS pathway (~30%) are most common (15) but no molecular aberration is specific of this entity, as they can be detected with different frequencies in other myeloid neoplasms (33).
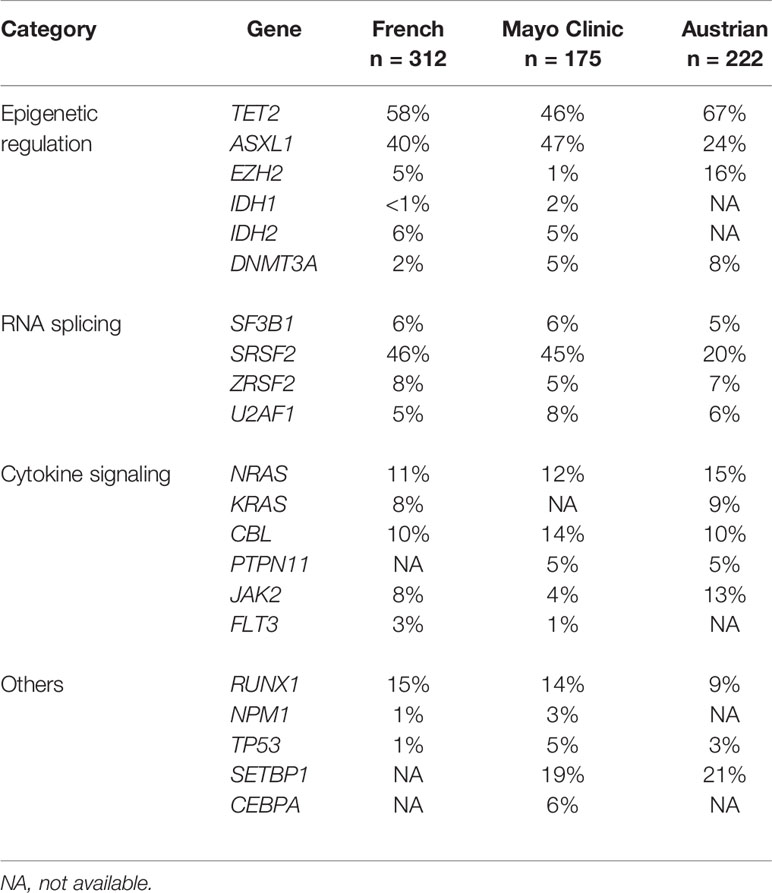
Table 1 Frequencies of molecular aberrations in different CMML cohorts.
In 1988 Geissler et al. have originally reported extensive in vitro formation of myelomonocytic colony forming units (CFU-GM) without addition of exogenous growth factors in a subset of patients with CMML (Table 2) (34). This spontaneous CFU-GM colony formation in CMML was markedly reduced by addition of anti-granulocyte/macrophage colony-stimulating factor (GM-CSF) antibodies, but not by antibodies against other growth factors, suggesting that this is a GM-CSF-dependent in vitro phenomenon (35) Figure 1. The biologic basis for this observation was later provided by Padron when he reported hypersensitivity of CMML progenitors using phospho-STAT5 flow cytometry (36). Moreover, the group in Vienna could show in a small retrospective study that CMML patients with high spontaneous CFU-GM growth (>100/105 PB mononuclear cells) have an inferior prognosis as compared to patients with low myeloid colony formation suggesting a clinical significance of the original observation (37). These results have been recently extended in a much larger CMML patient cohort indicating that spontaneous myeloid colony-formation was, compared to other single established prognostic factors, the strongest predictor regarding overall survival (OS) (38). This may indicate that in vitro cultures using unmanipulated mononuclear cells (MNC) may be a more global test that covers different aspects of malignancy better than any of the single parameters that are currently used to characterize the behavior of a tumor.

Table 2 Myeloid colony formation in patients with CMML.

Figure 1 Effect of anticytokine antibodies on spontaneous growth of CMML cells in 3 patients. PB MNC were cultured with medium alone or with antibodies against GM-CSF, G-CSF, IL-3, or IL-6, respectively. Data show a marked inhibition of spontaneous CFU-GM growth in the presence of anti-GM-CSF antibodies in all 3 patients indicating that autonomous colony formation is a GM-CSF dependent in vitro phenomenon [adapted from Geissler, K., et al., J Exp Med 1996 (35)].
There is also another in vitro phenomenon that seems to be characteristic for CMML patients. Semisolid in vitro cultures from PBMNC of normal individuals usually contain a higher concentration of erythroid colonies (BFU-E) as compared to myeloid colonies (CFU-GM). Skewed differentiation toward the myelomonocytic over erythroid commitment in patients, as indicated by an inverse BFU-E/CFU-GM ratio, is a common finding in CMML patients (39). Interestingly, the lack of myelomonocytic skewing separated patients with a particularly favorable prognosis and a minimal risk of transformation.
Myelomonocytic leukemias can be recapitulated by transplantation of mouse BM cells carrying an oncogenic mutation in the Nras locus (40–42). Interestingly, alterations of the other RASopathy genes including Kras, Cbl, Ptpn11 and Nf1 may also result in a similar phenotype in preclinical mouse models (Table 3) (44–47). In all these in vivo-models animals develop a myeloproliferative disorder with clonal expansion of the granulomonopoiesis.
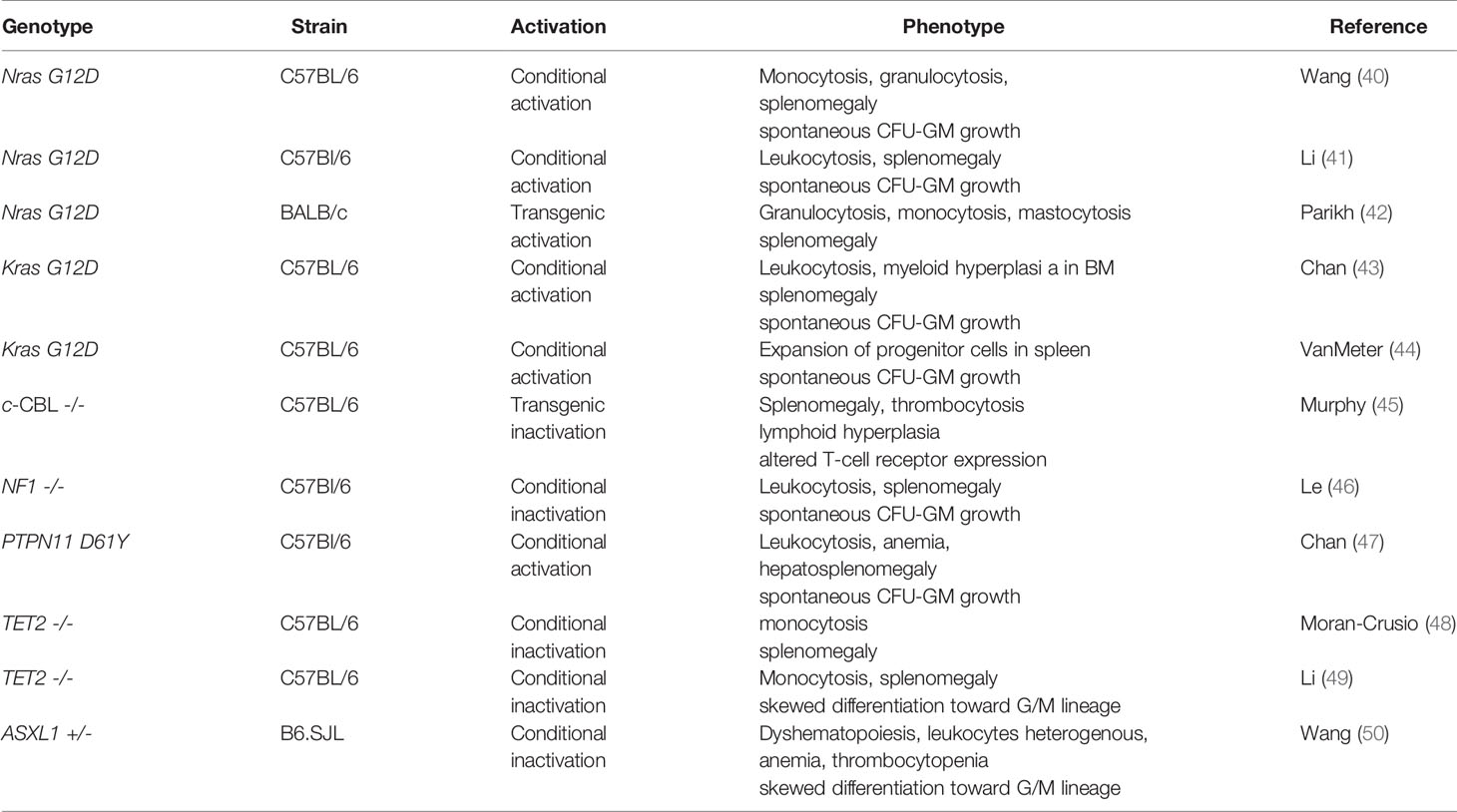
Table 3 Mouse models with CMML-like phenotype.
The effects of molecular aberrations in genes of the epigenetig machinery have been also studied in preclinical animal models (48–50). In a mouse model with complete functional deletion of Tet2 resulted in a progressive enlargement of the hematopoietic stem cell compartment and eventual myeloproliferation in vivo. Tet2 +/- mice displayed increased stem cell self-renewal and extramedullary hematopoiesis, indicating that Tet2 haploinsufficiency contributes to hematopoietic transformation in vivo (48). Importantly, one third of Tet2 -/- and 8% of Tet2 +/- mice died within 1 year of age because of the development of myeloid malignancies reminiscent of CMML indicating that Tet2 loss may represent a predisposition for the development of this malignancy. Moreover, it was shown that transplantation of Tet2 -/-, but not wild-type (WT) or Tet2 +/- BM cells, was associated with elevated white blood cell (WBC) counts, monocytosis and splenomegaly in WT recipient mice (49).
Recent evidence suggests that considering cancer only as a consequence of genetic aberrations is too simple (77). There is growing evidence that the complex nature of transformation from a normal to a cancer cell within different tissues is a result of the interplay among genetic and epigenetic events, tissue structure, exposure and the tissue microenvironment. Thus, molecular analysis of a tumor by NGS alone may be not sufficient to cover the biology of a tumor and emphasize the need for more comprehensive methods to characterize the biology of a tumour. By combining structural data, functional in vitro findings, an in vivo preclinical models a comprehensive view of pathogenesis of CMML is possible.
