
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Oncol. , 22 September 2021
Sec. Hematologic Malignancies
Volume 11 - 2021 | https://doi.org/10.3389/fonc.2021.720261
 Xiaolan Lai1
Xiaolan Lai1 Chuanqian Huang2
Chuanqian Huang2 Xuekun Nie3
Xuekun Nie3 Qi Chen1
Qi Chen1 Yirong Tang1
Yirong Tang1 Xianguo Fu4
Xianguo Fu4 Ying Lin1
Ying Lin1 Chengjun Nie1
Chengjun Nie1 Xinyu Xu1
Xinyu Xu1 Xiukang Wang1
Xiukang Wang1 Renli Chen1*
Renli Chen1* Zichun Chen3*
Zichun Chen3*Multiple myeloma (MM) is a malignant cancer with an increasing in incidence that can be alleviated through bortezomib (BTZ) treatment. Activating transcription factor 3 (ATF3) plays a major role in cancer development. Moreover, microRNAs (miRNAs) regulate carcinogenic pathways, apoptosis, and programmed necrotic cell death. However, the detailed mechanism by which ATF3 modulates BTZ drug sensitivity/resistance remains elusive. In the current study, expression of ATF3 was significantly increased under BTZ treatment in a dose-dependent manner in MM cell lines. In addition, ATF3 could regulate cell apoptosis under BTZ treatment. The effect of ATF3 was negatively regulated by its binding miRNA, miR-135a-5p. When either ATF3 was silenced or miR-135a-5p mimics were added to MM cells, they partially lost sensitivity to BTZ treatment. This was accompanied by low levels of Noxa, CHOP, and DR5, and a decrease in mitochondrial membrane potential. These results revealed the combinatorial regulatory patterns of ATF3 and miR-135a-5p in the regulatory protein interactome, which indicated a clinical significance of the miR-135a-5p-ATF3 protein interaction network in BTZ therapy. This study provides potential evidence for further investigation into BTZ resistance.
As a primary bone cancer, multiple myeloma (MM) commonly occurs in the elderly population (aged ≥70 years), which has an increasing incidence and can cause hematologic malignancy (1, 2). Currently, the therapeutic options for MM patients are mainly based on age, tumor stage, and comorbidities (3). A frequently used chemotherapeutic drug in MM is bortezomib (BTZ), it acts as a proteasome inhibitor and interferes with the degradation of misfolded proteins (4, 5). BTZ suppresses ubiquitin-dependent protein degradation and preferentially kills various tumor cells (6). BTZ also sensitizes cells to oxidative damage by the iron status to enhance proteasome inhibition (7, 8). Despite the unsurpassed anti-tumor activity of BTZ for MM (9), drug resistance and a lack of sensitivity to BTZ have restricted the clinical application and patient benefits (10). Thus, elucidation of the mechanism by which BTZ drug sensitivity/resistance occurs in MM would facilitate development of treatments for this disease.
Activating transcription factor (ATF) has been reported to be disrupted in BTZ-treated MM cells (5). After silencing ATF3 or ATF4 expression, MM cells partially lose sensitivity to BTZ treatment and have decreased levels of Noxa, C/EBP-homologous protein (CHOP), and Death receptor 5 (DR5) (11). Additionally, bioinformatics analysis of MM microarray data (GSE13591) has shown that ATF3 is deregulated in this disease (12). Overexpression of ATF3, ATF4, and ATF5 induced by BTZ treatment is associated with sensitivity to BTZ-induced apoptosis in B-lymphoma cells (13). Low levels of XBP1, ATF3, and ATF4 are observed in poor responders to BTZ treatment (14). Upregulation of chemokine (C-C motif) ligand 2 (CCL2) resulting from an enhanced interaction between c-Jun and ATF3 in the brain contributes to BTZ-induced mechanical allodynia (4). However, the detailed mechanism by which BTZ regulates ATF3 remains elusive.
