
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Oncol. , 02 December 2021
Sec. Molecular and Cellular Oncology
Volume 11 - 2021 | https://doi.org/10.3389/fonc.2021.738832
 Boning Cai1†
Boning Cai1† Xiaomo Li2†*
Xiaomo Li2†* Xiang Huang1
Xiang Huang1 Tonghui Ma2
Tonghui Ma2 Baolin Qu1Wei Yu1
Baolin Qu1Wei Yu1 Wei Yang1Pei Zhang1Jing Chen1
Wei Yang1Pei Zhang1Jing Chen1 Fang Liu1*
Fang Liu1*Epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKIs) are the standard of care for advanced non-small-cell lung cancer (NSCLC) patients. However, most patients will eventually develop resistance. For EGFR-TKI resistance mediated by MET amplification, the combination of EGFR and MET TKIs has shown promising results in early clinical trials. However, acquired resistance to MET inhibitors forms a formidable challenge to this dual blockade approach. Here, we presented an NSCLC patient with EGFR exon 19 deletion (ex19del) who was resistant to first-line erlotinib treatment but responded to chemotherapy. Given the finding of MET overexpression/amplification after disease progression, the patient received gefitinib plus crizotinib with a partial response. Her disease progressed again, and molecular testing revealed a novel MET Y1230H mutation and a PD-L1 TPS score of 75%. She received a salvage regime consisting of gefitinib, cabozantinib, and pembrolizumab with a partial response. Since we now know that EGFR ex19del NSCLC patients generally do not respond to PD-1 blockade therapy, this response is more likely the contribution from gefitinib plus cabozantinib. Therefore, sequential use of type I and II MET inhibitors in EGFR/MET dual blockade may be an effective therapeutic option for EGFR-mutant, MET-amplified NSCLC.
Epidermal growth factor receptor (EGFR) mutations are present in ~30-50% of non-small cell lung cancer (NSCLC) cases in East Asia and ~10% of cases in North America and Western Europe (1). EGFR tyrosine kinase inhibitors (EGFR-TKIs) are the standard of care for advanced EGFR-mutated NSCLC (2). As most NSCLC patients initially treated with EGFR TKIs will eventually acquire resistance, strategies to overcome EGFR TKI resistance are needed to improve patient outcomes. EGFR T790M mutation and MET amplification are the dominant on-target and off-target EGFR TKI resistance mechanisms, respectively (3). While the third-generation EGFR TKI osimertinib can overcome resistance mediated by EGFR T790M, resistance mediated by MET amplification remains a challenge (4).
Genetic alterations of MET are new therapeutic targets in NSCLC (4, 5). Three MET TKIs (capmatinib, tepotinib, and savolitinib) have been approved as first-line treatment for NSCLC patients with MET exon 14 skipping (METex14) mutations (6–8). Based on the mechanism of action, MET TKIs are divided into two groups, type I and type II, which bind to the active and inactive form of the ATP-pocket of MET, respectively (9). However, some acquired MET mutations such as Y1230H result in resistance to all type I MET inhibitors approved for NSCLC (10, 11). Furthermore, there is no approved targeted therapy for NSCLC patients with MET amplification, a less frequent driver in NSCLC compared with METex14 mutations (4). Therefore, the clinical management of MET-amplified NSCLC and acquired MET TKI resistance represent two clinical challenges.
To overcome EGFR TKI resistance mediated by MET amplification, clinicians are testing different EGFR/MET TKI combinations in clinical trials (12–15). Additionally, off-label use of type II MET TKI cabozantinib can be a feasible strategy to overcome acquired MET TKI resistance in METex14-NSCLC (4). Here, we described a metastatic EGFR-mutant NSCLC patient who developed EGFR TKI resistance mediated by MET overexpression/amplification and subsequently responded to gefitinib plus crizotinib. This patient then developed resistance to crizotinib due to an acquired MET Y1230H mutation, which was overcome by cabozantinib.
A 49-year-old Chinese female never-smoker without personal or family history was diagnosed with stage IV NSCLC (T4N3M1b) in June 2013 (Supplemental Table 1). PCR testing of the biopsy revealed the presence of EGFR exon 19 deletion (ex19del) mutation. The treatment timeline and molecular alterations are shown in Figure 1. The patient was administered erlotinib (150 mg daily). Progressive disease in the right lower lobe of lung and supraclavicular lymph nodes was noted after one month. Treatment was then changed to chemotherapy with cisplatin, pemetrexed, and bevacizumab for 6 cycles with partial response followed by 5 cycles of pemetrexed and bevacizumab maintenance.
