 Juan Zhang
Juan Zhang Xuefeng Gao
Xuefeng Gao Li Yu*
Li Yu*- Department of Hematology and Oncology, International Cancer Center, Shenzhen Key Laboratory of Precision Medicine for Hematological Malignancies, Shenzhen University General Hospital, Shenzhen University Clinical Medical Academy, Shenzhen University Health Science Center, Shenzhen, China
Accurate orchestration of gene expression is critical for the process of normal hematopoiesis, and dysregulation is closely associated with leukemogenesis. Epigenetic aberration is one of the major causes contributing to acute myeloid leukemia (AML), where chromosomal rearrangements are frequently found. Increasing evidences have shown the pivotal roles of histone deacetylases (HDACs) in chromatin remodeling, which are involved in stemness maintenance, cell fate determination, proliferation and differentiation, via mastering the transcriptional switch of key genes. In abnormal, these functions can be bloomed to elicit carcinogenesis. Presently, HDAC family members are appealing targets for drug exploration, many of which have been deployed to the AML treatment. As the majority of AML events are associated with chromosomal translocation resulting in oncogenic fusion proteins, it is valuable to comprehensively understand the mutual interactions between HDACs and oncogenic proteins. Therefore, we reviewed the process of leukemogenesis and roles of HDAC members acting in this progress, providing an insight for the target anchoring, investigation of hyperacetylated-agents, and how the current knowledge could be applied in AML treatment.
Introduction
Acute myeloid leukemia (AML) is characterized by genetic mutations and epigenetic alterations, marked by uncontrollable proliferation, blocked differentiation, and anti-apoptosis (1–3). And the majority of AML events are correlative with abnormal chromosomal translocations, which generates the oncogenic fusion genes. Mounting studies have demonstrated the central roles of fusion genes in initiating the leukemogenesis (4–6). And the successful strategies are paralleled by the decrease or degradation of chimeric proteins (7, 8). Commonly, the fusion partner in chimeric protein acts as a transcriptional protein interacting with the recruited corepressor complexes, which alters the expression of target genes that maintain the homeostasis of myeloid development, conferring the foundation of leukemic transformation (9, 10). Thereby, master the potential elements interacting with the fusion proteins is the prerequisite for targeting such oncogenic chimera.
Epigenetic modification has been acknowledged to paly crucial roles in the oncogenic transforming including AML (11, 12). Generally, epigenetic modification is not dedicated to some specific genes but serving for a vital regulator of transcriptional factors, which hold the specific capacity of DNA binding, whereby determining the potential transcriptional outcome (13–15). Thereby, the function of epigenetic modification is closely related to the cell-specific situation where the transcription factors are involved.
Accumulating evidences have been presented that epigenetic aberration prominently contribute to the leukemogenesis (16–18). As one of the major epigenetic regulators, histone deacetylases (HDACs) are indispensable in gene transcription. Dysregulation of HDACs has long been recognized as a crucial driver to hematological malignancies from initiation to metastasis, because they determine the fate of tumor cells, directing the cell to proliferate, differentiate, or be quiescent (13, 19). Therefore, the orchestration of HDACs is closely related to the cell development of both normal cells and tumor cells.
As acetyl group removers, HDACs control the accessibility of chromatin for transcription factors through switching the acetylated status, which finely tunes the transcriptional level of transcription factors and epigenetic modifiers, involving in development, cellular homeostasis, and carcinogenesis (20–22). And deregulated HDACs are associated with cell differentiation arrest, cell cycle disruption, DNA damage, and cell death (13, 23). Targeting the dysfunctional deacetylation in AML provides a promising strategy benefit for tumor treatment (24, 25). And experimental and clinical functions of HDAC inhibitors have been described by a number of reports (26–31), but the detailed mechanism acted by HDACs has not been elaborated. Comprehensively harness the roles of HDAC family members acting in leukemogenesis will provide us more precise prevision against such malignancy.
AML is frequently associated with chromatin rearrangement, including translocation and inversion, which generate oncogenic fusion proteins, among of which four most common chimeric proteins should be paid more attention, including AML1-ETO, PML-RARα, CBFβ-MYH11, and MLL-MLLT3 (4, 6, 32–34). Here we attempt to summarize the mutual interactions between HDACs and oncogenic fusion proteins involved in AML, providing a reference for the precise application of HDAC inhibitors and novel drug exploration against AML.
Acute Myeloid Leukemogenesis and Classification
Acute myeloid leukemogenesis is a complicated progress involved in genetic and epigenetic alterations, leading to uncontrolled proliferation, arrested differentiation, and myeloid dysfunction (1, 2). And the altered genes can be subdivided into five categories (Figure 1): Class I mutations, activators of tyrosine kinase, such as c-Kit, Flt3, and BCR-ABL, provide the hematopoietic progenitors with survival/proliferation advantage. Class II mutations, transcriptional factors such as NPM1, CEBPA, and TP53 as well as oncogenic fusion genes (e. g. AML1-ETO, PML-RARα, and CBFβ-MYH11), arrest the differentiation of hematopoietic cells. Mutations emerging in either class I or class II do not result in leukemogenesis until the both happen to mutate. When differentiation of hematopoietic cells is hindered by Class II mutations, Class I mutations would autonomously proliferate, initiating the leukemogenesis. Class III mutations, epigenetic regulatory molecules (e. g. TET2, IDH1 and IDH2, DNMT3A, and HDACs), silence/activate the tumor suppressor genes/pro-tumor genes. And the class IV mutations involve genes that alter cell adhesion and cell-cell interaction, leading to the flexible motility and migration. Class V mutation includes genes dysregulating DNA-repair (e.g.TP53 and NPM1) and RNA-splicing (35–40). We focus on the epigenetic abnormalities of histone modification in the progression of leukemogenesis.
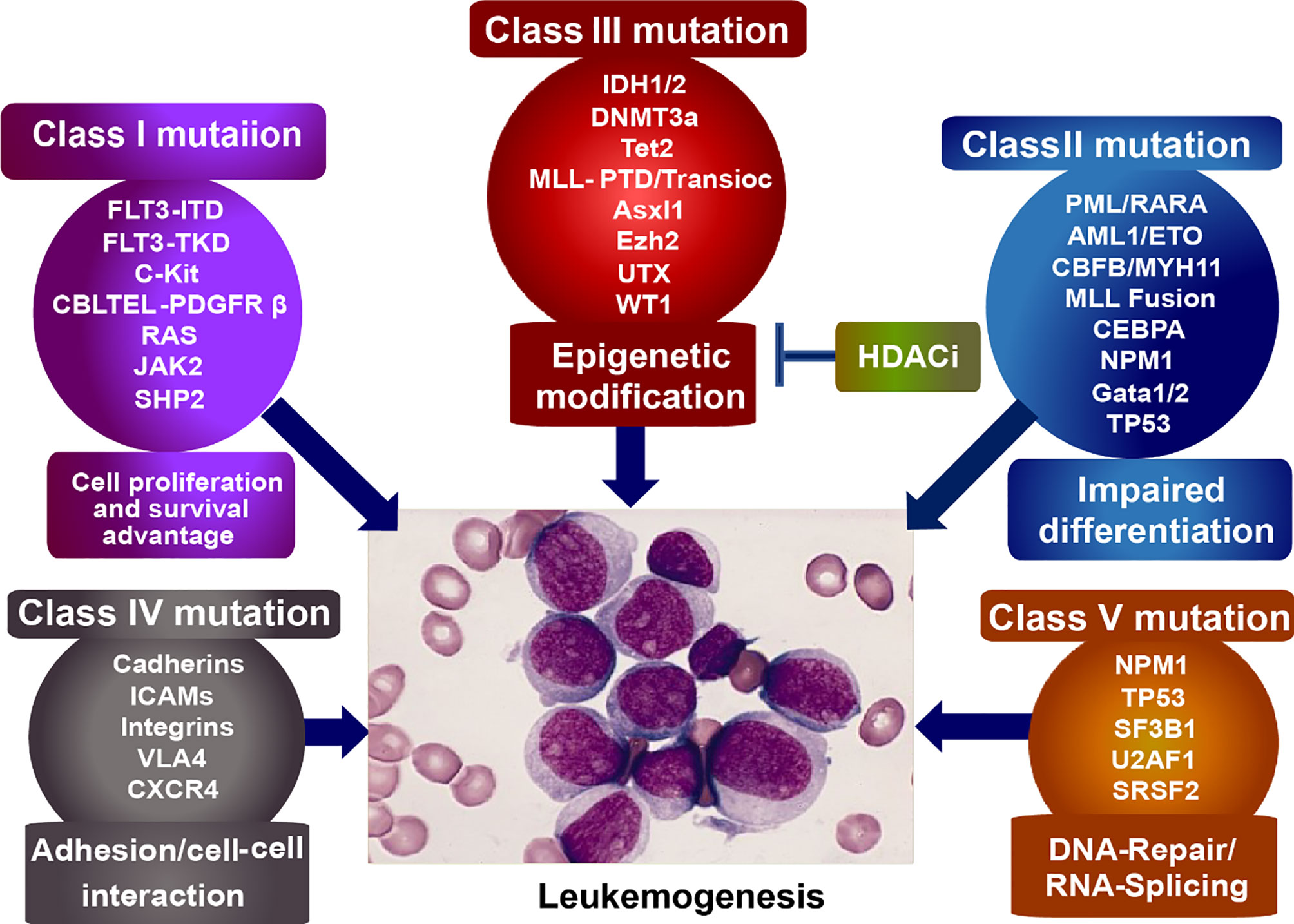
Figure 1 Classification of mutagenic genes eliciting leukemogenesis. Class I mutation, provides tumor cells with survival/proliferation advantages; Class II mutation, disturbs the cell differentiation; Class III mutation, epigenetically dysregulates the tumor suppressor/activator; Class IV mutation, alters cell adhesion and cell-cell interaction, leading to the flexible motility and migration. Class V mutation, dysregulates DNA-repair and RNA-splicing.
The subtypes of AML are majorly classified by two systems: French-American British (FAB) classification used earlier, and World Health Organization (WHO) classification, which has replaced the former (2). According to FAB classification (Table 1), AML can be grouped into eight subtypes from M0 to M7 based on the leukemic cell development and maturity. Among of them, M0 to M5 derived from the progenitors of white blood cells; M6 start with early forms of red blood cells; and M7 originates in the early forms of platelets (41–44).

Table 1 FAB subtype of AML.
According to WHO classification (45–50), AML is subdivided into 6 categories (Table 2): 1) AML with recurrent genetic abnormalities, involving in translocation, inversion, deletion, and mutation; 2) AML with myelodysplasia-related changes (MRC), a kind of multilineage dysplasia; 3) therapy-related myeloid neoplasms (t-MN), such as chemotherapy and radiation; 4) AML, not otherwise specified (NOS), including M0, 1, 2, 4, 5, 6, 7, acute basophilic leukemia, and acute panmyelosis with fibrosis; 5) myeloid sarcoma; 6) myeloid proliferations related to Down syndrome (DS). AML with recurrent genetic abnormalities contains balanced translocation/inversion, and mutation. The balanced translocations include t (8,21) (q22;q22.1) (AML1-ETO); inv (16) (p13.1q22)(CBFβ-MYH11); t (9,11)(p21.3;q23.3)(PML-RARα); t (6,9) (p23;q34.1) (KMT2A-MLLT3); inv (3)(q21.3q26.2)(DEK-NUP214); t (1,22)(p13.3; q13.1) (Gata2, Mecom); Rbm15-MKL1, and Bcr-Abl1. Here we will discuss the four most common fusion proteins involved in AML, focusing on the roles of HDACs functioning in the fusion proteins.