Aberrant expression of microRNAs (miRNAs) has been reported to be a molecular pathogenesis of MM. For example, miR-155 and miR-33b expression may elicit anti-MM activity, likely via proteasome inhibition (15, 16). A study shown that miR-22 negatively regulates the anti-apoptotic protein phosphoprotein enriched in diabetes (PED) and tumor necrosis factor-related apoptosis-inducing ligand-induced cell death in non-small cell lung cancer cells (17). The potential of miRNAs to participate in BTZ effectiveness and resistance was provided insights into promising therapeutic options for patients with MM (18). Combined overexpression of miR-520g and miR-520h exerts an inhibitive effect on BTZ-resistant MM tumor growth in vivo (19). MiR-200c is capable of enhancing BTZ-induced cancer cell death through the pro-apoptotic Bcl-2 family member Noxa (20). Furthermore, miR-29b has been reported to be a potentiator that synergistically promotes the anti-myeloma effect of BTZ (10).
Interestingly, expression of ATF3 is closely associated with miRNAs such as miR-488 (21). miR-342-3p directly targets ATF3 to modulate transcription of pro-osteogenic differentiation-associated genes (22). MiR-494 affects ATFs through binding to its 3′UTR (23). However, in MM, miRNAs-ATF3 regulatory protein interaction network in BTZ therapy is unclear. In this study, we investigated the detailed mechanisms by which BTZ modulates MM cells and demonstrate an interaction between ATF3 and miRNA in BTZ actions.
Human MM cell linea RPMI-8226 and U-266 were obtained from Xiamen Immocell Biotechnology Co. Ltd. (China). Cells were cultured in RPMI-1640 medium with 10% FBS (fetal bovine serum), 100 U/mL penicillin and streptomycin each, and 2 mM L-glutamine at 37°C in a 5% CO2 incubator.
A total of 3 × 104 cells/well were seeded in 96-well plates. After treatment with various concentrations of BTZ (S1013, Selleck) at 37°C for 24 h, cell proliferation was measured using an MTT cell proliferation assay kit (Beyotime, Shanghai). Absorbance was measured at 490 nm using a SpectraMax Absorbance Reader (Molecular Devices, San Francisco, CA, USA). The experiment was repeated three times.
RNA extracted from the cells was subjected to reverse transcription (RT) using Superscript III Reverse Transcriptase (Invitrogen, Thermo Fisher Scientific, Inc.) at 50°C for 45 min. qPCR was conducted using a Bio-Rad CFX96 system and ChamQ SYBR® qPCR Master Mix kit (Vazyme Biotech Co., Ltd.) to determine the mRNA or miRNA expression levels of the genes of interest. Quantitation was performed as described previously (24). The thermocycling conditions were 98°C for 2 min, followed by 30 cycles of 98°C for 10 s, 60°C for 25 s, and 72°C for 25 s. Each reaction was performed in triplicate. Expression levels were normalized to those of U6 or 18S ribosomal RNA. The primers used for qPCR are listed in Supplemental Table 1.
Cells were lysed in ice-cold radioimmunoprecipitation assay buffer (Sangon Biotech Co., Ltd.). The total protein was quantitated using a BCA Protein Assay kit (Abcam). Lysates (20 mg/sample) were resolved by 8%–12% denaturing SDS-PAGE and the proteins were transferred to a PVDF membrane (Roche Diagnostics GmbH). Tris-HCl buffer with 5% bovine serum albumin (BSA; Beijing Solarbio Science & Technology Co., Ltd.) was used to block the membranes at 28°C for 2 h. The membrane was probed with primary antibodies in Tris-HCl buffer with 5% BSA at 4°C overnight. Subsequently, the membranes were washed three times for 5 min each wash with Tris-HCl buffer that contained 0.1% Tween-20, followed by incubation with the corresponding secondary antibodies in Tris-HCl buffer at 28°C for 1 h. Finally, the membranes were washed three times for 5 min each wash, developed, and visualized using an enhanced chemiluminescence detection system (Thermo Fisher Scientific, Inc.). The antibodies used in this study were anti-ATF3 (1:500), anti-GAPDH (1:5,000; cat. no. YM3029; ImmunoWay Biotechnology), HRP-conjugated goat anti-rabbit IgG (1:1,000; cat. no. 7074; Cell Signaling Technology, Inc.), and HRP-conjugated rabbit anti-mouse IgG (1:1,000; cat. no. 7076; Cell Signaling Technology, Inc.). Detailed information on the antibodies is shown in Supplemental Table 2. Western blot densitometry was performed using ImageJ software.
