 Shenon Sethi
Shenon Sethi Zachary Epstein-Peterson2
Zachary Epstein-Peterson2 Caleb Ho
Caleb Ho
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 23 November 2021
Sec. Hematologic Malignancies
Volume 11 - 2021 | https://doi.org/10.3389/fonc.2021.739441
This article is part of the Research Topic Novel Diagnostic Tools and Biomarkers in Hematologic Malignancies View all 21 articles
Mantle Cell lymphoma (MCL) is a mature B-cell lymphoma with a well-known hallmark genetic alteration in most cases, t (11,14)(q13q32)/CCND1-IGH. However, our understanding of the genetic and epigenetic alterations in MCL has evolved over the years, and it is now known that translocations involving CCND2, or cryptic insertion of enhancer elements of IGK or IGL gene, can also lead to MCL. On a molecular level, MCL can be broadly classified into two subtypes, conventional MCL (cMCL) and non-nodal MCL (nnMCL), each with different postulated tumor cell origin, clinical presentation and behavior, mutational pattern as well as genomic complexity. This article reviews both the common and rare alterations in MCL on a gene mutational, chromosomal arm, and epigenetic level, in the context of their contribution to the lymphomagenesis and disease evolution in MCL. This article also summarizes the important prognostic factors, molecular diagnostic tools, and treatment options based on the most recent MCL literature.
Mantle cell lymphoma (MCL) is a relatively uncommon subtype of mature B-cell lymphoma with a heterogenous tumor behavior, with most behaving aggressively while others following an indolent clinical course. There have been a lot of advancements in the understanding of the genetics of MCL since the demonstration of t (11,14)(q13;q32)/CCND1-IGH as a hallmark feature of MCL in 1990s. The pathogenesis of MCL encompasses complex interactions between the tumor microenvironment, including stromal cells and T-cells, signaling via surface immunoglobulins, and tumor cell genetic alterations. Based on the proposed model of molecular pathogenesis, two subtypes of MCL have been recognized, which differ in their clinical and biologic behavior (1) (Table 1). The more common conventional MCL (cMCL) arises from expansion of pre-germinal center/naïve-like B cells that are characterized by frequent expression of the transcription factor SOX11, higher likelihood of unmutated immunoglobulin heavy chain variable region (IGHV), high genomic complexity, and an aggressive clinical behavior. The less common non-nodal (leukemic) variant (nnMCL), on the other hand, is derived from post-germinal center/memory-like B cells that are generally characterized by mutated IGHV, lack of SOX11 expression, low genomic complexity, and an indolent clinical behavior that is partially related to the lack of angiogenic or tumor invasive properties (2–4). The progress in unraveling the pathogenesis and genetic alterations of MCL has also propelled new treatment modalities including molecularly targeted therapies and immunotherapies. This review focuses on the recent developments in the understanding of the mutational-, methylation-, and chromosomal-level alterations seen in MCL and the currently available molecular diagnostic tools and therapy options.

Table 1 Two molecular subtypes of mantle cell lymphoma (MCL).
The translocation t (11,14)(q13;q32) is considered the primary oncogenic event in over 95% of MCL cases that results in the juxtaposition of immunoglobulin heavy-chain (IGH) enhancer region on 14q32 next to CCND1 on 11q13, resulting in its overexpression (1, 5, 6). Irrespective of the molecular subtype, in approximately 90% of the cases, CCND1 rearrangement (3) occurs in the pro/pre-B cell stage during IGH V(D)J recombination and is recombination-activating gene (RAG)-mediated, while in 10% of cases, the translocation occurs in mature B-cell stage during somatic hypermutation (SHM) or class switch recombination and is mediated by B cell-specific activation-induced cytidine deaminase (AID) machinery (3). The mechanism of CCND1 rearrangment has no apparent clinical or biologic impact. The most common breakpoint on IGH locus involves the region between the IGHD and IGHJ gene segments and occurs during the initial step of IGH V(D)J recombination (3, 7). In some cases, the breakpoint occurs during the second step of IGH V(D)J recombination involving the region between the IGHV and IGHD gene segments. The most common breakpoint on 11q32 is located upstream from the CCND1 gene within the major translocation cluster (MTC) (30% of cases), while in the remainder cases, the breakpoints are located either 5’ or 3’ to the MTC locus (3). Besides having IGH as a translocation partner, in a small number of cases, the translocation partner for CCND1 is the immunoglobulin light chain kappa (IGK) or lambda (IGL) gene (Figure 1A). Furthermore, another small subset of cases does not exhibit CCND1 translocation (so-called “Cyclin D1-Negative MCL”). Cases with these uncommon alterations will be discussed in the section Diagnostic Challenges below.
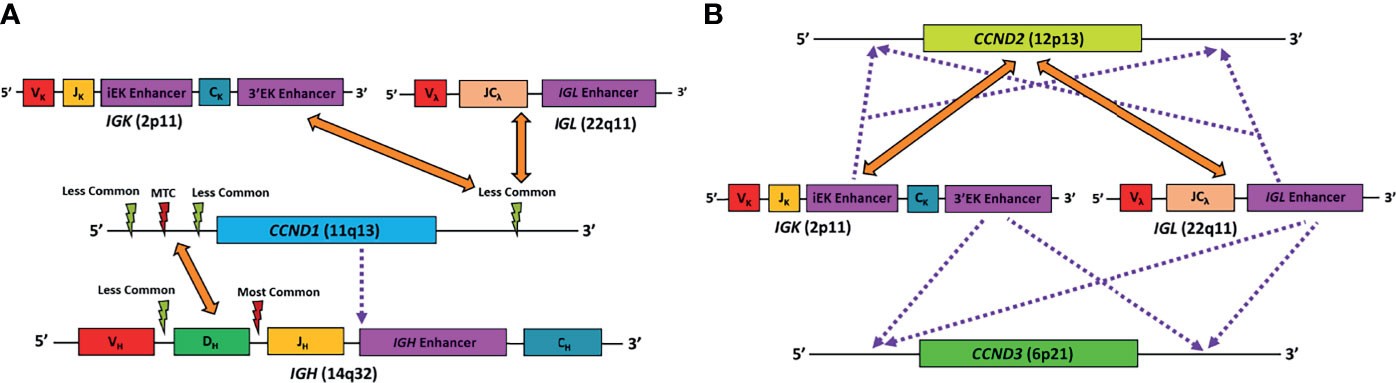
Figure 1 Schematic diagram representing the known major genetic alterations leading to MCL. (A) Alterations found in conventional MCL. Orange arrows point to gene translocation partners, red lightning symbol represents major translocation breakpoint, light green lightning symbol represents minor translocation breakpoints, purple dotted line represents cryptic insertion of elements. In CCND1-IGH, the IGH breakpoints are mostly between the IGHD and IGHJ segments, while others are located between the IGHV and IGHD segments. The CCND1 breakpoints are mostly in the region 5’ to the gene, particularly in the major translocation cluster (MTC). In very rare case, CCND1 coding region can be cryptically inserted into the IGH gene, resulting in CCND1 overexpression. Finally, CCND1 can sometimes pair with IGK or IGL as translocation partner. In these cases, the CCND1 breakpoints are more likely to be in the region 3’ to the gene. (B) Alterations found in cyclin D1-negative MCL. Orange arrows point to gene translocation partners, and purple dotted line represents cryptic insertion of elements. CCND2 mostly utilize IGK or IGL, rather than IGH, as translocation partners. On the other hand, conventional translocation of CCND3 has not been reported in MCL. Alternatively, IGK or IGL enhancer elements can be cryptically inserted into the vicinity of the CCND2 or CCND3 gene, resulting in overexpression of the corresponding gene. VH, VK, Vλ, V gene segments of the IGH, IGK, and IGL genes, respectively; DH, D gene segment of the IGH gene; JH, JK, J gene segments of the IGH and IGK genes, respectively; JCλ, J and C gene segments of the IGL gene.
All types of CCND1 translocations result in overexpression of CCND1, a proto-oncogene that regulates cell cycle transition from G1 to S phase and is often overexpressed or amplified in numerous cancers, including breast, lung, melanoma, and oral squamous cell carcinomas (8). Functions of cyclin D1 include control of cell growth, proliferation, transcription, DNA repair, and migration (9–11). Although Cyclin D1 is not essential for entry into cell cycle progression (12), its amplification/overexpression in human tumors is oncogenic as it allows cancer cells to proliferate independent of extracellular growth signaling cues (8). During lymphomagenesis, cyclin D1 requires cooperation of alterations in other genes, as supported by studies on transgenic mice, which showed that mice solely carrying CCND1 rearrangement did not develop spontaneous lymphomas (13). The other key alterations that play a role in MCL pathogenesis include deletion of CDKN2A locus that encodes p16, a member of the INK4 family of cyclin-dependent kinase inhibitor, amplification of BMI1 that inhibits CDKN2A, deregulation of TP53 via mutation or deletion, MDM2 overexpression, and ATM deletion (1, 2, 5).
Nearly all MCLs carry at least one known driver alteration beside CCND1 translocation, including gene mutations, copy number alterations (CNAs), and structural variants (SVs) (3). Although cMCL carry a significantly higher number of driver alterations as compared to nnMCL, the overall tumor mutational burden (which is calculated based on the number of point mutations) is similar in the two subtypes. Rather, the differences between the two primarily lie in the number of CNAs and SVs, which in general are higher in cMCL compared to nnMCL (3). Alterations in over 40 driver genes involving eight main pathways have been identified in MCL, namely: DNA damage response, proliferation, cell survival, chromatin remodeling, telomere maintenance, B-cell receptor/Toll-like receptor/NF-kB signaling, NOTCH and RNA regulation (3, 14). The most frequently mutated genes include: ATM, TP53, CCND1 (SHM or 3’UTR activation), KMT2D, RB1, BIRC3, CDKN2A, CDKN1B, BCOR, NOTCH1, and TERT alterations (promoter mutation, gain/amplification and translocations) (3, 14–17). More details are provided in Table 2. Most of the pathogenic mutations seen in MCL patients are somatic in nature. However, more recently, germline mutations in ATM and CHEK2 have also been seen, which raises the possibility that germline mutations in these genes may lead to genetic predisposition to the development of MCL (3, 18). Nonetheless, currently any causal relationship remains speculatory rather than proven.

Table 2 Major driver alterations in MCL.
While mutational profile is not necessarily specific to a particular type of lymphoma, mutations in CCND1, RB1, CTNNA2, NSD2, and to a lesser degree, certain types of mutations in ATM, are predominantly seen in MCL in comparison to other common mature B- and T-cell lymphomas (14, 15). The presence of these mutations may be a helpful clue in rare diagnostically-challenging cases of MCL.
