 Xiaochen Qi
Xiaochen Qi Qifei Wang
Qifei Wang Guangzhen Wu
Guangzhen Wu
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol., 15 September 2021
Sec. Cancer Metabolism
Volume 11 - 2021 | https://doi.org/10.3389/fonc.2021.727778
Kidney cancer is a cancer with an increasing incidence in recent years. Clear cell renal cell carcinoma (ccRCC) accounts for up to 80% of all kidney cancers. The understanding of the pathogenesis, tumor progression, and metastasis of renal carcinoma is not yet perfect. Kidney cancer has some characteristics that distinguish it from other cancers, and the metabolic aspect is the most obvious. The specificity of glucose and lipid metabolism in kidney cancer cells has also led to its being studied as a metabolic disease. As the most common type of kidney cancer, ccRCC has many characteristics that represent the specificity of kidney cancer. There are features that we are very concerned about, including the presence of lipid droplets in cells and the obesity paradox. These two points are closely related to glucose metabolism and lipid metabolism. Therefore, we hope to explore whether metabolic changes affect the occurrence and development of kidney cancer by looking for evidence of changes on expression at the genomic and protein levels in glucose metabolism and lipid metabolism in ccRCC. We begin with the representative phenomenon of abnormal cancer metabolism: the Warburg effect, through the collection of popular metabolic pathways and related genes in the last decade, as well as some research hotspots, including the role of ferroptosis and glutamine in cancer, systematically elaborated the factors affecting the incidence and metastasis of kidney cancer. This review also identifies the similarities and differences between kidney cancer and other cancers in order to lay a theoretical foundation and provide a valid hypothesis for future research.
According to the Global Cancer Statistics for 2020, renal cancer is a common cancer that accounts for 2.2% of the total cancer incidence and 1.8% of the total cancer mortality (1). In recent years, the incidence of renal cancer has been increasing annually, which is closely related to the improvement in the quality of life of people in modern society. Common carcinogenic factors such as cigarette smoking, hypertension, obesity, occupation, and radiation are the main causes of renal cancer. Clear cell renal cell carcinoma (ccRCC) is the most common type of renal cell carcinoma, accounting for 75%–85% of all renal cell carcinoma patients (2).
Currently, renal cell carcinoma (RCC), including ccRCC, is generally considered a metabolic disease. An obvious feature of renal cell carcinoma is the target gene mutation involved in metabolic pathways. These include mutations in the regulatory genes involved in aerobic glycolysis, fatty acid metabolism, and tryptophan glutamine utilization (3). With the discovery of the Warburg effect 80 years ago, studies on the abnormal metabolism of cancer cells began to increase. The Warburg effect indicates that abnormal aerobic glycolysis occurs in cancer cells, producing lactic acid and pyruvate. The Warburg effect has been proven to be significantly more obvious in ccRCC than in ordinary tissues (4). The end product of pyruvate in the body is acetyl-CoA, which is the raw material for cholesterol, a type of lipid (5). Although this energy rearrangement is not present in all cancers, renal cell carcinoma is thought to be driven by metabolic changes due to the high rate of mutations in genes that control metabolism, such as von Hippel-Lindau (VHL) in the hypoxia pathway, mammalian target of rapamycin (mTOR), phosphatase and tensin homolog deleted on chromosome ten (cf), and mesenchymal epithelial transition factor in the PI3K (phosphatidylinositol-3-kinase) -Akt (protein kinase B) -mTOR pathway (6). Therefore, abnormal lipid metabolism is known to occur in renal cancer cells. A large amount of lipid accumulation in ccRCC cells provide the evidence of the abnormal lipid metabolism. Studies have shown that the contents of cholesterol, cholesterol esters, and neutral lipids (triglycerides) in ccRCC cells are much higher than those in ordinary tissues (7).In addition to differences in lipid metabolism, there are other notable differences between ccRCC and other tumors, such as the obesity paradox (8), in which obese kidney cancer patients have a higher incidence, but with a better prognosis of kidney cancer than lean people (9). Furthermore,
Recent research has shown that there is a correlation between abnormal lipid metabolism and ferroptosis. This may be due to the fact that a mechanism of ferroptosis is under the action of iron divalent or ester oxygenase, catalyzing the highly expressed unsaturated fatty acids on the cell membrane, facilitating lipid peroxidation, and inducing cell death (10). Concurrently, glutamine metabolism has long been considered to be related to the development of cancer. As a substitute for cancer cells, glutamine provides carbon sources for the tricarboxylic acid cycle (TCA) cycle and provides a large amount of raw materials for the synthesis of abnormal lipids, we believe that it is associated with ccRCC (11). Glutathione, the metabolite of glutamine, is an important participant in the body’s antioxidant system (12). Is the abnormal metabolism of glutamine related to the occurrence and development of cancer cells?
All these abnormal phenomena point to the problem of lipid metabolism in ccRCC. Therefore, this review emphasizes the relationship and significance of lipid metabolism in ccRCC in the occurrence, development, and treatment of cancer.
Mutations in genes related to aerobic glycolysis and metabolism are major risk factors for renal cell carcinoma. For example, VHL and protein 53 (P53) have been found to have mutations in RCC and their role in promoting the progression of RCC is extremely obvious (13). The decreased expression of VHL in RCC can upregulate hypoxia-induced vascular endothelial growth factor (VEGF), and VEGF can guarantee the nutritional supply of tumor cells by promoting angiogenesis in tumors (14). The tumor protein 53 (TP53) mutation also resulted in fewer renal tumor cells dying from oxidative damage (15). Both genes regulate oxidative processes in the body and directly or indirectly affect the oxidation of glucose. Metabolic rearrangement takes place in cancer cells, including the production of lactic acid by cancer cells through aerobic glycolysis, which replaces the tricarboxylic acid cycle that uses glucose in normal cells (16). This change makes cancer cells use glucose much more efficiently than normal cells, a condition known as the Warburg effect and a well-established feature of cancer energy metabolism (17) (Figure 1).
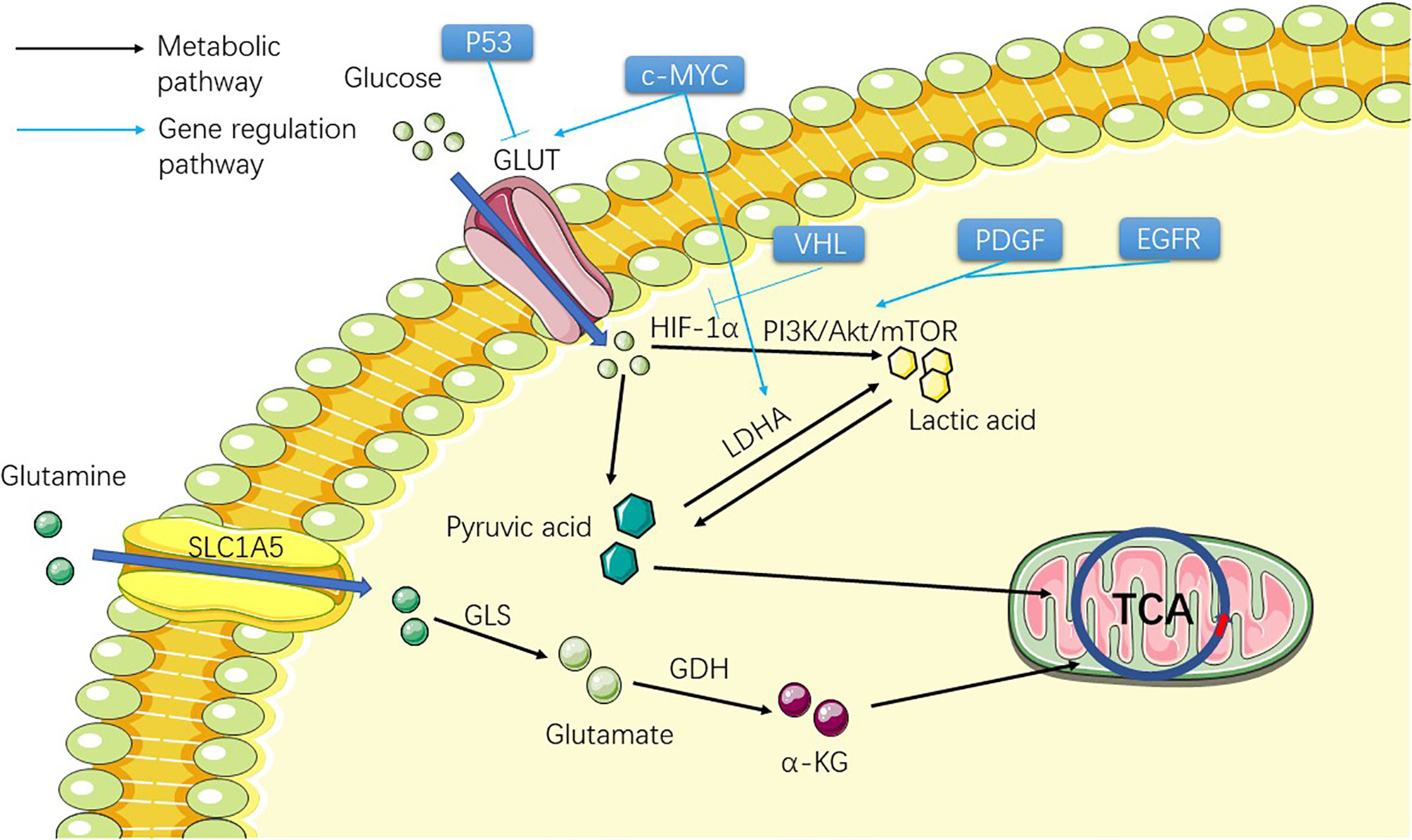
Figure 1 Figure shows the metabolic process of the substances involved in the Warburg effect in ccRCC cells before TCA, including two substances, glucose and glutamine. The picture shows the process of the two substances entering the cell from the outside of the cell and transforming them into substances that can participate in TCA through metabolism inside the cell, providing raw materials for the subsequent lipid metabolism.
Current studies have shown that this is particularly evident in ccRCC (4). Previous studies have shown a link between elevated lactic acid levels and the progression of cancer metastasis (18–20). Lactic acid also stimulates endothelial cells to produce VEGF (21). Meanwhile, lactic acid has been shown to promote cancer cell metastasis through the transforming growth factor-β2 (TGF-β2) pathway (22). Aerobic glycolysis inefficiently produces adenosine triphosphate (ATP), but confers many advantages to tumor cells. One of the products, lactic acid, can also be converted to pyruvate to provide a raw material for the synthesis of acetyl-CoA (23). This effect provides a large amount of fatty acids and cholesterol for cancer cells and a large number of raw materials that aid in cancer cells proliferation and metastasis. This process of cancer cells is regulated by many oncogenes or oncogenic pathways, such as MYC (myelocytomatosis oncogene), P53, hypoxia inducible factor-1α, 2α (HIF-1α, HIF-2α), AMPK (adenosine 5’-monophosphate-activated protein kinase)/mTOR signal pathway, and PI3K/Akt signaling pathway (24).