Similar to the in vitro phenomenon of spontaneous erythroid colony (78) and megakaryocyte colony formation (79) due to hypersensitivity to growth factors in patients with BCR/ABL negative MPN spontaneous myeloid colony formation seems to be an in vitro feature in a subset of patients with CMML. Molecular aberrations of RASopathy genes in murine hematopoietic cells induce growth-factor-independent CFU-GM formation in vitro due to hypersensitivity of granulomonocytic precursors to GM-CSF (40, 41, 43, 44, 46, 47). Moreover, in juvenile myelomonocytic leukemia (JMML) in which molecular aberrations are mainly restricted to RASopathy genes autonomous myeloid colony formation due to GM-CSF-specific hypersensitivity is a hallmark feature of disease, which has been included in the diagnostic criteria (80). In a small series of CMML patients who had in vitro cultures and molecular analyses Geissler et al. observed a close correlation between high spontaneous myeloid colony growth and the presence of RAS pathway mutations as shown in Figure 2 (81). This initial observation was later confirmed in a larger patient cohort including 100 CMML patients (82). The median number of spontaneously formed CFU-GM/105 MNC was 147.5 in RAS-mutated patients as compared with 2 in RAS-wildtype patients (p<0.00001). Unstimulated myeloid colony formation in RAS-mutated patients was also much higher than spontaneous formation of CFU-GM in normal individuals (median 4.8/105 PBMNC) which has been reported by this group previously (83). There was no significant difference regarding spontaneous CFU-GM formation in CMML patients with molecular aberrations in genes of epigenetic regulation and RNA-splicing, respectively. High spontaneous myeloid colony formation was also never observed in CMML patients with the JAK2 V617F mutation as the only molecular aberration in signaling pathways [Geissler et al., unpublished data]. All these findings, in mouse and human, indicate that hypersensitivity to GM-CSF, as manifested by growth factor-independent CFU-GM growth in vitro, is caused by molecular aberrations of the RAS-pathway which may be a major driver in CMML pathogenesis, in particular in MP-CMML. Moreover, it reveals high autonomous CFU-GM growth as a functional surrogate parameter of RAS pathway hyperactivation in CMML.
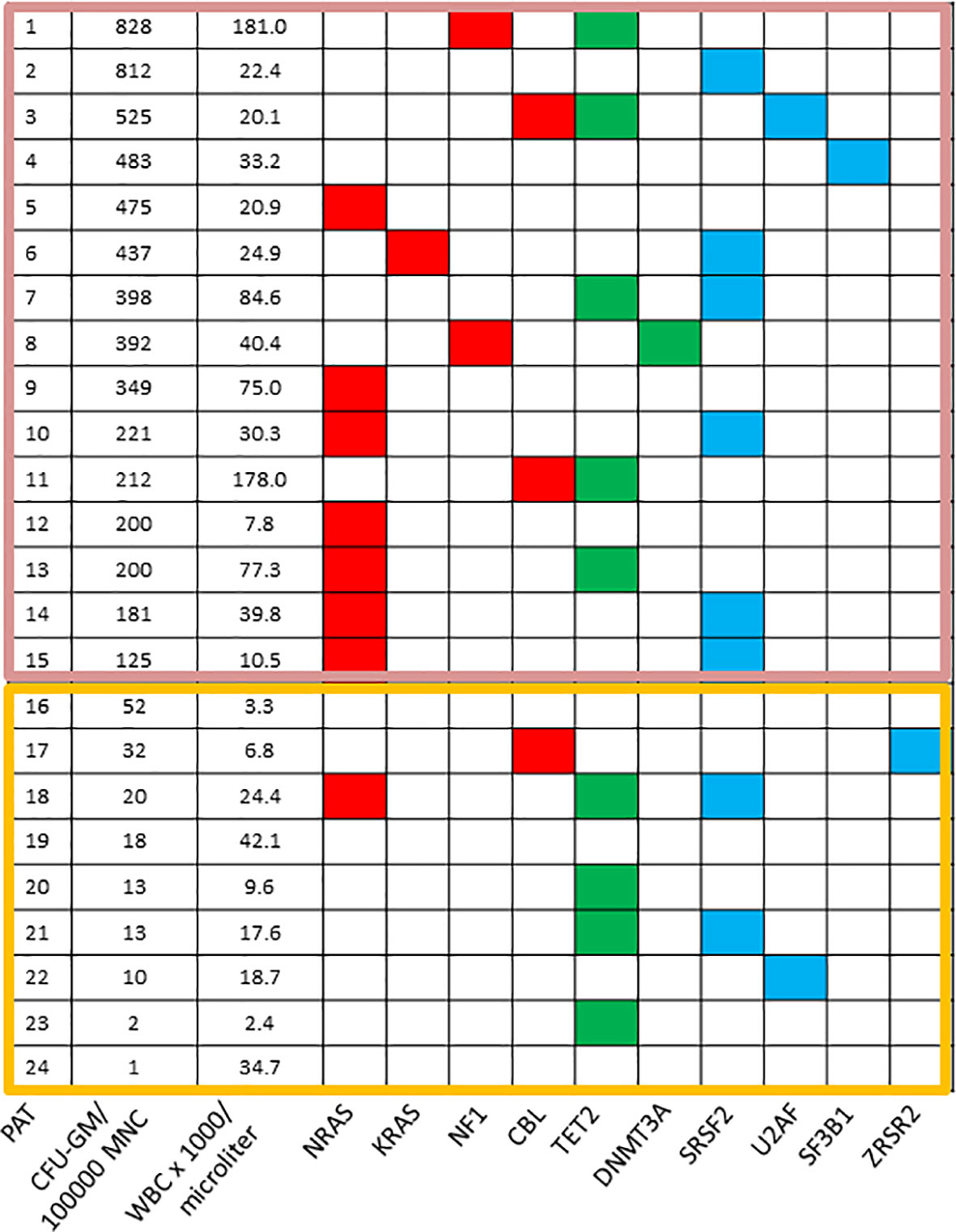
Figure 2 Mutation profiles in CMML according to spontaneous CFU-GM growth. Each row corresponds to one patient. The first column indicates the patient number, the second the number of CFU-GM per 105 peripheral blood mononuclear cells (PBMNC), the third the white blood cell (WBC) count and all other columns represent the status of genes. Colored squares indicate mutated, white squares wild-type genes. The clores of mutant genes indicate the most affected functional categories. Red, green, and blue indicate RAS-pathway, epigenetic factors, and splicing factors, respectively. Mutations in the components of the RAS-pathway were found in 12/15 (80%) CMML patients with high colony growth (≥100 CFU-GM/105 PBMNC) and in 2/9 (22%) patients with low spontaneous colony formation (<100 CFU-GM/105 PBMNC). [adapted from Geissler, K. MEMO 2016 (81)].
Myelomonocytic skewing has been proposed as a key phenomenon in the pathophysiology of CMML. In a seminal paper using mutation-specific discrimination analysis of single-cell-derived colonies in 28 patients with CMML, Itzykson et al. could show that the main features of this disease are early clonal dominance, arising at the CD34+/CD34- stage of hematopoiesis, and granulomonocytic differentiation skewing of multipotent and common myeloid progenitors (84). Geissler et al. could demonstrate that myelomonocytic skewing as determined by semisolid cultures can separate subgroups of CMML patients with a different phenotype, a different genotype and a different prognosis (39). The definitive link of this phenomenon to the pathophysiology of CMML comes from animal studies in which hematopoietic cells are genetically manipulated with molecular aberrations that are commonly found in CMML patients. Functional inactivation of TET2 in cord blood CD34+ cells skews progenitor differentiation toward the granulomonocytic lineage at the expense of lymphoid and erythroid lineages (85). In mice, deletion of Tet2 results in an increased hematopoietic repopulating capacity with an altered differentiation skewing towards the granulomonocytic lineage (49). Other epigenetic modifiers such as ASXL1 have also been shown to impact skewing of hematopoiesis. Asxl1 -/- mice had a reduced hematopoietic stem cell (HSC) pool, and Asxl1 -/- HSCs exhibited decreased hematopoietic repopulating capacity, with skewed cell differentiation favoring granulocytic lineage (50). Furthermore the splicing factors SRSF2 and U2AF1 seem to affect skewing. Mutations in both SRSF2 and U2AF1 are associated with abnormal differentiation by skewing granulomonocytic differentiation towards monocytes (86). Collectively, many molecular aberrations that can be found in CMML, induce myelomonocytic skewing in the preclinical mouse model providing the genetic basis for this key finding in patients.
Recent molecular analyses of large populations have indicated that somatic mutations in hematopoietic cells leading to clonal expansion are commonly acquired during human aging (87). Clonally restricted hematopiesis is associated with an increased risk of subsequent diagnosis of myeloid neoplasia. As some of the genes frequently mutated in age-related clonal hematopoesis such as TET2 and ASXL1 are also commonly mutated in CMML and aged hematopoiesis is characterized by a myelomonocytic differentiation bias it was recently hypothesized that CMML and aged hematopoiesis may be closely related (88). Analyses of the somatic mutation landscape of CMML by whole exome sequencing followed by gene-targeted validation indicated that most CMML patients (71%) had mutations in >2 age-related clonal hematopoiesis (ARCH) genes and 52% had >7 mutations overall (89). A higher mutation burden was associated with inferior survival. Considering age-adjusted population incidence and ARCH mutation rates one may speculate that CMML represents the leukemic conversion of the myelomonocytic-lineage-biased aged hematopoietic system.
There is now increasing evidence that mutations in TET2 are in fact an initial clonal driver in CMML (88, 90). This view is based on the high frequency (60%) of these mutations in CMML patients (9, 19, 21, 22), the fact that TET2 mutated clones can be detected in a small fraction of older subjects with clonal, but non-leukemic hematopoiesis (90–93), the competitive advantage of murine and human HSC invalidated for TET2 (48, 85) and the results of single-cell clonal tracking experiments indicating that a TET2 mutation, when present, is often the earliest recurrent genetic event in CMML (84). According to data from this study the preferred order of mutational accumulation is TET2 (or IDH1/2) or ASXL1 (EZH2) first, followed by molecular aberrations in spliceosome components (SRSF2, SF3B1, U2AF1, or ZRSR2). Mutations in the RAS-signaling pathway seem to be rather late events which induce GM-CSF hypersensitivity and myeloproliferation.