For ATF3 knockdown, human ATF3-targeting shRNA sequences were cloned into a plko.1 vector. primers for construction of plko.1-shATF3 plasmid were synthesized from Ribobio (Guangzhou, Guangdong, China) and the sequences were shown in Supplemental Table 3. A control shRNA unrelated to the human sequences was used as a negative control.
Genomic DNA of the cells was isolated using a FastPure Cell DNA Isolation Mini kit (cat. no. DC102; Vazyme Biotech Co., Ltd.). The 3′UTR of the human ATF3 gene was amplified by PCR using the genomic DNA with a 2X Phanta Master Mix kit (cat. no. P511-01; Vazyme Biotech Co., Ltd.). The PCR conditions were 98°C for 2 min, followed by 30 cycles of 98°C for 15 s, 60°C for 15 s, and 72°C for 60 s. ATF3, CHOP, DR5, and Noxa 3′UTR fragments were cloned downstream of the firefly luciferase reporter gene in the pmirGLO vector (Promega), to generate pmirGLO-ATF3, -CHOP, -DR5, and Noxa 3′UTR WT plasmids. Mutant ATF3, CHOP, DR5, and Noxa 3′UTRs were generated using a QuikChange II Site-Directed Mutagenesis kit (Agilent Technologies, Inc.) and cloned into the pmirGLO vector to generate the pmirGLO-3′UTR mutant plasmid. The primers for gene cloning are shown in Supplemental Table 3.
MM cells were seeded in 6-well plates at a density of 2 × 106/well. Four micrograms of plasmids (shcrtl or shATF3) or 200 pmol mimics (mimic NC or miR-135a-5p mimic) were transfected into cells using Lipofectamine® 3000 (Invitrogen; Thermo Fisher Scientific, Inc.). Six hours after transfection, RPMI-8226 and U-266 cells were treated with 10 or 20 nM BTZ at 37°C for 24 h. After treatment, the cells were harvested and used for reactive oxygen species (ROS) measurement, mitochondrial membrane potential (MMP) detection, and apoptosis assays.
The ROS level in cells was measured using a Cellular ROS Assay Kit (ab113851, Abcam) in accordance with the manufacturer’s instructions.
A JC-1 staining assay kit (C2006, Beyotime) was used to detect alterations in the mitochondrial membrane potential. Cells were resuspended in 500 μL RPMI-1640 medium and then mixed with 500 μL JC-1 working solution. After incubation for 20 min at 37°C while protect from light, the cells were washed twice with JC-1 staining buffer, and resuspended in 300 μL JC-1 staining buffer, and then analyzed by a NovoCyte FACS (Cat: 1300, ACEA, San Diego, CA, USA).
Cells were stained with Annexin V-fluorescein isothiocyanate and 30 mg/mL PI (Cat: A211-02, Vazyme, Nanjing, China) and were then analyzed by FACS and FlowJo software.
TargetScan (http://www.targetscan.org/vert_72/), miRPathDB (https://mpd.bioinf.uni-sb.de/), DIANA Tools (http://diana.imis.athenainnovation.gr/DianaTools/), and mirDIP (http://ophid.utoronto.ca/mirDIP/index.jsp) databases were used to screen ATF3 targets and binding. Statistical differences in survival rates were analyzed using the log-rank test.
Act D was purchased from Selleck (S9624, Shanghai, China). Cells were treated with Act D at a concentration in accordance with the IC50.