Glucose transporter (GLUT) mutation in ccRCC allows ccRCC cells to obtain large amounts of glucose from extracellular to support cell growth and metastasis (25). Therefore, many genes regulating GLUT family mutation are theoretically associated with the development of ccRCC. c-MYC (Table 1) (26) can directly transcriptionally activate glycolytic genes encoding glucose transporter 1 (GLUT1) (27) and lactate dehydrogenase A (LDHA) and promote abnormal glycolysis. Studies have suggested that ccRCC is related to the up-regulation of c-MYC (28). P53 (Table 1) (29) tumor suppressor directly inhibits the transcription of GLUT1 and GLUT4 genes, thereby inhibiting the uptake of grape and lactic acid production (Figure 1). The function of P53 also leads to its down-regulation related to ccRCC (30). HIF-1α (31) promotes glycolytic gene expression by binding to hypoxia response elements on glycolytic gene promoters. Under hypoxia, the degradation of the HIF-1α subunit is inhibited, and the active HIF-1 is transferred to the nucleus to regulate the transcription of many genes (Figure 1). These include many of the genes involved in tumorigenesis: VEGF, platelet-derived growth factor (PDGF), aldolase A, enolase 1, and LDHA, which regulate tumor angiogenesis and the process of abnormal glycolysis (32, 33). HIF-2α is involved in angiogenesis and several other processes and is thought to activate cancer progression (34). At present, some experiments have been conducted to inhibit ccRCC progression by inhibiting the angiogenesis of HIF-2α, and some results have been obtained (35) (Figure 1). In this study, the HIF-2 antagonist PT2399 was used to observe the expression of HIF-2 downstream proto-oncogenes (VEGF, LDHA, etc.) in tumor cells. The results showed that the expression of these downstream proto-oncogenes was significantly reduced. VHL (Table 1), which regulates HIF-1α and 2α and other types of cancer-related genes, has been shown to be extensively absent in renal cancer cells (36) (Figure 1). A review showed that 91% of patients with ccRCC had a deletion of VHL by methylation or mutation (6). Harlander et al. proved that VHL combined with TP53 and retinoblastoma susceptibility gene 1 mutation could lead to the occurrence of ccRCC in mice (13). The accumulation of HIF-α was caused by a loss of VHL. Nevertheless, HIF activation also affects ROS in two directions: 1. It affects the TCA in mitochondria to reduce ROS production; 2. Activation of downstream target gene 3 NADPH (nicotinamide adenine dinucleotide phosphate) oxidase (NOX) to increase ROS production, and increased ROS levels may also be a contributing factor to HIF stabilization during hypoxia and reoxygenation (37). A large number of HIF-α enters the nucleus and activates the transcription of HIF target genes, which affects several oncogenic pathways, including VEGF, GLUT1 (glucose transporter type I), and erythropoietin (38, 39). Hoefflin et al. demonstrated the oncogenic role of HIF-1α and HIF-2α in the initiation of ccRCC using a mouse ccRCC model and suggested that changes in the balance of HIF-1α and HIF-2α activity may influence different aspects of ccRCC biology and disease aggressiveness (40). Epidermal growth factor receptor (EGFR) (Table 1) (41), PDGF (39), and some of their downstream pathways have been shown to regulate the Warburg effect by affecting the PI3K/Akt/mTOR pathway (Figure 1). The AMPK (Table 1)/mTOR pathway is an important pathway for the regulation of cell metabolism and plays an important role in cancer cells (42, 43). Tuberous sclerosis complex subunit 1/2 (TSC1/TSC2) (Figure 2) is an important gene in this pathway, and the latter has been proven to be correlated with ccRCC through studies on the relationship between the tuberous sclerosis complex and ccRCC (44, 45). The products of aerobic glycolysis provide energy to cancer cells, while generating a large number of by-products and lactic acid (46). Glucose is broken down by glycolysis into pyruvate, which is eventually converted to acetyl-CoA (47). So far, everything seems to be consistent with what happens in normal cancer cells when a gene mutation occurs: the mutation genes that regulates GLUT causes a lot of glucose to enter the ccRCC cells. In the case of down-regulation or even disappearance of VHL expression, influenced by HIF and PI3K/Akt/mTOR pathway, glucose abnormal metabolism forms lactic acid and provides energy for cells. The outcome of lactic acid is generally to leave the cells. How does this provide raw material for the subsequent lipid metabolism.

Table 1 Upstream gene of lipid metabolism.
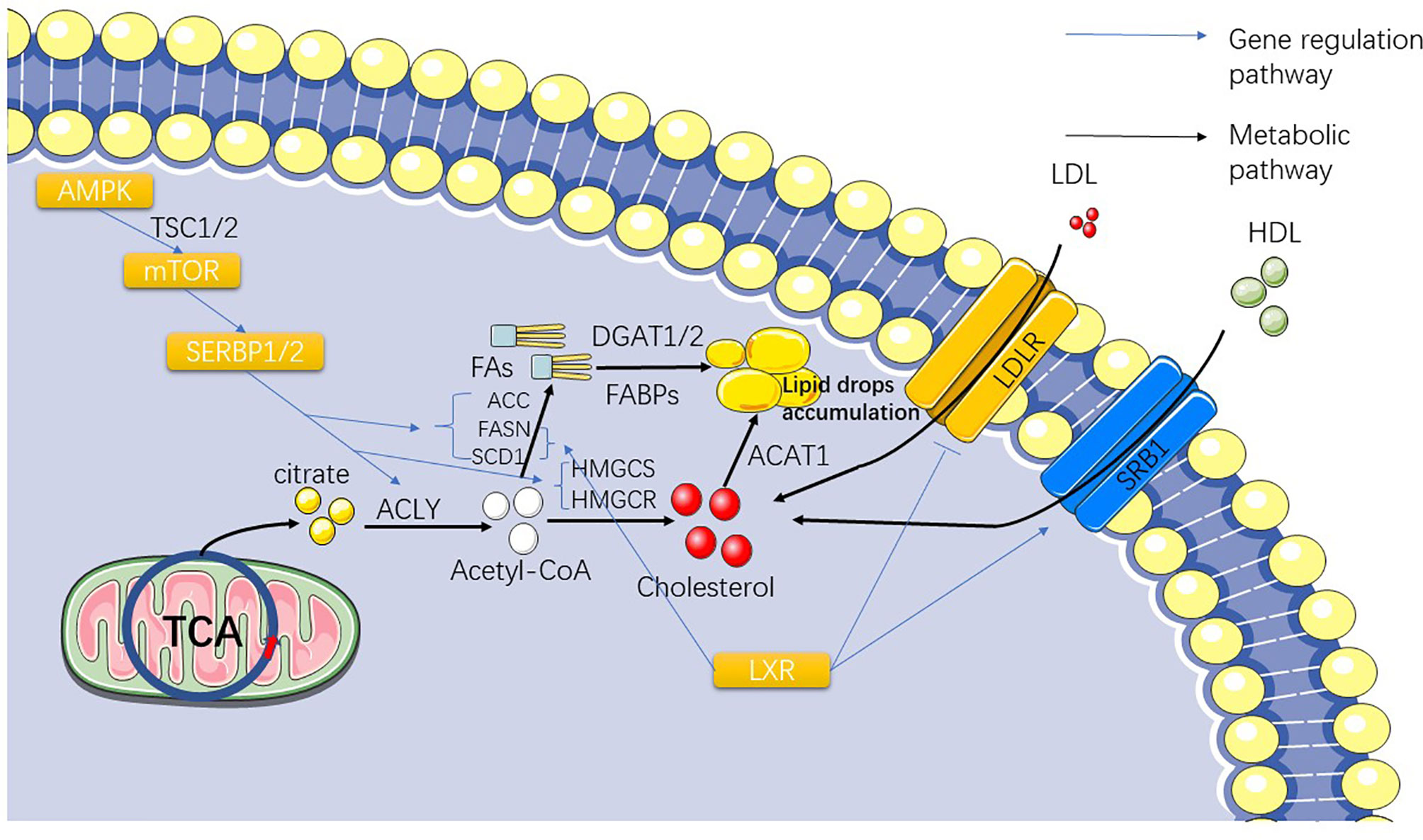
Figure 2 The figure shows the gradual conversion of citrate to lipid and cholesterol after TCA. The influence of the SERBP family and LXR on the synthesis and exocytosis of lipid acids and cholesterol are also presented.
There is a reciprocal transformation between lactic acid and pyruvic acid in ccRCC cells. LDHA catalyzes the conversion of pyruvate to lactic acid, which is a reversible reaction (Figure 1). The phosphorylation of LDHA is closely related to cancer development. Anoikis is a specific form of programmed cell death induced by the loss of contact between cells, the extracellular matrix, and other cells. It plays an important role in the homeostasis of developmental tissues in balancing disease development and tumor metastasis (48). A recent study sheds light on the role of LDHA in the development of anoikis, which leads to apoptosis in cancer cells (49). In this study, we used shRNA to inhibit LDHA activity to make cancer cells sensitive to anoikis induction. This leads to a decrease in cell invasiveness. One type of catalytic subtype of the LDHA mutant, Y10F, expresses phosphorus-deficient LDHA and has the same effect (49). Concurrently, LDHA knockout can also lead to increased mitochondrial respiration, which reduces the ability of cells to proliferate under hypoxic conditions and inhibits the occurrence of tumors (50). Previously, the influence of mitochondrial DNA copy number alteration on the occurrence and development of RCC has shown that there is higher expression of LDHA in RCC tissues than in the corresponding normal renal tissues (51), suggesting that LDHA promotes the occurrence and progression of RCC as an oncogene. Meanwhile, studies have applied LDHA inhibitors to the treatment of renal cell carcinoma and summarized the basic mechanism of treatment by observing the clinical indicators of the object. The abnormal expression of LDHA after siRNA treatment changed the expression of the cell cycle and apoptosis-related proteins and significantly reduced the migration and invasion ability of cancer cells (52). However, the results cannot directly explain the effect of LDHA on RCC. In terms of the Warburg effect of immune cells, a study on the relationship between T effector cells and Warburg in January of the same year pointed out that there is a positive feedback loop between the LDHA and the PI3K/Akt signaling pathway in the process of T effector cell maturation, which makes glycolytic ATP act as a rheostat to measure and modulate PI3K-dependent signals (53). LDHA has been shown to act as a downstream signal of oncogenes to regulate glycolysis and carcinogenesis in ccRCC (54). Therefore, its role in activating the Warburg effect in T effector cells is supported by some evidence. In view of the importance of LDHA in the Warburg effect, we believe that the orientation of LDHA in the catalytic conversion of pyruvate and lactic acid in renal cancer is the focus of current research. Both lactic acid and pyruvate are valuable for ccRCC. It includes the pathological characteristics of ccRCC cells and metastasis of ccRCC. Therefore, it is important to identify the advantages and disadvantages of the mutual transformation of the two, including early targeted therapy and the prevention of metastasis.
Notably, a new study in April 2021 offered a radically different view of cancer cell metabolism: cancer cells do not overconsume glucose (55). Glucose was found to be mainly supplied to the immune-infiltrating cells surrounding the cancer cells, while glutamine and lipids were largely delivered to the cancer cells. Fortunately, the study used ccRCC as subjects, and the results showed that the glucose supply in ccRCC was given priority to immune cells rather than cancer cells. This experiment revealed a contradictory view of the Warburg effect: there is no one type of cell that is receiving too much or too little of a nutrient; they are all programmed to take in different nutrients. The ideas are too new to be supported by other studies, but this does not mean that it cannot overturn what has been established about how cancer cells metabolize. We hope that similar studies will follow.
We know from the Warburg effect that an increased amount of acetyl-CoA is produced in cancer cells through aerobic glycolysis and how acetyl-CoA, a compound for cholesterol and lipid synthesis, turns into fatty acids and cholesterol.