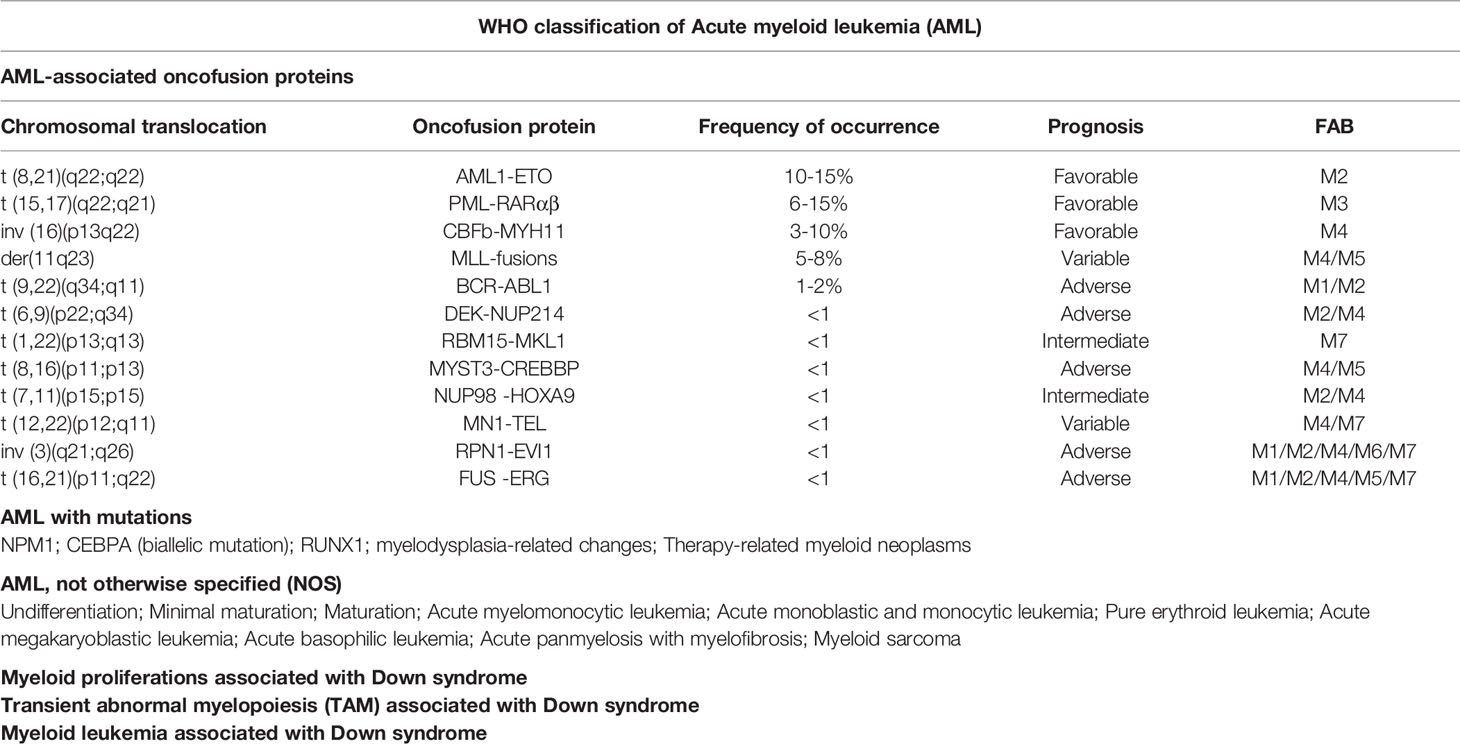
Table 2 WHO classification of AML.
HDACs Classification and Functions
Nucleosome, constituting the fundamental units of chromatin, is an octamer polymerized by four types of histones (H2A, H2B, H3, and H4), wrapped by 146 base-pair DNA. Each histone contains a structural domain and an unstructured tail of 25-40 amino acid residuals, which can be altered via post-translational modification, including acetylation, methylation, phosphorylation, and ubiquitination (51, 52). And the modification of histone residuals will determine the chromatin accessibility to transcription factors, keeping them activated or silent. Thereinto, the homeostasis of acetylation generally depends on the dynamic regulation of histone deacetylases (HDAC) and histone acetyltransferases (HAT) (53, 54).
HDAC and HAT play opposite roles in the epigenetic modification of chromatin, especially the histone proteins, where HATs allow the chromatin relaxed for gene transcription, and HDACs condense the chromatin making it inaccessible for transcriptional factors (Figure 2). HAT transfers the acetyl group from acetyl coenzyme A to lysine residual of histone N-terminal with positive charge, which binds to DNA strand with negative charge and prevents the chromatin from being condensed, thereby keeping the chromatin loosened available for the binding of transcription factors with DNA. Oppositely, HDACs favor to compact the chromatin, preventing the gene transcription. They remove the acetyl group from histone tail, and subsequently condense the chromatin, resulting in transcriptional inhibition (55–57). Therefore, the dysregulation is inevitable when the balance is disrupted between HDACs and HATs.
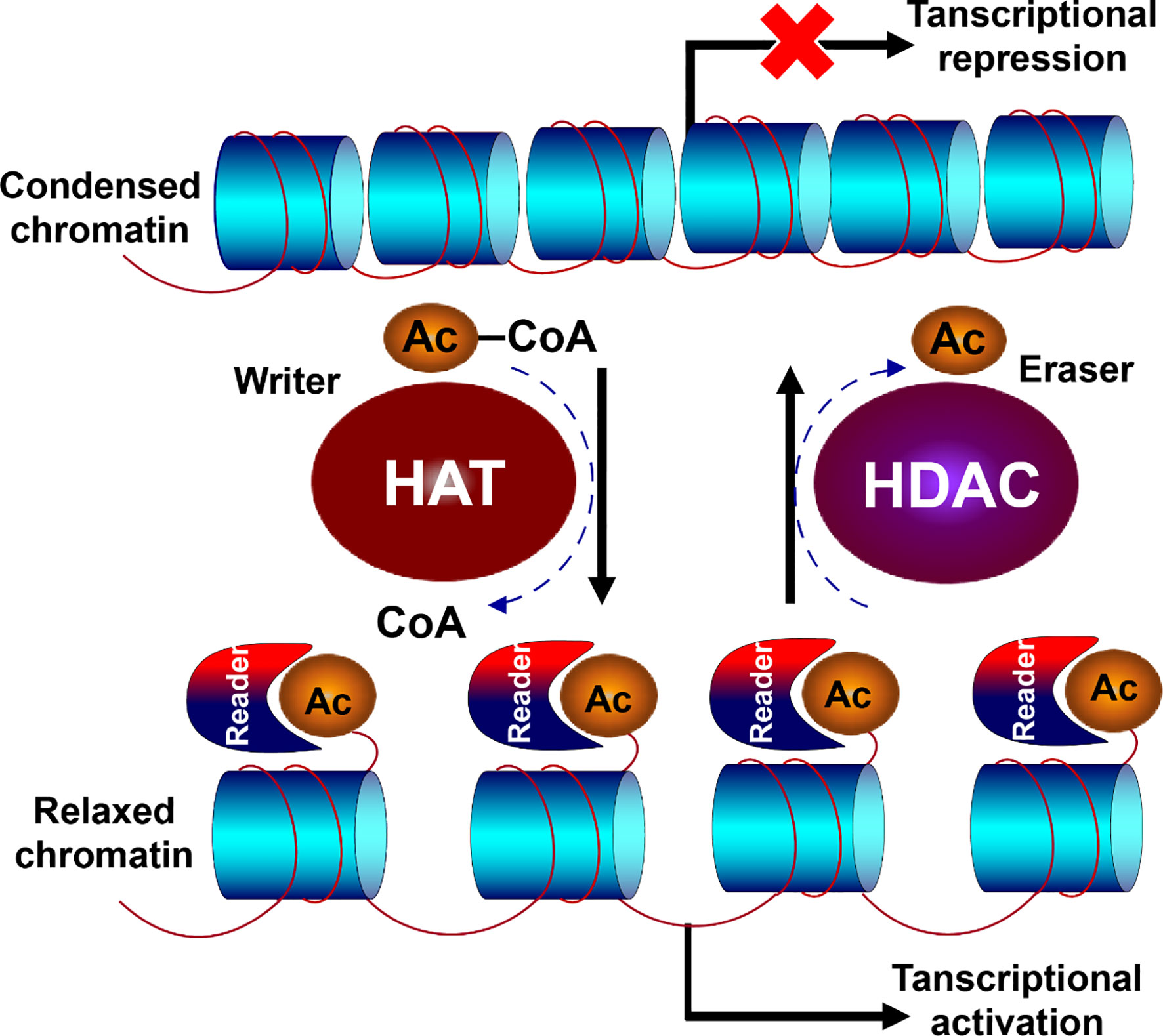
Figure 2 Opposite function of HDACs and HATs. HDAC and HAT play opposite roles in the epigenetic modification of chromatin. HAT transfers the acetyl group from acetyl coenzyme A to lysine residual of histone N-terminal with positive charge, which binds to DNA strand with negative charge and prevents the chromatin from being condensed, allowing the chromatin relaxed for gene transcription. Oppositely, HDACs remove the acetyl group from histone tail, and subsequently condenses the chromatin, resulting in transcriptional inhibition.
HDACs are universally spread in eukaryotes, which belong to a superfamily composed of 18 proteins with conserved deacetylase domain (21, 23). Based on the phylogenetic analysis, sequence homology to yeast protein, and domain organization, these proteins can be categorized into four families (class I, IIa, IIb, III and IV) (Figure 3). Three of them contain the Zn2+ dependent catalytic domain, which are referred to as classical HDACs, and class III members are NAD+-dependent, called sirtuins, which possesses deacetylase activity but is unrelated to HDACs, and will not be involved here. Distinguished by structure, enzymatic function, and localization, they display similar and specific functions during the regulation of gene expression (13, 21, 58).

Figure 3 Classification of HDACs. Based on the phylogenetic analysis, sequence homology to yeast protein, and domain organization, HDAC enzymes are categorized into four families (class I, IIa, IIb, III and IV). Three of them contain the Zn2+ dependent catalytic domain, which are referred to as classical HDACs, and class III members are NAD+-dependent, which were not involved in this description. Class I HDACs contains HDAC1, 2, 3, and 8, which majorly localize in nuclear; Class II HDACs include Class IIa (HDAC4, 5, 7, 9) and Class IIb (HDAC6, 10), which shuttle between nuclear and cytoplasm; and Class IV contains only HDAC11, shuttling between nuclear and cytoplasm.
Class I HDAC family is consist of HDAC1, 2, 3, and 8, which are homologous to yeast protein reduced potassium dependency-3 (Rpd3) (21, 59). They are chiefly expressed in nuclear, consisted by classic deacetylase domain, nuclear localization signal, showing high enzymatic activity to their substrates. Approximately 400 amino acids consist of each member, the catalytic domain contains two histidine residues, two aspartic acid residues and one tyrosine residue with Zn2+. And they generally function as gene transcriptional repressors. For instance, HDAC1 and HDAC2 bear closely identical structure and similar function, and usually work together in the repressive complexes, such as corepressor for element-1-silencing transcription factor (CoREST), nucleosome remodeling and deacetylase (NuRD), and transcription regulator family member switch-independent 3 (Sin3) complexes. HDAC3 generally emerges in another type of repressive complexes, such as N-CoR–SMRT complex. HDAC8 has been described to cooperate with SMAD3/4 complex, promoting the cell proliferation and migration (60–65).
According to the number of catalytic domains, Class II HDAC family can be subdivided into Class IIa (HDAC4, 5,7, 9) and Class IIb (HDAC6, 10), which can shuttle between nucleus and cytoplasm (66, 67). Class IIa HDAC members are grouped by a functionally important N-terminal domain, which mediates DNA-binding and nuclear-cytoplasmic shuttling. HDAC trafficking is regulated by nuclear export signal (NES) and binding sites for14-3-3 proteins. Upon 14-3-3 protein binding, cytoplasmatic retention or nuclear export of class IIa HDACs will be stimulated depending on the phosphorylation of 14-3-3 binding sites, which can be regulated by protein kinase-D, Ca2+/calmodulin-dependent kinases (CaMKs), and checkpoint kinase-1 (CHK1). And subsequently the transcriptional repressors will be regulated via binding with myocyte enhancer factor 2 (MEF2) binding domain, conferring signal responsiveness to downstream genes. When bound with Class IIa HDACs, MEF2 makes them a transcriptional repressor, whereas bound with HATs p300, MEF2 then converted them into a transcriptional activator. And the deregulated balance of HDAC and HAT will subsequently lead to diseases (68–71). Class IIb HDACs are atypical ones. HDAC6 contains two deacetylase domains and a C-terminal zinc-finger, which functions as a major cytoplasmic deacetylase targeting alpha-tubulin and HSP90, regulating cell motility, adhesion, and chaperone function (72, 73). Besides, binding with ubiquitin via zinc finger domain HDAC6 can regulates the aggresome formation, autophagy, heat shock factor-1 (HSF-1), and function of platelet derived growth factor (PDGF) (74, 75). HDAC10 holds single deacetylation domain and a leucine-rich domain. It possesses properties of immunoregulator, against the tolerogenic molecule PD-L1, implying an epigenetic target for immunotherapy. Overexpression of HDAC10 has been demonstrated to accelerate the progress of carcinogenesis. Deletion of HDAC10 in antigen-presenting cells (APCs) can increase the expression of MHC class II molecules and repress the transcription of PD-L1, which is associated with enhancement of immune system (76–79).
HDAC11, as the sole member of Class IV HDAC family, structurally similar to class I and II, mainly distributes in nucleus and acts as a repressor of IL-10 (80). It can regulate the dynamic balance between immune activation and tolerance. Upregulation of HDAC11 has been shown in various cancer cells (81, 82).
Besides, an increasing number of non-histone proteins have been identified as substrates of HDACs, such as p53, Stat3, Hsp90, GATA1, Tubulin, and β-catenin, which display vital roles during the progress of carcinogenesis (83–85). Via deacetylation, HDAC1 can affect the stability of tumor suppressor gene p53, arresting the interaction with DNA, inverting the function of p53. HDAC1 can also directly lead to the deacetylation of GATA1, repressing the gene transcription. HDAC6 is associated with the modulation of Akt and Stat3 signaling via regulation of Hsp90 acetylation in multiple myeloma cells. Deletion of HDAC6 will result in reducing phosphorylation of Stat3, which results in related genes inactivation (22, 86, 87).