MiR-135a-5p mimics and negative control mimics (mimics ctrl) were obtained from Guangzhou RiboBio Co., Ltd. The sequences were miR-135a-5p mimics 5′-UAUGGCUUUUUAUUCCUAUGUGA-3′ and mimics ctrl 5′-UUCUCCGAACGUGUCACGUTT-3′. Cells were seeded in a six-well plate (1×106 cells/well) and transfected with mimics ctrl or miR-135a-5p mimics (200 pmol/well), along with the pmirGLO-WT ATF3 3UTR (4 μg/well) or pmirGLO-Mutant ATF3 3′UTR plasmid (4 μg/well) using Lipofectamine® 3000 (Invitrogen; Thermo Fisher Scientific, Inc.) and incubated at 37°C. The cells were lysed at 50 h post-transfection and luciferase activity was measured using the Dual-Glo Luciferase Assay System (Promega) in accordance with the manufacturer’s instructions. Firefly luciferase activity was normalized to Renilla luciferase activity. Each treatment was carried out in triplicate.
All statistical analyses were conducted using SPSS version 22.0 (IBM) and GraphPad Prism version 8.0.2 (GraphPad Software, Inc.). Data are presented as the mean ± SD. Differences between two groups were analyzed using the Student’s t-test and one-way ANOVA followed by Tukey’s post-hoc test was used for multiple comparisons among three or more experimental groups. P < 0.05 was considered to indicate a significant difference.
To examine the dosage effects of BTZ on MM cell proliferation, the proliferation of MM cell lines RPMI-8226 and U-266 under various BTZ treatments was determined by MTT assays. There was a BTZ dose-dependent toxic effect by killing MM cells with an IC50 = 15.9 nM in RPMI-8226 cells and an IC50 = 7.1 nM in U-266 cells (Figure 1A). Additionally, the level of ATF3 mRNA was significantly increased under BTZ treatment in a dose-dependent manner in both MM cell lines (Figure 1B). Correspondingly, the protein level of ATF3 was also increased by BTZ (Figures 1C, D). These results indicated that BTZ inhibited cell proliferation by upregulating ATF3.