With increasing evidence that the phenotype of ccRCC (and to a lesser extent its genetic profile) is similar to that of adipocytes, the theoretical basis for targeting lipid synthesis pathways has taken root (56–58). The metabolic abnormalities of the whole cancer cell go from acetyl-CoA to abnormal lipid metabolism. Fatty acid synthesis depends on acetyl-CoA, and normal cells generated through the Krebs cycle can meet the demand of basic cellular functions, but mutations of stearoyl-CoA desaturase 1 (SCD1), fatty acid synthase (FASN), and acetyl-CoA carboxylase (ACC) in ccRCC can lead to the emergence of a large number of abnormal pathways for the synthesis of acetyl-CoA and synthetic fatty acid (FA), which are ultimately stored in the form of lipid droplets in the cytoplasm (24, 59, 60) (Figure 2). This also explains the existence of lipid droplets in ccRCC cells. Therefore, in order to understand abnormal lipid metabolism in ccRCC, it is necessary to identify the genes that control abnormal fatty acid synthesis.
SCD1 is an important regulator of lipid synthesis, mainly regulated by serpine mRNA binding protein-1 (SERBP-1) (Table 1) (61) (Figure 2). SCD1 regulates the transformation of saturated fatty acids into monounsaturated fatty acids in cells and is believed to be closely related to lipid metabolism in cancer (62, 63). Currently, it has been reported that SCD1, a downstream gene of the AMPK-mTOR-SERBP1 signaling pathway, is the target gene of sorafenib (64). Similar to the role of SCD1, FASN and ACC both play important regulatory roles in lipid synthesis in ccRCC cells (Figure 2). These three genes exhibit higher mRNA expression in ccRCC cells than in normal cells, and FASN has been associated with poor prognosis in patients (65, 66). FASN dominates the synthesis of lipids in cells, and it has been considered as an important target of anticancer targeted drugs as early as 2007 (61); however, subsequent drug-related experiments and clinical applications are not satisfactory (67, 68). Studies have shown that FASN is upregulated in ccRCC, which is significantly correlated with the presence of lipid droplets in ccRCC cells (69). The study also showed that FASN is an independent predictor of poor survival in ccRCC patients, which also indicates that the upregulation of FASN plays a promoting role in the progression of ccRCC (69). Acetyl-CoA carboxylase catalyzes ATP-dependent acetyl-CoA carboxylation, a rate-limiting step in fatty acid biosynthetic (70). In 2017, studies reported the use of TGFβ-activated kinase to phosphorylate ACC to prevent the metastasis and recurrence of breast cancer (71). In 2019, a study showed that inhibiting ACC expression in HCC cells by phosphorylation and the use of ND-654, an inhibitor of ACC, can slow down cancer progression (72). Theoretically, the carboxylation of acetyl-CoA related to lipid synthesis plays a decisive role in the occurrence and development of ccRCC. We also hypothesized that ACC is correlated with ccRCC. A 2013 article reported that ACC upregulation leads to worse outcomes in patients with RCC (73). Currently, there have been reports of in vitro inhibition of RCC cell lines (786-O) by TOFA, an inhibitor of ACC, which was speculated to be related to the inhibition of the PI3K/Akt/mTOR pathway (68). Currently, treatments for ACC have not been widely used in the treatment of RCC, but we believe that ACC-related treatments are still a useful tool for the treatment of RCC.
In addition to regulating the synthesis of fatty acids by acetyl-CoA, as the precursor of acetyl-CoA synthesis, citric acid depends on a very important enzyme, ATP citrate lyase (ACLY), which synthesizes the target product (74) (Figure 2). ACLY is a downstream target of SREBPs (75) and has been found to be upregulated in a variety of cancers, including breast cancer, non-small cell lung cancer, and hepatocellular carcinoma (76, 77). Nevertheless, a bioinformatics analysis of ccRCC-associated gene pathways suggests that ACLY is associated with favorable outcomes in ccRCC patients [p<0.001 (73)]. However, after finding more research reports related to the expression level of ACLY in ccRCC, we found that most of the studies indicated that ACLY was highly expressed in ccRCC (66, 78). This result confirms that the role of ACLY in patients with ccRCC is not simple. Early inhibitors targeting ACLY have been less active and have generally been less widely used in the clinic, but a new 2019 study has broadened the approach to ACLY inhibitors (79). They observed that an allosteric site at the citric acid binding site of the ACLY inhibitor NDI-091143 increased the efficacy of the ACLY inhibitor. Although the study of ACLY in ccRCC is limited to the comparison of its gene expression levels in different samples to prove the high expression of ACLY in ccRCC samples, several clues have proved that the treatment of ACLY can be applied to the clinical treatment of ccRCC. However, from the functional analysis of ACLY, acetyl-CoA can also be converted into oxaloacetic acid (80), which indirectly leads to the inhibition of the reaction of fatty acid synthesis and may be the cause of this contradiction. Acetyl-CoA synthetase 2 (ACSS2), a conservative ribozyme that converts acetic acid into acetyl-CoA, has attracted our attention (81). ACSS2 is associated with renal cancer metastasis and poor prognosis, which has been confirmed at the genetic level (82, 83) (Figure 2). Studies have shown that the upregulation of ACSS2 expression enhances the migration and invasion of RCC cells, resulting in a poor prognosis. This is mainly achieved through the PI3K/Akt pathway. Nonetheless, a study on the relationship between ACSS2 and cancer has shown that ACSS2-mediated histone acetylation plays an important role in maintaining cell homeostasis and tumor development, providing a clearer path for the research of ACSS2 (84). It is also a new idea to explore the therapy of ccRCC and other cancers that target this gene (85, 86). Based on the above study on the application value of ACLY and ACSS2 in cancer treatment, we believe that it is highly practical to develop new treatment methods based on these two genes.
Hydroxymethylglutaryl-CoA synthase (HMGCS) and hydroxymethylglutaryl-CoA reductase (HMGCR) are synthases of the cholesterol synthesis intermediate HMG-CoA, in which HMGCR is the target gene for statins (Figure 2). Statins are mainly used in the clinical prevention of coronary heart disease and other diseases related to lipid accumulation (87). A study showed that statins have a significant therapeutic effect on VHL-deficient ccRCC (88), but a population-based case-control study showed no significant effect of long-term use of statins on the prevention of renal cancer (89). If we pay attention to the mechanisms of the two genes and the major role of cholesterol in kidney cancer independently, we can immediately assume that they are present as a risk gene in kidney cancer cells, which is indeed the case in many other cancers (90). Interestingly, after collecting and analyzing the expression data of HMGCR and HMGCS from the TCGA database in the Pan-Cancer Project, we found that they actually exist in the form of protective genes in kidney cancer, which is completely different from other cancers (91). Our previous research on this situation sheds some light.
After understanding the synthesis of cholesterol and fatty acids in ccRCC, another important question arises: Do cholesterol and fatty acids in ccRCC depend solely on the intracellular synthesis? Is there a need for extracellular uptake? Liver X receptor (Table 1) (LXR) is a nuclear transcription factor receptor that regulates a number of key molecules involved in fatty acid synthesis and cholesterol transport. LXR regulates many downstream genes related to cholesterol and lipid metabolism, including SCD-1, FASN, SREBP, LDLR, and adenosine triphosphate binding cassette transporter A1 (ABCA1) and so on (92, 93) (Figure 2). By studying the agonists of LXR and the reverse agonists SR9243 and LXR623, we found that both of them can kill ccRCC cells (93). However, their mechanisms differ. SR9243, as an LXR inhibitor, downregulates SREBP-1c, ACC, FASN, and SCD1, while other FA synthesis genes inhibit the synthesis of FA in CCRCC cells. LXR623 as an inverse agonist that upregulates the cholesterol exogenous genes ABCA1 and MYLIP, which encodes one type of E3 ubiquitin ligase, resulting in the inability of cholesterol accumulation in cancer cells. We also found an interesting phenomenon in the experiment that the expression of genes regulating fatty acid synthesis such as FASN, SCD-1, and SREBP in ccRCC was relatively increased, but the expression of important genes regulating cholesterol synthesis, such as HMGCR and HMGCS, was downregulated. We hypothesized that this might represent the high presence of cholesterol in ccRCC cells due to exogenous uptake rather than autosynthesis. Later, we found that LXR could upregulate SRB1 (responsible for HDL absorption) and downregulate LDLR (responsible for LDL absorption). It was also speculated that ccRCC guaranteed intracellular cholesterol content through a large intake of high- density lipoprotein. A recent study on the risk reduction of ccRCC by LDLR variants also demonstrated the downregulation of LDLR gene expression in ccRCC cells (94). These experimental results also showed that the production and accumulation of fatty acids and cholesterol in ccRCC were variable, and we could not treat their beneficial or harmful side in isolation. We found that in renal cell carcinoma, the diversity and uncertainty of many related regulatory genes, such as those regulating cholesterol intake, synthesis, breakdown, and excretion, makes renal cell carcinoma very different from other cancers. Similar to cholesterol, fatty acid metabolism in ccRCC is also abnormal.
Acyl-CoA cholesterol acyltransferase1 (ACAT1/SOAT1) catalyzes the transfer of acyl groups from acyl-CoA to cholesterol to produce cholesterol ester, which is the main form of cholesterol when stored in cells and transported in plasma (95) (Figure 2). A study has shown that ACAT1 mRNA and protein levels are decreased in ccRCC, and this transcriptional inactivation is significantly (84) associated with advanced pathological staging and short survival time of ccRCC (96). Another study that analyzed ACAT1 by weighted co-expression network analysis in ccRCC concluded that ACAT1 expression was decreased in high-grade ccRCC, and aggressive tumors were unable to obtain sufficient energy from ketolysis and fatty acid oxidation to support their growth (97). Some studies have also shown that silencing ACAT1 or using ACAT1 inhibitors such as ATR-101, avasimibe can effectively inhibit tumor cell growth (98–100).
The obesity paradox has been found in many forms of cancer, including diabetic kidney disease (9, 101, 102). Obesity increases the risk of clear cell renal cell carcinoma, but obese ccRCC patients seem to live longer than non-obese patients, which is known as the obesity paradox (103). It is speculated to be related to the accumulation and metabolism of lipids in ccRCC cells (104). An epidemiological and genomic study of the obesity paradox in ccRCC found that the higher the body mass index (BMI), the lower the cancer-specific mortality. Moreover, a genome-wide survey using BMI found that the expression of metabolic genes and fatty acid genes are different, which further supports the obesity paradox (105). This study extended the principle of the obesity paradox to the genomic level and provided us with many new ideas on fat accumulation in ccRCC: Is lipid accumulation in ccRCC cells related to the good prognosis of obese patients? Obesity is a prognostic indicator of ccRCC. Can the obesity paradox be used to improve patient outcomes? We hypothesized that the development and prognosis of renal cancer is closely related to the obesity paradox because one of the characteristics of ccRCC cells is the accumulation of lipids, leading to the occurrence of a large number of lipid droplets in the cells.
As mentioned above, there are a large number of lipid droplets in the ccRCC cells. If these lipid droplets only exist in the cells, they will not be connected with the development of ccRCC. Therefore, we speculate that these lipid droplets play a role in disease progression.
Ferroptosis is a newly discovered form of iron-dependent oxidative cell death. We believe that this is closely related to lipid metabolism. Unlike traditional apoptosis and necrosis, it is characterized by the fatal accumulation of lipid reactive oxygen species (ROS), which suggests that genes expressing abnormal lipid metabolism play a very important role in the process of ferroptosis (106). This must be correlated with the accumulation of lipid droplets in ccRCC cells.
There are three main pathways of ferroptosis:
1. Regulation of the glutathione (GSH)/GPX4 (glutathione peroxidase 4) pathway, including sulfur metabolism and glutamine pathways.
