 Chen Chen
Chen Chen Yuying Guo1
Yuying Guo1 Zhenqiang Sun
Zhenqiang Sun
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 20 October 2021
Sec. Molecular and Cellular Oncology
Volume 11 - 2021 | https://doi.org/10.3389/fonc.2021.746789
This article is part of the Research Topic RNA and RNA Modification in the Pathogenesis, Diagnosis and Treatment of Cancers View all 27 articles
Cancer drug resistance has always been a major difficulty in cancer therapy. In the face of drug pressure, resistant cancer cells show complex molecular mechanisms including epigenetic changes to maintain survival. Studies prove that cancer cells exhibit abnormal m6A modification after acquiring drug resistance. m6A modification in the target RNA including non-coding RNA can be a controller to determine the fate and metabolism of RNA by regulating their stability, subcellular localization, or translation. In particular, m6A-modified non-coding RNA plays multiple roles in multiple drug-resistant cancer cells, which can be a target for cancer drug resistance. Here, we provide an overview of the complex regulatory mechanisms of m6A-modified non-coding RNA in cancer drug resistance, and we discuss its potential value and challenges in clinical applications.
RNA modification determines cell fate by regulating gene expression and responds to environmental pressures (1, 2). m6A methylation is the most common modification in mammalian RNAs. Many studies showed that m6A sites are highly conservative and prefer to the motif RRACH (R = G, A, or U; R = G or A; H = A, C, or U) in human, yeast, and mice RNA (3–5). In the early days, m6A modification was reported to be present in ribosomal RNA and tRNA. Subsequent studies have found that there are also abundant m6A sites depositing in mRNA and non-coding RNA (6–8). Among them, m6A modification could affect these types of RNA metabolism events, including RNA cleavage, processing, transportation, stability, and translation by embedding dynamic and reversible deposition of m6A sites (9, 10). Then, the dysregulated mRNA or non-coding RNA was reported to play an important role in physiological and pathologic activities, such as cancer (11–13).
Cancer, which has still endangered human health, kills approximately 600,000 people each year (14, 15). Therefore, it is an urgent problem that scientists are committed to solving. Currently, five mainstream approaches for oncotherapy are surgical resection, radiotherapy and chemotherapy, targeted therapy, and biological immunotherapy. Among them, chemotherapy and targeted therapy are an important leap in the development of cancer therapy. However, it is unsatisfactory that therapy pressure compels the cancer cell appearing drug resistance (16). In brief, a group of cancer cells are inherently resistant or becomes resistant cancer cells under drug treatment (17). The molecular mechanism of tumor resistance is extremely complex and changeable (18). Many studies have shown that epigenetic changes, such as differential non-coding RNA and dynamic m6A modification, can lead to cancer drug resistance (2, 19).
In this review, we focused on the important role of m6A modification in cancer drug resistance, summarized the molecular mechanism of m6A-modified RNA including non-coding RNA involved in cancer drug resistance, and discussed the application and clinical value of m6A modification in the prediction and treatment of cancer resistance.
Like other epigenetic modification, such as DNA methylation, m6A methylation is a dynamic and reversible phenomenon that embeds or removes m6A modification in RNA. This process is controlled by methylases and demethylases, which are vividly known as “writers” and “erasers” and then recognized by m6A “readers” (Figure 1). These m6A regulators affect multiple physiological and pathological activities such as cellular differentiation and cancer progression (10). The increasing rate suggested that m6A regulators decide RNA might play important roles in cancer progression (20).
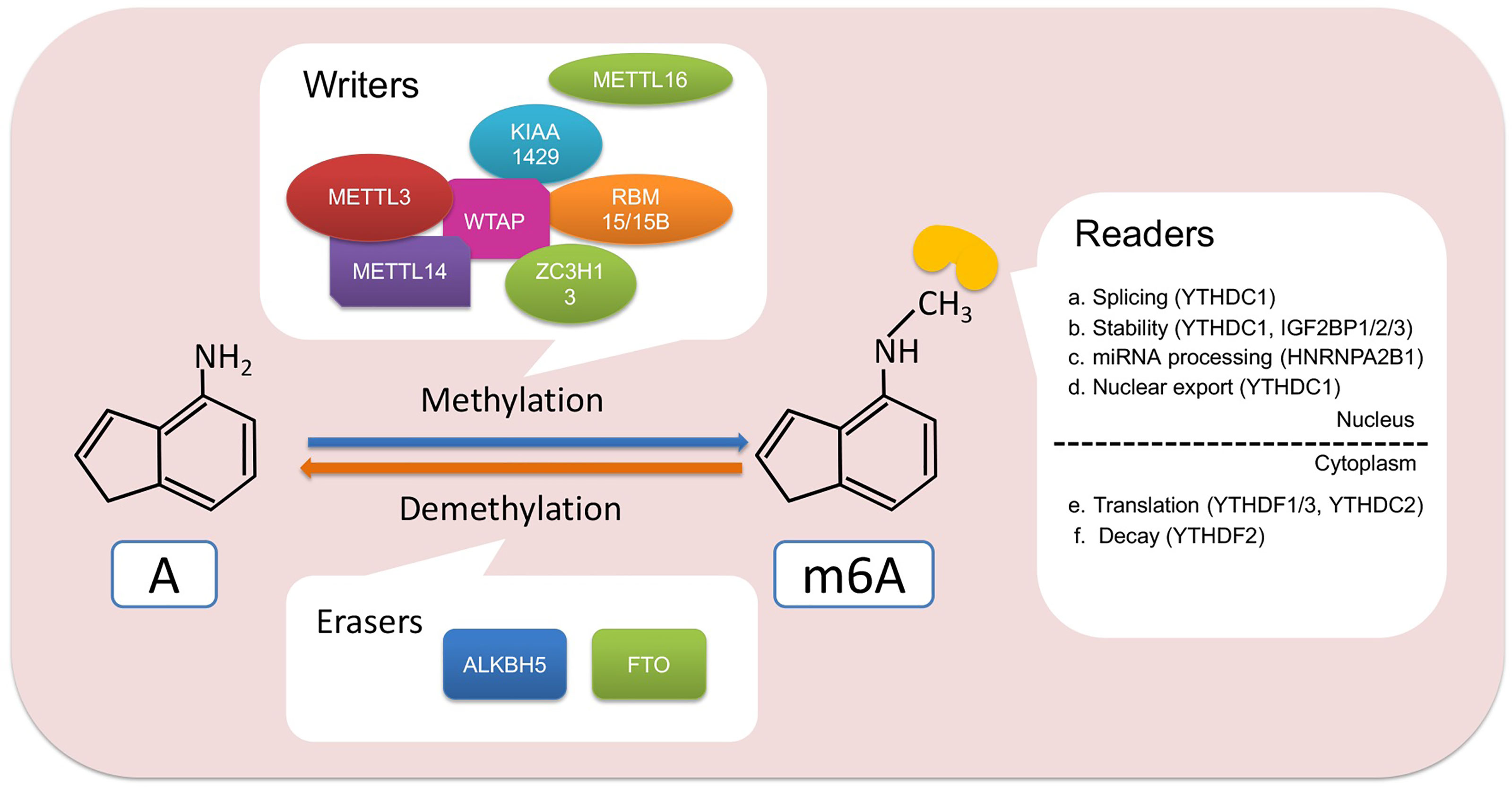
Figure 1 Regulators and functions involved in m6A modification. m6A writer, such as METTL3 and METTL14, binds to RNA to methylate adenine. Then, the m6A readers including YTHDC1/2, YTHDF1/2/3, IGF2BP1/2/3, and HNRNPA2B1 recognize the m6A site and participate in the process of RNA splicing, stability, nuclear transport, translation, degradation, or miRNA processing. In addition, m6A modifications can also be erased by FTO and ALKBH5, thereby affecting the fate of RNA. A, adenine.
Methyltransferase complexes, which consisted of multicomponents including methyltransferase-like 3/14/16 (METTL3/14/16), WT1-associated protein (WTAP), vir-like m6A methyltransferase-associated (VIRMA, another name: KIAA1429), zinc finger CCCH-type containing 13 (ZC3H13), and RNA-binding motif protein 15 (RBM15), is the key for catalyzing m6A sites in RNA. In the 1990s, an m6A complex containing three components was found in the mRNA of HeLa (21). Then, Joseph et al. found MT-A (MT-A70), which is also called METTL3, is the critical core of m6A methyltransferase complex and interacts with the methyl donor S-adenosylmethionine (SAM) for catalyzing methyl to partial RNA sequence (22–25). Moreover, another critical component of this complex, METTL14, could be as an RNA-binding platform stabilized METTL3 conformation when METTL3 is the catalytic core of this complex (23–25). WTAP, located at nuclear speckles, was another component that could interact with METTL3 and METTL14 for recruiting the m6A methyltransferase complex to RNA targets (26). Recently, Huang et al. reported that m6A deposition catalyzed by METTL3–METTL14–WTAP complex was regulated by histone H3 trimethylation at Lys36 (H3K36me3). Moreover, METTL14 could be a “reader” of H3K36me3 for promoting m6A deposition in transcribed nascent RNAs via binding adjacent RNA polymerase II (27). In addition, another m6A methylase component was identified in other types of RNAs, such as METTL16. Studies showed that m6A modification in U6 snRNA was catalyzed by METTL16 (28, 29). Moreover, METTL16 could efficiently induce splicing via binding 3′UTR of MAT2A, which encoded the SAM synthetase, a methyl donor (28). Removal of m6A modification in RNA was controlled by demethylase, FTO (obesity-associated protein), and alkB homolog 5 (ALKBH5). Initially, FTO was found to be correlated with childhood and adult obesity in early time (30). In 2011, FTO was firstly identified to have demethylation activity to decreased m6A amounts (31). Moreover, FTO could be as demethylase involved in physiology and pathology processes (32). ALKBH5 was identified as another demethylase in mammalian RNA and affects RNA export and RNA metabolism (33).
Gene expression on the post-transcriptional level, including RNA splicing, transport, stability, and translation, was closely mediated by m6A modification via recruiting reader protein or changing RNA structure (34–38).
The newly transcribed RNA is regulated by the m6A regulator to affect its splicing. The METTL3–METTL14–WTAP complex was in nuclear speckles to regulate alternative gene splicing (26). In addition, m6A demethyltransferase FTO regulated splicing factor serine/arginine-rich splicing factor 2 (SRSF2)-targeted exons to control RNA splicing (39). Nuclear m6A “reader” YTH domain-containing protein 1 (YTHDC1) was reported to recruit SRSF3 and blocked SRSF10 to promote exon inclusion for affecting RNA splicing (40). Moreover, YTHDC1 mediated RNA export from the nucleus to the cytoplasm in HeLa cells via interacting with SRSF3, which is a splicing factor and nuclear export adaptor protein (41).
The fate of RNA in the cytoplasm is to be translated into protein or be degraded. Increasing studies proved that m6A reader YTH domain-containing family proteins (YTHDFs) greatly contributed to RNA translation (42). YTHDF1 promoted RNA translation efficiency via interacting with translation initiation factor complex 3 (eIF3) (38). Moreover, RNAs that modified m6A in its 5′UTR could be translated in a cap-independent manner by directly binding eIF3 (43). RNA stability protected by its 5′ cap and 3′ poly A tail is the key for RNA to execute its roles in the vital movement. When RNA was redundant, the RNA degradation mechanism was initiated by the deadenylation-dependent decay pathway and/or deadenylation-independent decay pathway. It has been reported that m6A modification destabilized m6A-embedded RNA (44, 45). YTHDF2 could recruit CCR4-NOT deadenylase complex for destabilizing m6A-containing RNA. This binding is critical for m6A-containing RNA deadenylated by CAF1 and CCR4 (37). Furthermore, YTHDF3 interacted with YTHDF1 to promote protein synthesis and regulated decay of methylated RNA in a YTHDF2-dependent manner (46). In addition, the insulin-like growth factor 2 mRNA-binding protein (IGF2BP) family, including IGF2BP1/2/3, could be an m6A “reader” to promote RNA stability by interacting with ELAV-like RNA-binding protein 1 (ELAVL1), matrin 3 (MATR3), and poly(A) binding protein cytoplasmic 1 (PABPC1) for regulating RNA translation under normal and stress conditions (47). HnRNP A2/B1, which contains two RNA recognition motifs, was another reported m6A “reader” for promoting microRNA (miRNA) processing (48). The above shows that m6A modification decided the RNA destiny regulated by “writers,” “erasers,” and “reader”.
Increasing data showed that m6A modification playing as a decision maker contributed to cancer progression (13). For example, overexpressed FTO is a critical oncogene in hematologic malignancies, such as AML (49, 50). High expression of METTL3 has been reported to be an oncogene in multiple solid tumors, such as hepatocellular carcinoma (HCC) (51), non-small cell lung cancer (NSCLC) (52), gastric cancer (GC) (53), colorectal cancer (CRC) (54), and bladder cancer (BLC) (55). Deregulation of m6A regulators in cancer cells is involved in cell stemness, proliferation, apoptosis, metastasis, immune, and drug resistance (13). Chemotherapy and targeted therapy are the main methods for patients with hematologic tumors and solid tumors. Cancer cells that develop resistance to drug treatment usually cause tumor recurrence, leading to a bad clinical outcome (16). Complicated mechanisms including epigenetic change are crucial for cancer cell obtaining resistance (56). Emerging data indicated that the global m6A level was abundant and aberrant in drug resistance of cancer cells (Figure 2) (57–59). Moreover, m6A “writers,” “erasers,” and “readers” were dysregulated in multiple cancer cells, and these regulators played important roles in cancer cells resisting chemotherapeutic drugs and targeted drug (Table 1) (57, 61, 63).