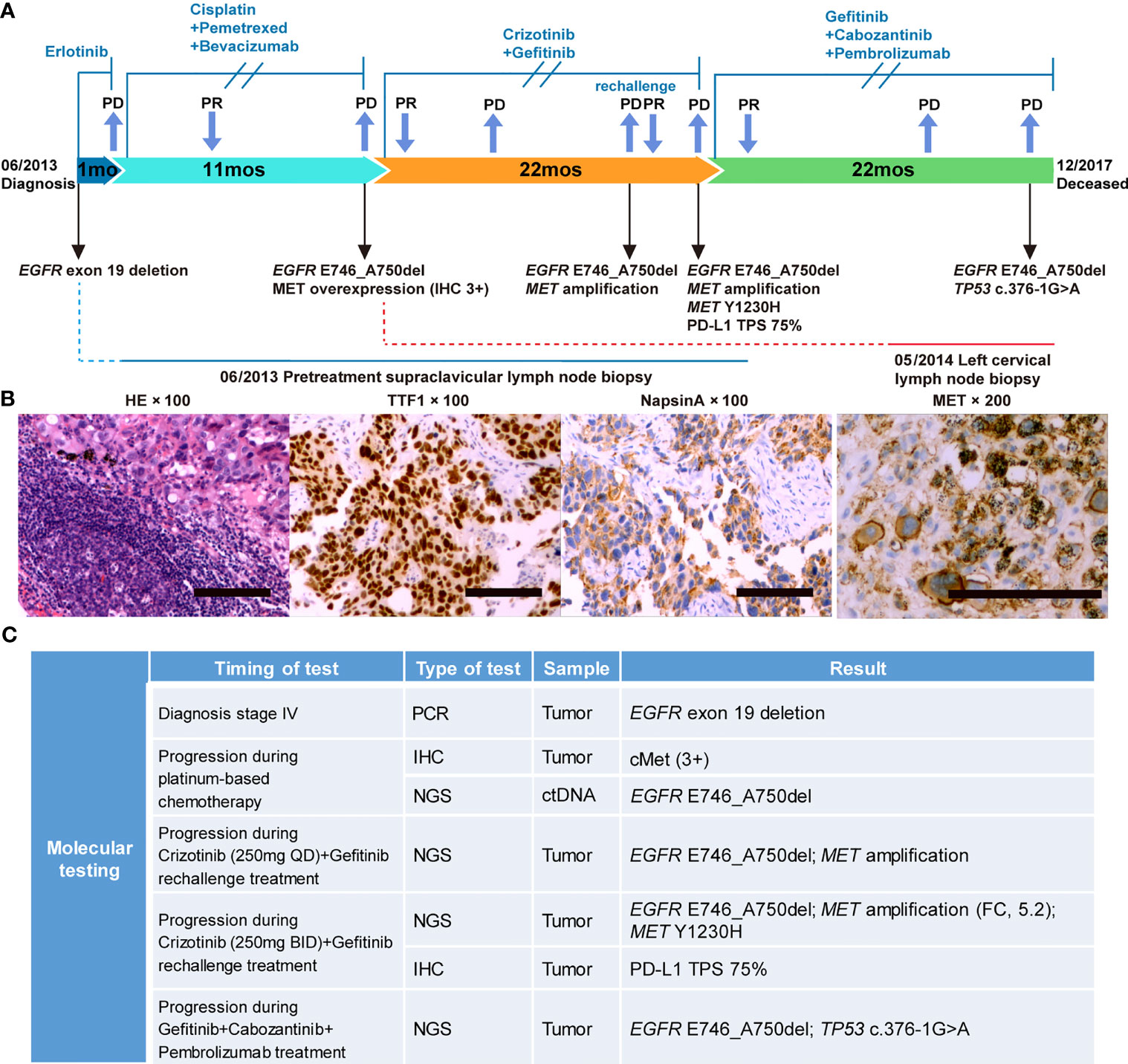
Figure 1 Case summary. (A) Summary of disease course, treatment timeline, and key molecular findings. (B) Hematoxylin-eosin staining, TTF1 and NapsinA positive immunohistochemical (IHC) staining before treatment; MET positive immunohistochemical staining during progression after platinum-based chemotherapy. Scale bars: 100 µm. (C) Detailed molecular alterations of tissue and liquid biopsy. FC, fold change; TPS, tumor proportion score.
In May 2014, the patient reported neck swelling, upper limb edema, dyspnea, and dysphagia with a PS score of 4. The barium swallow study demonstrated the presence of esophageal stricture. Imaging revealed new bilateral pulmonary nodules and supraclavicular lymph nodes enlargement. Her oxygen saturation values dropped to 70-85%, and she received supplemental oxygen by noninvasive ventilation. A biopsy revealed MET overexpression (IHC 3+), and ctDNA next-generation sequencing confirmed the EGFR E746_A750del mutation (Figures 1A, B). Crizotinib is an ALK, ROS1, and MET tyrosine kinases inhibitor approved for advanced ALK-positive lung cancer at that time (16). Crizotinib has shown antitumor activity in lung cancer patients with de novo MET amplification (17, 18), and was under validation in clinical trials (NCT01441128, NCT00585195). Therefore, treatment was changed to crizotinib (250 mg BID) plus gefitinib (250 mg QOD). On day 5, the patient’s neck swelling, upper limb edema, dyspnea, and dysphagia improved. Her oxygen saturation values improved to 85-95%, and ventilation was discontinued. After 16 days, computed tomography imaging showed an almost complete reduction of the target lesions (Figure 2). In March 2015, disease progression occurred, and she underwent radiotherapy (DT40Gy/20F) for metastatic lesions of the brain. In May 2015, treatment was changed to a combination of carboplatin, paclitaxel, and cetuximab for two cycles, and discontinued due to grade 4 myelosuppression. The patient reported dyspnea and chest pain with a PS score of 3. She was rechallenged with gefitinib plus crizotinib (reduced dose, 250 mg QD) for three months with stable disease. Her dyspnea improved, but chest pain remained. In November, imaging revealed new liver metastases and progressive disease in the lung. Genomic profiling of a biopsy confirmed the same EGFR ex19del mutation and MET amplification. Crizotinib was increased to 250 mg BID. On day 16, imaging showed a dramatic improvement of the target lesions (Figure 2).