Around 20% of CMML patients progress and transform to AML. Although the mechanisms behind are not known in detail, available data suggest that molecular aberrations in chromatin modelling as well in cell signaling may contribute to this process. Among genes of the epigenetic machinery, ASXL1 may have the most important impact on transformation. The ASXL1 gene regulates chromatin by interacting with the polycob-group repressive complex proteins (PRC1 and PRC2). Abdel-Wahab et al. reported that ASXL1 mutations resulted in loss of PRC2-mediated H3K27 trimethylation (94). In a study by Itzykson et al. in which the prognostic impact of different molecular aberrations in CMML patients was studied, only ASXL1 mutations retained their significant impact on AML-free survival in the multivariate analysis indicating the major role of this molecular aberration in the transformation process (9). Of these, only nonsense and frameshift ASXL1 mutations have been shown to negatively impact OS. The impact of mutations of RAS-pathway components on progression/transformation of CMML is more complex. The first indication of a potential role of NRAS aberrations in CMML evolution has been reported, at the molecular level, by Ricci et al. (95). In this study molecular analyses have been performed in 22 MD-CMML patients and in 18 MP-CMML patients. MP-CMML patients had a higher frequency of RAS mutations compared with MD-CMML. In two patients who progressed from MDS-CMML to MP-CMML, allele specific PCR showed low levels of the RAS mutations at the time of myelodysplastic disease which became predominant in the myeloproliferative phase, documenting for the first time the expansion of a RAS mutated clone in concomitance with CMML evolution. Other studies have confirmed that the MPN phenotye of CMML is a disease phase significantly associated with hyperactivation of the RAS-pathway. In a study reported by the Austrian study group MP-CMML as compared to MD-CMML patients had higher circulating blasts, LDH, RAS-pathway mutations, more often splenomegaly and higher growth-factor-independent myeloid colony growth in vitro (12). Recently, genetic differences were assessed between subtypes in 973 molecularly annotated Mayo Clinic-GFM-Austrian CMML patients. In this analysis NRAS mutations alone did not reach statistical significance as an independent factor impacting AML-free survival, however, the combined oncogenic RAS-pathway category including NRAS, KRAS and CBL was statistically significant in a model that only included genetic factors (74). Considering the fact, that spontaneous colony formation in CMML functionally covers the most frequent RASopathy gene mutations (38) these data are in line with findings in a small study which have been previously reported. In this study patients with CMML growth-factor-independent colony formation after transformation was significantly increased as compared to CFU-GM growth before transformation (37). Furthermore, a correlation of RAS-pathway mutations and spontaneous myeloid colony growth with progression and transformation could be demonstrated in a retrospective analysis of 337 CMML patients (96). Moreover, recent preclinical models also suggest that activating Nras mutations and somatic loss-of-function mutations in Tet2 exert cooperating effects and accelerate disease progression (97, 98). Altogether, these findings suggest that oncogenic RAS-pathway activation is a phenomenon associated with the MP-CMML phenotype, progressive disease and with transformation to AML.
The management of patients with CMML should be based on risk assessment. Several studies have shown that the percentage of PB and BM blasts is the most important factor determining survival (7, 99–104). Genetic alterations including gene mutations (7, 9, 10, 32) and chromosomal aberrations (105–107) further refine prognosis and have been included in different prognostic scoring systems. In the EHA guideline from 2018 five risk stratification systems are recommended (7, 9, 10, 106, 108, 109). Mutations in ASXL1 are included in all 3 molecularly based scoring systems whereas the molecular CMML-specific prognostic scoring system (CPSS-mol) also includes NRAS, SETBP1 and RUNX1 (10). A recent study validating different prognostic models demonstrated comparable performance with significant heterogeneity in predicting outcomes (110).
Traditionally many cancers have been treated with more or less unspecific treatments such as cytotoxic drugs in the past. In a molecular heterogenous malignancy this may have the advantage that many subclones may be impacted by one drug with the potential to improve symptoms associated with a high tumor mass and potentially improve survival. Unfortunately, these drugs often cause significant side effects due to the fact, that also normal cells from tissues with a high proliferation rate may be affected. Targeted drugs on the other hand may be of interest if they are able to specifically hit a cellular component which is critical for the pathophysiology of disease. Many examples from other cancers have shown that with targeted treatment we can expect higher efficacy and lower toxicity as compared to conventional therapy. Unfortunately, CMML is a clinically and molecularly heterogenous disease with sometimes multiple clones that may be pathophysiologically relevant. Theoretically, targeted treatment might offer clinical benefit only if these subclones contribute to inferior prognosis and/or symptoms in patients. Symptoms in patients with CMML are often the clinical consequence of a high tumor mass. Myeloproliferation in CMML is commonly associated with molecular aberrations in cytokine signaling. In particular, as mentioned before, molecular aberrations in components of the RAS signaling pathway are frequently found in these patients. On the other hand there are molecular markers that predict inferior survival. Targeting these components may have the potential to modify the biology of disease and to delay transformation to AML. For such concepts it would be important to know if targeted treatment, at all, will be beneficial in a complex disease such as CMML. Although this question cannot be answered for patients with CMML at the moment, there is some indication from other malignancies that treatment of subclones could be beneficial in patients. Patel et al. published a small series of patients with BCR/ABL negative MPN and a IDH2 mutation who were treated with the IDH2 inhibitor enasidenib which is approved for the treatment of patients with AML harboring this molecular aberration. Although IDH2 is often subclonal in this disease treatment with this IDH2 inhibitor resulted in clinically meaningful responses in these patients (111).
Etoposide (VP16) is a DNA-damaging molecule by inhibition of topoisomerase. Preliminary reports suggested that etposide could give good results in CMML, with true complete responses in some cases and in improvement rather than worsening of cytopenias (51) (Table 4). Hydroxyurea (HU), a potent ribonucleotide reductase inhibitor, acts as an S-phase-specific agent with inhibition of DNA synthesis. In a randomized phase III trial in patients with proliferative CMML, HU was more effective and achieved faster response than cytotoxic chemotherapy with VP16 (52). Interestingly, this study remains up to now the only randomized trial in a pure CMML patient population which demonstrated a survival benefit. Allogeneic stem cell transplantation which is the only curative therapy is rarely feasible because of age and/or comorbidities. While unresponsiveness to aggressive chemotherapy is a characteristic for most CMML patients, there may be subgroups that might benefit from more intensive chemotherapy. Although the presence of an NPM1 mutation, in contrast to AML patients, is an inferior prognostic parameter in CMML, CMML patients with this molecular aberration have shown relatively high response rates in a retrospective analysis (112).

Table 4 Potential molecular targets in CMML.
More targeted treatment with cytotoxic drugs can be expected by more detailed immunophenotypical characterization of surface proteins on CMML stem cells which could be used as potential target structures for antibody-drug conjugates (113). One example is the use of the IL-3 receptor as target structure for tagraxofusp, a CD123-directed cytotoxin consisting of human IL-3 fused to truncated diphtheria toxin. This antibody-drug conjugate has shown impressive activity in blastic plasmacytoid dendritic-cell neoplasm (BPDCN) that overexpresses CD123 (114). In an early clinical trial in patients with relapsed/refractory CMML 80% (8/10) of the patients receiving tagraxofusp showed ≥50% reduction in splenomegaly and three patients achieved bone marrow CR (53).
It is important to note that the approval of the hypomethylating agents (HMA) azacitidine and decitabine (DEC), respectively, was originally based on MDS studies which included only few patients with CMML. In a phase III clinical multicenter trial of 358 MDS patients including 11 patients with dysplastic CMML the median overall survival was 24.5 months in the azacitidine (AZA) group as compared to 15.0 months in the conventional care group leading to the FDA approval of AZA for this subtype of CMML (54). The approval of decitabine (DEC) for CMML was also based on a phase III clinical trial of 170 patients with MDS, 14 of them with CMML (55). The ORR was significantly higher in the DEC group versus supportive care (17% vs. 0%, p < 0.001), but the median OS was not significantly different between the two arms. Additional phase II studies confirmed the efficacy of hypomethylating agents in all subtypes of CMML and, therefore, these agents are considered commonly as standard of care for higher risk CMML (Table 3) (56–63). In the largest retrospective study with a pure CMML cohort patients were treated with AZA (n = 56) and DEC (n = 65) (115). The ORRs were 41% by the IWG MDS/MPN response criteria (AZA-56%, DEC-58%), with CR rates of <20% for both agents. No significant differences in response rates were seen between MP-CMML and MD-CMML. Similar findings were reported in a smaller, prospective phase II Italian study, with 43 CMML patients receiving DEC (63). The ORR after 6 cycles was 47.6%, with seven CRs (16.6%), eight marrow responses (19%), one partial response (2.4%) and four hematological improvements (9.5%). After a median follow-up of 51.5 months, median OS was 17 months, with responders having a significantly longer survival than non-responders. Despite some efficacy of HMA in CMML patients one has to keep in mind that this treatment does not alter mutational allele burden and disease biology (116).
Proof of efficacy but greatly variable response with HMA provide the rationale for searching biomarkers that predict response. Differentially methylated regions of DNA have been shown to separate DEC responders from non-responders by Meldi (117). Other predictors for response to HMA treatment were reported by Duchmann et al. (118). In a retrospective analysis of 174 CMML patients treated with HMA multivariate analysis mutations in ASXL1 predicted lower ORR, and RUNX1 mutations and CBL mutations predicted inferior OS. The combination of TET2 mutation and ASXL1 wildtype predicted higher CR and better OS. A multicenter retrospective study including 949 non-selected, consecutive CMML patients investigated whether HMA provide a benefit in subgroups of CMML patients (119). Adjusted median OS for patients treated with HU versus HMA was 15.6 months as compared to 20.7 months; (p=0.0002). In patients with MP-CMML, median OS was 12.6 months as compared to 17.6 months; (p=0.0027) for patients treated with HU versus HMA. HMA were not associated with an OS advantage for patients classified as having lower-risk disease (ie, MD-CMML with <10% blasts, CMML-0, or lower-risk CPSS). Considering all the caveats of a retrospective nonrandomized trial these data suggest HMA as the preferred treatment for patients with higher-risk CMML and those with MP-CMML. A recent European multicenter randomized phase III trial evaluating DEC +/- HU versus HU in advanced MP-CMML, however, did not show significant differences in outcome. Although HMA definitively play an important role in the management of CMML patients the need for newer, rationally derived therapies is apparent (120).
TET2 enzymes have been shown to provide a homeostatic link between intracellular metabolism and epigenetic gene regulation (121). These evolutionary conserved dioxygenases play a key role in the conversion of 5-methyl-cytosine (5-mC) to 5-hydoxymethyl-cytosine (5-hmC). TET dioxygenases require alpha-ketoglutarate, oxygen, Fe(II), and ascorbate for optimal activity (122). Isocitrate dehydrogenase (IDH) is a key enzyme for cellular respiration in the tricarboxylic acid (TCA) cycle. IDH mutations found in malignancies block normal cellular differentiation and promote tumorigenesis via the abnormal production of the oncometabolite 2-hydroxy-glutarate (2-hG). Recently, two inhibitors targeting IDH2 and IDH1 gene mutations, have become important components in AML management since molecular aberrations of IDH genes can be found in 20% of patients AML (123, 124). Although mutations involving IDH1 and IDH2 are uncommon in CMML (1% and 5-10%, respectively) IDH1/2 inhibitors are likely to present therapeutic options for these patients.
Loss-of-function mutations in TET2 occur in around 60% of CMML patients and are considered mutually exclusive with IDH1/2 mutations. Recently there has been accumulated significant preclinical evidence suggesting that ascorbate can restore dysfunctional TET2 activity. Agatocleous et al. generated mice lacking Gulo, the enzyme responsible for ascorbate synthesis. The resulting phenotype resembled mice carrying a homozygous Tet2 deletion (48). Indeed, ascorbate-depleted stem and progenitor cells showed decreased levels of 5-hmC, predominantly mediated by reduction of Tet2 function (125). On the other hand treatment with vitamin C mimicked Tet2 restoration in a reversible transgenic RNAi mouse model as described by Cimmino (64). Low ascorbate levels have been demonstrated in a subgroup of patients with hematologic malignancies (126). Although no beneficial effects of vitamin C intake regarding leukemia development have been seen in previous reports, these new preclinical data show that the possible impact of supra-physiological concentrations of vitamin C on leukemogenesis remains an interesting treatment concept, particular in CMML-patients harboring a partial or complete loss of TET2 function. In fact the is a current phase II trial which studies the effect of ascorbate in combination with AZA in patients with newly diagnosed AML, MDS, MDS/MPN with TET2 mutations (NCT03397173).