2. The pathway of iron metabolism which includes the autophagy related-protein - Atg7- nuclear receptor coactivator 1 pathway, p62 - Kelch-like ECH-associated Protein 1- nuclear factor erythroid-2 related factor 2 pathway related to ferritin metabolism.
3. Pathways related to lipid metabolism, such as p53-SAT1 (spermidine/spermine N1-Acetyltransferase 1) -ALOX15 (Arachidonate 15-Lipoxygenase), acyl-CoA synthetase long-chain family member 4 (ACSL4), and lysophosphatidylcholine acyltransferase 3 (LPCAT3), are responsible for lipid regulation and ferroptosis (107).
Because the ccRCC metabolic mechanism is highly dependent on glutamine, one of the most important pathways in ferroptosis, the GSH/GPX4 pathway directly affects the proliferation of ccRCC cells. ccRCC cells use the metabolite of glutamine and glutathione to combat ROS generated by lipid peroxidation (the mechanism of glutamine and glutathione in ccRCC will be mentioned later). In other words, glutathione is an important means of combating ferroptosis (12). Lipid metabolism pathways are also related to cancer progression. ACSL4 uses long-chain polyunsaturated omega-6 fatty acids to build cell membranes. This process plays a key role in the synthesis of cell membranes and is an important pathway for ferroptosis. Studies have shown that the regulation of ACSL4 gene expression in breast cancer cells is related to the sensitivity of cancer cells to ferroptosis (108). ALOX15 oxidizes polyunsaturated fatty acids to produce biologically active lipid metabolites, such as lipid peroxides. Therefore, it is a downstream enzyme of the p53-SAT1-ALOX15 pathway and one of the key enzymes responsible for ferroptosis. It has also been speculated that the expression level of ALOX15 is negatively correlated with renal cancer grade (109). LPCAT3 maintains the systemic lipid homeostasis by regulating intestinal lipid-absorbing lipoprotein secretion and lipogenesis in the liver (110). Studies have found that LPCAT3 gene expression is upregulated in ccRCC. This is mainly because ccRCC cells need to synthesize a large amount of phospholipids to build cell membranes and proliferate in large quantities (111). Therefore, we speculated that there is a strong correlation between ferroptosis and lipid metabolic pathways in ccRCC. The correlation between ferroptosis and lipid metabolic pathways requires further research. Erastin, considered an efficient inducer of ferroptosis, can mediate ferroptosis through multiple molecules, such as the cystine-glutamate transporter receptor and p53 (112). RCC cells are more susceptible to erastin-induced cell death than other cells. Further studies demonstrated that erastin induced RCC cell death in a manner characteristic of iron drape disease (increased lipid ROS production and decreased GPX4 expression) and was inhibited by antioxidants (10).
Current studies have found that ferroptosis is closely related to the death of RCC cells. A study on the correlation between Hippo-Yes Associated protein/TAZ (PDZ-binding motif) pathway and ferroptosis in renal cancer cells showed that TAZ knockout led to resistance to ferroptosis in RCC cells, while the overexpression of TAZS89A led to sensitivity of cells to ferroptosis (113).
We still cannot fully summarize the influence of ferroptosis on ccRCC, but we can be certain that it is one of the pathways that may be taken as an effect of lipid metabolism and glutamine metabolism in ccRCC.
As the most significant pathological feature of ccRCC, the existence of lipid droplet accumulation not only reveals the abnormal lipid metabolism of ccRCC, but also indicates some death pathways of ccRCC cells. It has been pointed out that lipid droplets in ccRCC cells can be used as bioenergy fuel and raw materials for cell membrane generation, and can also be involved in endoplasmic reticulum stress (114). Therefore, we are very interested in the formation and role of lipid droplet itself.
Fatty acids are substrates for the synthesis of various lipid types. Diacylglycerol acyltransferase (DGAT) catalyzes diacylglycerol to synthesize triacylglycerol (TAG) with FAs and plays an important role in lipid synthesis (115) (Figure 2). DGAT1 and DGAT2, two genes encoding DGAT, have been identified, and their main functions have been recognized in the past ten years. Researchers have used molecular tools to study the metabolic changes in mice lacking these two enzymes, revealing the function of DGAT1/2 (116). Currently, DGAT1/2 is known to play a positive regulatory role in many cancers related to lipid accumulation (117–120). Inhibitors targeting DGAT activity have also made significant progress (121–123). Related studies have tentatively confirmed the important role of DGAT in cancer, and we hope that more relevant studies can describe the detailed mechanism of DGAT in ccRCC, including other types of RCC.
Fatty acid binding protein (FABP) is responsible for transferring fatty acids to specific cell chambers and plays an important role in lipid binding and transport. Many types of FABP comprise the FABP family (124) (Figure 2). Studies have shown that the locations where FABPs perform overlapping functions are not limited to the synthesis and transport of lipids. They also exhibit unique characteristics in specific cells and tissues and their biochemical processes (125). By comparing the expression of FABP family genes in ccRCC samples and normal tissues, we found that the expression of FABP5, 6, and 7 in ccRCC samples was significantly upregulated, while the expression of FABP1 was significantly downregulated. This result shows that the influence of the FABP family on ccRCC is also diverse. Moreover, the prediction of overall survival or disease-free survival of ccRCC patients with high expression of FABP5/6/7 and low expression of FABP1 was significantly reduced (126).
When we focus back on upstream genes, we find that lipid droplet formation is consistently influenced by the VHL-HIF pathway. We discussed earlier that the loss of VHL expression leads to the intake of glucose, which has been reported to be the raw material for lipid synthesis and is mediated by HIF. Carnitine palmitoyltransferase 1 (CPT1A) can transport fatty acids to mitochondria and reduce intracellular accumulation, while increased HIF-1/2α caused by reduced VHL expression inhibits this step (127). The report also found that CPT1A may serve as a therapeutic target for ccRCC.
In the ferroptosis section we mentioned that the presence of lipid droplets in ccRCC cells acts through oxidative stress, but focused on the ferroptosis process, which ultimately leads to cell death. However, oxidative stress has two sides. ROS can increase genomic instability and promote the occurrence of cancer (128). Therefore, the content of lipid droplets in ccRCC should maintain a balance, which can not only make the gene mutation lead to cancer, but also cannot lead to the generation of ferroptosis due to the excess of ROS. As a result, lipid droplets are not only produced, but also removed. It has been reported that microtubule-associated protein family 1-s(MAP1S) is used to activate the autophagy system of cells to remove the lipid droplets, and the removal of lipid droplets reduces ROS content to a level that is insufficient to affect gene stability and expression (129). The report suggests that the rational use of this mechanism is a new approach to the treatment of ccRCC.
In summary, due to the double-sided nature of oxidative stress, the presence of lipid droplets in ccRCC also has different effects. The general idea of current research is to break the balance of oxidative stress, so that it is more likely to cause ccRCC cell death or reduce ccRCC cell generation.
Glutamine as the raw material of lipid droplets (114) has always played an important role in RCC, and although it has been reported that the formation of lipid droplets in ccRCC is not required by glutamine (127), inhibitors of glutamine metabolism do inhibit the development of ccRCC. Therefore, it is necessary to discuss the effects of glutamine on ccRCC.
In the TCA cycle, glutamine is converted to glutamate by glutaminase (GLS), whereas the conversion of glutamate to α-ketoglutarate (α-KG), an intermediate product of the tricarboxylic acid cycle, can be catalyzed by transaminase or glutamate dehydrogenase (GDH) (11, 130). In cancer cells, glutamine is mainly transported to the mitochondria by SLC1A5 (solute carrier family 1 member 5) to participate in TCA (Figure 1). SLC1A5 overexpression induced by HIF-2α-mediated hypoxia in cancer cells plays a key role in cancer metabolic reprogramming (131). A recent study identified a small-molecule drug candidate, IMD-0354, that targets the glutamine transporter SLC1A5 to inhibit the uptake of glutamine by cancer cells, blocking an important energy source for cancer cells and thus, slowing their growth (132). They also found that small-molecule drugs can be used in combination with LDHA inhibitors and GLS1 inhibitors to inhibit cancer development.
Experiments showed that cancer cells with the Warburg effect were dependent on glutamine as a carbon source for TCA (133). This also explains why citrate, an intermediate product of TCA, can be used in lipid synthesis pathways as well as other biosynthetic pathways (134). The synthesis of acetyl-CoA, the precursor of lipid synthesis in cancer cells, relies on glutamine to sustain it. Concurrently, related studies have shown that glutamine is related to the activation of mTORC1, which senses glutamine and leucine and is activated by glutamine decomposition and α-ketoglutaric acid production upstream of Rag (11). Bis-2-[5-phenylacetamido-1,2,4-thiadiazol-2-yl] ethyl solubility (BPTES), which inhibits glutaminase activity, reduces glutamate and α-KG levels, and increases the intermediates of glycolysis (135). As a GLS inhibitor, it inhibits oncogenic transformation in IDH1-mutated glioma cells (136, 137). mTORC1 signaling is often activated by glutamine decomposition. Upon inhibition of glutamine decomposition by GLS inhibitors (such as BPTES 21) or siRNA targeting GLS or GDH, mTORC1 activation can be blocked theoretically to inhibit the progression of cancer (11, 130, 138). Metabonomic studies of ccRCC have shown a high uptake of glutamine in renal cancer (139, 140). At present, glutamine metabolism in the treatment of RCC is believed to be related to sunitinib resistance, and the glutamine transporter SLC1A5 in this study was significantly overexpressed in sunitinib-resistant samples compared to the control group (141).
Glutathione (GSH), the final metabolite of glutamine, also plays an important role in ferroptosis. Glutathione expression is significantly reduced in ferroptotic cells (10). GSH and its metabolise-related metabolites have been shown to be increased in patients with advanced ccRCC (142). In ccRCC, GSH reacts with excess reactive ROS in the body to produce oxidized glutathione, which achieves the function of antioxidant protection against oxygen free radical attack of cancer cells. A large amount of lipid accumulates in ccRCC, leading to a large amount of ROS depending on excessive lipid production, which is the premise of ferroptosis (143). Therefore, we can reduce the generation of GSH, a feature of ccRCC, to promote ferroptosis. At present, there are reports of system Xc- inhibitors and GPx4 inhibitors that induce ferroptosis to achieve therapeutic effect (144, 145). In terms of ccRCC treatment, GPx1 was used as a biomarker to judge the prognosis of ccRCC (146), but there have been no reports on the application of ferroptosis in clinical treatment over a wide range. We believe that the research in this area has great potential and is a key direction for future drug development for ccRCC.
RCC is considered to be more prone to metabolic disease. Its pathogenesis is similar to that of many other cancers. For example, cell proliferation requires large quantities of cholesterol and fatty acids, as well as the formation of large amounts of lactic acid and aerobic glycolysis with the incomplete utilization of glucose. Nevertheless, RCC exhibits unique characteristics. Compared with other cancers, the lipid metabolism process remains specific, mainly due to the diversity of lipid synthesis, uptake, and transformation in RCC cells. In recent years, several research results have had an impact on the publicly recognized cancer mechanism, and even the widely recognized Warburg effect has strong evidence that it is cognitively wrong. Current research on lipid metabolism in RCC mainly involves the transport receptors of fatty acids and cholesterol and many of their precursors, the related synthetase and rate-limiting enzymes that control synthesis, and the upstream genes that regulate the expression of these enzymes and receptor proteins. Several studies are more inclined to conduct follow-up experiments by analogy and rely on the research results and foundation pertaining to other types of cancers, such as breast and prostate cancer. We believe that starting from lipid metabolism, assessing the differences between RCC and other cancers, including cholesterol intake and synthesis, iron death, obesity paradox, etc., we will be able to find more differences between RCC and other cancers and perform targeted research. These abnormal phenomena can help us better understand RCC and study the essence of RCC more objectively.