Taken together, HDACs participate in the regulation of key transcriptional factors involving in the gene transcription, cell apoptosis, cell cycles, and signal transduction, which depicts the pivotal roles of HDACs functioning in epigenetic modification and gene transcription. The histone modification determines the accessibility of chromatin, which will make genes activated or silent. Inevitably, dysregulated histone modification will lead to dysfunctional cell development, which is strongly associated with carcinogenesis. Disruption of specific HDACs usually associates with dysregulation of differentiation, proliferation, migration, chemotherapy resistance, and angiogenesis (Figure 4). Overexpression of HDAC usually emerges accompanying with leukemogenesis and the other tumor. They act to close the nucleosomes, inhibiting the expression of tumor suppressor genes. HDAC inhibitor, as an agonist of HDAC, can alter the abnormal hypoacetylation level of histone, and subsequently elicits cell differentiation and apoptosis, demonstrating the indispensable roles of HDACs in tumorigenesis (88–91). Harnessing the function of HDACs is the premise indicating to precisely target the master alterations.
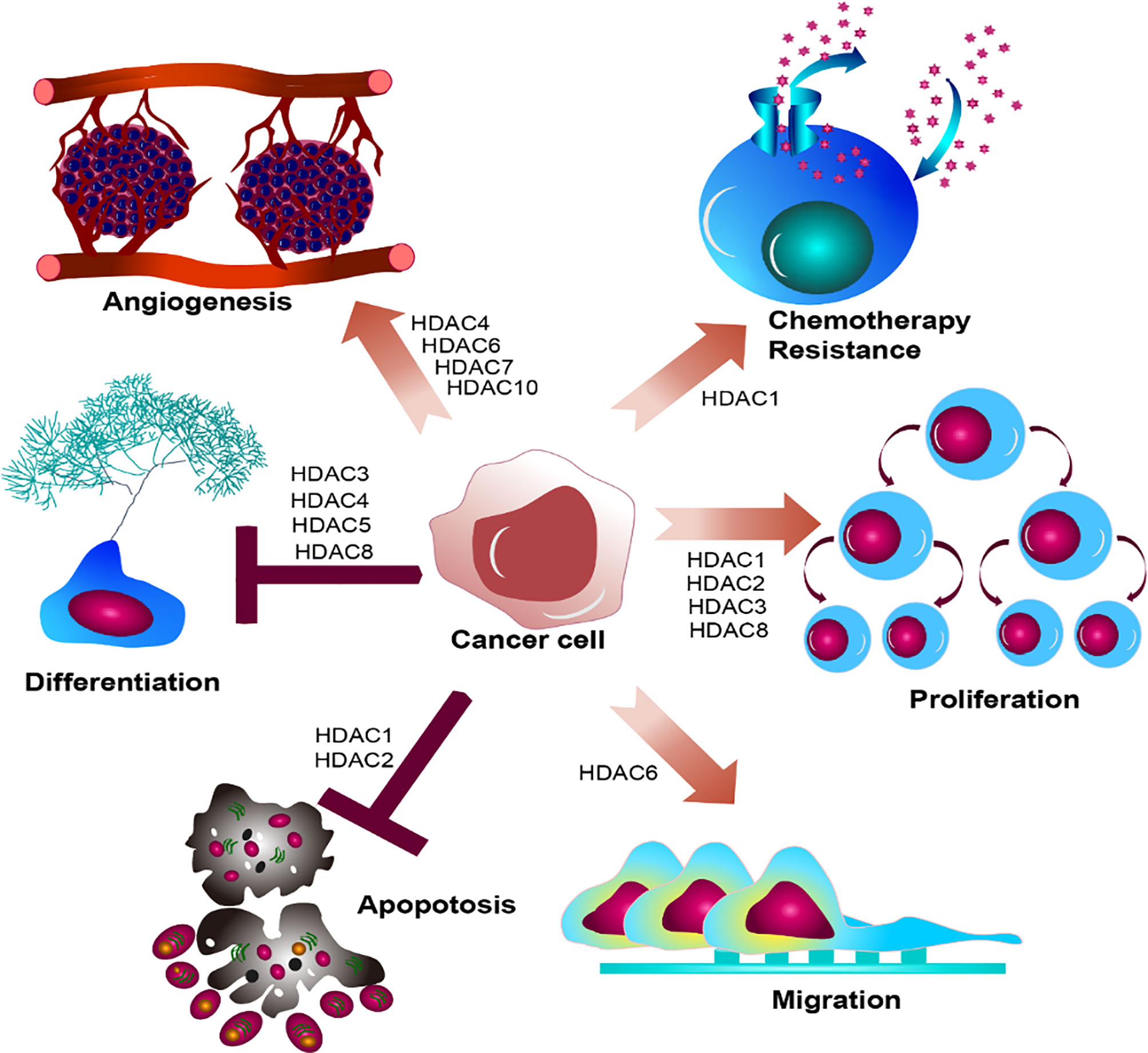
Figure 4 Summary of HDACs functioning in cancer cell. HDAC family members determine biological effect of oncogenic hallmarks emerging in cancer cell, disrupting the regular cell development in cancer cell, leading to dysregulated differentiation, proliferation, migration, chemotherapy resistance, and angiogenesis.
HDACs in Leukemogenesis
Although HDAC mutations in AML are relatively rare compared to solid tumors, HDAC proteins are abnormally recruited to oncogenic fusion proteins, such as AML1-ETO, CBFB-MYTH11, PML-RARα, and MLL-fusions, which function as vital roles in onsetting and promoting the progress of leukemogenesis (4, 13, 31). And HDAC inhibitors, as a series of compounds that neutralize the activities of HDACs, have long been utilized in treatment of AML for pre-clinical studies, which have some extend shown beneficial outcomes (26, 27, 30, 31). And the multiple functions of HDAC inhibitors have been discussed in numerous research articles and reviews (which will thereby not be included in this review). However, the mutual interaction between HDACs and AML has not been comprehensively described. And we choose the most frequent events of chromosomal translocation emerging in AML to elucidate the reciprocal functions of AML and HDACs.
HDACs in AML With AML1-ETO
One of the well-studied AML subtypes is t (8, 21) AML, which occurs in approximately 10-15% of total AML cases, and 18-40% of M2 AML (92–95). The translocation is generated by the fusion of AML1 gene (Runx1) on 21q22.1 and ETO gene (Runx1T1) on 8q22, leading to the forming of AML1-ETO fusion protein (5, 34, 96). It can invert the original function of AML1, performing opposite function during the leukemogenesis. The fusion protein AML1-ETO provides the DNA-binding domain via the hematopoietic master regulator AML1 and transcriptional domain via ETO, targeting the AML1 target genes. It substitutes the original function of AML1 and disrupts cellular processes involved in the myeloid proliferation, differentiation, and genome stability (95, 97, 98).
To understand the mutual interactions between AML1-ETO and HDACs in detail, we firstly figure down the functions of AML1 and ETO in normal condition and AML1-ETO in tumorigenic environment. AML1 functions as a master organizer, which in charge of regulating the hematopoietic specific promoters and enhancers. It widely spreads in hematopoietic system, cooperating with multiple lineage-specific transcriptional regulators, such as the driving of endothelial hematopoietic transition (99, 100). AML1 gene on 21q22 is composed of nine exons, with three breakpoint cluster regions (BCR) in intron5. The structure of AML1 is composed of conserved runt homology DNA binding domain (RHD), activation domain (AD), nuclear matrix-targeting signal (NMTS), proline-rich domain (PY), two inhibitory domains (ID), and an additional C-terminal motif with five amino acid (VWRPY), working as a recognition and recruitment signal for Groucho/TLE family. Besides, it contains two promoters: distal promoter P1 and proximal promoter P2. Both promoters include the AML1-binding sties, which can be regulated by itself and other AML1 transcriptional factors. RHD is in charge of recognizing and binding to DNA sequences, and localizing the AML1 transcriptional factors in nucleus. It also contributes to the binding of core biding factor β (CBFβ), which does not interact with DNA, but increases the α subunit affinity to DNA binding and stabilizes the complex (Figure 5A) (101–103).
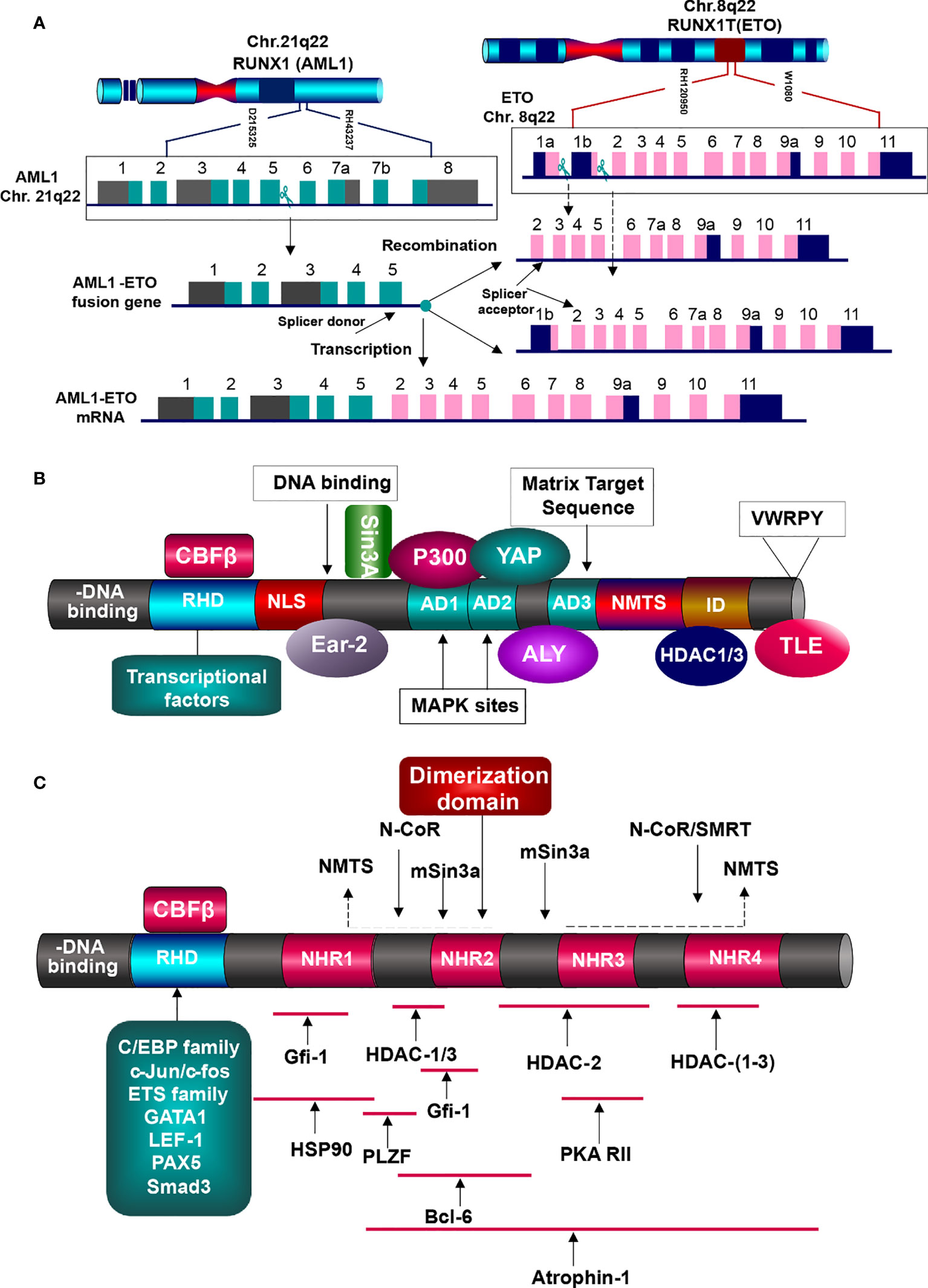
Figure 5 Generation and function of AML1-ETO fusion protein. (A) Generation of AML1-ETO. ETO gene on Chr.8q22 is consist of 13 exons, containing two breakpoints, but one splicer acceptor in exon2. AML1 gene is made up of nine exons with one breakpoint and one splicer donor. Absence of splicer acceptor in exon1b, the two genes generate the only fusion mRNA. (B) AML1 protein structures and partner proteins. AML1 is composed of DNA-binding domain (RHD) and other domains related to signal transduction, transcriptional factors binding, epigenetic modifiers interaction, and TLE co-repression, which can interact with HATs (MOZ, CBP, and p300) and HDACs (HDAC1, 2, and 3), resulting in gene activation or inhibition. HATs, histone acetyltransferases; HDACs, histone deaccetylases; RHD, runt homology domain; NLS, nuclear localization signal; AD, activation domain; NMTS, nuclear matrix targeting signal; ID, inhibitory signal; VWRPY sequence. (C) AML1-ETO fusion protein and the interacting partners. AML1 contributes the DNA-binding domain RHD, which binds with various transcription factors, but lacks of domains to elicit regular functions. And nearly whole of ETO structure is involved in the fusion, including the four NHRs. They interat with corepressive complexes, HDACs, and other molecules, initiating the oncogenesis.