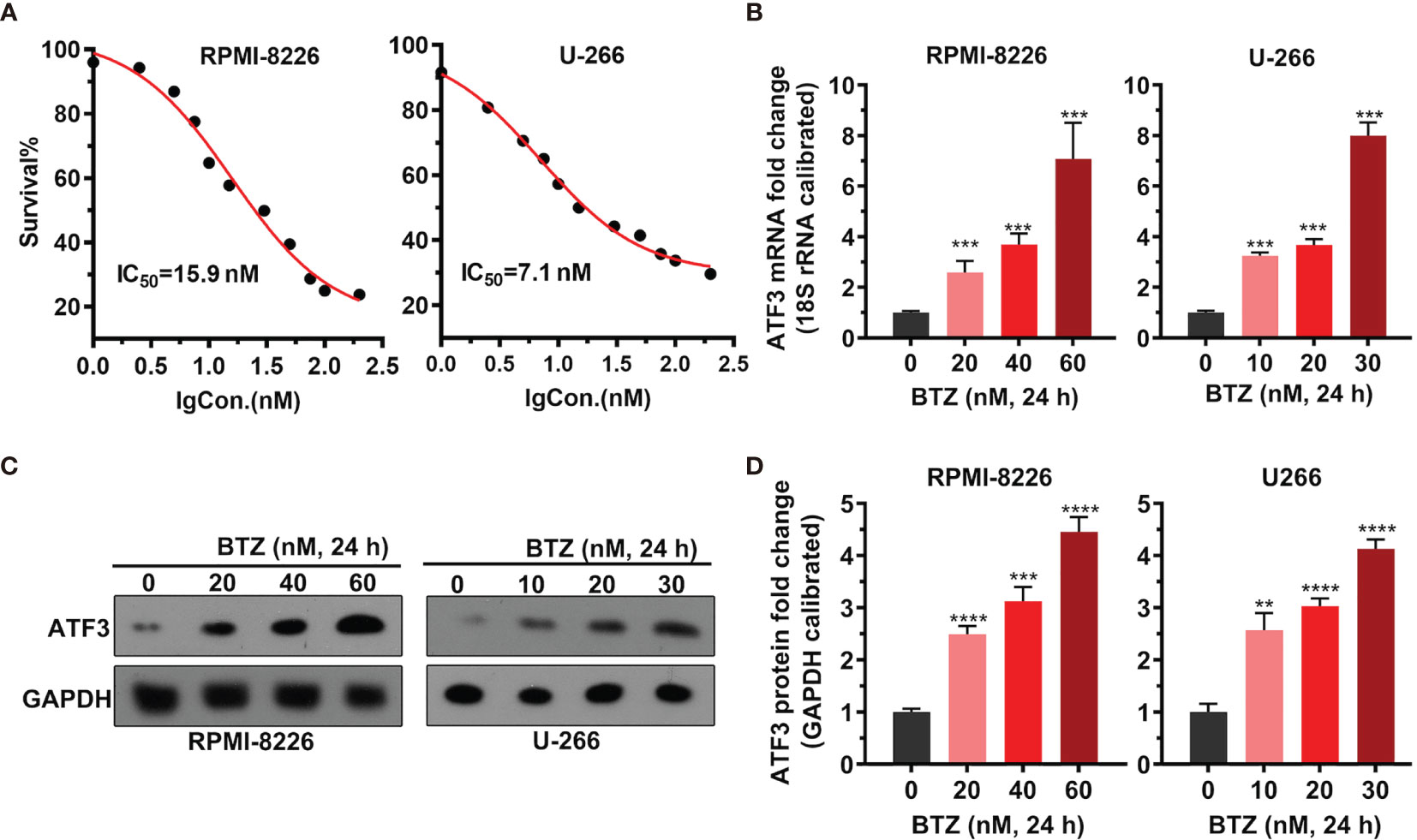
Figure 1 Effects of bortezomib (BTZ) on multiple myeloma (MM) cell proliferation and ATF3 expression. (A) MM cell proliferation was assessed by MTT assays. BTZ was used to treat RPMI-8226 and U-266 cells at various doses. (B) The ATF3 mRNA level was measured by RT-qPCR in RPMI-8226 and U-266 cells treated with various doses of BTZ. One-way ANOVA was used for statistical analysis. (C) The ATF3 protein level was measured by western blotting in RPMI-8226 and U-266 cells treated with various doses of BTZ. (D) Quantification of ATF3 band intensity, with values normalized to GAPDH. Data were analyzed by one-way ANOVA, followed by Tukey’s post-hoc test. **P < 0.01, ***P < 0.001, ****P < 0.0001 compared to 0 h.
To confirm the role of ATF3 during BTZ treatment in MM cells, ATF3 expression was modified by shRNA knockdown. ATF3 was successfully knocked down in RPMI-8226 and U-266 cells (Supplementary Figure 1). First, shATF3 significantly reduced the increased level of ATF3 mRNA and protein induced by 24 h of BTZ treatment using the IC50 dose (Figures 2A, B). Considering that the anti-tumor effect of BTZ may be associated with the apoptotic pathway, we measured apoptosis activity after BTZ treatment and the effect of ATF3 on apoptosis. A BTZ-induced increase in the ROS level was attenuated by ATF3 knockdown (Figures 2C, D). Similarly, in the JC-1 staining assay, a BTZ-induced elevation in the MMP was reduced by ATF3 knockdown (Figures 2E, F). Additionally, BTZ increased the number of apoptotic cells, which was significantly reversed by ATF3 knockdown in the Annexin-PI assay (Figures 2G, H).