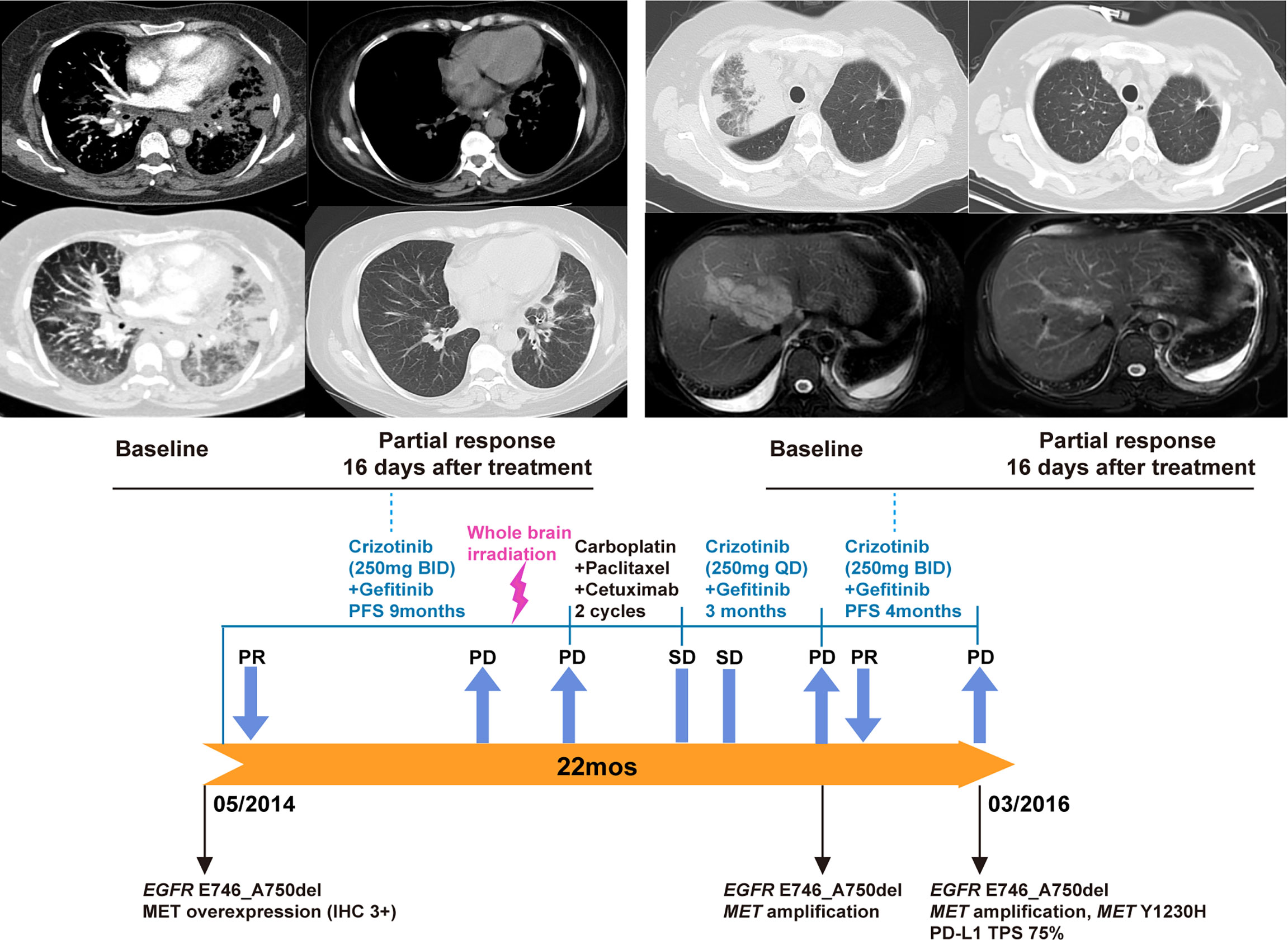
Figure 2 Gefitinib plus crizotinib treatment and rechallenge for EGFR-mutated NSCLC with MET overexpression/amplification. Based on the result of MET overexpression, the patient began gefitinib plus crizotinib. A partial response was observed 16 days after the initiation of this combination therapy (upper left panel). After disease progression, therapy was changed to chemotherapy plus cetuximab for two cycles. Due to chemotherapy-related toxicity, the patient was rechallenged with gefitinib plus crizotinib (reduced dose, 250 mg QD) with stable disease. Upon the development of new liver metastases, molecular testing of a biopsy confirmed concurrent EGFR mutation and MET amplification. Crizotinib dose was then increased to 250 mg BID (lower panel). Another partial response was observed 16 days after the crizotinib dose increase (upper right).
In February 2016, the patient developed progressive dyspnea, cough, left-sided limb edema, chest pain, swelling of her left breast, and multiple chest wall/pulmonary nodules. Analysis of her left breast biopsy revealed an acquired MET Y1230H mutation and a high PD-L1 expression level (TPS 75%). She received induction radiotherapy (DT18Gy/3F) for supraclavicular metastases. The function of MET Y1230H mutation in lung cancer was unknown at that time. One in vitro study demonstrated that Y1230H mutation resulted in resistance to type I but not type II MET inhibitors in BaF3 cells (19). We reasoned that a type II MET inhibitor might overcome this acquired resistance. Additionally, given the latest approval of PD-1 antibody pembrolizumab in NSCLC and its distinct mechanism of action, we believed that pembrolizumab could benefit this patient independent of targeted therapy. With the informed consent from the patient, she received a salvage therapy comprising of gefitinib (250 mg QOD), cabozantinib (40 mg QD), and pembrolizumab (100 mg every two weeks). At one-month follow-up, her chest wall nodules and left breast swelling regressed, and her dyspnea improved. Imaging demonstrated a dramatic radiographic response which lasted for 13 months (Figure 3). In April 2017, the patient developed new metastases in the left erector spinae muscle and posterior abdominal wall. Bevacizumab was added to the combination regime with stable disease. Unfortunately, the patient’s condition further deteriorated in October. Cognitive deficits and electroencephalograms (EEG) abnormalities were noted. Imaging revealed pleural effusion and new lesions in the liver. Both bevacizumab and pembrolizumab were discontinued. Genomic profiling of a biopsy revealed the original EGFR mutation, a TP53 c.376-1G>A splice site mutation, and the clearance of MET amplification/Y1230H mutation (Figure 1). The patient chose to continue gefitinib plus cabozantinib, and died of multiple organ dysfunction two months later. From the diagnosis of metastatic EGFR-mutant NSCLC in 2013, this patient achieved an overall survival of 54 months.