AML1 functioning as an activator or repressor is determined by its interaction with corresponding transcriptional factors and co-factors, rather than itself features (95, 104, 105). It has been shown to interact with various chromatin modifiers and remodelers (Figure 5B). For instance, its activation can be stimulated by binding with lysine acetyl transferase MOZ (MYST3), and the same to transcriptional co-activators P300 and CBP. They function as integrators, which bind with AML1 and other transcriptional activators driving the hematopoietic promoters (106). ALY expressed in nucleus can bind to the activation domain of AML1, forming multimers and bridging the interaction of AML1 and other transcription factors. c-Yes tyrosine kinase associated protein (YAP) binds to PPPY motif in the AML1 C-terminal activation domain, enhancing the activity of AML1 (105, 107, 108).
Furthermore, AML1 may function as a repressor of HDAC complex (15). Researches have demonstrated that it can inhibit the transcription of p21 via binding the promoter of p21 with AML1 VWRPY Groucho/TLE interaction domain. Through binding with co-repressors such as Sin3A and Groucho/TLE, it recruits HDACs to repress the transcription (15). And the HDAC inhibitor Trichostatin A can impair such suppression, demonstrating that HDACs contribute to AML1-mediated inhibition. It is also associated with HDAC1, 2, 3 and histone H3 lysine 9 methyltransferase SUV39H1 (KMT1A), leading to transcriptional suppression. In myeloid cells, AML1 binding with CCAAT/enhancer binding protein alpha (C/EBPα) and PU.1 can activated the macrophage colony-stimulating factor receptor (M-CSFR) expression (104, 109).
Meanwhile, AML1 can also been inhibited by corresponding transcription factors. For instance, bound with forkhead box P3 (FOXP3), it can suppress the expression of interleukin2 (IL2) and interferon gamma (IFN-γ) in T regulatory cells. It is multifunctional in the regulation of hematopoiesis, including cell differentiation, proliferation, and apoptosis. And the aberration of AML1 will speculatively deregulate the normal cellular development, which is involved in carcinogenesis (15, 109).
Eight-twenty-one (ETO) gene on 8q22 is consist of 13 exons, with one BCR in intron1a and three BCRs in intron1b, which generate different variants but create the same fusion gene AML1-ETO, because it supplies only one splice acceptor in exon2, as the exon1b lacks of splice acceptor (104, 110). ETO protein possesses three proline-serine-threonine (PST)-rich regions and four conserved nervy homology regions (NHR), involved in neuronal development of Drosophila embryos. The PST-rich domains contain multiple potential kinase phosphorylation sites (SP and TP). NHR1 is homologous to the Drosophila TATA-box-associated factors, including TAF110. NHR2 domain, containing a hydrophobic amino-acid (a.a.) heptad repeat, plays crucial role in the oligomerization between ETO family members, forming homo-/hetero-dimerization. NHR3 is with predicted coiled-coil structure. NHR4 is homologous to myeloid-Nervy-DEAF1 (MYND) homology domain, with two predicted zinc-finger motifs, which is required for the protein-protein interaction. For instance, ETO is associated to the co-repressors mSin3 and nuclear receptor corepressor (N-CoR), thyroid hormone receptor (SMRT), as wells as HDAC1, 2, 3. Via binding with NHR4 DNA binding domain, it can interact with the co-repressors N-Cor, SMRT, and mSin3A, which will then tether the DNA-binding proteins to HDACs, resulting in repressive transcription (105, 111). Researches have shown that ETO and AML1-ETO can pull-down by HDAC activity via Co-IP, bearing out the repressive role of AML1-ETO through the recruitment of HDACs to AML1 target genes (112). Such function may instead of AML1 complex which originally worked as an activator involving in the histone acetyl transferases p300/CBP. It is similar to the leukemogenic mechanism of APL. Fusion proteins PML/PLZF-RARα increase the affinity of RARα to co-repressors and RARα target genes (113). It also interacts with Atrophin-1, chaperon heat-shock protein (HSP90), PLZF, Gfi-1, and Bcl-6, functioning as a corepressor of transcriptional factors. Via NHR1 and NHR2, ETO can binds to Gfi-1 and Gfi-1b, contributing to the recruitment of HDACs, which subsequently repress the activity of Gfi-1/Gfi-1b proteins (111, 112, 114). Depending on the DNA binding site provided by RHD of AML1, AML1-ETO may perform as a repressor or activator of the AML1 target genes (115).
In AML1-ETO fusion protein, the important features of AML1 are lost: 1) the c-terminal activation domain interacting with co-activators; 2) domains in charge of binding with co-repressors such as Sin3 and TLE; NLS domain functioning as nuclear matrix-targeting signal. And such lost will subsequently result in the dysregulation of hematopoiesis. The AML1-ETO fusion protein can affect the expression of both AML1 target genes and other related genes. As a part of AML1-ETO, AML1 recruits HDACs to the promoter, which suppress the expression of relative target genes. In normal, these target genes are required for regulating cell growth and preventing hemopoietic cells from transformation. And abnormally, the target genes are suppressed and lose their control, leading to cell overgrowth (107).
In t (8,21) AML, a number of genes critical to normal hematopoiesis are up-regulated by AML1, while AML1-ETO disrupts such trans-activation. AML1–ETO fusion protein recruits various transcriptional factors, epigenetic modifiers such as HDACs, PRMT1, and p300, forming the first aberration vital for the t (8,21) AML onset (13). And then it can collaborate with the secondary mutations including c-Kit, FLT3, and RAS. Via recruiting the HDAC1, 2, 3, AML1-ETO can silence the target genes and block the cell differentiation and transformation (95, 116). AML1 contributes the DNA binding domain RHD to a number of transcriptional factors (such as Ets-1, LEF-1, C/EBPα, PU.1, MEF, Pax5, and GATA1) and epigenetic modifiers, but defaults the subsequential elements for activation, which are replaced by nearly entire ETO. The well-known binding protein of AML1 is CBFβ, which efficiently binds to RHD of AML1 and is required for the its full transcriptional activation (Figure 5B) (117, 118).
HDAC1 is a binding partner of AML1 that takes part in the forming of corepressor complex with nuclear receptor corepressor (N-CoR) and mammalian Sin3 (mSin3A and B) (119, 120). And ETO can bind to the central domain of N-CoR, generating the AML1-ETO/N-CoR/mSin3/HDAC1 complex, remodeling of chromatin structure and transcriptional suppression, dysregulating the normal hematopoiesis (26, 121). Additionally, the substrates of HDACs are not only histone but also non-histone proteins, such as oncogenes, tumor-suppressor genes, and chaperones. One of the presentative tumor suppressors is TP53. Specifically interacting with TP53, HDAC1 combined with the corepressor complex can mediated its deacetylation, and subsequent degradation. As a classical tumor suppressor, TP53 is crucial to the process of hematopoiesis. The alteration of TP53 is associated with the AML progress and therapy responsiveness, and generally predicts poor prognosis (9). Although its mutation frequency is relatively low in AML (less than 10% of de novo AML cases) compared to solid tumors (more than50% of cases), the function of TP53 in AML could not be ignored, as dysfunctional wild-type (WT) TP53 appears in various AML entities, implying a more attention to be paid (122).
HDAC2 are nearly identical to HDAC1, and usually work together in repressive complexes, such as nucleosome remodeling and deacetylase (NuRD), switch independent 3 (Sin3), and corepressor of RE1 silencing transcription factor (CoREST) complexes. Inhibition of HDAC1 and HDAC2 leads to down-regulation of RAD51, BRCA1, and CHK1, which are crucial for the DNA damage response (DDR) and subsequent DNA double-strand break and apoptosis in AML cell lines. And AML-1-ETO can bind with HDAC1, 2, and 3 to repress the AML1 target genes in t (8,21) AML (123). And HDAC6 deacetylates the chaperone Hsp90, eliciting the interaction with AML1-ETO protein, which can be dissociated by HDAC inhibitors that mediates the degradation of AML1-ETO protein.
HDAC11 may display a role in the immune system by regulating the immune cells. Antigen-presenting cells (APCs) plays critical role in T cell activation and tolerance, which is associated with HDAC11 (80). Up-regulation of HDAC11 can repress the expression of IL-10, and subsequently induce the APCs inflammation, which will prime naïve T cells and reactivate the response of tolerant CD4+ T cells. Meanwhile, down-regulation of HDAC11 in APCs promotes the expression of IL-10 and impairs the T cell response. Therefore, HDAC11 may act as a decider in the immune activation and tolerance, implying the substantial role of HDAC11 in the immunotherapy, involving in AML (80).
HDACs in AML With CBFβ-MYH11
The inv (16) translocation emerges in 8-10% of AML patients, which is associated with M4Eo AML. It is produced by the chromosomal breakpoints within core binding factor beta (CBFB) gene on 16q22 and smooth muscle myosin heavy chain gene (MYH11) gene on 16p13, encoding corresponding proteins: CBFβ and smooth muscle myosin heavy chain (SMMHC). And the oncogenic gene CBFB-MYH11 and fusion protein CBFβ-SMMHC will subsequently generated and arrest the differentiation of hematopoietic cells. Similar to AML with AML1-ETO, the original disorder of AML with CBFB-MYH11 derives from the disruption of hematopoietic function performed by the core binding factor (CBF).
CBF, as a heterodimer, is composed of CBFα (DNA-binding subunit) and CBFβ (partner of CBFα) (124). CBFα subunit is encoded by CBFA2 that is known as RUXN1 or AML1 gene. CBFβ does not directly bind with DNA, but enhances the affinity of CBFα to DNA, stabilizing the CBFα-DNA complex. CBFβ-SMMHC fusion protein displays a higher affinity to AML1 binding than wild type CBFβ. Additionally, it contains an additional AML1-binding domain in SMMHC portion. Therefore, AML1 is preferential to bind with CBFβ-SMMHC, which competes the RUNX1-binding site with CBFβ, resulting in the blocks of AML1 function and enhancement of the SMMHC activity. The dysregulation of CBFβ acts an indirect factor disrupting the function of AML1, whose pivotal functions in hematopoiesis has been described in t (8,21) AML. Both CBFA2 and CBFB genes are indispensable for the development of normal hematopoiesis, deletion of either gene will disrupt the definitive hematopoietic stem cells. CBFβ-SMMHC protein interacts with the pivotal transcription factor AML1, sequestering the normal essential hematopoietic function of AML1. It acts as a transcriptional repressor, interacting with transcriptional inhibitors and HDACs, repressing the transcription of corresponding genes.
HDAC1 is a binding partner of AML1. And further research showed that HDAC1 can bind to CBFβ-SMMHC complex, which colocalizes with the promoters of AML1 and CBFβ-SMMHC. As a key cofactor, HDAC1 participates in the forming of AML1: CBFβ-SMMHC complex, which is essential for the transcriptional activity of related genes, involving in leukemic cell differentiation block and pro-proliferation (125). Additionally, pharmacologic inhibition of HDAC1 contributes to the suppression of leukemogenesis with CBFβ-SMMHC (126, 127). And in vivo, it can decrease the mouse leukemic burden, showing an effective role of HDAC1 targeting the CBFβ-SMMHC protein (30).
HDAC8, as another member of class I HDAC, has been demonstrated to interact with CBFβ-SMMHC protein. Besides, it can also reduce the acetylation of P53, which is bound to CBFβ-SMMHC protein, and subsequently promote the transformation of CBFβ-SMMHC-related leukemic stem cells. And inhibition of HDAC8 will induce the apoptosis in inv (16) AML (128, 129).
HDACs in Acute Promyelocytic Leukemia With PML-RARA
The t (15,17) (q24;q21) translocation accounts for 10%-15% of acute promyelocytic leukemia (APL) issues. It is derived from the fusion of promyelocytic leukemia (PML) gene on 15q24 and retinoic acid receptor alpha (RARA) gene on 17q21, which is critical for the cellular transformation (130, 131).
PML gene is composed of nine exons that produces some alternative spliced transcripts variants, which share the N-terminal region, containing the RING-B-Box-Coiled-coil/tripartite motif (RBCC/TRIM) domain (132). Due to the alternative splicing, the isoforms of PML are different in central or C-terminal regions and the longest one is PML1, which harbors a nuclear export signal (NES) domain. In normal, PML mainly functions as a tumor suppressor. It can interact with over 170 proteins, most of which are mediated by the RBCC/TRIM domain leading to PML multimerization and organization or by other isoform-specific domains of PML. Conferred by these different binding interactions, PML is involved in proliferation and self-renewal of hematopoietic stem cells, epigenetic regulation in hematopoiesis, and p53-dependent/independent apoptosis and senescence (122). In addition, it is necessary for the formation of nuclear body (NB), which is associated with the protein release and sequestration, posttranscriptional modification, and promotion of nuclear issues (133).
RARA gene is consist of 10 exons producing two isoforms (RARA1 and RARA2) that are belonged to nuclear hormone receptor superfamily, acting as a nuclear transcriptional factor when retinoids are present, which is essential for the promyelocyte differentiation (130, 134). The RARA protein can interact with retinoid X receptor protein (RXRA), generating a heterodimer that acts as a transcription activator to bind with retinoic acid response elements (RARE). In the presence of ligand (all-trans retinoic acid (ATRA) or 9-cis retinoic acid), RARA binds to RXRA forming a heterodimer, which can interact with retinoic acid responsive elements (RARE). In the absence of ligand, RAR-RXR heterodimer recruits the transcriptional corepressors, such as HDACs, Sin3, SMRT, and N-CoR, keeping transcriptional repression, which can be dissociated when ligand emerges (135). In normal, RARA is ligand-dependent determining the transcriptional switch, which is critical for the differentiation of normal myeloid hematopoietic cells (134). In APL, the fusion protein PML-RARA alters the function of PML and RARA, disrupting the nuclear structure and blocking the cell differentiation. Additionally, PML-RARA provide leukemic cells with a survival and proliferative advantage, leading to the superiority accumulation of tumor cells in APL (130). Besides, through inducing the deacetylation of p53, PML-RARA fusion protein can directly suppress the activity of p53, conferring leukemic blasts to escape from p53-dependent cancer surveillance. And such phenomenon is realized by the recruitment of HDACs to PML-RARA complex, which can result in the deacetylation of p53 (136, 137).