Alterations of ATM, a gene involved in the DNA damage repair pathway, are associated with shorter telomere length in MCL in comparison to MCL with wild-type ATM, and consequently, chromosomal instability has been found to be significantly more in MCL with mutated ATM (3, 18). Apart from ATM, there are likely other genes that also play a role in chromosomal instability, as suggested by the observation that high chromosomal instability can be associated with the blastoid variants of MCL irrespective of the ATM gene mutation status. Interestingly, ATM mutations are seen mostly in cMCL but not nnMCL, and are commonly truncating mutations or missense mutations involving the PI3K domain (1, 18). In contrast, even though ATM mutations are also seen in chronic lymphocytic leukemia (CLL), they are present at a much lower frequency (10–15% in CLL vs 40–75% in MCL) and commonly are missense mutations distributed in different areas of the genes.
Among the other frequent mutations found in MCL, CCND1 SHM are predominantly seen in nnMCL (3, 14). Alterations in TP53 have been reported to be either equally distributed among the two subtypes or slightly more enriched in nnMCL, along with TERT alterations (3, 14, 19).
Overall, MCLs are characterized by frequent chromosomal arm-level abnormalities including gains of chromosomal arm 3q25-29, 7p22/CARD11, 8q24/MYC, 10p12/BMI1, 12q13/CDK4, 13q31/MIR17HG, and 18q21/BCL2 and losses of 1p32, 6q/TNFAIP3, 9p21/CDKN2A and CDKN2B, 9q, 11q22/ATM, and BIRC3, 13q14/RB1, and 17p/TP53 (20). Note that 13q14 loss is also common in CLL and not specific to MCL. Deletions involving 17p and 11q are often associated with TP53 and ATM mutations, respectively (3, 14). The genomic landscape of cMCL is more complex as they carry a significantly higher number of CNAs and SVs than nnMCL. Due to high chromosomal instability, complex alterations such as chromoplexy and chromothripsis are also more frequently observed in cMCL, while breakage–fusion–bridge (BFB) cycles have only been seen in cMCL. The following alterations are exclusively seen in cMCL and not nnMCL: deletions of 1p, 10p, and 19p; and gain of 7p (3). Table 3 summarizes the chromosomal arm-level abnormalities seen in MCL.
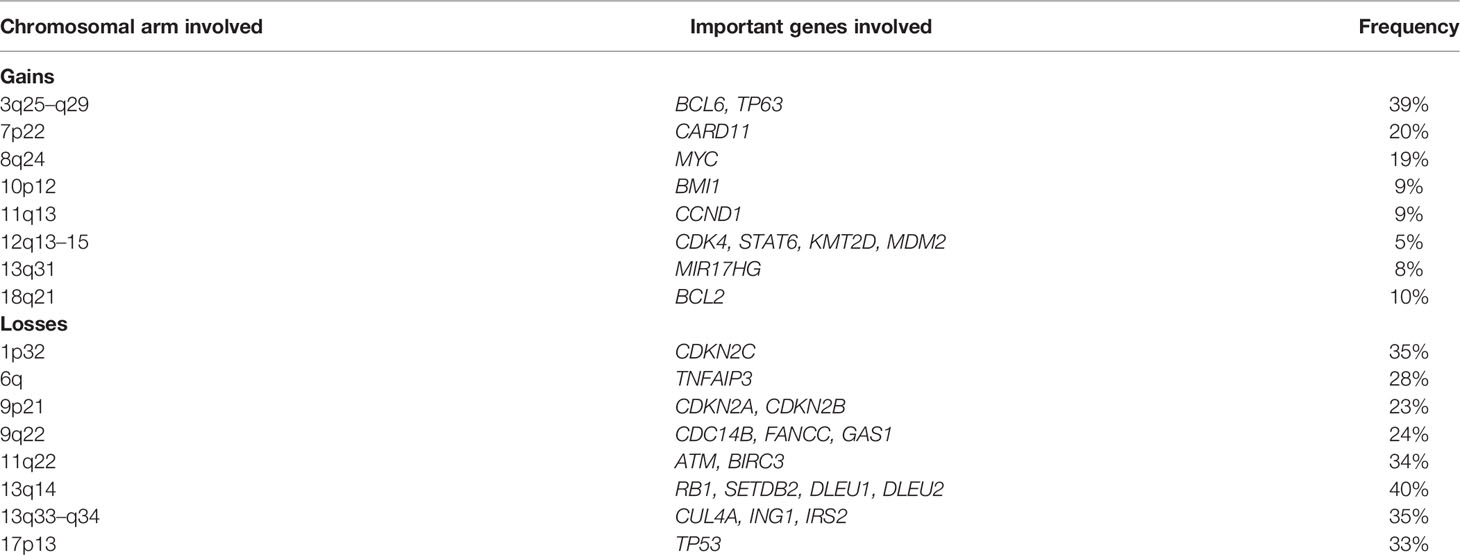
Table 3 Major chromosomal-level alterations in MCL.
Distinguishing between the two subtypes of MCL is important prognostically and therapeutically. Among the two molecular subtypes, there are also several genes that are differentially expressed on a mRNA level. HDGFRP3, FARP1, CSNK1E, SETMAR, HMGB3, LGALS3BP, PON2, CDK2AP1, DBN1, CNR1, CNN3, PLXNB1, DCHS1 NREP, MIML, FNBP1L, FHL1, and SOX11 are upregulated in cMCL, while CD200, BTLA, and SLAMF1 are upregulated in nnMCL (19, 21). Based on this differential gene expression pattern, Clot et al. developed a 16-gene assay (L-MCL16 assay) using the NanoString nCounter® platform (NanoString Technologies, Seattle, WA) that can utilize peripheral blood samples to distinguish between the two subtypes of MCL; however, one of the caveats is that it requires ≥60% tumor cell content (19), limiting the utility of this assay on a broader scale. It is also worth noting that a small number of patients in the validation cohort were in the “undetermined” category. As an alternative, a short 3-gene signature comprised of SOX11, HDGFRP3, and DBN1 can also be used reliably to distinguish cMCL from nnMCL in leukemic samples with at least 30% involvement, where low expression profile is associated with the nnMCL subtype (22). The RNA Gene Expression Profiling section under Important Molecular Diagnostic Tools for Genetic Alterations below provides some important caveats related to gene expression profiling and the nCounter® platform.
Besides looking at the gene expression profile and gene mutation pattern, the DNA methylation study represents another methodology that can shed insights on the tumor biology. Queiros et al. compared the DNA methylation profile of MCL cases and normal non-neoplastic B cells at different stages of maturation using principal component analysis of the first two main components. In the first principal component (PC), all MCL cases have been found to be more similar to germinal center-experienced B-cells, suggesting that MCL originates from cells with some degree of antigenic experience. However, the second PC showed that within MCL, the cases can be divided into two clusters that are biologically and clinically distinct. The first cluster represents mostly the cMCL, and as expected, has a pattern more resembling the germinal center-inexperienced cells, showing either the absence of or low but variable level of IGHV SHM. The second cluster represents most cases of nnMCL, and also as expected, has pattern more resembling the germinal center-experienced cells (3, 23).
One of the main distinguishing factors between cMCL and nnMCL is expression of SOX11, which is usually higher in the former and lower to absent in the latter. SOX11 expression alone is not reliable for classification, because there is a spectrum of SOX11 expression among cases in each subtype, and a subset of cMCL and nnMCL cases can have overlapping level of SOX11 expression (19). The mechanism of SOX11 expression is thought not to be due to gene mutation, but rather hypomethylation of a distant enhancer region of SOX11, leading to alteration of the 3-dimensional chromatin pattern with eventual activation of transcription of SOX11. This methylation change has been observed in cMCL, but is not seen in normal B-cells or most nnMCL (23). The precise role of SOX11 in MCL pathogenesis is still being explored. Mouse models overexpressing SOX11 have shown oligoclonal expansions of CD5+/CD23− B-cells, similar to MCL (24). SOX11 represses BCL6 expression, blocking the entry of B-cells into germinal center and thus may be integral for determining the cell of origin for MCL subtypes (25). Additionally, SOX11 promotes PAX5 expression, which in turn blocks plasmacytic differentiation, locking the cell in the mature B-cell stage, and has been previously implicated as an oncogenic mechanism in other B-cell lymphomas (26–28).
The role of tumor microenvironment in lymphomagenesis and drug resistance has been previously demonstrated in several B-cell malignancies. Recent studies show that the stromal interactions in MCL via adhesion molecules and cytokines influence activation of multiple pathways including B-cell receptor (BCR) and NF-kB, promoting cell proliferation and survival as well as trafficking to tumor supportive tissue microenvironments. When compared to peripheral blood, it has been observed that the lymph node microenvironment in MCL fosters BCR and NF-kB signaling (29). In the bone marrow, another common site of MCL involvement, stromal cells upregulate expression of focal adhesion kinase (FAK), CXCR4 and CXCR5 chemokine receptors, and VLA-4 adhesion molecules, likely resulting in downstream activation of the NF-kB and PI3K/AKT pathways (30, 31). SOX11 also interacts with the microenvironment via the FAK/PI3K/AKT pathway axis to promote cell growth, angiogenesis and cell migration (25, 26, 32).
When active, these pathways may serve as potential targets for inhibition, offering alternate treatment strategies from conventional chemotherapy agents. Discussion of all these targets is beyond the scope of this review article, but one class of therapy, small molecular inhibitor of Bruton tyrosine kinase (BTK), an essential component for BCR signaling, has gained a lot of attention in recent years as a treatment for various B-cell lymphomas. One irreversible inhibitor in this class, ibrutinib, has shown efficacy in MCL, as well as in CLL and activated B-cell subtype of diffuse large B-cell lymphoma (33–37). Nevertheless, a subset of patient showed either intrinsic resistance or developed acquired resistance to ibrutinib. Other approved BTK inhibitors include zanubrutinib and acalabrutinib. Newer generation of BTK inhibitors are under development, and may potentially overcome the therapy resistance observed with ibrutinib. These were will be discussed more in the Current Treatment Options section below.
At most institutions, the diagnosis of MCL relies on demonstrating the classic histopathologic features and tumor cell immunophenotype (CD5+, cyclin D1+, CD23−, SOX11+/−), but many also perform IGH/CCND1 dual-color dual-fusion fluorescence in situ hybridization (FISH) and/or CCND1 break-apart probe studies to confirm the diagnosis. However, it is important to keep in mind that very rare cases of MCL with cyclin D1 overexpression detectable at the protein level, may have cryptic CCND1 insertional event into the IGH locus that escapes detection by conventional FISH probes or karyotype analysis (38) (Figure 1A). These cryptic rearrangements may be more easily detected by whole genome sequencing (WGS) instead.