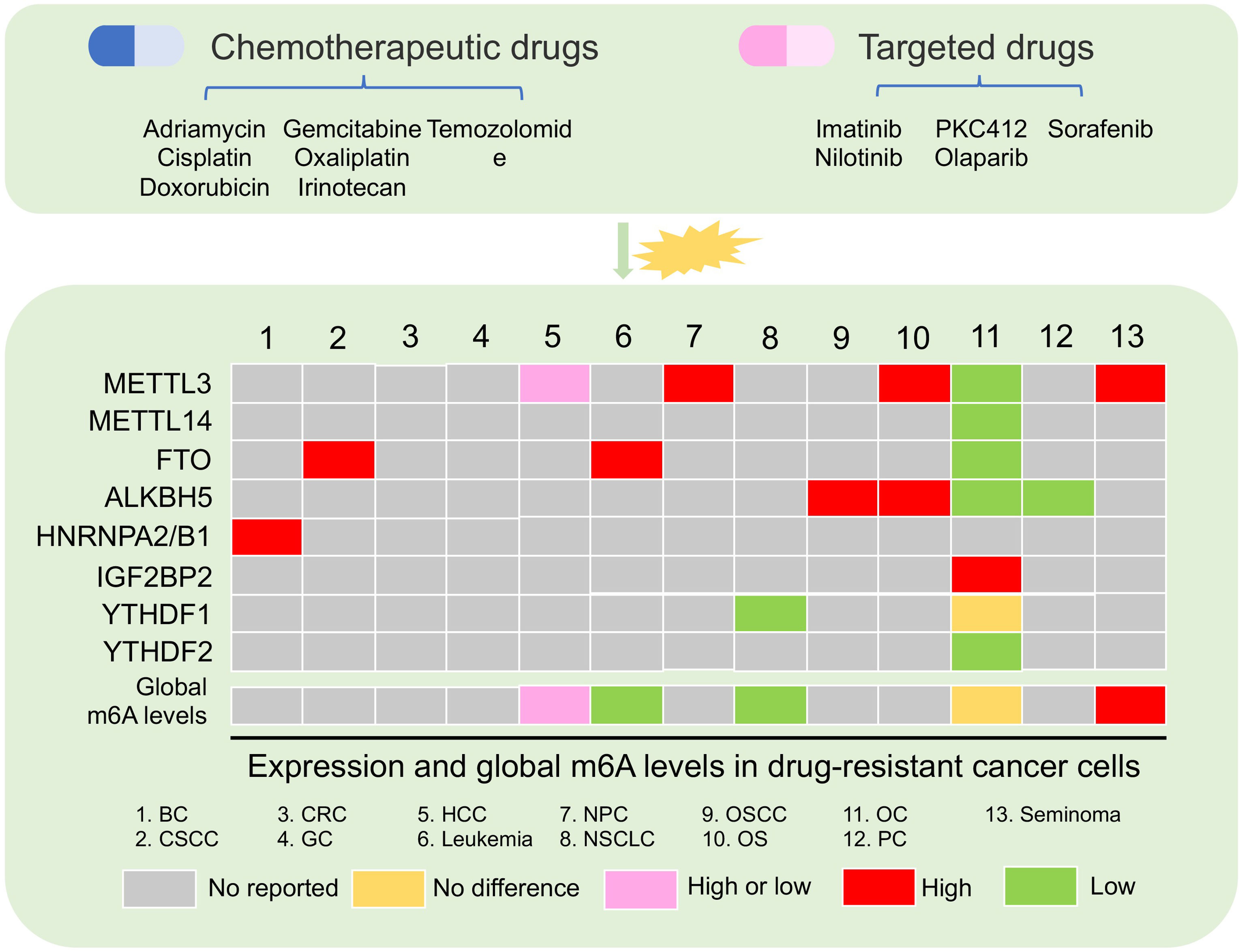
Figure 2 Aberrant m6A regulator and global m6A levels in drug-resistant cancer. The changes in m6A levels and the expression of m6A regulators were present after chemotherapy and targeted drug treatment in multiple cancer. BC, breast cancer; CSCC, cervical squamous cell carcinoma; CRC, colon cancer; GC, gastric cancer; HCC, hepatocellular carcinoma; NPC, nasopharyngeal carcinoma; NSCLC, non-small cell lung cancer; OSCC, oral squamous cell carcinoma; OS, osteosarcoma; OC, ovarian cancer; PC, pancreatic cancer.
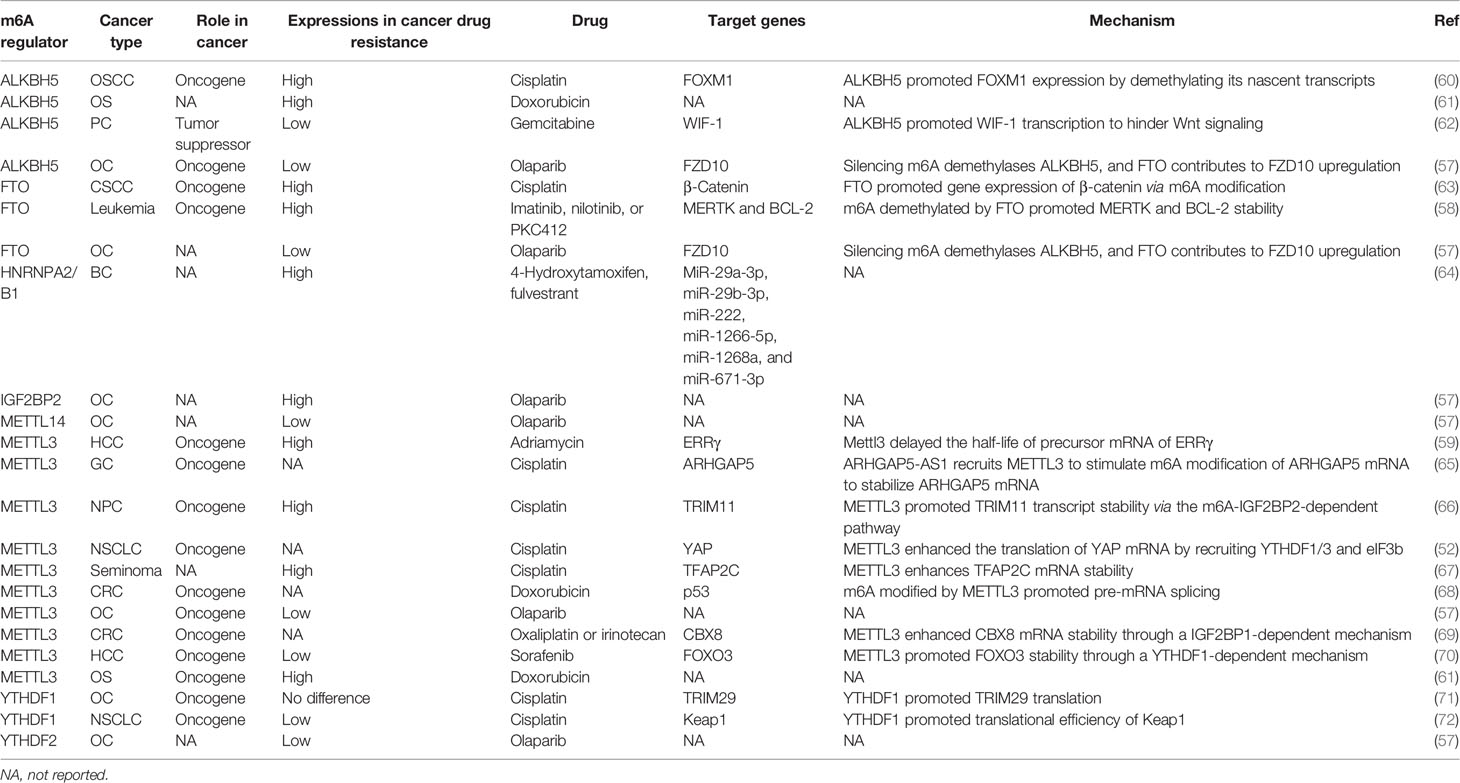
Table 1 The role and regulatory mechanism of m6A regulator in cancer drug resistance.
Increasing data showed that the global m6A level in cancer tissues was different from that in normal tissues (73, 74). Aberrant and abundant global m6A levels have also been reported in cancer drug resistance (57–59). In leukemia, researchers found that the development of resistance phenotype during tyrosine kinase inhibitor (TKI) treatment depended on the reduction of m6A detected by m6A dot-blotting due to FTO overexpression in leukemia cells. Furthermore, these results lead to the upregulation of proliferation/anti-apoptosis-related genes (58). Not only in blood tumors, but also in solid tumors, were there abnormal m6A levels in the drug-resistant cancer cell. For example, in HCC, low m6A global level cause by downregulated METTL3 was found in HepG2 cells resisting sorafenib therapy. This situation induced the increased autophagosomes of HepG2 cells to fight sorafenib therapy (70). Conversely, a high m6A global level in HepG2 cells and breast cancer cells with adriamycin resistance was detected by liquid chromatography coupled to tandem MS (LC-MS/MS) compared with parental cells. A high m6A level induced by upregulated METTL3 was responsible for the upregulation of ERRγ with metabolic reprogramming in chemoresistant cancer cells (59). In NSCLC, m6A but not the gene expression level in cisplatin-resistant A549 cells was significantly increased compared with A549 cells (72).
However, there are few studies that showed that the expression of m6A regulator was abnormal in the drug-resistant cancer cells, but the overall m6A level had no perturbation. In ovarian cancer, the global m6A level was not significantly changed in parental and cancer cells with olaparib and poly(ADP-ribose) polymerase inhibitor (PARPi) resistance. No change of global m6A levels in ovarian cancer resistant cells might be due to downregulated FTO, ALKBH5, METTL3, and METTL14. Furthermore, a high m6A level in 3′UTR region of the FZD10 mRNA due to the decrease of FTO and ALKBH5 was found to activate the Wnt signaling pathway, leading to PARPi resistance (57). These evidence indicated that the global m6A level is very complicated under different types of tumor cells and different therapy resistance. This complexity is due to the abnormal expression of different m6A regulators under resistant conditions. Therefore, it is necessary to acquaint entirely the molecular mechanism of abnormal m6A level and m6A regulators under the drug resistance pressure.
The global m6A level was regulated at different layers by “writers,” “erasers,” and “readers,” which showed irregular expression and played important roles in drug resistance of multiple cancers (Table 1).
Dysregulated m6A “writers” were found in multiple cancers after drug treatment (57, 59, 61, 66, 70). It has been reported that METTL3 and METTL14 were downregulated in ovarian carcinoma cells (57). METTL3, as a critical methylase, was reported to play an essential oncogene in 2017 (75). It was not until recent years that increasing fact about the oncogene effect of METTL3 and its dysregulation in cancer drug resistance was displayed (76). In osteosarcoma (OS), METTL3 was found to have a critical role in OS cells through promoting cell proliferation, migration, and invasion ability (77). When OS cells showed doxorubicin resistance, an increasing METTL3 expression was detected (61). Similarly, highly expressed METTL3 in drug-resistant cell lines was also found in nasopharyngeal carcinoma (NPC) (61). A recent study showed that overexpression of METTL3 in NPC tissues promoted EMT process via m6A-modified Snail (78). Studies showed that high METTL3 expression in recurrent NPC tissue was associated with a bad prognosis of NPC patients (78, 79). After cisplatin induction, METTL3 was upregulated in drug-resistant NPC cell lines, where it could promote the viability of cell culture with a series of doses of cisplatin via regulating TRIM11 transcript (66). In HCC, METTL3 was found to promote HCC progression via YTHDF2-dependent silencing of SOCS2 on the post-transcriptional level (51). Interestingly, the role of METTL3 in HCC drug resistance was also complicated. Chen et al. reported that high METTL3 expression was detected in HepG2 cells with adriamycin resistance (59). Deleting METTL3 leads to the increase of dox sensitivity in adriamycin resistant HepG2 cells by regulating pre-mRNA of ERRγ. However, low METTL3 expression was found in human sorafenib-resistant HCC (70). Different from adriamycin, sorafenib is a multi-target drug that inhibited cancer cell proliferation and angiogenesis. This interesting result showed that METTL3 could be a tumor suppressor when its knockout could enhance sorafenib resistance, promote angiogenesis-associated genes expression, and activate pathway-associated autophagy in HCC cells under hypoxia condition (70). It is worth noting that METTL3 not only was involved in tumor progression but also plays an important role in tumor resistance, especially solid tumors.
As m6A demethylase, FTO and ALKBH5 were reported to play critical roles in multiple cancers and are involved in cancer drug resistance (58). FTO has been firstly reported m6A demethylase, which subsequently proved its oncogene role in AML (31, 50). Through reducing m6A abundance of ASB2 and RARA for destabilizing its transcripts, high FTO expression could enhance cell transformation and leukemogenesis, while it could inhibit all-trans-retinoic acid (ATRA)-induced AML cell differentiation (50). Moreover, high FTO expression was found in AMLs with multiple mutations including t(11q23)/MLL rearrangements, t (15, 17)/PML-RARA, FLT3-ITD, and/or NPM1. TKIs were considered for leukemia clinic treatment with these mutations (80, 81). However, rapidly acquiring resistance to TKIs was the main reason for the failure of leukemia treatment. Yan et al. found that FTO expression was increased in leukemia nilotinib-resistant cells. Upregulated FTO assisted leukemia cells to display more TKI resistant ability, and higher rates of cell growth in vivo via enhance mRNA stability of MERTK and BCL-2 (58). Moreover, FTO overexpression in cervical squamous cell carcinoma (CSCC) was proved to be related to chemo-radiotherapy resistance in vitro and in vivo by decreasing demethylation of β-catenin for promoting its expression (63). In addition, another m6A demethylase, ALKBH5, was upregulated in OS cell lines with doxorubicin resistance and doxorubicin resistance oral squamous cell carcinoma (OSCC) lines (60, 61). However, downregulated ALKBH5 expression was reported in a patient-derived xenograft (PDX) model treated with gemcitabine in pancreatic ductal adenocarcinoma (PDAC) (62). When ALKBH5 was overexpressed, PDAC cells were sensitive to chemotherapy due to a decrease of methylation in WIF-1 (62). Moreover, in ovarian cancer, downregulated FTO and ALKBH5 in PARPi resistance cancer cells also could be decreased PARPi sensitivity (57).