Geissler et al. have shown that growth factor-independent in vitro myeloid colony formation by CMML cells can be inhibited by the addition of anti-GM-CSF antibodies, but not by addition of antibodies against IL-6, Il-3, or G-CSF indicating that GM-CSF signaling may play an important role in the pathophysiology of CMML (35). Because of its cytokine synthesis-inhibiting effects IL-10 was studied on CMML cell growth in vitro. The addition of IL-10 revealed a profound and dose dependent inhibitory effect on spontaneous in vitro growth of CMML cells (35). It was shown that IL-10 induced suppression of CMML cell proliferation was associated with reduced GM-CSF production by leukemic cells, both at the mRNA and protein level. Therefore it was concluded that the inhibitory effect of IL-10 in vitro is most likely through suppression of endogenous GM-CSF release. Based on these findings a small pilot trial was initiated in which five patients with CMML were treated with 4µg/kg/day recombinant human IL-10 sc for 1 month and with 8 µg/kg/day for another month (65). Although no meaningful effects of IL-10 treatment was seen on the WBC counts in any of the five patients, one out of three patients with histologically confirmed skin infiltration markedly improved during IL-10 therapy. IL-10 administration was associated with a decline in lysozyme serum levels, a biomarker of the monocytic cell lineage, and downregulation of CD86 which has been shown to be upregulated by GM-CSF and downregulated by IL-10 in vitro. Interestingly, the clinical impact of IL-10 war recently supported by a study in which cytokine profiles were analyzed using cryopreserved PB plasma samples from 215 CMML patients (127). CMML patients with decreased IL-10 expression were found to have a poor OS when compared to CMML patients with increased IL-10 expression (P = 0.017), even when adjusted for other prognostic features including ASXL1.
Lenzilumab is a monoclonal antibody with high affinity for human GM-CSF. Based on data showing that anti-GM-CSF antibodies significantly inhibited the growth factor independent myeloid in vitro colony formation from primary CMML patient samples (35) and a study reporting that 90% of primary CMML samples demonstrated GM-CSF-dependent STAT5 hypersensitivity (36) lenzilumab was studied in CMML patients. In this early clinical trial of 15 CMML patients the antibody was well tolerated and effective with a durable ORR of 33.3% (66).
The clinical management of FLT3-mutated AML has been changed by the development of FLT3 inhibitors such as midostaurin and gilteritinib which are now in use in the frontline and relapsed/refractory settings in patients with a FLT3 mutation (128, 129). FLT3 aberrations have been reported in 1-3% of CMML patients (9, 32). Although these aberrations are uncommon in CMML FLT3 signaling may also occur in wildtype FLT3 malignancies. Thus, mice carrying a mutation in the RING finger domain of c-CBL develop a myeloproliferative disease involving hematopoietic progenitors that show increased FLT3 signaling (67). The incidence of molecular aberrations of the CBL gene has been reported from 10-14% (9, 11, 32) and thus is more common than that of the FLT3 gene. Therefore, CMML patients with mutations in the CBL gene could be potential candidates for studies with FLT3 inhibitors. In an ongoing phase I/II trial the FLT3 inhibitor quizartinib in combination with AZA is investigated in patients with untreated or HMA-refractory MDS, MDS/MPN with FLT3 or CBL mutations (NCT04493138).
Mutated RAS proteins have been deemed “undruggable” for a long time due to their high affinity for GTP and lack of accessible binding pockets. However, the discovery by Ostrem et al. of compounds that covalently bind to the switch II pocket of KRAS G12C provided the rationale for the development of inhibitors suitable for clinical testing (130). At the moment this concept does not play an important role in the treatment concepts for CMML, since the KRAS G12C mutation is extremely rare in CMML.
RAS proteins require post-translational farnesylation by the enzyme farnesyltransferase to become functionally active. Therefore, inhibitors of this enzyme have been considered as potential candidates for RAS-pathway inhibition. In a clinical phase III trial 85 patients with newly diagnosed JMML, a RAS pathway driven disease, were enrolled between 2001 and 2006 (131). 47 patients received the FTI tipifarnib alone in a phase II window before proceeding to HSCT. Tipifarnib as a single agent was safe and achieved a response rate of 51%, but failed to reduce relapse rates or improve long-term overall survival in the phase III trial. In a preliminary report of a phase II trial in CMML patients tipifarnib was well tolerated, however, had only limited efficacy (132).
The elucidation of the RAS/MEK/ERK signaling pathway in regulating cell proliferation has stimulated the development of selective MEK inhibitors (MEKi). These molecules represent promising therapies for RAS-driven neoplasias and RASopathies associated with hyperactivated RAS signaling. Preclinically, the MEKi PD0325901 was highly effective in reversing the CMML-like phenotype in a Kras G12D and in a NF1 -/- mouse model (68, 69). In a phase II study in patients with in Neurofibromatosis 1 (NF1) which is a prototypic RASopathy the MEKi selumetinib resulted in at least 20% reduction in the size of plexiform neurofibromas (pNF) from baseline in 71% of patients and was associated with clinically meaningful improvements (133). On the basis of this clinical benefit, selumetinib received FDA approval for children 2 years of age and older with inoperable, symptomatic pNF. In another phase II trial trametinib, another MEKi, was studied in patients with relapsed/refractory leukemias (70). Cohort 1 included patients with relapsed/refractory AML or high-risk MDS with NRAS or KRAS mutations, cohort 2 patients with AML, MDS, or CMML with a RAS wild-type mutation or an unknown mutation status, and cohort 3 patients with CMML with an NRAS or KRAS mutation. The recommended dose for trametinib was 2 mg orally daily. The overall response rates for cohorts 1, 2, and 3 were 20%, 3%, and 27%, respectively, with a preferential activity among myeloid malignancies with RAS mutations. Repeated cycles of trametinib were well tolerated with manageable or reversible toxicities. Thus, some therapeutic potential of trametinib was demonstrated in myeloid malignancies, particularly in RAS-pathway mutated CMML.
There is some evidence of activity or JAK inhibitors in CMML patients. In a study by Geissler the specific JAK2 inhibitor TG101209 was found to either block or strongly inhibit spontaneous CFU-GM growth in vitro in all 10 CMML patients tested (71). Among these 10 patients 6 were tested by NGS and, in 5 of them, RAS-pathway hyperactivation was documented due to mutations in NRAS (n=3) or PTPN11 (n=2), respectively. In a NRAS-mutant CMML patient who was treated with the JAK1/2 inhibitor ruxolitinib off label, spleen response and the disappearance of constitutional symptoms was associated with a decrease of autonomous CFU-GM formation ex vivo. Thus, therapeutic potential of inhibition of the JAK2/STAT5 pathway by ruxolitinib in CMML is suggested. In a phase I/II clinical trial of ruxolitinib in 20 CMML patients the recommended dose of ruxolitinib was 20 mg twice daily and the ORR of 35%, 5/9 spleen responses, and 10/11 symptom responses were seen (72). Correlative analysis demonstrated a downregulation in inflammatory cytokines and GM-CSF-dependent STAT5 phosphorylation in responders. Further studies are required to demonstrate a potential disease modifying effect of ruxolitinib in CMML.
Biological crosstalk is a phenomenon in which one component of a signal transduction pathway can affects another pathway. Thus, the PI3 Kinase-pathway may be aberrantly activated in CMML without molecular aberration in it. Treatment with inhibitors of this aberrantly activated signaling could have the potential to impact malignant cell growth. Using the class I PIK3 inhibitor pictilisib this approach has been successfully applied in a Kras G12D and in a NF1 -/- mouse model with a CMML-like phenotype (73). In this models, pictilisib attenuated activation of both PI3K/AKT and RAS/MEK/ERK pathways in primary hematopoietic cells. Several PI3K inhibitors have now received regulatory approval for the treatment of breast cancer and B-cell malignancies suggesting that the treatment concept of PI3K-pathway inhibition comes into the clinic (134, 135). Thus, based on some crosstalk between the RAS-signaling and the PI3K/AKT-pathway PI3K inhibitors could be important molecules for the design of future therapeutic strategies for patients with CMML.
In MP-CMML RAS-pathway mutations are associated with a unique gene expression profile enriched in mitotic kinases including polo-like kinase 1 (PLK1) (74) as shown in a study using a multiomics platform and biochemical and molecular analyses. In this study unmutated MLL regulated PLK1 transcript levels via promoter monomethylation of lysine 4 of histone 3. In the preclinical mouse model pharmacologic inhibition of PLK1 was effective in RAS-mutant patient-derived xenografts providing the rationale for a new biomarker-driven therapeutic approach in patients with proliferative CMML. Currently the administration of the PLK4 inhibitor CFI-400945 with or without HMA is tested in a phase II trial in patients with relapsed/refractory or untreated AML, MDS, or CMML (NCT04730258).
The inflammasome is a multimeric protein complex including NLRP3, NLRC4, AIM2 and NLRP6 that initiates an inflammatory form of cell death (pyroptosis) and triggers the release of proinflammatory cytokines (136). Recently, a functional link between oncogenic Kras G12D and inflammasome activation was reported in a preclinical model (75). In this mouse model Kras driven myeloproliferation and cytopenia was reversed by functional inactivation of NLRP3. A similar phenotypic improvement was seen with therapeutic IL-1-receptor blockade. Importantly, Kras activation induced the production of reactive oxygen species suggesting that KRAS not only has an ongogenic driver function but also activates the proinflammatory machinery. These findings open a new therapeutic opportunity for Kras mediated malignancies including CMML. Interestingly, there is a current phase II study, in which the anti-IL1ß inhibitor canakinumab is studied in ESA or HMA-refractory low risk-MDS or CMML (NCT04239157).
Given the complexity of CMML one can expect, that combinations of molecules impacting different pathways may yield better efficacy. At least in preclinical models this seems to be true (76). In Nras hyperactive mouse models mimicking MP-CMML inhibition of the MEK-pathway alone was only partially effective to improve disease associated features. Despite MEK inhibitor treatment 60% of Nras G12D expressing mice died within 20 weeks and surviving animals continued to retain their MP-CMML phenotype. Combined inhibition of the MEK and JAK/STAT signaling, however, greatly inhibited human and mouse CMML cell growth in vitro, rescued mutant Nras G12D-expressing HSC function in vivo, and promoted long-term survival without evident disease manifestation in animals with RAS-pathway driven MP-CMML. Still much work has to be done to address optimal ways to target these pathways in patients with CMML to improve clinical outcome.
The author confirms being the sole contributor of this work and has approved it for publication.
This article was supported by the “Gesellschaft zur Erforschung der Biologie und Therapie von Tumorkrankheiten” – ABCMML-112015”.
