 Carlos Cuesta-Mateos
Carlos Cuesta-Mateos Fernando Terrón2,3
Fernando Terrón2,3 Marco Herling
Marco Herling- 1Immunology Department, Hospital Universitario de La Princesa, Instituto de Investigación Sanitaria- Instituto la Princesa (IIS-IP), Madrid, Spain
- 2Immunological and Medicinal Products (IMMED S.L.), Madrid, Spain
- 3Catapult Therapeutics BV, Lelystad, Netherlands
- 4Clinic of Hematology and Cellular Therapy, University of Leipzig, Leipzig, Germany
According to the classical paradigm, CCR7 is a homing chemokine receptor that grants normal lymphocytes access to secondary lymphoid tissues such as lymph nodes or spleen. As such, in most lymphoproliferative disorders, CCR7 expression correlates with nodal or spleen involvement. Nonetheless, recent evidence suggests that CCR7 is more than a facilitator of lymphatic spread of tumor cells. Here, we review published data to catalogue CCR7 expression across blood cancers and appraise which classical and novel roles are attributed to this receptor in the pathogenesis of specific hematologic neoplasms. We outline why novel therapeutic strategies targeting CCR7 might provide clinical benefits to patients with CCR7-positive hematopoietic tumors.
1 Introduction
Lymph nodes (LN) function as major immunological hubs, being essential for immune homeostasis and the generation of effective immune responses (1). LNs are also fundamental sites-of-origin in disease development and progression as well as in treatment failure of several hematological malignancies. Growing evidence suggests that cell trafficking orchestrated by the C-C chemokine receptor 7 (CCR7) plays a critical role in the pathophysiology of several leukemias and lymphomas. This receptor assists malignant cells in access to niches that provide proliferating cues and enable escape from therapy-induced death, hence, promoting disease progression and resistance. In this review we provide a summary of insights towards a better understanding in which blood cancers, particularly B-cell, T-cell, and myeloid-cell malignancies, CCR7 mediates which pathogenetic functions. We further appraise how this chemokine receptor is of great potential for the development of rational and effective therapies in some of these conditions.
2 CCR7: A Single Receptor Linking Innate and the Adaptive Immunity in the LN
The homeostatic chemokine receptor CCR7 (also known as Epstein–Barr virus-induced gene 1 (EBI1), Burkitt’s lymphoma receptor 2 (BLR2), or CD197) is a G-protein coupled receptor (GPCR) (2–4). CCR7 is expressed by various immune cells including double negative (DN) and single positive (SP) thymocytes, naïve, central memory, and regulatory T-cells (TN, TCM, TREG) as well as naïve B-cells, CD56+CD16- regulatory NK cells, and semi-mature and fully mature dendritic cells (DC) (5–7). Generally, T-cell subsets and mature B-cells constitutively express CCR7 whereas NK cells and DC acquire CCR7 expression upon pathogen encounter (5). In homeostasis CCR7 specifically drives cell homing into LN and other secondary lymphoid organs (SLO) (8–10). This single GPCR orchestrates efficient interactions between different CCR7-expressing cell types that belong to both the innate and the adaptive functional axis of immunity and which immigrated to the LN from different peripheral environments. As part of this, CCR7 directs central aspects of immune cell migration into the LN: cell trafficking, firm arrest at the endothelium, extravasation, positioning within SLO, activation, differentiation, survival, and egress. All these processes mediated by CCR7 take place upon binding to either of its two cognate ligands, the chemokines CCL19 (also known as ELC or MIP-3β) and CCL21 (also known as SLC or 6CK), which are constitutively expressed by stroma cells in SLO and which are present on lymphatic vessels, high-endothelial venules (HEVs), and fibroblastic reticular cells (FBR) of the T-cell zones (5, 6, 11). Different signaling pathways downstream of CCR7 and several mechanisms differentially ignited by CCL19 or CCL21 signaling determine the overall outcome of CCR7 engagement in each cell type. A detailed review on those mechanisms is provided by Hauser et al. (11).
3 CCR7 Expression and Functions in Distinct Blood Cancers
Most of CCR7’s roles in homeostasis (e.g. cell trafficking, interstitial migration, or survival) are particularly relevant for leukemias and lymphomas, which very often express CCR7 because of their lymphoid origin and/or maturation stage. In this section we will appraise the current knowledge on CCR7 biology in several B-cell and T-cell cancers and in selected myeloid neoplasms. In addition, we will review the evidence that associates expression profiles of CCR7 with functionality and pathological findings such as LN infiltration or spread to the central nervous system (CNS).
3.1 B-Cell Malignancies
B-cell malignancies consist of distinct diseases that can arise throughout the developmental lifespan of a B-cell. From pro-B-cells in the bone marrow (BM), through circulating mature memory B-cells, each stage of B-cell development is prone to oncogenic alterations and transformation. The corresponding entities carry characteristic protein profiles, including differential expression of CCR7. In some diseases, expression can differ between malignant cells and the corresponding normal ontogenetic counterpart. In others, tumor-associated CCR7 expression can be unaltered, but may trigger different cellular functions.
3.1.1 B-Cell Acute Lymphoblastic Leukemia (B-ALL)
B-ALL is the most common childhood malignancy and represents the leading cause of cancer-related deaths in children and young adults (12). B-ALL arises from a monoclonal or oligoclonal expansion of malignant B-cell precursors in the BM. Normally, CCR7 is not expressed by precursor B-cells (6, 13) and scant information is available on CCR7 expression and function in childhood and adult B-ALL. Indeed, reports are somehow controversial since gene expression profiles showed both unchanged (14, 15) or upregulated CCR7 mRNA (especially in pediatric B-ALL) (16, 17). Similarly, some protein studies performed in a low number of cases showed no CCR7 in B-ALL (18, 19) while, in others series, expression was detected in specific subtypes of B-ALL, mainly in pediatric Burkitt´s-like B-ALL and in one third of pediatric pro-B, early-pre-B, and pre-B ALL (20). In most cases, the surface levels of CCR7 tested by flow cytometry were low-to-moderate. In our hands, adult B-ALL showed detectable CCR7 in only a minor tumor cell fraction of 10-40% (13, 21).
Related to differential CCR7 functionality, isolated early pre-B-ALL cells showed spontaneous migration towards CCL19 (20) whereas normal pre-B and pro-B-cells showed chemotactic responses to this ligand only after a previous exposure to soluble recombinant CD40 ligand (CD40L). In fact, engagement of CD40 seems a common mechanism to up-regulate CCR7 in lymphoblastic cells from patients potentiating the migration towards CCL19 (22, 23). Interestingly, this phenomenon seems highly specific to CCR7 since pre-incubation with CD40L did not affect chemotaxis mediated by other chemokine receptors (e.g. CXCR4) (20). Nonetheless, robust expression data confirm that, in general, CCR7 is absent or found at variably low levels in B-ALL suggesting a rather low impact in mediating migration of this malignancy into LN or other lymphoid niches. This is in accordance with the low incidence of lymphadenopathy in B-ALL. However, CCR7 may provide competitive advantages to the minor fraction of leukemic cells that express this receptor, potentially enabling them to escape to non-lymphoid protective tissues. Indeed, a recent study on a cohort of 160 B-ALL could associate expression of CCR7 and of zeta-chain-associated protein kinase 70 (ZAP-70) protein with enhanced migration (24). These authors also showed that CCR7 expression at diagnosis correlated with cerebral manifestation, which led to the hypothesis that CCR7 is involved in preferred CNS homing in the first phases of the disease. Notably, similar mechanisms have been previously proposed for T-ALL (25). We will address the more established contribution of the CCR7/CCL19 axis in CNS infiltration and survival of T-ALL cells below.
Once at their protective niches, the minor fraction of homed CCR7-expressing B-ALL cells could utilize CCR7 also as a mediator of survival signals. In this context, synergisms between CCR7/CCL19 and CXCR5/CXCL13 were shown to mediate resistance of B-ALL cells to tumor necrosis factor alpha (TNF-α)-mediated apoptosis through activation of paternally expressed gene 10 (PEG10) (26, 27). Moreover, both ligands also synergistically regulated CD40-CD40L crosstalk between B-ALL cells and CD8+ T-cells leading to a PEG10-mediated enhanced production of IL-10 in CD40-activated leukemic cells, which impaired tumor-specific cytotoxic T-cell (CTL) responses (28). Similarly, CD40-acivated B-ALL cells can deplete IL-12 from the local milieu and block the differentiation process of CCR7-expressing naïve T-cells towards active TH1 effectors (22). Therefore, CCR7 is likely involved in the creation of tolerogenic niches and its expression might confer escape of B-ALL cells from immune surveillance.
3.1.2 Chronic Lymphocytic Leukemia (CLL)
CLL is the most common leukemia in Western countries (29). It is characterized by the clonal proliferation and accumulation of mature, typically CD5-positive B-cells within the peripheral blood (PB), BM, LN, and spleen. Typically, blood circulating CLL cells are arrested in the G0/G1 cell-cycle phase, whereas leukemic cells within LN proliferate and receive protective anti-apoptotic signals (30, 31). CCR7 overexpression, as mRNA (14, 32–34) and as surface protein (in comparison to normal pan-B-cells and CD5+CD19+ B-cells), is consistently found in virtually all CLL, irrespective whether sampled from PB, BM, or LN (13, 19, 21, 35–48). In agreement, migration in response to CCR7 ligands is enhanced in CLL cells as compared to its normal cell counterparts (13, 35, 40, 46) and both expression and functionality have been associated with nodal disease involvement (13, 27, 35, 36, 38, 46, 47).
In CLL, genetic factors or polymorphisms involved in the overexpression of CCR7 remain uncovered but one single-nucleotide polymorphism (SNP) in the CCR7 gene was strongly associated with the risk of acquiring CLL. Out of 6 tested SNPs (including rs11574665, rs2023906, rs2290065, rs3136685, rs3136687, and rs588019) (49, 50), the major G allele in the SNP rs3136687, which is located at the first intron and is in linkage disequilibrium with rs11574665, was associated with a higher risk towards CLL whereas the minor A allele resulted in a protective effect (49). The authors found no differences in CCR7 expression for such allelic variants. This lack of association between CCR7 polymorphisms and receptor over-expression suggests that other proteins might ultimately determine different signaling pathways controlling CCR7 gene transcription and/or surface protein expression. Accordingly, activating mutations at Notch1 intracellular domain were shown to repress the dual specificity protein phosphatase 22 (DUSP22) tumor suppressor gene that encodes a phosphatase that dephosphorylates STAT3 (51). Because of this, STAT3 is constitutively activated resulting in increased CCR7 levels in CLL cells. Another STAT family member, STAT-4, which is profoundly reduced in CLL cells (52), was implicated in in vivo down-regulation of CCR7 in T-cells (53). Different B-cell receptor (BCR) signaling pathways have been implicated as well in the aberrant up-regulation of CCR7. For instance, after BCR engagement, the ZAP-70 protein has been shown to up-regulate CCR7 through an extracellular signal-regulated kinase 1/2 (ERK1/2)-dependent mechanism (42). Similarly, the transcription factors NFATC1 (nuclear factor of activated T-cells), NF-κB (nuclear factor kappa B), and AP-1 (activator protein 1) are known to regulate CCR7 expression in CLL following activation via the BCR or other receptors (54–56).
In CLL, the normal LN architecture is effaced by the malignant infiltrate (57). Different studies confirm CCR7 as the main receptor involved in nodal entry of CLL cells. Mechanistically, binding to CCL21 on the surface of HEVs activates α4β1 (CD49d/CD29; VLA-4) and αLβ2 (CD11a/CD18; LFA-1) (35, 58), which respectively bind to ICAM-1 and VCAM-1. Whether β1 and β2 integrins are equally relevant in CCR7-mediated homing of CLL cells is still unknown. By one hand, Till et al. showed spontaneous active conformation (without chemokine-induced clustering) of LFA-1 on CLL cells (59). On the other hand, circulating CLL cells usually express low levels of these integrins (60). Therefore, few CLL cells are able to arrest in ICAM-1 expressing endothelium in vitro and to migrate to lymph nodes of NOD/SCID mice in vivo (60). However, a significantly higher expression of both types of integrins (thus facilitated access to LN) is detected in CLL cells derived from high-risk patients with unfavorable cytogenetic abnormalities such as deletion 17p, deletion 11q and, specially, with trisomy 12 (47, 61, 62). Recently, Legler et al. have shown on trisomy-12 carrying OSU-CLL cells that CCR7-mediated inside-out signaling to the β1 integrinVLA-4 and the β2 integrin LFA-1 is controlled by Src and ZAP-70 kinases (58, 63). This process is critical for static and dynamic cell adhesions to endothelium and subsequent migration, but did not seem to impact the speed of migration velocity, and was dispensable for chemokine-mediated crawling and diapedesis. Further studies are needed to know which are the molecular mechanisms driving the latter processes.
Activation of integrins promotes the production and release of MMP-9 (64) and the subsequent transmigration of CLL cells through the endothelial cell wall into the LN [transendothelial migration (TEM)]. This ability of CLL cells to eventually accumulate at these sites may be determined by the genetic background of CLL. The more aggressive immunoglobulin heavy variable chain (IGHV) gene unmutated CLL subset displays higher CCR7 expression (35, 36, 43, 46, 65). In support, the presence of adverse factors such as IGHV unmutated status or expression of CD38 or ZAP-70 was shown to be associated with increased responsiveness of CLL cells to CCR7 ligands in both chemotactic and TEM assays (38, 47, 60, 66, 67).
There is data that indicate CCR7 to drive interstitial migration within the lymphoid tissue and to facilitate the positioning of leukemic cells close to accessory CD40L+CD4+ TH cells, follicular DC (FDC), and stromal cells (e.g. stromal-like cells and nurse-like cells), which are all known to promote the survival and growth of the malignant clone (9, 38, 68, 69). This crosstalk with accessory cells can induce the release of high levels of CCL19 and CCL21, which among others, has two functional consequences. First, it causes the establishment of a self-enhancing loop that recruits more CCR7-expressing tumor and accessory cells favoring the creation of a protective and tolerogenic microenvironment. Secondly, CCR7 ligands directly promote survival of CLL cells. In these scenarios CCL19 and CCL21 can act as single factors that activate mitogen-activated protein kinase (MAPK) and phosphatidylinositol-3-kinase (PI3K)-AKT signaling (40, 70) or in a cooperative fashion with CXCL13 which contributes to resistance of CLL cells (but not normal CD5+ B-cells) to TNF-α-mediated apoptosis through up-regulation of PEG10 which in turn stabilizes caspases-3 and -8 (26, 27).
Finally, besides its role in homing and survival within SLO, CCR7 along with sphingosine-1-phosphate receptor 1 (S1P1) are crucial molecules in the egress of lymphocytes from lymphoid organs to PB. As shown in CCR7-deficient mice, cells lacking CCR7 left LN quicker than wild-type cells. In contrast, overexpression of CCR7 retarded emigration from LN to PB (71). Evidence implicates the characteristic high CCR7 expression in CLL alongside the low expression of the egress receptor S1P1 to contribute to nodal retention (43, 46, 72), driving a scenario of lymphadenopathy that favors homing and accumulation in SLO. Remarkably, driving leukemic cells out of LN into PB induces death by deprivation of milieu-derived signals and is one of the modes of action (MOA) of a very efficient treatment option in CLL, ibrutinib, the first approved Bruton´s tyrosine kinase (BTK) inhibitor with activities on multiple other kinases (73). Recent work suggests ibrutinib to down-modulate CCR7 expression and function in CLL (e.g. integrin activation and receptor recycling) and by that to restore the balance between CCR7 and S1P1 and to enforce nodal egress of leukemic cells (44, 46).
3.1.3 Mantle Cell Lymphoma (MCL)
MCL is a B-cell tumor that originates from the clonal expansion of a naïve CD5+ B-cell localized in the mantle cell zone surrounding the germinal center (GC) in secondary follicles of the LN (74). Lymphadenopathy and BM infiltration are the most common clinical manifestations, followed by splenomegaly and PB lymphocytosis. Gastrointestinal and CNS involvement have been reported as well (75–77). This preferential pattern of dissemination can be attributed to the high surface expression of CCR7 by MCL cells as per flow cytometry, second to those levels observed in CLL (13, 21, 78, 79). In most of these studies, MCL cells from LN, PB, BM, and pleural effusions expressed higher levels of CCR7 than their proposed normal counterparts. The underlying causes of this overexpression are largely unknown. Comparative transcript analysis between MCL and normal B-cells have shown CCR7 mRNA to be significantly up-regulated in lymphoma cells (32). The fact that CCR7 was not among the top differentially regulated RNAs in MCL (80) suggests that additional mechanisms such as altered protein turn-over (46) are responsible for overexpression of surface CCR7 in MCL. Nevertheless, MCL and normal B-cells differ in their migratory behavior towards CCR7 ligands. In chemotaxis assays, MCL cells, but not their normal counterparts, migrated in response to CCL19, which was selectively potentiated by pre-exposure to CXCL12 (78). These results suggested CCR7-driven migration to be of relevance in the dissemination pattern seen in MCL patients. We corroborated this hypothesis in pre-clinical in vivo models in which the inhibition of CCR7 by anti-CCR7 antibodies abrogated infiltration of CCR7-expressing MCL cell lines into LN, spleen, lung, or CNS, all of them tissues in which CCR7 ligands are found (21). Moreover, this neutralization of the CCR7 axis also induced a strong reduction in viability of lymphoma cells within tumor masses, confirming that in MCL CCR7 overexpression is not only involved in orchestrating migration, but also in directly promotion of survival.
3.1.4 Follicular Lymphoma (FL)
FL is the second most common type of non-Hodgkin´s lymphoma (NHL) and despite its indolent nature, it is essentially incurable (81). FL encompasses lymphomas emerging from a GC B-cell, which can vary in presentation from indolent to aggressive courses (82). Similar to normal GC lymphocytes, which physiologically down-regulate CCR7 and up-regulate CXCR5 (83), FL cells express low to moderate levels of CCR7. Moreover, the proportions of CCR7-expressing cells were reported to be low and, in some patients, no expression of CCR7 was detected at all (13, 19, 21, 79). In agreement, mRNA levels in FL cells did not differ from those of their normal counterparts (32, 84–87). The genetic variants of CCR7 rs2023906, rs2290065, rs3136685, and rs588019, were not associated with differential expression or with clinical course in FL (50). The fact that CCR7 is not prominently found in most FL suggests that it has a limited role in the pathophysiology of this lymphoma, which is supported by recent evidence. In fact, comparative analyses of LN from FL versus reactive LN revealed that CCL21 and CXCL12 were neither over- nor differentially expressed, whereas FL-LN nearly lacked expression of CCL19. In addition, in FL lymphoid tissues in which both CCR7 ligands were detected, they were preferentially found in HEVs and in lymphatic vessels of T-cell zones, but on average at 3-fold lower levels than in reactive LN (88). Conceivably, the reduced abundance of CCL19/CCL21 in LN of FL is lymphoma instructed and contributes to evasion from anti-tumor immunity. Accordingly, FL progression may be associated with reduced numbers of perifollicular CCR7+ gamma-delta T-lymphocytes due to a shortage of attracting CCR7 ligands (88).
3.1.5 Burkitt`s Lymphoma (BL)
CCR7 was first characterized in EBV infected BL cell lines, hence the initially coined term Epstein-Barr-induced 1 (EBI-1) for CCR7 (4). CCR7 upregulation was shown to rely on the viral transactivator EBV nuclear antigen 2 (EBNA-2), which after binding to centromere-binding factor 1 (CBF-1, also known as RBP-jκ), a highly conserved cellular DNA binding repressor, gains access to regulatory regions of CCR7 target genes and activates transcription in infected (previously EBV‐negative) BL4 BL cells (4, 89). Information on CCR7 expression in primary BL material is scarce. No upregulated mRNA levels were seen in 22 patient samples (32, 84) and, to our knowledge, only one work studied CCR7 expression by flow cytometry in another 9 patients (79). The receptor was found in all cases, but in a fraction of ~53% of tumor cells per sample with no disclosed results on receptor surface levels. Interestingly, in the NC37 BL cell line, in vitro chemotaxis and TEM was modulated by a cooperative activity of CXCL12 with CCL19 or CCL21, suggesting that the CCR7 axis is involved in BL cell homing to LN (90). Accordingly, in the syngeneic Eμ-Myc mouse model of BL, CCR7 was found necessary for lymphoma cells to home to LN (9). These results also indicated that CCR7 guides tumor cells to distinct microanatomic sites in spleen and LN, especially to their T-cell zones. Cross-talk with resident stromal and accessory cells at these sites contributed to the creation and preservation of pro-tumor niches that conferred a survival advantage to CCR7-positive lymphoma cells over CCR7-deficient lymphoma cells (9). In a proposed model, stromal cells (e.g. fibroblastic reticular cells, FRC, and HEV endothelial cells) secrete CCL21 through which CCR7-expressing lymphoma cells home through HEV into the LN (or spleen) and migrate towards FRC in the T-cell region. Upon interaction with FRC, lymphoma cells secrete lymphotoxin through which they stimulate lymphotoxin-β-receptor expressing FRC. In turn, BL cells receive survival signals, presumably including Indian hedgehog (Ihh) secreted by FRC and CD40 stimulation through CD40 ligand-expressing CD4+ T-cells located in the T-zones. The importance of this cross-talk was demonstrated by showing that genetic deletion of CCR7 impaired lymphoma growth (9). Therefore, this model not only established the basis for a better understanding of the pathogenic role of CCR7 in BL, but also in many other blood cancers with a high dependence on the nodal or splenic microenvironments.
3.1.6 Subsets of Diffuse Large B-Cell Lymphoma (DLBCL)
DLBCL, the most common type of malignant lymphoma, accounts for ~30% of adult NHL (91). DLBCL can arise at multiple anatomical sites and comprises two major groups: activated B-cell like (ABC) and GC B-cell like (GCB) DLBCL. Therefore, it is not surprising that chemokine receptor (CKR) expression varies between these subtypes and in association with disease location (91, 92). Up to 62% of DLBCL express CCR7, both in analyses of flow cytometry and immunohistochemistry (IHC), with a preferential mRNA and protein expression in the non-GCB subtypes, especially in patients with both LN and BM involvement (79, 85, 93, 94). In EBV-positive DLBCL of the elderly, in primary effusion lymphoma, in gastric extranodal DLBCL, and in transformation of gastric mucosa-associated lymphoid tissue (MALT) lymphomas to gastric extranodal DLBCL, up-regulation of CCR7 mRNA, among other CKR, was reported (32, 91, 94–96). Notably, in EBV-associated DLBCL recurrent mutations in the CCR7 gene are found in 11% of patients (94). These alterations seem exclusive to this subtype and could enable homing of tumor cells to SLOs where the virus in turn propagates infection or establishes latency, thereby driving lymphomagenesis (97). In other related primary lymphomas such as intravascular large B-cell lymphoma and mediastinal large B-cell lymphoma a characteristic decrease in immunodetected CCR7 was described (32, 98, 99). As these types typically show sparing of LN manifestation, this corroborates the role of CCR7 in nodal homing. The genetic polymorphisms in CCR7 that were disclosed in FL did not associate with the risk of acquiring DLBCL (50). CCR7 expression, both at mRNA and protein levels, was an independent prognostic factor for disease progression, advanced clinical stages, shorter median survival times, and poorer survival rates in GC and ABC DLBCL (93, 100). First functional data on CCR7 in DLBCL indicate that receptor expression facilitates CCR7‐mediated in vitro migration in EBV‐positive DLBCL cell lines with functional analyses on primary samples still missing (91).