GW conceived the review. QL and XC undertook the initial research. XQ was involved in writing and plotting. QW reviewed the manuscript. All authors contributed to the article and approved the submitted version.
The PhD Start-up Fund of Liaoning Province from GW (2021-BS-209, Liaoning Province, 30000 CNY).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
ccRCC: clear cell renal cell carcinoma
VHL: von Hippel-Lindau
PI3K: phosphatidylinositol-3-kinase
Akt: protein kinase B
mTOR: mammalian target of rapamycin
TCA: tricarboxylic acid cycle
P53: protein 53
VEGF: vascular endothelial growth factor
TGF-β2: transforming growth factor-β2
ATP: adenosine triphosphate
MYC: myelocytomatosis oncogene
AMPK: adenosine 5’-monophosphate-activated protein kinase
HIF: hypoxia-inducible factor
LDLR: low-density lipoprotein receptor
GLUT: glucose transporter
LDHA: lactate dehydrogenase A
PDGF: platelet-derived growth factor
NADPH: nicotinamide adenine dinucleotide phosphate
NOX: NADPH oxidase
EGFR: epidermal growth factor receptor (EGFR)
TSC: tuberous sclerosis complex subunit
SCD1: stearoyl-CoA desaturase 1
FASN: fatty acid synthase
ACC: acetyl-CoA carboxylase
FA: fatty acid
SERBP: serpine mRNA binding protein
ACLY: ATP citrate lyase
ACSS2: acetyl-CoA synthetase 2
HMGCS: hydroxymethylglutaryl-CoA synthase
HMGCR: hydroxymethylglutaryl-CoA reductase
LXR: liver X receptor
ABCA1: adenosine triphosphate binding cassette transporter A-1
ACAT1: acyl-CoA cholesterol acyltransferase-1
BMI: body mass index
ROS: reactive oxygen species
GSH: glutathione
GPX4: glutathione peroxidase 4
SAT1: spermidine/spermine N1-Acetyltransferase 1
ALOX15: arachidonate 15-Lipoxygenase
ACSL4: acyl-CoA synthetase long-chain family member 4
LPCAT3: lysophosphatidylcholine acyltransferase 3
DGAT: diacylglycerol acyltransferase
TAG: triacylglycerol
FABP: fatty acid binding protein
CPT1A: carnitine palmitoyltransferase 1
MAP1S: microtubule-associated protein family 1-s
GLS: glutaminase
α-KG: α-ketoglutarate
GDH: glutamate dehydrogenase
SLC1A5: solute carrier family 1 member 5
GSH: glutathione
1. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin (2021) 71(3):209–49. doi: 10.3322/caac.21660
2. Cohen HT, McGovern FJ. Renal-Cell Carcinoma. N Engl J Med (2005) 353:2477–90. doi: 10.1056/NEJMra043172
3. Lucarelli G, Loizzo D, Franzin R, Battaglia S, Ferro M, Cantiello F, et al. Metabolomic Insights Into Pathophysiological Mechanisms and Biomarker Discovery in Clear Cell Renal Cell Carcinoma. Expert Rev Mol Diagn (2019) 19:397–407. doi: 10.1080/14737159.2019.1607729
4. Courtney KD, Bezwada D, Mashimo T, Pichumani K, Vemireddy V, Funk AM, et al. Isotope Tracing of Human Clear Cell Renal Cell Carcinomas Demonstrates Suppressed Glucose Oxidation In Vivo. Cell Metab (2018) 28(5):793–800.e2.
5. Vaupel P, Schmidberger H, Mayer A. The Warburg Effect: Essential Part of Metabolic Reprogramming and Central Contributor to Cancer Progression. Int J Radiat Biol (2019) 95:912–9. doi: 10.1080/09553002.2019.1589653
6. Linehan WM, Srinivasan R, Schmidt LS. The Genetic Basis of Kidney Cancer: A Metabolic Disease. Nat Rev Urol (2010) 7:277–85. doi: 10.1038/nrurol.2010.47
7. Saito K, Arai E, Maekawa K, Ishikawa M, Fujimoto H, Taguchi R, et al. Lipidomic Signatures and Associated Transcriptomic Profiles of Clear Cell Renal Cell Carcinoma. Sci Rep (2016) 6:28932. doi: 10.1038/srep28932.
8. Park Y, Peterson LL, Colditz GA. The Plausibility of Obesity Paradox in Cancer-Point. Cancer Res (2018) 78:1898–903. doi: 10.1158/0008-5472.CAN-17-3043
9. Lennon H, Sperrin M, Badrick E, Renehan AG. The Obesity Paradox in Cancer: A Review. Curr Oncol Rep (2016) 18:56. doi: 10.1007/s11912-016-0539-4
10. Yu H, Guo P, Xie X, Wang Y, Chen G. Ferroptosis, A New Form of Cell Death, and Its Relationships With Tumourous Diseases. J Cell Mol Med (2017) 21:648–57. doi: 10.1111/jcmm.13008
11. Durán RV, Oppliger W, Robitaille AM, Heiserich L, Skendaj R, Gottlieb E, et al. Glutaminolysis Activates Rag-Mtorc1 Signaling. Mol Cell (2012) 47(3):349–58. doi: 10.1016/j.molcel.2012.05.043
12. Miess H, Dankworth B, Gouw AM, Rosenfeldt M, Schmitz W, Jiang M, et al. The Glutathione Redox System Is Essential to Prevent Ferroptosis Caused by Impaired Lipid Metabolism in Clear Cell Renal Cell Carcinoma. Oncogene (2018) 37(40):5435–50. doi: 10.1038/s41388-018-0315-z
13. Harlander S, Schönenberger D, Toussaint NC, Prummer M, Catalano A, Brandt L, et al. Combined Mutation in Vhl, Trp53 and Rb1 Causes Clear Cell Renal Cell Carcinoma in Mice. Nat Med (2017) 23(7):869–77. doi: 10.1038/nm.4343
14. Hsieh JJ, Purdue MP, Signoretti S, Swanton C, Albiges L, Schmidinger M, et al. Renal Cell Carcinoma. Nat Rev Dis Primers (2017) 3:17009. doi: 10.1038/nrdp.2017.9
15. Yu S, Dai J, Ma M, Xu T, Kong Y, Cui C, et al. RBCK1 Promotes P53 Degradation via Ubiquitination in Renal Cell Carcinoma. Cell Death Dis (2019) 10(4):254. doi: 10.1038/s41419-019-1488-2
16. Samec M, Liskova A, Koklesova L, Samuel SM, Zhai K, Buhrmann C, et al. Flavonoids Against the Warburg Phenotype—Concepts of Predictive, Preventive and Personalised Medicine to Cut the Gordian Knot of Cancer Cell Metabolism. EPMA J (2020) 11(3):377–98. doi: 10.1007/s13167-020-00217-y
17. Ben-Shmuel A, Biber G, Barda-Saad M. Unleashing Natural Killer Cells in the Tumor Microenvironment–The Next Generation of Immunotherapy? Front Immunol (2020) 11:275. doi: 10.3389/fimmu.2020.00275
18. Doherty JR, Cleveland JL. Targeting Lactate Metabolism for Cancer Therapeutics. J Clin Invest (2013) 123:3685–92. doi: 10.1172/JCI69741
19. Bonuccelli G, Tsirigos A, Whitaker-Menezes D, Pavlides S, Pestell RG, Chiavarina B, et al. Ketones and Lactate ‘Fuel’ Tumor Growth and Metastasis: Evidence That Epithelial Cancer Cells Use Oxidative Mitochondrial Metabolism. Cell Cycle (2010) 9(17):3506–14. doi: 10.4161/cc.9.17.12731
20. Zhao D, Zou SW, Liu Y, Zhou X, Mo Y, Wang P, et al. Lysine-5 Acetylation Negatively Regulates Lactate Dehydrogenase A and Is Decreased in Pancreatic Cancer. Cancer Cell (2013) 23(4):464–76. doi: 10.1016/j.ccr.2013.02.005
21. Beckert S, Farrahi F, Aslam RS, Scheuenstuhl H, Königsrainer A, Hussain MZ, et al. Lactate Stimulates Endothelial Cell Migration. Wound Repair Regener (2006) 14(3):321–4. doi: 10.1111/j.1743-6109.2006.00127.x
22. Baumann F, Leukel P, Doerfelt A, Beier CP, Dettmer K, Oefner PJ, et al. Lactate Promotes Glioma Migration by TGF-Beta2-Dependent Regulation of Matrix Metalloproteinase-2. Neuro Oncol (2009) 11(4):368–80. doi: 10.1215/15228517-2008-106
23. Buchsteiner M, Quek L-E, Gray P, Nielsen LK. Improving Culture Performance and Antibody Production in CHO Cell Culture Processes by Reducing the Warburg Effect. Biotechnol Bioeng (2018) 115:2315–27. doi: 10.1002/bit.26724
24. Cheng C, Geng F, Cheng X, Guo D. Lipid Metabolism Reprogramming and its Potential Targets in Cancer. Cancer Commun (Lond) (2018) 38(1):27. doi: 10.1186/s40880-018-0301-4
25. Suganuma N, Segade F, Matsuzu K, Bowden DW. Differential Expression of Facilitative Glucose Transporters in Normal and Tumour Kidney Tissues. BJU Int (2007) 99:1143–9. doi: 10.1111/j.1464-410X.2007.06765.x
26. Uh W, Birzele F, Nopora A. microRNAs Promoting Growth of Gastric Cancer Xenografts and Correlation to Clinical Prognosis. Cancer Genomics Proteomics (2021) 18(1):1–15. doi: 10.21873/cgp.20237.
27. Garg AD, Maes H, Vliet ARv, Agostinis P. Targeting the Hallmarks of Cancer With Therapy-Induced Endoplasmic Reticulum (ER) Stress. Mol Cell Oncol (2015) 2(1):e975089. doi: 10.4161/23723556.2014.975089
28. Song W, Li L, He D, Xie H, Chen J, Yeh CR, et al. Infiltrating Neutrophils Promote Renal Cell Carcinoma (RCC) Proliferation via Modulating Androgen Receptor (AR) → c-Myc Signals. Cancer Lett (2015) 368(1):71–8. doi: 10.1016/j.canlet.2015.07.027
29. Zhang C, Liu J, Liang Y, Wu R, Zhao Y, Hong X, et al. Tumour-Associated Mutant P53 Drives the Warburg Effect. Nat Commun (2013) 4:2935. doi: 10.1038/ncomms3935
30. Feng X, Yan N, Sun W, Zheng S, Jiang S, Wang J, et al. miR-4521-FAM129A Axial Regulation on ccRCC Progression Through TIMP-1/MMP2/MMP9 and MDM2/p53/Bcl2/Bax Pathways. Cell Death Discovery (2019) 5:89. doi: 10.1038/s41420-019-0167-5
31. Yang F, Zhang H, Mei Y, Wu M. Reciprocal Regulation of HIF-1α and lincRNA-P21 Modulates the Warburg Effect. Mol Cell (2014) 53:88–100. doi: 10.1016/j.molcel.2013.11.004
32. Marín-Hernández A, Gallardo-Pérez JC, Ralph SJ, Rodríguez-Enríquez S, Moreno-Sánchez R. HIF-1alpha Modulates Energy Metabolism in Cancer Cells by Inducing Over-Expression of Specific Glycolytic Isoforms. Mini Rev Med Chem (2009) 9:1084–101. doi: 10.2174/138955709788922610
33. Wierenga ATJ, Cunningham A, Erdem A, Lopera NV, Brouwers-Vos AZ, Pruis M, et al. HIF1/2-Exerted Control Over Glycolytic Gene Expression is Not Functionally Relevant for Glycolysis in Human Leukemic Stem/Progenitor Cells. Cancer Metab (2019) 7:11. doi: 10.1186/s40170-019-0206-y
34. Befani C, Liakos P. The Role of Hypoxia-Inducible Factor-2 Alpha in Angiogenesis. J Cell Physiol (2018) 233:9087–98. doi: 10.1002/jcp.26805
35. Chen W, Hill H, Christie A, Kim MS, Holloman E, Pavia-Jimenez A, et al. Targeting Renal Cell Carcinoma With a HIF-2 Antagonist. Nature (2016) 539(7627):112–7. doi: 10.1038/nature19796
36. Zhang J, Zhang Q. VHL and Hypoxia Signaling: Beyond HIF in Cancer. Biomedicines (2018) 6(1):35. doi: 10.3390/biomedicines6010035
37. Chen R, Lai UH, Zhu L, Singh A, Ahmed M, Forsyth NR. Reactive Oxygen Species Formation in the Brain at Different Oxygen Levels: The Role of Hypoxia Inducible Factors. Front Cell Dev Biol (2018) 6:132. doi: 10.3389/fcell.2018.00132
38. Gossage L, Eisen T, Maher ER. VHL, the Story of a Tumour Suppressor Gene. Nat Rev Cancer (2015) 15:55–64. doi: 10.1038/nrc3844
39. Yang H, Kaelin WG Jr. Molecular Pathogenesis of the Von Hippel-Lindau Hereditary Cancer Syndrome: Implications for Oxygen Sensing. Cell Growth Differ (2001) 12(9):447–55.