The process that m6A modification mediated RNA metabolism always needs the participation of m6A “readers” to decide RNA fates. In cancer, aberrant reader proteins, such as YTHDF1, could lead to the disorders of RNA metabolism that contributed to tumor progression (82). The role of YTHDF1 was reported in various cancer types, and it plays a critical role in cancer (72, 83, 84). Cancer stem cells (CSCs) are cell populations with stem-like characteristics, which are considered to be the main cause of tumor chemoresistance and recurrence (85). A recent study reported that YTHDF1 promoted TRIM29 translation in cisplatin-resistant ovarian cancer cells. Decreasing YTHDF1 inhibited the CSC-like characteristics subsequently rescued by overexpressed TRIM29 (71). However, there is no difference of YTHDF1 in the cisplatin-resistant cells compared with the parental control cells, which need more clinical samples to be proved. Another study displayed that YTHDF1 was upregulated in NSCLC tissues compared with paracancerous tissues. Deletion YTHDF1 in vitro impeded cancer cell proliferation and inhibited cancer progression in vivo (72). Moreover, recent studies reported that YTHDF1 was involved not only in the glycolysis of cancer cells by promoting mRNA stability of PDK4 but also in cancer drug resistance (72, 86). A study showed that YTHDF1 expression was downregulated in cisplatin-resistant A549 cells. Silencing YTHDF1 rendered cancer cells resistant to cisplatin treatment, which showed a bad clinical outcome (72). In addition, decreased YTHDF2 expression and increased IGF2BP2 expression were found in resistant ovarian cancer cells, which might be contributing to FZD10 upregulation for promoting cancer drug resistance (57).
Increasing studies showed that dysregulation of m6A-deposited RNA significantly contributed to cancer progression (13). Recently, abundant m6A modification was found in 3′UTR, 5′UTR, and/or CDS in drug tolerance of cancer cells, such as ovarian cancer cell (57). m6A methyl embedded in different areas of RNA could influence RNA splicing, RNA stability, and translation in cancer drug resistance (Figure 3) (52, 58, 68).
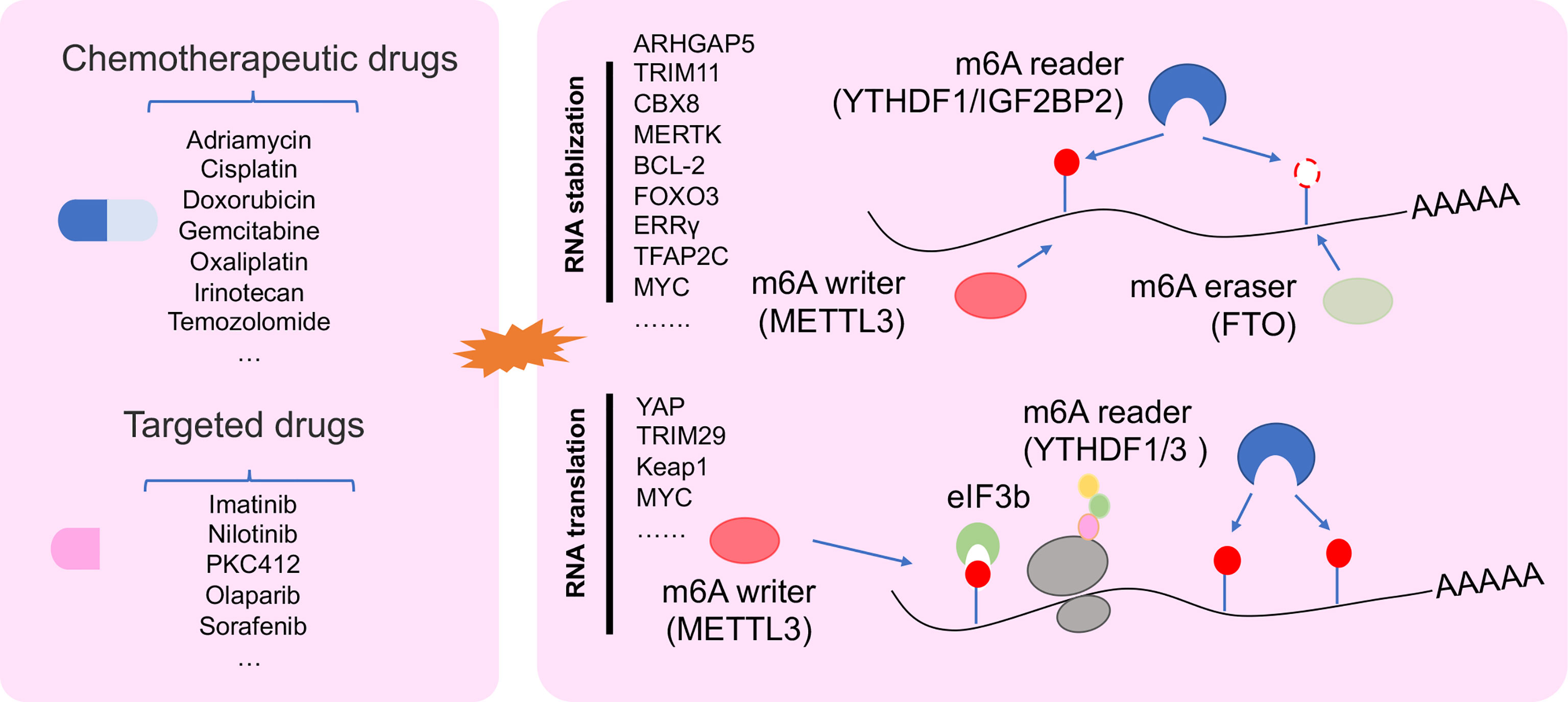
Figure 3 The regulatory mechanism of m6A regulator in drug resistant cancer. m6A modification in drug resistant cancer cell was involved in changing RNA stability, and RNA translation.
Transcript stability is a very important character for RNA to execute its function. Changes in RNA stability are the inevitable result of tumor progression (87). Increasing data showed that m6A participated in RNA stability for regulating cancer drug resistance mechanism. In drug-resistant NPC cells, METTL3 promoted TRIM11 transcript stability via the m6A-IGF2BP2-dependent pathway. m6A-marked TRIM11 could promote multidrug resistance and suppressed apoptosis by activating the β-catenin axis (66). Moreover, METLL3 enhanced RNA stability of CBX8, which could promote stemness and inhibit chemosensitivity by activating LGR5 transcription in colon cancer (CC) cells (69). Not only mRNA but also METTL3 could change the stability of pre-mRNA. A study reported that METTL3 could delay the half-life of pre-mRNA of ERRγ to enhance chemoresistance by upregulating ABCB1 and metabolic reprogramming in HCC (59). Demethylation of RNA regulated by ALKBH5 was found in cisplatin-resistant OSCC lines (60). A study showed that DDX3 could enhance CSC population by demethylation of FOXM1 and NANOG nascent transcript regulated by AKBH5 in chemoresistant cells (60). Other demethylase FTO could enhance RNA stability of MERTK and BCL-2 to control intrinsic and acquired resistance of CSCC via TKI therapy (58).
Dysregulation RNA translation was found in multiple diseases, including cancer (88, 89). Tumor cells trigger cellular stress under drug stress, such as genotoxic, oxidative, metabolic, and protein toxic stress. These stress responses lead to the plasticity of translational control, thereby changing tumor behavior that tend to drug resistance (90). m6A modification as a regulator could promote RNA translation via YTHDF1 interacting with initiation factor eIF3 under a selective regulation mechanism (38). Recent work reported that the deficiency of YTHDF1 inhibited translational efficiency of Keap1 and activated the antioxidant reactive oxygen species (ROS) clearance system (Nrf2-AKR1C1) under DDP therapeutic burden, which leads to a bad prognosis of NSCLC patients (72). Furthermore, METTL3 could be binding with YTHDF3, YTHDF1, and eIF3b to increase YAP1 translation for inducing DDP resistance and metastasis in NSCLC (52).
After drug therapy, cancer cells always accumulate DNA damage and increase the probability of gene mutations, which leads to a small number of tumor cells evolving drug resistance (91). Increasing or decreasing m6A level in RNA transcripts always leads to cancer progression. Recently, a study reported that m6A site mutation in transcripts may change m6A deposition and play an important role in cancer (92). TP53 mutation is a high-frequency mutation in 12 types of cancer and indicated close relation with clinical outcome (93). An interesting result showed that an m6A-modified mutation site found in the transited codon 273 of p53 pre-mRNA could increase the expression of R273H mutant protein and lead to multiple drug resistance of CRC (68). This showed that m6A site mutations in transcript greatly contributed to cancer drug resistance and could be a method to indicate patient prognosis in cancer therapy.
Non-coding RNAs are a class of RNAs that are not translated into protein and regulate the epigenetic change of genes. MiRNAs, long non-coding RNAs (lncRNAs), and circular RNAs (circRNAs) are three common non-coding RNAs that were reported to play important roles in cancer progression (94, 95). For example, highly expressed miR-21 was found to target PTEN for promoting HCC growth (96). LncTCF7 promoted self-renewal of HCC stem cells via triggering Wnt signaling (97). As the sponge of miR-9, circMTO1 promoted p21 expression for hindering HCC progression (98). Moreover, dysregulation of non-coding RNA in multiple cancers may be the primary cause of drug resistance (99–101). Linc00152, which promoted CRC progression, conferred resistance of oxaliplatin (L-OHP) that induced cancer cell apoptosis via the AKT pathway (102). In ovarian cancer cells, downregulated miR-29a/b/c enhanced the ability of cells to escape cell apoptosis induced by cisplatin via targeting collagen type I alpha 1 (COL1A1) (103). Furthermore, upregulated lncRNA UCA1 contributed to multiple drug resistance of GC via sponging miR-27b (104). Collectively, these findings displayed important roles of non-coding RNAs in one or more drug resistance of cancer. Interestingly, m6A modification commonly existed in not only mRNA but also non-coding RNA (9). More and more findings suggested a novel and complicated pattern of m6A regulator with non-coding RNA in cancer drug resistance.
MiRNAs are a group of highly abundant small RNAs (21–25 nucleotides) involved in post-transcriptional control via targeting 3′UTR of mRNA (105). It has been reported that miRNAs play a crucial role in cancer drug resistance (106). In NSCLC resistant to EGFR TKIs, exogenous miR-146b-5p in EGFR TKI-resistant cells could promote the cell apoptosis induced by EGFR TKIs via regulating the IRAK1/NF-κB pathway (107). In addition, miR-675-3p, which was from GC-secreted extracellular vesicles (GC-EVs), could enhance cisplatin resistance in vivo via targeting CXXC4 (108). These works displayed the potential role of miRNAs in cancer drug resistance.
Recently, m6A modification was found in long primary miRNAs (pri-miRNAs) to affect miRNA processing. It is well known that pri-miRNAs are firstly transcribed in the nucleus. Subsequently, pri-miRNAs are transported into cytoplasm to be processed into precursor miRNAs (pre-miRNAs) via the double-stranded RNA-binding protein (RBP) DGCR8, which is the critical component of miRNA microprocessor complex binding with RNase III endonuclease DROSHA (109). METTL3 marked pri-miRNA for recognition and processing by DGCR8. Deleting METTL3 leads to the reduction of global miRNA and accumulation of unprocessed pri-miRNA. Gain of METTL3 reversed this change of global miRNA in a non-cell-type-specific manner, which suggested that m6A methylation plays a key post-transcriptional modification to promote miRNA mature (110). Moreover, m6A “reader” protein, heterogeneous nuclear ribonucleoprotein A2/B1 (hnRNPA2/B1), and hnRNPC could recognize m6A site in pri-miRNA and subsequently interact with DGCR8 to promote miRNA mature (48, 111). Interestingly, this new mechanism was found in the progression of multiple cancers (112–114). In BLC cells, METTL3 recruited DGCR8 to promote miR-221/222 maturation via decreasing PTEN expression for tumor growth (57). Another m6A complex component, METTL14, inhibited HCC metastasis via recognizing and binding DGCR8 to promote pri-miR-126 processing (115). Furthermore, m6A regulator–miRNA model was found to play an important role in cancer drug resistance (Figure 4A). In breast cancer cells, hnRNPA2/B1–miRNA model was reported to be involved in endocrine resistance. Upregulated hnRNPA2/B1 in endocrine-resistant breast cancer cells changed miRNA transcriptome including 148 upregulated miRNAs and 88 downregulated miRNAs. Moreover, overexpressed hnRNPA2/B1 decreased the sensitivity of cancer cells to 4-hydroxytamoxifen and fulvestrant (64). In addition, m6A regulators are involved in cancer drug resistance by changing miRNA expression by an indirect mechanism. MYC as a transcription factor is an oncogene by accelerating cell proliferation (116). A study reported that miRNA-155 (miR-155) and the miRNA-23a~27a~24-2 cluster (miR-23a cluster) were induced by MYC to promote tumorigenesis in glioma cells. Interestingly, FTO increased MYC stability and translation efficiency via wiping m6A modification, and its downregulation reduced the primary and mature transcripts of the transcripts of miR-155-5p, miR-24-3p, and miR-27a-3p (117). Furthermore, FTO inhibitor and MA2 increased the antitumor effect of temozolomide on decreasing the viability of glioma cells (117).