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be constructed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Bennett JM, Catovsky D, Daniel M-T, Flandrin G, Galton DAG, Gralnick HR, et al. Proposals for the Classification of the Acute Leukaemias French-American-British (FAB) Co-Operative Group. Br J Haematol (1976) 33(4):451–8. doi: 10.1111/j.1365-2141.1976.tb03563.x
2. Bennett JM, Catovsky D, Daniel MT, Flandrin G, Galton DA, Gralnick HR, et al. Proposals for the Classification of the Myelodysplastic Syndromes. Br J Haematol (1982) 51(2):189–99. doi: 10.1111/j.1365-2141.1982.tb08475.x
3. Vardiman JW, Thiele J, Arber DA, Brunning RD, Borowitz MJ, Porwit A, et al. The 2008 Revision of the World Health Organization (WHO) Classification of Myeloid Neoplasms and Acute Leukemia: Rationale and Important Changes. Blood (2009) 114(5):937–51. doi: 10.1182/blood-2009-03-209262
4. Bennett JM, Catovsky D, Daniel MT, Flandrin G, Galton DAG, Gralnick H, et al. The Chronic Myeloid Leukaemias: Guidelines for Distinguishing Chronic Granulocytic, Atypical Chronic Myeloid, and Chronic Myelomonocytic Leukaemia: Proposals by the French - American - British Cooperative Leukaemia Group. Br J Haematol (1994) 87(4):746–54. doi: 10.1111/j.1365-2141.1994.tb06734.x
5. Vardiman JW, Harris NL, Brunning RD. The World Health Organization (WHO) Classification of the Myeloid Neoplasms. Blood (2002) 100(7):2292–302. doi: 10.1182/blood-2002-04-1199
6. Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Beau L, et al. The 2016 Revision to the World Health Organization Classification of Myeloid Neoplasms and Acute Leukemia. Blood (2016) 127(20):2391–405. doi: 10.1182/blood-2016-03-643544
7. Onida F, Kantarjian HM, Smith TL, Ball G, Keating MJ, Estey EH, et al. Prognostic Factors and Scoring Systems in Chronic Myelomonocytic Leukemia: A Retrospective Analysis of 213 Patients. Blood (2002) 99(3):840–9. doi: 10.1182/blood.v99.3.840
8. Patnaik MM, Padron E, LaBorde RR, Lasho TL, Finke CM, Hanson CA, et al. Mayo Prognostic Model for WHO-Defined Chronic Myelomonocytic Leukemia: ASXL1 and Spliceosome Component Mutations and Outcomes. Leukemia (2013) 27(7):1504–10. doi: 10.1038/leu.2013.88
9. Itzykson R, Kosmider O, Renneville A, Gelsi-Boyer V, Meggendorfer M, Morabito M, et al. Prognostic Score Including Gene Mutations in Chronic Myelomonocytic Leukemia. J Clin Oncol (2013) 31(19):2428–36. doi: 10.1200/JCO.2012.47.3314
10. Elena C, Gallì A, Such E, Meggendorfer M, Germing U, Rizzo E, et al. Integrating Clinical Features and Genetic Lesions in the Risk Assessment of Patients With Chronic Myelomonocytic Leukemia. Blood (2016) 128(10):1408–17. doi: 10.1182/blood-2016-05-714030
11. Machherndl-Spandl S, Jäger E, Barna A, Gurbisz M, Marschon R, Graf T, et al. Impact of Age on the Cumulative Risk of Transformation in Patients With Chronic Myelomonocytic Leukaemia. Eur J Haematol (2021) 107(2):265–74. doi: 10.1111/ejh.13647
12. Geissler K, Jäger E, Barna A, Gurbisz M, Marschon R, Graf T, et al. The Austrian Biodatabase for Chronic Myelomonocytic Leukemia (ABCMML): A Representative and Useful Real-Life Data Source for Further Biomedical Research. Wien Klin Wochenschr (2019) 131(17–18):410–8. doi: 10.1007/s00508-019-1526-1
13. Janssen JW, Steenvoorden AC, Lyons J, Anger B, Bohlke JU, Bos JL, et al. RAS Gene Mutations in Acute and Chronic Myelocytic Leukemias, Chronic Myeloproliferative Disorders, and Myelodysplastic Syndromes. Proc Natl Acad Sci (1987) 84(24):9228–32. doi: 10.1073/pnas.84.24.9228
14. Hirsch-Ginsberg C, LeMaistre AC, Kantarjian H, Talpaz M, Cork A, Freireich EJ, et al. RAS Mutations are Rare Events in Philadelphia Chromosome-Negative/Bcr Gene Rearrangement-Negative Chronic Myelogenous Leukemia, But Are Prevalent in Chronic Myelomonocytic Leukemia. Blood (1990) 76(6):1214–9. doi: 10.1182/blood.V76.6.1214.1214
15. Patnaik MM, Tefferi A. Chronic Myelomonocytic Leukemia: 2020 Update on Diagnosis, Risk Stratification and Management. Am J Hematol (2020) 95(1):97–115. doi: 10.1002/ajh.25684
16. Abdel-Wahab O, Pardanani A, Patel J, Wadleigh M, Lasho T, Heguy A, et al. Concomitant Analysis of EZH2 and ASXL1 Mutations in Myelofibrosis, Chronic Myelomonocytic Leukemia and Blast-Phase Myeloproliferative Neoplasms. Leukemia (2011) 25(7):1200–2. doi: 10.1038/leu.2011.58
17. Ernst T, Chase AJ, Score J, Hidalgo-Curtis CE, Bryant C, Jones AV, et al. Inactivating Mutations of the Histone Methyltransferase Gene EZH2 in Myeloid Disorders. Nat Genet (2010) 42(8):722–6. doi: 10.1038/ng.621
18. Gelsi-Boyer V, Trouplin V, Adélaïde J, Bonansea J, Cervera N, Carbuccia N, et al. Mutations of Polycomb-Associated Gene ASXL1 in Myelodysplastic Syndromes and Chronic Myelomonocytic Leukaemia. Br J Haematol (2009) 145(6):788–800. doi: 10.1111/j.1365-2141.2009.07697.x
19. Smith AE, Mohamedali AM, Kulasekararaj A, Lim Z, Gaken J, Lea NC, et al. Next-Generation Sequencing of the TET2 Gene in 355 MDS and CMML Patients Reveals Low-Abundance Mutant Clones With Early Origins, But Indicates No Definite Prognostic Value. Blood (2010) 116(19):3923–32. doi: 10.1182/blood-2010-03-274704
20. Jankowska AM, Makishima H, Tiu RV, Szpurka H, Huang Y, Traina F, et al. Mutational Spectrum Analysis of Chronic Myelomonocytic Leukemia Includes Genes Associated With Epigenetic Regulation: UTX, EZH2, and DNMT3A. Blood (2011) 118(14):3932–41. doi: 10.1182/blood-2010-10-311019
21. Grossmann V, Kohlmann A, Eder C, Haferlach C, Kern W, Cross NCP, et al. Molecular Profiling of Chronic Myelomonocytic Leukemia Reveals Diverse Mutations in <80% of Patients With TET2 and EZH2 Being of High Prognostic Relevance. Leukemia (2011) 25(5):877–9. doi: 10.1038/leu.2011.10
22. Tefferi A, Lim K-H, Abdel-Wahab O, Lasho TL, Patel J, Patnaik MM, et al. Detection of Mutant TET2 in Myeloid Malignancies Other Than Myeloproliferative Neoplasms: CMML, MDS, MDS/MPN and AML. Leukemia (2009) 23(7):1343–5. doi: 10.1038/leu.2009.59
23. Meggendorfer M, Roller A, Haferlach T, Eder C, Dicker F, Grossmann V, et al. SRSF2 Mutations in 275 Cases With Chronic Myelomonocytic Leukemia (CMML). Blood (2012) 120(15):3080–8. doi: 10.1182/blood-2012-01-404863
24. Patnaik MM, Lasho TL, Finke CM, Hanson CA, Hodnefield JM, Knudson RA, et al. Spliceosome Mutations Involving SRSF2, SF3B1, and U2AF35 in Chronic Myelomonocytic Leukemia: Prevalence, Clinical Correlates, and Prognostic Relevance. Am J Hematol (2013) 88(3):201–6. doi: 10.1002/ajh.23373
25. Gelsi-Boyer V, Trouplin V, Adélaïde J, Aceto N, Remy V, Pinson S, et al. Genome Profiling of Chronic Myelomonocytic Leukemia: Frequent Alterations of RAS and RUNX1 Genes. BMC Cancer (2008) 8:299. doi: 10.1186/1471-2407-8-299
26. Kohlmann A, Grossmann V, Klein H-U, Schindela S, Weiss T, Kazak B, et al. Next-Generation Sequencing Technology Reveals a Characteristic Pattern of Molecular Mutations in 72.8% of Chronic Myelomonocytic Leukemia by Detecting Frequent Alterations in TET2, CBL, RAS, and RUNX1. J Clin Oncol (2010) 28(24):3858–65. doi: 10.1200/JCO.2009.27.1361
27. Levine RL, Wadleigh M, Cools J, Ebert BL, Wernig G, Huntly BJP, et al. Activating Mutation in the Tyrosine Kinase JAK2 in Polycythemia Vera, Essential Thrombocythemia, and Myeloid Metaplasia With Myelofibrosis. Cancer Cell (2005) 7(4):387–97. doi: 10.1016/j.ccr.2005.03.023
28. Makishima H, Cazzolli H, Szpurka H, Dunbar A, Tiu R, Huh J, et al. Mutations of E3 Ubiquitin Ligase Cbl Family Members Constitute a Novel Common Pathogenic Lesion in Myeloid Malignancies. J Clin Oncol (2009) 27(36):6109–16. doi: 10.1200/JCO.2009.23.7503
29. Daver N, Strati P, Jabbour E, Kadia T, Luthra R, Wang S, et al. FLT3 Mutations in Myelodysplastic Syndrome and Chronic Myelomonocytic Leukemia. Am J Hematol (2013) 88(1):56–9. doi: 10.1002/ajh.23345
30. Laborde RR, Patnaik MM, Lasho TL, Finke CM, Hanson CA, Knudson RA, et al. SETBP1 Mutations in 415 Patients With Primary Myelofibrosis or Chronic Myelomonocytic Leukemia: Independent Prognostic Impact in CMML. Leukemia (2013) 27(10):2100–2. doi: 10.1038/leu.2013.97
31. Damm F, Itzykson R, Kosmider O, Droin N, Renneville A, Chesnais V, et al. SETBP1 Mutations in 658 Patients With Myelodysplastic Syndromes, Chronic Myelomonocytic Leukemia and Secondary Acute Myeloid Leukemias. Leukemia (2013) 27(6):1401–3. doi: 10.1038/leu.2013.35