3.1.7 Primary Central Nervous System Lymphomas (PCNSL)
Immunohistochemical staining of PCNSL and secondary CNS lymphoma (sCNSL) showed these disorders to present CKR profiles that were different from those of systemic DLBCL. CCR7 was detected in the malignant B-cells of specimens of PCNSL (101) and in CNS relapses of DLBCL (102). However, and opposed to lymphomas with peripheral involvement, CCR7 was present in the cytoplasm rather than at the cell surface indicating that the receptor may not respond to its corresponding ligands in the same conventional fashion (101). It is tempting to associate this loss of surface CCR7 with the absence of nodal involvement, which defines PCNSL (103). However, one should also take into account that the restricted intracellular CCR7 expression pattern may in part be a consequence of a milieu that is highly enriched in CCR7 ligands, especially in CCL19, the most potent inducer of CCR7 endocytosis (11). In agreement, a recent study addressing the role of gliosis in lymphoma cell retention in the CNS found that astrocyte-derived CCL19 was required for gliosis-promoted CNSL via enhancing parenchymal retention of lymphoma cells (104).
3.1.8 Marginal Zone Lymphoma (MZL)
MZL comprises three entities that arise from the marginal zone surrounding the follicular GC of the LN: extranodal MZL or MALT lymphoma, splenic MZL, and nodal MZL. Analyses on MALT lymphoma samples showed more than 50% of malignant cells to express CCR7 (79). In extragastric MALT lymphoma or malignant transformation from Helycobacter pylori-associated gastritis to MALT lymphoma, up-regulation of CCR7 mRNA, among other CKRs, was a common finding (91, 95, 96). In splenic MZL, no changes were seen at the mRNA level (87) whereas flow cytometry revealed significantly reduced expression of CCR7 as compared to normal B-cells (13, 19, 21, 79). The low CCR7 expression in extranodal or splenic MZL suggest a minor role of this receptor in their pathobiology. This in turn might explain the minimal lymphadenopathy seen in these types of MZL (13) and reports on CCR7 expression in nodal MZL are still missing. Accordingly, one study in salivary gland MALT lymphoma samples selectively implicated the chemokine CCL21 in the organization of ectopic reactive lymphoid tissue whereas no significant expression of the ligand was detected in the malignant lymphoid aggregate (105). The authors concluded that CCR7 plays no major role in the infiltration of the epithelium or in the regulation of malignant cell survival.
3.1.9 Hairy Cell Leukemia (HCL)
HCL is an indolent, rare disease that accounts for approximately 2% of leukemias and is typically defined by the B-raf kinase mutation pV600E (106). Cell surface expression of CCR7 is low (or absent) in HCL cells when compared to normal B-cells (13, 19, 21, 107). Similarly, CCR7 transcription in HCL samples is reduced (32). This would explain why nodal dissemination is not a key feature in this disease.
3.1.10 Lymphoplasmacytic Lymphoma (LPL) and Multiple Myeloma (MM)
LPL and its subgroup Waldenstroem´s macroglobulinemia (WM) are rare and indolent lymphomas that arise from terminally differentiated B-cells that physiologically do not express CCR7 (82). Analyses on very few clinical samples did not shed light on the expression profile of CCR7 in LPL, as some cases did express the receptor (21) while others did not (13). Available evidence does not link CCR7 to an altered genetic profile in plasma cell leukemia (PCL) (108). Normal plasma cells and those of MM do not typically express surface CCR7, and if expressed, it is found in a minor proportion of cells as demonstrated in samples from BM or extramedullary sites (PB and pleural effusions) (13, 21, 109, 110). Curiously, in non-Hispanic Caucasian subjects the genetic CCR7 variant s3136685 was reported to be associated with an elevated risk for MM (110). Since this genotype was not associated with MM in further permutation-based tests and since previous evidence did not link CCR7 to MM (108, 111), these findings should be interpreted with caution and further investigation is required to clarify this issue. Finally, gene expression profiles of monoclonal gammopathy of undetermined significance (MGUS) and of smoldering myeloma seem to discard a prominent role of CCR7 in these conditions as well (108, 111, 112).
3.1.11 Hodgkin Lymphoma (HL)
HL is a unique type of B-cell lymphoma characterized by the presence of a minority (<1%) of neoplastic cells in a background of infiltrating reactive cells (113). The microenvironment is considered to be shaped by the malignant cells and provides survival signals and protection against anti-tumor immune responses (114). Based on differences in histopathology, HL is classified in two subgroups: the classical form (cHL) that accounts for 95% of all HL cases and the nodular lymphocyte predominant variant (NLPHL) that represents only 5% of all cases (113). Tumor cells in cHL are termed Reed/Sternberg (RS) cells, which generally express CD30, whereas tumor cells in NLPHL are called lymphocytic and histiocytic (L&H) cells and lack CD30.
CCR7 expression has been observed in cHL-derived tumor cell lines and in primary tissue. In the majority of cell lines expression was moderate-high and CCR7 was functional in inducing migration towards both of its ligands (115). In patient samples, IHC revealed a differential expression of CCR7 between cHL and NLPHL. The classical form, located in the interfollicular zones, showed strong CCR7 expression whereas NLPHL, regularly associated to follicles, was shown to be CCR7 negative (115). Accordingly, mRNA levels were highly expressed in cHL when compared to NLPHL and normal B-cells (84, 116). Moreover, CCL19/CCL21 were found in tumor infiltrates of cHL, whereas the tumor nodules in NLPHD almost completely lacked these chemokines (115).
In cHL, CCR7 upregulation might be a consequence of two, or more, altered pathways that are partially interconnected. For example, the CCR7 gene contains binding sites in its promoter region for the transcription factors AP‐1 and NF‐κB (3), and both axes have been shown to be constitutively active in cHL and to upregulate CCR7, individually or cooperatively (3, 117). Notably, the combined constitutive activation of AP‐1 and NF‐κB mimics a state of chronic inflammation that involves the production of cytokines by RS cells. Whether CCR7 up-regulation is part of the prominent NF‐κB program in cHL, as it is the case for CD30 expression (115, 118), remains to be investigated.
At the functional level, constitutively active WNT signaling in cHL is important in CCR7-mediated migration and generation of protumorigeneic milieus. Binding of the WNT protein to the low-density lipoprotein receptor-related protein 5/6 (LRP5/6) regulates CCL19-guided chemotaxis through the β-catenin and lymphocyte enhancer-binding factor-1 (LEF-1) pathways (119). WNT signaling is commonly involved in metastasis and angiogenesis in various tumors (120). In tumor cells of cHL canonical WNT/β-catenin/LEF-1 signaling is also required to secrete vascular endothelial growth factor A (VEGF-A), and by that, to attract endothelial cells as well as to enhance their migration, sprouting and tube formation. Therefore, canonical WNT signaling is a regulator of the endothelium-lymphoma interplay. WNT is a prerequisite for secretion of VGEF-A by cHL cells which stimulates biogenesis of vascular endothelium which in turn presents CCR7 ligands that direct movement of cHL cells towards vascular niches. Thus homing and interstitial movement of tumor cells within the affected LN is facilitated by constitutive WNT. Moreover, CCR7’s ligand, CCL21, was shown to be absent on RS cells, but was detected on the majority of small vessels (including HEV) with a luminal membranous localization (121).
The CCR7 axis not only seems to play a pathogenic role by recruiting cHL tumor cells, it is also implicated in recruiting pro-tumorigenic CCR7-expressing immune cells from the circulation. Within infiltrating immune cells in cHL, CCR7 (and the related homing markers CD62 and lymphocyte function-associated antigen 1, LFA1) were demonstrated to be expressed on a large proportion (~33%) of reactive T-cells, which showed receptor-mediated chemotaxis that was similar to PBMC from healthy donors (121). Notably, in cHL the infiltrate is commonly enriched by CCR7+ TREG and activated T-cells (122–126). In contrast, in NLPHL these T-cell subsets are less abundant and are found outside the tumor area (115, 127). Together, these findings suggest different immune escape mechanisms in both subtypes of HL that may be related to the different expression profiles of CCR7 in tumor-associated cells and of CCR7 ligands in the surrounding tissue.
3.2 CCR7 in T-Cell Malignancies
As described for B-cell malignancies, T-cell neoplasms consist of multiple entities that are thought to arise from particular stages of T-cell development. For instance, T-cell acute lymphoblastic leukemia (T-ALL) originates from thymic stages of T-cell evolution while peripheral (post-thymic) T-cell neoplasms show features of mature T-cells with distinct phenotypes of differentiation, e.g. T-cell prolymphocytic leukemia (T-PLL) mostly resembling TCM or unconventional transitional stages between TN and TCM; adult T-cell leukemia/lymphoma (ATLL) resembling TREG; Mycosis fungoides (MF) resembling TEM; Sézary syndrome (SS) resembling TCM; or T-cell large granular lymphocytic leukemia (T-LGL) resembling activated cytotoxic T-cells (113, 128–130). Based on this phenotypic characterization, CCR7 expression would follow its physiological T-lineage pattern and be highest in those diseases resembling a DN or SP thymocyte, TN, TREG, or TCM and to show LN or CNS involvement. A low number of studies limits the knowledge on the role of CCR7 in some of these disorders. However, in light of the restricted armamentarium of available efficient therapies for T-cell malignancies, such insights are highly desired. In this section we will address our current knowledge on CCR7 biology in several T-cell cancers and will try to associate reported expression profiles with CCR7 functionality and pathological findings.
3.2.1 T-Cell Acute Lymphoblastic Leukemia (T-ALL)
T-ALL mainly afflicts children and adolescents. It presents with increased white blood cell counts and often with hepatosplenomegaly. At relapse, there is an increased incidence of CNS manifestations (131, 132). A seminal report showed CCR7 to be a functional receptor that is highly expressed in 4 of 5 T-ALL cell lines and in PB tumor cells of 8 of 11 T-ALL patients (25). A recent study in a larger cohort of 130 patients (24) and unpublished data from our laboratory confirm these results. In T-ALL, CCR7 expression is controlled by the activity of the oncogene Notch1. Significantly up-regulated CCR7 was found in human T-ALL cells that harbor Notch1-activating mutations while receptor expression was repressed by Notch1-specific γ-secretase inhibitors (DBZ or compound E), both at mRNA and at protein levels (25). Mechanistically, Notch receptor engagement initiated the PI3K/mammalian target of rapamycin complex 2 (mTORC2) pathway, which transmitted through NF-κB to regulate expression of the CCR7 gene in leukemic cells (25, 133). Notably, in pre-clinical in vivo T-ALL models generated by overexpression of the intracellular cleaved form of Notch1 (ICN1), CCR7 overexpression led to enhanced chemotaxis and invasion into different tissues, especially to leptomeningeal spaces of brain and spinal cord, in which endothelial cells were shown to produce CCL19 (25). This CCR7-driven homing into CNS facilitated leukemic cell survival and was associated with reduced animal survival. Similarly, CCL19 promoted T-ALL cell invasion of spleen in syngeneic in vivo models and shortened host survival (134). Inside cerebral or spleen parenchyma, cross-talk between stromal cells and leukemic cells mediates the production of higher levels of tissue CCL19 (25, 134, 135). In CNS, these positive loops and the concomitant alteration of drainage from cerebrospinal fluid facilitated lymphoblastic meningeal infiltration (25). Nonetheless, it is likely that CCR7 is not the sole mediator of this process as meningeal infiltration is also detected in ICN1-induced T-ALL with CCR7-deficient hematopoietic progenitors (135). A recent study suggested that CNS infiltration in xenograft models is regulated by ZAP-70 which positively correlated with the overexpression of both CCR7 and CXCR4 and with migratory abilities towards CCL19 and CXCL12 (24). This study also confirmed, in a large cohort of 130 T-ALL patients, the positive correlation between ZAP-70 and CCR7 expression and, importantly, high CCR7 expression in tumor cells from BM biopsies at diagnosis was associated with a significant 11-fold increased risk of CNS involvement (24). Together, despite some T-ALL patients showing low or absent expression of CCR7 in their tumor cells from BM (135), the herein presented evidence supports CCR7 as a key element responsible of high-risk features such as CNS infiltration.
3.2.2 T-Cell Prolymphocytic Leukemia (T-PLL)
Although being the most frequent mature T-cell leukemia in Western countries, T-PLL represents only ~3% of all T-cell malignancies (136–138). Its clinical course is typically aggressive with poor responses to conventional chemotherapies resulting in a median overall survival (OS) of usually <2-3 years (139, 140). An inevitably rapid proliferation of mostly CD4+ prolymphocytes involves the PB, BM, spleen, liver, LN as well as skin and effusions (136, 137, 141). Not uncommon are CNS involvements (136, 137, 142). This pattern of dissemination suggests chemokine receptors to play an important role in T-PLL, however, little is known about their relevance and the role of their ligands in the pathophysiology of T-PLL (143). Although previous evidence did not show overexpression of CCR7 mRNA in six primary T-PLL samples (144), a recent study by our groups focused on CCR7 in T-PLL biology and its interventional potential (130). We assayed CCR7 surface levels at diagnosis by flow cytometry in 109 patients and found that receptor overexpression in malignant cells is seen in a very high proportion of cases (86.5%). CCR7 expression profiles were also instrumental in assigning T-PLL to stages of memory T-cell differentiation (130, 145). The proportion of CCR7-expressing T-PLL cells in PB at diagnosis was associated with a shorter OS and a higher risk of death within an 8-year follow-up period (130). CCR7 was a fully functional receptor upon CCL19 and CCL21 binding and its downstream signaling pathways activated PI3K and ERK (130), two axes that have shown to be relevant in T-PLL pathogenesis (145–147). We further showed that receptor activation triggered chemotaxis, invasion trough biological matrices or endothelial cells, and T-PLL cell survival (130). In in vivo pre-clinical studies, we confirmed CCR7 to play critical roles in enabling tumor cells to access tumor microenvironments in CNS and lymphoid organs, especially in LN (130). In agreement, prominent HEV are often infiltrated by neoplastic cells in T-PLL (148), which suggests CCL21 as a major route for homing into lymphoid tissues and in mediating the dissemination of T-PLL cells to different organs. Our results demonstrated CCR7 to promote a rapid niche colonization as well as survival and proliferation in these environments.
3.2.3 Adult T-Cell Leukemia/Lymphoma (ATLL)
ATLL is an aggressive peripheral T-cell malignancy associated with human T-cell leukemia virus, type 1 (HTLV-1) infection and predominantly occurs in HTLV-1 endemic areas such as South-Western Japan, the Caribbean Islands, Central and South America, intertropical Africa, and the Middle East (149, 150). The prognosis of ATLL is very poor with a 4-year OS rate of 11%, 16%, 36%, and 52%, in the subtypes of acute, lymphoma, chronic, and smoldering ATLL, respectively (151, 152). In the majority of cases, ATLL cells express CD4 and CD25 and often lack CD7 (152–154). Forkhead box P3 (FoxP3) expression is detected and led to concepts of ATLL cells to resemble TREG (155) which, however, remains a subject of debate (156). The malignant cells of ATLL express surface CCR7 (157) and up-regulated CCR7 transcripts are associated with the aggressive acute ATLL subset, which distinguishes these cases from the less aggressive chronic ATLL (158, 159). Studies in larger cohorts confirmed upregulated CCR7 mRNA and protein, especially in patients with acute, progressive, or treatment refractory acute disease. These reports also associated higher CCR7 expression levels with a poor prognosis and nodal involvement (154, 160, 161). Accordingly, ATLL cells from patients with lymphadenopathy and splenomegaly showed enhanced ability to adhere to surfaces coated with intercellular adhesion molecule 1 (ICAM-1) and to migrate towards CCL19 or CCL21 (157). Recent whole-exome sequencing studies revealed gain-of-function mutations in the receptor (162, 163). The CCR7 gene was recurrently and significantly affected in 11% of ATLL with a majority of cases harboring mutations that led to truncated protein forms at the C-terminal cytoplasmic domain, which regulates multiple biological processes. Of special interest were the mutations at CCR7 Trp355, which prevented receptor turn-over and internalization upon ligand stimulation resulting in increased surface receptor expression. These mutations led to an enhanced ligand-induced chemotaxis and PI3K/AKT signaling (162, 163). More recently, CCR7 gene mutations were mutually associated with mutations at phospholipase C gamma 1 (PLCG1) and caspase recruitment domain family member 11 (CARD11) genes, which are frequent alterations in TCR/NF-ĸB signaling (164). The pathological implications of this coexistence in ATLL remain unaddressed.
3.2.4 Mycosis Fungoides (MF)
MF is the most common type of cutaneous T-cell lymphoma (CTCL), in which a protracted clonal expansion of atypical dermatotropic CD3+CD4+ T-lymphocytes underlies a chronic cutaneous manifestation (165). The majority of patients with early-stage (i.e. limited patch/plaque) disease have a normal life expectancy, while in advanced (i.e. ubiquitous, tumor, nodal) stages survival is drastically reduced, which in addition to a marked symptomatology requires multimodal treatments (166–168). Available data emphasize a complementary, prominent pro-tumorigenic role of distinct factors present within the skin or LN milieus of CTCL, such as chemokines (CCL21 or CXCL12) (169), cytokines (IL-13) (170) or antigens able to entertain chronic T-cell receptor stimulation (171).
Expression of CCR7 has been considered a marker of advanced MF and a component involved in the spread of cutaneous lesions to lymphoid tissues. Indeed, single-cell RNA sequencing of skin biopsies from one patient with aggressive disease showed that malignant clones in PB and LN displayed a transcriptional program reminiscent of a more central CCR7+ memory-like phenotype, while retaining tissue-homing receptors (i.e. CLA, CCR10) (172). Nonetheless, evidence of CCR7 protein expression in MF samples is scarce. Kallinich et al. analyzed expression of several CKRs in MF (165). They studied CCR7 expression in skin biopsies from six patients with early disease and six patients at the tumor stage. Using IHC, they found no expression of CCR7 in any tested sample, however, two skin samples of advanced disease showed strong and uniform expression of CCR7 on tumor cells by flow cytometry. A more recent study reported CCR7 expression in 62% (13/21) of specimens as per IHC, and indicated that CCR7 expression strongly correlated with subcutaneous extension of lymphoma cells (173). The CCR7-expressing MF cell line MyLa shows enhanced in vitro migration towards CCL21 in an mTOR-dependent manner (161) and through up-regulation of metastasis-associated lung adenocarcinoma transcript 1 (MALAT1) (174), a long noncoding RNA that is also associated with migration of several solid tumor types (175). These authors also concluded that CCR7 promotes subcutaneous involvement of MF. In agreement, total RNAs from skin biopsies of epidermis versus those from involved dermis of MF associated the presence of tumor cells in the dermis with the CCR7/CCL21 axis (161). Accompanying IHC analyses confirmed that expression of CCR7 was high in infiltrating lymphoma cells. It also demonstrated CCL21 in the cytoplasm of epidermal keratinocytes and to be diffusely distributed in the dermal extracellular matrix.
3.2.5 Sézary Syndrome (SS)
SS is a mature systemic T-cell malignancy in which skin-homing T-lymphocytes also accumulate in PB and LN. Patients are highly symptomatic (e.g. pruritus, staphylococcal infections) and prognosis is poor with a median of survival of 63 months (166, 167). Despite increasingly better knowledge on disease biology, currently applied therapies show short-lived responses (168). By convention, SS has been regarded as a systemic variant of MF based on identical cytologic and immunophenotypic features. In addition, long-standing MF may subsequently develop into a secondary SS-like disease that exhibits circulating neoplastic cells, indistinguishable from those of primary de-novo SS. Nonetheless, several studies provide clues to consider SS and MF as two separate entities. First, patients with primary SS typically experience a more aggressive disease course, characterized by frequent involvement of LN (166–168). Second, MF and SS tumor cells show different molecular and CKR profiles (176–178). SS is thought to arise from expansions of mature long-lived CD4+CD7- T-cells with a CD45RO+CD27+CD62L+ TCM phenotype accompanied by a consistently high CCR7 mRNA and protein expression, whereas CCR7 expression in the predominantly TEM cells from cutaneous MF lesions is controversial (165, 178–182). Admittedly, biases by the different sources of sampling, e.g. skin preferentially for MF versus blood and LN for SS might attribute to the observed differences.
Although CCR7 gene expression could not be significantly correlated with lymphoid organ involvement or patient survival in SS (179), it appears plausible that production of CCL19 and CCL21 by stromal and endothelial cells in lymphoid tissues contributes to the lymphotropism of SS cells. In support, the chemokine CXCL13, mainly produced in lymphoid tissues, promotes a synergistic CCR7-mediated migration that was of higher efficiency in SS cells than in normal T-cells (183). Additionally, CCR7 activation enhanced invasion by modulating adhesion and secretion of metalloproteases in clinical samples and in SS cell lines (183, 184). CCR7-induced integrin activation and metalloprotease secretion are processes that in other CCR7-expressing blood cancers are known to be required for CCR7-mediated TEM and for homing (35, 130).
Epidermotropism and tumor growth within the skin environment of SS are features that had been attributed to CCR7 function, but the exact mechanisms are poorly characterized. A first report found no function for CCR7 in promoting in vitro survival or proliferation of primary SS tumor cells (183). Another study on a cohort of 43 SS cases found contradictory results (169). This more recent investigation demonstrated that overexpressed CCL21 (and CXCL12) in skin tissue induced activation of PI3K/AKT/mTORC1 signaling in skin-resident SS cells. SS samples frequently show a recurrent loss of the phosphatase PTEN (phosphatase and tensin homolog) and the liver kinase B1 (LKB1) (169), two proteins that under normal conditions attenuate upstream activation of mTORC1 in low energy conditions (185). Therefore, these defects might result in a constitutive TORC1 activation that promotes protein translation and a metabolic shift from oxidative phosphorylation (mainly observed in quiescent/memory lymphocytes) toward aerobic glycolysis (typically observed in activated lymphocytes) (185). This increase in glucose demand (also known as Warburg effect) might be energetically beneficial during the recruitment of SS cells to skin and/or LN by CCL21, with the latter being able to further enhance mTORC1 activation and by that SS cell growth (169). Indeed, among other, in part stronger stimuli such as IL-2/IL-7, CCL21-mTORC1 also promoted up-regulation of the Ki67 proliferative protein in SS-derived cell lines and in primary SS cells (169).