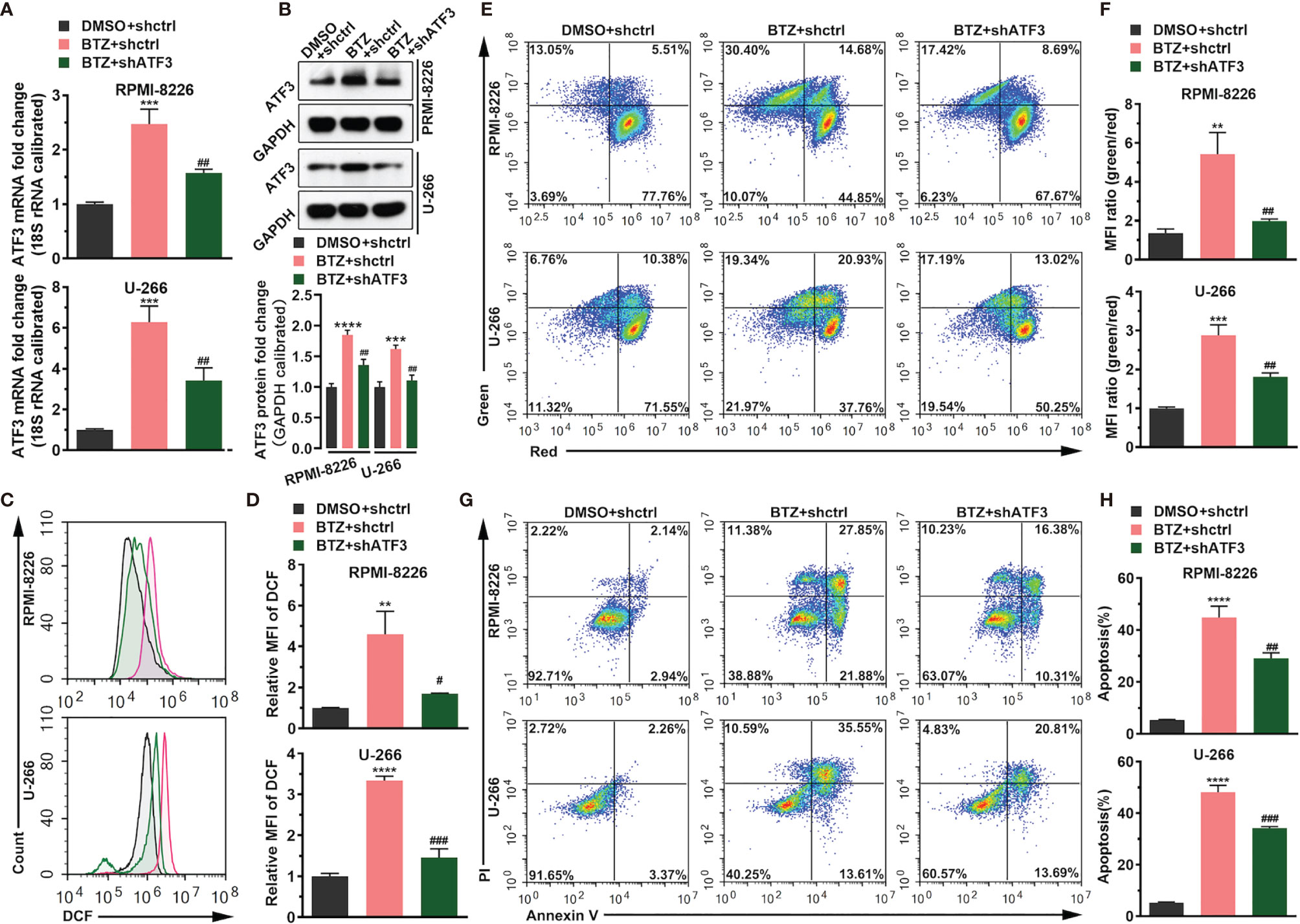
Figure 2 BTZ modulates cell apoptosis via ATF3. Apoptosis was analyzed in three groups: (A) DMSO + shRNA ctrl; (B) bortezomib (BTZ) + shRNA ctrl; (C) BTZ + shATF3. (A) The ATF3 mRNA level was analyzed by RT-qPCR in RPMI-8226 and U-266 cells of the three groups. (B) The ATF3 protein level was analyzed in the three groups. Quantification of ATF3 band intensity, with values normalized to GAPDH. (C) The reactive oxygen species (ROS) level was measured by the Cellular ROS Assay Kit in the three groups. 2′,7′-Dichlorofluorescein (DCF) was detected by FACS. (D) Quantitation of the ROS level indicated by DCF. (E) The mitochondrial membrane potential (MMP) was analyzed by the JC-1 assay. (F) Quantitation of the MMP level indicated by the mean fluorescence intensity (MFI). (G) Apoptosis was analyzed by annexin-V PI staining. (H) Quantitation of the percentage of apoptotic cells. Data were analyzed by one-way ANOVA, followed by Tukey’s post-hoc test. **P < 0.01, ***P < 0.001, and ****P < 0.0001 compared with the BTZ + shctrl group; #P < 0.05, ##P < 0.01, and, ###P value < 0.001 compared with the BTZ + shATF3 group.