There are rare reports of MCL with translocations involving CCND1 and IGK (chromosome 2) or IGL (chromosome 22) instead of IGH (chromosome 14), analogous to IGK/MYC or IGL/MYC translocations seen in Burkitt lymphoma (39–43) (Figure 1A). A case of MCL with t(11, 12) (q13;p11.2) has also been reported (44). The 11q32 breakpoints for these variant translocations are mostly located in the 3’ region, which contrasts with the breakpoints associated with conventional IGH/CCND1 translocation that are centromeric (5’) from the CCND1 gene (39, 43). These cases would lack the expected fusion signal pattern by IGH/CCND1 dual-color dual-fusion FISH, but a helpful clue in such cases would be the presence of an extra signal for CCND1 as a consequence of one intact and one split CCND1 signal. Use of CCND1 break-apart probe, IGK/IGL break-apart probes or IGK/CCND1 or IGL/CCND1 dual-color dual-fusion probes could help prove the presence of these variant translocations, and aid in the diagnosis of MCL (Figure 2). While very rare, instead of conventional gene translocation involving CCND1, cryptic insertion of IGK and IGL enhancers in the vicinity of CCND1 can occur, which also leads to cyclin D1 overexpression and MCL phenotype, and would be missed by CCND1 break-apart probe (45). Such cases may be detectable only with IGK/IGL break-apart probes, IGK/CCND1 or IGL/CCND1 dual-color dual-fusion probes, or WGS assays.
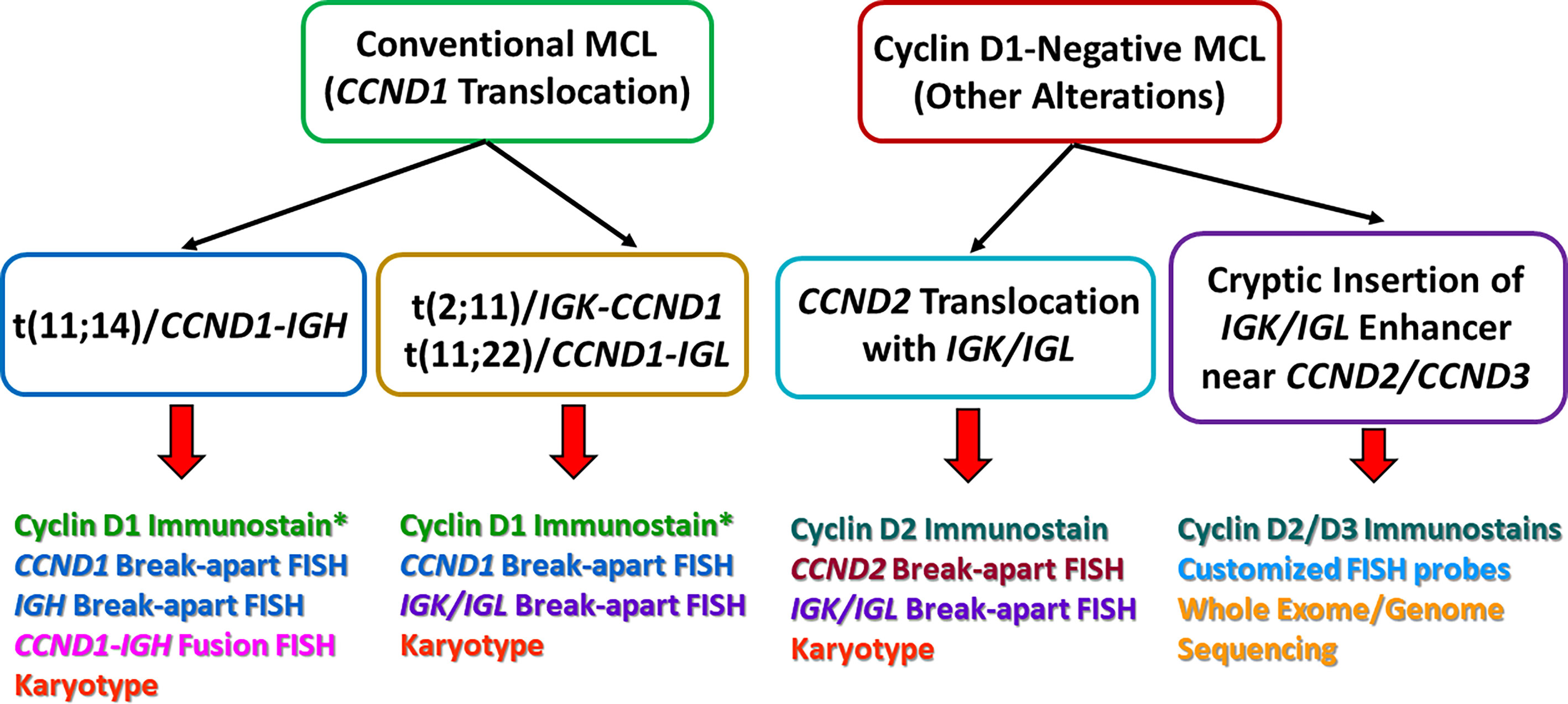
Figure 2 A comparison of the major genetic alterations between conventional mantle cell lymphoma (MCL) and cyclin D1-negative MCL. The bottom of the picture also listed some of the common methods for detection. *In rare cases, MCL can harbor the CCND1-IGH translocation, but cyclin D1 immunostain can be negative. This can be due to poor viability of tumor cells, technical issues, as well as mutations within the CCND1 gene that alter the immunostain antibody epitope.
A minor subgroup of MCL lacks CCND1 translocation as well as cyclin D1 expression (so-called “cyclin D1-negative MCL”). This subgroup has otherwise similar morphologic, phenotypic (including expression of SOX11) and genomic profiles as the cyclin D1-positive MCL (46–49). Nearly all of these cases show overexpression of either cyclin D2 or cyclin D3. Immunohistochemistry for cyclin D2 or cyclin D3 may serve as a screening tool for such cases. The major alteration in these cases involves the translocation of CCND2 (55–70% of cyclin D1-negative MCL) with an immunoglobulin gene, preferably with IGK and IGL rather than IGH. Most of the CCND2 translocations can be detected by break-apart probes (CCND2 or IGK/IGL). In the remaining cases, the lymphomas harbor cryptic insertion of enhancer elements of IGK or IGL into the vicinity of the CCND2 or CCND3 genes (Figure 1B). Due to the small size of the inserted enhancer element, such cases will be missed by conventional break-apart probes and require the use of either special customized IGK enhancer-CCND2/CCND3 fusion probes or WGS for detection (46, 47) (Figure 2). Older studies describing cyclin D1-negative MCL, confirmed by gene expression signature, and demonstrating cyclin D2 or D3 overexpression but lacking demonstrable rearrangement by conventional break-apart probes, possibly represented such cases (49).
Even more rare are cyclin D1-negative cases (<10%) lacking protein expression of cyclin D1, cyclin D2 and cyclin D3. Such cases show concomitant upregulation of CCNE1 and CCNE2, but lack a demonstrable relevant structural rearrangement (46). These cases show high genomic complexity and are associated with blastoid morphology (46).
Immunohistochemical (IHC) stains for SOX11, cyclin D2 and cyclin D3 may be helpful as part of the secondary panel in cyclin D1-negative lymphoid malignancies that have other features suggestive of MCL. Unlike cyclin D1, neither SOX11, cyclin D2 nor cyclin D3 expression is specific for MCL. Cyclin D2 or D3 expression can also be seen in CLL, follicular lymphoma or splenic marginal zone lymphoma. SOX11 expression has been reported in T-cell and B-cell lymphoblastic lymphoma/leukemia, Burkitt lymphoma, T-cell prolymphocytic leukemia, and rarely in classic Hodgkin lymphoma. Nevertheless, as compared to other B-cell lymphomas, with the exception for some cases of Burkitt lymphomas, the SOX11 mRNA levels in MCL is much higher (48).
Finally, an uncommon but potential pitfall in the diagnosis of MCL is the lack of cyclin D1 positivity by IHC stain despite the presence of t (11,14) detected by genetic studies, along with high CCND1 mRNA expression and SOX11 expression. Some potential explanations include mutations in CCND1 that significantly alter the 3D protein structure of the IHC antibody-binding epitope, suboptimal staining due to technical issues with instrumentation or IHC antibody, and pre-analytical factors such as poor formalin fixation, or low tumor cell viability. One of the most commonly-used commercially-available cyclin D1 IHC antibodies (clone SP4) is a monoclonal antibody that binds to the C-terminus portion of cyclin D1. Mutations that alter the 3’ end of CCND1, such as CCND1 p.D292P, can alter the C-terminus portion of the cyclin D1 protein, and can impair binding of this antibody, resulting in a false negative IHC result (50). In such cases, the use of alternative antibodies that bind to the N-terminus of cyclin D1 will circumvent the problem.
Another mechanism for cyclin D1 IHC false negativity is related to mutations that can affect selected isoforms of CCND1. Alternative splicing of CCND1 produces two major isoforms, the long isoform (cyclin D1a) and the short isoform (cyclin D1b). The short isoform derives from a splicing event that skips exon 5 and includes part of intron 4. Consequentially, cyclin D1b lacks the epitope for the C-terminal-binding cyclin D1 antibody. A defect in the expression of cyclin D1a isoform, which has been seen in association with the CCND1 p.L6P mutation, has been found to be another reason for cyclin D1 immunostain false negativity, due to the presence of predominantly cyclin D1b isoform that lacks the appropriate antibody epitope (50).
For practical purposes, the morphologic spectrum of MCL can be broadly divided into the classic and aggressive histologic variants. MCLs with blastoid and pleomorphic morphology are considered aggressive, as they exhibit inferior survival and response to chemotherapy (51, 52). These aggressive variants may arise de novo or from progression of an underlying classic variant of MCL, and are associated with high degree of aneuploidy and mutation burden, including KMT2D and KMT2B mutations (53). Jain et al. performed whole-exome sequencing (WES) in 183 patients of MCL with aggressive histology and found that mutations in NOTCH2, NOTCH3, and UBR5 were exclusive to the blastoid and pleomorphic variants. Additionally, they reported that aggressive histology MCL with Ki-67 proliferation index ≥50% have exclusive mutations in CCND1, NOTCH1, TP53, SPEN, SMARCA4, RANBP2, KMT2C, NOTCH2, NOTCH3, and NSD2 (53). It is also worth noting that since treatments for lymphomas can alter the tumor cell morphology, morphologic features may not be as reliable in predicting aggressive behavior and genomic complexity in the setting of post-treatment relapsed/refractory (R/R) MCL.