3.2.6 T-Cell Large Granular Lymphocytic Leukemia (T-LGL)
T-LGL is characterized by the chronic low-level expansion of mostly CD8+ T-cells in blood, BM, and spleen. Nodal disease is infrequent. T-LGL cells express pan-T-antigens, programmed cell death 1 (PD-1), some NK-cell associated molecules, cytotoxic granules (containing perforin and granzymes) and lack the CD28 co-stimulatory receptor. Detection of CD45RA and further markers of T-cell differentiation suggest a terminally differentiated effector memory (TEM-RA) phenotype (128, 186–188). TEM-RA cells are featured by the absence of CCR7, and accordingly, most T-LGL cases in these studies did not show tumor cell expression of CCR7.
3.2.7 Other T-Cell Malignancies
In other types of T-cell neoplasms such as peripheral T-cell lymphoma not otherwise specified (PTCL-NOS), extra-nodal NK/T-cell lymphoma (ENKTL), anaplastic large cell lymphoma (ALCL), and angioimmunoblastic T-cell lymphoma (AITL) expression of CCR7 remains poorly studied and controversial (including the source of expression, namely tumor cells versus local bystander cells). Two studies in a total of 41 ALCL patient samples and in 7 ALCL cell lines found the anaplastic-lymphoma kinase (ALK)-negative ALCL variant to overexpress CCR7 genes (compared to ALK-positive or primary cutaneous ALCL) (116, 189) while another series on LN biopsies associated ALCL to a CD4+CD45RO+CD27- TEM phenotype that lacks CCR7 (190). In contrast, PTCL-NOS showed CCR7 expression as part of its TCM signature (190). However, gene expression profiles showed no upregulation of CCR7 transcripts in PTCL-NOS as compared to normal T-cells; as also observed for AITL (191). A subsequent study reported expression of CCR7 by IHC in an overall of 83% of samples that contained PTCL-NOS, ENKTL, ALCL, and AILT, but without disclosed resolution for the proportions of CCR7 positive cases per entity (184). Despite this shortcoming, this work corroborated the expression of CCR7 in a high proportion of mature T-cell malignancies and, importantly, significantly associated CCR7 staining with lymphatic or hematogeneous dissemination as well as with clinical stage. As in B-cell malignancies, constitutive activation of the transcription factor AP-1 is a proposed mechanism underlying CCR7 overexpression and CCR7-mediated cell survival in some of these conditions, particularly ALCL (117).
In other, very rare T-cell lymphomas, studies on CCR7 have been sporadically reported. In a case of primary cutaneous aggressive epidermotropic CD8+ T-cell lymphoma transformation from an indolent to an aggressive phase was accompanied by a shift to CCR7 expression (177). A case of enteropathy-associated T-cell lymphoma (EATL) showed no lymphoma-cell associated CCR7 (178), fitting its cytotoxic T-cell nature, similar to CCR7-negative T-LGL.
3.2.8 Natural Killer (NK) Cell-Type Lymphoproliferative Diseases
NK-cell cancers can be subdivided into aggressive NK cell leukemia (ANKL) and indolent chronic NK cell lymphocytosis (CNKL), both characterized by leukemic infiltration into multiple organs (192). In a cohort composed of PB samples of nine ANKL and six CNKL cases several CKR were investigated by flow cytometry (193). In both types of leukemia, CCR7 was detected in a small proportion of tumorous NK-cells (<25%), a lower proportion than the relative number of CCR7-positive NK-cells the authors found in six healthy controls. Together, these results suggested that CCR7 might not play an important role in the pathophysiology of ANKL or CNKL.
3.3 CCR7 in Myeloid-Cell Malignancies
Description of CCR7 in myeloid-cell derived cancers is anecdotal and, as opposed to lymphoid disorders, myeloid neoplasms seem to be mainly characterized by downregulated CCR7, although this aspect still remains controversial.
3.3.1 Myelodysplastic Syndrome (MDS)
MDS constitutes a heterogeneous group of clonal hematopoietic stem cell diseases that share ineffective hematopoiesis, increased risk of developing acute myeloid leukemia (AML), and augmented prevalence of immune deregulation. To our knowledge no studies have addressed the expression or functions of CCR7 in myeloid cells from MDS patients. A comparative study of a cohort of 33 MDS, a condition with a known prominent inflammasome, patients with healthy controls found that in MDS CD8+ T-cells exhibited decreased levels of CCR7 and a concomitant upregulation of CCR3, CCR5, or CX3CR1 (194). Hence, a central pathogenic relevance of CCR7 and other CKR in MDS still has to be shown.
3.3.2 Acute Myeloid Leukemia (AML)
AML is a heterogeneous group of aggressive proliferations with variable genetic make-up and differential responses to therapy (195). In clinical practice, CCR7 is sporadically detected by flow cytometry in AML samples from PB and BM in a small proportion of tumor cells (unpublished data). In agreement, CCR7 mRNA is not highly abundant in AML in several transcriptome analyses (14, 16, 196, 197). Only one study reported CCR7 transcript over-expression (~3-fold) in 148 human AML samples as compared to 12 samples of normal cord blood-derived CD34+CD45RA- cells (198). Protein expression to confirm high membrane levels of CCR7 was not studied. Potential associations of CCR7 mRNA with the most frequent genetic aberrations were also not investigated (198).
3.3.3 Blastic Plasmacytoid Dendritic Cell Neoplasm (BPDCN)
BPDCN is a rare and clinically aggressive hematologic tumor derived from cells of immature PDC differentiation (199). The clinical course of BPDCN shows progressive systemic expansion, partially attributed to the local production of chemokine ligands of CKR expressed by the tumor cells (CXCR3, CXCR4, CCR6, CCR7) (200). Beyond expression data, no clues are available on the pathogenic roles of CCR7 in BPDCN.
3.3.4 Langerhans Cell Histiocytosis (LCH)
In LCH pathological CD207+ DC show constitutively activated MAPK pathway signaling. In in vivo and in vitro models, the B-raf V600E activating mutation impaired the Raf/ERK-mediated CCR7-induced migration of DC (201). This in turn caused their retention in the tissue lesions and, by promoting expression of BCL2-like protein 1 (BCL2L1), this resulted in enhanced resistance to apoptosis.
3.3.5 Myeloproliferative Disorders
Chronic myeloid leukemia (CML) is a clonal disease characterized by premature release of aberrant cells from the BM alongside their substantial accumulation in PB, spleen, and BM (202). In CML, the presence of the Philadelphia chromosome and its oncogenic product, the fusion oncoprotein BCR/ABL, is directly linked to multiple pathways involved in cell survival, growth promotion, and disease progression (203, 204). Similarly to LCH, an impaired adhesion and motility towards CCR7 was first reported for CML cells (14, 205) though this effect remains controversial since more recent reports showed in vitro and in vivo how a positive activation loop between BCR-ABL and the signal-transducing adaptor protein-2 (STAP-2) led to enhanced ERK signaling resulting in overexpression of CCR7, LN enlargement, and hepatosplenomegaly (203, 204). Whether these contradictory outcomes are a result of differential in vitro versus in vivo settings, or a consequence of artifacts associated to the use of cell lines versus primary tumor cells, needs clarification.
4 Pathophysiological Role of CCR7 in Hematologic Malignancies
Chemokines obey Stephen Paget’s ‘seed and soil’ paradigm, proposing that the microenvironments of different organs are different from each other, and that certain tumor cells have specific attraction to the milieu of specific organs (206). As reviewed above, CCR7 is a single receptor driving immune cells into LN, and for this reason this receptor assumes a central role in the pathogenesis of many leukemia and lymphomas, which very often express CCR7 due to their lymphoid or myeloid origin (Table 1).

Table 1 Summary of blood cancers with reported CCR7 expression studies (following 2016 WHO classification of blood neoplasms) (207).
In lymphoid malignancies, the role of CCR7 in hallmark deregulations of cancer such as enhanced migration or death resistance, can be associated to functional differences between CCR7-expressing normal and malignant cells. In some cases, gain-of-function in CCR7 is a consequence of an upregulated transcription and/or protein translation (Figure 1). For example, tonic signaling through the BCR or CD40 activates transcription factors such as NFATC1, NF-κB, and AP-1, which target the CCR7 gene, a mechanism found in CLL and B-ALL (22, 23, 42, 54–56, 208, 211). Similarly, CD30 down-stream signaling seems to increase CCR7 gene transcription in cHL and ALCL likely through NF-κB and AP-1 (3, 115, 117). Moreover, constitutive activation of the Notch1 oncoprotein increases CCR7 expression in T-ALL through the mTORC2/NF-κB cascade (25, 133), or in CLL through down-modulation of the DUSP22 phosphatase levels and the subsequent increase in STAT3 activation (51). In other instances, CCR7 up-regulation is promoted by a viral machinery that suppresses CCR7 gene repressor factors like CBF-1. This is described for the viral transactivator EBNA-2 in BL and DLBCL (89, 91) and it could be hypothesized that a similar mechanism governs HTLV-1-induced transformation in ATLL. Notably, downregulation of CCR7 expression and reduction of associated chemotaxis during viral infections, have been reported (11, 212). The EBV (213), the murine lymphocytic choriomeningitis virus (LCMV) (214), the human immunodeficiency virus type 1(HIV-1) (215), or the influenza virus (216) are examples of CCR7-downmodulating viruses. In other cases, e.g. during HIV-1 infection, primary CD4+ T-cells showed and enhancement of CCR7-mediated motility, leading to efficient propagation of HIV-1 (217, 218). Based on this, one might be tempted to associate these changes of the CKR expression profile to particular needs of each virus’ cycle. Therefore, discrepancies between outcomes in CCR7 expression after viral infections might be also a consequence of distinct cell-to-cell aspects such as the time elapsed after cell infection, the cell development stage at which the infection takes place, or baseline CCR7 expression by host cell. For example, the impact of in vitro EBV infection on CCR7 expression was very different between tonsillar or PB B-cells, being milder (if at all) in the last cell type (213). Moreover, the presence of additional tumorigeneic events in the infected tumor cells may synergize with the viral machinery to induce CCR7 gene expression (219, 220). In agreement, expression of CKR and chemokines in immortalized cell lines differs from that of EBV-infected PB B-cells (89, 213) and different growth requirements, such as the oestrogens, are known to positively regulate viral factors like EBNA2, which subsequently activate CCR7 gene expression (4, 213).
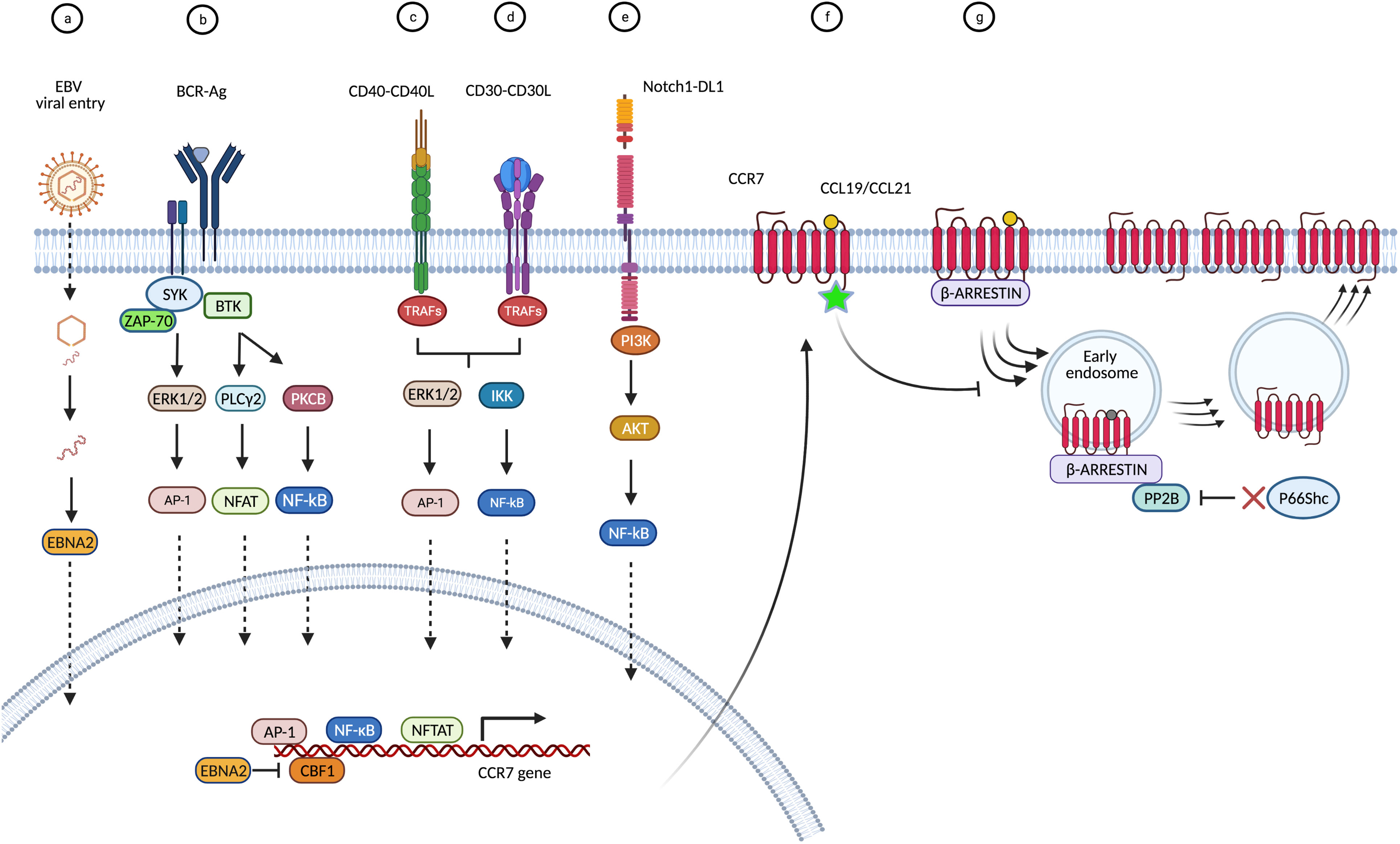
Figure 1 Causes and modes that underlie CCR7 overexpression in blood cancers. Overview of proposed signaling cascades with reported data. (A) CCR7 upregulation may be promoted by EBV. In BL and DLBCL cells, following viral endocytosis, the virion and packaged proteins are released into the cytoplasm. The viral transactivator EBNA-2 binds to and inhibits the CCR7 gene repressor factor CBF-1 (also known as RBP-Jκ) thus promoting CCR7 gene transcription (4, 89, 91). (B) In B-cell malignancies (CLL, B-ALL) tonic signaling through the BCR activates transcription factors such as NFATC1, NF-κB, and AP-1, which target the CCR7 gene (42, 54, 56). (C) When engaged by CD40L, the receptor CD40 recruits tumor necrosis factor receptor-associated proteins (TRAF) to the membrane, which initiate different signaling pathways leading to activation of NFκB or AP-1 (22, 23, 208, 209). (D) In cHL and ALCL, binding of CD30L (CD153) or sCD30 to CD30 can result in trimerization and signal mediation through TRAF proteins to stimulate the NFκB pathway resulting in CCR7 gene expression (3, 210). In addition, CD30 can signal through MAPK pathways, including ERK1/2 and the nuclear transcription factor AP-1, all leading to enhanced CCR7 transcription (3, 115, 117). (E) In T-ALL cells, release of intracellular Notch1 (ICN1) from membrane-tethered heterodimeric Notch1 protein upregulates PI3K/mTORC2/NF-κB pathways and activation of the CCR7 gene (25, 133). In CLL cells, activating mutations in Notch1 intracellular domain favor the downmodulation of DUSP22 phosphatase thus facilitating the accumulation of activated STAT3 which mediates CCR7 gene transcription (51). (F) Mutations in the C-terminal amino acid Trp355, located in the cytoplasmic region of CCR7, impair internalization upon ligand stimulation resulting in increased expression of the surface receptor in ATLL cells (162, 163). (G) Dysregulation of the endocytic machinery of CCR7, e.g. in CLL, impacts receptor turn-over and increases CCR7 membrane expression. Deficiency of the cytoplasmic p66Shc protein causes enhanced activity of the PP2B/calcineurin phosphatase on the endosomal CCR7 pool, which enhances its recycling back to the plasma membrane (43, 46).
Finally, CCR7 up-regulation may be also promoted by mutations in the C-terminal cytoplasmic region of CCR7 or dysregulation of its endocytic machinery both affect receptor turn-over. For example, in ATLL or CLL cells impaired internalization upon ligand stimulation results in increased surface receptor expression (43, 46, 162, 163). Whatever the underlying reasons for the upregulation of CCR7, in the majority of diseases that are reviewed here, all these events lead to increased numbers of functional receptors at the surface of the tumor cells, which endows them with an increased migratory capacity (13, 20, 35, 40, 46, 99, 115, 173). CCR7-mediated migratory abilities can be selectively potentiated in leukemia/lymphoma cells (as opposed to their normal counterparts) by pre-exposure or co-incubation with other homeostatic chemokines like CXCL12 or CXCL13, as demonstrated in MCL, BL, or SS (78, 90, 183). Although in many entities the molecular mechanisms of CCR7 upregulation remain unknown, it is consistently found across various diseases, e.g. B-ALL, T-ALL, or CLL. Therein, its overexpression is associated with the presence of adverse prognostic factors, e.g. ZAP-70, which seem to directly cooperate with CCR7 towards facilitation of homing to survival niches such as LN or CNS (24, 38, 47). In fact, in CLL ZAP-70 has been shown to govern integrin activation upon CCR7 stimulation, in a G-protein independent fashion and through oligomerization of four CCR7 molecules (58, 63). Nevertheless, some associations of CCR7 expression with markers of disease subsets, hence aggressiveness or outcome, might be of indirect nature and just represent indicators of different inherent cellular programs (e.g. higher migratory potential), as suggested for the histogenetic subsets of CLL with unmutated IGHV and/or with trisomy 12 that show higher responsiveness to CCR7 ligands (35, 36, 38, 46, 47, 66).
Generally, in most blood cancers, CCR7 expression correlates with nodal or spleen involvement. In the conditions of B-ALL, MCL, T-ALL, or T-PLL it is also associated with infiltration of the CNS and in CTCL it correlates with the degree of epidermotropism (21, 24, 25, 130, 134, 161, 169, 221, 222). Therefore, it is consistently proposed that overexpression of CCR7 confers an invasive phenotype that contributes to lymphatic and hematogenous spread and promotes homing into target tissues (Figure 2). This CCR7+ transmigrating phenotype is further characterized by activation of α4β1 and αLβ2 integrins that facilitate adhesion of malignant cells to HEV or stromal proteins, and that promote the secretion of matrix metalloproteases MMP-2 and/or MMP-9, which degrade extracellular matrix (35, 58, 64, 130, 157, 183, 184). Both events result in transmigration of tumor cells into protective niches at which, particularly in T-zones, CCR7 contributes to disease progression in four major ways:
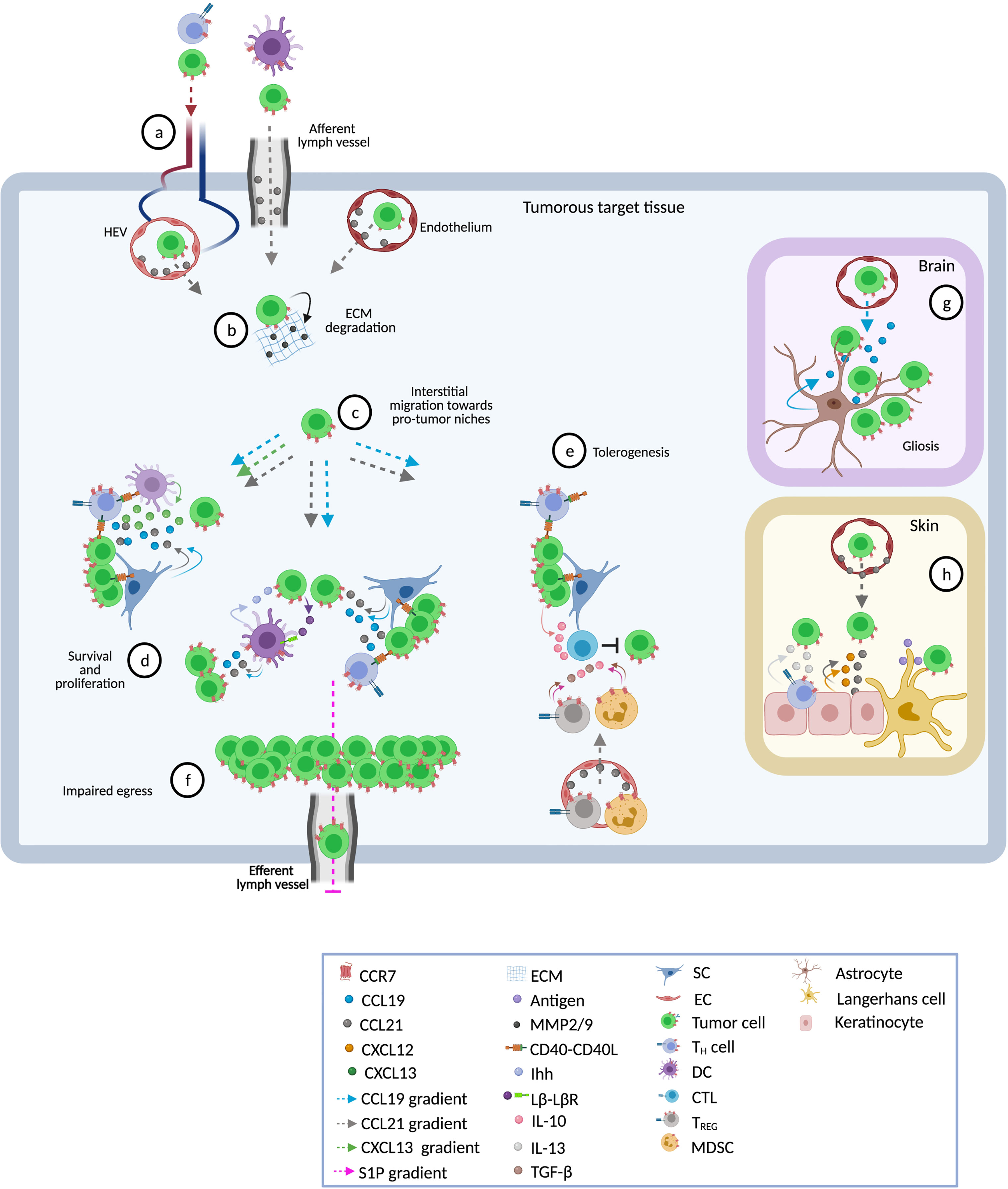
Figure 2 Pathophysiological roles of CCR7 in blood cancers. Shown are different tissues and ways in which the receptor contributes to disease progression. (A) Following CCL21 gradients, CCR7-expressing tumor and accessory cells enter into target tissues through high endothelial venules (HEV, in LN and other SLO), afferent lymphatic vessels (in LN), or endothelial cells (BM, spleen, skin, CNS) (8–10, 24, 25, 104, 130, 134). CCL21 on the surface of endothelial cells (EC) activates α4β1 and αLβ2 integrins thus facilitating transmigration (35, 58, 64, 130, 157). (B) Subsequently, activated CCR7 promotes invasive phenotypes that secrete metalloproteinases 2 and 9 (MMP-2/9) and promotes extracellular matrix (ECM) degradation (64, 130, 183, 184). (C) Within the tissue, CCR7 drives interstitial migration of tumor cells to distinct sites such as T-zones or B-zones in spleen and LN (9), or to lymphoid-like (tertiary) structures in skin or CNS (104, 169). Some tumor cells have inherent abilities to migrate towards CCL19/CCL21 gradients whereas others need cooperative signaling by other chemokines (e.g. CXCL12 or CXCL13) or a previous stimulus, such as exposure to CD40L, to initiate this process (20, 22, 23, 78, 90, 183, 209). (D) CCR7-driven interstitial migration assists tumor cells in their right positioning adjacent to accessory cells such as CD40L+CD4+ TH cells, DC, and stromal cells (SC) which foster growth and resistance to spontaneous or drug-induced cell death (9, 68). Cross-talk with accessory cells induces the release of CCL19 and CCL21 (9, 25, 134) directly promoting survival and proliferation of tumor cells via MAP-kinase and PI3K signaling pathways (9, 40, 70, 223). CCL19 can also act in cooperation with CXCL13 to confer resistance to TNF-α-mediated apoptosis via up-regulation of PEG10 (26, 27). Other pro-tumor factors delivered by accessory cells include the Indian hedgehog (Ihh) secreted by fibroblastic reticular cells (FRC) and CD40 stimulation through CD40 ligand-expressing CD4+ TH cells (9). In turn, malignant cells secrete factors that stimulate and protect accessory cells. One of these factors is lymphotoxin through which tumor cells stimulate lymphotoxin-β-receptor (LβR) expressing FRC (9). Finally, in these niches enhanced production of CCR7 ligands establishes a self-enhancing loop that recruits more tumor and accessory CCR7-expressing cells favoring the continuation of pro-tumor microenvironments (9, 25, 134, 161, 169, 221). (E) CCR7 ligands may attract CCR7+ immunosuppressive cells such as TREG and myeloid-derived suppressor cells (MDSC) (224, 225). These suppressor cells inhibit anti-tumor effector cells (e.g. CTL) through cell-cell interactions or by creating a tolerant milieu enriched in suppressor cytokines like IL-10 and tumor growth factor beta (TGF-β) (22, 28, 225, 226). Similarly, CCL19 and CXCL13 may synergistically regulate CD40-CD40L cross-talk between cancer cells and CD8+ T-cells leading to a PEG10-mediated enhanced production of IL-10 in CD40-activated tumor cells that inhibits tumor-specific CTL (28). Together, these CCR7-induced mechanisms facilitate permissive milieus within tumor target tissues. (F) CCR7 also prolongs the time of residence of CCR7-expressing cells in lymphoid tissues (i.e. LN) thus favoring proliferative cycles and providing niches of escape from systemic therapies. In the steady state, internalization of CCR7 activates the transcription and surface expression of S1P1, facilitating the egress of lymph-node-homed immune cells through efferent lymphatic vessels (71). Tumor-associated overexpression of CCR7 impairs S1P1 upregulation thus retaining tumor cells within the LNs, reducing the egress and causing lymphadenopathy (43, 46, 72). (G) In CNS, astrocyte-derived CCL19 attracts tumor cells and enhances their parenchymal retention thus contributing to gliosis (24, 25, 104). Inside cerebral or spleen parenchyma, cross-talk between stromal cells and leukemic cells mediates the production of higher levels of tissue CCL19 (25, 134) facilitating the infiltration of tumor cells. (H) In the skin, CCL21 in the cytoplasm of epidermal keratinocytes and to be diffusely distributed in the dermal extracellular matrix may lead tumor cells to milieus enriched in growth factors such as CXCL12, IL-13 or antigens able to entertain chronic T-cell receptor stimulation (169).