Figure 3 shows that BTZ reversed Act-D (potent inhibitor of transcription) inhibitory effects on ATF3 mRNA expression (Figure 3A). Additionally, the luciferase assay showed that BTZ increased the promoter activity of ATF3 (Figure 3B). To determine potential miRNAs that acted as master regulators in this process and miRNAs that targeted ATF3, we screened miRNAs that directly targeted ATF3 mRNA in TargetScan, miRDB, mirDIP, and DIANA databases. As shown in the Venn plot, miR-135a-5p was a common miRNA that targeted ATF3 (Figure 3C). Furthermore, the expression level of miR-135a-5p was downregulated in the MM cell lines under BTZ treatment, which was inversely associated with ATF3 mRNA expression (Figure 3D). Furthermore, we found conserved binding sites between the ATF3 3′UTR region and miR-135a-5p (Figure 3E). Additionally, the dual luciferase reporter assay revealed a significant decrease in luciferase activity in MM cells that were cotransfected with miR-135a-5p and the ATF3-3′UTR luciferase reporter vector compared with the luciferase activity in cells cotransfected with the negative control (mimics ctrl) (Figure 3F). The ATF3-3UTR mutant abolished the interaction between miR-135a-5p and the ATF3 3′UTR region in both RPMI-8226 and U-266 cell lines (Figure 3F). Furthermore, miR-135a-5p mimics increased miR-135a-5p expression (Figure 3G). Accordingly, the levels of ATF3 mRNA and protein were reduced by miR-135a-5p mimic-transfected in cell lines (Figures 3H, I). Taken together, the overall action of BTZ in MM cells was associated with ATF3 and miR-135a-5p expression.
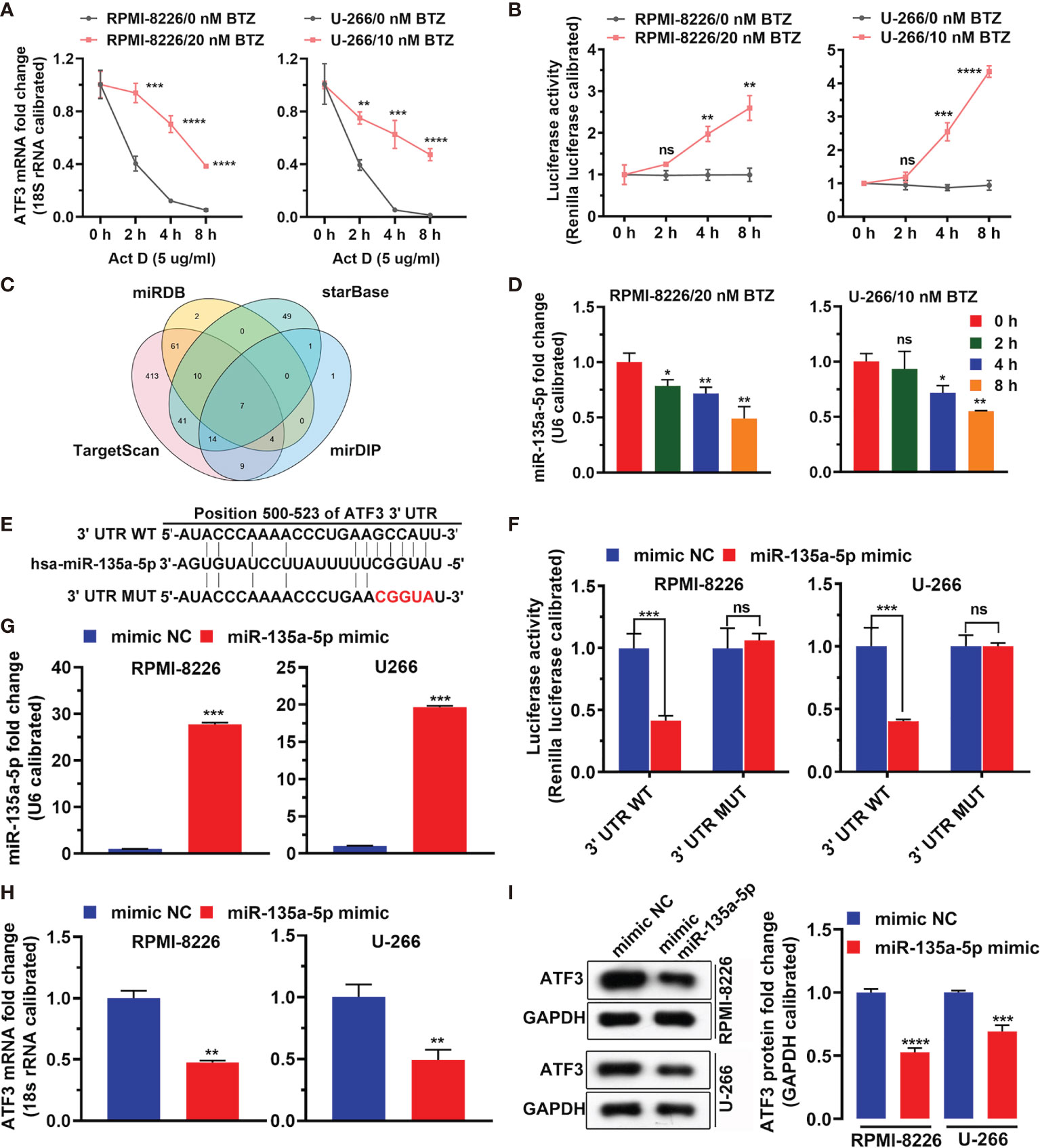
Figure 3 MiRNA regulates ATF3 after BTZ treatment. (A) Dynamic level of the ATF3 mRNA change after actinomycin D (Act D) treatment at various doses. Data was analyzed by one-way ANOVA, followed by Tukey’s post-hoc test. **P < 0.01, ***P < 0.001, and ****P < 0.0001. (B) Luciferase activity of ATF3 