40. Hoefflin R, Harlander S, Schäfer S, Metzger P, Kuo F, Schönenberger D, et al. HIF-1α and HIF-2α Differently Regulate Tumour Development and Inflammation of Clear Cell Renal Cell Carcinoma in Mice. Nat Commun (2020) 11(1):4111. doi: 10.1038/s41467-020-17873-3
41. Lee J-H, Liu R, Li J, Wang Y, Tan L, Li XJ, et al. EGFR-Phosphorylated Platelet Isoform of Phosphofructokinase 1 Promotes PI3K Activation. Mol Cell (2018) 70(2):197–210.e7. doi: 10.1016/j.molcel.2018.03.018
42. Wang Z, Wang N, Liu P, Xie X. AMPK and Cancer. Exp Suppl (2016) 107:203–26. doi: 10.1007/978-3-319-43589-3_9
43. Tamargo-Gómez I, Mariño G. AMPK: Regulation of Metabolic Dynamics in the Context of Autophagy. Int J Mol Sci (2018) 19(12):3812. doi: 10.3390/ijms19123812
44. Crino PB, Nathanson KL, Henske EP. The Tuberous Sclerosis Complex. N Engl J Med (2006) 355:1345–56. doi: 10.1056/NEJMra055323
45. Bjornsson J, Short MP, Kwiatkowski DJ, Henske EP. Tuberous Sclerosis-Associated Renal Cell Carcinoma. Clinical, Pathological, and Genetic Features. Am J Pathol (1996) 149(4):1201–8.
46. Xiao Y, Peng H, Hong C, Chen Z, Deng X, Wang A, et al. PDGF Promotes the Warburg Effect in Pulmonary Arterial Smooth Muscle Cells via Activation of the PI3K/AKT/mTOR/HIF-1α Signaling Pathway. Cell Physiol Biochem (2017) 42(4):1603–13. doi: 10.1159/000479401
47. Kostyuk AI, Kokova AD, Podgorny OV, Kelmanson IV, Fetisova ES, Belousov VV, et al. Genetically Encoded Tools for Research of Cell Signaling and Metabolism Under Brain Hypoxia. Antioxid (Basel Switzerland) (2020) 9(6):516. doi: 10.3390/antiox9060516
48. Ashrafizadeh M, Mohammadinejad R, Tavakol S, Ahmadi Z, Roomiani S, Katebi M. Autophagy, Anoikis, Ferroptosis, Necroptosis, and Endoplasmic Reticulum Stress: Potential Applications in Melanoma Therapy. J Cell Physiol (2019) 234(11):19471–9. doi: 10.1002/jcp.28740
49. Jin L, Chun J, Pan C, Alesi GN, Li D, Magliocca KR, et al. Phosphorylation-Mediated Activation of LDHA Promotes Cancer Cell Invasion and Tumour Metastasis. Oncogene (2017) 36:3797–806. doi: 10.1038/onc.2017.6
50. Fantin VR, St-Pierre J, Leder P. Attenuation of LDH-A Expression Uncovers a Link Between Glycolysis, Mitochondrial Physiology, and Tumor Maintenance. Cancer Cell (2006) 9:425–34. doi: 10.1016/j.ccr.2006.04.023
51. Lin C-S, Lee HT, Lee MH, Pan SC, Ke CY, Chiu AW, et al. Role of Mitochondrial DNA Copy Number Alteration in Human Renal Cell Carcinoma. Int J Mol Sci (2016) 17(6):814. doi: 10.3390/ijms17060814
52. Wang X, Xu L, Wu Q, Liu M, Tang F, Cai Y, et al. Inhibition of LDHA Deliver Potential Anticancer Performance in Renal Cell Carcinoma. Urol Int (2017) 99(2):237–44. doi: 10.1159/000445125
53. Xu K, Xu L, Wu Q, Liu M, Tang F, Cai Y, et al. Glycolysis Fuels Phosphoinositide 3-Kinase Signaling to Bolster T Cell Immunity. Science (2021) 371(6527):405–10. doi: 10.1126/science.abb2683
54. Shi L, An S, Liu Y, Liu J, Wang F. PCK1 Regulates Glycolysis and Tumor Progression in Clear Cell Renal Cell Carcinoma Through LDHA. Onco Targets Ther (2020) 13:2613–27. doi: 10.2147/OTT.S241717
55. Reinfeld BI, Madden MZ, Wolf MM, Chytil A, Bader JE, Patterson AR, et al. Cell-Programmed Nutrient Partitioning in the Tumour Microenvironment. Nature (2021) 593(7858):282–88. doi: 10.1038/s41586-021-03442-1
56. Yan L, Liu Z, Wang G, Huang Y, Liu Y, Yu Y, et al. Angiomyolipoma With Minimal Fat: Differentiation From Clear Cell Renal Cell Carcinoma and Papillary Renal Cell Carcinoma by Texture Analysis on CT Images. Acad Radiol (2015) 22(9):1115–21. doi: 10.1016/j.acra.2015.04.004
57. Wettersten HI, Aboud OA, Lara PN, Weiss RH. Metabolic Reprogramming in Clear Cell Renal Cell Carcinoma. Nat Rev Nephrol (2017) 13:410–9. doi: 10.1038/nrneph.2017.59
58. Linehan WM, Ricketts CJ. The Cancer Genome Atlas of Renal Cell Carcinoma: Findings and Clinical Implications. Nat Rev Urol (2019) 16:539–52. doi: 10.1038/s41585-019-0211-5
59. Sajnani K, Islam F, Smith RA, Gopalan V, Lam AK-Y. Genetic Alterations in Krebs Cycle and its Impact on Cancer Pathogenesis. Biochimie (2017) 135:164–72. doi: 10.1016/j.biochi.2017.02.008
60. Maan M, Peters JM, Dutta M, Patterson AD. Lipid Metabolism and Lipophagy in Cancer. Biochem Biophys Res Commun (2018) 504:582–9. doi: 10.1016/j.bbrc.2018.02.097
61. Guo D, Reinitz F, Youssef M, Hong C, Nathanson D, Akhavan D, et al. An LXR Agonist Promotes Glioblastoma Cell Death Through Inhibition of an EGFR/AKT/SREBP-1/LDLR-Dependent Pathway. Cancer Discovery (2011) 1(5):442–56. doi: 10.1158/2159-8290.CD-11-0102
62. McCauley C, Anang V, Cole B, Simmons GE Jr. Potential Links Between YB-1 and Fatty Acid Synthesis in Clear Cell Renal Carcinoma. Med Res Arch (2020) 8(10):10.18103/mra.v8i10.2273. doi: 10.18103/mra.v8i10.2273.