1) In ‘homing’ tumor cells, CCR7 drives interstitial migration within the tissue and assists in optimal positioning, e.g. adjacent to accessory cells such as CD40L+CD4+ TH cells, DC, and stromal cells, which foster growth and resistance to spontaneous or drug-induced cell death. As described, this positioning can be controlled by local factors including gradients of CCR7 ligands (9, 69, 104, 121), CD40-CD40L interactions (9, 68), BCR/ZAP-70 signal transduction (38, 42), or canonical WNT signaling (119).
2) Crosstalk with accessory cells can induce the release of high levels of the chemokines CCL19 and CCL21, which engage and activate CCR7 to provide pro-survival signals (9, 25, 134). In addition, stromal cells can produce CCR7 ligands in a constitutive manner (35, 46, 121, 161, 169, 223). Whatever the source of CCR7 ligands, they promote survival, e.g. by acting as trophic factors that induce MAPK and PI3K-AKT signaling (9, 25, 40, 70, 134, 223). They can also act in cooperation with CXCL13, which contributes to resistance to TNFα-mediated apoptosis preferentially in malignant over normal B-cells, via upregulation of PEG10 and stabilization of caspases-3 and -8 (26, 27). In some entities, e.g. MCL, T-PLL, or SS, CCR7 ligands have also shown to trigger proliferation (21, 130, 169).
3) Besides promoting influx, CCR7 (together with S1P1) also contributes to LN enlargement by regulating egress from lymphoid tissues, hence accumulation. It prolongs the time of residence of CCR7-expressing cells in lymphoid tissues, by that favoring proliferative cycles and providing niches of escape from systemic therapies. In CLL, the characteristic high expression of CCR7 can be attributed to abnormalities in the surface membrane recycling machinery as a consequence of abnormally low production of the cytoplasmatic p66Shc protein (43, 46). Deficiency of the cytoplasmic p66Shc protein causes enhanced activity of the PP2B/calcineurin phosphatase on the endosomal CCR7 pool, which enhances its recycling back to the plasma membrane. Likewise, ATLL cells carry mutations in the C-terminal cytoplasmic domain of CCR7 that impairs proper recycling (162, 163).
4) CCR7 participates in the creation of permissive tumor microenvironments within tumorous target tissues (SLOs, CNS, or skin). As part of the involved tissue interactions between tumor cells and stromal or other accessory cells, an increased production of CCR7 ligands is stimulated (9, 25, 134). For example, cooperative CCR7/WNT signaling is needed for secretion of VGEF-A by cHL cells, which leads to de novo generation of vascular endothelium which, in turn, presents CCR7 ligands that direct movement of cHL towards vascular niches (119). Similarly, BL cells can secrete lymphotoxin through which they stimulate lymphotoxin-β-receptor expressing gp38+ FRC that secrete the survival factors Ihh and CCL21 (9). These patterns and higher amounts of chemokines establish a feed-forward loop that not only recruits additional CCR7-expressing malignant cells, but also further supportive bystander cells (9, 25, 105, 134, 161, 221). Moreover, in B-ALL CCR7 ligands potentiate secretion of immunosuppressive IL-10 by tumor cells leading to impaired specific anti-tumor CTL responses (28). CCR7 can also recruit regulatory cells that hamper anti-tumor immunity. For example, an increase of functional TREG has been established for patients with blood cancers (226). Expansion of TREG is needed to generate and sustain a tolerogenic TME (227). Indeed, higher numbers of TREG associate with progressive disease in cHL, CLL, MGUS, MM, or DLBCL (228–232).
5 Biased Signaling of CCR7 Ligands in Blood Cancers
In the field of GPCRs, knowledge on the diversity in signaling pathways has promoted the concept of “biased signaling”, which involves a context-specific preference for one intracellular signaling pathway over another (233). This concept can be considered as either receptor bias (the same ligand has different actions through different receptors), ligand bias (more than one or naturally modified ligands act on the same receptor and induce different outcomes), or tissue bias (the cellular effect depends on the tissue/cell type) (234). How biased signaling enables different downstream pathways that eventually will determine the overall outcome of CCR7 engagement in different immune cell types has been recently, and deeply, reviewed by Hauser et al. (11). The role(s) of biased signaling of CCR7 in the pathophysiology of blood cancers is not clear, although a scarce number of studies comparing CCR7 activation in healthy versus malignant cells indicate that a differential regulation is plausible. For example, in CLL we demonstrated that PI3K and ROCK, but not MAPK, were involved in migration of CLL cells toward CCL19 and CCL21, whereas normal B-cells relied more on PI3K, ROCK, and p38-SAPK pathways (40). Moreover, while CLL cells showed an enhanced, similar migratory response to both CCR7 ligands, normal B-cells showed a moderate response, and preferentially towards CCL21 (13, 35, 40, 46). Likewise, CLL and normal B-cells showed a different signaling to migrate through the endothelium (35, 235). Finally, it is worth mentioning that in the last years CCR7 has been shown to form heterodimers with CXCR4 (217). This process affects CCR7 signaling and might explain a poorly understood, but important, mechanism of chemokine biology that allows synergistic and/or inhibitory outputs produced by simultaneous activation or inhibition of multiple CKRs. Interestingly, CCR7/CXCR4 dimers may enhance tumor B-cell homing to LN by potentiating the TEM upon simultaneous exposure to CXCL12, CCL19, and CCL21 (90) while in healthy, mature B-cells CCR7 acts as a selective allosteric modulator that inactivates CXCR4 thus impairing retention in the BM (236). Together, these findings support the existence of a functional and phenotypic diversity as a result of a biased signaling of CCR7 in homeostasis and blood cancers. Nonetheless, other studies show identical mechanisms in both healthy and neoplastic cells. For example, CCL19-specific translation of S1P1 is mediated by ERK-5/Krüppel-like factor-2 in the HuT78 SS cell line and healthy primary T-cells (237). Therefore, additional comparative studies addressing biased signaling in both healthy and tumor tissues are mandatory to better understand the pathophysiological roles of these regulatory mechanisms in CCR7 and other CKRs.
6 CCR7 as a Therapeutic Target in Blood Cancers
The circumstantial and direct evidence presented in this review suggests that the tumor-associated CCR7-ligand interaction is an actionable vulnerability. At CCR7-ligand permissive sites, e.g. LN or brain, malignant cells evade spontaneous or drug-induced apoptosis as well as escape immune cell control or proliferate, all in a CCR7-mediated manner. Therefore, interfering in CCR7-signalling promises to be of therapeutic potential in many CCR7 expressing and CCR7 promoted blood cancers. Nonetheless, targeting CCR7 in cancer has the potential downsite of activating and/or potentiating alternative pathways that would eventually allow homing of tumor cells to protective niches. For instance, to adhere to HEVs of peripheral and mesenteric LNs T-cells rely on CCR7 and partially on CXCL12/CXCR4 whereas normal B-cells can exploit the CXCR4 and CXCL13/CXCR5 axes to induce integrin-mediated arrest on HEVs and homing to the LN (238–243). To avoid this scenario, it is desirable that anti-CCR7 drugs feature a double MOA including both, neutralization of the target and tumoricidal capacities. In this regard, we and others have demonstrated that approaches based on we and others have demonstrated that approaches based on blocking (non-activating) monoclonal antibodies (mAbs) that target CCR7 or its ligands, are highly effective in in vitro and in vivo preclinical models, including B-ALL (24), CLL (37, 40, 45), MCL (21), T-ALL (24, 25), or T-PLL (130). In these studies, such anti-CCR7 therapies reduced tumor cell migration and infiltration into CCR7-specific environments and additionally impaired survival/proliferation. Overall, the combined neutralizing and killing activities of anti-CCR7 mAbs led to retarded tumor implantation, reduced tumor burden, and significantly extended host survival in in vivo models.
Taken together, there is ample data on target expression and mechanistic rationales as well as sufficient proof of principle and feasibility data that strongly encourage the therapeutic application of anti-CCR7 therapies in blood cancers. Consequently, first clinical-grade anti-CCR7 antibodies have been developed during the last years. Novartis is enrolling patients into a phase-I trial with JBH492 an antibody-drug conjugate (ADC) targeting CCR7 (NCT042140704). Moreover, Catapult Therapeutics presented first pre-clinical results of an antagonist mAb called CAP-100 that will be evaluated in first-in-human clinical trials in 2021 (NCT04704323) (244). In preclinical settings, both compounds have shown to be highly effective as a single agent and at least CAP-100 revealed the potential for combinations with current standard-of-care drugs (245). Owing to their particular MOA, antagonisticanti-CCR7 mAbs may be likely combined with other standard-of-care drugs to obtain additive or synergistic effects while reducing the likelihood of treatment resistances. For example, by blocking ligand-receptor interactions, anti-CCR7 therapies may displace tumor cells out of protective niches, forcing them to accumulate in blood where they may become more accessible to other cytotoxic drugs such antibodies against established targets (e.g. CD20, CD30, CCR4, etc), or chemotherapeutics (e.g. fludarabine), or small molecule inhibitors (e.g. BCL2 inhibitors). Along with the BTK inhibitor ibrutinib or with the PI3Kδ inhibitor idelalisib, anti-CCR7 mAbs would additively or synergistically target CCR7-mediated adhesion to lymphoid stroma or endothelium, thus favoring an enhanced cell egress from lymphoid tissues into circulation (44, 46, 48, 246). In fact, we have recently demonstrated that CCR7 expression and functionality is not impaired during ibrutinib treatment in CLL patients and that the anti-CCR7 CAP-100 and ibrutinib show complementary activities (245). Moreover, while the antibody would block recirculation and loops of LN homing, ibrutinib would also interfere with CXCR4- and CXCR5-mediated signaling and with the production of chemokines (CXCL12, CXCL13, CCL19) by myeloid stroma cells (44, 247), thus acting against potential redundant chemotactic pathways. Finally, it is worth mentioning that immune checkpoint blockade and CAR-T-cells have revolutionized the field of cancer therapy during the last decades. Whether anti-CCR7 therapy may complement such treatments is uncertain since for avoiding negative interactions it seems necessary that therapeutic T-cells to express an effector or effector memory CCR7-negative phenotype.
Given the various roles in tumor development and progression, adhesion molecules are promising targets to block the access of tumor cells to tumor-permissive niches like the LN (248, 249). For instance, LFA-1 and VLA-4 are involved in the development of hematological malignancies and tumor cells require their expression to migrate into lymphoid tissues (250, 251). Therefore, it is plausible to speculate that targeting these leukocyte adhesion molecules might be an alternative way to target the CCR7 axis. Like anti-CCR7 assets do, targeting cell adhesion exerts direct effects to the tumor cell (e.g. reduction of motility, invasiveness, and proliferation) that may impair homing to SLO (252, 253). In addition, cell adhesion molecules are common downstream players activated by several CKRs, including CCR7 (254), hence, their inhibition would potentially inhibit CCR7 along with several other receptors, thus overcoming the redundancy of CKRs (255). Yanguas et al. showed in murine melanoma models that an increased number of intra-tumorally injected tumor-specific T-cells migrated into the draining LN when treated with anti-ICAM-1 or anti-LFA-1 mAbs (256). This indicates that specific approaches, such as anti-CCR7 mAbs, are needed to block LN homing. Moreover, since integrins play diverse roles in immunity and anti-tumor responses, targeting the function of these molecules in vivo may be a difficult task in cancer therapy (254, 257, 258). Accordingly, multiple clinical trials that involve the targeting of αV or β1 integrins have shown disappointing results with low therapeutic efficacies (249), while anti-LFA-1 strategies have been associated with an increased risk of malignancies, infections, and rare, but severe, systemic adverse events such as immune-mediated thrombocytopenia and hemolytic anemia (259, 260). To overcome these deleterious effects, novel approaches aiming to target the integrin and/or ligands on tumor cells or tumor vessels, but not in immune cells, are needed. Bispecific antibodies simultaneously directed against LFA-1 and a tumor specific antigen may contribute to specifically block LFA-1-mediated tumor cell adhesion without affecting immune responses, as shown in mice (261). Since targeting CCR7 shares both overlapping and differentiating MOAs with such therapies that target adhesion, combining both of these strategies could provide clinical benefits and needs to be further investigated.
7 Safety of Novel Therapies Targeting CCR7 in Blood Cancers
Currently, two clinical trials aim to validate anti-CCR7 approaches in hematologic diseases with an urgent need for more rationally based and efficient therapies. These studies will also allow us to learn the real risks that are associated with blocking CCR7 and/or depleting CCR7-expressing immune cell subsets as this receptor is critical for activation steps in the adaptive immune system and for the homeostasis of TREG, which limit self-reactive events and autoimmunity (262). In one hand, several mouse models revealed that deficiency of CCR7 signaling was not a life-threatening condition, as it was associated with a moderate impact on immunity by retarded, but preserved, T-cell and B-cell responses (8, 240, 263–265), especially against infections with replicating antigens (266–269). However, in different vaccinations approaches (e.g. HIV, HSV, or HCV) adjuvant CCL19 was relevant for augmenting the trafficking of T-cells and DC (270–272). Therefore, anti-CCR7 therapy may reduce priming of antigen-specific T-cells and the production of Abs in a virus-dependent manner. From pre-clinical models, we know that the use of anti-CCR7 mAbs selectively inhibited and/or depleted tumor cells while sparing healthy counterparts (37, 45, 244, 273, 274). Notably, CD4+ TN and TCM cells were preferentially impacted while other CCR7-expressing subsets such as DC or B-cells were not. Lower target density in non-tumor cells or lower affinity of these antibodies for CCR7 expressed in these cell types could explain these observations which also suggests that anti-CCR7 therapy might impair new immunization processes dependent on TN cells, but not memory effector responses against infections (273–275). In this regard, CCR7-negative TEFF and TEM rather than CCR7-expressing TN or TCM are necessary for effective anti-tumour responses (276). Moreover, naïve tumor-specific CD8+ T-cells, which seem less susceptible to anti-CCR7 therapy (244, 273, 274), can also become activated and gain effector-cell phenotypes directly at the tumor site, suggesting that cross-presenting DC are also able to prime CD8+ T-cells within the tumor (277). These results indicate that DC migration into LN may not even be completely necessary for DC-mediated anti-tumor responses.
Targeting CCR7 could also affect B-cell homing during antigen-dependent and independent B-cell differentiation; however, CCR7-deficient mice show splenic B-cell responses upon bacterial challenge (240). In addition, normal B-cells are less dependent on CCR7 than leukemic cells for the arrest on HEVs and for homing (71, 238, 278) while BM B-cell precursors and plasma cells lack CCR7 (13), suggesting that CCR7 therapy would not suppress B-cell lymphopoiesis nor immunoglobulin secretory function (8, 240).
Related to unwanted autoimmune side-effects, lack of CCR7 signaling in TREG hampered central and peripheral tolerance (224, 279–282) and led to generalized multi-organ autoimmunity. Whether anti-CCR7 therapies will resemble phenotypes in CCR7-deficient animals will remain unknown until first evidence in patients becomes available. Until then, clinical results with the therapeutic antibody mogamulizumab (which removes CCR4+ T-cell subsets, including TREG) (283) allow us to speculate that anti-CCR7 can be safe and well-tolerated. In line with this, anti-CCR7 therapy in pre-clinical syngeneic mouse models of cancer, autoimmunity, GVHD, or inflammation did not uncover treatment-associated side effects (225, 273, 275) and CAP-100 toxicology studies in NHP did not reveal overt toxicities or autoimmune disease, all indicating superior tolerability of this novel therapy (244). In the coming months, first data in patients receiving a chronic administration of an anti-CCR7 mAb will be available and, hopefully, results from clinical studies will shed light into the safety and utility of targeting CCR7, and more importantly, will validate anti-CCR7 approaches in hematologic diseases with an urgent need for more rationally based and efficient therapies.
8 Conclusions
Classically, the pathogenic role of CCR7 as a cancer-associated receptor in hematology has been attributed to its unique ability to drive tumor cells into the LN and other SLO. Accordingly, CCR7 expression has been strongly linked to bulky disease in these lymphoid tissues. Nonetheless, this canonical (and somehow narrow) view of CCR7 as a migratory receptor is changing thanks to recent evidence that supports additional pathogenic functions of CCR7. Beyond cancer cell lymphotropism, we have disclosed that CCR7 expression is also associated to neurotropism and epidermotropism, to interstitial migration within tumor tissues, to juxta-positioning to accessory cells, and to cell survival and proliferation. Moreover, CCR7 also guides different accessory cell types that are needed to create and preserve pro-tumor niches and to protect cancer cells from spontaneous or drug-induced apoptosis. Likewise, immunosuppressive cells take advantage of CCR7 to locate themselves close to innate or adaptative anti-tumor immune cells, thus facilitating their tolerogenic or their inhibitory participation in the TME.
However, our knowledge on CCR7 biology in blood cancers is still scant and additional efforts are needed to solve relevant questions such as around the major mechanisms regulating CCR7 (over)expression, how CCR7 contributes to tumor growth during the first tumorigeneic events, or what the exact contributions of CCR7 in accessory cells in early cancer events or during different stages of target tissue colonization are.
CCR7 is currently postulated as a potential therapeutic target for some blood cancers and novel antibody(conjugate)-based strategies targeting CCR7 are being evaluated in early-phase clinical trials. It is also tempting to speculate that modulation of CCR7 expression and signaling in therapeutic lymphocytes might allow manipulation of the performance (e.g. migratory potential, longevity) of T- or NK-cells carrying chimeric-antigen receptors. If such direct or indirect modulation of tumor-cell or milieu-derived CCR7-signaling stands out as a promising approach it is likely that in the coming years an extensive collection of novel evidence will help to better understand its biology and to refine CCR7-based translational applications.
Author Contributions
CC-M conceived the manuscript, wrote the manuscript, and conceived and created the figures and tables. FT co-wrote the manuscript. MH co-wrote the manuscript, conceived and edited tables and figures. All authors contributed to the article and approved the submitted version.
Conflict of Interest
CC-M is an employee of Catapult Therapeutics and of Immunological and Medical Products (IMMED S.L.), and a shareholder in this last company. FT declares that he is CEO and a shareholder in the same companies.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors would like to thank Marty Wulferink and Wim Mol for language editing and proofreading of the manuscript. In addition, we would like to thank BioRender.com for figure editing.