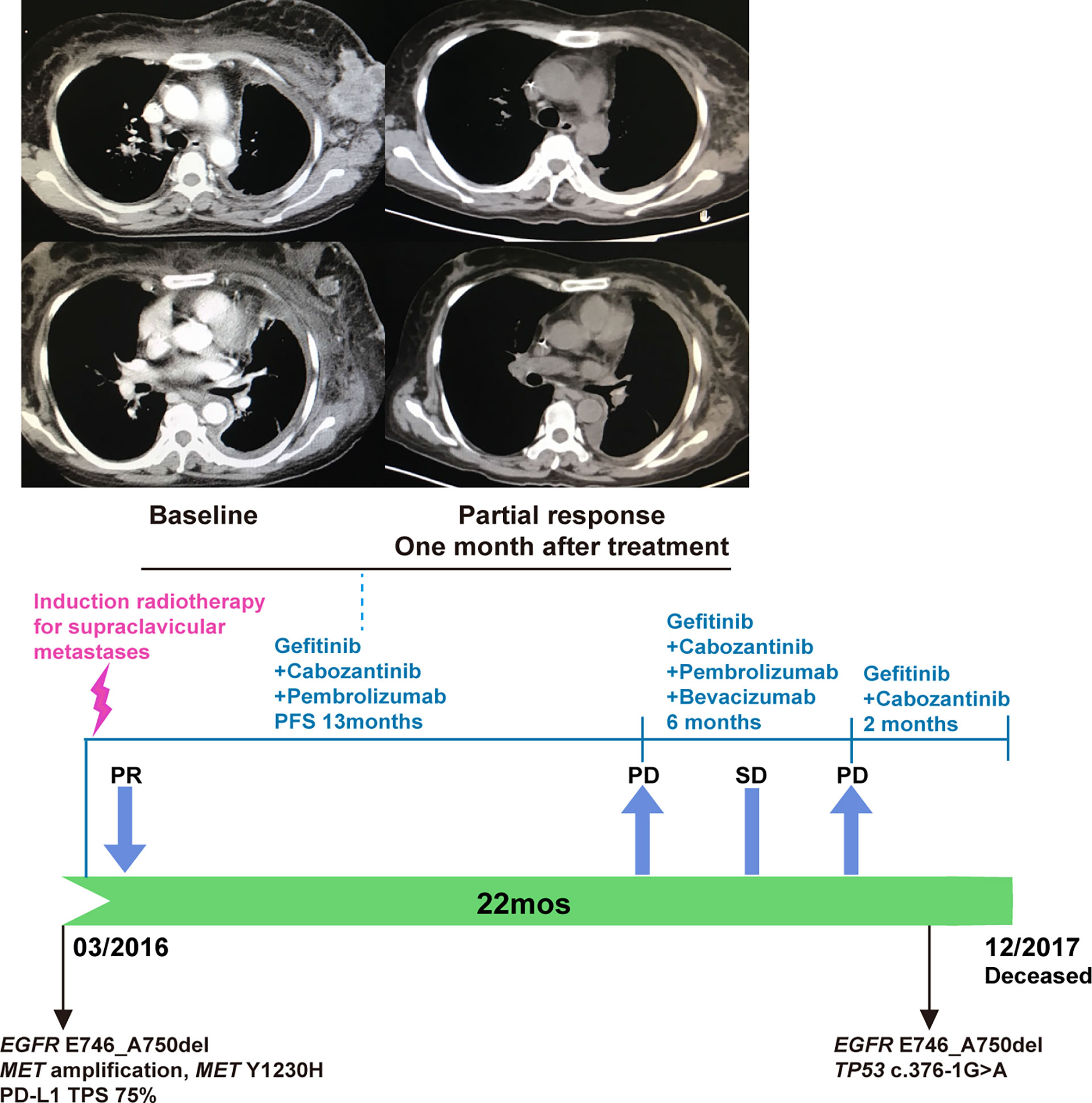
Figure 3 The combination of gefitinib, cabozantinib, and pembrolizumab for PD-L1-positive, EGFR-mutated, MET-amplified, MET Y1230H NSCLC. A partial response was observed one month after the initiation of this triplet regime (upper panel). Bevacizumab was added when the patient developed new metastases in April 2017. Both bevacizumab and pembrolizumab were stopped in October 2017 due to acute patient deterioration (lower panel).
In the past decade, precision therapy has gradually become the standard of care for metastatic NSCLC patients with actionable biomarkers (20). These predictive biomarkers include immune biomarker PD-L1 and targetable driver mutations such as EGFR L858R/ex19del mutations. While next-generation sequencing and PD-L1 testing have significantly improved the treatment decision-making process for NSCLC, oncologists need to realize that the optimal treatment for patients with multiple actionable biomarkers requires evidence-based biological rationale and up-to-date knowledge of clinical trial results.
The EGFR-mutant NSCLC case we reported involved the interplay of four biomarkers: EGFR exon 19 deletion (ex19del), PD-L1, MET amplification (MET-amp), and MET Y1230H mutation. To make the discussion more relevant to today’s clinical practice, we constructed three scenarios related to this case: 1. EGFR ex19del mutation and high PD-L1 expression in untreated NSCLC patients; 2. acquired MET amplification in EGFR ex19del NSCLC after first-line EGFR TKI therapy; 3. acquired MET Y1230H mutation in MET-driven NSCLC after first-line MET TKI therapy.
For the first scenario, EGFR ex19del NSCLC patients with high PD-L1 expression levels, three treatment choices are available: chemotherapy, EGFR-targeted therapy, and immunotherapy. The decision-making process for this scenario can be simplified by a review of NSCLC NCCN guidelines and literature. The 2017 NSCLC NCCN guideline stated that EGFR TKIs resulted in longer PFS and fewer toxicities than chemotherapy in patients with sensitizing EGFR mutations (21). Additionally, pembrolizumab did not show any response in the first 11 patients enrolled in a phase 2 trial for EGFR-mutant, PD-L1-positive NSCLC, including 8 patients with PD-L1 expression more than 50% (22). In a multicenter, retrospective study involving 171 EGFR-mutant NSCLC patients treated with immunotherapy, subgroup analysis demonstrated that the response rate of PD(L)-1 antibodies was very low in EGFR ex19del NSCLC irrespective of the PD-L1 status (23). Furthermore, the combination of EGFR TKIs and immunotherapy had significantly higher toxicities than either alone (24). For these reasons, Calles et al. commented in the 2020 ASCO educational book that immunotherapy alone or combined with EGFR TKI is not recommended to EGFR-mutant NSCLC (25). For the first scenario, EGFR TKI should be the first-line therapy choice.