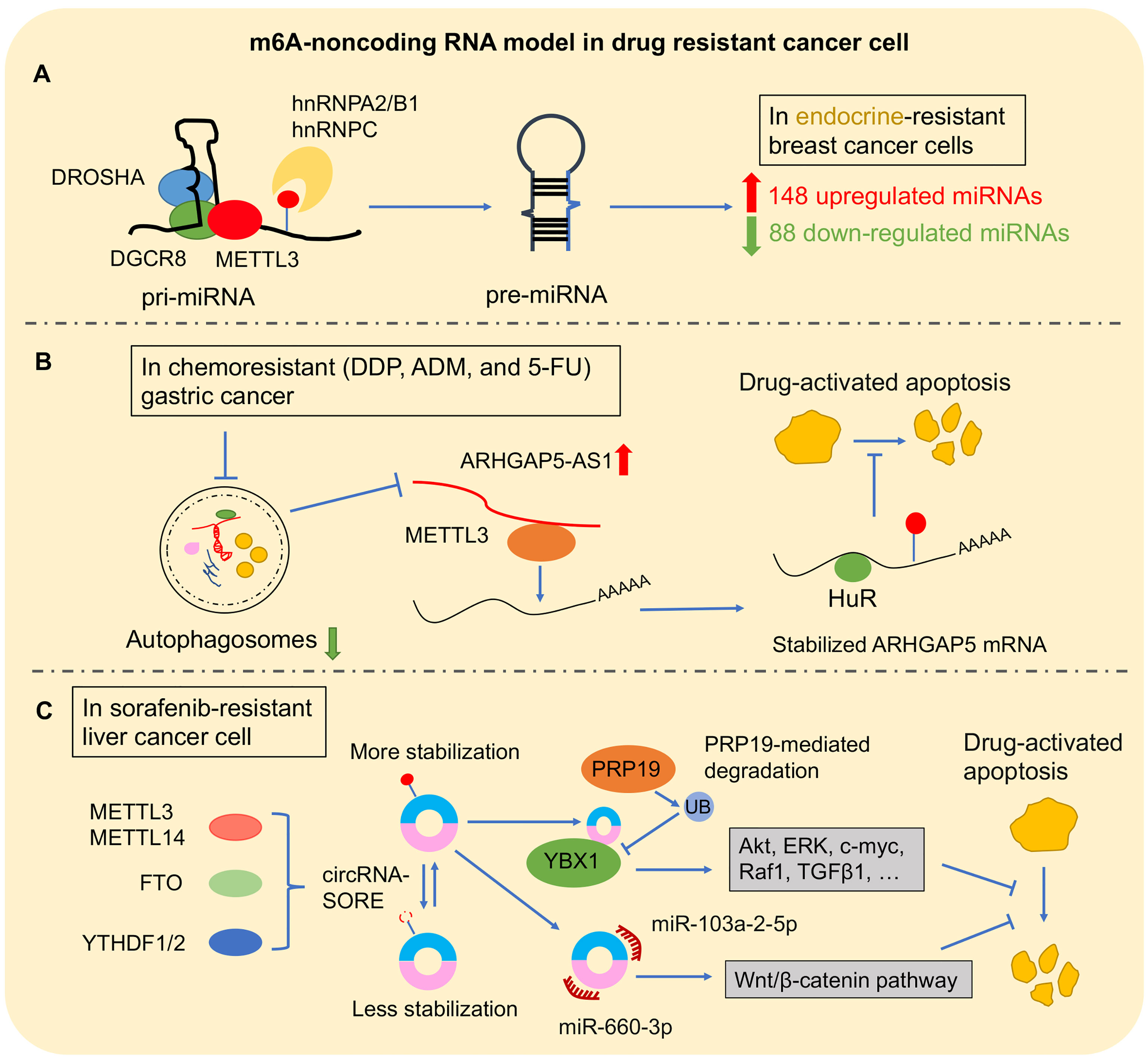
Figure 4 The m6A–non-coding RNA model in drug-resistant cancer. (A) m6A–miRNA model: m6A modification directly affects the pre-miRNA splicing, resulting in abnormal miRNA level changes in cancer drug resistance. (B) m6A–lncRNA model: ARHGAP5-AS1 recruits METTL3 to promote the stability of ARHGAP5 for inhibiting drug-induced apoptosis. (C) In sorafenib-resistant liver cancer cell, m6A modification promoted circRNA-SORE stabilization. Then, circRNA-SORE could directly bind YBX1 protein or sponge miR-660-3p and miR-103a-2-5p for arresting drug-activated apoptosis.
Compared with miRNA, lncRNAs are a class of non-coding RNA with its length more than 200 nucleotides defecting coding protein function (118). LncRNA generally contributed to gene regulation via binding with miRNA, interacting with RBPs, or chromatin remodeling (119). In gemcitabine-resistant cells, GSTM3TV2 promoted the drug resistance by sponging let-7 for upregulating L-type amino acid transporter 2 (LAT2) and oxidized low-density lipoprotein receptor 1 (OLR1) in pancreatic cancer cells (120). Upregulated H19 in tamoxifen-resistant breast cancer cell line promoted autophagy by inhibiting the binding of DNMT3B to Beclin-1 (121). These studies showed that lncRNA could be a controller to regulate cancer drug resistance.
It has been observed to conserve m6A modification in lncRNA (122). Subsequently, increasing findings showed that m6A modification as “switches” regulated lncRNA–protein interaction contributed to the lncRNA–miRNA interaction or influenced lncRNA stability (52, 123, 124). Metastasis-associated lung adenocarcinoma transcript 1 (MALAT1) was a conserved lncRNA that was upregulated in multiple types of cancers (125). In NSCLC, MALAT1 promoted cancer cell migration in vitro and tumor formation and growth in vivo, and a high expression level of MALAT1 is associated with poor prognosis (126, 127). Recently, high m6A modification in transcripts of MALAT1 was observed (8). METTL3/YTHDF3 complex increased the stability of MALAT1, which sponged miR-1914-3p to promote the invasion and metastasis of NSCLC and enhanced sensitivity to DDP via regulating YAP1 (52). On the other hand, lncRNA was also involved in cancers when it was coupled with m6A regulators. Upregulated ALKBH5 in glioblastoma stem-like cells (GSC) demethylated FOXM1 to promote its expression. Interestingly, as a lncRNA antisense to FOXM1, FOXM1-AS accelerated the interaction between ALKBH5 with FOXM1 for promoting GSC proliferation and tumorigenesis (128). This interaction of lncRNA assistance to m6A regulators was also found in cancer drug resistance (Figure 4B). In chemoresistant GC cells, overexpressed ARHGAP5 antisense RNA 1 (ARHGAP5-AS1) promoted the cancer cell resistance to chemotherapeutic drugs including DDP, ADM, and 5-FU; decreased drug-activated apoptosis; and reduced intracellular drug concentration. ARHGAP5-AS1 could stabilize ARHGAP5 via recruiting METTL3 for m6A modification (65).
CircRNAs are a class of non-coding RNA that has a special continuous loop by back splicing. The reported biological function of circRNA seems to bind miRNA or RBPs, regulate transcription, interfere with splicing, or translate peptide fragments and played important roles in cancer drug resistance (129, 130). In GC, circCUL2 could affect cisplatin sensitivity by inhibiting the autophagy activation mediated by miR-142-3p/ROCK2 (131). In glioblastoma cells that acquired temozolomide resistance, circASAP1 was markedly upregulated. Its overexpression promoted cell proliferation and temozolomide resistance by sponging miR-502-5p in glioblastoma (132). These studies showed that dysregulated circRNAs play a significant role in cancer drug resistance.
CircRNAs are widely modified by m6A and exhibited cell-type-specific methylation, which suggested the potentially important role of m6A modification in modulating circRNA biogenesis, transport, degradation, and translation (133, 134). It is well known that circRNAs are the product of back splicing, which is an alternative to linear splicing. Recently, m6A modification at specific sites was found to significantly modulate these two splicing ways. Furthermore, these specific m6A sites affected by METTL3–YTHDC1 were proved to command circRNA biogenesis, which offers a novel reason for circRNA biogenesis (135). CircRNA location is the key to determining the regulatory mechanisms of circRNA, especially ceRNA model in cytoplasm. However, few data illustrated the mechanisms of circRNA localization or nuclear export. It has been found that Drosophila DExH/D-box helicase Hel25E and human UAP49/56 act as key factors in the nuclear export of circRNAs depending on its length (136). Interestingly, m6A reader YTHDC1 was reported to accelerate cytoplasmic export of circNSUN2 in an m6A-dependent manner (137). The degradation of circRNA is another important part of understanding its function and expression, but the understanding of related mechanisms is still very limited. It has been reported that YTHDF2 interacting with HRSP12 acted as a guide of RNase P/MRP endoribonucleases for identifying and degrading m6A-modified circRNAs (138). In translation initiation, different from mRNA (cap-dependent pathway), circRNA lacking dissociative 5′ end was translated by cap-independent pathways, such as IRES-dependent pathway and m6A-dependent pathway (139–141). Yang et al. found that m6A-modified circRNAs have sufficient translation ability regulated by YTHDF3 and the translation initiation factors eIF4G2 and eIF3A (141). Recently, increasing findings prove the important role of the m6A-circRNA model in cancer progression. In CRC, m6A reader YTHDC1 expedited cytoplasmic export of circNSUN2 to promote cancer liver metastasis via stabilizing HMGA2, which acted as a driver of cancer metastasis via enhancing EMT process (137, 142, 143). In cervical cancer cells, an interesting study found that circE7 derived from human papilloma virus 16 (HPV16) translated into E7 protein to promote cancer cell proliferation in an m6A-dependent manner, which provided a basis for the diagnosis of high-risk HPV infection (144).
A recent study showed a significant role of m6A-circRNA model in HCC resistance with sorafenib, which is the first-line chemotherapeutic therapy for advanced HCC (Figure 4C). After sorafenib therapy, HCC cell with drug resistance showed high expression of circRNA-SORE in which depletion could enhance the cell-killing ability of sorafenib by stabilizing YBX1 (145). Another study showed that circRNA-SORE inhibited the efficacy of sorafenib-induced apoptosis in HCC cells. Interestingly, m6A modification resulted in the increased RNA stability of circRNA-SORE, which could sponge miR-103a-2-5p and miR-660-3p to activate the Wnt/β-catenin pathway (146).
Tumor drug resistance is often the result of a combination of multiple mechanisms (147). Urgently, we need more effective assessment methods to judge whether patients have better treatment effects to improve patient prognosis. Researchers have found that methods such as gene sequencing for analysis of mutant genes, tumor-derived organoids for evaluating drug efficacy, or network-based machine learning can be used to predict tumor resistance (148, 149). Tiriac et al. reported that next-generation sequencing (NSG) technology combined with patient-derived organoid (PDO) drug classification can predict the response of pancreatic cancer patients and provide a basis for the selection of treatment options (150).
Abnormal and abundant global m6A levels frequently appeared in multiple cancer drug resistance. The global m6A level was decreased in leukemia cell line that obtained nilotinib and PKC412 resistance and HepG2 resisting sorafenib therapy, inversely upregulated in breast cancer cell with adriamycin resistance (58, 59). Therefore, the detection of m6A levels has the potential to be used as a method to predict whether tumor cells are/will be resistant. At present, the main technical methods used to detect the overall level of m6A are LC-MS/MS, colorimetry, and dot blotting (151). Based on liquid mass spectrometry, LC-MS/MS uses tandem MS to obtain molecular ion peaks and fragment ion peaks. LC-MS/MS can simultaneously perform qualitative and quantitative analyses of bases. With the use of a method similar to ELISA to suppress competitive immunity, the colorimetry uses m6A standards to quantify the m6A level of samples. Dot blotting uses m6A antibody to detect m6A level after binding RNA to a nylon membrane. Compared with the cumbersome operation of LC-MS/MS, colorimetry or dot blotting using a kit is simpler, faster, and more sensitive. These methods could have a potential application for detecting cancer drug resistance. Increasing data suggested that abnormally expressed m6A regulators can also be used as a detection strategy for drug response of multiple cancer. In NPC, m6A-modified TRIM11 stabilized by METTL3 could inhibit cancer cell apoptosis to promote multidrug resistance and enhance cisplatin resistance in vivo (66). Similar in GC, high METTL3 group tumors showed more sensitivity to mTOR inhibitor (everolimus) compared with the low METTL3 group (152). In ovarian cancer, downregulated FTO and ALKBH5 induced FZD10 upregulation, which led to reducing PARPi sensitivity (57). Moreover, in CSCC, FTO reduced m6A level of β-catenin to promote its expression for enhancing the chemo-radiotherapy resistance both in vitro and in vivo (63). Oppositely, FTO upregulation in leukemia showed more TKI tolerance via enhancing MERTK and BCL-2 stability (58).