As mentioned previously, the two molecular subtypes of MCL, cMCL and nnMCL differ in IGHV SHM status, gene expression profile and genomic complexity, and are usually associated with aggressive and indolent behavior, respectively (3, 54, 55). Time to treatment and overall survival (OS) from the time of diagnosis is significantly longer for nnMCL patients than cMCL (19, 22). The literature showed that a 5-year OS for cMCL ranged from 32 to 40%, and for nnMCL ranged from 59 to75% (22, 54). Nevertheless, nnMCL may acquire additional genetic alterations and undergo transformation to aggressive variants that confer worse prognoses (see Secondary Genetic Events below).
Although the total number of mutated driver genes does not have an impact on prognosis, chromosomal instability in the form of BFB and chromothripsis or high number of CNA and SV are associated with a shorter OS in MCL[3]. The number of CNA (>7), or presence of BFB shows independent prognostic value in multivariate analysis (3). This is also in keeping with the observation that the more aggressive blastoid variant of MCL has higher number of copy number gains and losses (56) (see Histologic Subtypes of MCL above).
Immunohistochemical evaluation of Ki-67 proliferative index is part of routine evaluation of MCL as per the European Mantle Cell Lymphoma Pathology Panel, because it is a strong prognostic factor for OS and progression-free survival (PFS) independent of the Mantle Cell Lymphoma International Prognostic Index (MIPI). Consequently, a combination of Ki-67 and MIPI (biologic MIPI) provides a better risk stratification into four groups with significantly different prognoses (51). A Ki-67 >30% is the currently accepted cut-off for high-risk behavior (57).
Tumor cell proliferation can also be assessed on a gene expression level. A gene signature comprised of 20 genes that has been identified as a strong predictor of survival. It is highly expressed in proliferating cells (such as CDC2, ASPM, tubulin α, etc.) and correlates with mitotic index, further validating the role of proliferation rate in determining the clinical course in MCL (58). A similar proliferation assay (MCL35), developed by Scott et al., utilizes the NanoString nCounter® platform (NanoString Technologies, Seattle, WA) to assess expression of 17 genes on MCL formalin-fixed paraffin-embedded (FFPE) tissue (expression of 18 other housekeeping genes was also assessed for normalization purposes, for a total of 35 genes assessed). This assay classifies patients treated with R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone) into the high-risk, standard-risk, and low-risk groups, and the stratification was thought to have independent prognostication value from the MIPI score (59). The RNA Gene Expression Profiling section under Important Molecular Diagnostic Tools for Genetic Alterations below provides some important caveats related to gene expression profiling and the nCounter® platform.
Finally, the strength of a proliferation signature, which is predictive of inferior survival in MCL, correlates with the strength of the BCR signaling as well (29). The highly proliferative MCLs show high levels of cyclin D1 mRNA, with the short, truncated variant of cyclin D1a isoform preferentially expressed. This short isoform has a longer half-life and consequently prolongs the oncogenic effect of cyclin D1 (60). The proliferative activity also determines the DNA methylation burden, which has been found to be an independent prognostic factor in MCL. High DNA methylation burden has stronger prognostic value than that of IGHV mutation rate and patient’s age, and is associated with a worse clinical outcome (3, 23).
Even in the generally indolent subtype of MCL, nnMCL, the tumors may acquire additional genetic alterations, such as TP53 mutations and 17p deletions, that may impart an aggressive behavior (22, 54). TP53 alterations have been shown to be an independent adverse risk factor in MCL irrespective of high Ki-67, high MIPI score or blastoid morphology (14, 61). Other known chromosomal- and mutational-level alterations associated with shorter OS include: loss of CDKN2A, loss of RB1, loss of 13q33-q34, loss of 9q22-q31, rearrangement of MYC, gain of 18q21-q22, and SP140 mutations (3). In a recent study, MYC gene rearrangements, but not extra copies of MYC, were found to have a negative impact on OS in multivariate analysis (62).
From a therapeutic target standpoint, some of the secondary genetic alterations involving the BCR signaling, PIK3-AKT, canonical and non-canonical NF-kB pathways such as NSD2, NOTCH2, UBR5, BIRC3, TRAF2, MAP2K14, KMT2D, CARD11, SMARCA4, and BTK, have been found to be associated with ibrutinib resistance (63). These alterations pose therapeutic challenges, and their detection can help identify patients in need of alternative treatment regimens.
On the other hand, the prognostic role of other secondary genetic events are either more controversial or less established due to limited data available. For example, some studies have reported a negative impact of NOTCH1 mutations in univariate analysis (14, 64) and in a multivariate Cox regression model that also included IPI and histology (16), while in others, NOTCH1 mutations did not carry an independent prognostic value due to co-occurrence with TP53 mutations (61). NOTCH2 mutations occur independently of NOTCH1 mutations, and have been found to have significantly lower 3-year OS than the non-mutated cases (0 vs 62%, P = 0.0002 in NOTCH2-mutated and wild-type cases, respectively). In one prospective randomized control study of young patients, KMT2D mutations were found to be an independent prognostic marker of OS and PFS despite intensive immunochemotherapy and autologous stem cell transplant (61).
Most of the existing literature thus far have focused on the role of TP53 alterations in MCL. In current clinical practice, in the context of genetic alterations, only TP53 abnormalities influence treatment decisions (see Frontline Treatment for MCL section under Current Treatment Options below). Also, a major limitation in many of the existing studies is that it remains unclear if subclonal/low-level alterations in the genes have the same prognostic impact as having the alterations in the entire disease clone. The therapeutic implications of gene alterations other than TP53 certainly deserve further and more thorough evaluations.
Overall, due to rarity of cases and treatment heterogeneity of MCL with variant translocations or cryptic enhancer insertions reported in the literature, any conclusion regarding their prognoses should be interpreted with caution. The few reported cases of MCL with variant CCND1 translocations involving IGK or IGL instead of IGH mostly presented as nnMCL and followed a relatively indolent clinical course (39–43). However, no statistical difference in OS has been found among CCND1 translocated and non-CCND1 translocated cases. Furthermore, cases with CCND2 translocations or cryptic insertions of IGK/IGL enhancer elements near CCND2/CCND3 are associated with a similar OS as the CCND1-translocated cases (46).
Although not part of current routine management strategies, laboratory techniques for assessing MRD may serve as alternatives to 18 fluorodeoxyglucose positron emission tomography/computed tomography (18FDG PET-CT) for lymphoma response assessment and clinical decision making. In the context of MCL, measurement of molecular MRD is usually performed on peripheral blood or bone marrow, using methodology such as high sensitivity flow cytometry, allele-specific oligonucleotide polymerase chain reaction (PCR) or next-generation sequencing (NGS)-based IGH clonal rearrangement assay. Each of these can serve as a highly sensitive tool for monitoring tumor response to therapy at all time points during induction and consolidation. These diagnostic tools have been particularly helpful in the design of clinical trials to assess the efficacy of existing and novel drug regimens, and aid in tailoring the intensity and combinations of various frontline treatments. MRD assessment by molecular assays on peripheral blood or bone marrow samples, following initial immunochemotherapy, has been shown to be a strong independent prognostic factor and predictor of sustained clinical response and progression-free survival. However, MRD in MCL is still an emerging concept and prospective randomized clinical trials (such as NCT03267433) are warranted to design and evaluate MRD-guided treatment strategies (65–68). Further details on molecular MRD assays can be found in the section IGHV Somatic Hypermutation (SHM) Assessment and NGS-Based MRD Assay below.
Based on the most recent National Comprehensive Cancer Network (NCCN) guidelines (v.5.2021), besides relevant cell marker assessment by flow cytometry/IHC and cytogenetics karyotype/FISH studies for confirming a diagnosis of MCL, the only molecular testing considered essential is TP53 gene sequencing, for patients with expected aggressive clinical course (69) (see Frontline Treatment for MCL section under Current Treatment Options below). IGHV sequencing for determination of SHM status is considered helpful under certain circumstances, particularly for determination of clinically indolent MCL. Otherwise, additional molecular studies are considered optional. Other studies such as RNA gene expression and DNA methylation profiling are not performed routinely for clinical purposes, but can aid in the understanding of tumor pathogenesis.
Detection of mutations in tumor cells can be performed by a variety of methodology, such as allele-specific PCR for detection of specific known mutations, Sanger sequencing for detection of mutation within a specific region, and NGS assays (also known as massive parallel sequencing). Different sequencing platforms and assay kits are available commercially, and discussion of each will be beyond the scope of this article. However, for the purpose of detection of TP53 mutations, which are distributed throughout the gene rather than being confined to a few codons, Sanger sequencing and NGS assays are usually utilized, rather than allele-specific PCR. Sanger sequencing can be helpful for confirmation of findings, but in general it has worse technical detection sensitivity, is more costly, and inefficient in processing a large number of samples, as compared to the widely-used NGS assays (70).
For NGS assays, the sequencing strategy can be broadly classified into: 1. Targeted sequencing—enrichment of specific genes/exons by amplicon (PCR-based) or hybrid-capture techniques; 2. WES—enrichment of all exonic and surrounding region; 3. WGS—covering the entire genome, including the noncoding and intronic regions. In general, WGS yields the greatest breadth of information, is best suited for detecting larger gene insertions/deletions and SVs, and less prone to certain technical artifacts seen in targeted sequencing and WES. However, it is currently the most costly and time-consuming method for analyses, and due to lower overall coverage, may miss variants in samples with relatively low tumor content, or subclonal, low-level variants (70). Targeted sequencing has been favored in some clinical laboratories, due to the ability to detect variants at lower allele frequencies, lower cost, and the shorter amount of time needed for analyses, which has translated to faster report generation and sample turnaround time. If a targeted sequencing assay is chosen, it should be confirmed that important driver genes in MCL are covered by the assay (See Table 2).
Regardless of the breadth of sequencing regions, mutational profiling could be performed by sequencing of the tumor sample only, or paired sequencing of a normal control with the tumor sample. Analysis of a patient’s paired normal and tumor sample allows most germline variants to be identified and filtered out during analyses (71). The main advantage of this approach is that only somatic variants, which are more likely to be pathogenic, are eventually included in the clinical reports. In addition, separate analysis can be performed on the sequencing data from the normal sample for detection of clinically significant germline/inherited variants (72). For paired tumor-normal sequencing of MCL, since there is a possibility of circulating tumor cells, caution should be made if blood sample is used as normal control, which is commonly used in the sequencing of non-hematologic neoplasms. Rather, alternative normal control, such as saliva or buccal swab, should be considered instead.