The second scenario, EGFR TKI resistance mediated by MET amplification, is a frontier currently under intensive investigation (26). There are at least three possible strategies to overcome this challenge: EGFR TKI plus MET TKI, EGFR-MET bispecific antibodies, and EGFR TKI plus EGFR-MET bispecific antibodies. The EGFR/MET TKI combinations evaluated in early-stage clinical trials include osimertinib/savolitinib, gefitinib/savolitinib, gefitinib/capmatinib, and gefitinib/tepotinib. In the phase 1b TATTON study, osimertinib plus savolitinib achieved an ORR of 64% in osimertinib-naive, EGFR T790M-negative patients and 48% in a mixed pool of osimertinib-treated, osimertinib-naive/T790M-positive, and osimertinib-naive/T790M-negative patients (15). A phase 1b trial of gefitinib plus savolitinib showed an ORR of 52% (12/23), 9% (2/23), and 40% (2/5) in EGFR T790M-negative, -positive, and -unknown patients, respectively (12). In a phase 1b/2 trial, gefitinib plus capmatinib reached an ORR of 47% in patients with high MET-amplification (MET gene copy number ≥ 6) and 27% overall (14). Similarly, in an early-terminated phase 2 trial (INSIGHT), subgroup analysis revealed that gefitinib plus tepotinib resulted in longer PFS/OS than platinum duplet chemotherapy control in 34 patients with high MET overexpression (IHC3+) (mPFS 8.3 vs 4.4 months, HR 0.35; mOS 37.3 vs 17.9 months, HR 0.33) as well as 19 patients with high MET amplification (mean gene copy number ≥5 or MET to the centromere of chromosome 7 ratio ≥2) (mPFS 16.6 vs 4.2 months, HR 0.13; mOS 37.3 vs 13.1 months, HR 0.08) (13). Our patient had high MET overexpression (IHC 3+) and high MET amplification (MET gene copy number = 5.2). Although she received different gefitinib/MET TKI combination therapies (gefitinib/crizotinib and gefitinib/cabozantinib), her PFS and OS are similar to the results of the INSIGHT study (gefitinib/tepotinib).
In May 2021, FDA granted the accelerated approval of an EGFR-MET bispecific antibody amivantamab as the first targeted therapy for NSCLC patients with EGFR exon 20 insertions (ex20ins) based on the phase 1 CHRYSALIS study (27). Amivantamab inhibits EGFR/MET signaling through three mechanisms: internalization and degradation of EGFR/MET receptors, blocking ligand-dependent receptor activation, and antibody-dependent cell-mediated cytotoxicity (ADCC) (28). In a preclinical model of EGFR-mutant, MET-amplified NSCLC, amivantamab showed superior antitumor activity than the combination of erlotinib and crizotinib (29). Interestingly, results of the CHRYSALIS trial demonstrated that amivantamab alone or combined with a third-generation EGFR TKI lazertinib had antitumor activity in patients with EGFR-mutant, MET-amplified NSCLC (30). The subgroup analysis showed that amivantamab plus lazertinib had a 90% (9/10) and 10% (1/10) response rate in MET IHC-high and MET IHC-low patients, respectively (31). Therefore, amivantamab could be the game-changer for this specific patient population.
The third scenario, MET TKI resistance mediated by MET Y1230H mutation, is associated with METex14 and MET-amplified NSCLC. Currently, the approval of capmatinib, tepotinib, and salvotinib are limited to METex14, which is the only MET alteration included in the 2021 NSCLC NCCN guideline (2). According to the TCGA data, METex14 and MET amplification were the first and second most frequent MET alterations in NSCLC, respectively (32). In agreement, Y1230H mutation was more frequently observed in METex14 NSCLC than MET-amplified NSCLC (33, 34). Works from Engstrom and others confirmed that MET Y1230H mutant was resistant to type I MET TKI capmatinib, salvotinib, and crizotinib but remained sensitive to type II MET TKI glesatinib (10). As type I and II MET TKIs bind the ATP-pocket of MET differently, they have distinct inhibition capacities to MET mutants. The IC50 values of METex14 single mutant for capmatinib, crizotinib, and glesatinib in NIH/3T3 cells were 2.4 nM, 28.9 nM, and 80.6 nM, respectively. In contrast, the IC50 values of METex14/Y1230H double mutant for capmatinib, crizotinib, and glesatinib in NIH/3T3 cells were >3,000 nM, 278 nM, and 56 nM, respectively (10). Similarly, the IC50 values of MET Y1230H single mutant for capmatinib, savolitinib, crizotinib, glesatinib, cabozantinib, merestinib in Ba/F3 cells were 401 nM, >1,000 nM, 216 nM, 19 nM, 20 nM, and 8.2 nM, respectively (35). These results predict that acquired MET Y1230H mutation will confer resistance to all MET TKIs currently approved for NSCLC.
Given the fact that cabozantinib is the only approved type II MET inhibitor, the combination of cabozantinib and EGFR TKI is a reasonable and feasible strategy to combat acquired resistance to type I MET TKI. Bahcall et al. reported acquired D1228V mutation resulted in resistance to osimertinib plus salvotinib in an EGFR ex19del/MET-amp NSCLC patient, who subsequently responded to erlotinib plus cabozantinib (36). In another similar case, plasma genotyping revealed four acquired MET mutations (D1228H/N/Y and Y1230C) in an EGFR ex19del/MET-amp NSCLC patient who became resistant to osimertinib plus salvotinib (37). Treatment was changed to osimertinib plus cabozantinib with stable disease and clearance of Y1230C mutation. However, cabozantinib was soon discontinued due to toxicities, and the patient died three months later. These clinical observations are consistent with the report that MET D1228X mutations are more resistant to cabozantinib than Y1230X mutations (38).