PML–RARA recruits HDACs leading to RARs suppress the transcription of RA target genes, which displays a central role in the oncogenic transformation of APL (132). The aberrant recruitment of HDACs induced by PML–RARA contributes to the differentiation blocks and accumulation of APL blasts, because it inappropriately represses the RAR target genes. RA functioning as a therapeutic agent is based on the mechanism that RA can lead to the dissociation of PML–RAR/HDAC complex and degradation of such fusion protein (138, 139). Furthermore, ATRA resistance can be neutralized by HDAC inhibitors (140), which should have been paid more attention. Deregulated HDAC3 acts as a crucial role in the progress of acute promyelocytic leukemia (APL) with PML-RARα fusion protein. HDAC4 can interact with the PLZF-RARα fusion protein, mediating the differentiation arrest (141, 142).
HDACs in AML With MLL-MLLT3
The t (9,11) AML presents in 3-5% of AML events, generated by the fusion of mixed lineage leukemia (MLL) gene on 11q23 and mixed lineage leukemia translocated to chromosome 3 (MLLT3) gene on 9p22, producing the fusion protein MLL-MLLT3 (143, 144).
MLL gene is made up of 14 exons, encoding the histone lysine methyltransferase whereby it is also called KMT2A, which harbors powerful transforming potential associated with neoplastic diseases assisted by specific partners, such as AF9 (MLLT3), AF4, and ENL (MLLT1) (145). It orchestrates various facets of cell development, including cell fate determine, stem cell maintenance, and embryogenesis. MLL protein contains multiple conserved domains with specific functions: 1) three AT hooks domains in the N-terminal of MLL mediating itself to bind with minor groove of DNA with AT-rich; 2) a transcriptional repressive domain that is composed of cysteine-rich CXXC DNMT (DNA methyltransferase1) homology region, which can bind to unmethylated CpG islands; 3) four plant homeodomain (PHD) fingers that mediate the protein-protein interactions; 4) a transactivation domain that is employed to interact with CBP/p300 complex; 5) SET [Su(var)3–9, enhancer of zeste, and trithorax] domain in C-terminal, serving as a histone H3 methyltransferase. Carrying along with such multiple domains, MLL can generate complexes with various partners, such as tumor suppressor Menin (multiple endocrine neoplasia), cell cycle regulator E2Fs, and HDACs (146, 147).
Overexpression of HDAC1, 2, and 3 is frequently found in leukemia (13). They can interact with MLL fusion protein leading to dysregulated chromatin remodeling, which could be neutralized by chidamide (148). Targeting MLL dysfunction by HDAC inhibitors such as vorinostat and panobinostat may counteract the aggressive resistance in MLL-fusion leukemia (149). And mocetinostat, a class I HDAC inhibitor, can inhibit the HOXA9 expression in AML with MLL-AF9 (147). Researchers have purified the stable MLL complex, where HDAC1 and HDAC2 were found. Additionally, they have also demonstrated that the repressive domain of MLL can specifically bind with HDAC1 and HDAC2, which can be partially released by HDAC inhibitor TSA but not RD1domain, which implies that additional cofactors are involved in the complex to fully perform the repressive function. And through binding to PHD fingers, Cyp33 can increase the affinity of MLL to HDAC1. Hypoacetylated histone in chromatin is frequently involved in transcriptionally repressive status (148, 150, 151).
Concluding Remarks
HDACs serving as the pivotal epigenetic modifier of chromatin determine the chromatin accessibility to transcriptional factors, which is essential for specific gene transcription and oncogenic transformation. And the same to hematopoiesis, function of HDACs is indispensable, which determines the fate of hematopoietic cells, going through self-renewal, proliferation, differentiation, or apoptosis, terminating in various cell lineages (13, 14, 20). Thereby, dysregulation of HDACs inevitably leads to disruption of hematopoiesis (25, 91). It is necessary to concentrate on the investigation of HDACs functions.
The vital function of HDACs has long been acknowledged in the process of normal hematopoietic cell development and leukemogenesis, and numerous HDAC inhibitors have been applied in the treatment of various tumors but the mechanism of HDAC inhibitors serving in AML is elusive (20, 21). As the studies of HDACs function in AML increasing, we summarized the predominant importance in AML.
AML, with disrupted hematopoietic system, is usually hallmarked by oncogenic fusion proteins, majorly centralizing on AML1-ETO, CBFB-MYH11, PML-RARA, and MLL-AF9 (32, 33). HDAC inhibitors, the hyperacetylated agents, theoretically gear toward the alteration of the aberrant hypoacetylated status, providing a reasonable strategy against AML. They own the theoretical feasibility but practical hinderance, which provoked us to explore the precise function of HDAC, contributing to the utilization of HDAC inhibitors (152, 153). And mounting researches and reviews have demonstrated the roles of HDAC inhibitors in the treatment of AML. However, the function of HDACs in oncogenic molecules is rarely described (15, 26). Although the relative material is of shortage, it is meaningful to elucidate the potential function of HDACs in AML, focusing on the oncogenic fusion proteins that provides a directing target against specific types of AML.
Besides, HDACs display immunoregulatory properties in integral level, which overall regulates the progress of leukemogenesis through modulating the master elements of immune system such as PD-L1, CTLA-4, Treg, and cytotoxic T lymphocyte (CTL), and antigen-presenting cell (APC) (154–156). For instance, expression of HDAC10 is associated with the presentation of MHC class II molecules in antigen presentation cells (157, 158). Members of HDACs participate in the different stages of T cell development, including CD4+ T cell-mediated immunity (154, 159). That is to say, HDACs not only function with specific fusion proteins but also do regulate the entirety level of immune system which is involved in tumor microenvironment.
Attentions paid on HDACs usually focus on the HDAC inhibitors in the process of carcinogenesis, whereas the roles of HDACs have not got enough attention. It is necessary to harness the interaction between HDACs and leukemogenesis, which would precisely direct the investigation of novel HDAC inhibitors. Here, we summarized the current knowledge of HDACs functioning in leukemogenesis with oncogenic fusion proteins. They are closely associated with the suppression of oncogenic fusion genes, and can be blocked by HDAC inhibitors. However, pan-inhibitors presented various side effects and it can be improved by the specific HDAC inhibitors. And the searching of special targets is based on harnessing the traits of each HDAC member functioning in the epigenetic modification. The review summarized the functional properties of HDAC members, which may be useful for the exploration of specific HDAC inhibitors. Furthermore, HDACs is involved in the regulation of immune system, which may benefit to the investigation of novel agents or combinational drugs.
Author Contributions
JZ drafted the manuscript. XG modified the manuscript. LY provided valuable advices, supervised, and approved the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by grants National Natural Science Foundation of China (82030076, 82000161, 82070161, 81970151, 81670162, and 81870134), China Postdoctoral Science Foundation (2018M640824), Chinese National Major Project for New Drug Innovation (2019ZX09201002003), Shenzhen Science and Technology Foundation (JCYJ20190808163601776, JCYJ20200109113810154). Shenzhen Key Laboratory Foundation (ZDSYS20200811143757022). Sanming Project of Shenzhen (SZSM202111004).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Kantarjian H, Kadia T, DiNardo C, Daver N, Borthakur G, Jabbou E, et al. Acute Myeloid Leukemia: Current Progress and Future Directions. Blood Cancer J (2021) 11(2):41. doi: 10.1038/s41408-021-00425-3
2. Estey EH. Acute Myeloid Leukemia: 2021 Update On Risk-Stratification and Management. Am J Hematol (2020) 95(11):1368–98. doi: 10.1002/ajh.25975
3. De Kouchkovsky I, Abdul-Hay M. Acute Myeloid Leukemia: A Comprehensive Review and 2016 update'. Blood Cancer J (2016) 6(7):e441. doi: 10.1038/bcj.2016.50
4. Martens JH, Stunnenberg HG. The Molecular Signature of Oncofusion Proteins In Acute Myeloid Leukemia. FEBS Lett (2010) 584(12):2662–9. doi: 10.1016/j.febslet.2010.04.002
5. Tijchon E, Yi G, Mandoli A, Smits JGA, Ferrari F, Heuts BMH, et al. The Acute Myeloid Leukemia Associated AML1-ETO Fusion Protein Alters The Transcriptome and Cellular Progression In A Single-Oncogene Expressing In Vitro Induced Pluripotent Stem Cell Based Granulocyte Differentiation Model. PLoS One (2019) 14(12):e0226435. doi: 10.1371/journal.pone.0226435
6. Matsukawa T, Aplan PD. Clinical and Molecular Consequences of Fusion Genes in Myeloid Malignancies. Stem Cells (2020) 38(11):1366–74. doi: 10.1002/stem.3263
7. Pikman Y, Stegmaier K. Targeted Therapy for Fusion-Driven High-Risk Acute Leukemia. Blood (2018) 132(12):1241–7. doi: 10.1182/blood-2018-04-784157
8. Brien GL, Stegmaier K, Armstrong SA. Targeting Chromatin Complexes in Fusion Protein-Driven Malignancies. Nat Rev Cancer (2019) 19(5):255–69. doi: 10.1038/s41568-019-0132-x
9. Al-Harbi S, Aljurf M, Mohty M, Almohareb F, Ahmed SOA. An Update on the Molecular Pathogenesis and Potential Therapeutic Targeting of AML with t ((q22;q22.1);RUNX1-RUNX1T1. Blood Adv (2020) 4(1):229–38. doi: 10.1182/bloodadvances.2019000168
10. van Dijk AD, de Bont E, Kornblau SM. Targeted Therapy in Acute Myeloid Leukemia: Current Status and New Insights From a Proteomic Perspective. Expert Rev Proteomics (2020) 17(1):1–10. doi: 10.1080/14789450.2020.1717951
11. Goldman SL, Hassan C, Khunte M, Soldatenko A, Jong Y, Afshinnekoo K, et al. Epigenetic Modifications in Acute Myeloid Leukemia: Prognosis, Treatment, and Heterogeneity. Front Genet (2019) 10:133. doi: 10.3389/fgene.2019.00133
12. Dhall A, Zee BM, Yan F, Blanco MA. Intersection of Epigenetic and Metabolic Regulation of Histone Modifications in Acute Myeloid Leukemia. Front Oncol (2019) 9:432. doi: 10.3389/fonc.2019.00432
13. Wang P, Wang Z, Liu J. Role of HDACs in Normal and Malignant Hematopoiesis. Mol Cancer (2020) 19(1):5. doi: 10.1186/s12943-020-01182-w
14. Ropero S, Esteller M. The Role of Histone Deacetylases (HDACs) in Human Cancer. Mol Oncol (2007) 1(1):19–25. doi: 10.1016/j.molonc.2007.01.001
15. Li Y, Seto E. HDACs and HDAC Inhibitors in Cancer Development and Therapy. Cold Spring Harb Perspect Med (2016) 6(10):1–34. doi: 10.1101/cshperspect.a026831
16. Hiller JK, Schmoor C, Gaidzik VI, Schmidt-Salzmann C, Yalcin A, Abdelkarim M, et al. Evaluating the Impact of Genetic and Epigenetic Aberrations on Survival and Response in Acute Myeloid Leukemia Patients Receiving Epigenetic Therapy. Ann Hematol (2017) 96(4):559–65. doi: 10.1007/s00277-016-2912-7
17. Eriksson A, Lennartsson A, Lehmann S. Epigenetic Aberrations in Acute Myeloid Leukemia: Early Key Events During Leukemogenesis. Exp Hematol (2015) 43(8):609–24. doi: 10.1016/j.exphem.2015.05.009