References
1. Riedel A, Shorthouse D, Haas L, Hall BA, Shields J. Tumor-Induced Stromal Reprogramming Drives Lymph Node Transformation. Nat Immunol (2016) 17(9):1118–27. doi: 10.1038/ni.3492
2. Birkenbach M, Josefsen K, Yalamanchili R, Lenoir G, Kieff E. Epstein-Barr Virus-Induced Genes: First Lymphocyte-Specific G Protein-Coupled Peptide Receptors. J Virol (1993) 67(4):2209–20. doi: 10.1128/jvi.67.4.2209-2220.1993
3. Schweickart VL, Raport CJ, Godiska R, Byers MG, Eddy RL Jr., Shows TB, et al. Cloning of Human and Mouse EBI1, a Lymphoid-Specific G-Protein-Coupled Receptor Encoded on Human Chromosome 17q12-Q21.2. Genomics (1994) 23(3):643–50. doi: 10.1006/geno.1994.1553
4. Burgstahler R, Kempkes B, Steube K, Lipp M. Expression of the Chemokine Receptor BLR2/EBI1 is Specifically Transactivated by Epstein-Barr Virus Nuclear Antigen 2. Biochem Biophys Res Commun (1995) 215(2):737–43. doi: 10.1006/bbrc.1995.2525
5. Comerford I, Harata-Lee Y, Bunting MD, Gregor C, Kara EE, McColl SR. A Myriad of Functions and Complex Regulation of the CCR7/CCL19/CCL21 Chemokine Axis in the Adaptive Immune System. Cytokine Growth Factor Rev (2013) 24(3):269–83. doi: 10.1016/j.cytogfr.2013.03.001
6. Forster R, Davalos-Misslitz AC, Rot A. CCR7 and its Ligands: Balancing Immunity and Tolerance. Nat Rev Immunol (2008) 8(5):362–71. doi: 10.1038/nri2297
7. Legler DF, Uetz-von Allmen E, Hauser MA. CCR7: Roles in Cancer Cell Dissemination, Migration and Metastasis Formation. Int J Biochem Cell Biol (2014) 54:78–82. doi: 10.1016/j.biocel.2014.07.002
8. Forster R, Schubel A, Breitfeld D, Kremmer E, Renner-Muller I, Wolf E, et al. CCR7 Coordinates the Primary Immune Response by Establishing Functional Microenvironments in Secondary Lymphoid Organs. Cell (1999) 99(1):23–33. doi: 10.1016/S0092-8674(00)80059-8
9. Rehm A, Mensen A, Schradi K, Gerlach K, Wittstock S, Winter S, et al. Cooperative Function of CCR7 and Lymphotoxin in the Formation of a Lymphoma-Permissive Niche Within Murine Secondary Lymphoid Organs. Blood (2011) 118(4):1020–33. doi: 10.1182/blood-2010-11-321265
10. Wiley HE, Gonzalez EB, Maki W, Wu MT, Hwang ST. Expression of CC Chemokine Receptor-7 and Regional Lymph Node Metastasis of B16 Murine Melanoma. J Natl Cancer Inst (2001) 93(21):1638–43. doi: 10.1093/jnci/93.21.1638
11. Hauser MA, Legler DF. Common and Biased Signaling Pathways of the Chemokine Receptor CCR7 Elicited by its Ligands CCL19 and CCL21 in Leukocytes. J Leukoc Biol (2016) 99(6):869–82. doi: 10.1189/jlb.2MR0815-380R
12. Pui CH, Robison LL, Look AT. Acute Lymphoblastic Leukaemia. Lancet (2008) 371(9617):1030–43. doi: 10.1016/S0140-6736(08)60457-2
13. Lopez-Giral S, Quintana NE, Cabrerizo M, Alfonso-Perez M, Sala-Valdes M, De Soria VG, et al. Chemokine Receptors That Mediate B Cell Homing to Secondary Lymphoid Tissues Are Highly Expressed in B Cell Chronic Lymphocytic Leukemia and Non-Hodgkin Lymphomas With Widespread Nodular Dissemination. J Leukoc Biol (2004) 76(2):462–71. doi: 10.1189/jlb.1203652
14. Haferlach T, Kohlmann A, Wieczorek L, Basso G, Kronnie GT, Béné M-C, et al. Clinical Utility of Microarray-Based Gene Expression Profiling in the Diagnosis and Subclassification of Leukemia: Report From the International Microarray Innovations in Leukemia Study Group. J Clin Oncol Off J Am Soc Clin Oncol (2010) 28(15):2529–37. doi: 10.1200/JCO.2009.23.4732
15. Maia S, Haining WN, Ansén S, Xia Z, Armstrong SA, Seth NP, et al. Gene Expression Profiling Identifies BAX-Delta as a Novel Tumor Antigen in Acute Lymphoblastic Leukemia. Cancer Res (2005) 65(21):10050–8. doi: 10.1158/0008-5472.CAN-05-1574
16. Andersson A, Ritz C, Lindgren D, Edén P, Lassen C, Heldrup J, et al. Microarray-Based Classification of a Consecutive Series of 121 Childhood Acute Leukemias: Prediction of Leukemic and Genetic Subtype as Well as of Minimal Residual Disease Status. Leukemia (2007) 21(6):1198–203. doi: 10.1038/sj.leu.2404688
17. Coustan-Smith E, Song G, Clark C, Key L, Liu P, Mehrpooya M, et al. New Markers for Minimal Residual Disease Detection in Acute Lymphoblastic Leukemia. Blood (2011) 117(23):6267–76. doi: 10.1182/blood-2010-12-324004
18. Dürig J, Schmucker U, Duhrsen U. Differential Expression of Chemokine Receptors in B Cell Malignancies. Leukemia (2001) 15(5):752–6. doi: 10.1038/sj.leu.2402107
19. Wong S, Fulcher D. Chemokine Receptor Expression in B-Cell Lymphoproliferative Disorders. Leuk Lymphoma (2004) 45(12):2491–6. doi: 10.1080/10428190410001723449
20. Corcione A, Arduino N, Ferretti E, Pistorio A, Spinelli M, Ottonello L, et al. Chemokine Receptor Expression and Function in Childhood Acute Lymphoblastic Leukemia of B-Lineage. Leuk Res (2006) 30(4):365–72. doi: 10.1016/j.leukres.2005.07.009
21. Somovilla-Crespo B, Alfonso-Perez M, Cuesta-Mateos C, Carballo-de Dios C, Beltran AE, Terron F, et al. Anti-CCR7 Therapy Exerts a Potent Anti-Tumor Activity in a Xenograft Model of Human Mantle Cell Lymphoma. J Hematol Oncol (2013) 6:89. doi: 10.1186/1756-8722-6-89
22. D’Amico G, Vulcano M, Bugarin C, Bianchi G, Pirovano G, Bonamino M, et al. CD40 Activation of BCP-ALL Cells Generates IL-10-Producing, IL-12-Defective APCs That Induce Allogeneic T-Cell Anergy. Blood (2004) 104(3):744–51. doi: 10.1182/blood-2003-11-3762
23. Luczynski W, Ilendo E, Kovalchuk O, Krawczuk-Rybak M, Malinowska I, Koltan A, et al. Acute Lymphoblastic Leukaemia Cells Express CCR7 But Not Higher Amounts of IL-10 After CD40 Ligation. Scand J Clin Lab Invest (2006) 66(8):695–703. doi: 10.1080/00365510600931098
24. Alsadeq A, Fedders H, Vokuhl C, Belau NM, Zimmermann M, Wirbelauer T, et al. The Role of ZAP70 Kinase in Acute Lymphoblastic Leukemia Infiltration Into the Central Nervous System. Haematologica (2017) 102(2):346–55. doi: 10.3324/haematol.2016.147744
25. Buonamici S, Trimarchi T, Ruocco MG, Reavie L, Cathelin S, Mar BG, et al. CCR7 Signalling as an Essential Regulator of CNS Infiltration in T-Cell Leukaemia. Nature (2009) 459(7249):1000–4. doi: 10.1038/nature08020
26. Chunsong H, Yuling H, Li W, Jie X, Gang Z, Qiuping Z, et al. CXC Chemokine Ligand 13 and CC Chemokine Ligand 19 Cooperatively Render Resistance to Apoptosis in B Cell Lineage Acute and Chronic Lymphocytic Leukemia CD23+CD5+ B Cells. J Immunol (2006) 177(10):6713–22. doi: 10.4049/jimmunol.177.10.6713
27. Hu C, Xiong J, Zhang L, Huang B, Zhang Q, Li Q, et al. PEG10 Activation by Co-Stimulation of CXCR5 and CCR7 Essentially Contributes to Resistance to Apoptosis in CD19+CD34+ B Cells From Patients With B Cell Lineage Acute and Chronic Lymphocytic Leukemia. Cell Mol Immunol (2004) 1(4):280–94.
28. Wang X, Yuling H, Yanping J, Xinti T, Yaofang Y, Feng Y, et al. CCL19 and CXCL13 Synergistically Regulate Interaction Between B Cell Acute Lymphocytic Leukemia CD23+CD5+ B Cells and CD8+ T Cells. J Immunol (Baltimore Md 1950) (2007) 179(5):2880–8. doi: 10.4049/jimmunol.179.5.2880
29. Hallek M, Cheson BD, Catovsky D, Caligaris-Cappio F, Dighiero G, Dohner H, et al. iwCLL Guidelines for Diagnosis, Indications for Treatment, Response Assessment, and Supportive Management of CLL. Blood (2018) 131(25):2745–60. doi: 10.1182/blood-2017-09-806398
30. Hayden RE, Pratt G, Roberts C, Drayson MT, Bunce CM. Treatment of Chronic Lymphocytic Leukemia Requires Targeting of the Protective Lymph Node Environment With Novel Therapeutic Approaches. Leuk Lymphoma (2012) 53(4):537–49. doi: 10.3109/10428194.2011.610014
31. Ponzoni M, Doglioni C, Caligaris-Cappio F. Chronic Lymphocytic Leukemia: The Pathologist’s View of Lymph Node Microenvironment. Semin Diagn Pathol (2011) 28(2):161–6. doi: 10.1053/j.semdp.2011.02.014
32. Basso K, Margolin AA, Stolovitzky G, Klein U, Dalla-Favera R, Califano A. Reverse Engineering of Regulatory Networks in Human B Cells. Nat Genet (2005) 37(4):382–90. doi: 10.1038/ng1532
33. Rosenwald A, Wright G, Chan WC, Connors JM, Campo E, Fisher RI, et al. The Use of Molecular Profiling to Predict Survival After Chemotherapy for Diffuse Large-B-Cell Lymphoma. N Engl J Med (2002) 346(25):1937–47. doi: 10.1056/NEJMoa012914
34. Haslinger C, Schweifer N, Stilgenbauer S, Döhner H, Lichter P, Kraut N, et al. Microarray Gene Expression Profiling of B-Cell Chronic Lymphocytic Leukemia Subgroups Defined by Genomic Aberrations and VH Mutation Status. J Clin Oncol Off J Am Soc Clin Oncol (2004) 22(19):3937–49. doi: 10.1200/JCO.2004.12.133
35. Till KJ, Lin K, Zuzel M, Cawley JC. The Chemokine Receptor CCR7 and Alpha4 Integrin Are Important for Migration of Chronic Lymphocytic Leukemia Cells Into Lymph Nodes. Blood (2002) 99(8):2977–84. doi: 10.1182/blood.V99.8.2977
36. Ghobrial IM, Bone ND, Stenson MJ, Novak A, Hedin KE, Kay NE, et al. Expression of the Chemokine Receptors CXCR4 and CCR7 and Disease Progression in B-Cell Chronic Lymphocytic Leukemia/ Small Lymphocytic Lymphoma. Mayo Clin Proc (2004) 79(3):318–25. doi: 10.4065/79.3.318
37. Alfonso-Perez M, Lopez-Giral S, Quintana NE, Loscertales J, Martin-Jimenez P, Munoz C. Anti-CCR7 Monoclonal Antibodies as a Novel Tool for the Treatment of Chronic Lymphocyte Leukemia. J Leukoc Biol (2006) 79(6):1157–65. doi: 10.1189/jlb.1105623
38. Richardson SJ, Matthews C, Catherwood MA, Alexander HD, Carey BS, Farrugia J, et al. ZAP-70 Expression is Associated With Enhanced Ability to Respond to Migratory and Survival Signals in B-Cell Chronic Lymphocytic Leukemia (B-CLL). Blood (2006) 107(9):3584–92. doi: 10.1182/blood-2005-04-1718
39. Calissano C, Damle RN, Hayes G, Murphy EJ, Hellerstein MK, Moreno C, et al. In Vivo Intraclonal and Interclonal Kinetic Heterogeneity in B-Cell Chronic Lymphocytic Leukemia. Blood (2009) 114(23):4832–42. doi: 10.1182/blood-2009-05-219634
40. Cuesta-Mateos C, Lopez-Giral S, Alfonso-Perez M, de Soria VG, Loscertales J, Guasch-Vidal S, et al. Analysis of Migratory and Prosurvival Pathways Induced by the Homeostatic Chemokines CCL19 and CCL21 in B-Cell Chronic Lymphocytic Leukemia. Exp Hematol (2010) 38(9):756–64, 64 e1-4. doi: 10.1016/j.exphem.2010.05.003
41. Calissano C, Damle RN, Marsilio S, Yan XJ, Yancopoulos S, Hayes G, et al. Intraclonal Complexity in Chronic Lymphocytic Leukemia: Fractions Enriched in Recently Born/Divided and Older/Quiescent Cells. Mol Med (2011) 17(11-12):1374–82. doi: 10.2119/molmed.2011.00360
42. Calpe E, Codony C, Baptista MJ, Abrisqueta P, Carpio C, Purroy N, et al. ZAP-70 Enhances Migration of Malignant B Lymphocytes Toward CCL21 by Inducing CCR7 Expression via IgM-ERK1/2 Activation. Blood (2011) 118(16):4401–10. doi: 10.1182/blood-2011-01-333682
43. Capitani N, Patrussi L, Trentin L, Lucherini OM, Cannizzaro E, Migliaccio E, et al. S1P1 Expression is Controlled by the Pro-Oxidant Activity of p66Shc and is Impaired in B-CLL Patients With Unfavorable Prognosis. Blood (2012) 120(22):4391–9. doi: 10.1182/blood-2012-04-425959
44. de Rooij MF, Kuil A, Geest CR, Eldering E, Chang BY, Buggy JJ, et al. The Clinically Active BTK Inhibitor PCI-32765 Targets B-Cell Receptor- and Chemokine-Controlled Adhesion and Migration in Chronic Lymphocytic Leukemia. Blood (2012) 119(11):2590–4. doi: 10.1182/blood-2011-11-390989
45. Cuesta-Mateos C, Loscertales J, Kreutzman A, Colom-Fernandez B, Portero-Sainz I, Perez-Villar JJ, et al. Preclinical Activity of Anti-CCR7 Immunotherapy in Patients With High-Risk Chronic Lymphocytic Leukemia. Cancer Immunol Immunother (2015) 64(6):665–76. doi: 10.1007/s00262-015-1670-z
46. Patrussi L, Capitani N, Martini V, Pizzi M, Trimarco V, Frezzato F, et al. Enhanced Chemokine Receptor Recycling and Impaired S1P1 Expression Promote Leukemic Cell Infiltration of Lymph Nodes in Chronic Lymphocytic Leukemia. Cancer Res (2015) 75(19):4153–63. doi: 10.1158/0008-5472.CAN-15-0986
47. Ganghammer S, Hutterer E, Hinterseer E, Brachtl G, Asslaber D, Krenn PW, et al. CXCL12-Induced VLA-4 Activation is Impaired in Trisomy 12 Chronic Lymphocytic Leukemia Cells: A Role for CCL21. Oncotarget (2015) 6(14):12048–60. doi: 10.18632/oncotarget.3660
48. Patrussi L, Capitani N, Cattaneo F, Manganaro N, Gamberucci A, Frezzato F, et al. p66Shc Deficiency Enhances CXCR4 and CCR7 Recycling in CLL B Cells by Facilitating Their Dephosphorylation-Dependent Release From Beta-Arrestin at Early Endosomes. Oncogene (2018) 37(11):1534–50. doi: 10.1038/s41388-017-0066-2
49. Enjuanes A, Benavente Y, Bosch F, Martin-Guerrero I, Colomer D, Perez-Alvarez S, et al. Genetic Variants in Apoptosis and Immunoregulation-Related Genes are Associated With Risk of Chronic Lymphocytic Leukemia. Cancer Res (2008) 68(24):10178–86. doi: 10.1158/0008-5472.CAN-08-2221
50. Chen Q, Zheng T, Lan Q, Lerro C, Zhao N, Qin Q, et al. Single-Nucleotide Polymorphisms in Genes Encoding for CC Chemokines Were Not Associated With the Risk of Non-Hodgkin Lymphoma. Cancer Epidemiology Biomarkers Prev Publ Am Assoc Cancer Research cosponsored by Am Soc Prev Oncol (2013) 22(7):1332–5. doi: 10.1158/1055-9965.EPI-13-0328
51. Arruga F, Gizdic B, Bologna C, Cignetto S, Buonincontri R, Serra S, et al. Mutations in NOTCH1 PEST Domain Orchestrate CCL19-Driven Homing of Chronic Lymphocytic Leukemia Cells by Modulating the Tumor Suppressor Gene DUSP22. Leukemia (2017) 31(9):1882–93. doi: 10.1038/leu.2016.383
52. Cattaneo F, Patrussi L, Capitani N, Frezzato F, D’Elios MM, Trentin L, et al. Expression of the p66Shc Protein Adaptor is Regulated by the Activator of Transcription STAT4 in Normal and Chronic Lymphocytic Leukemia B Cells. Oncotarget (2016) 7(35):57086–98. doi: 10.18632/oncotarget.10977
53. Kim SH, Gunst KV, Sarvetnick N. STAT4/6-Dependent Differential Regulation of Chemokine Receptors. Clin Immunol (2006) 118(2-3):250–7. doi: 10.1016/j.clim.2003.10.002
54. Wolf C, Garding A, Filarsky K, Bahlo J, Robrecht S, Becker N, et al. NFATC1 Activation by DNA Hypomethylation in Chronic Lymphocytic Leukemia Correlates With Clinical Staging and can be Inhibited by Ibrutinib. Int J Cancer (2017) 2017(18):31057. doi: 10.1002/ijc.31057
55. Macian F, Lopez-Rodriguez C, Rao A. Partners in Transcription: NFAT and AP-1. Oncogene (2001) 20(19):2476–89. doi: 10.1038/sj.onc.1204386
56. Rodriguez A, Martinez N, Camacho FI, Ruiz-Ballesteros E, Algara P, Garcia JF, et al. Variability in the Degree of Expression of Phosphorylated IkappaBalpha in Chronic Lymphocytic Leukemia Cases With Nodal Involvement. Clin Cancer Res (2004) 10(20):6796–806. doi: 10.1158/1078-0432.CCR-04-0753
57. Faguet GB. Chronic Lymphocytic Leukemia: An Updated Review. J Clin Oncol (1994) 12(9):1974–90. doi: 10.1200/JCO.1994.12.9.1974
58. Laufer JM, Lyck R, Legler DF. ZAP70 Expression Enhances Chemokine-Driven Chronic Lymphocytic Leukemia Cell Migration and Arrest by Valency Regulation of Integrins. FASEB J (2018) 32(9):4824–35. doi: 10.1096/fj.201701452RR
59. Till KJ, Harris RJ, Linford A, Spiller DG, Zuzel M, Cawley JC. Cell Motility in Chronic Lymphocytic Leukemia: Defective Rap1 and Alphalbeta2 Activation by Chemokine. Cancer Res (2008) 68(20):8429–36. doi: 10.1158/0008-5472.CAN-08-1758
60. Hartmann TN, Grabovsky V, Wang W, Desch P, Rubenzer G, Wollner S, et al. Circulating B-Cell Chronic Lymphocytic Leukemia Cells Display Impaired Migration to Lymph Nodes and Bone Marrow. Cancer Res (2009) 69(7):3121–30. doi: 10.1158/0008-5472.CAN-08-4136
61. Riches JC, O’Donovan CJ, Kingdon SJ, McClanahan F, Clear AJ, Neuberg DS, et al. Trisomy 12 Chronic Lymphocytic Leukemia Cells Exhibit Upregulation of Integrin Signaling That is Modulated by NOTCH1 Mutations. Blood (2014) 123(26):4101–10. doi: 10.1182/blood-2014-01-552307
62. Hutterer E, Asslaber D, Caldana C, Krenn PW, Zucchetto A, Gattei V, et al. CD18 (ITGB2) Expression in Chronic Lymphocytic Leukaemia is Regulated by DNA Methylation-Dependent and -Independent Mechanisms. Br J haematology (2015) 169(2):286–9. doi: 10.1111/bjh.13188
63. Hauser MA, Schaeuble K, Kindinger I, Impellizzieri D, Krueger WA, Hauck CR, et al. Inflammation-Induced CCR7 Oligomers Form Scaffolds to Integrate Distinct Signaling Pathways for Efficient Cell Migration. Immunity (2016) 44(1):59–72. doi: 10.1016/j.immuni.2015.12.010
64. Redondo-Munoz J, Jose Terol M, Garcia-Marco JA, Garcia-Pardo A. Matrix Metalloproteinase-9 is Up-Regulated by CCL21/CCR7 Interaction via Extracellular Signal-Regulated Kinase-1/2 Signaling and is Involved in CCL21-Driven B-Cell Chronic Lymphocytic Leukemia Cell Invasion and Migration. Blood (2008) 111(1):383–6. doi: 10.1182/blood-2007-08-107300
65. Zucchetto A, Cattarossi I, Nanni P, Zaina E, Prato G, Gilestro M, et al. Cluster Analysis of Immunophenotypic Data: The Example of Chronic Lymphocytic Leukemia. Immunol Lett (2011) 134(2):137–44. doi: 10.1016/j.imlet.2010.09.017
66. Zucchetto A, Caldana C, Benedetti D, Tissino E, Rossi FM, Hutterer E, et al. CD49d is Overexpressed by Trisomy 12 Chronic Lymphocytic Leukemia Cells: Evidence for a Methylation-Dependent Regulation Mechanism. Blood (2013) 122(19):3317–21. doi: 10.1182/blood-2013-06-507335
67. Brachtl G, Sahakyan K, Denk U, Girbl T, Alinger B, Hofbauer SW, et al. Differential Bone Marrow Homing Capacity of VLA-4 and CD38 High Expressing Chronic Lymphocytic Leukemia Cells. PloS One (2011) 6(8):e23758. doi: 10.1371/journal.pone.0023758
68. Girbl T, Hinterseer E, Grossinger EM, Asslaber D, Oberascher K, Weiss L, et al. CD40-Mediated Activation of Chronic Lymphocytic Leukemia Cells Promotes Their CD44-Dependent Adhesion to Hyaluronan and Restricts CCL21-Induced Motility. Cancer Res (2013) 73(2):561–70. doi: 10.1158/0008-5472.CAN-12-2749
69. Catusse J, Leick M, Groch M, Clark DJ, Buchner MV, Zirlik K, et al. Role of the Atypical Chemoattractant Receptor CRAM in Regulating CCL19 Induced CCR7 Responses in B-Cell Chronic Lymphocytic Leukemia. Mol Cancer (2010) 9:297. doi: 10.1186/1476-4598-9-297
70. Ticchioni M, Essafi M, Jeandel PY, Davi F, Cassuto JP, Deckert M, et al. Homeostatic Chemokines Increase Survival of B-Chronic Lymphocytic Leukemia Cells Through Inactivation of Transcription Factor FOXO3a. Oncogene (2007) 26(50):7081–91. doi: 10.1038/sj.onc.1210519
71. Pham TH, Okada T, Matloubian M, Lo CG, Cyster JG. S1P1 Receptor Signaling Overrides Retention Mediated by G Alpha I-Coupled Receptors to Promote T Cell Egress. Immunity (2008) 28(1):122–33. doi: 10.1016/j.immuni.2007.11.017
72. Borge M, Remes Lenicov F, Nannini PR, de los Rios Alicandu MM, Podaza E, Ceballos A, et al. The Expression of Sphingosine-1 Phosphate Receptor-1 in Chronic Lymphocytic Leukemia Cells is Impaired by Tumor Microenvironmental Signals and Enhanced by Piceatannol and R406. J Immunol (2014) 193(6):3165–74. doi: 10.4049/jimmunol.1400547
73. Cameron F, Sanford M. Ibrutinib: First Global Approval. Drugs (2014) 74(2):263–71. doi: 10.1007/s40265-014-0178-8
74. Herrmann A, Hoster E, Zwingers T, Brittinger G, Engelhard M, Meusers P, et al. Improvement of Overall Survival in Advanced Stage Mantle Cell Lymphoma. J Clin Oncol (2009) 27(4):511–8. doi: 10.1200/JCO.2008.16.8435
75. Barista I, Romaguera JE, Cabanillas F. Mantle-Cell Lymphoma. Lancet Oncol (2001) 2(3):141–8. doi: 10.1016/S1470-2045(00)00255-2
76. Argatoff LH, Connors JM, Klasa RJ, Horsman DE, Gascoyne RD. Mantle Cell Lymphoma: A Clinicopathologic Study of 80 Cases. Blood (1997) 89(6):2067–78. doi: 10.1182/blood.V89.6.2067
77. Perez-Galan P, Dreyling M, Wiestner A. Mantle Cell Lymphoma: Biology, Pathogenesis, and the Molecular Basis of Treatment in the Genomic Era. Blood (2011) 117(1):26–38. doi: 10.1182/blood-2010-04-189977
78. Corcione A, Arduino N, Ferretti E, Raffaghello L, Roncella S, Rossi D, et al. CCL19 and CXCL12 Trigger In Vitro Chemotaxis of Human Mantle Cell Lymphoma B Cells. Clin Cancer Res (2004) 10(3):964–71. doi: 10.1158/1078-0432.CCR-1182-3
79. Bryson MS. The Role of Chemokines and Their Receptors in Non-Hodgkin’s Lymphoma. Glasgow, Scotland, United Kingdom: PhD Thesis University of Glasgow (2011).