Lastly, we would like to discuss our treatment decision-making for crizotinib resistance mediated by MET Y1230H mutation. In February 2016, we could not find reports of this mutation in lung cancer except for two preclinical MET TKI resistance studies (19, 39). Tiedt et al. conducted a drug resistance screen in BaF3 TPR-MET cells with type I MET inhibitor NVP-BVU972 and type II MET inhibitor AMG-458 (19). Most NVP-BVU972-resistant clones carry missense mutations in Y1230 and D1228. Structure study revealed that NVP-BVU972 interacts with the aromatic side chain of Y1230. Therefore, a mutation in this residue will disrupt NVP-BVU972 binding and result in drug resistance. The Y1230 mutation was not detected in the AMG-458 screen. Biochemical assay results demonstrated that MET Y1230H mutant was sensitive to AMG-458 but not NVP-BVU972 (IC50 value: 1.6 nM vs. >127 nM). Similarly, Funakoshi et al. conducted a screen in a MET-amplified gastric cell line MKN45 with a type I MET inhibitor PHA665752 and a type II MET inhibitor GSK1363089/XL880/foretinib (39). MET Y1230H mutation was only identified in PHA665752-resistant clones but not foretinib-resistant clones. This result was expected as the IC50 value of Y1230H mutant for XL880/foretinib was only 0.7 nM (19). Based on these results, we reasoned that the crizotinib resistance seen in our case was likely mediated by Y1230H, a MET mutation sensitive to type II MET inhibitors. Therefore, we replaced type I MET TKI crizotinib with type II MET TKI cabozantinib in our EGFR/MET dual blockade regime.
In summary, we presented the efficacy of gefitinib plus crizotinib in an EGFR-mutant NSCLC patient with high-level MET overexpression/amplification and resistance to erlotinib. Because crizotinib is more accessible and affordable than capmatinib, tepotinib, and salvotinib, this combination provides a feasible treatment option for EGFR-mutant, MET-amplified NSCLC patients who can not assess or afford MET-specific TKIs. Furthermore, the switch from type I MET TKI to type II MET TKI cabozantinib can be an effective strategy to overcome acquired type I MET TKI resistance in NSCLC. Given the recent approval of EGFR-MET bispecific antibody amivantamab, future investigations are required to explore the safety and efficacy of TKI-based and antibody-based EGFR/MET dual blockade therapy in EGFR-mutant, MET-amplified NSCLC.
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding authors.
Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
FL: conceptualization, methodology, supervision, and writing-reviewing and editing. BC and XL: visualization, investigation, and writing-original draft preparation. XH, BQ, WYu, WYang, PZ, and JC: investigation, data curation, and validation. TM and BQ: writing-reviewing and editing. All authors contributed to the article and approved the submitted version.
XL and TM are employees of Genetron Health (Beijing) Technology, Co. Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The authors would like to thank Dr. Yi Cai for his valuable and constructive suggestions during the preparation of this manuscript.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2021.738832/full#supplementary-material
1. Sharma SV, Bell DW, Settleman J, Haber DA. Epidermal Growth Factor Receptor Mutations in Lung Cancer. Nat Rev Cancer (2007) 7:169–81. doi: 10.1038/nrc2088
2. Ettinger DS, Wood DE, Aisner DL, Akerley W, Bauman JR, Bharat A, et al. NCCN Guidelines Insights: Non-Small Cell Lung Cancer, Version 2.2021. J Natl Compr Cancer Netw JNCCN (2021) 19:254–66. doi: 10.6004/jnccn.2021.0013
3. Westover D, Zugazagoitia J, Cho BC, Lovly CM, Paz-Ares L. Mechanisms of Acquired Resistance to First- and Second-Generation EGFR Tyrosine Kinase Inhibitors. Ann Oncol Off J Eur Soc Med Oncol (2018) 29:i10–9. doi: 10.1093/annonc/mdx703
4. Guo R, Luo J, Chang J, Rekhtman N, Arcila M, Drilon A. MET-Dependent Solid Tumours - Molecular Diagnosis and Targeted Therapy. Nat Rev Clin Oncol (2020) 17:569–87. doi: 10.1038/s41571-020-0377-z
5. Mathieu LN, Larkins E, Akinboro O, Roy P, Amatya AK, Fiero MH, et al. FDA Approval Summary: Capmatinib and Tepotinib for the Treatment of Metastatic NSCLC Harboring MET Exon 14 Skipping Mutations or Alterations. Clin Cancer Res Off J Am Assoc Cancer Res (2021). doi: 10.1158/1078-0432.Ccr-21-1566
6. Wolf J, Seto T, Han JY, Reguart N, Garon EB, Groen HJM, et al. Capmatinib in MET Exon 14-Mutated or MET-Amplified Non-Small-Cell Lung Cancer. N Engl J Med (2020) 383:944–57. doi: 10.1056/NEJMoa2002787
7. Paik PK, Felip E, Veillon R, Sakai H, Cortot AB, Garassino MC, et al. Tepotinib in Non-Small-Cell Lung Cancer With MET Exon 14 Skipping Mutations. N Engl J Med (2020) 383:931–43. doi: 10.1056/NEJMoa2004407