Finally, it is worth pointing out that the terms “mutation” and “variant” should not be used interchangeably. “Variant” is a broader term that describes nucleotide changes as compared to the reference sequence, and does not imply pathogenicity of the change. The American College of Medical Genetics and Genomics (ACMG) and the Association for Molecular Pathology (AMP) advocated for a 5-tier classification of variants, which include: pathogenic, likely pathogenic, uncertain significance, likely benign, or benign (73). The classification of variants is particularly important for tumor-only sequencing assays, in which germline benign variants (some of which are also known as polymorphisms) can be detected. In the absence of paired tumor-normal sequencing, to help distinguish between somatic and germline benign variants, the variant allele frequencies can be used in conjunction with information from publicly-available databases of somatic variants (e.g., COSMIC: https://cancer.sanger.ac.uk/cosmic), and several population-based databases of germline variants (e.g., dbSNP: https://www.ncbi.nlm.nih.gov/snp; gnomAD: https://gnomad.broadinstitute.org; The International Genome Sample Resource/The 1000 Genomes Project: https://www.internationalgenome.org). As a general rule, due to the clinical implication of assigning a variant as pathogenic, it is recommended to do so only when there is a reasonably high level of confidence in it being so (e.g., listing in multiple reliable databases of known pathogenic variants, available literature on functional studies on that particular variant).
Historically, much of the knowledge of IGHV SHM assessment arose from the data for CLL, where the IGHV SHM status has served as a key prognostic marker for the past two decades (74). In CLL as well as MCL, the IGHV SHM status also appears to be stable over time, regardless of therapy, and has traditionally been assessed by Sanger sequencing of the IGH gene VDJ segments. As clinical laboratories increasingly incorporate NGS-based assays into their workflow, and the corresponding sequencing instruments in their laboratories, IGHV sequencing by NGS method has also gained in popularity as a replacement for Sanger sequencing.
In terms of IGHV sequencing by NGS method, several commercial assays are available, and the same assay is often used for IGH clonal rearrangement (B-cell clonality) detection, as well as MRD detection of the known IGH disease clone in a post-treatment or follow-up sample. Different assays employ different strategies, but the basic principles remain the same: the use of PCR primer sets to flank and amplify the IGH gene VDJ segments [usually the forward primer set is either upstream to or within the framework 1, 2, or 3 (FR1, FR2 or FR3) region of the V segment, or the D segment, while the reverse primer set is in the J gene segment] (75). Some assays may employ multiple primer sets to different regions of the IGH gene to overcome the problem of poor primer annealing that can result from high-level somatic mutations within the IGH gene. After sequencing, the disease-associated clonal sequence is identified and compared to a reference database, to determine the % of SHM as compared to the germline sequence. The patient- and disease-specific clonal sequence can also be saved, and searched for at very low level in subsequent samples, which form the basis of MRD detection. Besides commercial assays, in recent years, the European Research Initiative on CLL (ERIC) and the EuroClonality-NGS Working Group have collaborated to develop the appropriate primer sets and analysis pipeline for clonal sequence characterization and SHM determination from a NGS-based IGHV sequencing assay (74).
Somewhat analogous to the different DNA sequencing strategies mentioned above, global RNA gene expression can be performed to provide a global picture of the different alterations as compared to normal control/non-neoplastic tissue, but is mostly done in research settings. Targeted RNA gene expression profile is more feasible in the clinical setting, although in the case of MCL, it is not currently widely used for routine cases. This may be partially related to the need for RNA extraction from FFPE or blood, which is not performed at some smaller laboratories. For testing on archival FFPE samples, RNA is also not as well-preserved over time, compared to DNA.
Both MCL35 (59) and L-MCL16 (19) assays mentioned previously were designed using the NanoString nCounter® platform and require specific instrumentation from the manufacturer. This is in contrast to the sequencing instrumentation for NGS-based assays, where the same instruments can potentially be adapted for a variety of assays and serve multiple purposes (e.g., targeted mutation profiling assays using reagents/kits from different manufacturers, as well as IGHV sequencing by NGS method).
SNP array as a method of assessing CNAs has several advantages over conventional karyotype. First of all, while conventional karyotype requires fresh cell cultures, SNP array is a DNA-based assay, and therefore can be performed on existing extracted DNA, or on DNA extracted from FFPE tissue. With routine, conventional G-band karyotyping, chromosomal change >10 Mb can be detected; on the other hand, with newer SNP array assays, the average inter-marker distance is 680 bp, allowing a much higher resolution in detecting small areas of chromosomal-level gain/loss (76). Furthermore, SNP array can detect copy neutral-loss of heterozygosity (CN-LOH), as well as provide information on the level of copy number gain/loss, which are not possible by karyotyping. Nevertheless, unlike karyotype, SNP array cannot detect balanced translocation with no net loss of chromosomal material, and certainly not the partners involved in a translocation. Compared to FISH studies, which are usually much more targeted, and each set of probes assess alterations in a single chromosome, gene, or specific translocation, SNP array has the advantage of providing a broad overview of CNAs across all the chromosomes. On the flip side, since FISH studies assess for alterations in individual cells, in the context of fresh tissue studies, FISH studies can detect abnormalities at a much lower level than SNP array. This can be a consideration for samples with low tumor content, where the extracted DNA originated from a mixture of tumor and non-neoplastic cells, hampering the ability for SNP array to detect abnormalities.
The treatment approach and therapeutic landscape for patients with newly diagnosed and R/R MCL has changed substantially in recent years, largely due to pivotal new therapies and deepened understanding into the molecular alterations in MCL to inform their use. Besides conventional immunochemotherapy (IC), current treatments also include molecularly targeted therapies such as ibrutinib (BTK inhibitor) and venetoclax (BCL-2 inhibitor) and more recently, immune effector cells such as the chimeric antigen receptor (CAR)-modified T-cell product brexucabtagene autoleucel directed against CD19. Of note, clinical trial participation at any stage of treatment is generally encouraged given the rarity of MCL and the pressing need to improve outcomes in this disease.
A number of considerations inform the therapeutic recommendations for patients with newly diagnosed MCL who require treatments. Of note, variability exists across treatment centers in this initial treatment approach. In current practice, the greatest patient- and tumor-specific factors of interest are the patient’s age, frailty/comorbidity, and goals for care, as well as factors alluded to in the Prognostic Factors section above, namely clinical and biologic features of the disease, including tumor TP53 alteration status.
Standard frontline treatments include IC or rituximab-lenalidomide (77). Younger, fit patients seeking maximal response duration generally receive multiagent IC followed by autologous stem cell rescue (HDT/ASCR). Conventional IC regimens include R-DHAP (78) (rituximab, dexamethasone, cytarabine, platinum), alternating R-CHOP/R-DHAP (52), and dose-intensified R-CHOP plus cytarabine (79). Maintenance rituximab is typically given post-HDT/ASCR based on data from Le Gouill et al. demonstrating improvements in PFS, event-free survival, and OS (78) with this approach. Of note, given uncertainty regarding the effect of HDT/ASCR on OS, an ongoing randomized study (NCT03267433) is evaluating a MRD-based transplant approach wherein MRD-negative patients following IC are randomized to either rituximab maintenance or HDT/ASCR. Details of MRD molecular testing have been discussed in earlier sections of this review.
Transplant-ineligible patients or those seeking less intensive treatments commonly receive rituximab plus bendamustine (80); in select cases (for example, averse to IC or especially frail or comorbid), patients can also receive rituximab plus the immunomodulatory agent lenalidomide (77).
The clinical importance of TP53 status is delineated in data from the Nordic Lymphoma Group (64) who evaluated the impact of TP53 alterations in a cohort of patients who underwent intensive IC followed by HDT/ASCR. They demonstrated poor outcomes in these patients, with a median OS of less than 2 years and a cumulative incidence of relapse of 50% at 1 year. Other groups have demonstrated similar findings (61, 81) to reinforce the prognostic importance of TP53 alterations. Based on these findings, IC + HDT/ASCR consolidation is not recommended; rather, treatment with novel agents in the context of a clinical trial is favored for upfront treatment of patients with TP53-altered MCL.
For patients with R/R MCL, the treatment recommendations are highly individualized, and are primarily driven by the extent of disease/symptomatology; time to relapse; response, tolerability, and category/number of prior therapies received; and patient age/frailty/comorbidity and goals for care. Clinically, R/R MCL is characterized by progressive shortening of response duration with subsequent lines of treatment (82) and represents an unmet clinical need. Patients who have experienced favorable PFS with upfront IC may be treated with second-line IC, given the potential for achieving long responses again (83). More commonly, patients are treated with targeted agents. Approved agents (frequently combined with rituximab) include the BTK inhibitors ibrutinib, zanubrutinib, and acalabrutinib (33), the Bcl-2 inhibitor venetoclax (84), and the immunomodulatory agent lenalidomide (Table 4) (85). Additionally, many of these agents are being used in combination based on noteworthy preliminary data from ongoing trials [ibrutinib + lenalidomide + rituximab (35) and ibrutinib + venetoclax (37)]. The recent approval of brexucabtagene autoleucel and promising early data for lisocabtagene maraleucel are major landmarks in treating MCL given promising efficacy and tolerability in early results from trials (86, 87). For example, among 32 patients who received lisocabtagene maraleucel (median number prior therapies = 3), the response rate was 84%, including 59% complete response (87). Longer follow-up data are awaited to define the durability of responses with these agents and to inform proper sequencing with other agents in the R/R setting. Future investigations will likely investigate the use of CAR-T cells earlier in the care of patients with MCL (currently only approved for patients with R/R MCL), potentially targeting higher-risk MCL subtypes such as TP53-altered or blastoid morphology. Finally, for eligible patients with multiply relapsed MCL, there are supportive data for allogeneic stem cell transplantation, however, the role for this approach along with other therapies will likely evolve given the promise of CAR T-cell therapy.
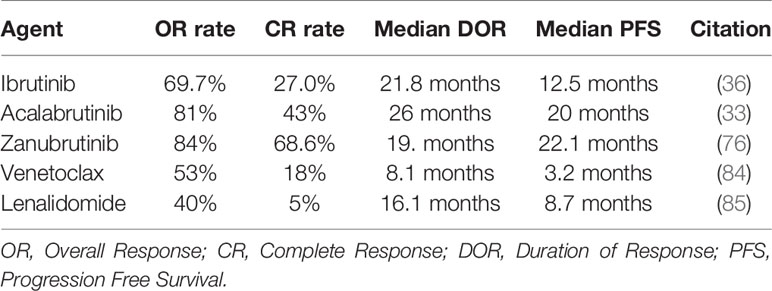
Table 4 Approved targeted therapies for MCL.