80. Ek S, Hogerkorp CM, Dictor M, Ehinger M, Borrebaeck CA. Mantle Cell Lymphomas Express a Distinct Genetic Signature Affecting Lymphocyte Trafficking and Growth Regulation as Compared With Subpopulations of Normal Human B Cells. Cancer Res (2002) 62(15):4398–405.
81. Friedberg JW, Taylor MD, Cerhan JR, Flowers CR, Dillon H, Farber CM, et al. Follicular Lymphoma in the United States: First Report of the National LymphoCare Study. J Clin Oncol (2009) 27(8):1202–8. doi: 10.1200/JCO.2008.18.1495
82. Nugent A, Proia RL. The Role of G Protein-Coupled Receptors in Lymphoid Malignancies. Cell Signal (2017) 39:95–107. doi: 10.1016/j.cellsig.2017.08.002
83. Bowman EP, Campbell JJ, Soler D, Dong Z, Manlongat N, Picarella D, et al. Developmental Switches in Chemokine Response Profiles During B Cell Differentiation and Maturation. J Exp Med (2000) 191(8):1303–18. doi: 10.1084/jem.191.8.1303
84. Brune V, Tiacci E, Pfeil I, Döring C, Eckerle S, van Noesel CJM, et al. Origin and Pathogenesis of Nodular Lymphocyte-Predominant Hodgkin Lymphoma as Revealed by Global Gene Expression Analysis. J Exp Med (2008) 205(10):2251–68. doi: 10.1084/jem.20080809
85. Compagno M, Lim WK, Grunn A, Nandula SV, Brahmachary M, Shen Q, et al. Mutations of Multiple Genes Cause Deregulation of NF-kappaB in Diffuse Large B-Cell Lymphoma. Nature (2009) 459(7247):717–21. doi: 10.1038/nature07968
86. Rosenwald A, Alizadeh AA, Widhopf G, Simon R, Davis RE, Yu X, et al. Relation of Gene Expression Phenotype to Immunoglobulin Mutation Genotype in B Cell Chronic Lymphocytic Leukemia. J Exp Med (2001) 194(11):1639–47. doi: 10.1084/jem.194.11.1639
87. Storz MN, van de Rijn M, Kim YH, Mraz-Gernhard S, Hoppe RT, Kohler S. Gene Expression Profiles of Cutaneous B Cell Lymphoma. J Invest Dermatol (2003) 120(5):865–70. doi: 10.1046/j.1523-1747.2003.12142.x
88. Braza MS, Caraux A, Rousset T, Lafaye de Micheaux S, Sicard H, Squiban P, et al. Gammadelta T Lymphocytes Count is Normal and Expandable in Peripheral Blood of Patients With Follicular Lymphoma, Whereas it is Decreased in Tumor Lymph Nodes Compared With Inflammatory Lymph Nodes. J Immunol (Baltimore Md 1950) (2010) 184(1):134–40. doi: 10.4049/jimmunol.0901980
89. Maier S, Santak M, Mantik A, Grabusic K, Kremmer E, Hammerschmidt W, et al. A Somatic Knockout of CBF1 in a Human B-Cell Line Reveals That Induction of CD21 and CCR7 by EBNA-2 is Strictly CBF1 Dependent and That Downregulation of Immunoglobulin M is Partially CBF1 Independent. J Virol (2005) 79(14):8784–92. doi: 10.1128/JVI.79.14.8784-8792.2005
90. Zabel BA, Lewén S, Berahovich RD, Jaén JC, Schall TJ. The Novel Chemokine Receptor CXCR7 Regulates Trans-Endothelial Migration of Cancer Cells. Mol Cancer (2011) 10:73. doi: 10.1186/1476-4598-10-73
91. Wu L, Ehlin-Henriksson B, Zhou X, Zhu H, Ernberg I, Kis LL, et al. Epstein-Barr Virus (EBV) Provides Survival Factors to EBV(+) Diffuse Large B-Cell Lymphoma (DLBCL) Lines and Modulates Cytokine Induced Specific Chemotaxis in EBV(+) DLBCL. Immunology (2017) 152(4):562–73. doi: 10.1111/imm.12792
92. Alizadeh AA, Eisen MB, Davis RE, Ma C, Lossos IS, Rosenwald A, et al. Distinct Types of Diffuse Large B-Cell Lymphoma Identified by Gene Expression Profiling. Nature (2000) 403(6769):503–11. doi: 10.1038/35000501
93. Du H, Zhang L, Li G, Liu W, Tang W, Zhang H, et al. CXCR4 and CCR7 Expression in Primary Nodal Diffuse Large B-Cell Lymphoma-A Clinical and Immunohistochemical Study. Am J Med Sci (2019) 357(4):302–10. doi: 10.1016/j.amjms.2019.01.008
94. Gebauer N, Künstner A, Ketzer J, Witte HM, Rausch T, Benes V, et al. Genomic Insights Into the Pathogenesis of Epstein-Barr Virus-Associated Diffuse Large B-Cell Lymphoma by Whole-Genome and Targeted Amplicon Sequencing. Blood Cancer J (2021) 11(5):102. doi: 10.1038/s41408-021-00493-5
95. Deutsch AJ, Aigelsreiter A, Steinbauer E, Fruhwirth M, Kerl H, Beham-Schmid C, et al. Distinct Signatures of B-Cell Homeostatic and Activation-Dependent Chemokine Receptors in the Development and Progression of Extragastric MALT Lymphomas. J Pathol (2008) 215(4):431–44. doi: 10.1002/path.2372
96. Deutsch AJ, Steinbauer E, Hofmann NA, Strunk D, Gerlza T, Beham-Schmid C, et al. Chemokine Receptors in Gastric MALT Lymphoma: Loss of CXCR4 and Upregulation of CXCR7 is Associated With Progression to Diffuse Large B-Cell Lymphoma. Mod Pathol (2013) 26(2):182–94. doi: 10.1038/modpathol.2012.134
97. Kocks JR, Adler H, Danzer H, Hoffmann K, Jonigk D, Lehmann U, et al. Chemokine Receptor CCR7 Contributes to a Rapid and Efficient Clearance of Lytic Murine Gamma-Herpes Virus 68 From the Lung, Whereas Bronchus-Associated Lymphoid Tissue Harbors Virus During Latency. J Immunol (2009) 182(11):6861–9. doi: 10.4049/jimmunol.0801826
98. Kasuya A, Fujiyama T, Shirahama S, Hashizume H, Tokura Y. Decreased Expression of Homeostatic Chemokine Receptors in Intravascular Large B-Cell Lymphoma. Eur J Dermatol (2012) 22(2):272–3. doi: 10.1684/ejd.2012.1639
99. Rehm A, Anagnostopoulos I, Gerlach K, Broemer M, Scheidereit C, Johrens K, et al. Identification of a Chemokine Receptor Profile Characteristic for Mediastinal Large B-Cell Lymphoma. Int J Cancer (2009) 125(10):2367–74. doi: 10.1002/ijc.24652
100. Li S, Huang S, Peng SB. Overexpression of G Protein-Coupled Receptors in Cancer Cells: Involvement in Tumor Progression. Int J Oncol (2005) 27(5):1329–39. doi: 10.3892/ijo.27.5.1329
101. Jahnke K, Coupland SE, Na IK, Loddenkemper C, Keilholz U, Korfel A, et al. Expression of the Chemokine Receptors CXCR4, CXCR5, and CCR7 in Primary Central Nervous System Lymphoma. Blood (2005) 106(1):384–5. doi: 10.1182/blood-2005-01-0324
102. Lemma SA, Pasanen AK, Haapasaari K-M, Sippola A, Sormunen R, Soini Y, et al. Similar Chemokine Receptor Profiles in Lymphomas With Central Nervous System Involvement - Possible Biomarkers for Patient Selection for Central Nervous System Prophylaxis, a Retrospective Study. Eur J haematology (2016) 96(5):492–501. doi: 10.1111/ejh.12626
103. Han CH, Batchelor TT. Diagnosis and Management of Primary Central Nervous System Lymphoma. Cancer (2017) 123(22):4314–24. doi: 10.1002/cncr.30965
104. O’Connor T, Zhou X, Kosla J, Adili A, Garcia Beccaria M, Kotsiliti E, et al. Age-Related Gliosis Promotes Central Nervous System Lymphoma Through CCL19-Mediated Tumor Cell Retention. Cancer Cell (2019) 36(3):250–67.e9. doi: 10.1016/j.ccell.2019.08.001
105. Barone F, Bombardieri M, Rosado MM, Morgan PR, Challacombe SJ, De Vita S, et al. CXCL13, CCL21, and CXCL12 Expression in Salivary Glands of Patients With Sjogren’s Syndrome and MALT Lymphoma: Association With Reactive and Malignant Areas of Lymphoid Organization. J Immunol (Baltimore Md 1950) (2008) 180(7):5130–40. doi: 10.4049/jimmunol.180.7.5130
106. Grever MR, Blachly JS, Andritsos LA. Hairy Cell Leukemia: Update on Molecular Profiling and Therapeutic Advances. Blood Rev (2014) 28(5):197–203. doi: 10.1016/j.blre.2014.06.003
107. Basso K, Liso A, Tiacci E, Benedetti R, Pulsoni A, Foa R, et al. Gene Expression Profiling of Hairy Cell Leukemia Reveals a Phenotype Related to Memory B Cells With Altered Expression of Chemokine and Adhesion Receptors. J Exp Med (2004) 199(1):59–68. doi: 10.1084/jem.20031175
108. Agnelli L, Mosca L, Fabris S, Lionetti M, Andronache A, Kwee I, et al. A SNP Microarray and FISH-Based Procedure to Detect Allelic Imbalances in Multiple Myeloma: An Integrated Genomics Approach Reveals a Wide Gene Dosage Effect. Genes Chromosomes Cancer (2009) 48(7):603–14. doi: 10.1002/gcc.20668
109. Trentin L, Miorin M, Facco M, Baesso I, Carraro S, Cabrelle A, et al. Multiple Myeloma Plasma Cells Show Different Chemokine Receptor Profiles at Sites of Disease Activity. Br J Haematol (2007) 138(5):594–602. doi: 10.1111/j.1365-2141.2007.06686.x
110. Purdue MP, Lan Q, Menashe I, Zheng T, Zhang Y, Yeager M, et al. Variation in Innate Immunity Genes and Risk of Multiple Myeloma. Hematol Oncol (2011) 29(1):42–6. doi: 10.1002/hon.954
111. Zhan F, Hardin J, Kordsmeier B, Bumm K, Zheng M, Tian E, et al. Global Gene Expression Profiling of Multiple Myeloma, Monoclonal Gammopathy of Undetermined Significance, and Normal Bone Marrow Plasma Cells. Blood (2002) 99(5):1745–57. doi: 10.1182/blood.V99.5.1745
112. Zhan F, Barlogie B, Arzoumanian V, Huang Y, Williams DR, Hollmig K, et al. Gene-Expression Signature of Benign Monoclonal Gammopathy Evident in Multiple Myeloma is Linked to Good Prognosis. Blood (2007) 109(4):1692–700. doi: 10.1182/blood-2006-07-037077
113. Swerdlow SHC, E., Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J, et al. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th ed Vol. 2008. Geneva, Switzerland: World Health Organization (2008) p. 323–5.
114. Liu Y, Sattarzadeh A, Diepstra A, Visser L, van den Berg A. The Microenvironment in Classical Hodgkin Lymphoma: An Actively Shaped and Essential Tumor Component. Semin Cancer Biol (2014) 24:15–22. doi: 10.1016/j.semcancer.2013.07.002
115. Hopken UE, Foss HD, Meyer D, Hinz M, Leder K, Stein H, et al. Up-Regulation of the Chemokine Receptor CCR7 in Classical But Not in Lymphocyte-Predominant Hodgkin Disease Correlates With Distinct Dissemination of Neoplastic Cells in Lymphoid Organs. Blood (2002) 99(4):1109–16. doi: 10.1182/blood.V99.4.1109
116. Eckerle S, Brune V, Döring C, Tiacci E, Bohle V, Sundström C, et al. Gene Expression Profiling of Isolated Tumour Cells From Anaplastic Large Cell Lymphomas: Insights Into its Cellular Origin, Pathogenesis and Relation to Hodgkin Lymphoma. Leukemia (2009) 23(11):2129–38. doi: 10.1038/leu.2009.161
117. Mathas S, Hinz M, Anagnostopoulos I, Krappmann D, Lietz A, Jundt F, et al. Aberrantly Expressed C-Jun and JunB Are a Hallmark of Hodgkin Lymphoma Cells, Stimulate Proliferation and Synergize With NF-Kappa B. EMBO J (2002) 21(15):4104–13. doi: 10.1093/emboj/cdf389
118. Bleul CC, Wu L, Hoxie JA, Springer TA, Mackay CR. The HIV Coreceptors CXCR4 and CCR5 Are Differentially Expressed and Regulated on Human T Lymphocytes. Proc Natl Acad Sci USA (1997) 94(5):1925–30. doi: 10.1073/pnas.94.5.1925
119. Linke F, Harenberg M, Nietert MM, Zaunig S, von Bonin F, Arlt A, et al. Microenvironmental Interactions Between Endothelial and Lymphoma Cells: A Role for the Canonical WNT Pathway in Hodgkin Lymphoma. Leukemia (2017) 31(2):361–72. doi: 10.1038/leu.2016.232
120. Zhong Z, Yu J, Virshup DM, Madan B. Wnts and the Hallmarks of Cancer. Cancer Metastasis Rev (2020) 39(3):625–45. doi: 10.1007/s10555-020-09887-6
121. Machado L, Jarrett R, Morgan S, Murray P, Hunter B, Hamilton E, et al. Expression and Function of T Cell Homing Molecules in Hodgkin’s Lymphoma. Cancer Immunol Immunother (2009) 58(1):85–94. doi: 10.1007/s00262-008-0528-z
122. Atayar C, Poppema S, Visser L, van den Berg A. Cytokine Gene Expression Profile Distinguishes CD4+/CD57+ T Cells of the Nodular Lymphocyte Predominance Type of Hodgkin’s Lymphoma From Their Tonsillar Counterparts. J Pathol (2006) 208(3):423–30. doi: 10.1002/path.1894
123. Bosler DS, Douglas-Nikitin VK, Harris VN, Smith MD. Detection of T-Regulatory Cells has a Potential Role in the Diagnosis of Classical Hodgkin Lymphoma. Cytometry B Clin Cytom (2008) 74(4):227–35. doi: 10.1002/cyto.b.20407
124. Quesada AE, Assylbekova B, Jabcuga CE, Zhang R, Covinsky M, Rios A, et al. Expression of Sirt1 and FoxP3 in Classical Hodgkin Lymphoma and Tumor Infiltrating Lymphocytes: Implications for Immune Dysregulation, Prognosis and Potential Therapeutic Targeting. Int J Clin Exp Pathol (2015) 8(10):13241–8.