Increasing data displayed that m6A regulators play an important role in drug resistance of cancer, which suggested that m6A regulators may be a potential target to restore the drug sensitivity of tumor cells. In cisplatin-resistant seminoma cells, METTL3 promoted cellular viability by enhancing TFAP2C stability, which was identified by m6A reader, IGF2BP1. Encouragingly, more rapid tumor growth due to overexpressed METTL3 was inhibited by IGF2BP1 inhibition under cisplatin treatment in vivo (67). In NSCLC, METTL3 inhibition observably arrested tumor growth and enhanced sensitivity to cisplatin in vivo by reducing YAP1 expression (52). In addition, a significant antitumor drug effect was also found in drug-treated cancer cells combined with FTO inhibitors. In glioma, MA2, which could be an FTO inhibitor, notably inhibited cell proliferation compared with a single treatment group (117). Regarding the targeting of m6A-modified molecules, a variety of drugs have entered clinical research. For example, a competitive 2OG inhibitor can target and inhibit the activation of FTO. But it also inhibits other molecules, such as m1A demethylase ALKBH3, causing adverse side effects (153, 154). Many kinds of literature have confirmed that m6A modification participates in the regulation mechanism of tumor resistance. In detail, under the pressure of drugs, tumor cells cleverly change the modification level of m6A in RNA, thereby affecting the fate of RNA, especially non-coding RNA. Therefore, compared with looking for highly specific inhibitors of m6A regulatory molecules, directly targeting its downstream RNA molecules is also an ideal tumor drug resistance treatment policy. Non-coding RNA that interacts with m6A regulators has been reported to regulate the sensitivity of drug-resistant cells. A large number of studies have found that many non-coding RNAs can be used as targets or partners for m6A regulators in drug-resistant tumor cells. METTL3 coupled with lncRNA ARHGAP5-AS1 stabilized ARHGAP5, which promoted chemoresistance in GC (65). In addition, in vivo shRNA delivery of m6A-modified circRNA-SORE could enhance sorafenib efficacy in animal models (146). These studies suggested that m6A regulator–non-coding RNA model plays important roles in cancer drug resistance. In general, the combined strategy of using m6A targeted drugs with chemotherapy or its related non-coding RNA may provide new ideas for avoiding drug resistance in clinical practice.
Tumor immunotherapy is currently one of the mainstream methods of cancer treatment. However, the effects of immunotherapy in some tumors, such as lung cancer, are not satisfactory. The main reason is that the tumor behaved innately or acquired resistance to immunotherapy. Therefore, it is urgent to find a breakthrough in resistance to immunotherapy. Recently, most works implied m6A regulators have huge potential in PD-1 therapy. m6A regulators were reported to regulate PD-L1 expression, which implied its potential implication in immunotherapy. In intrahepatic cholangiocarcinoma (ICC), Qiu et al. found that tumor-intrinsic ALKBH5 stopped T-cell expansion and cytotoxicity by maintaining PD-L1 expression in tumor cells. Moreover, the results of clinical sample analysis from patients receiving anti-PD1 immunotherapy showed that strong nuclear expression patterns of ALKBH5 are more sensitive to anti-PD1 immunotherapy (155). Knockdown YTHDF1/2 in NSCLC cells could upregulate PD-L1 expression and multiple immune-related genes. High YTHDF1/2 expression showed a good prognostic outcome of NSCLC patients (156). Most notably, Han et al. found that the YTHDF1 could be a potential therapeutic target in immunotherapy because the absence of YTHDF1 can enhance the therapeutic effect of PD-L1 in Ythdf1−/− mice (157). Interestingly, non-coding RNA with m6A modification represented promising therapeutic targets in improving immunotherapeutic efficacy. m6A-modified circIGF2BP3 inhibited the activity of T cells in vitro and restrain antitumor immunity in vivo by upregulating PKP3 to elevate PD-L1 abundance. Furthermore, the inhibition of circIGF2BP3/PKP3 enhanced the effects of anti-PD-1 therapy in a Lewis lung carcinoma mouse model (158). These works suggested that m6A regulators or m6A-modified non-coding RNA could be a therapeutic target for immunotherapy in combination with PD-L1 inhibitors.
m6A modification is the most common modification in higher organisms. Many studies have confirmed that m6A modification plays important biological functions in mammals, such as regulating RNA stability, positioning, transportation, splicing, and translation at the transcriptional level. We have introduced some work that found that tumor cells exhibit abnormal m6A levels after drug resistance, and further control RNA stability, RNA translation, or regulation of non-coding RNA to avoid drug-induced apoptosis and escape successfully. These papers about cancer drug resistance displayed a part of the important role of m6A modification. Recent studies indicated that m6A modification was regulated by other factors. NADP, an enzyme-regulated metabolite, could directly bind FTO for improving its catalytic activity and adipogenesis (159). It indicated that some key regulators could be a potential strategy for regulating m6A modification, and we need more work to explore the mechanism that triggered abnormal global m6A level or differential expression of m6A regulators in the future.
At present, more and more m6A regulators are involved in clinical research for oncotherapy. However, their disadvantages cannot be ignored, such as poor specificity and side effects. Therefore, it is urgent to find another way to change the dilemma of m6A modification in the application of tumor resistance. Although m6A–non-coding RNA has positive prospects in clinical applications, the current situation shows that there are still shortcomings in the study of m6A–non-coding RNA mode. Firstly, m6A has fewer potential targets and partners in different drug resistance mechanisms of different tumors. Therefore, the regulatory mechanism of m6A modification in the common drug resistance of a variety of tumor cells still needs to be further explored more comprehensively. Secondly, there are fewer methods to detect the methylation level at the m6A site on the non-coding RNA sequence. Therefore, the method detecting more high resolution of m6A in non-coding RNA sequence needs to be exploited and widely apply in cancer drug treatment. Finally, the drug development of non-coding RNA related to m6A drug should be considered in the future.
In the review, we summarized the research progress of m6A modification in cancer drug resistance. We show that the overall m6A level and m6A regulatory molecules in tumor cells are changed after different drug-resistant multiple tumors. For example, in adriamycin-resistant liver cancer cells, the expression of METL3 was significantly upregulated, resulting in a significant increase in overall m6A levels. However, in sorafenib-resistant liver cells, the expression of METL3 was significantly downregulated, resulting in a significant decrease in overall m6A levels. These contradictory results showed the complexity of m6A modification in drug-resistant cancer cells. In addition, we also summarized the complex regulatory mechanism of m6A modification in tumor resistance. Abnormal m6A regulators in tumor cells that have acquired drug resistance can maintain tumor cells against drug therapy by changing RNA stability, RNA translation, and catalytic mutation site m6A modification. Here, we focus on the important role of m6A modification that interacted with non-coding RNA in cancer drug resistance. Sorafenib is the first-line chemotherapeutic agent for the treatment of liver cancer. It is worth noting that circRNA-SORE stabilized by m6A modification can inhibit drug-activated apoptosis through a variety of ways, which elucidates that m6A regulator–non-coding RNA model plays an important role in cancer drug resistance. Therefore, targeting m6A-modified non-coding RNA has potential as a combined strategy to overcome therapeutic resistance. But the current situation is that the molecular understanding of m6A modification is still in its infancy. Therefore, more research is still needed to realize the application of m6A modification combined with non-coding RNA in cancer drug therapy.
Conceptualization, writing, and editing: all authors. All authors contributed to the article and approved the submitted version.
This study was supported by the National Natural Science Foundation of China (81972663, U2004112); Key Scientific Research Projects of Institutions of Higher Education in Henan Province (19A310024); the National Natural Science Foundation of Henan Province (182300410342); and The Health Commission Technology Talents Overseas Training Project of Henan Province (2018140).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Roundtree IA, Evans ME, Pan T, He C. Dynamic RNA Modifications in Gene Expression Regulation. Cell (2017) 169(7):1187–200. doi: 10.1016/j.cell.2017.05.045
2. Delaunay S, Frye M. RNA Modifications Regulating Cell Fate in Cancer. Nat Cell Biol (2019) 21(5):552–9. doi: 10.1038/s41556-019-0319-0
3. Schwartz S, Agarwala SD, Mumbach MR, Jovanovic M, Mertins P, Shishkin A, et al. High-Resolution Mapping Reveals a Conserved, Widespread, Dynamic mRNA Methylation Program in Yeast Meiosis. Cell (2013) 155(6):1409–21. doi: 10.1016/j.cell.2013.10.047
4. Dominissini D, Moshitch-Moshkovitz S, Schwartz S, Salmon-Divon M, Ungar L, Osenberg S, et al. Topology of the Human and Mouse M6a RNA Methylomes Revealed by M6a-Seq. Nature (2012) 485(7397):201–6. doi: 10.1038/nature11112
5. Meyer KD, Saletore Y, Zumbo P, Elemento O, Mason CE, Jaffrey SR. Comprehensive Analysis of mRNA Methylation Reveals Enrichment in 3’ UTRs and Near Stop Codons. Cell (2012) 149(7):1635–46. doi: 10.1016/j.cell.2012.05.003
6. Dubin DT, Taylor RH. The Methylation State of Poly A-Containing Messenger RNA From Cultured Hamster Cells. Nucleic Acids Res (1975) 2(10):1653–68. doi: 10.1093/nar/2.10.1653
7. Perry RP, Kelley DE, Friderici K, Rottman F. The Methylated Constituents of L Cell Messenger RNA: Evidence for an Unusual Cluster at the 5’ Terminus. Cell (1975) 4(4):387–94. doi: 10.1016/0092-8674(75)90159-2
8. Liu N, Parisien M, Dai Q, Zheng G, He C, Pan T. Probing N6-Methyladenosine RNA Modification Status at Single Nucleotide Resolution in mRNA and Long Noncoding RNA. RNA (2013) 19(12):1848–56. doi: 10.1261/rna.041178.113
9. Ma S, Chen C, Ji X, Liu J, Zhou Q, Wang G, et al. The Interplay Between M6a RNA Methylation and Noncoding RNA in Cancer. J Hematol Oncol (2019) 12(1):121. doi: 10.1186/s13045-019-0805-7
10. Zaccara S, Ries RJ, Jaffrey SR. Reading, Writing and Erasing mRNA Methylation. Nat Rev Mol Cell Biol (2019) 20(10):608–24. doi: 10.1038/s41580-019-0168-5
11. Livneh I, Moshitch-Moshkovitz S, Amariglio N, Rechavi G, Dominissini D. The M6a Epitranscriptome: Transcriptome Plasticity in Brain Development and Function. Nat Rev Neurosci (2020) 21(1):36–51. doi: 10.1038/s41583-019-0244-z
12. Vu LP, Cheng Y, Kharas MG. The Biology of mA RNA Methylation in Normal and Malignant Hematopoiesis. Cancer Discov (2019) 9(1):25–33. doi: 10.1158/2159-8290.CD-18-0959
13. Huang H, Weng H, Chen J. M6a Modification in Coding and Non-Coding RNAs: Roles and Therapeutic Implications in Cancer. Cancer Cell (2020) 37(3):270–88. doi: 10.1016/j.ccell.2020.02.004
14. Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2020. CA Cancer J Clin (2020) 70(1):7–30. doi: 10.3322/caac.21590
15. Cronin KA, Lake AJ, Scott S, Sherman RL, Noone A-M, Howlader N, et al. Annual Report to the Nation on the Status of Cancer, Part I: National Cancer Statistics. Cancer (2018) 124(13):2785–800. doi: 10.1002/cncr.31551
16. Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG. Cancer Drug Resistance: An Evolving Paradigm. Nat Rev Cancer (2013) 13(10):714–26. doi: 10.1038/nrc3599
17. Kalbasi A, Ribas A. Tumour-Intrinsic Resistance to Immune Checkpoint Blockade. Nat Rev Immunol (2020) 20(1):25–39. doi: 10.1038/s41577-019-0218-4
18. Gottesman MM. Mechanisms of Cancer Drug Resistance. Annu Rev Med (2002) 53:615–27. doi: 10.1146/annurev.med.53.082901.103929
19. Wei L, Wang X, Lv L, Liu J, Xing H, Song Y, et al. The Emerging Role of microRNAs and Long Noncoding RNAs in Drug Resistance of Hepatocellular Carcinoma. Mol Cancer (2019) 18(1):147. doi: 10.1186/s12943-019-1086-z
20. Liu J, Harada BT, He C. Regulation of Gene Expression by N6-Methyladenosine in Cancer. Trends Cell Biol (2019) 29(6):487–99. doi: 10.1016/j.tcb.2019.02.008
21. Bokar JA, Rath-Shambaugh ME, Ludwiczak R, Narayan P, Rottman F. Characterization and Partial Purification of mRNA N6-Adenosine Methyltransferase From HeLa Cell Nuclei. Internal mRNA Methylation Requires a Multisubunit Complex. J Biol Chem (1994) 269(26):17697–704. doi: 10.1016/S0021-9258(17)32497-3
22. Bokar JA, Shambaugh ME, Polayes D, Matera AG, Rottman FM. Purification and cDNA Cloning of the AdoMet-Binding Subunit of the Human mRNA (N6-Adenosine)-Methyltransferase. RNA (1997) 3(11):1233–47.