Although MCLs are characterized in most cases by t (11,14)(q13q32)/CCND1-IGH, other variant translocations and cryptic insertion of enhancer elements can also lead to MCL. There are certain common gene mutations and chromosomal-level alterations for MCLs. Although specific alterations may be more common in either the cMCL or nnMCL subtype, cMCL as a group has higher genomic complexity and higher average number of CNAs on a chromosomal level. As these alterations are accumulated over time during tumor progression, they can affect tumor aggressiveness and prognostication. Many alterations can be detected using widely-available clinical molecular diagnostic tools, including SNP array and targeted/WES assays. On the other hand, studies such as methylation profile assays, are currently mostly performed in the research settings. Nevertheless, detection of relevant genetic alterations is crucial for prognostication and therapy selection. Different frontline and refractory/relapsed treatment options are available for MCL, including immunochemotherapies, targeted therapies, immune effector cell therapy, and stem cell transplantation.
SS performed the literature review and wrote the manuscript for this review article. ZE-P and AK researched and wrote the Current Treatment Options section. CH researched and wrote the Important Molecular Diagnostic Tools for Genetic Alterations section, as well as oversaw the writing of the manuscript in the capacity of the senior author. All authors contributed to the article and approved the submitted version.
The authors have been supported by the Comprehensive Cancer Center Core Grant (P30 CA008748) at Memorial Sloan Kettering Cancer Center (MSKCC) from the National Institute of Health, USA (Grant recipient: CT).
ZE-P received salary support through the AACR-AstraZeneca Lymphoma Research Fellowship and the Lymphoma Research Foundation LSRMP. AK has stocks and other ownership interests in Bridgebio Pharma, consulting or advisory role for Celgene, Kite Pharma (a Gilead company), AstraZeneca, and research funding from: AbbVie/Genetech, Adaptive Biotechnologies, Celgene, Seattle Genetics, AstraZeneca, Pharmacyclics. CH serves on the Hematopathology advisory board for Blueprint Medicines, received honorarium from Invivoscribe, Inc. and Maryland Society of Pathologists, and is a current employee of Loxo Oncology at Lilly.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, et al. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Revised 4th Edition. Lyon, France: IARC (2017).
2. Veloza L, Ribera-Cortadam I, Campom E. Mantle Cell Lymphoma Pathology Update in the 2016 WHO Classification. Ann Lymphoma (2019) 3:3–3. doi: 10.21037/aol.2019.03.01
3. Nadeu F, Martin-Garcia D, Clot G, Diaz-Navarro A, Duran-Ferrer M, Navarro A, et al. Genomic and Epigenomic Insights Into the Origin, Pathogenesis, and Clinical Behavior of Mantle Cell Lymphoma Subtypes. Blood (2020) 136(12):1419–32. doi: 10.1182/blood.2020005289
4. Puente XS, Jares P, Campo E. Chronic Lymphocytic Leukemia and Mantle Cell Lymphoma: Crossroads of Genetic and Microenvironment Interactions. Blood (2018) 131(21):2283–96. doi: 10.1182/blood-2017-10-764373
5. Jares P, Colomer D, Campo E. Molecular Pathogenesis of Mantle Cell Lymphoma. J Clin Invest (2012) 122(10):3416–23. doi: 10.1172/JCI61272
6. Royo C, Salaverria I, Hartmann EM, Rosenwald A, Campo E, Bea S. The Complex Landscape of Genetic Alterations in Mantle Cell Lymphoma. Semin Cancer Biol (2011) 21(5):322–34. doi: 10.1016/j.semcancer.2011.09.007
7. Kuppers R, Dalla-Favera R. Mechanisms of Chromosomal Translocations in B Cell Lymphomas. Oncogene (2001) 20(40):5580–94. doi: 10.1038/sj.onc.1204640
8. Santarius T, Shipley J, Brewer D, Stratton MR, Cooper CS. A Census of Amplified and Overexpressed Human Cancer Genes. Nat Rev Cancer (2010) 10(1):59–64. doi: 10.1038/nrc2771
9. Meyerson M, Harlow E. Identification of G1 Kinase Activity for Cdk6, a Novel Cyclin D Partner. Mol Cell Biol (1994) 14(3):2077–86. doi: 10.1128/MCB.14.3.2077
10. Resnitzky D, Gossen M, Bujard H, Reed SI. Acceleration of the G1/S Phase Transition by Expression of Cyclins D1 and E With an Inducible System. Mol Cell Biol (1994) 14(3):1669–79. doi: 10.1128/MCB.14.3.1669
11. Musgrove EA, Caldon CE, Barraclough J, Stone A, Sutherland RL. Cyclin D as a Therapeutic Target in Cancer. Nat Rev Cancer (2011) 11(8):558–72. doi: 10.1038/nrc3090
12. Kozar K, Ciemerych MA, Rebel VI, Shigematsu H, Zagozdzon A, Sicinska E, et al. Mouse Development and Cell Proliferation in the Absence of D-Cyclins. Cell (2004) 118(4):477–91. doi: 10.1016/j.cell.2004.07.025
13. Lovec H, Grzeschiczek A, Kowalski MD, Möröy T. Cyclin D1/bcl-1 Cooperates With Myc Genes in the Generation of B-Cell Lymphoma in Transgenic Mice. EMBO J (1994) 13(15):3487–95. doi: 10.1002/j.1460-2075.1994.tb06655.x
14. Bea S, Valdes-Mas R, Navarro A, Salaverria I, Martin-Garcia D, Jares P, et al. Landscape of Somatic Mutations and Clonal Evolution in Mantle Cell Lymphoma. Proc Natl Acad Sci USA (2013) 110(45):18250–5. doi: 10.1073/pnas.1314608110
15. Zhang J, Jima D, Moffitt AB, Liu Q, Czader M, Hsi ED, et al. The Genomic Landscape of Mantle Cell Lymphoma Is Related to the Epigenetically Determined Chromatin State of Normal B Cells. Blood (2014) 123(19):2988–96. doi: 10.1182/blood-2013-07-517177
16. Kridel R, Meissner B, Rogic S, Boyle M, Telenius A, Woolcock B, et al. Whole Transcriptome Sequencing Reveals Recurrent NOTCH1 Mutations in Mantle Cell Lymphoma. Blood (2012) 119(9):1963–71. doi: 10.1182/blood-2011-11-391474
17. Meissner B, Kridel R, Lim RS, Rogic S, Tse K, Scott DW, et al. The E3 Ubiquitin Ligase UBR5 Is Recurrently Mutated in Mantle Cell Lymphoma. Blood (2013) 121(16):3161–4. doi: 10.1182/blood-2013-01-478834
18. Camacho E, Hernandez L, Hernandez S, Tort F, Bellosillo B, Bea S, et al. ATM Gene Inactivation in Mantle Cell Lymphoma Mainly Occurs by Truncating Mutations and Missense Mutations Involving the Phosphatidylinositol-3 Kinase Domain and Is Associated With Increasing Numbers of Chromosomal Imbalances. Blood (2002) 99(1):238–44. doi: 10.1182/blood.V99.1.238
19. Clot G, Jares P, Gine E, Navarro A, Royo C, Pinyol M, et al. A Gene Signature That Distinguishes Conventional and Leukemic Nonnodal Mantle Cell Lymphoma Helps Predict Outcome. Blood (2018) 132(4):413–22. doi: 10.1182/blood-2018-03-838136
20. Stilgenbauer S, Nickolenko J, Wilhelm J, Wolf S, Weitz S, Döhner K, et al. Expressed Sequences as Candidates for a Novel Tumor Suppressor Gene at Band 13q14 in B-Cell Chronic Lymphocytic Leukemia and Mantle Cell Lymphoma. Oncogene (1998) 16(14):1891–7. doi: 10.1038/sj.onc.1201764
21. Fernandez V, Salamero O, Espinet B, Sole F, Royo C, Navarro A, et al. Genomic and Gene Expression Profiling Defines Indolent Forms of Mantle Cell Lymphoma. Cancer Res (2010) 70(4):1408–18. doi: 10.1158/0008-5472.CAN-09-3419
22. Royo C, Navarro A, Clot G, Salaverria I, Gine E, Jares P, et al. Non-Nodal Type of Mantle Cell Lymphoma Is a Specific Biological and Clinical Subgroup of the Disease. Leukemia (2012) 26(8):1895–8. doi: 10.1038/leu.2012.72
23. Queiros AC, Beekman R, Vilarrasa-Blasi R, Duran-Ferrer M, Clot G, Merkel A, et al. Decoding the DNA Methylome of Mantle Cell Lymphoma in the Light of the Entire B Cell Lineage. Cancer Cell (2016) 30(5):806–21. doi: 10.1016/j.ccell.2016.09.014
24. Kuo PY, Jatiani SS, Rahman AH, Edwards D, Jiang Z, Ahr K, et al. SOX11 Augments BCR Signaling to Drive MCL-Like Tumor Development. Blood (2018) 131(20):2247–55. doi: 10.1182/blood-2018-02-832535
25. Palomero J, Vegliante MC, Eguileor A, Rodriguez ML, Balsas P, Martinez D, et al. SOX11 Defines Two Different Subtypes of Mantle Cell Lymphoma Through Transcriptional Regulation of BCL6. Leukemia (2016) 30(7):1596–9. doi: 10.1038/leu.2015.355
26. Vegliante MC, Palomero J, Perez-Galan P, Roue G, Castellano G, Navarro A, et al. SOX11 Regulates PAX5 Expression and Blocks Terminal B-Cell Differentiation in Aggressive Mantle Cell Lymphoma. Blood (2013) 121(12):2175–85. doi: 10.1182/blood-2012-06-438937
27. Morrison AM, Jäger U, Chott A, Schebesta M, Haas OA, Busslinger M. Deregulated PAX-5 Transcription From a Translocated IgH Promoter in Marginal Zone Lymphoma. Blood (1998) 92(10):3865–78. doi: 10.1182/blood.V92.10.3865