32. Patnaik MM, Lasho TL, Vijayvargiya P, Finke CM, Hanson CA, Ketterling RP, et al. Prognostic Interaction Between ASXL1 and TET2 Mutations in Chronic Myelomonocytic Leukemia. Blood Cancer J (2016) 6(1):e385–5. doi: 10.1038/bcj.2015.113
33. Padron E. Surveying the Landscape of MDS/MPN Research: Overlap Among the Overlap Syndromes? Hematology (2015) 2015(1):349–54. doi: 10.1182/asheducation-2015.1.349
34. Geissler K, Hinterberger W, Bettelheim P, Haas O, Lechner K. Colony Growth Characteristics in Chronic Myelomonocytic Leukemia. Leuk Res (1988) 12(5):373–7. doi: 10.1016/0145-2126(88)90055-0
35. Geissler K, Ohler L, Födinger M, Virgolini I, Leimer M, Kabrna E, et al. Interleukin 10 Inhibits Growth and Granulocyte/Macrophage Colony-Stimulating Factor Production in Chronic Myelomonocytic Leukemia Cells. J Exp Med (1996) 184(4):1377–84. doi: 10.1084/jem.184.4.1377
36. Padron E, Painter JS, Kunigal S, Mailloux AW, McGraw K, McDaniel JM, et al. GM-CSF-Dependent Pstat5 Sensitivity Is a Feature With Therapeutic Potential in Chronic Myelomonocytic Leukemia. Blood (2013) 121(25):5068–77. doi: 10.1182/blood-2012-10-460170
37. Sagaster V, Ohler L, Berer A, Kabrna E, Ofner P, Lechner K, et al. High Spontaneous Colony Growth in Chronic Myelomonocytic Leukemia Correlates With Increased Disease Activity and Is a Novel Prognostic Factor for Predicting Short Survival. Ann Hematol (2004) 83(1):9–13. doi: 10.1007/s00277-003-0743-9
38. Geissler K, Jäger E, Barna A, Gurbisz M, Graf T, Graf E, et al. Molecular Basis and Clinical Application of Growth-Factor-Independent In Vitro Myeloid Colony Formation in Chronic Myelomonocytic Leukemia. Int J Mol Sci (2020) 21(17):6057. doi: 10.3390/ijms21176057
39. Geissler K, Jäger E, Barna A, Graf T, Graf E, Öhler L, et al. Myelomonocytic Skewing in Chronic Myelomonocytic Leukemia: Phenotypic, Molecular and Biologic Features and Impact on Survival. Eur J Haematol (2021) 106(5):627–33. doi: 10.1111/ejh.13577
40. Wang J, Liu Y, Li Z, Du J, Ryu M-J, Taylor PR, et al. Endogenous Oncogenic Nras Mutation Promotes Aberrant GM-CSF Signaling in Granulocytic/Monocytic Precursors in a Murine Model of Chronic Myelomonocytic Leukemia. Blood (2010) 116(26):5991–6002. doi: 10.1182/blood-2010-04-281527
41. Li Q, Haigis KM, McDaniel A, Harding-Theobald E, Kogan SC, Akagi K, et al. Hematopoiesis and Leukemogenesis in Mice Expressing Oncogenic NrasG12D From the Endogenous Locus. Blood (2011) 117(6):2022–32. doi: 10.1182/blood-2010-04-280750
42. Parikh C. Oncogenic NRAS Rapidly and Efficiently Induces CMML- and AML-Like Diseases in Mice. Blood (2006) 108(7):2349–57. doi: 10.1182/blood-2004-08-009498
43. Chan IT, Kutok JL, Williams IR, Cohen S, Kelly L, Shigematsu H, et al. Conditional Expression of Oncogenic K-Ras From Its Endogenous Promoter Induces a Myeloproliferative Disease. J Clin Invest (2004) 113(4):528–38. doi: 10.1172/JCI20476
44. Van Meter MEM, Diaz-Flores E, Archard JA, Passegue E, Irish JM, Kotecha N, et al. K-RasG12D Expression Induces Hyperproliferation and Aberrant Signaling in Primary Hematopoietic Stem/Progenitor Cells. Blood (2007) 109(9):3945–52. doi: 10.1182/blood-2006-09-047530
45. Murphy MA, Schnall RG, Venter DJ, Barnett L, Bertoncello I, Thien CB, et al. Tissue Hyperplasia and Enhanced T-Cell Signalling via ZAP-70 in C-Cbl-Deficient Mice. Mol Cell Biol (1998) 18(8):4872–82. doi: 10.1128/MCB.18.8.4872
46. Le DT. Somatic Inactivation of Nf1 in Hematopoietic Cells Results in a Progressive Myeloproliferative Disorder. Blood (2004) 103(11):4243–50. doi: 10.1182/blood-2003-08-2650
47. Chan G, Kalaitzidis D, Usenko T, Kutok JL, Yang W, Mohi MG, et al. Leukemogenic Ptpn11 Causes Fatal Myeloproliferative Disorder via Cell-Autonomous Effects on Multiple Stages of Hematopoiesis. Blood (2009) 113(18):4414–24. doi: 10.1182/blood-2008-10-182626
48. Moran-Crusio K, Reavie L, Shih A, Abdel-Wahab O, Ndiaye-Lobry D, Lobry C, et al. Tet2 Loss Leads to Increased Hematopoietic Stem Cell Self-Renewal and Myeloid Transformation. Cancer Cell (2011) 20(1):11–24. doi: 10.1016/j.ccr.2011.06.001
49. Li Z, Cai X, Cai C-L, Wang J, Zhang W, Petersen BE, et al. Deletion of Tet2 in Mice Leads to Dysregulated Hematopoietic Stem Cells and Subsequent Development of Myeloid Malignancies. Blood (2011) 118(17):4509–18. doi: 10.1182/blood-2010-12-325241
50. Wang J, Li Z, He Y, Pan F, Chen S, Rhodes S, et al. Loss of Asxl1 Leads to Myelodysplastic Syndrome–Like Disease in Mice. Blood (2014) 123(4):541–53. doi: 10.1182/blood-2013-05-500272
51. Oscier DG, Worsley A, Hamblin TJ, Mufti GJ. Treatment of Chronic Myelomonocytic Leukaemia with Low Dose Etoposide. Br J Haematol (1989) 72(3):468–71. doi: 10.1111/j.1365-2141.1989.tb07735.x
52. Wattel E, Guerci A, Hecquet B, Economopoulos T, Copplestone A, Mahé B, et al. A Randomized Trial of Hydroxyurea Versus VP16 in Adult Chronic Myelomonocytic Leukemia. Groupe Français Des Myélodysplasies and European CMML Group. Blood (1996) 88(7):2480–7.
53. Patnaik MM, Ali H, Gupta V, Schiller GJ, Lee S, Yacoub A, et al. Results From Ongoing Phase 1/2 Clinical Trial of Tagraxofusp (SL-401) in Patients With Relapsed/Refractory Chronic Myelomonocytic Leukemia (CMML). Blood (2018) 132:1821. doi: 10.1182/blood-2018-99-119302
54. Fenaux P, Mufti GJ, Hellstrom-Lindberg E, Santini V, Finelli C, Giagounidis A, et al. Efficacy of Azacitidine Compared With That of Conventional Care Regimens in the Treatment of Higher-Risk Myelodysplastic Syndromes: A Randomised, Open-Label, Phase III Study. Lancet Oncol (2009) 10(3):223–32. doi: 10.1016/S1470-2045(09)70003-8
55. Kantarjian H, Oki Y, Garcia-Manero G, Huang X, O’Brien S, Cortes J, et al. Results of a Randomized Study of 3 Schedules of Low-Dose Decitabine in Higher-Risk Myelodysplastic Syndrome and Chronic Myelomonocytic Leukemia. Blood (2007) 109(1):52–7. doi: 10.1182/blood-2006-05-021162
56. Aribi A, Borthakur G, Ravandi F, Shan J, Davisson J, Cortes J, et al. Activity of Decitabine, a Hypomethylating Agent, in Chronic Myelomonocytic Leukemia. Cancer (2007) 109(4):713–7. doi: 10.1002/cncr.22457
57. Wijermans PW, Rüter B, Baer MR, Slack JL, Saba HI, Lübbert M. Efficacy of Decitabine in the Treatment of Patients With Chronic Myelomonocytic Leukemia (CMML). Leuk Res (2008) 32(4):587–91. doi: 10.1016/j.leukres.2007.08.004
58. Costa R, Abdulhaq H, Haq B, Shadduck RK, Latsko J, Zenati M, et al. Activity of Azacitidine in Chronic Myelomonocytic Leukemia. Cancer (2011) 117(12):2690–6. doi: 10.1002/cncr.25759
59. Braun T, Itzykson R, Renneville A, de Renzis B, Dreyfus F, Laribi K, et al. Molecular Predictors of Response to Decitabine in Advanced Chronic Myelomonocytic Leukemia: A Phase 2 Trial. Blood (2011) 118(14):3824–31. doi: 10.1182/blood-2011-05-352039
60. Thorpe M, Montalvão A, Pierdomenico F, Moita F, Almeida A. Treatment of Chronic Myelomonocytic Leukemia With 5-Azacitidine: A Case Series and Literature Review. Leuk Res (2012) 36(8):1071–3. doi: 10.1016/j.leukres.2012.04.024
61. Adès L, Sekeres MA, Wolfromm A, Teichman ML, Tiu RV, Itzykson R, et al. Predictive Factors of Response and Survival Among Chronic Myelomonocytic Leukemia Patients Treated With Azacitidine. Leuk Res (2013) 37(6):609–13. doi: 10.1016/j.leukres.2013.01.004
62. Pleyer L, Germing U, Sperr WR, Linkesch W, Burgstaller S, Stauder R, et al. Azacitidine in CMML: Matched-Pair Analyses of Daily-Life Patients Reveal Modest Effects on Clinical Course and Survival. Leuk Res (2014) 38(4):475–83. doi: 10.1016/j.leukres.2014.01.006
63. Santini V, Allione B, Zini G, Gioia D, Lunghi M, Poloni A, et al. Multicentre Trial of Decitabine in Higher-Risk Chronic Myelomonocytic Leukemia. Leukemia (2018) 32(2):413–8. doi: 10.1038/leu.2017.186
64. Cimmino L, Dolgalev I, Wang Y, Yoshimi A, Martin GH, Wang J, et al. Restoration of TET2 Function Blocks Aberrant Self-Renewal and Leukemia Progression. Cell (2017) 170(6):1079–95.e20. doi: 10.1016/j.cell.2017.07.032
65. Pöchlauer S, Jäger E, Jäger U, Geissler K. Recombinant Human Interleukin-10 in Patients With Chronic Myelomonocytic Leukemia. Ann Hematol (2014) 93(10):1775–6. doi: 10.1007/s00277-014-2012-5