23. Śledź P, Jinek M. Structural Insights Into the Molecular Mechanism of the M(6)A Writer Complex. Elife (2016) 5:e18434. doi: 10.7554/eLife.18434
24. Wang P, Doxtader KA, Nam Y. Structural Basis for Cooperative Function of Mettl3 and Mettl14 Methyltransferases. Mol Cell (2016) 63(2):306–17. doi: 10.1016/j.molcel.2016.05.041
25. Wang X, Feng J, Xue Y, Guan Z, Zhang D, Liu Z, et al. Structural Basis of N(6)-Adenosine Methylation by the METTL3-METTL14 Complex. Nature (2016) 534(7608):575–8. doi: 10.1038/nature18298
26. Ping X-L, Sun B-F, Wang L, Xiao W, Yang X, Wang W-J, et al. Mammalian WTAP Is a Regulatory Subunit of the RNA N6-Methyladenosine Methyltransferase. Cell Res (2014) 24(2):177–89. doi: 10.1038/cr.2014.3
27. Huang H, Weng H, Zhou K, Wu T, Zhao BS, Sun M, et al. Histone H3 Trimethylation at Lysine 36 Guides mA RNA Modification Co-Transcriptionally. Nature (2019) 567(7748):414–9. doi: 10.1038/s41586-019-1016-7
28. Pendleton KE, Chen B, Liu K, Hunter OV, Xie Y, Tu BP, et al. The U6 snRNA mA Methyltransferase METTL16 Regulates SAM Synthetase Intron Retention. Cell (2017) 169(5):824–835.e14. doi: 10.1016/j.cell.2017.05.003
29. Warda AS, Kretschmer J, Hackert P, Lenz C, Urlaub H, Höbartner C, et al. Human METTL16 Is a -Methyladenosine (Ma) Methyltransferase That Targets pre-mRNAs and Various Non-Coding RNAs. EMBO Rep (2017) 18(11):2004–14. doi: 10.15252/embr.201744940
30. Frayling TM, Timpson NJ, Weedon MN, Zeggini E, Freathy RM, Lindgren CM, et al. A Common Variant in the FTO Gene Is Associated With Body Mass Index and Predisposes to Childhood and Adult Obesity. Science (2007) 316(5826):889–94. doi: 10.1126/science.1141634
31. Jia G, Fu Y, Zhao X, Dai Q, Zheng G, Yang Y, et al. N6-Methyladenosine in Nuclear RNA Is a Major Substrate of the Obesity-Associated FTO. Nat Chem Biol (2011) 7(12):885–7. doi: 10.1038/nchembio.687
32. Chen J, Du B. Novel Positioning From Obesity to Cancer: FTO, an mA RNA Demethylase, Regulates Tumour Progression. J Cancer Res Clin Oncol (2019) 145(1):19–29. doi: 10.1007/s00432-018-2796-0
33. Zheng G, Dahl JA, Niu Y, Fedorcsak P, Huang C-M, Li CJ, et al. ALKBH5 Is a Mammalian RNA Demethylase That Impacts RNA Metabolism and Mouse Fertility. Mol Cell (2013) 49(1):18–29. doi: 10.1016/j.molcel.2012.10.015
34. Shi H, Wei J, He C. Where, When, and How: Context-Dependent Functions of RNA Methylation Writers, Readers, and Erasers. Mol Cell (2019) 74(4):640–50. doi: 10.1016/j.molcel.2019.04.025
35. Bartosovic M, Molares HC, Gregorova P, Hrossova D, Kudla G, Vanacova S. N6-Methyladenosine Demethylase FTO Targets pre-mRNAs and Regulates Alternative Splicing and 3’-End Processing. Nucleic Acids Res (2017) 45(19):11356–70. doi: 10.1093/nar/gkx778
36. Roundtree IA, Luo G-Z, Zhang Z, Wang X, Zhou T, Cui Y, et al. YTHDC1 Mediates Nuclear Export of N-Methyladenosine Methylated mRNAs. Elife (2017) 6:e31311. doi: 10.7554/eLife.31311
37. Du H, Zhao Y, He J, Zhang Y, Xi H, Liu M, et al. YTHDF2 Destabilizes M(6)A-Containing RNA Through Direct Recruitment of the CCR4-NOT Deadenylase Complex. Nat Commun (2016) 7:12626. doi: 10.1038/ncomms12626
38. Wang X, Zhao BS, Roundtree IA, Lu Z, Han D, Ma H, et al. N(6)-Methyladenosine Modulates Messenger RNA Translation Efficiency. Cell (2015) 161(6):1388–99. doi: 10.1016/j.cell.2015.05.014
39. Zhao X, Yang Y, Sun BF, Shi Y, Yang X, Xiao W, et al. FTO-Dependent Demethylation of N6-Methyladenosine Regulates mRNA Splicing and Is Required for Adipogenesis. Cell Res (2014) 24(12):1403–19. doi: 10.1038/cr.2014.151
40. Xiao W, Adhikari S, Dahal U, Chen YS, Hao YJ, Sun BF, et al. Nuclear M(6)A Reader YTHDC1 Regulates mRNA Splicing. Mol Cell (2016) 61(4):507–19. doi: 10.1016/j.molcel.2016.01.012
41. Roundtree IA, Luo GZ, Zhang ZJ, Wang X, Zhou T, Cui YQ, et al. YTHDC1 Mediates Nuclear Export of N-6 - Methyladenosine Methylated mRNAs. Elife (2017) 6:28. doi: 10.7554/eLife.31311
42. Liao S, Sun H, Xu C. YTH Domain: A Family of N-6-Methyladenosine (M(6)A) Readers. Genom Proteom Bioinform (2018) 16(2):99–107. doi: 10.1016/j.gpb.2018.04.002
43. Meyer KD, Patil DP, Zhou J, Zinoviev A, Skabkin MA, Elemento O, et al. 5 ’ UTR M(6)A Promotes Cap-Independent Translation. Cell (2015) 163(4):999–1010. doi: 10.1016/j.cell.2015.10.012
44. Wang X, Lu Z, Gomez A, Hon GC, Yue Y, Han D, et al. N6-Methyladenosine-Dependent Regulation of Messenger RNA Stability. Nature (2014) 505(7481):117–20. doi: 10.1038/nature12730
45. Mauer J, Luo X, Blanjoie A, Jiao X, Grozhik AV, Patil DP, et al. Reversible Methylation of mA in the 5’ Cap Controls mRNA Stability. Nature (2017) 541(7637):371–5. doi: 10.1038/nature21022
46. Shi HL, Wang X, Lu ZK, Zhao BXS, Ma HH, Hsu PJ, et al. YTHDF3 Facilitates Translation and Decay of N-6-Methyladenosine-Modified RNA. Cell Res (2017) 27(3):315–28. doi: 10.1038/cr.2017.15
47. Huang HL, Weng HY, Sun WJ, Qin X, Shi HL, Wu HZ, et al. Recognition of RNA N-6- Methyladenosine by IGF2BP Proteins Enhances mRNA Stability and Translation. Nat Cell Biol (2018) 20(3):285–95. doi: 10.1038/s41556-018-0045-z
48. Alarcon CR, Goodarzi H, Lee H, Liu X, Tavazoie S, Tavazoie SF. HNRNPA2B1 Is a Mediator of M(6)A-Dependent Nuclear RNA Processing Events. Cell (2015) 162(6):1299–308. doi: 10.1016/j.cell.2015.08.011
49. Huang Y, Su R, Sheng Y, Dong L, Dong Z, Xu H, et al. Small-Molecule Targeting of Oncogenic FTO Demethylase in Acute Myeloid Leukemia. Cancer Cell (2019) 35(4):677–91. doi: 10.1016/j.ccell.2019.03.006
50. Li Z, Weng H, Su R, Weng X, Zuo Z, Li C, et al. FTO Plays an Oncogenic Role in Acute Myeloid Leukemia as a N-Methyladenosine RNA Demethylase. Cancer Cell (2017) 31(1):127–41. doi: 10.1016/j.ccell.2016.11.017
51. Chen M, Wei L, Law C-T, Tsang FH-C, Shen J, Cheng CL-H, et al. RNA N6-Methyladenosine Methyltransferase-Like 3 Promotes Liver Cancer Progression Through YTHDF2-Dependent Posttranscriptional Silencing of SOCS2. Hepatology (2018) 67(6):2254–70. doi: 10.1002/hep.29683
52. Jin D, Guo J, Wu Y, Du J, Yang L, Wang X, et al. M6a mRNA Methylation Initiated by METTL3 Directly Promotes YAP Translation and Increases YAP Activity by Regulating the MALAT1-miR-1914-3p-YAP Axis to Induce NSCLC Drug Resistance and Metastasis. J Hematol Oncol (2019) 12(1):135. doi: 10.1186/s13045-019-0830-6
53. Wang Q, Chen C, Ding Q, Zhao Y, Wang Z, Chen J, et al. METTL3-Mediated mA Modification of HDGF mRNA Promotes Gastric Cancer Progression and has Prognostic Significance. Gut (2020) 69(7):1193–205. doi: 10.1136/gutjnl-2019-319639
54. Li T, Hu P-S, Zuo Z, Lin J-F, Li X, Wu Q-N, et al. METTL3 Facilitates Tumor Progression via an mA-IGF2BP2-Dependent Mechanism in Colorectal Carcinoma. Mol Cancer (2019) 18(1):112. doi: 10.1186/s12943-019-1038-7
55. Han J, Wang J-Z, Yang X, Yu H, Zhou R, Lu H-C, et al. METTL3 Promote Tumor Proliferation of Bladder Cancer by Accelerating Pri-Mir221/222 Maturation in M6a-Dependent Manner. Mol Cancer (2019) 18(1):110. doi: 10.1186/s12943-019-1036-9
56. Brown R, Curry E, Magnani L, Wilhelm-Benartzi CS, Borley J. Poised Epigenetic States and Acquired Drug Resistance in Cancer. Nat Rev Cancer (2014) 14(11):747–53. doi: 10.1038/nrc3819
57. Fukumoto T, Zhu H, Nacarelli T, Karakashev S, Fatkhutdinov N, Wu S, et al. N6-Methylation of Adenosine of FZD10 mRNA Contributes to PARP Inhibitor Resistance. Cancer Res (2019) 79(11):2812–20. doi: 10.1158/0008-5472.CAN-18-3592
58. Yan F, Al-Kali A, Zhang Z, Liu J, Pang J, Zhao N, et al. A Dynamic N6-Methyladenosine Methylome Regulates Intrinsic and Acquired Resistance to Tyrosine Kinase Inhibitors. Cell Res (2018) 28(11):1062–76. doi: 10.1038/s41422-018-0097-4
59. Chen Z, Wu L, Zhou J, Lin X, Peng Y, Ge L, et al. N6-Methyladenosine-Induced Errγ Triggers Chemoresistance of Cancer Cells Through Upregulation of ABCB1 and Metabolic Reprogramming. Theranostics (2020) 10(8):3382–96. doi: 10.7150/thno.40144
60. Shriwas O, Priyadarshini M, Samal SK, Rath R, Panda S, Das Majumdar SK, et al. DDX3 Modulates Cisplatin Resistance in OSCC Through ALKBH5-Mediated mA-Demethylation of FOXM1 and NANOG. Apoptosis (2020) 25(3-4):233–46. doi: 10.1007/s10495-020-01591-8
61. Wang Y, Zeng L, Liang C, Zan R, Ji W, Zhang Z, et al. Integrated Analysis of Transcriptome-Wide mA Methylome of Osteosarcoma Stem Cells Enriched by Chemotherapy. Epigenomics (2019) 11(15):1693–715. doi: 10.2217/epi-2019-0262
62. Tang B, Yang Y, Kang M, Wang Y, Wang Y, Bi Y, et al. mA Demethylase ALKBH5 Inhibits Pancreatic Cancer Tumorigenesis by Decreasing WIF-1 RNA Methylation and Mediating Wnt Signaling. Mol Cancer (2020) 19(1):3. doi: 10.1186/s12943-019-1128-6
63. Zhou S, Bai Z-L, Xia D, Zhao Z-J, Zhao R, Wang Y-Y, et al. FTO Regulates the Chemo-Radiotherapy Resistance of Cervical Squamous Cell Carcinoma (CSCC) by Targeting β-Catenin Through mRNA Demethylation. Mol Carcinog (2018) 57(5):590–7. doi: 10.1002/mc.22782
64. Klinge CM, Piell KM, Tooley CS, Rouchka EC. HNRNPA2/B1 Is Upregulated in Endocrine-Resistant LCC9 Breast Cancer Cells and Alters the miRNA Transcriptome When Overexpressed in MCF-7 Cells. Sci Rep (2019) 9:22. doi: 10.1038/s41598-019-45636-8
65. Zhu L, Zhu Y, Han S, Chen M, Song P, Dai D, et al. Impaired Autophagic Degradation of lncRNA ARHGAP5-AS1 Promotes Chemoresistance in Gastric Cancer. Cell Death Dis (2019) 10(6):383. doi: 10.1038/s41419-019-1585-2
66. Zhang R, Li S-W, Liu L, Yang J, Huang G, Sang Y. TRIM11 Facilitates Chemoresistance in Nasopharyngeal Carcinoma by Activating the β-Catenin/ABCC9 Axis via P62-Selective Autophagic Degradation of Daple. Oncogenesis (2020) 9(5):45. doi: 10.1038/s41389-020-0229-9
67. Wei J, Yin Y, Zhou J, Chen H, Peng J, Yang J, et al. METTL3 Potentiates Resistance to Cisplatin Through M(6)A Modification of TFAP2C in Seminoma. J Cell Mol Med (2020) 24(19):11366–80. doi: 10.1111/jcmm.15738
68. Uddin MB, Roy KR, Hosain SB, Khiste SK, Hill RA, Jois SD, et al. An N-Methyladenosine at the Transited Codon 273 of P53 pre-mRNA Promotes the Expression of R273H Mutant Protein and Drug Resistance of Cancer Cells. Biochem Pharmacol (2019) 160:134–45. doi: 10.1016/j.bcp.2018.12.014
69. Zhang Y, Kang M, Zhang B, Meng F, Song J, Kaneko H, et al. M6a Modification-Mediated CBX8 Induction Regulates Stemness and Chemosensitivity of Colon Cancer via Upregulation of LGR5. Mol Cancer (2019) 18(1):185. doi: 10.1186/s12943-019-1116-x
70. Lin Z, Niu Y, Wan A, Chen D, Liang H, Chen X, et al. RNA M6a Methylation Regulates Sorafenib Resistance in Liver Cancer Through FOXO3-Mediated Autophagy. EMBO J (2020) 39(12):e103181. doi: 10.15252/embj.2019103181
71. Hao L, Wang J-M, Liu B-Q, Yan J, Li C, Jiang J-Y, et al. M6a-YTHDF1-Mediated TRIM29 Upregulation Facilitates the Stem Cell-Like Phenotype of Cisplatin-Resistant Ovarian Cancer Cells. Biochim Biophys Acta Mol Cell Res (2020) 1868(1):118878–8. doi: 10.1016/j.bbamcr.2020.118878
72. Shi Y, Fan S, Wu M, Zuo Z, Li X, Jiang L, et al. YTHDF1 Links Hypoxia Adaptation and Non-Small Cell Lung Cancer Progression. Nat Commun (2019) 10(1):4892. doi: 10.1038/s41467-019-12801-6
73. Yang S, Wei J, Cui Y-H, Park G, Shah P, Deng Y, et al. M6a mRNA Demethylase FTO Regulates Melanoma Tumorigenicity and Response to Anti-PD-1 Blockade. Nat Commun (2019) 10(1):2782. doi: 10.1038/s41467-019-10669-0
74. Gu C, Wang Z, Zhou N, Li G, Kou Y, Luo Y, et al. Mettl14 Inhibits Bladder TIC Self-Renewal and Bladder Tumorigenesis Through N-Methyladenosine of Notch1. Mol Cancer (2019) 18(1):168. doi: 10.1186/s12943-019-1084-1
75. Barbieri I, Tzelepis K, Pandolfini L, Shi J, Millán-Zambrano G, Robson SC, et al. Promoter-Bound METTL3 Maintains Myeloid Leukaemia by mA-Dependent Translation Control. Nature (2017) 552(7683):126–31. doi: 10.1038/nature24678
76. Chen X-Y, Zhang J, Zhu J-S. The Role of mA RNA Methylation in Human Cancer. Mol Cancer (2019) 18(1):103. doi: 10.1186/s12943-019-1033-z
77. Miao W, Chen J, Jia L, Ma J, Song D. The M6a Methyltransferase METTL3 Promotes Osteosarcoma Progression by Regulating the M6a Level of LEF1. Biochem Biophys Res Commun (2019) 516(3):719–25. doi: 10.1016/j.bbrc.2019.06.128
78. Yu X, Zhao H, Cao Z. The M6a Methyltransferase METTL3 Aggravates the Progression of Nasopharyngeal Carcinoma Through Inducing EMT by M6a-Modified Snail mRNA. Minerva Med (2020). doi: 10.23736/S0026-4806.20.06653-7
79. Meng QZ, Cong CH, Li XJ, Zhu F, Zhao X, Chen FW. METTL3 Promotes the Progression of Nasopharyngeal Carcinoma Through Mediating M6A Modification of EZH2. Eur Rev Med Pharmacol Sci (2020) 24(8):4328–36. doi: 10.26355/eurrev_202004_21014
80. Carter TA, Wodicka LM, Shah NP, Velasco AM, Fabian MA, Treiber DK, et al. Inhibition of Drug-Resistant Mutants of ABL, KIT, and EGF Receptor Kinases. Proc Natl Acad Sci USA (2005) 102(31):11011–6. doi: 10.1073/pnas.0504952102
81. O’Hare T, Shakespeare WC, Zhu X, Eide CA, Rivera VM, Wang F, et al. AP24534, A Pan-BCR-ABL Inhibitor for Chronic Myeloid Leukemia, Potently Inhibits the T315I Mutant and Overcomes Mutation-Based Resistance. Cancer Cell (2009) 16(5):401–12. doi: 10.1016/j.ccr.2009.09.028
82. Liu S, Li G, Li Q, Zhang Q, Zhuo L, Chen X, et al. The Roles and Mechanisms of YTH Domain-Containing Proteins in Cancer Development and Progression. Am J Cancer Res (2020) 10(4):1068–84.