28. Busslinger M, Klix N, Pfeffer P, Graninger PG, Kozmik Z. Deregulation of PAX-5 by Translocation of the Emu Enhancer of the IgH Locus Adjacent to Two Alternative PAX-5 Promoters in a Diffuse Large-Cell Lymphoma. Proc Natl Acad Sci USA (1996) 93(12):6129–34. doi: 10.1073/pnas.93.12.6129
29. Saba NS, Liu D, Herman SEM, Underbayev C, Tian X, Behrend D, et al. Pathogenic Role of B-Cell Receptor Signaling and Canonical NF-Kb Activation in Mantle Cell Lymphoma. Blood (2016) 128(1):82–92. doi: 10.1182/blood-2015-11-681460
30. Kurtova AV, Tamayo AT, Ford RJ, Burger JA. Mantle Cell Lymphoma Cells Express High Levels of CXCR4, CXCR5, and VLA-4 (CD49d): Importance for Interactions With the Stromal Microenvironment and Specific Targeting. Blood (2009) 113(19):4604–13. doi: 10.1182/blood-2008-10-185827
31. Rudelius M, Rosenfeldt MT, Leich E, Rauert-Wunderlich H, Solimando AG, Beilhack A, et al. Inhibition of Focal Adhesion Kinase Overcomes Resistance of Mantle Cell Lymphoma to Ibrutinib in the Bone Marrow Microenvironment. Haematologica (2018) 103(1):116–25. doi: 10.3324/haematol.2017.177162
32. Balsas P, Palomero J, Eguileor A, Rodriguez ML, Vegliante MC, Planas-Rigol E, et al. SOX11 Promotes Tumor Protective Microenvironment Interactions Through CXCR4 and FAK Regulation in Mantle Cell Lymphoma. Blood (2017) 130(4):501–13. doi: 10.1182/blood-2017-04-776740
33. Wang M, Rule S, Zinzani PL, Goy A, Casasnovas O, Smith SD, et al. Durable Response With Single-Agent Acalabrutinib in Patients With Relapsed or Refractory Mantle Cell Lymphoma. Leukemia (2019) 33(11):2762–6. doi: 10.1038/s41375-019-0575-9
34. Agarwal R, Chan YC, Tam CS, Hunter T, Vassiliadis D, Teh CE, et al. Dynamic Molecular Monitoring Reveals That SWI-SNF Mutations Mediate Resistance to Ibrutinib Plus Venetoclax in Mantle Cell Lymphoma. Nat Med (2019) 25(1):119–29. doi: 10.1038/s41591-018-0243-z
35. Jerkeman M, Eskelund CW, Hutchings M, Räty R, Wader KF, Laurell A, et al. Ibrutinib, Lenalidomide, and Rituximab in Relapsed or Refractory Mantle Cell Lymphoma (PHILEMON): A Multicentre, Open-Label, Single-Arm, Phase 2 Trial. Lancet Haematol (2018) 5(3):e109–16. doi: 10.1016/S2352-3026(18)30018-8
36. Rule S, Dreyling M, Goy A, Hess G, Auer R, Kahl B, et al. Ibrutinib for the Treatment of Relapsed/Refractory Mantle Cell Lymphoma: Extended 3.5-Year Follow Up From a Pooled Analysis. Haematologica (2019) 104(5):e211–4. doi: 10.3324/haematol.2018.205229
37. Tam CS, Anderson MA, Pott C, Agarwal R, Handunnetti S, Hicks RJ, et al. Ibrutinib Plus Venetoclax for the Treatment of Mantle-Cell Lymphoma. New Engl J Med (2018) 378(13):1211–23. doi: 10.1056/NEJMoa1715519
38. Peterson JF, Baughn LB, Ketterling RP, Pitel BA, Smoley SA, Vasmatzis G, et al. Characterization of a Cryptic IGH/CCND1 Rearrangement in a Case of Mantle Cell Lymphoma With Negative CCND1 FISH Studies. Blood Adv (2019) 3(8):1298–302. doi: 10.1182/bloodadvances.2019031450
39. Wlodarska I, Meeus P, Stul M, Thienpont L, Wouters E, Marcelis L, et al. Variant T(2,11)(P11;Q13) Associated With the IgK-CCND1 Rearrangement Is a Recurrent Translocation in Leukemic Small-Cell B-Non-Hodgkin Lymphoma. Leukemia (2004) 18(10):1705–10. doi: 10.1038/sj.leu.2403459
40. Marrero WD, Cruz-Chacon A, Cabanillas F. Mantle Cell Lymphoma With t(11,22) (Q13;Q11.2) an Indolent Clinical Variant? Leuk Lymphoma (2018) 59(10):2509–11. doi: 10.1080/10428194.2018.1427863
41. Espinet B, Salaverria I, Bea S, Ruiz-Xiville N, Balague O, Salido M, et al. Incidence and Prognostic Impact of Secondary Cytogenetic Aberrations in a Series of 145 Patients With Mantle Cell Lymphoma. Genes Chromosomes Cancer (2010) 49(5):439–51. doi: 10.1002/gcc.20754
42. Rocha CK, Praulich I, Gehrke I, Hallek M, Kreuzer KA. A Rare Case of t(11,22) in a Mantle Cell Lymphoma Like B-Cell Neoplasia Resulting in a Fusion of IGL and CCND1: Case Report. Mol Cytogenet (2011) 4(1):8. doi: 10.1186/1755-8166-4-8
43. Komatsu H, Iida S, Yamamoto K, Mikuni C, Nitta M, Takahashi T, et al. A Variant Chromosome Translocation at 11q13 Identifying PRAD1/cyclin D1 as the BCL-1 Gene. Blood (1994) 84(4):1226–31. doi: 10.1182/blood.V84.4.1226.1226
44. Menke JR, Vasmatzis G, Murphy S, Yang L, Menke DM, Tun HW, et al. Mantle Cell Lymphoma With a Novel T(11,12)(Q13;P11.2): A Proposed Alternative Mechanism of CCND1 Up-Regulation. Hum Pathol (2017) 64:207–12. doi: 10.1016/j.humpath.2017.01.001
45. Fuster C, Martin-Garcia D, Balague O, Navarro A, Nadeu F, Costa D, et al. Cryptic Insertions of the Immunoglobulin Light Chain Enhancer Region Near CCND1 in T (11,14)-Negative Mantle Cell Lymphoma. Haematologica (2020) 105(8):e408–11. doi: 10.3324/haematol.2019.237073
46. Martin-Garcia D, Navarro A, Valdes-Mas R, Clot G, Gutierrez-Abril J, Prieto M, et al. CCND2 and CCND3 Hijack Immunoglobulin Light-Chain Enhancers in Cyclin D1(-) Mantle Cell Lymphoma. Blood (2019) 133(9):940–51. doi: 10.1182/blood-2018-07-862151
47. Salaverria I, Royo C, Carvajal-Cuenca A, Clot G, Navarro A, Valera A, et al. CCND2 Rearrangements Are the Most Frequent Genetic Events in Cyclin D1(-) Mantle Cell Lymphoma. Blood (2013) 121(8):1394–402. doi: 10.1182/blood-2012-08-452284
48. Mozos A, Royo C, Hartmann E, De Jong D, Baro C, Valera A, et al. SOX11 Expression Is Highly Specific for Mantle Cell Lymphoma and Identifies the Cyclin D1-Negative Subtype. Haematologica (2009) 94(11):1555–62. doi: 10.3324/haematol.2009.010264
49. Fu K, Weisenburger DD, Greiner TC, Dave S, Wright G, Rosenwald A, et al. Cyclin D1-Negative Mantle Cell Lymphoma: A Clinicopathologic Study Based on Gene Expression Profiling. Blood (2005) 106(13):4315–21. doi: 10.1182/blood-2005-04-1753
50. Iaccarino I, Afify L, Aukema SM, Reddemann K, Schütt P, Flür M, et al. t(11,14)-Positive Mantle Cell Lymphomas Lacking Cyclin D1 (CCND1) Immunostaining Because of a CCND1 Mutation or Exclusive Expression of the CCND1b Isoform. Haematologica (2018) 103(9):e432–5. doi: 10.3324/haematol.2018.192435
51. Hoster E, Rosenwald A, Berger F, Bernd HW, Hartmann S, Loddenkemper C, et al. Prognostic Value of Ki-67 Index, Cytology, and Growth Pattern in Mantle-Cell Lymphoma: Results From Randomized Trials of the European Mantle Cell Lymphoma Network. J Clin Oncol (2016) 34(12):1386–94. doi: 10.1200/JCO.2015.63.8387
52. Hermine O, Hoster E, Walewski J, Bosly A, Stilgenbauer S, Thieblemont C, et al. Addition of High-Dose Cytarabine to Immunochemotherapy Before Autologous Stem-Cell Transplantation in Patients Aged 65 Years or Younger With Mantle Cell Lymphoma (MCL Younger): A Randomised, Open-Label, Phase 3 Trial of the European Mantle Cell Lymphoma Network. Lancet (2016) 388(10044):565–75. doi: 10.1016/S0140-6736(16)00739-X
53. Jain P, Zhang S, Kanagal-Shamanna R, Ok CY, Nomie K, Nogueras-Gonzalez G, et al. Genomic Profiles and Clinical Outcomes of De Novo Blastoid/Pleomorphic MCL Are Distinct From Those of Transformed MCL. Blood Adv (2020) 4(6):1038–50. doi: 10.1182/bloodadvances.2019001396
54. Navarro A, Clot G, Royo C, Jares P, Hadzidimitriou A, Agathangelidis A, et al. Molecular Subsets of Mantle Cell Lymphoma Defined by the IGHV Mutational Status and SOX11 Expression Have Distinct Biologic and Clinical Features. Cancer Res (2012) 72(20):5307–16. doi: 10.1158/0008-5472.CAN-12-1615
55. Orchard J, Garand R, Davis Z, Babbage G, Sahota S, Matutes E, et al. A Subset of T (11,14) Lymphoma With Mantle Cell Features Displays Mutated IgVH Genes and Includes Patients With Good Prognosis, Nonnodal Disease. Blood (2003) 101(12):4975–81. doi: 10.1182/blood-2002-06-1864
56. Bea S, Ribas M, Hernandez JM, Bosch F, Pinyol M, Hernandez L, et al. Increased Number of Chromosomal Imbalances and High-Level DNA Amplifications in Mantle Cell Lymphoma Are Associated With Blastoid Variants. Blood (1999) 93(12):4365–74. doi: 10.1182/blood.V93.12.4365
57. Determann O, Hoster E, Ott G, Bernd HW, Loddenkemper C, Hansmann ML, et al. Ki-67 Predicts Outcome in Advanced-Stage Mantle Cell Lymphoma Patients Treated With Anti-CD20 Immunochemotherapy: Results From Randomized Trials of the European MCL Network and the German Low Grade Lymphoma Study Group. Blood (2008) 111(4):2385–7. doi: 10.1182/blood-2007-10-117010