18. Nebbioso A, Tambaro FP, Dell'Aversana C, Altucci L. Cancer epigenetics: Moving forward. PLoS Genet (2018) 14(6):e1007362. doi: 10.1371/journal.pgen.1007362
19. Hu D, Shilatifard A. Epigenetics of Hematopoiesis and Hematological Malignancies. Genes Dev (2016) 30(18):2021–41. doi: 10.1101/gad.284109.116
20. Gallinari P, Di Marco S, Jones P, Pallaoro M, Steinkuhler C. HDACs, Histone Deacetylation and Gene Transcription: From Molecular Biology to Cancer Therapeutics. Cell Res (2007) 17(3):195–211. doi: 10.1038/sj.cr.7310149
21. Milazzo G, Mercatelli D, Di Muzio G, Triboli L, De Rosa P, Perini G, et al. Histone Deacetylases (HDACs): Evolution, Specificity, Role in Transcriptional Complexes, and Pharmacological Actionability. Genes (Basel) (2020) 11(5):1–49. doi: 10.3390/genes11050556
22. Xia C, Tao Y, Li M, Che T, Qu J. Protein Acetylation and Deacetylation: An Important Regulatory Modification in Gene Transcription (Review). Exp Ther Med (2020) 20(4):2923–40. doi: 10.3892/etm.2020.9073
23. Lawlor L, Yang XB. Harnessing the HDAC-Histone Deacetylase Enzymes, Inhibitors and How These Can be Utilised In Tissue Engineering. Int J Oral Sci (2019) 11(2):20. doi: 10.1038/s41368-019-0053-2
24. Mithraprabhu S, Kalff A, Chow A, Khong T, Spencer A. Dysregulated Class I Histone Deacetylases Are Indicators of Poor Prognosis In Multiple Myeloma. Epigenetics (2014) 9(11):1511–20. doi: 10.4161/15592294.2014.983367
25. Patra S, Panigrahi DP, Praharaj PP, Bhol CS, Mahapatra KK, Mishra SR, et al. Dysregulation of Histone Deacetylases in Carcinogenesis and Tumor Progression: A Possible Link to Apoptosis And Autophagy. Cell Mol Life Sci (2019) 76(17):3263–82. doi: 10.1007/s00018-019-03098-1
26. Xu QY, Yu L. Epigenetic Therapies in Acute Myeloid Leukemia: The Role of Hypomethylating Agents, Histone Deacetylase Inhibitors and the Combination of Hypomethylating Agents With Histone Deacetylase Inhibitors. Chin Med J (Engl) (2020) 6):699–715. doi: 10.1097/CM9.0000000000000685
27. Blagitko-Dorfs N, Schlosser P, Greve G, Pfeifer D, Meier R, Baude A, et al. Combination Treatment of Acute Myeloid Leukemia Cells With DNMT and HDAC Inhibitors: Predominant Synergistic Gene Downregulation Associated With Gene Body Demethylation. Leukemia (2019) 33(4):945–56. doi: 10.1038/s41375-018-0293-8
28. Li X, Su Y, Madlambayan G, Edwards H, Polin L, Kushner J, et al. Antileukemic Activity and Mechanism of Action of the Novel PI3K and Histone Deacetylase Dual Inhibitor CUDC-907 in Acute Myeloid Leukemia. Haematologica (2019) 104(11):2225–40. doi: 10.3324/haematol.2018.201343
29. He Y, Xu L, Feng J, Wu K, Zhao Y, Huang H, et al. HDAC Inhibitor LBH589 Suppresses the Proliferation but Enhances the Antileukemic Effect of Human gammadeltaT Cells. Mol Ther Oncolytics (2020) 18:623–30. doi: 10.1016/j.omto.2020.08.003
30. Yu X, Li H, Hu P, Qing Y, Wang X, Zhu M, et al. Natural HDAC-1/8 Inhibitor Baicalein Exerts Therapeutic Effect in CBF-AML. Clin Transl Med (2020) 10(4):e154. doi: 10.1002/ctm2.154
31. San Jose-Eneriz E, Gimenez-Camino N, Agirre X, Prosper F. HDAC Inhibitors in Acute Myeloid Leukemia. Cancers (Basel) (2019) 11(11):1–24. doi: 10.3390/cancers11111794
32. Rapin N, Porse BT. Oncogenic Fusion Proteins Expressed in Immature Hematopoietic Cells Fail to Recapitulate the Transcriptional Changes Observed in Human AML. Oncogenesis (2014) 3:e106. doi: 10.1038/oncsis.2014.22
33. Lu C. Decoding the Function of an Oncogenic Transcription Factor: Finding the First Responders. Mol Cell (2021) 81(3):418–20. doi: 10.1016/j.molcel.2021.01.008
34. Singh AA, Mandoli A, Prange KH, Laakso M, Martens JH. AML Associated Oncofusion Proteins PML-RARA, AML1-ETO and CBFB-MYH11 Target RUNX/ETS-Factor Binding Sites to Modulate H3ac Levels and Drive Leukemogenesis. Oncotarget (2017) 8(8):12855–65. doi: 10.18632/oncotarget.14150
35. Panuzzo C, Signorino E, Calabrese C, Ali MS, Petiti J, Bracco E, et al. Landscape of Tumor Suppressor Mutations in Acute Myeloid Leukemia. J Clin Med (2020) 9(3):1–25. doi: 10.3390/jcm9030802
36. DiNardo CD, Cortes JE. Mutations in AML: Prognostic and Therapeutic Implications. Hematol Am Soc Hematol Educ Program (2016) 2016(1):348–55. doi: 10.1182/asheducation-2016.1.348
37. Wang RQ, Chen CJ, Jing Y, Qin JY, Li Y, Chen GF, et al. Characteristics and Prognostic Significance of Genetic Mutations in Acute Myeloid Leukemia Based on a Targeted Next-Generation Sequencing Technique. Cancer Med (2020) 9(22):8457–67. doi: 10.1002/cam4.3467
38. Dombret H. Gene Mutation and AML Pathogenesis. Blood (2011) 118(20):5366–7. doi: 10.1182/blood-2011-09-379081
39. Lauber C, Correia N, Trumpp A, Rieger MA, Dolnik A, Bullinger L, et al. Survival Differences and Associated Molecular Signatures of DNMT3A-Mutant Acute Myeloid Leukemia Patients. Sci Rep (2020) 10(1):12761. doi: 10.1038/s41598-020-69691-8
40. Li Y, Lv X, Ge X, Yuan D, Ding M, Zhen C, et al. Mutational Spectrum and Associations With Clinical Features in Patients With Acute Myeloid Leukaemia Based on Nextgeneration Sequencing. Mol Med Rep (2019) 19(5):4147–58. doi: 10.3892/mmr.2019.10081
41. Walter RB, Othus M, Burnett AK, Lowenberg B, Kantarjian HM, Ossenkoppele GJ, et al. Significance of FAB subclassification of "Acute Myeloid Leukemia, NOS" in the 2008 WHO Classification: Analysis of 5848 Newly Diagnosed Patients. Blood (2013) 121(13):2424–31. doi: 10.1182/blood-2012-10-462440
42. Schoch C, Schnittger S, Kern W, Dugas M, Hiddemann W, Haferlach T. Acute Myeloid Leukemia With Recurring Chromosome Abnormalities as Defined by the WHO-Classification: Incidence of Subgroups, Additional Genetic Abnormalities, FAB Subtypes and Age Distribution in an Unselected Series of 1,897 Patients With Acute Myeloid Leukemia. Haematologica (2003) 88(3):351–2.
43. Mrozek K, Eisfeld AK, Kohlschmidt J, Carroll AJ, Walker CJ, Nicolet D, et al. Complex Karyotype in De Novo Acute Myeloid Leukemia: Typical and Atypical Subtypes Differ Molecularly and Clinically. Leukemia (2019) 33(7):1620–34. doi: 10.1038/s41375-019-0390-3
44. Hassan K, Qureshi M, Shafi S, Ikram N, Akhtar MJ. Acute Myeloid Leukemia-FAB Classification and Its Correlation With Clinico-Haematological Features. J Pak Med Assoc (1993) 43(10):200–3.
45. Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, et al. The 2016 Revision to the World Health Organization Classification of Myeloid Neoplasms and Acute Leukemia. Blood (2016) 127(20):2391–405. doi: 10.1182/blood-2016-03-643544
46. Zhang RD, Wang LY, Zheng HY. [Interpretation of the 2016 Revision to the World Health Organization Classification of Acute Leukemia]. Zhonghua Er Ke Za Zhi (2017) 55(1):15–8. doi: 10.3760/cma.j.issn.0578-1310.2017.01.003
47. Arber DA. The 2016 WHO Classification of Acute Myeloid Leukemia: What the Practicing Clinician Needs to Know. Semin Hematol (2019) 56(2):90–5. doi: 10.1053/j.seminhematol.2018.08.002
48. Hong M, He G. 2016 Revision to the WHO Classification of Acute Myeloid Leukemia. J Transl Int Med (2017) 5(2):69–71. doi: 10.1515/jtim-2016-0041
49. Jung J, Cho BS, Kim HJ, Han E, Jang W, Han K, et al. Reclassification of Acute Myeloid Leukemia According to the 2016 WHO Classification. Ann Lab Med (2019) 39(3):311–6. doi: 10.3343/alm.2019.39.3.311
50. Pourrajab F, Zare-Khormizi MR, Hashemi AS, Hekmatimoghaddam S. Genetic Characterization and Risk Stratification of Acute Myeloid Leukemia. Cancer Manag Res (2020) 12:2231–53. doi: 10.2147/CMAR.S242479
51. Marino-Ramirez L, Kann MG, Shoemaker BA, Landsman D. Histone Structure and Nucleosome Stability. Expert Rev Proteomics (2005) 2(5):719–29. doi: 10.1586/14789450.2.5.719
52. Vaughan RM, Kupai A, Rothbart SB. Chromatin Regulation Through Ubiquitin and Ubiquitin-like Histone Modifications. Trends Biochem Sci (2021) 46(4):258–69. doi: 10.1016/j.tibs.2020.11.005
53. Morgan MAJ, Shilatifard A. Reevaluating the Roles of Histone-Modifying Enzymes and Their Associated Chromatin Modifications in Transcriptional Regulation. Nat Genet (2020) 52(12):1271–81. doi: 10.1038/s41588-020-00736-4
54. Binder H, Steiner L, Przybilla J, Rohlf T, Prohaska S, Galle J. Transcriptional Regulation by Histone Modifications: Towards a Theory of Chromatin Re-Organization During Stem Cell Differentiation. Phys Biol (2013) 10(2):026006. doi: 10.1088/1478-3975/10/2/026006
55. Tessarz P, Kouzarides T. Histone core modifications regulating nucleosome structure and dynamics. Nat Rev Mol Cell Biol (2014) 15(11):703–8. doi: 10.1038/nrm3890
56. Kurumizaka H, Kujirai T, Takizawa Y. Contributions of Histone Variants in Nucleosome Structure and Function. J Mol Biol (2021) 433(6):166678. doi: 10.1016/j.jmb.2020.10.012
57. Zhao Z, Shilatifard A. Epigenetic Modifications of Histones in Cancer. Genome Biol (2019) 20(1):245. doi: 10.1186/s13059-019-1870-5
58. Seto E, Yoshida M. Erasers of Histone Acetylation: The Histone Deacetylase Enzymes. Cold Spring Harb Perspect Biol (2014) 6(4):a018713. doi: 10.1101/cshperspect.a018713
59. Kee HJ, Kook H. Roles and Targets of Class I and IIa Histone Deacetylases in Cardiac Hypertrophy. J BioMed Biotechnol (2011) 2011:928326. doi: 10.1155/2011/928326
60. Tang X, Li G, Su F, Cai Y, Shi L, Meng Y, et al. HDAC8 Cooperates With SMAD3/4 Complex to Suppress SIRT7 and Promote Cell Survival and Migration. Nucleic Acids Res (2020) 48(6):2912–23. doi: 10.1093/nar/gkaa039
61. Delcuve GP, Khan DH, Davie JR. Roles of Histone Deacetylases in Epigenetic Regulation: Emerging Paradigms From Studies With Inhibitors. Clin Epigenet (2012) 4(1):5. doi: 10.1186/1868-7083-4-5
62. Rajan A, Shi H, Xue B. Class I and II Histone Deacetylase Inhibitors Differentially Regulate Thermogenic Gene Expression in Brown Adipocytes. Sci Rep (2018) 8(1):13072. doi: 10.1038/s41598-018-31560-w
63. Chakrabarti A, Oehme I, Witt O, Oliveira G, Sippl W, Romier C, et al. HDAC8: A Multifaceted Target For Therapeutic Interventions. Trends Pharmacol Sci (2015) 36(7):481–92. doi: 10.1016/j.tips.2015.04.013
64. Millard CJ, Watson PJ, Fairall L, Schwabe JWR. Targeting Class I Histone Deacetylases in a "Complex" Environment. Trends Pharmacol Sci (2017) 38(4):363–77. doi: 10.1016/j.tips.2016.12.006
65. Millard CJ, Watson PJ, Celardo I, Gordiyenko Y, Cowley SM, Robinson CV, et al. Class I HDACs Share a Common Mechanism of Regulation by Inositol Phosphates. Mol Cell (2013) 51(1):57–67. doi: 10.1016/j.molcel.2013.05.020
66. Park SY, Kim JS. A Short Guide to Histone Deacetylases Including Recent Progress on Class II Enzymes. Exp Mol Med (2020) 52(2):204–12. doi: 10.1038/s12276-020-0382-4
67. Clocchiatti A, Florean C, Brancolini C. Class IIa HDACs: From Important Roles in Differentiation to Possible Implications in Tumourigenesis. J Cell Mol Med (2011) 15(9):1833–46. doi: 10.1111/j.1582-4934.2011.01321.x
68. Haberland M, Montgomery RL, Olson EN. The Many Roles of Histone Deacetylases in Development and Physiology: Implications For Disease and Therapy. Nat Rev Genet (2009) 10(1):32–42. doi: 10.1038/nrg2485
69. Di Giorgio E, Dalla E, Franforte E, Paluvai H, Minisini M, Trevisanut M, et al. Different Class IIa HDACs Repressive Complexes Regulate Specific Epigenetic Responses Related to Cell Survival in Leiomyosarcoma Cells. Nucleic Acids Res (2020) 48(2):646–64.