66. Patnaik MM, Sallman DA, Mangaonkar AA, Heuer R, Hirvela J, Zblewski D, et al. A Phase 1 Study of Lenzilumab, a Humaneered Recombinant Anti-Human Granulocyte-Macrophage Colony- Stimulating Factor (Anti-hGM-CSF) Antibody, for Chronic Myelomonocytic Leukemia (CMML). Blood (2019) 134(Suppl_1):4234–4. doi: 10.1182/blood-2019-131181
67. Rathinam C, Thien CBF, Flavell RA, Langdon WY. Myeloid Leukemia Development in C-Cbl RING Finger Mutant Mice Is Dependent on FLT3 Signaling. Cancer Cell (2010) 18(4):341–52. doi: 10.1016/j.ccr.2010.09.008
68. Lyubynska N, Gorman MF, Lauchle JO, Hong WX, Akutagawa JK, Shannon K, et al. A MEK Inhibitor Abrogates Myeloproliferative Disease in Kras Mutant Mice. Sci Trans Med (2011) 3(76):76ra27. doi: 10.1126/scitranslmed.3001069
69. Chang T, Krisman K, Theobald EH, Xu J, Akutagawa J, Lauchle JO, et al. Sustained MEK Inhibition Abrogates Myeloproliferative Disease in Nf1 Mutant Mice. J Clin Invest (2013) 123(1):335–9. doi: 10.1172/JCI63193
70. Borthakur G, Popplewell L, Boyiadzis M, Foran J, Platzbecker U, Vey N, et al. Activity of the Oral Mitogen-Activated Protein Kinase Kinase Inhibitor Trametinib in RAS-Mutant Relapsed or Refractory Myeloid Malignancies. Cancer (2016) 122(12):1871–9. doi: 10.1002/cncr.29986
71. Geissler K, Jäger E, Barna A, Sliwa T, Knöbl P, Schwarzinger I, et al. In Vitro and In Vivo Effects of JAK2 Inhibition in Chronic Myelomonocytic Leukemia. Eur J Haematol (2016) 97(6):562–7. doi: 10.1111/ejh.12773
72. Padron E, Dezern A, Andrade-Campos M, Vaddi K, Scherle P, Zhang Q, et al. A Multi-Institution Phase I Trial of Ruxolitinib in Patients With Chronic Myelomonocytic Leukemia (CMML). Clin Cancer Res (2016) 22(15):3746–54. doi: 10.1158/1078-0432.CCR-15-2781
73. Akutagawa J, Huang TQ, Epstein I, Chang T, Quirindongo-Crespo M, Cottonham CL, et al. Targeting the PI3K/Akt Pathway in Murine MDS/MPN Driven by Hyperactive Ras. Leukemia (2016) 30(6):1335–43. doi: 10.1038/leu.2016.14
74. Carr RM, Vorobyev D, Lasho T, Marks DL, Tolosa EJ, Vedder A, et al. RAS Mutations Drive Proliferative Chronic Myelomonocytic Leukemia via a KMT2A-PLK1 Axis. Nat Commun (2021) 12(1):2901. doi: 10.1038/s41467-021-23186-w
75. Hamarsheh S, Osswald L, Saller BS, Unger S, De Feo D, Vinnakota JM, et al. Oncogenic KrasG12D Causes Myeloproliferation via NLRP3 Inflammasome Activation. Nat Commun (2020) 11(1):1659. doi: 10.1038/s41467-020-15497-1
76. Kong G, Wunderlich M, Yang D, Ranheim EA, Young KH, Wang J, et al. Combined MEK and JAK Inhibition Abrogates Murine Myeloproliferative Neoplasm. J Clin Invest (2014) 124(6):2762–73. doi: 10.1172/JCI74182
77. Yizhak K, Aguet F, Kim J, Hess JM, Kübler K, Grimsby J, et al. RNA Sequence Analysis Reveals Macroscopic Somatic Clonal Expansion Across Normal Tissues. Science (2019) 364(6444):eaaw0726. doi: 10.1126/science.aaw0726
78. Prchal JF, Adamson JW, Murphy S, Steinmann L, Fialkow PJ. Polycythemia Vera. The In Vitro Response of Normal and Abnormal Stem Cell Lines to Erythropoietin. J Clin Invest (1978) 61(4):1044–7. doi: 10.1172/JCI109003
79. Juvonen E, Partanen S, Ruutu T. Colony Formation by Megakaryocytic Progenitors in Essential Thrombocythaemia. Br J Haematol (1987) 66(2):161–4. doi: 10.1111/j.1365-2141.1987.tb01293.x
80. Estrov Z, Grunberger T, Chan HS, Freedman MH. Juvenile Chronic Myelogenous Leukemia: Characterization of the Disease Using Cell Cultures. Blood (1986) 67(5):1382–7. doi: 10.1182/blood.V67.5.1382.1382
81. Geissler K. Molecular Pathogenesis of Chronic Myelomonocytic Leukemia. Memo - Mag Eur Med Oncol (2016) 9(4):172–7. doi: 10.1007/s12254-016-0295-z
82. Geissler K, Jäger E, Barna A, Alendar T, Ljubuncic E, Sliwa T, et al. Chronic Myelomonocytic Leukemia Patients With RAS Pathway Mutations Show High In Vitro Myeloid Colony Formation in the Absence of Exogenous Growth Factors. Leukemia (2016) 30(11):2280–1. doi: 10.1038/leu.2016.235
83. Oehler L, Foedinger M, Koeller M, Kollars M, Reiter E, Bohle B, et al. Interleukin-10 Inhibits Spontaneous Colony-Forming Unit-Granulocyte-Macrophage Growth From Human Peripheral Blood Mononuclear Cells by Suppression of Endogenous Granulocyte-Macrophage Colony-Stimulating Factor Release. Blood (1997) 89(4):1147–53. doi: 10.1182/blood.V89.4.1147
84. Itzykson R, Kosmider O, Renneville A, Morabito M, Preudhomme C, Berthon C, et al. Clonal Architecture of Chronic Myelomonocytic Leukemias. Blood (2013) 121(12):2186–98. doi: 10.1182/blood-2012-06-440347
85. Pronier E, Almire C, Mokrani H, Vasanthakumar A, Simon A, da Costa Reis Monte Mor B, et al. Inhibition of TET2-Mediated Conversion of 5-Methylcytosine to 5-Hydroxymethylcytosine Disturbs Erythroid and Granulomonocytic Differentiation of Human Hematopoietic Progenitors. Blood (2011) 118(9):2551–5. doi: 10.1182/blood-2010-12-324707
86. Bapat A, Keita N, Martelly W, Kang P, Seet C, Jacobsen JR, et al. Myeloid Disease Mutations of Splicing Factor SRSF2 Cause G2-M Arrest and Skewed Differentiation of Human Hematopoietic Stem and Progenitor Cells. Stem Cells (Dayton Ohio) (2018) 36(11):1663–75. doi: 10.1002/stem.2885
87. Steensma DP, Bejar R, Jaiswal S, Lindsley RC, Sekeres MA, Hasserjian RP, et al. Clonal Hematopoiesis of Indeterminate Potential and Its Distinction From Myelodysplastic Syndromes. Blood (2015) 126(1):9–16. doi: 10.1182/blood-2015-03-631747
88. Itzykson R, Solary E. An Evolutionary Perspective on Chronic Myelomonocytic Leukemia. Leukemia (2013) 27(7):1441–50. doi: 10.1038/leu.2013.100
89. Mason CC, Khorashad JS, Tantravahi SK, Kelley TW, Zabriskie MS, Yan D, et al. Age-Related Mutations and Chronic Myelomonocytic Leukemia. Leukemia (2016) 30(4):906–13. doi: 10.1038/leu.2015.337
90. Busque L, Patel JP, Figueroa ME, Vasanthakumar A, Provost S, Hamilou Z, et al. Recurrent Somatic TET2 Mutations in Normal Elderly Individuals With Clonal Hematopoiesis. Nat Genet (2012) 44(11):1179–81. doi: 10.1038/ng.2413
91. Xie M, Lu C, Wang J, McLellan MD, Johnson KJ, Wendl MC, et al. Age-Related Mutations Associated With Clonal Hematopoietic Expansion and Malignancies. Nat Med (2014) 20(12):1472–8. doi: 10.1038/nm.3733
92. Genovese G, Kähler AK, Handsaker RE, Lindberg J, Rose SA, Bakhoum SF, et al. Clonal Hematopoiesis and Blood-Cancer Risk Inferred From Blood DNA Sequence. N Engl J Med (2014) 371(26):2477–87. doi: 10.1056/NEJMoa1409405
93. Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, et al. Age-Related Clonal Hematopoiesis Associated With Adverse Outcomes. N Engl J Med (2014) 371(26):2488–98. doi: 10.1056/NEJMoa1408617
94. Abdel-Wahab O, Adli M, LaFave LM, Gao J, Hricik T, Shih AH, et al. ASXL1 Mutations Promote Myeloid Transformation Through Loss of PRC2-Mediated Gene Repression. Cancer Cell (2012) 22(2):180–93. doi: 10.1016/j.ccr.2012.06.032
95. Ricci C, Fermo E, Corti S, Molteni M, Faricciotti A, Cortelezzi A, et al. RAS Mutations Contribute to Evolution of Chronic Myelomonocytic Leukemia to the Proliferative Variant. Clin Cancer Res (2010) 16(8):2246–56. doi: 10.1158/1078-0432.CCR-09-2112
96. Geissler K, Jäger E, Barna A, Gurbisz M, Graf T, Graf E, et al. Correlation of RAS-Pathway Mutations and Spontaneous Myeloid Colony Growth With Progression and Transformation in Chronic Myelomonocytic Leukemia—A Retrospective Analysis in 337 Patients. Int J Mol Sci (2020) 21(8):3025. doi: 10.3390/ijms21083025
97. Kunimoto H, Meydan C, Nazir A, Whitfield J, Shank K, Rapaport F, et al. Cooperative Epigenetic Remodeling by TET2 Loss and NRAS Mutation Drives Myeloid Transformation and MEK Inhibitor Sensitivity. Cancer Cell (2018) 33(1):44–59.e8. doi: 10.1016/j.ccell.2017.11.012
98. Jin X, Qin T, Zhao M, Bailey N, Liu L, Yang K, et al. Oncogenic N-Ras and Tet2 Haploinsufficiency Collaborate to Dysregulate Hematopoietic Stem and Progenitor Cells. Blood Adv (2018) 2(11):1259–71. doi: 10.1182/bloodadvances.2018017400
99. Fenaux P, Beuscart R, Lai JL, Jouet JP, Bauters F. Prognostic Factors in Adult Chronic Myelomonocytic Leukemia: An Analysis of 107 Cases. J Clin Oncol: Off J Am Soc Clin Oncol (1988) 6(9):1417–24. doi: 10.1200/JCO.1988.6.9.1417
100. Germing U, Strupp C, Aivado M, Gattermann N. New Prognostic Parameters for Chronic Myelomonocytic Leukemia. Blood (2002) 100(2):731–2; author reply 732-733. doi: 10.1182/blood-2002-01-0330
101. Storniolo AM, Moloney WC, Rosenthal DS, Cox C, Bennett JM. Chronic Myelomonocytic Leukemia. Leukemia (1990) 4(11):766–70.