125. Greaves P, Clear A, Owen A, Iqbal S, Lee A, Matthews J, et al. Defining Characteristics of Classical Hodgkin Lymphoma Microenvironment T-Helper Cells. Blood (2013) 122(16):2856–63. doi: 10.1182/blood-2013-06-508044
126. Wu R, Sattarzadeh A, Rutgers B, Diepstra A, van den Berg A, Visser L. The Microenvironment of Classical Hodgkin Lymphoma: Heterogeneity by Epstein-Barr Virus Presence and Location Within the Tumor. Blood Cancer J (2016) 6:e417. doi: 10.1038/bcj.2016.26
127. Visser L, Wu R, Rutgers B, Diepstra A, van den Berg A. Characterization of the Microenvironment of Nodular Lymphocyte Predominant Hodgkin Lymphoma. Int J Mol Sci (2016) 17(12):pii:E2127. doi: 10.3390/ijms17122127
128. Moura J, Rodrigues J, Santos AH, Teixeira Mdos A, Queiros ML, Santos M, et al. Chemokine Receptor Repertoire Reflects Mature T-Cell Lymphoproliferative Disorder Clinical Presentation. Blood Cells Mol Dis (2009) 42(1):57–63. doi: 10.1016/j.bcmd.2008.08.002
129. Foucar K. Mature T-Cell Leukemias Including T-Prolymphocytic Leukemia, Adult T-Cell Leukemia/Lymphoma, and Sezary Syndrome. Am J Clin Pathol (2007) 127(4):496–510. doi: 10.1309/KWJYBCCGTB90B6AE
130. Cuesta-Mateos C, Fuentes P, Schrader A, Juárez-Sánchez R, Loscertales J, Mateu-Albero T, et al. CCR7 as a Novel Therapeutic Target in T-Cell PROLYMPHOCYTIC Leukemia. Biomark Res (2020) 8:54. doi: 10.1186/s40364-020-00234-z
131. Aifantis I, Raetz E, Buonamici S. Molecular Pathogenesis of T-Cell Leukaemia and Lymphoma. Nat Rev Immunol (2008) 8(5):380–90. doi: 10.1038/nri2304
132. Pui CH, Howard SC. Current Management and Challenges of Malignant Disease in the CNS in Paediatric Leukaemia. Lancet Oncol (2008) 9(3):257–68. doi: 10.1016/S1470-2045(08)70070-6
133. Lee K, Nam KT, Cho SH, Gudapati P, Hwang Y, Park D-S, et al. Vital Roles of mTOR Complex 2 in Notch-Driven Thymocyte Differentiation and Leukemia. J Exp Med (2012) 209(4):713–28. doi: 10.1084/jem.20111470
134. Ma S, Shi Y, Pang Y, Dong F, Cheng H, Hao S, et al. Notch1-Induced T Cell Leukemia can be Potentiated by Microenvironmental Cues in the Spleen. J Hematol Oncol (2014) 7:71. doi: 10.1186/s13045-014-0071-7
135. Jost TR, Borga C, Radaelli E, Romagnani A, Perruzza L, Omodho L, et al. Role of CXCR4-Mediated Bone Marrow Colonization in CNS Infiltration by T Cell Acute Lymphoblastic Leukemia. J Leukoc Biol (2016) 99(6):1077–87. doi: 10.1189/jlb.5MA0915-394R
136. Matutes E, Brito-Babapulle V, Swansbury J, Ellis J, Morilla R, Dearden C, et al. Clinical and Laboratory Features of 78 Cases of T-Prolymphocytic Leukemia. Blood (1991) 78(12):3269–74. doi: 10.1182/blood.V78.12.3269.3269
137. Dearden C. How I Treat Prolymphocytic Leukemia. Blood (2012) 120(3):538–51. doi: 10.1182/blood-2012-01-380139
138. Bellone M, Svensson AM, Zaslav AL, Spitzer S, Golightly M, Celiker M, et al. Pediatric T-Cell Prolymphocytic Leukemia With an Isolated 12(P13) Deletion and Aberrant CD117 Expression. Exp Hematol Oncol (2012) 1(1):7. doi: 10.1186/2162-3619-1-7
139. Hopfinger G, Busch R, Pflug N, Weit N, Westermann A, Fink AM, et al. Sequential Chemoimmunotherapy of Fludarabine, Mitoxantrone, and Cyclophosphamide Induction Followed by Alemtuzumab Consolidation is Effective in T-Cell Prolymphocytic Leukemia. Cancer (2013) 119(12):2258–67. doi: 10.1002/cncr.27972
140. Ravandi F, O’Brien S, Jones D, Lerner S, Faderl S, Ferrajoli A, et al. T-Cell Prolymphocytic Leukemia: A Single-Institution Experience. Clin Lymphoma myeloma (2005) 6(3):234–9. doi: 10.3816/CLM.2005.n.051
141. Herling M, Khoury JD, Washington LT, Duvic M, Keating MJ, Jones D. A Systematic Approach to Diagnosis of Mature T-Cell Leukemias Reveals Heterogeneity Among WHO Categories. Blood (2004) 104(2):328–35. doi: 10.1182/blood-2004-01-0002
142. Bennett JM, Catovsky D, Daniel MT, Flandrin G, Galton DA, Gralnick HR, et al. Proposals for the Classification of Chronic (Mature) B and T Lymphoid Leukaemias. French-American-British (FAB) Cooperative Group. J Clin Pathol (1989) 42(6):567–84. doi: 10.1136/jcp.42.6.567
143. Schrader A, Crispatzu G, Oberbeck S, Mayer P, Putzer S, von Jan J, et al. Actionable Perturbations of Damage Responses by TCL1/ATM and Epigenetic Lesions Form the Basis of T-PLL. Nat Commun (2018) 9(1):697. doi: 10.1038/s41467-017-02688-6
144. Dürig J, Bug S, Klein-Hitpass L, Boes T, Jöns T, Martin-Subero JI, et al. Combined Single Nucleotide Polymorphism-Based Genomic Mapping and Global Gene Expression Profiling Identifies Novel Chromosomal Imbalances, Mechanisms and Candidate Genes Important in the Pathogenesis of T-Cell Prolymphocytic Leukemia With Inv(14)(Q11q32). Leukemia (2007) 21(10):2153–63. doi: 10.1038/sj.leu.2404877
145. Oberbeck S, Schrader A, Warner K, Jungherz D, Crispatzu G, von Jan J, et al. Non-Canonical Effector Functions of the T-Memory-Like T-PLL Cell Are Shaped by Cooperative TCL1A and TCR Signaling. Blood (2020) 107(10):2786–08. doi: 10.1182/blood.2019003348
146. Despouy G, Joiner M, Le Toriellec E, Weil R, Stern MH. The TCL1 Oncoprotein Inhibits Activation-Induced Cell Death by Impairing PKCtheta and ERK Pathways. Blood (2007) 110(13):4406–16. doi: 10.1182/blood-2006-11-059501
147. Herling M, Patel KA, Teitell MA, Konopleva M, Ravandi F, Kobayashi R, et al. High TCL1 Expression and Intact T-Cell Receptor Signaling Define a Hyperproliferative Subset of T-Cell Prolymphocytic Leukemia. Blood (2008) 111(1):328–37. doi: 10.1182/blood-2007-07-101519
148. Graham RL, Cooper B, Krause JR. T-Cell Prolymphocytic Leukemia. Proc (Bayl Univ Med Cent) (2013) 26(1):19–21. doi: 10.1080/08998280.2013.11928902
149. Iwanaga M, Watanabe T, Yamaguchi K. Adult T-Cell Leukemia: A Review of Epidemiological Evidence. Front Microbiol (2012) 3:322. doi: 10.3389/fmicb.2012.00322
150. Ishitsuka K, Tamura K. Human T-Cell Leukaemia Virus Type I and Adult T-Cell Leukaemia-Lymphoma. Lancet Oncol (2014) 15(11):e517–26. doi: 10.1016/S470-2045(14)70202-5[doi]LID-S1470-2045(14)-5[pii
151. Katsuya H, Ishitsuka K, Utsunomiya A, Hanada S, Eto T, Moriuchi Y, et al. Treatment and Survival Among 1594 Patients With ATL. Blood (2015) 126(24):2570–7. doi: 10.1182/blood-2015-03-632489
152. Watanabe T. Adult T-Cell Leukemia: Molecular Basis for Clonal Expansion and Transformation of HTLV-1-Infected T Cells. Blood (2017) 129(9):1071–81. doi: 10.1182/blood-2016-09-692574
153. Dahmoush L, Hijazi Y, Barnes E, Stetler-Stevenson M, Abati A. Adult T-Cell Leukemia/Lymphoma: A Cytopathologic, Immunocytochemical, and Flow Cytometric Study. Cancer (2002) 96(2):110–6. doi: 10.1002/cncr.10480
154. Kagdi HH, Demontis MA, Fields PA, Ramos JC, Bangham CR, Taylor GP. Risk Stratification of Adult T-Cell Leukemia/Lymphoma Using Immunophenotyping. Cancer Med (2017) 6(1):298–309. doi: 10.1002/cam4.928
155. Yoshie O, Fujisawa R, Nakayama T, Harasawa H, Tago H, Izawa D, et al. Frequent Expression of CCR4 in Adult T-Cell Leukemia and Human T-Cell Leukemia Virus Type 1-Transformed T Cells. Blood (2002) 99(5):1505–11. doi: 10.1182/blood.V99.5.1505
156. Toulza F, Nosaka K, Takiguchi M, Pagliuca T, Mitsuya H, Tanaka Y, et al. FoxP3+ Regulatory T Cells Are Distinct From Leukemia Cells in HTLV-1-Associated Adult T-Cell Leukemia. Int J Cancer (2009) 125(10):2375–82. doi: 10.1002/ijc.24664
157. Hasegawa H, Nomura T, Kohno M, Tateishi N, Suzuki Y, Maeda N, et al. Increased Chemokine Receptor CCR7/EBI1 Expression Enhances the Infiltration of Lymphoid Organs by Adult T-Cell Leukemia Cells. Blood (2000) 95(1):30–8. doi: 10.1182/blood.V95.1.30.001k09_30_38
158. Kohno T, Moriuchi R, Katamine S, Yamada Y, Tomonaga M, Matsuyama T. Identification of Genes Associated With the Progression of Adult T Cell Leukemia (ATL). Jpn J Cancer Res (2000) 91(11):1103–10. doi: 10.1111/j.1349-7006.2000.tb00892.x
159. Choi YL, Tsukasaki K, O’Neill MC, Yamada Y, Onimaru Y, Matsumoto K, et al. A Genomic Analysis of Adult T-Cell Leukemia. Oncogene (2007) 26(8):1245–55. doi: 10.1038/sj.onc.1209898
160. Harasawa H, Yamada Y, Hieshima K, Jin Z, Nakayama T, Yoshie O, et al. Survey of Chemokine Receptor Expression Reveals Frequent Co-Expression of Skin-Homing CCR4 and CCR10 in Adult T-Cell Leukemia/Lymphoma. Leuk Lymphoma (2006) 47(10):2163–73. doi: 10.1080/10428190600775599
161. Hashikawa K, Yasumoto S, Nakashima K, Arakawa F, Kiyasu J, Kimura Y, et al. Microarray Analysis of Gene Expression by Microdissected Epidermis and Dermis in Mycosis Fungoides and Adult T-Cell Leukemia/Lymphoma. Int J Oncol (2014) 45(3):1200–8. doi: 10.3892/ijo.2014.2524
162. Kataoka K, Nagata Y, Kitanaka A, Shiraishi Y, Shimamura T, Yasunaga J, et al. Integrated Molecular Analysis of Adult T Cell Leukemia/Lymphoma. Nat Genet (2015) 47(11):1304–15. doi: 10.1038/ng.3415
163. Nakagawa M, Schmitz R, Xiao W, Goldman CK, Xu W, Yang Y, et al. Gain-Of-Function CCR4 Mutations in Adult T Cell Leukemia/Lymphoma. J Exp Med (2014) 211(13):2497–505. doi: 10.1084/jem.20140987
164. Sakihama S, Morichika K, Saito R, Miyara M, Miyagi T, Hayashi M, et al. Genetic Profile of Adult T-Cell Leukemia/Lymphoma in Okinawa: Association With Prognosis, Ethnicity, and HTLV-1 Strains. Cancer Sci (2021) 112(3):1300–9. doi: 10.1111/cas.14806
165. Kallinich T, Muche JM, Qin S, Sterry W, Audring H, Kroczek RA. Chemokine Receptor Expression on Neoplastic and Reactive T Cells in the Skin at Different Stages of Mycosis Fungoides. J Invest Dermatol (2003) 121(5):1045–52. doi: 10.1046/j.1523-1747.2003.12555.x
166. Scarisbrick JJ, Prince HM, Vermeer MH, Quaglino P, Horwitz S, Porcu P, et al. Cutaneous Lymphoma International Consortium Study of Outcome in Advanced Stages of Mycosis Fungoides and Sézary Syndrome: Effect of Specific Prognostic Markers on Survival and Development of a Prognostic Model. J Clin Oncol Off J Am Soc Clin Oncol (2015) 33(32):3766–73. doi: 10.1200/JCO.2015.61.7142
167. Olsen EA, Whittaker S, Kim YH, Duvic M, Prince HM, Lessin SR, et al. Clinical End Points and Response Criteria in Mycosis Fungoides and Sézary Syndrome: A Consensus Statement of the International Society for Cutaneous Lymphomas, the United States Cutaneous Lymphoma Consortium, and the Cutaneous Lymphoma Task Force of the European Organisation for Research and Treatment of Cancer. J Clin Oncol Off J Am Soc Clin Oncol (2011) 29(18):2598–607. doi: 10.1200/JCO.2010.32.0630
168. Wilcox RA. Cutaneous T-Cell Lymphoma: 2017 Update on Diagnosis, Risk-Stratification, and Management. Am J Hematol (2017) 92(10):1085–102. doi: 10.1002/ajh.24876
169. Cristofoletti C, Bresin A, Picozza M, Picchio MC, Monzo F, Helmer Citterich M, et al. Blood and Skin-Derived Sezary Cells: Differences in Proliferation-Index, Activation of PI3K/AKT/mTORC1 Pathway and its Prognostic Relevance. Leukemia (2019) 33(5):1231–42. doi: 10.1038/s41375-018-0305-8
170. Geskin LJ, Viragova S, Stolz DB, Fuschiotti P. Interleukin-13 is Overexpressed in Cutaneous T-Cell Lymphoma Cells and Regulates Their Proliferation. Blood (2015) 125(18):2798–805. doi: 10.1182/blood-2014-07-590398
171. Wang T, Lu Y, Polk A, Chowdhury P, Murga-Zamalloa C, Fujiwara H, et al. T-Cell Receptor Signaling Activates an ITK/NF-κb/GATA-3 Axis in T-Cell Lymphomas Facilitating Resistance to Chemotherapy. Clin Cancer Res an Off J Am Assoc Cancer Res (2017) 23(10):2506–15. doi: 10.1158/1078-0432.CCR-16-1996
172. Rindler K, Bauer WM, Jonak C, Wielscher M, Shaw LE, Rojahn TB, et al. Single-Cell RNA Sequencing Reveals Tissue Compartment-Specific Plasticity of Mycosis Fungoides Tumor Cells. Front Immunol (2021) 12:666935. doi: 10.3389/fimmu.2021.666935
173. Hu SC, Lin CL, Hong CH, Yu HS, Chen GS, Lee CH. CCR7 Expression Correlates With Subcutaneous Involvement in Mycosis Fungoides Skin Lesions and Promotes Migration of Mycosis Fungoides Cells (MyLa) Through mTOR Activation. J Dermatol Sci (2016) 74(1):31–8. doi: 10.1016/j.jdermsci.2013.12.003
174. Hong C-H, Lin S-H, Lee C-H. CCL21 Induces mTOR-Dependent MALAT1 Expression, Leading to Cell Migration in Cutaneous T-Cell Lymphoma. In Vivo (Athens Greece) (2019) 33(3):793–800. doi: 10.21873/invivo.11541
175. Zhou Y, Xu X, Lv H, Wen Q, Li J, Tan L, et al. The Long Noncoding RNA MALAT-1 Is Highly Expressed in Ovarian Cancer and Induces Cell Growth and Migration. PloS One (2016) 11(5):e0155250. doi: 10.1371/journal.pone.0155250
176. Shin J, Monti S, Aires DJ, Duvic M, Golub T, Jones DA, et al. Lesional Gene Expression Profiling in Cutaneous T-Cell Lymphoma Reveals Natural Clusters Associated With Disease Outcome. Blood (2007) 110(8):3015–27. doi: 10.1182/blood-2006-12-061507
177. van Doorn R, van Kester MS, Dijkman R, Vermeer MH, Mulder AA, Szuhai K, et al. Oncogenomic Analysis of Mycosis Fungoides Reveals Major Differences With Sezary Syndrome. Blood (2009) 113(1):127–36. doi: 10.1182/blood-2008-04-153031
178. Campbell JJ, Clark RA, Watanabe R, Kupper TS. Sezary Syndrome and Mycosis Fungoides Arise From Distinct T-Cell Subsets: A Biologic Rationale for Their Distinct Clinical Behaviors. Blood (2010) 116(5):767–71. doi: 10.1182/blood-2009-11-251926
179. Capriotti E, Vonderheid EC, Thoburn CJ, Bright EC, Hess AD. Chemokine Receptor Expression by Leukemic T Cells of Cutaneous T-Cell Lymphoma: Clinical and Histopathological Correlations. J Invest Dermatol (2007) 127(12):2882–92. doi: 10.1038/sj.jid.5700916
180. Morice WG, Katzmann JA, Pittelkow MR, el-Azhary RA, Gibson LE, Hanson CA. A Comparison of Morphologic Features, Flow Cytometry, TCR-Vbeta Analysis, and TCR-PCR in Qualitative and Quantitative Assessment of Peripheral Blood Involvement by Sezary Syndrome. Am J Clin Pathol (2006) 125(3):364–74. doi: 10.1309/25E9Y7RRAY84HTAT
181. Sokolowska-Wojdylo M, Wenzel J, Gaffal E, Lenz J, Speuser P, Erdmann S, et al. Circulating Clonal CLA(+) and CD4(+) T Cells in Sezary Syndrome Express the Skin-Homing Chemokine Receptors CCR4 and CCR10 as Well as the Lymph Node-Homing Chemokine Receptor CCR7. Br J Dermatol (2005) 152(2):258–64. doi: 10.1111/j.1365-2133.2004.06325.x
182. Narducci MG, Scala E, Bresin A, Caprini E, Picchio MC, Remotti D, et al. Skin Homing of Sezary Cells Involves SDF-1-CXCR4 Signaling and Down-Regulation of CD26/dipeptidylpeptidase Iv. Blood (2006) 107(3):1108–15. doi: 10.1182/blood-2005-04-1492
183. Picchio MC, Scala E, Pomponi D, Caprini E, Frontani M, Angelucci I, et al. CXCL13 is Highly Produced by Sezary Cells and Enhances Their Migratory Ability via a Synergistic Mechanism Involving CCL19 and CCL21 Chemokines. Cancer Res (2008) 68(17):7137–46. doi: 10.1158/0008-5472.CAN-08-0602
184. Yang J, Wang S, Zhao G, Sun B. Effect of Chemokine Receptors CCR7 on Disseminated Behavior of Human T Cell Lymphoma: Clinical and Experimental Study. J Exp Clin Cancer Res (2011) 30:51. doi: 10.1186/1756-9966-30-51
185. Zeng H, Chi H. mTOR and Lymphocyte Metabolism. Curr Opin Immunol (2013) 25(3):347–55. doi: 10.1016/j.coi.2013.05.002
186. Lima M, Almeida J, Dos Anjos Teixeira M, Alguero Md Mdel C, Santos AH, Balanzategui A, et al. TCRalphabeta+/CD4+ Large Granular Lymphocytosis: A New Clonal T-Cell Lymphoproliferative Disorder. Am J Pathol (2003) 163(2):763–71. doi: 10.1016/S0002-9440(10)63703-0
187. Wlodarski MW, Schade AE, Maciejewski JP. T-Large Granular Lymphocyte Leukemia: Current Molecular Concepts. Hematology (2006) 11(4):245–56. doi: 10.1080/10245330600774793
188. Caperton C, Agrawal S, Gupta S. Good Syndrome Presenting With CD8⁺ T-Cell Large Granular Lymphocyte Leukemia. Oncotarget (2015) 6(34):36577–86. doi: 10.18632/oncotarget.5369
189. Lamant L, de Reynies A, Duplantier MM, Rickman DS, Sabourdy F, Giuriato S, et al. Gene-Expression Profiling of Systemic Anaplastic Large-Cell Lymphoma Reveals Differences Based on ALK Status and Two Distinct Morphologic ALK+ Subtypes. Blood (2007) 109(5):2156–64. doi: 10.1182/blood-2006-06-028969
190. Geissinger E, Bonzheim I, Krenacs L, Roth S, Reimer P, Wilhelm M, et al. Nodal Peripheral T-Cell Lymphomas Correspond to Distinct Mature T-Cell Populations. J Pathol (2006) 210(2):172–80. doi: 10.1002/path.2046
191. Piccaluga PP, Agostinelli C, Califano A, Rossi M, Basso K, Zupo S, et al. Gene Expression Analysis of Peripheral T Cell Lymphoma, Unspecified, Reveals Distinct Profiles and New Potential Therapeutic Targets. J Clin Invest (2007) 117(3):823–34. doi: 10.1172/JCI26833
192. Suzuki R, Suzumiya J, Nakamura S, Aoki S, Notoya A, Ozaki S, et al. Aggressive Natural Killer-Cell Leukemia Revisited: Large Granular Lymphocyte Leukemia of Cytotoxic NK Cells. Leukemia (2004) 18(4):763–70. doi: 10.1038/sj.leu.2403262
193. Makishima H, Ito T, Asano N, Nakazawa H, Shimodaira S, Kamijo Y, et al. Significance of Chemokine Receptor Expression in Aggressive NK Cell Leukemia. Leukemia (2005) 19(7):1169–74. doi: 10.1038/sj.leu.2403732
194. Sand KE, Rye KP, Mannsåker B, Bruserud O, Kittang AO. Expression Patterns of Chemokine Receptors on Circulating T Cells From Myelodysplastic Syndrome Patients. Oncoimmunology (2013) 2(2):e23138. doi: 10.4161/onci.23138
195. Voso MT, Ottone T, Lavorgna S, Venditti A, Maurillo L, Lo-Coco F, et al. MRD in AML: The Role of New Techniques. Front Oncol (2019) 9:655. doi: 10.3389/fonc.2019.00655
196. Stegmaier K, Ross KN, Colavito SA, O’Malley S, Stockwell BR, Golub TR. Gene Expression-Based High-Throughput Screening(GE-HTS) and Application to Leukemia Differentiation. Nat Genet (2004) 36(3):257–63. doi: 10.1038/ng1305
197. Valk PJM, Verhaak RGW, Beijen MA, Erpelinck CAJ, Barjesteh van Waalwijk van Doorn-Khosrovani S, Boer JM, et al. Prognostically Useful Gene-Expression Profiles in Acute Myeloid Leukemia. N Engl J Med (2004) 350(16):1617–28. doi: 10.1056/NEJMoa040465
198. Maiga A, Lemieux S, Pabst C, Lavallée VP, Bouvier M, Sauvageau G, et al. Transcriptome Analysis of G Protein-Coupled Receptors in Distinct Genetic Subgroups of Acute Myeloid Leukemia: Identification of Potential Disease-Specific Targets. Blood Cancer J (2016) 6(6):e431. doi: 10.1038/bcj.2016.36
199. Falcone U, Sibai H, Deotare U. A Critical Review of Treatment Modalities for Blastic Plasmacytoid Dendritic Cell Neoplasm. Crit Rev Oncol/Hematol (2016) 107:156–62. doi: 10.1016/j.critrevonc.2016.09.003
200. Bendriss-Vermare N, Chaperot L, Peoc’h M, Vanbervliet B, Jacob MC, Briere F, et al. In Situ Leukemic Plasmacytoid Dendritic Cells Pattern of Chemokine Receptors Expression and In Vitro Migratory Response. Leukemia (2004) 18(9):1491–8. doi: 10.1038/sj.leu.2403452
201. Hogstad B, Berres M-L, Chakraborty R, Tang J, Bigenwald C, Serasinghe M, et al. RAF/MEK/extracellular Signal-Related Kinase Pathway Suppresses Dendritic Cell Migration and Traps Dendritic Cells in Langerhans Cell Histiocytosis Lesions. J Exp Med (2018) 215(1):319–36. doi: 10.1084/jem.20161881
202. Hehlmann R. Chronic Myeloid Leukemia in 2020. Hemasphere (2020) 4(5):e468. doi: 10.1097/HS9.0000000000000468
203. Kubo K, Iwakami M, Muromoto R, Inagaki T, Kitai Y, Kon S, et al. CCR7 is Involved in BCR-ABL/STAP-2-Mediated Cell Growth in Hematopoietic Ba/F3 Cells. Biochem Biophys Res Commun (2015) 463(4):825–31. doi: 10.1016/j.bbrc.2015.06.020
204. Sekine Y, Ikeda O, Mizushima A, Ueno Y, Muromoto R, Yoshimura A, et al. STAP-2 Interacts With and Modulates BCR-ABL-Mediated Tumorigenesis. Oncogene (2012) 31(40):4384–96. doi: 10.1038/onc.2011.604
205. Jongen-Lavrencic M, Salesse S, Delwel R, Verfaillie CM. BCR/ABL-Mediated Downregulation of Genes Implicated in Cell Adhesion and Motility Leads to Impaired Migration Toward CCR7 Ligands CCL19 and CCL21 in Primary BCR/ABL-Positive Cells. Leukemia (2005) 19(3):373–80. doi: 10.1038/sj.leu.2403626
206. Ben-Baruch A. Organ Selectivity in Metastasis: Regulation by Chemokines and Their Receptors. Clin Exp Metastasis (2008) 25(4):345–56. doi: 10.1007/s10585-007-9097-3
207. Swerdlow SH, Campo E, Pileri SA, Harris NL, Stein H, Siebert R, et al. The 2016 Revision of the World Health Organization Classification of Lymphoid Neoplasms. Blood (2016) 127(20):2375–90. doi: 10.1182/blood-2016-01-643569
208. D’Amico G, Frascaroli G, Bianchi G, Transidico P, Doni A, Vecchi A, et al. Uncoupling of Inflammatory Chemokine Receptors by IL-10: Generation of Functional Decoys. Nat Immunol (2000) 1(5):387–91. doi: 10.1038/80819
209. Luczynski W, Stasiak-Barmuta A, Piszcz J, Ilendo E, Kowalczuk O, Krawczuk-Rybak M. B-Cell Chronic Lymphocytic Leukemia-Derived Dendritic Cells Stimulate Allogeneic T-Cell Response and Express Chemokines Involved in T-Cell Migration. Neoplasma (2007) 54(6):527–35.