83. Liu T, Wei Q, Jin J, Luo Q, Liu Y, Yang Y, et al. The M6a Reader YTHDF1 Promotes Ovarian Cancer Progression via Augmenting EIF3C Translation. Nucleic Acids Res (2020) 48(7):3816–31. doi: 10.1093/nar/gkaa048
84. Bai Y, Yang C, Wu R, Huang L, Song S, Li W, et al. YTHDF1 Regulates Tumorigenicity and Cancer Stem Cell-Like Activity in Human Colorectal Carcinoma. Front Oncol (2019) 9:332. doi: 10.3389/fonc.2019.00332
85. Dean M, Fojo T, Bates S. Tumour Stem Cells and Drug Resistance. Nat Rev Cancer (2005) 5(4):275–84. doi: 10.1038/nrc1590
86. Li Z, Peng Y, Li J, Chen Z, Chen F, Tu J, et al. N6-Methyladenosine Regulates Glycolysis of Cancer Cells Through PDK4. Nat Commun (2020) 11(1):2578. doi: 10.1038/s41467-020-16306-5
87. Benjamin D, Moroni C. mRNA Stability and Cancer: An Emerging Link? Expert Opin Biol Ther (2007) 7(10):1515–29. doi: 10.1517/14712598.7.10.1515
88. Tahmasebi S, Khoutorsky A, Mathews MB, Sonenberg N. Translation Deregulation in Human Disease. Nat Rev Mol Cell Biol (2018) 19(12):791–807. doi: 10.1038/s41580-018-0034-x
89. Bhat M, Robichaud N, Hulea L, Sonenberg N, Pelletier J, Topisirovic I. Targeting the Translation Machinery in Cancer. Nat Rev Drug Discov (2015) 14(4):261–78. doi: 10.1038/nrd4505
90. Fabbri L, Chakraborty A, Robert C, Vagner S. The Plasticity of mRNA Translation During Cancer Progression and Therapy Resistance. Nat Rev Cancer (2021) 21(9):558–77. doi: 10.1038/s41568-021-00380-y
91. Fitzgerald DM, Hastings PJ, Rosenberg SM. Stress-Induced Mutagenesis: Implications in Cancer and Drug Resistance. Annu Rev Cancer Biol (2017) 1:119–40. doi: 10.1146/annurev-cancerbio-050216-121919
92. Tian J, Ying P, Ke J, Zhu Y, Yang Y, Gong Y, et al. ANKLE1 N6-Methyladenosine-Related Variant Is Associated With Colorectal Cancer Risk by Maintaining the Genomic Stability. Int J Cancer (2020) 146(12):3281–93. doi: 10.1002/ijc.32677
93. Bykov VJN, Eriksson SE, Bianchi J, Wiman KG. Targeting Mutant P53 for Efficient Cancer Therapy. Nat Rev Cancer (2018) 18(2):89–102. doi: 10.1038/nrc.2017.109
94. Chan JJ, Tay Y. Noncoding RNA:RNA Regulatory Networks in Cancer. Int J Mol Sci (2018) 19(5):1310. doi: 10.3390/ijms19051310
95. George J, Patel T. Noncoding RNA as Therapeutic Targets for Hepatocellular Carcinoma. Semin Liver Dis (2015) 35(1):63–74. doi: 10.1055/s-0034-1397350
96. Meng FY, Henson R, Wehbe-Janek H, Ghoshal K, Jacob ST, Patel T. MicroRNA-21 Regulates Expression of the PTEN Tumor Suppressor Gene in Human Hepatocellular Cancer. Gastroenterology (2007) 133(2):647–58. doi: 10.1053/j.gastro.2007.05.022
97. Wang YY, He L, Du Y, Zhu PP, Huang GL, Luo JJ, et al. The Long Noncoding RNA IncTCF7 Promotes Self-Renewal of Human Liver Cancer Stem Cells Through Activation of Wnt Signaling. Cell Stem Cell (2015) 16(4):413–25. doi: 10.1016/j.stem.2015.03.003
98. Han D, Li JX, Wang HM, Su XP, Hou J, Gu Y, et al. Circular RNA Circmto1 Acts as the Sponge of MicroRNA-9 to Suppress Hepatocellular Carcinoma Progression. Hepatology (2017) 66(4):1151–64. doi: 10.1002/hep.29270
99. Bhat AA, Younes SN, Raza SS, Zarif L, Nisar S, Ahmed I, et al. Role of Non-Coding RNA Networks in Leukemia Progression, Metastasis and Drug Resistance. Mol Cancer (2020) 19(1):21. doi: 10.1186/s12943-020-01175-9
100. Wei L, Wang XW, Lv LY, Zheng Y, Zhang NS, Yang M. The Emerging Role of Noncoding RNAs in Colorectal Cancer Chemoresistance. Cell Oncol (2019) 42(6):757–68. doi: 10.1007/s13402-019-00466-8
101. Wei L, Sun JJ, Zhang NS, Zheng Y, Wang XW, Lv LY, et al. Noncoding RNAs in Gastric Cancer: Implications for Drug Resistance. Mol Cancer (2020) 19(1):62. doi: 10.1186/s12943-020-01185-7
102. Yue B, Cai DL, Liu CC, Fang CY, Yan DW. Linc00152 Functions as a Competing Endogenous RNA to Confer Oxaliplatin Resistance and Holds Prognostic Values in Colon Cancer. Mol Ther (2016) 24(12):2064–77. doi: 10.1038/mt.2016.180
103. Yu PN, Yan MD, Lai HC, Huang RL, Chou YC, Lin WC, et al. Downregulation of miR-29 Contributes to Cisplatin Resistance of Ovarian Cancer Cells. Int J Cancer (2014) 134(3):542–51. doi: 10.1002/ijc.28399
104. Fang Q, Chen XY, Zhi XT. Long Non-Coding RNA (LncRNA) Urothelial Carcinoma Associated 1 (UCA1) Increases Multi-Drug Resistance of Gastric Cancer via Downregulating miR-27b. Med Sci Monit (2016) 22:8. doi: 10.12659/msm.900688
105. Bartel DP. MicroRNAs: Target Recognition and Regulatory Functions. Cell (2009) 136(2):215–33. doi: 10.1016/j.cell.2009.01.002
106. Si W, Shen J, Zheng H, Fan W. The Role and Mechanisms of Action of microRNAs in Cancer Drug Resistance. Clin Epigenet (2019) 11(1):25. doi: 10.1186/s13148-018-0587-8
107. Liu Y-N, Tsai M-F, Wu S-G, Chang T-H, Tsai T-H, Gow C-H, et al. miR-146b-5p Enhances the Sensitivity of NSCLC to EGFR Tyrosine Kinase Inhibitors by Regulating the IRAK1/NF-κb Pathway. Mol Ther Nucleic Acids (2020) 22:471–83. doi: 10.1016/j.omtn.2020.09.015
108. Li P, Luo X, Xie Y, Li P, Hu F, Chu J, et al. GC-Derived EVs Enriched With MicroRNA-675-3p Contribute to the MAPK/PD-L1-Mediated Tumor Immune Escape by Targeting Cxxc4. Mol Ther Nucleic Acids (2020) 22:615–26. doi: 10.1016/j.omtn.2020.08.020
109. Han JJ, Lee Y, Yeom KH, Kim YK, Jin H, Kim VN. The Drosha-DGCR8 Complex in Primary microRNA Processing. Genes Dev (2004) 18(24):3016–27. doi: 10.1101/gad.1262504
110. Alarcon CR, Lee H, Goodarzi H, Halberg N, Tavazoie SF. N6-Methyladenosine Marks Primary microRNAs for Processing. Nature (2015) 519(7544):482–5. doi: 10.1038/nature14281
111. Park YM, Hwang SJ, Masuda K, Choi KM, Jeong MR, Nam DH, et al. Heterogeneous Nuclear Ribonucleoprotein C1/C2 Controls the Metastatic Potential of Glioblastoma by Regulating Pdcd4. Mol Cell Biol (2012) 32(20):4237–44. doi: 10.1128/mcb.00443-12
112. Cui X, Wang Z, Li J, Zhu J, Ren Z, Zhang D, et al. Cross Talk Between RNA N6-Methyladenosine Methyltransferase-Like 3 and miR-186 Regulates Hepatoblastoma Progression Through Wnt/β-Catenin Signalling Pathway. Cell Prolif (2020) 53(3):e12768. doi: 10.1111/cpr.12768
113. Yi D, Wang R, Shi X, Xu L, Yilihamu Ye, Sang J. METTL14 Promotes the Migration and Invasion of Breast Cancer Cells by Modulating N6−methyladenosine and Hsa−Mir−146a−5p Expression. Oncol Rep (2020) 43(5):1375–86. doi: 10.3892/or.2020.7515
114. Zhang BY, Han L, Tang YF, Zhang GX, Fan XL, Zhang JJ, et al. METTL14 Regulates M6A Methylation-Modified Primary miR-19a to Promote Cardiovascular Endothelial Cell Proliferation and Invasion. Eur Rev Med Pharmacol Sci (2020) 24(12):7015–23. doi: 10.26355/eurrev_202006_21694
115. Ma JZ, Yang F, Zhou CC, Liu F, Yuan JH, Wang F, et al. METTL14 Suppresses the Metastatic Potential of Hepatocellular Carcinoma by Modulating N-6-Methyladenosine- Dependent Primary MicroRNA Processing. Hepatology (2017) 65(2):529–43. doi: 10.1002/hep.28885
116. Chen H, Liu H, Qing G. Targeting Oncogenic Myc as a Strategy for Cancer Treatment. Signal Transduct Target Ther (2018) 3:5. doi: 10.1038/s41392-018-0008-7
117. Xiao L, Li X, Mu Z, Zhou J, Zhou P, Xie C, et al. FTO Inhibition Enhances the Antitumor Effect of Temozolomide by Targeting MYC-miR-155/23a Cluster-MXI1 Feedback Circuit in Glioma. Cancer Res (2020) 80(18):3945–58. doi: 10.1158/0008-5472.Can-20-0132
118. Ponting CP, Oliver PL, Reik W. Evolution and Functions of Long Noncoding RNAs. Cell (2009) 136(4):629–41. doi: 10.1016/j.cell.2009.02.006
119. Wang KC, Chang HY. Molecular Mechanisms of Long Noncoding RNAs. Mol Cell (2011) 43(6):904–14. doi: 10.1016/j.molcel.2011.08.018
120. Xiong G, Liu C, Yang G, Feng M, Xu J, Zhao F, et al. Long Noncoding RNA GSTM3TV2 Upregulates LAT2 and OLR1 by Competitively Sponging Let-7 to Promote Gemcitabine Resistance in Pancreatic Cancer. J Hematol Oncol (2019) 12(1):97. doi: 10.1186/s13045-019-0777-7
121. Wang J, Xie S, Yang J, Xiong H, Jia Y, Zhou Y, et al. The Long Noncoding RNA H19 Promotes Tamoxifen Resistance in Breast Cancer via Autophagy. J Hematol Oncol (2019) 12(1):81. doi: 10.1186/s13045-019-0747-0
122. Batista PJ, Molinie B, Wang JK, Qu K, Zhang JJ, Li LJ, et al. M(6)A RNA Modification Controls Cell Fate Transition in Mammalian Embryonic Stem Cells. Cell Stem Cell (2014) 15(6):707–19. doi: 10.1016/j.stem.2014.09.019
123. Liu N, Dai Q, Zheng G, He C, Parisien M, Pan T. N-6-Methyladenosine-Dependent RNA Structural Switches Regulate RNA-Protein Interactions. Nature (2015) 518(7540):560–4. doi: 10.1038/nature14234
124. Yang D, Qiao J, Wang G, Lan Y, Li G, Guo X, et al. N-6-Methyladenosine Modification of lincRNA 1281 Is Critically Required for mESC Differentiation Potential. Nucleic Acids Res (2018) 46(8):3906–20. doi: 10.1093/nar/gky130