102. Schuler E, Schroeder M, Neukirchen J, Strupp C, Xicoy B, Kündgen A, et al. Refined Medullary Blast and White Blood Cell Count Based Classification of Chronic Myelomonocytic Leukemias. Leuk Res (2014) 38(12):1413–9. doi: 10.1016/j.leukres.2014.09.003
103. Tefferi A, Hoagland HC, Therneau TM, Pierre RV. Chronic Myelomonocytic Leukemia: Natural History and Prognostic Determinants. Mayo Clin Proc (1989) 64(10):1246–54. doi: 10.1016/s0025-6196(12)61287-7
104. Worsley A, Oscier DG, Stevens J, Darlow S, Figes A, Mufti GJ, et al. Prognostic Features of Chronic Myelomonocytic Leukaemia: A Modified Bournemouth Score Gives the Best Prediction of Survival. Br J Haematol (1988) 68(1):17–21. doi: 10.1111/j.1365-2141.1988.tb04173.x
105. Such E, Cervera J, Costa D, Solé F, Vallespí T, Luño E, et al. Cytogenetic Risk Stratification in Chronic Myelomonocytic Leukemia. Haematologica (2011) 96(3):375–83. doi: 10.3324/haematol.2010.030957
106. Such E, Germing U, Malcovati L, Cervera J, Kuendgen A, Della Porta MG, et al. Development and Validation of a Prognostic Scoring System for Patients With Chronic Myelomonocytic Leukemia. Blood (2013) 121(15):3005–15. doi: 10.1182/blood-2012-08-452938
107. Wassie EA, Itzykson R, Lasho TL, Kosmider O, Finke CM, Hanson CA, et al. Molecular and Prognostic Correlates of Cytogenetic Abnormalities in Chronic Myelomonocytic Leukemia: A Mayo Clinic-French Consortium Study. Am J Hematol (2014) 89(12):1111–5. doi: 10.1002/ajh.23846
108. Itzykson R, Fenaux P, Bowen D, Cross NCP, Cortes J, De Witte T, et al. Diagnosis and Treatment of Chronic Myelomonocytic Leukemias in Adults: Recommendations From the European Hematology Association and the European LeukemiaNet. HemaSphere (2018) 1. doi: 10.1097/HS9.0000000000000150
109. Patnaik MM, Itzykson R, Lasho TL, Kosmider O, Finke CM, Hanson CA, et al. ASXL1 and SETBP1 Mutations and Their Prognostic Contribution in Chronic Myelomonocytic Leukemia: A Two-Center Study of 466 Patients. Leukemia (2014) 28(11):2206–12. doi: 10.1038/leu.2014.125
110. Padron E, Garcia-Manero G, Patnaik MM, Itzykson R, Lasho T, Nazha A, et al. An International Data Set for CMML Validates Prognostic Scoring Systems and Demonstrates a Need for Novel Prognostication Strategies. Blood Cancer J (2015) 5(7):e333. doi: 10.1038/bcj.2015.53
111. Patel AA, Cahill K, Charnot-Katsikas A, Liu H, Gurbuxani S, Thirman M, et al. Clinical Outcomes of IDH2 -Mutated Advanced-Phase Ph-Negative Myeloproliferative Neoplasms Treated With Enasidenib. Br J Haematol (2020) 190(1):e48–51. doi: 10.1111/bjh.16709
112. Montalban-Bravo G, Kanagal-Shamanna R, Sasaki K, Patel K, Ganan-Gomez I, Jabbour E, et al. NPM1 Mutations Define a Specific Subgroup of MDS and MDS/MPN Patients With Favorable Outcomes With Intensive Chemotherapy. Blood Adv (2019) 3(6):922–33. doi: 10.1182/bloodadvances.2018026989
113. Eisenwort G, Sadovnik I, Keller A, Ivanov D, Peter B, Berger D, et al. Phenotypic Characterization of Leukemia-Initiating Stem Cells in Chronic Myelomonocytic Leukemia. Leukemia (2021). doi: 10.1038/s41375-021-01227-z
114. Pemmaraju N, Lane AA, Sweet KL, Stein AS, Vasu S, Blum W, et al. Tagraxofusp in Blastic Plasmacytoid Dendritic-Cell Neoplasm. N Engl J Med (2019) 380(17):1628–37. doi: 10.1056/NEJMoa1815105
115. Coston T, Pophali P, Vallapureddy R, Lasho TL, Finke CM, Ketterling RP, et al. Suboptimal Response Rates to Hypomethylating Agent Therapy in Chronic Myelomonocytic Leukemia; a Single Institutional Study of 121. Am J Hematol (2019). doi: 10.1002/ajh.25488
116. Merlevede J, Droin N, Qin T, Meldi K, Yoshida K, Morabito M, et al. Mutation Allele Burden Remains Unchanged in Chronic Myelomonocytic Leukaemia Responding to Hypomethylating Agents. Nat Commun (2016) 7(1):10767. doi: 10.1038/ncomms10767
117. Meldi K, Qin T, Buchi F, Droin N, Sotzen J, Micol J-B, et al. Specific Molecular Signatures Predict Decitabine Response in Chronic Myelomonocytic Leukemia. J Clin Invest (2015) 125(5):1857–72. doi: 10.1172/JCI78752
118. Duchmann M, Yalniz FF, Sanna A, Sallman D, Coombs CC, Renneville A, et al. Prognostic Role of Gene Mutations in Chronic Myelomonocytic Leukemia Patients Treated With Hypomethylating Agents. EBioMedicine (2018) 31:174–81. doi: 10.1016/j.ebiom.2018.04.018
119. Pleyer L, Leisch M, Kourakli A, Padron E, Maciejewski JP, Xicoy Cirici B, et al. Outcomes of Patients With Chronic Myelomonocytic Leukaemia Treated With Non-Curative Therapies: A Retrospective Cohort Study. Lancet Haematol (2021) 8(2):e135–48. doi: 10.1016/S2352-3026(20)30374-4
120. Itzykson R, Santini V, Chaffaut C, Lionel A, Thepot S, Giagounidis A, et al. Decitabine Versus Hydroxyurea for Advanced Proliferative CMML: Results of the Emsco Randomized Phase 3 Dacota Trial. Blood (2020) 136(Suppl 1):53–4. doi: 10.1182/blood-2020-138680
121. Kaelin WG, McKnight SL. Influence of Metabolism on Epigenetics and Disease. Cell (2013) 153(1):56–69. doi: 10.1016/j.cell.2013.03.004
122. Blaschke K, Ebata KT, Karimi MM, Zepeda-Martínez JA, Goyal P, Mahapatra S, et al. Vitamin C Induces Tet-Dependent DNA Demethylation and a Blastocyst-Like State in ES Cells. Nature (2013) 500(7461):222–6. doi: 10.1038/nature12362
123. DiNardo CD, Stein EM, de Botton S, Roboz GJ, Altman JK, Mims AS, et al. Durable Remissions With Ivosidenib in IDH1 -Mutated Relapsed or Refractory AML. N Engl J Med (2018) 378(25):2386–98. doi: 10.1056/NEJMoa1716984
124. Stein EM, DiNardo CD, Pollyea DA, Fathi AT, Roboz GJ, Altman JK, et al. Enasidenib in Mutant IDH2 Relapsed or Refractory Acute Myeloid Leukemia. Blood (2017) 130(6):722–31. doi: 10.1182/blood-2017-04-779405
125. Agathocleous M, Meacham CE, Burgess RJ, Piskounova E, Zhao Z, Crane GM, et al. Ascorbate Regulates Haematopoietic Stem Cell Function and Leukaemogenesis. Nature (2017) 549(7673):476–81. doi: 10.1038/nature23876
126. Huijskens MJAJ, Wodzig WKWH, Walczak M, Germeraad WTV, Bos GMJ. Ascorbic Acid Serum Levels are Reduced in Patients With Hematological Malignancies. Results Immunol (2016) 6:8–10. doi: 10.1016/j.rinim.2016.01.001
127. Niyongere S, Lucas N, Zhou J-M, Sansil S, Pomicter AD, Balasis ME, et al. Heterogeneous Expression of Cytokines Accounts for Clinical Diversity and Refines Prognostication in CMML. Leukemia (2019) 33(1):205–16. doi: 10.1038/s41375-018-0203-0
128. Stone RM, Mandrekar SJ, Sanford BL, Laumann K, Geyer S, Bloomfield C, et al. Midostaurin Plus Chemotherapy for Acute Myeloid Leukemia With a FLT3 Mutation. N Engl J Med (2017) 377(5):454–64. doi: 10.1056/NEJMoa1614359
129. Perl AE, Martinelli G, Cortes JE, Neubauer A, Berman E, Paolini S, et al. Gilteritinib or Chemotherapy for Relapsed or Refractory FLT3 -Mutated AML. N Engl J Med (2019) 381(18):1728–40. doi: 10.1056/NEJMoa1902688
130. Ostrem JM, Peters U, Sos ML, Wells JA, Shokat KM. K-Ras(G12C) Inhibitors Allosterically Control GTP Affinity and Effector Interactions. Nature (2013) 503(7477):548–51. doi: 10.1038/nature12796
131. Stieglitz E, Ward AF, Gerbing RB, Alonzo TA, Arceci RJ, Liu YL, et al. Phase II/III Trial of a Pre-Transplant Farnesyl Transferase Inhibitor in Juvenile Myelomonocytic Leukemia: A Report From the Children’s Oncology Group: Pre-HSCT Farnesyl Transferase Inhibition in JMML. Pediatr Blood Cancer (2015) 62(4):629–36. doi: 10.1002/pbc.25342
132. Patnaik MM, Sallman DA, Sekeres MA, Luger S, Bejar R, Hobbs GS, et al. Preliminary Results From an Open-Label, Phase 2 Study of Tipifarnib in Chronic Myelomonocytic Leukemia (CMML). Blood (2017) 130:2963.
133. Gross AM, Wolters PL, Dombi E, Baldwin A, Whitcomb P, Fisher MJ, et al. Selumetinib in Children With Inoperable Plexiform Neurofibromas. N Engl J Med (2020) 382(15):1430–42. doi: 10.1056/NEJMoa1912735
134. Turner NC, Ro J, André F, Loi S, Verma S, Iwata H, et al. Palbociclib in Hormone-Receptor–Positive Advanced Breast Cancer. N Engl J Med (2015) 373(3):209–19. doi: 10.1056/NEJMoa1505270
135. Furman RR, Sharman JP, Coutre SE, Cheson BD, Pagel JM, Hillmen P, et al. Idelalisib and Rituximab in Relapsed Chronic Lymphocytic Leukemia. N Engl J Med (2014) 370(11):997–1007. doi: 10.1056/NEJMoa1315226
Keywords: CMML, chronic myelomonocytic leukemia, molecular, pathogenesis, targets, treatment
Citation: Geissler K (2021) Molecular Pathogenesis of Chronic Myelomonocytic Leukemia and Potential Molecular Targets for Treatment Approaches. Front. Oncol. 11:751668. doi: 10.3389/fonc.2021.751668
Received: 01 August 2021; Accepted: 26 August 2021;
Published: 30 September 2021.
Edited by:
Argiris Symeonidis, University of Patras, GreeceReviewed by:
Haifa Al-Ali, University Hospital in Halle, GermanyCopyright © 2021 Geissler. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Klaus Geissler, Z2Vpc3NsZXIua2xhdXNAZ214LmF0
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.