9. Recondo G, Bahcall M, Spurr LF, Che J, Ricciuti B, Leonardi GC, et al. Molecular Mechanisms of Acquired Resistance to MET Tyrosine Kinase Inhibitors in Patients With MET Exon 14-Mutant NSCLC. Clin Cancer Res Off J Am Assoc Cancer Res (2020) 26:2615–25. doi: 10.1158/1078-0432.Ccr-19-3608
10. Engstrom LD, Aranda R, Lee M, Tovar EA, Essenburg CJ, Madaj Z, et al. Glesatinib Exhibits Antitumor Activity in Lung Cancer Models and Patients Harboring MET Exon 14 Mutations and Overcomes Mutation-Mediated Resistance to Type I MET Inhibitors in Nonclinical Models. Clin Cancer Res Off J Am Assoc Cancer Res (2017) 23:6661–72. doi: 10.1158/1078-0432.Ccr-17-1192
11. Shen B, Wu F, Ye J, Liang R, Wang R, Yu R, et al. Crizotinib-Resistant MET Mutations in Gastric Cancer Patients Are Sensitive to Type II Tyrosine Kinase Inhibitors. Future Oncol (London England) (2019) 15:2585–93. doi: 10.2217/fon-2019-0140
12. Yang JJ, Fang J, Shu YQ, Chang JH, Chen GY, He JX, et al. A Phase Ib Study of the Highly Selective MET-TKI Savolitinib Plus Gefitinib in Patients With EGFR-Mutated, MET-Amplified Advanced Non-Small-Cell Lung Cancer. Invest New Drugs (2021) 39:477–87. doi: 10.1007/s10637-020-01010-4
13. Wu YL, Cheng Y, Zhou J, Lu S, Zhang Y, Zhao J, et al. Tepotinib Plus Gefitinib in Patients With EGFR-Mutant Non-Small-Cell Lung Cancer With MET Overexpression or MET Amplification and Acquired Resistance to Previous EGFR Inhibitor (INSIGHT Study): An Open-Label, Phase 1b/2, Multicentre, Randomised Trial. Lancet Respir Med (2020) 8:1132–43. doi: 10.1016/s2213-2600(20)30154-5
14. Wu YL, Zhang L, Kim DW, Liu X, Lee DH, Yang JC, et al. Phase Ib/II Study of Capmatinib (INC280) Plus Gefitinib After Failure of Epidermal Growth Factor Receptor (EGFR) Inhibitor Therapy in Patients With EGFR-Mutated, MET Factor-Dysregulated Non-Small-Cell Lung Cancer. J Clin Oncol Off J Am Soc Clin Oncol (2018) 36:3101–9. doi: 10.1200/jco.2018.77.7326
15. Sequist LV, Han JY, Ahn MJ, Cho BC, Yu H, Kim SW, et al. Osimertinib Plus Savolitinib in Patients With EGFR Mutation-Positive, MET-Amplified, Non-Small-Cell Lung Cancer After Progression on EGFR Tyrosine Kinase Inhibitors: Interim Results From a Multicentre, Open-Label, Phase 1b Study. Lancet Oncol (2020) 21:373–86. doi: 10.1016/s1470-2045(19)30785-5
16. Shaw AT, Kim DW, Nakagawa K, Seto T, Crinó L, Ahn MJ, et al. Crizotinib Versus Chemotherapy in Advanced ALK-Positive Lung Cancer. New Engl J Med (2013) 368:2385–94. doi: 10.1056/NEJMoa1214886
17. Ou SH, Kwak EL, Siwak-Tapp C, Dy J, Bergethon K, Clark JW, et al. Activity of Crizotinib (PF02341066), a Dual Mesenchymal-Epithelial Transition (MET) and Anaplastic Lymphoma Kinase (ALK) Inhibitor, in a Non-Small Cell Lung Cancer Patient With De Novo MET Amplification. J Thorac Oncol Off Publ Int Assoc Study Lung Cancer (2011) 6:942–6. doi: 10.1097/JTO.0b013e31821528d3
18. Schwab R, Petak I, Kollar M, Pinter F, Varkondi E, Kohanka A, et al. Major Partial Response to Crizotinib, a Dual MET/ALK Inhibitor, in a Squamous Cell Lung (SCC) Carcinoma Patient With De Novo C-MET Amplification in the Absence of ALK Rearrangement. Lung Cancer (Amsterdam Netherlands) (2014) 83:109–11. doi: 10.1016/j.lungcan.2013.10.006
19. Tiedt R, Degenkolbe E, Furet P, Appleton BA, Wagner S, Schoepfer J, et al. A Drug Resistance Screen Using a Selective MET Inhibitor Reveals a Spectrum of Mutations That Partially Overlap With Activating Mutations Found in Cancer Patients. Cancer Res (2011) 71:5255–64. doi: 10.1158/0008-5472.Can-10-4433
20. Camidge DR, Doebele RC, Kerr KM. Comparing and Contrasting Predictive Biomarkers for Immunotherapy and Targeted Therapy of NSCLC. Nat Rev Clin Oncol (2019) 16:341–55. doi: 10.1038/s41571-019-0173-9
21. Ettinger DS, Wood DE, Aisner DL, Akerley W, Bauman J, Chirieac LR, et al. Non-Small Cell Lung Cancer, Version 5.2017, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Cancer Netw JNCCN (2017) 15:504–35. doi: 10.6004/jnccn.2017.0050
22. Lisberg A, Cummings A, Goldman JW, Bornazyan K, Reese N, Wang T, et al. A Phase II Study of Pembrolizumab in EGFR-Mutant, PD-L1+, Tyrosine Kinase Inhibitor Naïve Patients With Advanced NSCLC. J Thorac Oncol Off Publ Int Assoc Study Lung Cancer (2018) 13:1138–45. doi: 10.1016/j.jtho.2018.03.035
23. Hastings K, Yu HA, Wei W, Sanchez-Vega F, DeVeaux M, Choi J, et al. EGFR Mutation Subtypes and Response to Immune Checkpoint Blockade Treatment in Non-Small-Cell Lung Cancer. Ann Oncol Off J Eur Soc Med Oncol (2019) 30:1311–20. doi: 10.1093/annonc/mdz141