210. Xie P, Kraus ZJ, Stunz LL, Bishop GA. Roles of TRAF Molecules in B Lymphocyte Function. Cytokine Growth Factor Rev (2008) 19(3-4):199–207. doi: 10.1016/j.cytogfr.2008.04.002
211. Pei L, Choi JH, Liu J, Lee EJ, McCarthy B, Wilson JM, et al. Genome-Wide DNA Methylation Analysis Reveals Novel Epigenetic Changes in Chronic Lymphocytic Leukemia. Epigenetics (2012) 7(6):567–78. doi: 10.4161/epi.20237
212. Yan Y, Chen R, Wang X, Hu K, Huang L, Lu M, et al. CCL19 and CCR7 Expression, Signaling Pathways, and Adjuvant Functions in Viral Infection and Prevention. Front Cell Dev Biol (2019) 7:212. doi: 10.3389/fcell.2019.00212
213. Ehlin-Henriksson B, Liang W, Cagigi A, Mowafi F, Klein G, Nilsson A. Changes in Chemokines and Chemokine Receptor Expression on Tonsillar B Cells Upon Epstein-Barr Virus Infection. Immunology (2009) 127(4):549–57. doi: 10.1111/j.1365-2567.2008.03029.x
214. Potsch C, Vohringer D, Pircher H. Distinct Migration Patterns of Naive and Effector CD8 T Cells in the Spleen: Correlation With CCR7 Receptor Expression and Chemokine Reactivity. Eur J Immunol (1999) 29(11):3562–70. doi: 10.1002/(SICI)1521-4141(199911)29:11<3562::AID-IMMU3562>3.0.CO;2-R
215. Ramirez PW, Famiglietti M, Sowrirajan B, DePaula-Silva AB, Rodesch C, Barker E, et al. Downmodulation of CCR7 by HIV-1 Vpu Results in Impaired Migration and Chemotactic Signaling Within CD4⁺ T Cells. Cell Rep (2014) 7(6):2019–30. doi: 10.1016/j.celrep.2014.05.015
216. Debes GF, Bonhagen K, Wolff T, Kretschmer U, Krautwald S, Kamradt T, et al. CC Chemokine Receptor 7 Expression by Effector/Memory CD4+ T Cells Depends on Antigen Specificity and Tissue Localization During Influenza A Virus Infection. J Virol (2004) 78(14):7528–35. doi: 10.1128/JVI.78.14.7528-7535.2004
217. Hayasaka H, Kobayashi D, Yoshimura H, Nakayama EE, Shioda T, Miyasaka M. The HIV-1 Gp120/CXCR4 Axis Promotes CCR7 Ligand-Dependent CD4 T Cell Migration: CCR7 Homo- and CCR7/CXCR4 Hetero-Oligomer Formation as a Possible Mechanism for Up-Regulation of Functional CCR7. PloS One (2015) 10(2):e0117454. doi: 10.1371/journal.pone.0117454
218. Saleh S, Lu HK, Evans V, Harisson D, Zhou J, Jaworowski A, et al. HIV Integration and the Establishment of Latency in CCL19-Treated Resting CD4(+) T Cells Require Activation of NF-κb. Retrovirology (2016) 13(1):49. doi: 10.1186/s12977-016-0284-7
219. Spender LC, Lucchesi W, Bodelon G, Bilancio A, Karstegl CE, Asano T, et al. Cell Target Genes of Epstein-Barr Virus Transcription Factor EBNA-2: Induction of the P55alpha Regulatory Subunit of PI3-Kinase and its Role in Survival of EREB2.5 Cells. J Gen Virol (2006) 87(Pt 10):2859–67. doi: 10.1099/vir.0.82128-0
220. Nakayama T, Fujisawa R, Izawa D, Hieshima K, Takada K, Yoshie O. Human B Cells Immortalized With Epstein-Barr Virus Upregulate CCR6 and CCR10 and Downregulate CXCR4 and CXCR5. J Virol (2002) 76(6):3072–7. doi: 10.1128/JVI.76.6.3072-3077.2002
221. Hopken UE, Rehm A. Homeostatic Chemokines Guide Lymphoma Cells to Tumor Growth-Promoting Niches Within Secondary Lymphoid Organs. J Mol Med (Berl) (2012) 90(11):1237–45. doi: 10.1007/s00109-012-0906-z
222. Pals ST, de Gorter DJ, Spaargaren M. Lymphoma Dissemination: The Other Face of Lymphocyte Homing. Blood (2007) 110(9):3102–11. doi: 10.1182/blood-2007-05-075176
223. Link A, Vogt TK, Favre S, Britschgi MR, Acha-Orbea H, Hinz B, et al. Fibroblastic Reticular Cells in Lymph Nodes Regulate the Homeostasis of Naive T Cells. Nat Immunol (2007) 8(11):1255–65. doi: 10.1038/ni1513
224. Schneider MA, Meingassner JG, Lipp M, Moore HD, Rot A. CCR7 Is Required for the In Vivo Function of CD4+ CD25+ Regulatory T Cells. J Exp Med (2007) 204(4):735–45. doi: 10.1084/jem.20061405
225. Shields JD, Kourtis IC, Tomei AA, Roberts JM, Swartz MA. Induction of Lymphoidlike Stroma and Immune Escape by Tumors That Express the Chemokine CCL21. Science (2010) 328(5979):749–52. doi: 10.1126/science.1185837
226. Yang ZZ, Novak AJ, Stenson MJ, Witzig TE, Ansell SM. Intratumoral CD4+CD25+ Regulatory T-Cell-Mediated Suppression of Infiltrating CD4+ T Cells in B-Cell Non-Hodgkin Lymphoma. Blood (2006) 107(9):3639–46. doi: 10.1182/blood-2005-08-3376
227. Strauss L, Bergmann C, Szczepanski M, Gooding W, Johnson JT, Whiteside TL. A Unique Subset of CD4+CD25highFoxp3+ T Cells Secreting Interleukin-10 and Transforming Growth Factor-Beta1 Mediates Suppression in the Tumor Microenvironment. Clin Cancer Res (2007) 13(15 Pt 1):4345–54. doi: 10.1158/1078-0432.CCR-07-0472
228. Alvaro T, Lejeune M, Salvado MT, Bosch R, Garcia JF, Jaen J, et al. Outcome in Hodgkin’s Lymphoma can be Predicted From the Presence of Accompanying Cytotoxic and Regulatory T Cells. Clin Cancer Res (2005) 11(4):1467–73. doi: 10.1158/1078-0432.CCR-04-1869
229. Beyer M, Kochanek M, Darabi K, Popov A, Jensen M, Endl E, et al. Reduced Frequencies and Suppressive Function of CD4+CD25hi Regulatory T Cells in Patients With Chronic Lymphocytic Leukemia After Therapy With Fludarabine. Blood (2005) 106(6):2018–25. doi: 10.1182/blood-2005-02-0642
230. Beyer M, Kochanek M, Giese T, Endl E, Weihrauch MR, Knolle PA, et al. In Vivo Peripheral Expansion of Naive CD4+CD25high FOXP3+ Regulatory T Cells in Patients With Multiple Myeloma. Blood (2006) 107(10):3940–9. doi: 10.1182/blood-2005-09-3671
231. Mittal S, Marshall NA, Duncan L, Culligan DJ, Barker RN, Vickers MA. Local and Systemic Induction of CD4+CD25+ Regulatory T-Cell Population by Non-Hodgkin Lymphoma. Blood (2008) 111(11):5359–70. doi: 10.1182/blood-2007-08-105395
232. Lee NR, Song EK, Jang KY, Choi HN, Moon WS, Kwon K, et al. Prognostic Impact of Tumor Infiltrating FOXP3 Positive Regulatory T Cells in Diffuse Large B-Cell Lymphoma at Diagnosis. Leuk Lymphoma (2008) 49(2):247–56. doi: 10.1080/10428190701824536
233. Lefkowitz RJ, Shenoy SK. Transduction of Receptor Signals by Beta-Arrestins. Science (2005) 308(5721):512–7. doi: 10.1126/science.1109237
234. Steen A, Larsen O, Thiele S, Rosenkilde MM. Biased and G Protein-Independent Signaling of Chemokine Receptors. Front Immunol (2014) 5:277. doi: 10.3389/fimmu.2014.00277
235. Till KJ, Spiller DG, Harris RJ, Chen H, Zuzel M, Cawley JC. CLL. But Not Normal, B Cells Are Dependent on Autocrine VEGF and Alpha4beta1 Integrin for Chemokine-Induced Motility on and Through Endothelium. Blood (2005) 105(12):4813–9. doi: 10.1182/blood-2004-10-4054
236. McHeik S, Van Eeckhout N, De Poorter C, Galés C, Parmentier M, Springael J-Y. Coexpression of CCR7 and CXCR4 During B Cell Development Controls CXCR4 Responsiveness and Bone Marrow Homing. Front Immunol (2019) 10:2970. doi: 10.3389/fimmu.2019.02970
237. Shannon LA, McBurney TM, Wells MA, Roth ME, Calloway PA, Bill CA, et al. CCR7/CCL19 Controls Expression of EDG-1 in T Cells. J Biol Chem (2012) 287(15):11656–64. doi: 10.1074/jbc.M111.310045
238. Okada T, Ngo VN, Ekland EH, Forster R, Lipp M, Littman DR, et al. Chemokine Requirements for B Cell Entry to Lymph Nodes and Peyer’s Patches. J Exp Med (2002) 196(1):65–75. doi: 10.1084/jem.20020201
239. Muller G, Lipp M. Shaping Up Adaptive Immunity: The Impact of CCR7 and CXCR5 on Lymphocyte Trafficking. Microcirculation (2003) 10(3-4):325–34. doi: 10.1080/mic.10.3-4.325.334
240. Hopken UE, Achtman AH, Kruger K, Lipp M. Distinct and Overlapping Roles of CXCR5 and CCR7 in B-1 Cell Homing and Early Immunity Against Bacterial Pathogens. J Leukoc Biol (2004) 76(3):709–18. doi: 10.1189/jlb.1203643
241. Velaga S, Herbrand H, Friedrichsen M, Jiong T, Dorsch M, Hoffmann MW, et al. Chemokine Receptor CXCR5 Supports Solitary Intestinal Lymphoid Tissue Formation, B Cell Homing, and Induction of Intestinal IgA Responses. J Immunol (2009) 182(5):2610–9. doi: 10.4049/jimmunol.0801141
242. Warnock RA, Campbell JJ, Dorf ME, Matsuzawa A, McEvoy LM, Butcher EC. The Role of Chemokines in the Microenvironmental Control of T Versus B Cell Arrest in Peyer’s Patch High Endothelial Venules. J Exp Med (2000) 191(1):77–88. doi: 10.1084/jem.191.1.77
243. Ebisuno Y, Tanaka T, Kanemitsu N, Kanda H, Yamaguchi K, Kaisho T, et al. Cutting Edge: The B Cell Chemokine CXC Chemokine Ligand 13/B Lymphocyte Chemoattractant is Expressed in the High Endothelial Venules of Lymph Nodes and Peyer’s Patches and Affects B Cell Trafficking Across High Endothelial Venules. J Immunol (2003) 171(4):1642–6. doi: 10.4049/jimmunol.171.4.1642
244. Cuesta-Mateos C, Juárez-Sánchez R, Mateu-Albero T, Loscertales J, Mol W, Terrón F, et al. Targeting Cancer Homing Into the Lymph Node With a Novel Anti-CCR7 Therapeutic Antibody: The Paradigm of CLL. mAbs (2021) 13(1):1917484. doi: 10.1080/19420862.2021.1917484
245. Mateu-Albero T, Juárez-Sánchez R, Loscertales J, Mol W, Terrón F, Muñoz-Calleja C, et al. Effect of Ibrutinib on CCR7 Expression and Functionality in Chronic Lymphocytic Leukemia and its Implication for the Activity of CAP-100, a Novel Therapeutic Anti-CCR7 Antibody. Cancer Immunol Immunother CII. (2021). doi: 10.1007/s00262-021-03014-2
246. Fiorcari S, Brown WS, McIntyre BW, Estrov Z, Maffei R, O’Brien S, et al. The PI3-Kinase Delta Inhibitor Idelalisib (GS-1101) Targets Integrin-Mediated Adhesion of Chronic Lymphocytic Leukemia (CLL) Cell to Endothelial and Marrow Stromal Cells. PloS One (2013) 8(12):e83830. doi: 10.1371/journal.pone.0083830
247. Ping L, Ding N, Shi Y, Feng L, Li J, Liu Y, et al. The Bruton’s Tyrosine Kinase Inhibitor Ibrutinib Exerts Immunomodulatory Effects Through Regulation of Tumor-Infiltrating Macrophages. Oncotarget (2017) 8(24):39218–29. doi: 10.18632/oncotarget.16836
248. Lafouresse F, Groom JR. A Task Force Against Local Inflammation and Cancer: Lymphocyte Trafficking to and Within the Skin. Front Immunol (2018) 9:2454. doi: 10.3389/fimmu.2018.02454
249. Harjunpää H, Llort Asens M, Guenther C, Fagerholm SC. Cell Adhesion Molecules and Their Roles and Regulation in the Immune and Tumor Microenvironment. Front Immunol (2019) 10:1078. doi: 10.3389/fimmu.2019.01078
250. Reina M, Espel E. Role of LFA-1 and ICAM-1 in Cancer. Cancers (Basel) (2017) 9(11):153. doi: 10.20944/preprints201709.0146.v1
251. San Sebastián E, Zimmerman T, Zubia A, Vara Y, Martin E, Sirockin F, et al. Design, Synthesis, and Functional Evaluation of Leukocyte Function Associated Antigen-1 Antagonists in Early and Late Stages of Cancer Development. J Medicinal Chem (2013) 56(3):735–47. doi: 10.1021/jm3016848
252. Lin Y-C, Shun C-T, Wu M-S, Chen C-C. A Novel Anticancer Effect of Thalidomide: Inhibition of Intercellular Adhesion Molecule-1-Mediated Cell Invasion and Metastasis Through Suppression of Nuclear Factor-Kappab. Clin Cancer Res an Off J Am Assoc Cancer Res (2006) 12(23):7165–73. doi: 10.1158/1078-0432.CCR-06-1393
253. Yu JA, Sadaria MR, Meng X, Mitra S, Ao L, Fullerton DA, et al. Lung Cancer Cell Invasion and Expression of Intercellular Adhesion Molecule-1 (ICAM-1) Are Attenuated by Secretory Phospholipase A₂ Inhibition. J Thorac Cardiovasc Surg (2012) 143(2):405–11. doi: 10.1016/j.jtcvs.2011.10.026
254. Laufer JM, Legler DF. Beyond Migration-Chemokines in Lymphocyte Priming, Differentiation, and Modulating Effector Functions. J Leukoc Biol (2018) 104(2):301–12. doi: 10.1002/JLB.2MR1217-494R
255. Ma J, Wang JH, Guo YJ, Sy MS, Bigby M. In Vivo Treatment With Anti-ICAM-1 and Anti-LFA-1 Antibodies Inhibits Contact Sensitization-Induced Migration of Epidermal Langerhans Cells to Regional Lymph Nodes. Cell Immunol (1994) 158(2):389–99. doi: 10.1006/cimm.1994.1285
256. Yanguas A, Garasa S, Teijeira Á, Aubá C, Melero I, Rouzaut A. ICAM-1-LFA-1 Dependent CD8+ T-Lymphocyte Aggregation in Tumor Tissue Prevents Recirculation to Draining Lymph Nodes. Front Immunol (2018) 9:2084. doi: 10.3389/fimmu.2018.02084
257. Schmits R, Kündig TM, Baker DM, Shumaker G, Simard JJ, Duncan G, et al. LFA-1-Deficient Mice Show Normal CTL Responses to Virus But Fail to Reject Immunogenic Tumor. J Exp Med (1996) 183(4):1415–26. doi: 10.1084/jem.183.4.1415
258. Mukai S, Kagamu H, Shu S, Plautz GE. Critical Role of CD11a (LFA-1) in Therapeutic Efficacy of Systemically Transferred Antitumor Effector T Cells. Cell Immunol (1999) 192(2):122–32. doi: 10.1006/cimm.1998.1439
259. Seminara NM, Gelfand JM. Assessing Long-Term Drug Safety: Lessons (Re) Learned From Raptiva. Semin Cutaneous Med Surg (2010) 29(1):16–9. doi: 10.1016/j.sder.2010.01.001
260. Talamonti M, Spallone G, Di Stefani A, Costanzo A, Chimenti S. Efalizumab. Expert Opin Drug Saf (2011) 10(2):239–51. doi: 10.1517/14740338.2011.524925
261. Cohen S, Haimovich J, Hollander N. Anti-Idiotype X Anti-LFA-1 Bispecific Antibodies Inhibit Metastasis of B Cell Lymphoma. J Immunol (Baltimore Md 1950) (2003) 170(5):2695–701. doi: 10.4049/jimmunol.170.5.2695
262. Moschovakis GL, Forster R. Multifaceted Activities of CCR7 Regulate T-Cell Homeostasis in Health and Disease. Eur J Immunol (2012) 42(8):1949–55. doi: 10.1002/eji.201242614
263. Moschovakis GL, Bubke A, Dittrich-Breiholz O, Braun A, Prinz I, Kremmer E, et al. Deficient CCR7 Signaling Promotes TH2 Polarization and B-Cell Activation In Vivo. Eur J Immunol (2012) 42(1):48–57. doi: 10.1002/eji.201141753
264. Vassileva G, Soto H, Zlotnik A, Nakano H, Kakiuchi T, Hedrick JA, et al. The Reduced Expression of 6Ckine in the Plt Mouse Results From the Deletion of One of Two 6Ckine Genes. J Exp Med (1999) 190(8):1183–8. doi: 10.1084/jem.190.8.1183
265. Luther SA, Tang HL, Hyman PL, Farr AG, Cyster JG. Coexpression of the Chemokines ELC and SLC by T Zone Stromal Cells and Deletion of the ELC Gene in the Plt/Plt Mouse. Proc Natl Acad Sci USA (2000) 97(23):12694–9. doi: 10.1073/pnas.97.23.12694
266. Junt T, Scandella E, Forster R, Krebs P, Krautwald S, Lipp M, et al. Impact of CCR7 on Priming and Distribution of Antiviral Effector and Memory CTL. J Immunol (2004) 173(11):6684–93. doi: 10.4049/jimmunol.173.11.6684
267. Junt T, Nakano H, Dumrese T, Kakiuchi T, Odermatt B, Zinkernagel RM, et al. Antiviral Immune Responses in the Absence of Organized Lymphoid T Cell Zones in Plt/Plt Mice. J Immunol (2002) 168(12):6032–40. doi: 10.4049/jimmunol.168.12.6032
268. Kursar M, Hopken UE, Koch M, Kohler A, Lipp M, Kaufmann SH, et al. Differential Requirements for the Chemokine Receptor CCR7 in T Cell Activation During Listeria Monocytogenes Infection. J Exp Med (2005) 201(9):1447–57. doi: 10.1084/jem.20041204
269. Scandella E, Fink K, Junt T, Senn BM, Lattmann E, Forster R, et al. Dendritic Cell-Independent B Cell Activation During Acute Virus Infection: A Role for Early CCR7-Driven B-T Helper Cell Collaboration. J Immunol (2007) 178(3):1468–76. doi: 10.4049/jimmunol.178.3.1468
270. Aritomi K, Kuwabara T, Tanaka Y, Nakano H, Yasuda T, Ishikawa F, et al. Altered Antibody Production and Helper T Cell Function in Mice Lacking Chemokines CCL19 and CCL21-Ser. Microbiol Immunol (2010) 54(11):691–701. doi: 10.1111/j.1348-0421.2010.00266.x
271. Hartoonian C, Sepehrizadeh Z, Tabatabai Yazdi M, Jang YS, Langroudi L, Amir Kalvanagh P, et al. Enhancement of Immune Responses by Co-Delivery of CCL19/MIP-3beta Chemokine Plasmid With HCV Core DNA/Protein Immunization. Hepat Mon (2014) 14(3):e14611. doi: 10.5812/hepatmon.14611
272. Hu K, Luo S, Tong L, Huang X, Jin W, Huang W, et al. CCL19 and CCL28 Augment Mucosal and Systemic Immune Responses to HIV-1 Gp140 by Mobilizing Responsive Immunocytes Into Secondary Lymph Nodes and Mucosal Tissue. J Immunol (Baltimore Md 1950) (2013) 191(4):1935–47. doi: 10.4049/jimmunol.1300120
273. Moschovakis GL, Bubke A, Friedrichsen M, Ristenpart J, Back JW, Falk CS, et al. The Chemokine Receptor CCR7 is a Promising Target for Rheumatoid Arthritis Therapy. Cell Mol Immunol (2019) 16(10):791–9. doi: 10.1038/s41423-018-0056-5
274. Cuesta-Mateos C, Portero-Sainz I, García-Peydró M, Alcain J, Fuentes P, Juárez-Sánchez R, et al. Evaluation of Therapeutic Targeting of CCR7 in Acute Graft-Versus-Host Disease. Bone Marrow Transplant (2020) 55(10):1935–45. doi: 10.1038/s41409-020-0830-8
275. Fowler KA, Vasilieva V, Ivanova E, Rimkevich O, Sokolov A, Abbasova S, et al. R707, a Fully Human Antibody Directed Against CC-Chemokine Receptor 7, Attenuates Xenogeneic Acute Graft-Versus-Host Disease. Am J Transplant. (2019) 19(7):1941–54. doi: 10.1111/ajt.15298
276. Soudja SM, Henri S, Mello M, Chasson L, Mas A, Wehbe M, et al. Disrupted Lymph Node and Splenic Stroma in Mice With Induced Inflammatory Melanomas is Associated With Impaired Recruitment of T and Dendritic Cells. PloS One (2011) 6(7):e22639. doi: 10.1371/journal.pone.0022639
277. Thompson ED, Enriquez HL, Fu YX, Engelhard VH. Tumor Masses Support Naive T Cell Infiltration, Activation, and Differentiation Into Effectors. J Exp Med (2010) 207(8):1791–804. doi: 10.1084/jem.20092454
278. Forster R, Mattis AE, Kremmer E, Wolf E, Brem G, Lipp M. A Putative Chemokine Receptor, BLR1, Directs B Cell Migration to Defined Lymphoid Organs and Specific Anatomic Compartments of the Spleen. Cell (1996) 87(6):1037–47. doi: 10.1016/S0092-8674(00)81798-5
279. Hintzen G, Ohl L, del Rio ML, Rodriguez-Barbosa JI, Pabst O, Kocks JR, et al. Induction of Tolerance to Innocuous Inhaled Antigen Relies on a CCR7-Dependent Dendritic Cell-Mediated Antigen Transport to the Bronchial Lymph Node. J Immunol (2006) 177(10):7346–54. doi: 10.4049/jimmunol.177.10.7346
280. Worbs T, Bode U, Yan S, Hoffmann MW, Hintzen G, Bernhardt G, et al. Oral Tolerance Originates in the Intestinal Immune System and Relies on Antigen Carriage by Dendritic Cells. J Exp Med (2006) 203(3):519–27. doi: 10.1084/jem.20052016
281. Davalos-Misslitz AC, Rieckenberg J, Willenzon S, Worbs T, Kremmer E, Bernhardt G, et al. Generalized Multi-Organ Autoimmunity in CCR7-Deficient Mice. Eur J Immunol (2007) 37(3):613–22. doi: 10.1002/eji.200636656
282. Kurobe H, Liu C, Ueno T, Saito F, Ohigashi I, Seach N, et al. CCR7-Dependent Cortex-to-Medulla Migration of Positively Selected Thymocytes is Essential for Establishing Central Tolerance. Immunity (2006) 24(2):165–77. doi: 10.1016/j.immuni.2005.12.011
Keywords: CCR7, lymph node, lymphoma, leukemia, immunotherapy
Citation: Cuesta-Mateos C, Terrón F and Herling M (2021) CCR7 in Blood Cancers – Review of Its Pathophysiological Roles and the Potential as a Therapeutic Target. Front. Oncol. 11:736758. doi: 10.3389/fonc.2021.736758
Received: 05 July 2021; Accepted: 12 October 2021;
Published: 29 October 2021.
Edited by:
Mohamed A. Yassin, Hamad Medical Corporation, QatarReviewed by:
Emilia Jaskula, Hirszfeld Institute of Immunology and Experimental Therapy (PAN), PolandChristine Moussion, Genentech, Inc., United States
Lydia Scarfò, Vita-Salute San Raffaele University, Italy
Copyright © 2021 Cuesta-Mateos, Terrón and Herling. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Carlos Cuesta-Mateos, Y2FybG9zLmN1ZXN0YUBzYWx1ZC5tYWRyaWQub3Jn
†These authors have contributed equally to this work