125. Gutschner T, Haemmerle M, Diederichs S. MALAT1- a Paradigm for Long Noncoding RNA Function in Cancer. J Mol Med (2013) 91(7):791–801. doi: 10.1007/s00109-013-1028-y
126. Schmidt LH, Spieker T, Koschmieder S, Humberg J, Jungen D, Bulk E, et al. The Long Noncoding MALAT-1 RNA Indicates a Poor Prognosis in Non-Small Cell Lung Cancer and Induces Migration and Tumor Growth. J Thorac Oncol (2011) 6(12):1984–92. doi: 10.1097/JTO.0b013e3182307eac
127. Zhu L, Liu J, Ma S, Zhang S. Long Noncoding RNA MALAT-1 Can Predict Metastasis and a Poor Prognosis: A Meta-Analysis. Pathol Oncol Res (2015) 21(4):1259–64. doi: 10.1007/s12253-015-9960-5
128. Zhang S, Zhao BS, Zhou A, Lin K, Zheng S, Lu Z, et al. M(6)A Demethylase ALKBH5 Maintains Tumorigenicity of Glioblastoma Stem-Like Cells by Sustaining FOXM1 Expression and Cell Proliferation Program. Cancer Cell (2017) 31(4):591–606.e6. doi: 10.1016/j.ccell.2017.02.013
129. Meng S, Zhou H, Feng Z, Xu Z, Tang Y, Li P, et al. CircRNA: Functions and Properties of a Novel Potential Biomarker for Cancer. Mol Cancer (2017) 16(1):94. doi: 10.1186/s12943-017-0663-2
130. Xu T, Wang M, Jiang L, Ma L, Wan L, Chen Q, et al. CircRNAs in Anticancer Drug Resistance: Recent Advances and Future Potential. Mol Cancer (2020) 19(1):127. doi: 10.1186/s12943-020-01240-3
131. Peng L, Sang H, Wei S, Li Y, Jin D, Zhu X, et al. Circcul2 Regulates Gastric Cancer Malignant Transformation and Cisplatin Resistance by Modulating Autophagy Activation via miR-142-3p/ROCK2. Mol Cancer (2020) 19(1):156. doi: 10.1186/s12943-020-01270-x
132. Wei Y, Lu C, Zhou P, Zhao L, Lyu X, Yin J, et al. EIF4A3-Induced Circular RNA ASAP1(circASAP1) Promotes Tumorigenesis and Temozolomide Resistance of Glioblastoma via NRAS/MEK1/ERK1/2 Signaling. Neuro Oncol (2020) 23(4):611–24. doi: 10.1093/neuonc/noaa214
133. Zhou C, Molinie B, Daneshvar K, Pondick JV, Wang J, Van Wittenberghe N, et al. Genome-Wide Maps of M6a circRNAs Identify Widespread and Cell-Type-Specific Methylation Patterns That Are Distinct From mRNAs. Cell Rep (2017) 20(9):2262–76. doi: 10.1016/j.celrep.2017.08.027
134. Zhang L, Hou C, Chen C, Guo Y, Yuan W, Yin D, et al. The Role of N-6-Methyladenosine (M(6)A) Modification in the Regulation of circRNAs. Mol Cancer (2020) 19(1):105. doi: 10.1186/s12943-020-01224-3
135. Di Timoteo G, Dattilo D, Centron-Broco A, Colantoni A, Guarnacci M, Rossi F, et al. Modulation of circRNA Metabolism by M(6)A Modification. Cell Rep (2020) 31(6):107641. doi: 10.1016/j.celrep.2020.107641
136. Huang C, Liang DM, Tatomer DC, Wilusz JE. A Length-Dependent Evolutionarily Conserved Pathway Controls Nuclear Export of Circular RNAs. Genes Dev (2018) 32(9-10):639–44. doi: 10.1101/gad.314856.118
137. Chen RX, Chen X, Xia LP, Zhang JX, Pan ZZ, Ma XD, et al. N-6-Methyladenosine Modification of Circnsun2 Facilitates Cytoplasmic Export and Stabilizes HMGA2 to Promote Colorectal Liver Metastasis. Nat Commun (2019) 10:15. doi: 10.1038/s41467-019-12651-2
138. Park OH, Ha H, Lee Y, Boo SH, Kwon DH, Song HK, et al. Endoribonucleolytic Cleavage of M(6)A-Containing RNAs by RNase P/MRP Complex. Mol Cell (2019) 74(3):494–507.e8. doi: 10.1016/j.molcel.2019.02.034
139. Zhang M, Huang N, Yang X, Luo J, Yan S, Xiao F, et al. A Novel Protein Encoded by the Circular Form of the SHPRH Gene Suppresses Glioma Tumorigenesis. Oncogene (2018) 37(13):1805–14. doi: 10.1038/s41388-017-0019-9
140. Granados-Riveron JT, Aquino-Jarquin G. The Complexity of the Translation Ability of circRNAs. Biochim Biophys Acta Gene Regul Mech (2016) 1859(10):1245–51. doi: 10.1016/j.bbagrm.2016.07.009
141. Yang Y, Fan X, Mao M, Song X, Wu P, Zhang Y, et al. Extensive Translation of Circular RNAs Driven by N-6-Methyladenosine. Cell Res (2017) 27(5):626–41. doi: 10.1038/cr.2017.31
142. Zha L, Zhang J, Tang W, Zhang N, He M, Guo Y, et al. HMGA2 Elicits EMT by Activating the Wnt/beta-Catenin Pathway in Gastric Cancer. Dig Dis Sci (2013) 58(3):724–33. doi: 10.1007/s10620-012-2399-6
143. Wang X, Liu X, Li AY-J, Chen L, Lai L, Lin HH, et al. Overexpression of HMGA2 Promotes Metastasis and Impacts Survival of Colorectal Cancers. Clin Cancer Res (2011) 17(8):2570–80. doi: 10.1158/1078-0432.Ccr-10-2542
144. Zhao J, Lee EE, Kim J, Yang R, Chamseddin B, Ni C, et al. Transforming Activity of an Oncoprotein-Encoding Circular RNA From Human Papillomavirus. Nat Commun (2019) 10(1):2300. doi: 10.1038/s41467-019-10246-5
145. Xu J, Ji L, Liang Y, Wan Z, Zheng W, Song X, et al. CircRNA-SORE Mediates Sorafenib Resistance in Hepatocellular Carcinoma by Stabilizing YBX1. Signal Transduct Target Ther (2020) 5(1):298. doi: 10.1038/s41392-020-00375-5
146. Xu J, Wan Z, Tang M, Lin Z, Jiang S, Ji L, et al. N6-Methyladenosine-Modified CircRNA-SORE Sustains Sorafenib Resistance in Hepatocellular Carcinoma by Regulating β-Catenin Signaling. Mol Cancer (2020) 19(1):163. doi: 10.1186/s12943-020-01281-8
147. Kartal-Yandim M, Adan-Gokbulut A, Baran Y. Molecular Mechanisms of Drug Resistance and Its Reversal in Cancer. Crit Rev Biotechnol (2016) 36(4):716–26. doi: 10.3109/07388551.2015.1015957
148. Drost J, Clevers H. Organoids in Cancer Research. Nat Rev Cancer (2018) 18(7):407–18. doi: 10.1038/s41568-018-0007-6
149. Kong J, Lee H, Kim D, Han SK, Ha D, Shin K, et al. Network-Based Machine Learning in Colorectal and Bladder Organoid Models Predicts Anti-Cancer Drug Efficacy in Patients. Nat Commun (2020) 11(1):5485. doi: 10.1038/s41467-020-19313-8
150. Tiriac H, Belleau P, Engle DD, Plenker D, Deschênes A, Somerville TDD, et al. Organoid Profiling Identifies Common Responders to Chemotherapy in Pancreatic Cancer. Cancer Discov (2018) 8(9):1112–29. doi: 10.1158/2159-8290.CD-18-0349
151. Mishima E, Jinno D, Akiyama Y, Itoh K, Nankumo S, Shima H, et al. Immuno-Northern Blotting: Detection of RNA Modifications by Using Antibodies Against Modified Nucleosides. PloS One (2015) 10(11):e0143756. doi: 10.1371/journal.pone.0143756
152. Sun Y, Li S, Yu W, Zhao Z, Gao J, Chen C, et al. N6-Methyladenosine-Dependent pri-miR-17-92 Maturation Suppresses PTEN/TMEM127 and Promotes Sensitivity to Everolimus in Gastric Cancer. Cell Death Dis (2020) 11(10):836. doi: 10.1038/s41419-020-03049-w
153. Herr CQ, Hausinger RP. Amazing Diversity in Biochemical Roles of Fe(II)/2-Oxoglutarate Oxygenases. Trends Biochem Sci (2018) 43(7):517–32. doi: 10.1016/j.tibs.2018.04.002
154. Toh JDW, Sun L, Lau LZM, Tan J, Low JJA, Tang CWQ, et al. A Strategy Based on Nucleotide Specificity Leads to a Subfamily-Selective and Cell-Active Inhibitor of -Methyladenosine Demethylase FTO. Chem Sci (2015) 6(1):112–22. doi: 10.1039/c4sc02554g
155. Qiu X, Yang S, Wang S, Wu J, Zheng B, Wang K, et al. M6A Demethylase ALKBH5 Regulates PD-L1 Expression and Tumor Immunoenvironment in Intrahepatic Cholangiocarcinoma. Cancer Res (2021) 81(18):4778–93. doi: 10.1158/0008-5472.CAN-21-0468
156. Tsuchiya K, Yoshimura K, Inoue Y, Iwashita Y, Yamada H, Kawase A, et al. YTHDF1 and YTHDF2 Are Associated With Better Patient Survival and an Inflamed Tumor-Immune Microenvironment in Non-Small-Cell Lung Cancer. Oncoimmunology (2021) 10(1):1962656. doi: 10.1080/2162402X.2021.1962656
157. Han D, Liu J, Chen C, Dong L, Liu Y, Chang R, et al. Anti-Tumour Immunity Controlled Through mRNA mA Methylation and YTHDF1 in Dendritic Cells. Nature (2019) 566(7743):270–4. doi: 10.1038/s41586-019-0916-x
158. Liu Z, Wang T, She Y, Wu K, Gu S, Li L, et al. N-Methyladenosine-Modified Circigf2bp3 Inhibits CD8 T-Cell Responses to Facilitate Tumor Immune Evasion by Promoting the Deubiquitination of PD-L1 in Non-Small Cell Lung Cancer. Mol Cancer (2021) 20(1):105. doi: 10.1186/s12943-021-01398-4
Keywords: cancer drug resistance, m6A modification, miRNA, lncRNA, circRNA
Citation: Chen C, Guo Y, Guo Y, Wu X, Si C, Xu Y, Kang Q and Sun Z (2021) m6A Modification in Non-Coding RNA: The Role in Cancer Drug Resistance. Front. Oncol. 11:746789. doi: 10.3389/fonc.2021.746789
Received: 24 July 2021; Accepted: 27 September 2021;
Published: 20 October 2021.
Edited by:
Tanmoy Mondal, University of Gothenburg, SwedenReviewed by:
Wei Yang, Southern Medical University, ChinaCopyright © 2021 Chen, Guo, Guo, Wu, Si, Xu, Kang and Sun. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhenqiang Sun, ZmNjc3VuenFAenp1LmVkdS5jbg==; Qiaozhen Kang, cXprYW5nQHp6dS5lZHUuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.