58. Rosenwald A, Wright G, Wiestner A, Chan WC, Connors JM, Campo E, et al. The Proliferation Gene Expression Signature Is a Quantitative Integrator of Oncogenic Events That Predicts Survival in Mantle Cell Lymphoma. Cancer Cell (2003) 3(2):185–97. doi: 10.1016/S1535-6108(03)00028-X
59. Scott DW, Abrisqueta P, Wright GW, Slack GW, Mottok A, Villa D, et al. New Molecular Assay for the Proliferation Signature in Mantle Cell Lymphoma Applicable to Formalin-Fixed Paraffin-Embedded Biopsies. J Clin Oncol (2017) 35(15):1668–77. doi: 10.1200/JCO.2016.70.7901
60. Wiestner A, Tehrani M, Chiorazzi M, Wright G, Gibellini F, Nakayama K, et al. Point Mutations and Genomic Deletions in CCND1 Create Stable Truncated Cyclin D1 mRNAs That Are Associated With Increased Proliferation Rate and Shorter Survival. Blood (2007) 109(11):4599–606. doi: 10.1182/blood-2006-08-039859
61. Ferrero S, Rossi D, Rinaldi A, Bruscaggin A, Spina V, Eskelund CW, et al. KMT2D Mutations and TP53 Disruptions Are Poor Prognostic Biomarkers in Mantle Cell Lymphoma Receiving High-Dose Therapy: A FIL Study. Haematologica (2020) 105(6):1604–12. doi: 10.3324/haematol.2018.214056
62. Wang L, Tang G, Medeiros LJ, Xu J, Huang W, Yin CC, et al. MYC Rearrangement But Not Extra MYC Copies Is an Independent Prognostic Factor in Patients With Mantle Cell Lymphoma. Haematologica (2020) 106(5):1381–9. doi: 10.3324/haematol.2019.243071
63. Hershkovitz-Rokah O, Pulver D, Lenz G, Shpilberg O. Ibrutinib Resistance in Mantle Cell Lymphoma: Clinical, Molecular and Treatment Aspects. Br J Haematol (2018) 181(3):306–19. doi: 10.1111/bjh.15108
64. Eskelund CW, Dahl C, Hansen JW, Westman M, Kolstad A, Pedersen LB, et al. TP53 Mutations Identify Younger Mantle Cell Lymphoma Patients Who Do Not Benefi T From Intensive Chemoimmunotherapy. Blood J Am Soc Hematol (2017) 130(17):1903–10. doi: 10.1182/blood-2017-04-779736
65. Kumar A, Bantilan KS, Jacob AP, Park A, Schoninger SF, Sauter C, et al. Noninvasive Monitoring of Mantle Cell Lymphoma by Immunoglobulin Gene Next-Generation Sequencing in a Phase 2 Study of Sequential Chemoradioimmunotherapy Followed by Autologous Stem-Cell Rescue. Clin Lymphoma Myeloma Leukemia (2021) 21(4):230–7.e12. doi: 10.1016/j.clml.2020.09.007
66. Merryman RW, Edwin N, Redd R, Bsat J, Chase M, LaCasce A, et al. Rituximab/bendamustine and Rituximab/Cytarabine Induction Therapy for Transplant-Eligible Mantle Cell Lymphoma. Blood Adv (2020) 4(5):858–67. doi: 10.1182/bloodadvances.2019001355
67. Hoster E, Pott C. Minimal Residual Disease in Mantle Cell Lymphoma: Insights Into Biology and Impact on Treatment. Hematology (2016) 2016(1):437–45. doi: 10.1182/asheducation-2016.1.437
68. Pott C, Hoster E, Delfau-Larue MH, Beldjord K, Böttcher S, Asnafi V, et al. Molecular Remission Is an Independent Predictor of Clinical Outcome in Patients With Mantle Cell Lymphoma After Combined Immunochemotherapy: A European MCL Intergroup Study. Blood (2010) 115(16):3215–23. doi: 10.1182/blood-2009-06-230250
69. National Comprehensive Cancer Network (NCCN). Clinical Practice Guidelines in Oncology (NCCN Guidelines). B-Cell Lymphomas (2021) 4. Available at: https://www.nccn.org/guidelines/guidelines-detail?category=1&id=1480.
70. Kumar KR, Cowley MJ, Davis RL. Next-Generation Sequencing and Emerging Technologies. Semin Thromb Hemost (2019) 45(7):661–73. doi: 10.1055/s-0039-1688446
71. Cheng DT, Mitchell TN, Zehir A, Shah RH, Benayed R, Syed A, et al. Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): A Hybridization Capture-Based Next-Generation Sequencing Clinical Assay for Solid Tumor Molecular Oncology. J Mol Diagn (2015) 17(3):251–64. doi: 10.1016/j.jmoldx.2014.12.006
72. Mandelker D, Zhang L, Kemel Y, Stadler ZK, Joseph V, Zehir A, et al. Mutation Detection in Patients With Advanced Cancer by Universal Sequencing of Cancer-Related Genes in Tumor and Normal DNA vs Guideline-Based Germline Testing. JAMA (2017) 318(9):825–35. doi: 10.1001/jama.2017.11137
73. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med (2015) 17(5):405–24. doi: 10.1038/gim.2015.30
74. Davi F, Langerak AW, Langlois de Septenville A, Kolijn PM, Hengeveld PJ, Chatzidimitriou A, et al. Immunoglobulin Gene Analysis in Chronic Lymphocytic Leukemia in the Era of Next Generation Sequencing. Leukemia (2020) 34(10):2545–51. doi: 10.1038/s41375-020-0923-9
75. Ho C, Arcila ME. Minimal Residual Disease Detection of Myeloma Using Sequencing of Immunoglobulin Heavy Chain Gene VDJ Regions. Semin Hematol (2018) 55(1):13–8. doi: 10.1053/j.seminhematol.2018.02.007
76. Song Y, Zhou K, Zou D, Zhou J, Hu J, Yang H, et al. Treatment of Patients With Relapsed or Refractory Mantle–Cell Lymphoma With Zanubrutinib, a Selective Inhibitor of Bruton's Tyrosine Kinase. Clin Cancer Res (2020) 26(16):4216–24. doi: 10.1158/1078-0432.CCR-19-3703
77. Ruan J, Martin P, Shah B, Schuster SJ, Smith SM, Furman RR, et al. Lenalidomide Plus Rituximab as Initial Treatment for Mantle-Cell Lymphoma. N Engl J Med (2015) 373(19):1835–44. doi: 10.1056/NEJMoa1505237
78. Le Gouill S, Thieblemont C, Oberic L, Moreau A, Bouabdallah K, Dartigeas C, et al. Rituximab After Autologous Stem-Cell Transplantation in Mantle-Cell Lymphoma. N Engl J Med (2017) 377(13):1250–60. doi: 10.1056/NEJMoa1701769
79. Eskelund CW, Kolstad A, Jerkeman M, Räty R, Laurell A, Eloranta S, et al. 15-Year Follow-Up of the Second Nordic Mantle Cell Lymphoma Trial (MCL 2): Prolonged Remissions Without Survival Plateau. Br J Haematol (2016) 175(3):410–8. doi: 10.1111/bjh.14241
80. Villa D, Sehn LH, Savage KJ, Toze CL, Song K, den Brok WD, et al. Bendamustine and Rituximab as Induction Therapy in Both Transplant-Eligible and-Ineligible Patients With Mantle Cell Lymphoma. Blood Adv (2020) 4(15):3486–94. doi: 10.1182/bloodadvances.2020002068
81. Delfau-Larue M-H, Klapper W, Berger F, Jardin F, Briere J, Salles G, et al. High-Dose Cytarabine Does Not Overcome the Adverse Prognostic Value of CDKN2A and TP53 Deletions in Mantle Cell Lymphoma. Blood J Am Soc Hematol (2015) 126(5):604–11. doi: 10.1182/blood-2015-02-628792
82. Kumar A, Sha F, Toure A, Dogan A, Ni A, Batlevi CL, et al. Patterns of Survival in Patients With Recurrent Mantle Cell Lymphoma in the Modern Era: Progressive Shortening in Response Duration and Survival After Each Relapse. Blood Cancer J (2019) 9(6):1–10. doi: 10.1038/s41408-019-0209-5
83. Czuczman MS, Goy A, Lamonica D, Graf DA, Munteanu MC, van der Jagt RH. Phase II Study of Bendamustine Combined With Rituximab in Relapsed/Refractory Mantle Cell Lymphoma: Efficacy, Tolerability, and Safety Findings. Ann Hematol (2015) 94(12):2025–32. doi: 10.1007/s00277-015-2478-9
84. Eyre TA, Walter HS, Iyengar S, Follows G, Cross M, Fox CP, et al. Efficacy of Venetoclax Monotherapy in Patients With Relapsed, Refractory Mantle Cell Lymphoma After Bruton Tyrosine Kinase Inhibitor Therapy. Haematologica (2019) 104(2):e68. doi: 10.3324/haematol.2018.198812
85. Trněný M, Lamy T, Walewski J, Belada D, Mayer J, Radford J, et al. Lenalidomide Versus Investigator's Choice in Relapsed or Refractory Mantle Cell Lymphoma (MCL-002; SPRINT): A Phase 2, Randomised, Multicentre Trial. Lancet Oncol (2016) 17(3):319–31. doi: 10.1016/S1470-2045(15)00559-8
86. Wang M, Munoz J, Goy A, Locke FL, Jacobson CA, Hill BT, et al. KTE-X19 CAR T-Cell Therapy in Relapsed or Refractory Mantle-Cell Lymphoma. N Engl J Med (2020) 382(14):1331–42. doi: 10.1056/NEJMoa1914347
Keywords: mantle cell lymphoma, genetic, epigenetic, molecular diagnostics, immunochemotherapy, targeted therapy
Citation: Sethi S, Epstein-Peterson Z, Kumar A and Ho C (2021) Current Knowledge in Genetics, Molecular Diagnostic Tools, and Treatments for Mantle Cell Lymphomas. Front. Oncol. 11:739441. doi: 10.3389/fonc.2021.739441
Received: 10 July 2021; Accepted: 29 October 2021;
Published: 23 November 2021.
Edited by:
Mina Luqing Xu, Yale University, United StatesReviewed by:
Susanna Akiki, Hamad Medical Corporation, QatarCopyright © 2021 Sethi, Epstein-Peterson, Kumar and Ho. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shenon Sethi, c2V0aGlzQG1za2NjLm9yZw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.