70. Bertos NR, Wang AH, Yang XJ. Class II Histone Deacetylases: Structure, Function, and Regulation. Biochem Cell Biol (2001) 79(3):243–52. doi: 10.1139/o01-032
71. Cante-Barrett K, Pieters R, Meijerink JP. Myocyte Enhancer Factor 2C in Hematopoiesis and Leukemia. Oncogene (2014) 33(4):403–10. doi: 10.1038/onc.2013.56
72. Kovacs JJ, Murphy PJ, Gaillard S, Zhao X, Wu JT, Nicchitta CV, et al. HDAC6 Regulates Hsp90 Acetylation and Chaperone-Dependent Activation of Glucocorticoid Receptor. Mol Cell (2005) 18(5):601–7. doi: 10.1016/j.molcel.2005.04.021
73. Pinzi L, Benedetti R, Altucci L, Rastelli G. Design of Dual Inhibitors of Histone Deacetylase 6 and Heat Shock Protein 90. ACS Omega (2020) 5(20):11473–80. doi: 10.1021/acsomega.0c00559
74. Kuta R, Larochelle N, Fernandez M, Pal A, Minotti S, Tibshirani M, et al. Depending on the Stress, Histone Deacetylase Inhibitors Act as Heat Shock Protein Co-Inducers in Motor Neurons and Potentiate Arimoclomol, Exerting Neuroprotection Through Multiple Mechanisms in ALS Models. Cell Stress Chaperones (2020) 25(1):173–91. doi: 10.1007/s12192-019-01064-1
75. Pernet L, Faure V, Gilquin B, Dufour-Guerin S, Khochbin S, Vourc'h C. HDAC6-Ubiquitin Interaction Controls the Duration of HSF1 Activation After Heat Shock. Mol Biol Cell (2014) 25(25):4187–94. doi: 10.1091/mbc.e14-06-1032
76. Liu X, Wang Y, Zhang R, Jin T, Qu L, Jin Q, et al. HDAC10 Is Positively Associated With PD-L1 Expression and Poor Prognosis in Patients With NSCLC. Front Oncol (2020) 10:485. doi: 10.3389/fonc.2020.00485
77. Wang L, Zheng S, Zhang L, Xiao H, Gan H, Chen H, et al. Histone Deacetylation 10 Alleviates Inflammation After Intracerebral Hemorrhage via the PTPN22/NLRP3 Pathway in Rats. Neuroscience (2020) 432:247–59. doi: 10.1016/j.neuroscience.2020.02.027
78. Powers J, Lienlaf M, Perez-Villarroel P, Deng S, Knox T, Villagra A, et al. Expression and Function of Histone Deacetylase 10 (HDAC10) in B Cell Malignancies. Methods Mol Biol (2016) 1436:129–45. doi: 10.1007/978-1-4939-3667-0_10
79. Boltz TA, Khuri S, Wuchty S. Promoter Conservation in HDACs Points to Functional Implications. BMC Genomics (2019) 20(1):613. doi: 10.1186/s12864-019-5973-x
80. Villagra A, Cheng F, Wang H, Suarez I, Glozak M, Maurin M, et al. The Histone Deacetylase HDAC11 Regulates The Expression of Interleukin 10 and Immune Tolerance. Nat Immunol (2009) 10(1):92–100. doi: 10.1038/ni.1673
81. Liu SS, Wu F, Jin YM, Chang WQ, Xu TM. HDAC11: A Rising Star in Epigenetics. BioMed Pharmacother (2020) 131:110607. doi: 10.1016/j.biopha.2020.110607
82. Yue L, Sharma V, Horvat NP, Akuffo AA, Beatty MS, Murdun C, et al. HDAC11 Deficiency Disrupts Oncogene-Induced Hematopoiesis in Myeloproliferative Neoplasms. Blood (2020) 135(3):191–207. doi: 10.1182/blood.2019895326
83. Kim E, Bisson WH, Lohr CV, Williams DE, Ho E, Dashwood RH, et al. Histone and Non-Histone Targets of Dietary Deacetylase Inhibitors. Curr Top Med Chem (2016) 16(7):714–31. doi: 10.2174/1568026615666150825125857
84. Peng L, Seto E. Deacetylation of Nonhistone Proteins by HDACs and the Implications in Cancer. Handb Exp Pharmacol (2011) 206:39–56. doi: 10.1007/978-3-642-21631-2_3
85. Toro TB, Watt TJ. Critical Review of Non-Histone Human Substrates of Metal-Dependent Lysine Deacetylases. FASEB J (2020) 34(10):13140–55. doi: 10.1096/fj.202001301RR
86. Keremu A, Aimaiti A, Liang Z, Zou X. Role of the HDAC6/STAT3 Pathway in Regulating PD-L1 Expression in Osteosarcoma Cell Lines. Cancer Chemother Pharmacol (2019) 83(2):255–64. doi: 10.1007/s00280-018-3721-6
87. Kumar S, Attrish D, Srivastava A, Banerjee J, Tripathi M, Chandra PS, et al. Non-Histone Substrates of Histone Deacetylases as Potential Therapeutic Targets in Epilepsy. Expert Opin Ther Targets (2021) 25(1):75–85. doi: 10.1080/14728222.2021.1860016
88. Feng Y, Endo M, Sugiyama H. Nucleosomes and Epigenetics from a Chemical Perspective. Chembiochem (2021) 22(4):595–612. doi: 10.1002/cbic.202000332
89. Wang ZA, Millard CJ, Lin CL, Gurnett JE, Wu M, Lee K, et al. Diverse Nucleosome Site-Selectivity Among Histone Deacetylase Complexes. Elife (2020) 9:1–19. doi: 10.7554/eLife.57663
90. Suraweera A, O'Byrne KJ, Richard DJ. Combination Therapy With Histone Deacetylase Inhibitors (HDACi) for the Treatment of Cancer: Achieving the Full Therapeutic Potential of HDACi. Front Oncol (2018) 8:92. doi: 10.3389/fonc.2018.00092
91. Sanaei M, Kavoosi F. Histone Deacetylases and Histone Deacetylase Inhibitors: Molecular Mechanisms of Action in Various Cancers. Adv BioMed Res (2019) 8:63. doi: 10.4103/abr.abr_142_19
92. Klaus M, Haferlach T, Schnittger S, Kern W, Hiddemann W, Schoch C. Cytogenetic Profile in De Novo Acute Myeloid Leukemia With FAB Subtypes M0, M1, and M2: A Study Based on 652 Cases Analyzed With Morphology, Cytogenetics, and Fluorescence In Situ Hybridization. Cancer Genet Cytogenet (2004) 155(1):47–56. doi: 10.1016/j.cancergencyto.2004.03.008
93. Reikvam H, Hatfield KJ, Kittang AO, Hovland R, Bruserud O. Acute Myeloid Leukemia With the t (8,21) Translocation: Clinical Consequences and Biological Implications. J BioMed Biotechnol (2011) 2011:104631. doi: 10.1155/2011/104631
94. Nishii K, Usui E, Katayama N, Lorenzo FT, Nakase K, Kobayashi T, et al. Characteristics of t (8,21) Acute Myeloid Leukemia (AML) with Additional Chromosomal Abnormality: Concomitant Trisomy 4 May Constitute a Distinctive Subtype of t (8,21) AML. Leukemia (2003) 17(4):731–7. doi: 10.1038/sj.leu.2402871
95. Kellaway S, Chin PS, Barneh F, Bonifer C, Heidenreich O. t(8,21) Acute Myeloid Leukemia as a Paradigm for the Understanding of Leukemogenesis at the Level of Gene Regulation and Chromatin Programming. Cells (2020) 9(12):1–13. doi: 10.3390/cells9122681
96. Licht JD. AML1 and the AML1-ETO Fusion Protein in the Pathogenesis of t (8,21) AML. Oncogene (2001) 20(40):5660–79. doi: 10.1038/sj.onc.1204593
97. Li X, Xu YB, Wang Q, Lu Y, Zheng YC, Wang YC, et al. Leukemogenic AML1-ETO Fusion Protein Upregulates Expression of Connexin 43: The Role in AML 1-ETO-Induced Growth Arrest in Leukemic Cells. J Cell Physiol (2006) 208(3):594–601. doi: 10.1002/jcp.20695
98. DeKelver RC, Yan M, Ahn EY, Shia WJ, Speck NA, Zhang DE, et al. Attenuation of AML1-ETO Cellular Dysregulation Correlates With Increased Leukemogenic Potential. Blood (2013) 121(18):3714–7. doi: 10.1182/blood-2012-11-465641
99. Ichikawa M, Goyama S, Asai T, Kawazu M, Nakagawa M, Takeshita M, et al. AML1/Runx1 Negatively Regulates Quiescent Hematopoietic Stem Cells in Adult Hematopoiesis. J Immunol (2008) 180(7):4402–8. doi: 10.4049/jimmunol.180.7.4402
100. Okuda T, Nishimura M, Nakao M, Fujita Y. RUNX1/AML1: A Central Player in Hematopoiesis. Int J Hematol (2001) 74(3):252–7. doi: 10.1007/BF02982057
101. Tahirov TH, Inoue-Bungo T, Morii H, Fujikawa A, Sasaki M, Kimura K, et al. Structural Analyses of DNA Recognition by the AML1/Runx-1 Runt Domain and Its Allosteric Control by CBFbeta. Cell (2001) 104(5):755–67. doi: 10.1016/S0092-8674(01)00271-9
102. Yan M, Ahn EY, Hiebert SW, Zhang DE. RUNX1/AML1 DNA-Binding Domain and ETO/MTG8 NHR2-Dimerization Domain Are Critical to AML1-ETO9a Leukemogenesis. Blood (2009) 113(4):883–6. doi: 10.1182/blood-2008-04-153742
103. Elagib KE, Goldfarb AN. Oncogenic Pathways of AML1-ETO in Acute Myeloid Leukemia: Multifaceted Manipulation of Marrow Maturation. Cancer Lett (2007) 251(2):179–86. doi: 10.1016/j.canlet.2006.10.010
104. Ito Y, Bae SC, Chuang LS. The RUNX Family: Developmental Regulators in Cancer. Nat Rev Cancer (2015) 15(2):81–95. doi: 10.1038/nrc3877
105. Otalora-Otalora BA, Henriquez B, Lopez-Kleine L, Rojas A. RUNX Family: Oncogenes or Tumor Suppressors (Review). Oncol Rep (2019) 42(1):3–19. doi: 10.3892/or.2019.7149
106. Samarakkody AS, Shin NY, Cantor AB. Role of RUNX Family Transcription Factors in DNA Damage Response. Mol Cells (2020) 43(2):99–106. doi: 10.14348/molcells.2019.0304
107. Bonifer C, Levantini E, Kouskoff V, Lacaud G. Runx1 Structure and Function in Blood Cell Development. Adv Exp Med Biol (2017) 962:65–81. doi: 10.1007/978-981-10-3233-2_5
108. Mottis A, Mouchiroud L, Auwerx J. Emerging Roles of the Corepressors NCoR1 and SMRT in Homeostasis. Genes Dev (2013) 27(8):819–35. doi: 10.1101/gad.214023.113
109. Guenther MG, Barak O, Lazar MA. The SMRT and N-CoR Corepressors Are Activating Cofactors For Histone Deacetylase 3. Mol Cell Biol (2001) 21(18):6091–101. doi: 10.1128/MCB.21.18.6091-6101.2001
110. Chen-Wichmann L, Shvartsman M, Preiss C, Hockings C, Windisch R, Redondo Monte E, et al. Compatibility of RUNX1/ETO Fusion Protein Modules Driving CD34+ Human Progenitor Cell Expansion. Oncogene (2019) 38(2):261–72. doi: 10.1038/s41388-018-0441-7
111. van der Kouwe E, Staber PB. RUNX1-ETO: Attacking the Epigenome for Genomic Instable Leukemia. Int J Mol Sci (2019) 20(2):1–17. doi: 10.3390/ijms20020350
112. Hug BA, Lazar MA. ETO Interacting Proteins. Oncogene (2004) 23(24):4270–4. doi: 10.1038/sj.onc.1207674
113. Okumura AJ, Peterson LF, Okumura F, Boyapati A, Zhang DE. t (8,21)(q22;q22) Fusion Proteins Preferentially Bind to Duplicated AML1/RUNX1 DNA-Binding Sequences to Differentially Regulate Gene Expression. Blood (2008) 112(4):1392–401. doi: 10.1182/blood-2007-11-124735
114. Xiao Y, Liu Y. Recent Advances in the Discovery of Novel HSP90 Inhibitors: An Update From 2014. Curr Drug Targets (2020) 21(3):302–17. doi: 10.2174/1389450120666190829162544
115. Peterson LF, Zhang DE. The 8;21 Translocation in Leukemogenesis. Oncogene (2004) 23(24):4255–62. doi: 10.1038/sj.onc.1207727
116. Rulina AV, Spirin PV, Prassolov VS. Activated Leukemic Oncogenes AML1-ETO and c-kit: Role in Development of Acute Myeloid Leukemia and Current Approaches for Their Inhibition. Biochem (Mosc) (2010) 75(13):1650–66. doi: 10.1134/S0006297910130092