63. Igal RA. Stearoyl-CoA Desaturase-1: A Novel Key Player in the Mechanisms of Cell Proliferation, Programmed Cell Death and Transformation to Cancer. Carcinogenesis (2010) 31:1509–15. doi: 10.1093/carcin/bgq131
64. Liu G, Kuang S, Cao R, Wang J, Peng Q, Sun C. Sorafenib Kills Liver Cancer Cells by Disrupting SCD1-Mediated Synthesis of Monounsaturated Fatty Acids via the ATP-AMPK-mTOR-SREBP1 Signaling Pathway. FASEB J (2019) 33(9):10089–103. doi: 10.1096/fj.201802619RR
65. Takahashi M, Rhodes DR, Furge KA, Kanayama H, Kagawa S, Haab BB, et al. Gene Expression Profiling of Clear Cell Renal Cell Carcinoma: Gene Identification and Prognostic Classification. Proc Natl Acad Sci USA (2001) 98(17):9754–9. doi: 10.1073/pnas.171209998
66. Zhao Z, Liu Y, Liu Q, Wu F, Liu X, Qu H, et al. The mRNA Expression Signature and Prognostic Analysis of Multiple Fatty Acid Metabolic Enzymes in Clear Cell Renal Cell Carcinoma. J Cancer (2019) 10(26):6599–607. doi: 10.7150/jca.33024
67. Menendez JA, Lupu R. Fatty Acid Synthase (FASN) as a Therapeutic Target in Breast Cancer. Expert Opin Ther Targets (2017) 21:1001–16. doi: 10.1080/14728222.2017.1381087
68. Wu X, Qin L, Fako V, Zhang J-T. Molecular Mechanisms of Fatty Acid Synthase (FASN)-Mediated Resistance to Anti-Cancer Treatments. Adv Biol Regul (2014) 54:214–21. doi: 10.1016/j.jbior.2013.09.004
69. Yuan Y, Yang X, Li Y, Liu Q, Wu F, Qu H, et al. Expression and Prognostic Significance of Fatty Acid Synthase in Clear Cell Renal Cell Carcinoma. Pathol Res Pract (2020) 216(1):153227. doi: 10.1016/j.prp.2020.153227
70. Hunkeler M, Hagmann A, Stuttfeld E, Chami M, Guri Y, Stahlberg H, et al. Structural Basis for Regulation of Human Acetyl-CoA Carboxylase. Nature (2018) 558(7710):470–4. doi: 10.1038/s41586-018-0201-4
71. Rios Garcia M, Steinbauer B, Srivastava K, Singhal M, Mattijssen F, Maida A, et al. Acetyl-CoA Carboxylase 1-Dependent Protein Acetylation Controls Breast Cancer Metastasis and Recurrence. Cell Metab (2017) 26(6):842–55.e5. doi: 10.1016/j.cmet.2017.09.018
72. Lally JSV, Ghoshal S, DePeralta DK, Moaven O, Wei L, Masia R, et al. Inhibition of Acetyl-CoA Carboxylase by Phosphorylation or the Inhibitor ND-654 Suppresses Lipogenesis and Hepatocellular Carcinoma. Cell Metab (2019) 29(1):174–82.e5. doi: 10.1016/j.cmet.2018.08.020
73. Cancer Genome Atlas Research Network. Comprehensive Molecular Characterization of Clear Cell Renal Cell Carcinoma. Nature (2013) 499:43–9. doi: 10.1038/nature12222
74. Zhao S, Torres A, Henry RA, Trefely S, Wallace M, Lee JV, et al. ATP-Citrate Lyase Controls a Glucose-To-Acetate Metabolic Switch. Cell Rep (2016) 17(4):1037–52. doi: 10.1016/j.celrep.2016.09.069
75. Moon YA, Lee JJ, Park SW, Ahn YH, Kim KS. The Roles of Sterol Regulatory Element-Binding Proteins in the Transactivation of the Rat ATP Citrate-Lyase Promoter. J Biol Chem (2000) 275:30280–6. doi: 10.1074/jbc.M001066200
76. Khwairakpam AD, Shyamananda MS, Sailo BL, Rathnakaram SR, Padmavathi G, Kotoky J, et al. ATP Citrate Lyase (ACLY): A Promising Target for Cancer Prevention and Treatment. Curr Drug Targets (2015) 16(2):156–63. doi: 10.2174/1389450115666141224125117
77. Csanadi A, Kayser C, Donauer M, Gumpp V, Aumann K, Rawluk J, et al. Prognostic Value of Malic Enzyme and ATP-Citrate Lyase in Non-Small Cell Lung Cancer of the Young and the Elderly. PloS One (2015) 10(5):e0126357. doi: 10.1371/journal.pone.0126357
78. Zhao Q, Kun D, Hong B, Deng X, Guo S, Tang X, et al. Identification of Novel Proteins Interacting With Vascular Endothelial Growth Inhibitor 174 in Renal Cell Carcinoma. Anticancer Res (2017) 37(8):4379–88. doi: 10.21873/anticanres.11832
79. Wei J, Leit S, Kuai J, Therrien E, Rafi S, Harwood HJ Jr, et al. An Allosteric Mechanism for Potent Inhibition of Human ATP-Citrate Lyase. Nature (2019) 568(7753):566–70. doi: 10.1038/s41586-019-1094-6
80. Icard P, Wu Z, Fournel L, Coquerel A, Lincet H, Alifano M. ATP Citrate Lyase: A Central Metabolic Enzyme in Cancer. Cancer Lett (2020) 471:125–34. doi: 10.1016/j.canlet.2019.12.010
81. Huang Z, Zhang M, Plec AA, Estill SJ, Cai L, Repa JJ, et al. ACSS2 Promotes Systemic Fat Storage and Utilization Through Selective Regulation of Genes Involved in Lipid Metabolism. Proc Natl Acad Sci USA (2018) 115(40):E9499–506. doi: 10.1073/pnas.1806635115
82. Zhang S, He J, Jia Z, Yan Z, Yang J. Acetyl-CoA Synthetase 2 Enhances Tumorigenesis and is Indicative of a Poor Prognosis for Patients With Renal Cell Carcinoma. Urol Oncol (2018) 36:243.e9–20. doi: 10.1016/j.urolonc.2018.01.013
83. Yao L, Guo X, Gui Y. Acetyl-CoA Synthetase 2 Promotes Cell Migration and Invasion of Renal Cell Carcinoma by Upregulating Lysosomal-Associated Membrane Protein 1 Expression. Cell Physiol Biochem (2018) 45:984–92. doi: 10.1159/000487293
84. Li X, Yu W, Qian X, Xia Y, Zheng Y, Lee JH, et al. Nucleus-Translocated ACSS2 Promotes Gene Transcription for Lysosomal Biogenesis and Autophagy. Mol Cell (2017) 66(5):684–97.e9.
85. Miller KD, Pniewski K, Perry CE, Papp SB, Shaffer JD, Velasco-Silva JN, et al. Targeting ACSS2 With a Transition-State Mimetic Inhibits Triple-Negative Breast Cancer Growth. Cancer Res (2021) 81(5):1252–64. doi: 10.1158/0008-5472.CAN-20-1847
86. Yao L, Jiang L, Zhang F, Li M, Yang B, Zhang F, et al. Acetate Promotes SNAI1 Expression by ACSS2-Mediated Histone Acetylation Under Glucose Limitation in Renal Cell Carcinoma Cell. Biosci Rep (2020) 40(6):BSR20200382. doi: 10.1042/BSR20200382
87. Sirtori CR. The Pharmacology of Statins. Pharmacol Res (2014) 88:3–11. doi: 10.1016/j.phrs.2014.03.002
88. Thompson JM, Alvarez A, Singha MK, Pavesic MW, Nguyen QH, Nelson LJ, et al. Targeting the Mevalonate Pathway Suppresses VHL-Deficient CC-RCC Through a HIF-Dependent Mechanism. Mol Cancer Ther (2018) 17(8):1781–92. doi: 10.1158/1535-7163.MCT-17-1076
89. Pottegård A, Clark P, Friis S, Hallas J, Lund L. Long-Term Use of Statins and Risk of Renal Cell Carcinoma: A Population-Based Case-Control Study. Eur Urol (2016) 69:877–82. doi: 10.1016/j.eururo.2015.10.020
90. Xue L, Zhang H, Ding L, Huang Q, Zhao D, Wu BJ, Li X, et al. Targeting SREBP-2-Regulated Mevalonate Metabolism for Cancer Therapy. Front Oncol (2020) 10:1510. doi: 10.3389/fonc.2020.01510
91. Wu G, Wang Q, Xu Y, Li Q, Cheng L. A New Survival Model Based on Ferroptosis-Related Genes for Prognostic Prediction in Clear Cell Renal Cell Carcinoma. Aging (Albany NY) (2020) 12:14933–48. doi: 10.18632/aging.103553
92. Bovenga F, Sabbà C, Moschetta A. Uncoupling Nuclear Receptor LXR and Cholesterol Metabolism in Cancer. Cell Metab (2015) 21:517–26. doi: 10.1016/j.cmet.2015.03.002
93. Wu G, Wang Q, Xu Y, Li J, Zhang H, Qi G, et al. Targeting the Transcription Factor Receptor LXR to Treat Clear Cell Renal Cell Carcinoma: Agonist or Inverse Agonist? Cell Death Dis (2019) 10(6):416. doi: 10.1038/s41419-019-1654-6
94. Zhang G-M, Wang MY, Liu YN, Zhu Y, Wan FN, Wei QY, et al. Functional Variants in the Low-Density Lipoprotein Receptor Gene Are Associated With Clear Cell Renal Cell Carcinoma Susceptibility. Carcinogenesis (2017) 38(12):1241–8. doi: 10.1093/carcin/bgx098
95. Qian H, Zhao X, Yan R, Yao X, Gao S, Sun X, et al. Structural Basis for Catalysis and Substrate Specificity of Human ACAT1. Nature (2020) 581(7808):333–8. doi: 10.1038/s41586-020-2290-0
96. Cui W, Luo W, Zhou X, Lu Y, Xu W, Zhong S, et al. Dysregulation of Ketone Body Metabolism Is Associated With Poor Prognosis for Clear Cell Renal Cell Carcinoma Patients. Front Oncol (2019) 9:1422. doi: 10.3389/fonc.2019.01422.
97. Chen L, Peng T, Luo Y, Zhou F, Wang G, Qian K, et al. ACAT1 and Metabolism-Related Pathways Are Essential for the Progression of Clear Cell Renal Cell Carcinoma (ccRCC), as Determined by Co-Expression Network Analysis. Front Oncol (2019) 9:957. doi: 10.3389/fonc.2019.00957.
98. Geng F, Cheng X, Wu X, Yoo JY, Cheng C, Guo JY, et al. Inhibition of SOAT1 Suppresses Glioblastoma Growth via Blocking SREBP-1-Mediated Lipogenesis. Clin Cancer Res (2016) 22(21):5337–48. doi: 10.1158/1078-0432.CCR-15-2973
99. LaPensee CR, Mann JE, Rainey WE, Crudo V, Hunt SW 3rd, Hammer GD. ATR-101, a Selective and Potent Inhibitor of Acyl-CoA Acyltransferase 1, Induces Apoptosis in H295R Adrenocortical Cells and in the Adrenal Cortex of Dogs. Endocrinology (2016) 157(5):1775–88. doi: 10.1210/en.2015-2052
100. Bemlih S, Poirier M-D, El Andaloussi A. Acyl-Coenzyme A: Cholesterol Acyltransferase Inhibitor Avasimibe Affect Survival and Proliferation of Glioma Tumor Cell Lines. Cancer Biol Ther (2010) 9:1025–32. doi: 10.4161/cbt.9.12.11875
101. Carnethon MR, De Chavez PJ, Biggs ML, Lewis CE, Pankow JS, Bertoni AG, et al. Association of Weight Status With Mortality in Adults With Incident Diabetes. JAMA (2012) 308(20):581–90. doi: 10.1001/jama.2012.9282
102. Kalantar-Zadeh K, Streja E, Kovesdy CP, Oreopoulos A, Noori N, Jing J, et al. The Obesity Paradox and Mortality Associated With Surrogates of Body Size and Muscle Mass in Patients Receiving Hemodialysis. Mayo Clin Proc (2010) 85(11):991–1001. doi: 10.4065/mcp.2010.0336
103. Wang Q, Tu H, Zhu M, Liang D, Ye Y, Chang DW, Long Y, Wu X. Circulating Obesity-Driven Biomarkers are Associated With Risk of Clear Cell Renal Cell Carcinoma: A Two-Stage, Case-Control Study. Carcinogenesis (2019) 40(10):1191–7. doi: 10.1093/carcin/bgz074
104. Stone L. Improving Understanding of the Obesity Paradox in ccRCC. Nat Rev Urol (2020) 17:63. doi: 10.1038/s41585-020-0285-0
105. Hakimi AA, Furberg H, Zabor EC, Jacobsen A, Schultz N, Ciriello G, et al. An Epidemiologic and Genomic Investigation Into the Obesity Paradox in Renal Cell Carcinoma. J Natl Cancer Inst (2013) 105(24):1862–70. doi: 10.1093/jnci/djt310