24. Oshima Y, Tanimoto T, Yuji K, Tojo A. EGFR-TKI-Associated Interstitial Pneumonitis in Nivolumab-Treated Patients With Non-Small Cell Lung Cancer. JAMA Oncol (2018) 4:1112–5. doi: 10.1001/jamaoncol.2017.4526
25. Calles A, Riess JW, Brahmer JR. Checkpoint Blockade in Lung Cancer With Driver Mutation: Choose the Road Wisely. American Society of Clinical Oncology Educational Book. Am Soc Clin Oncol Annu Meeting (2020) 40:372–84. doi: 10.1200/edbk_280795
26. Melosky B, Wheatley-Price P, Juergens RA, Sacher A, Leighl NB, Tsao MS, et al. The Rapidly Evolving Landscape of Novel Targeted Therapies in Advanced Non-Small Cell Lung Cancer. Lung Cancer (Amsterdam Netherlands) (2021) 160:136–51. doi: 10.1016/j.lungcan.2021.06.002
27. Park K, Haura EB, Leighl NB, Mitchell P, Shu CA, Girard N, et al. Amivantamab in EGFR Exon 20 Insertion-Mutated Non-Small-Cell Lung Cancer Progressing on Platinum Chemotherapy: Initial Results From the CHRYSALIS Phase I Study. J Clin Oncol Off J Am Soc Clin Oncol (2021) 39:3391–402. doi: 10.1200/jco.21.00662
28. Yun J, Lee SH, Kim SY, Jeong SY, Kim JH, Pyo KH, et al. Antitumor Activity of Amivantamab (JNJ-61186372), an EGFR-MET Bispecific Antibody, in Diverse Models of EGFR Exon 20 Insertion-Driven NSCLC. Cancer Discovery (2020) 10:1194–209. doi: 10.1158/2159-8290.Cd-20-0116
29. Neijssen J, Cardoso RMF, Chevalier KM, Wiegman L, Valerius T, Anderson GM, et al. Discovery of Amivantamab (JNJ-61186372), a Bispecific Antibody Targeting EGFR and MET. J Biol Chem (2021) 296:100641. doi: 10.1016/j.jbc.2021.100641
30. Köhler J, Jänne PA. Amivantamab: Treating EGFR Exon 20-Mutant Cancers With Bispecific Antibody-Mediated Receptor Degradation. J Clin Oncol Off J Am Soc Clin Oncol (2021) 39:3403–6. doi: 10.1200/jco.21.01494
31. Bauml J, Cho BC, Park K, Lee KH, CHO EK, Kim D-W, et al. Amivantamab in Combination With Lazertinib for the Treatment of Osimertinib-Relapsed, Chemotherapy-Naïve EGFR Mutant (EGFRm) Non-Small Cell Lung Cancer (NSCLC) and Potential Biomarkers for Response. J Clin Oncol (2021) 39:9006–6. doi: 10.1200/JCO.2021.39.15_suppl.9006
32. Cancer Genome Atlas Research Network. Comprehensive Molecular Profiling of Lung Adenocarcinoma. Nature (2014) 511:543–50. doi: 10.1038/nature13385
33. Heist RS, Sequist LV, Borger D, Gainor JF, Arellano RS, Le LP, et al. Acquired Resistance to Crizotinib in NSCLC With MET Exon 14 Skipping. J Thorac Oncol Off Publ Int Assoc Study Lung Cancer (2016) 11:1242–5. doi: 10.1016/j.jtho.2016.06.013
34. Ou SI, Young L, Schrock AB, Johnson A, Klempner SJ, Zhu VW, et al. Emergence of Preexisting MET Y1230C Mutation as a Resistance Mechanism to Crizotinib in NSCLC With MET Exon 14 Skipping. J Thorac Oncol Off Publ Int Assoc Study Lung Cancer (2017) 12:137–40. doi: 10.1016/j.jtho.2016.09.119
35. Fujino T, Kobayashi Y, Suda K, Koga T, Nishino M, Ohara S, et al. Sensitivity and Resistance of MET Exon 14 Mutations in Lung Cancer to Eight MET Tyrosine Kinase Inhibitors In Vitro. J Thorac Oncol Off Publ Int Assoc Study Lung Cancer (2019) 14:1753–65. doi: 10.1016/j.jtho.2019.06.023
36. Bahcall M, Sim T, Paweletz CP, Patel JD, Alden RS, Kuang Y, et al. Acquired METD1228V Mutation and Resistance to MET Inhibition in Lung Cancer. Cancer Discovery (2016) 6:1334–41. doi: 10.1158/2159-8290.Cd-16-0686
37. Piper-Vallillo AJ, Halbert BT, Rangachari D, Kobayashi SS, Costa DB. Acquired Resistance to Osimertinib Plus Savolitinib Is Mediated by MET-D1228 and MET-Y1230 Mutations in EGFR-Mutated MET-Amplified Lung Cancer. JTO Clin Res Rep (2020) 1:100071. doi: 10.1016/j.jtocrr.2020.100071
38. Fujino T, Mitsudomi T. Acquired Resistance Mechanism for MET Tyrosine Kinase Inhibitor. JTO Clin Res Rep (2021) 2:100134. doi: 10.1016/j.jtocrr.2020.100134
Keywords: non-small cell lung cancer (NSCLC), EGFR mutation, targeted therapy resistance, MET amplification, case report
Citation: Cai B, Li X, Huang X, Ma T, Qu B, Yu W, Yang W, Zhang P, Chen J and Liu F (2021) Case Report: Sequential Combination Targeted Therapy With Type I and II MET Inhibitors in a Metastatic EGFR-Mutated, MET-Amplified NSCLC Patient With Acquired MET Y1230H Mutation. Front. Oncol. 11:738832. doi: 10.3389/fonc.2021.738832
Received: 09 July 2021; Accepted: 17 November 2021;
Published: 02 December 2021.
Edited by:
Kenneth K. W. To, The Chinese University of Hong Kong, ChinaReviewed by:
Neha Nanda, Johns Hopkins University School of Medicine, United StatesCopyright © 2021 Cai, Li, Huang, Ma, Qu, Yu, Yang, Zhang, Chen and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fang Liu, bGl1ZmFuZ2ZzcUAxNjMuY29t; Xiaomo Li, eGlhb21vLmxpQGdlbmV0cm9uaGVhbHRoLmNvbQ==
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.