117. Roudaia L, Cheney MD, Manuylova E, Chen W, Morrow M, Park S, et al. CBFbeta is Critical for AML1-ETO and TEL-AML1 Activity. Blood (2009) 113(13):3070–9. doi: 10.1182/blood-2008-03-147207
118. Thiel VN, Giaimo BD, Schwarz P, Soller K, Vas V, Bartkuhn M, et al. Heterodimerization of AML1/ETO With CBFbeta is Required for Leukemogenesis But Not for Myeloproliferation. Leukemia (2017) 31(11):2491–502. doi: 10.1038/leu.2017.105
119. Liu S, Klisovic RB, Vukosavljevic T, Yu J, Paschka P, Huynh L, et al. Targeting AML1/ETO-Histone Deacetylase Repressor Complex: A Novel Mechanism For Valproic Acid-Mediated Gene Expression and Cellular Differentiation in AML1/ETO-Positive Acute Myeloid Leukemia Cells. J Pharmacol Exp Ther (2007) 321(3):953–60. doi: 10.1124/jpet.106.118406
120. Huang Y, Chen J, Lu C, Han J, Wang G, Song C, et al. HDAC1 and Klf4 Interplay Critically Regulates Human Myeloid Leukemia Cell Proliferation. Cell Death Dis (2014) 5:e1491. doi: 10.1038/cddis.2014.433
121. Wingelhofer B, Somervaille TCP. Emerging Epigenetic Therapeutic Targets in Acute Myeloid Leukemia. Front Oncol (2019) 9:850. doi: 10.3389/fonc.2019.00850
122. Prokocimer M, Molchadsky A, Rotter V. Dysfunctional Diversity of p53 Proteins in Adult Acute Myeloid Leukemia: Projections on Diagnostic Workup and Therapy. Blood (2017) 130(6):699–712. doi: 10.1182/blood-2017-02-763086
123. Guo C, Li J, Steinauer N, Wong M, Wu B, Dickson A, et al. Histone Deacetylase 3 Preferentially Binds and Collaborates With the Transcription Factor RUNX1 to Repress AML1-ETO-Dependent Transcription in t (8,21) AML. J Biol Chem (2020) 295(13):4212–23. doi: 10.1074/jbc.RA119.010707
124. Beghini A. Core Binding Factor Leukemia: Chromatin Remodeling Moves Towards Oncogenic Transcription. Cancers (Basel) (2019) 11(12):1–21. doi: 10.3390/cancers11121973
125. Richter LE, Wang Y, Becker ME, Coburn RA, Williams JT, Amador C, et al. HDAC1 Is a Required Cofactor of CBFbeta-SMMHC and a Potential Therapeutic Target in Inversion 16 Acute Myeloid Leukemia. Mol Cancer Res (2019) 17(6):1241–52. doi: 10.1158/1541-7786.MCR-18-0922
126. Biernacki MA, Foster KA, Woodward KB, Coon ME, Cummings C, Cunningham TM, et al. CBFB-MYH11 Fusion Neoantigen Enables T Cell Recognition and Killing of Acute Myeloid Leukemia. J Clin Invest (2020) 130(10):5127–41. doi: 10.1172/JCI137723
127. Cordonnier G, Mandoli A, Cagnard N, Hypolite G, Lhermitte L, Verhoeyen E, et al. CBFbeta-SMMHC Affects Genome-wide Polycomb Repressive Complex 1 Activity in Acute Myeloid Leukemia. Cell Rep (2020) 30(2):299–307.e293. doi: 10.1016/j.celrep.2019.12.026
128. Qi J, Singh S, Hua WK, Cai Q, Chao SW, Li L, et al. HDAC8 Inhibition Specifically Targets Inv(16) Acute Myeloid Leukemic Stem Cells by Restoring p53 Acetylation. Cell Stem Cell (2015) 17(5):597–610. doi: 10.1016/j.stem.2015.08.004
129. Spreafico M, et al. HDAC8: A Promising Therapeutic Target for Acute Myeloid Leukemia. Front Cell Dev Biol (2020) 8:844. doi: 10.3389/fcell.2020.00844
130. Liquori A, Ibanez M, Sargas C, Sanz MA, Barragan E, Cervera J. Acute Promyelocytic Leukemia: A Constellation of Molecular Events around a Single PML-RARA Fusion Gene. Cancers (Basel) (2020) 12(3). doi: 10.3390/cancers12030624
131. Geoffroy MC, de The H. Classic and Variants APLs, as Viewed from a Therapy Response. Cancers (Basel) (2020) 12(4):1–22. doi: 10.3390/cancers12040967
132. Martens JH, Brinkman AB, Simmer F, Francoijs KJ, Nebbioso A, Ferrara F, et al. PML-RARalpha/RXR Alters the Epigenetic Landscape in Acute Promyelocytic Leukemia. Cancer Cell (2010) 17(2):173–85. doi: 10.1016/j.ccr.2009.12.042
133. Guan D, Kao HY. The function, regulation and therapeutic implications of the tumor suppressor protein, PML. Cell Biosci (2015) 5:60. doi: 10.1186/s13578-015-0051-9
134. Saeed S, Logie C, Stunnenberg HG, Martens JH. Genome-Wide Functions of PML-RARalpha in Acute Promyelocytic Leukaemia. Br J Cancer (2011) 104(4):554–8. doi: 10.1038/sj.bjc.6606095
135. Liu XL, Liu HQ, Li J, Mao CY, He JT, Zhao X. Role of Epigenetic in Leukemia: From Mechanism to Therapy. Chem Biol Interact (2020) 317:108963. doi: 10.1016/j.cbi.2020.108963
136. Insinga A, Monestiroli S, Ronzoni S, Carbone R, Pearson M, Pruneri G, et al. Impairment of p53 Acetylation, Stability and Function By an Oncogenic Transcription Factor. EMBO J (2004) 23(5):1144–54. doi: 10.1038/sj.emboj.7600109
137. Molica M, Mazzone C, Niscola P, de Fabritiis P. TP53 Mutations in Acute Myeloid Leukemia: Still a Daunting Challenge? Front Oncol (2020) 10:610820. doi: 10.3389/fonc.2020.610820
138. Arteaga MF, Mikesch JH, Fung TK, So CW. Epigenetics in Acute Promyelocytic Leukaemia Pathogenesis and Treatment Response: A TRAnsition to Targeted Therapies. Br J Cancer (2015) 112(3):413–8. doi: 10.1038/bjc.2014.374
139. Singh AA, Petraglia F, Nebbioso A, Yi G, Conte M, Valente S, et al. Multi-Omics Profiling Reveals a Distinctive Epigenome Signature For High-Risk Acute Promyelocytic Leukemia. Oncotarget (2018) 9(39):25647–60. doi: 10.18632/oncotarget.25429
140. Leiva M, Moretti S, Soilihi H, Pallavicini I, Peres L, Mercurio C, et al. Valproic Acid Induces Differentiation and Transient Tumor Regression, But Spares Leukemia-Initiating Activity in Mouse Models of APL. Leukemia (2012) 26(7):1630–7. doi: 10.1038/leu.2012.39
141. Mehdipour P, Santoro F, Botrugno OA, Romanenghi M, Pagliuca C, Matthews GM, et al. HDAC3 Activity is Required for Initiation of Leukemogenesis in Acute Promyelocytic Leukemia. Leukemia (2017) 31(4):995–7. doi: 10.1038/leu.2017.3
142. Chauchereau A, Mathieu M, de Saintignon J, Ferreira R, Pritchard LL, Mishal Z, et al. HDAC4 Mediates Transcriptional Repression by the Acute Promyelocytic Leukaemia-Associated Protein PLZF. Oncogene (2004) 23(54):8777–84. doi: 10.1038/sj.onc.1208128
143. Biswas D, Milne TA, Basrur V, Kim J, Elenitoba-Johnson KS, Allis CD, et al. Function of Leukemogenic Mixed Lineage Leukemia 1 (MLL) Fusion Proteins Through Distinct Partner Protein Complexes. Proc Natl Acad Sci U S A (2011) 108(38):15751–6. doi: 10.1073/pnas.1111498108
144. Slany RK. MLL Fusion Proteins and Transcriptional Control. Biochim Biophys Acta Gene Regul Mech (2020) 1863(3):194503. doi: 10.1016/j.bbagrm.2020.194503
145. Marschalek R. Systematic Classification of Mixed-Lineage Leukemia Fusion Partners Predicts Additional Cancer Pathways. Ann Lab Med (2016) 36(2):85–100. doi: 10.3343/alm.2016.36.2.85
146. Ney Garcia DR, Liehr T, Emerenciano M, Meyer C, Marschalek R, Pombode-Oliveira Mdo S, et al. Molecular Studies Reveal a MLL-MLLT3 Gene Fusion Displaced in a Case of Childhood Acute Lymphoblastic Leukemia With Complex Karyotype. Cancer Genet (2015) 208(4):143–7. doi: 10.1016/j.cancergen.2015.02.002
147. Lillico R, Lawrence CK, Lakowski TM. Selective DOT1L, LSD1, and HDAC Class I Inhibitors Reduce HOXA9 Expression in MLL-AF9 Rearranged Leukemia Cells, But Dysregulate the Expression of Many Histone-Modifying Enzymes. J Proteome Res (2018) 17(8):2657–67. doi: 10.1021/acs.jproteome.8b00118
148. Ye J, Zha J, Shi Y, Li Y, Yuan D, Chen Q, et al. Co-inhibition of HDAC and MLL-Menin Interaction Targets MLL-Rearranged Acute Myeloid Leukemia Cells via Disruption of DNA Damage Checkpoint and DNA Repair. Clin Epigenet (2019) 11(1):137. doi: 10.1186/s13148-019-0723-0
149. Ahmad K, Katryniok C, Scholz B, Merkens J, Loscher D, Marschalek R, et al. Inhibition of Class I HDACs Abrogates the Dominant Effect of MLL-AF4 by Activation Of Wild-Type MLL. Oncogenesis (2014) 3:e127. doi: 10.1038/oncsis.2014.39
150. Xia ZB, Anderson M, Diaz MO, Zeleznik-Le NJ. MLL Repression Domain Interacts With Histone Deacetylases, the Polycomb Group Proteins HPC2 and BMI-1, and the Corepressor C-Terminal-Binding Protein. Proc Natl Acad Sci U S A (2003) 100(14):8342–7. doi: 10.1073/pnas.1436338100
151. Lenard A, Xie HM, Pastuer T, Shank T, Libbrecht C, Kingsley M, et al. Epigenetic Regulation of Protein Translation in KMT2A-Rearranged AML. Exp Hematol (2020) 85:57–69. doi: 10.1016/j.exphem.2020.04.007
152. Li S, Mason CE, Melnick A. Genetic and Epigenetic Heterogeneity in Acute Myeloid Leukemia. Curr Opin Genet Dev (2016) 36:100–6. doi: 10.1016/j.gde.2016.03.011
153. Wouters BJ, Delwel R. Epigenetics and Approaches to Targeted Epigenetic Therapy in Acute Myeloid Leukemia. Blood (2016) 127(1):42–52. doi: 10.1182/blood-2015-07-604512
154. Ellmeier W, Seiser C. Histone Deacetylase Function in CD4(+) T Cells. Nat Rev Immunol (2018) 18(10):617–34. doi: 10.1038/s41577-018-0037-z
155. Yang Y, Wang Y. Role of Epigenetic Regulation in Plasticity of Tumor Immune Microenvironment. Front Immunol (2021) 12:640369. doi: 10.3389/fimmu.2021.640369
156. Boila LD, Sengupta A. Evolving Insights on Histone Methylome Regulation in Human Acute Myeloid Leukemia Pathogenesis and Targeted Therapy. Exp Hematol (2020) 92:19–31. doi: 10.1016/j.exphem.2020.09.189
157. Dahiya S, Beier UH, Wang L, Han R, Jiao J, Akimova T, et al. HDAC10 Deletion Promotes Foxp3(+) T-Regulatory Cell Function. Sci Rep (2020) 10(1):424. doi: 10.1038/s41598-019-57294-x
158. Banik D, Moufarrij S, Villagra A. Immunoepigenetics Combination Therapies: An Overview of the Role of HDACs in Cancer Immunotherapy. Int J Mol Sci (2019) 20(9):1–29. doi: 10.3390/ijms20092241
Keywords: HDACs, AML, leukemogenesis, epigenetic modification, oncogenic fusion protein, chromosomal translocation
Citation: Zhang J, Gao X and Yu L (2021) Roles of Histone Deacetylases in Acute Myeloid Leukemia With Fusion Proteins. Front. Oncol. 11:741746. doi: 10.3389/fonc.2021.741746
Received: 15 July 2021; Accepted: 11 August 2021;
Published: 01 September 2021.
Edited by:
Shujun Liu, University of Minnesota Twin Cities, United StatesReviewed by:
Yushi Yao, Zhejiang University, ChinaLin Fu, The Second Affiliated Hospital of Guangzhou Medical University, China
Copyright © 2021 Zhang, Gao and Yu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Li Yu, eXVsaUBzenUuZWR1LmNu; Xuefeng Gao, eGZnYW9Ac3p1LmVkdS5jbg==