106. Liang C, Zhang X, Yang M, Dong X. Recent Progress in Ferroptosis Inducers for Cancer Therapy. Adv Mater (2019) 31:e1904197. doi: 10.1002/adma.201904197
107. Li J, Cao F, Yin HL, Huang ZJ, Lin ZT, Mao N, et al. Ferroptosis: Past, Present and Future. Cell Death Dis (2020) 11(2):88. doi: 10.1038/s41419-020-2298-2
108. Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingold I, et al. Acsl4 Dictates Ferroptosis Sensitivity by Shaping Cellular Lipid Composition. Nat Chem Biol (2017) 13(1):91–8. doi: 10.1038/nchembio.2239
109. Gohara A, Eltaki N, Sabry D, Murtagh D Jr, Jankun J, Selman SH, et al. Human 5-, 12- and 15-Lipoxygenase-1 Coexist in Kidney But Show Opposite Trends and Their Balance Changes in Cancer. Oncol Rep (2012) 28(4):1275–82. doi: 10.3892/or.2012.1924
110. Wang B, Tontonoz P. Phospholipid Remodeling in Physiology and Disease. Annu Rev Physiol (2019) 81:165–88. doi: 10.1146/annurev-physiol-020518-114444
111. Du Y, Wang Q, Zhang X, Wang X, Qin C, Sheng Z, et al. Lysophosphatidylcholine Acyltransferase 1 Upregulation and Concomitant Phospholipid Alterations in Clear Cell Renal Cell Carcinoma. J Exp Clin Cancer Res (2017) 36(1):66. doi: 10.1186/s13046-017-0525-1
112. Zhao Y, Li Y, Zhang R, Wang F, Wang T, Jiao Y. The Role of Erastin in Ferroptosis and Its Prospects in Cancer Therapy. Onco Targets Ther (2020) 13:5429–41. doi: 10.2147/OTT.S254995
113. Yang W-H, Ding CC, Sun T, Rupprecht G, Lin CC, Hsu D, et al. The Hippo Pathway Effector TAZ Regulates Ferroptosis in Renal Cell Carcinoma. Cell Rep (2019) 28(10):2501–8.e4. doi: 10.1016/j.celrep.2019.07.107
114. van der Mijn JC, Fu L, Khani F, Zhang T, Molina AM, Barbieri CE, et al. Combined Metabolomics and Genome-Wide Transcriptomics Analyses Show Multiple Hif1α-Induced Changes in Lipid Metabolism in Early Stage Clear Cell Renal Cell Carcinoma. Trans Oncol (2020) 13(2):177–85. doi: 10.1016/j.tranon.2019.10.015
115. Bhatt-Wessel B, Jordan TW, Miller JH, Peng L. Role of DGAT Enzymes in Triacylglycerol Metabolism. Arch Biochem Biophys (2018) 655:1–11. doi: 10.1016/j.abb.2018.08.001
116. Yen C-LE, Stone SJ, Koliwad S, Harris C, Farese RV. DGAT Enzymes and Triacylglycerol Biosynthesis. J Lipid Res (2008) 49:2283–301. doi: 10.1194/jlr.R800018-JLR200
117. Casaschi A, Rubio BK, Maiyoh GK, Theriault AG. Inhibitory Activity of Diacylglycerol Acyltransferase (DGAT) and Microsomal Triglyceride Transfer Protein (MTP) by the Flavonoid, Taxifolin, in HepG2 Cells: Potential Role in the Regulation of Apolipoprotein B Secretion. Atherosclerosis (2004) 176:247–53. doi: 10.1016/j.atherosclerosis.2004.05.020
118. Balaban S, Nassar ZD, Zhang AY, Hosseini-Beheshti E, Centenera MM, Schreuder M, et al. Extracellular Fatty Acids Are the Major Contributor to Lipid Synthesis in Prostate Cancer. Mol Cancer Res (2019) 17(4):949–62. doi: 10.1158/1541-7786.MCR-18-0347
119. Balaban S, Lee LS, Varney B, Aishah A, Gao Q, Shearer RF, et al. Heterogeneity of Fatty Acid Metabolism in Breast Cancer Cells Underlies Differential Sensitivity to Palmitate-Induced Apoptosis. Mol Oncol (2018) 12(9):1623–38. doi: 10.1002/1878-0261.12368
120. Jones DT, Valli A, Haider S, Zhang Q, Smethurst EA, Schug ZT, et al. 3d Growth of Cancer Cells Elicits Sensitivity to Kinase Inhibitors But Not Lipid Metabolism Modifiers. Mol Cancer Ther (2019) 18:(2)376–88. doi: 10.1158/1535-7163.MCT-17-0857
121. Lin X, Li BB, Zhang L, Li HZ, Meng X, Jiang YY, et al. Four New Compounds Isolated From Psoralea Corylifolia and Their Diacylglycerol Acyltransferase (DGAT) Inhibitory Activity. Fitoterapia (2018) 128:130–4. doi: 10.1016/j.fitote.2018.05.004
122. Zambre VP, Khamkar SM, Gavhane DD, Khedkar SC, Chavan MR, Pandey MM, et al. Patent Landscape for Discovery of Promising Acyltransferase DGAT and MGAT Inhibitors. Expert Opin Ther Pat (2020) 30(11):873–96. doi: 10.1080/13543776.2020.1815707
123. Naik R, Obiang-Obounou BW, Kim M, Choi Y, Lee HS, Lee K. Therapeutic Strategies for Metabolic Diseases: Small-Molecule Diacylglycerol Acyltransferase (DGAT) Inhibitors. ChemMedChem (2014) 9(11):2410–24. doi: 10.1002/cmdc.201402069
124. Wang Y-T, Liu C-H, Zhu H-L. Fatty Acid Binding Protein (FABP) Inhibitors: A Patent Review (2012-2015). Expert Opin Ther Pat (2016) 26:767–76. doi: 10.1080/13543776.2016.1182500
125. Li B, Hao J, Zeng J, Sauter ER. SnapShot: FABP Functions. Cell (2020) 182(4):1066–66.e1. doi: 10.1016/j.cell.2020.07.027
126. Wu G, Zhang Z, Tang Q, Liu L, Liu W, Li Q, et al. Study of FABP’s Interactome and Detecting New Molecular Targets in Clear Cell Renal Cell Carcinoma. J Cell Physiol (2020) 235(4):3776–89. doi: 10.1002/jcp.29272
127. Du W, Zhang L, Brett-Morris A, Aguila B, Kerner J, Hoppel CL, et al. HIF Drives Lipid Deposition and Cancer in ccRCC via Repression of Fatty Acid Metabolism. Nat Commun (2017) 8(1):1769. doi: 10.1038/s41467-017-01965-8
128. Liu L, Trimarchi JR, Smith PJS, Keefe DL. Mitochondrial Dysfunction Leads to Telomere Attrition and Genomic Instability. Aging Cell (2002) 1:40–6. doi: 10.1046/j.1474-9728.2002.00004.x
129. Xu G, Jiang Y, Xiao Y, Liu XD, Yue F, Li W, et al. Fast Clearance of Lipid Droplets Through MAP1S-Activated Autophagy Suppresses Clear Cell Renal Cell Carcinomas and Promotes Patient Survival. Oncotarget (2015) 7(5):6255–65. doi: 10.18632/oncotarget.6669
130. Zhao Y, Butler EB, Tan M. Targeting Cellular Metabolism to Improve Cancer Therapeutics. Cell Death Dis (2013) 4:e532. doi: 10.1038/cddis.2013.60
131. Yoo HC, Park SJ, Nam M, Kang J, Kim K, Yeo JH, et al. A Variant of SLC1A5 Is a Mitochondrial Glutamine Transporter for Metabolic Reprogramming in Cancer Cells. Cell Metab (2020) 31(2):267–83.e12. doi: 10.1016/j.cmet.2019.11.020
132. Feng Y, Pathria G, Heynen-Genel S, Jackson M, James B, Yin J, et al. Identification and Characterization of IMD-0354 as a Glutamine Carrier Protein Inhibitor in Melanoma. Mol Cancer Ther (2021) 20(5):816–32. doi: 10.1158/1535-7163.MCT-20-0354
133. DeBerardinis RJ, Mancuso A, Daikhin E, Nissim I, Yudkoff M, Wehrli S, et al. Beyond Aerobic Glycolysis: Transformed Cells Can Engage in Glutamine Metabolism That Exceeds the Requirement for Protein and Nucleotide Synthesis. Proc Natl Acad Sci USA (2007) 104(49):19345–50. doi: 10.1073/pnas.0709747104
134. Deberardinis RJ, Sayed N, Ditsworth D, Thompson CB. Brick by Brick: Metabolism and Tumor Cell Growth. Curr Opin Genet Dev (2008) 18:54–61. doi: 10.1016/j.gde.2008.02.003
135. Johmura Y, Yamanaka T, Omori S, Wang TW, Sugiura Y, Matsumoto M, et al. Senolysis by Glutaminolysis Inhibition Ameliorates Various Age-Associated Disorders. Science (2021) 371(6526):265–70. doi: 10.1126/science.abb5916
136. Seltzer MJ, Bennett BD, Joshi AD, Gao P, Thomas AG, Ferraris DV, et al. Inhibition of Glutaminase Preferentially Slows Growth of Glioma Cells With Mutant IDH1. Cancer Res (2010) 70(22):8981–7. doi: 10.1158/0008-5472.CAN-10-1666
137. Robinson MM, McBryant SJ, Tsukamoto T, Rojas C, Ferraris DV, Hamilton SK, et al. Novel Mechanism of Inhibition of Rat Kidney-Type Glutaminase by Bis-2-(5-Phenylacetamido-1,2,4-Thiadiazol-2-Yl)Ethyl Sulfide (BPTES). Biochem J (2007) 406(3):407–14. doi: 10.1042/BJ20070039
138. Chiarini F, Grimaldi C, Ricci F, Tazzari PL, Evangelisti C, Ognibene A, et al. Activity of the Novel Dual Phosphatidylinositol 3-Kinase/Mammalian Target of Rapamycin Inhibitor NVP-BEZ235 Against T-Cell Acute Lymphoblastic Leukemia. Cancer Res (2010) 70(20):8097–107. doi: 10.1158/0008-5472.CAN-10-1814
139. Weiss RH. Metabolomics and Metabolic Reprogramming in Kidney Cancer. Semin Nephrol (2018) 38:175–82. doi: 10.1016/j.semnephrol.2018.01.006
140. Wettersten HI, Hakimi AA, Morin D, Bianchi C, Johnstone ME, Donohoe DR, et al. Grade-Dependent Metabolic Reprogramming in Kidney Cancer Revealed by Combined Proteomics and Metabolomics Analysis. Cancer Res (2015) 75(12):2541–52. doi: 10.1158/0008-5472.CAN-14-1703
141. Sato T, Kawasaki Y, Maekawa M, Takasaki S, Morozumi K, Sato M, et al. Metabolomic Analysis to Elucidate Mechanisms of Sunitinib Resistance in Renal Cell Carcinoma. Metabolites (2020) 11(1):1. doi: 10.3390/metabo11010001
142. Hakimi AA, Reznik E, Lee CH, Creighton CJ, Brannon AR, Luna A, et al. An Integrated Metabolic Atlas of Clear Cell Renal Cell Carcinoma. Cancer Cell (2016) 29(1):104–16. doi: 10.1016/j.ccell.2015.12.004
143. Xiao Y, Meierhofer D. Glutathione Metabolism in Renal Cell Carcinoma Progression and Implications for Therapies. Int J Mol Sci (2019) 20(15):3672. doi: 10.3390/ijms20153672
144. Murphy TH, Miyamoto M, Sastre A, Schnaar RL, Coyle JT. Glutamate Toxicity in a Neuronal Cell Line Involves Inhibition of Cystine Transport Leading to Oxidative Stress. Neuron (1989) 2:1547–58. doi: 10.1016/0896-6273(89)90043-3
145. Yang WS, Shimada K, Delva D, Patel M, Ode E, Skouta R, et al. Identification of Simple Compounds With Microtubule-Binding Activity That Inhibit Cancer Cell Growth With High Potency. ACS Med Chem Lett (2012) 3(1):35–8. doi: 10.1021/ml200195s
Keywords: lipid metabolism, clear cell renal cell carcinoma, Warburg effect, glucose metabolism, cholesterol
Citation: Qi X, Li Q, Che X, Wang Q and Wu G (2021) The Uniqueness of Clear Cell Renal Cell Carcinoma: Summary of the Process and Abnormality of Glucose Metabolism and Lipid Metabolism in ccRCC. Front. Oncol. 11:727778. doi: 10.3389/fonc.2021.727778
Received: 22 June 2021; Accepted: 10 August 2021;
Published: 15 September 2021.
Edited by:
Katarína Smolková, Academy of Sciences of the Czech Republic (ASCR), CzechiaReviewed by:
Eric Hervouet, Université Bourgogne Franche-Comté, FranceCopyright © 2021 Qi, Li, Che, Wang and Wu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Guangzhen Wu, d3VndWFuZzA2MTNAaG90bWFpbC5jb20=; Qifei Wang, d2FuZ3FpZmVpNjAwOEBob3RtYWlsLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.