after Act D treatment at various doses. Data was analyzed by one-way ANOVA, followed by Tukey’s post-hoc test. **P < 0.01, ***P < 0.001, and ****P < 0.0001. (C) Venn plot of common miRNAs that target ATF3 in TargetScan, miRDB, mirDIP, and DIANA databases. (D) Expression of miR-135a-5p was measured by RT-qPCR in cells treated with BTZ. Data were analyzed by one-way ANOVA, followed by Tukey’s post-hoc test. Ns, no significance; *P < 0.05, and **P < 0.01 compared with 0 h (E) Conserved and mutant binding sites between the ATF3 3′UTR and miR-135a-5p. (F) Dual luciferase reporter assay showing reduced luciferase reporter activity in MM cells that contained the ATF3 3′UTR WT fragment. (G) MiR-135a-5p mimics increased miR-135a-5p expression assessed by RT-qPCR. (H) The ATF3 mRNA level was reduced by miR-135a-5p mimic transfection as assessed by RT-qPCR. (I) Left panel: The ATF3 protein level was reduced by miR-135a-5p mimic transfection in cell lines as measured by western blotting. Right panel: Quantification of ATF3 band intensity, with values normalized to GAPDH. Data are all presented as the mean ± SD. Differences between two groups were analyzed by the Student’s t-test. *P < 0.05, **P < 0.01, ***P < 0.001, ***P < 0.0001. ns, no significance. NC, negative control.
Next, we examined the effects of miR-135a-5p on BTZ-induced MM cell apoptosis. The result shows that BTZ reduced the miR-135a-5p level, and this inhibitory effect was rescued by miR-135a-5p overexpression (Figure 4A). In addition, we observed that miR-135a-5p mimics resulted in reversing BTZ induced ATF3 mRNA and protein expression levels (Figures 4B, C). Furthermore, BTZ-induced increases in ROS levels were reversed by the miR-135a-5p mimics (Figures 4D, E). The enhancing effect of BTZ on the MMP was reduced by miR-135a-5p mimics (Figure 4F). Moreover, overexpression of miR-135a-5p reversed the BTZ-induced increase in apoptosis (Figure 4G).
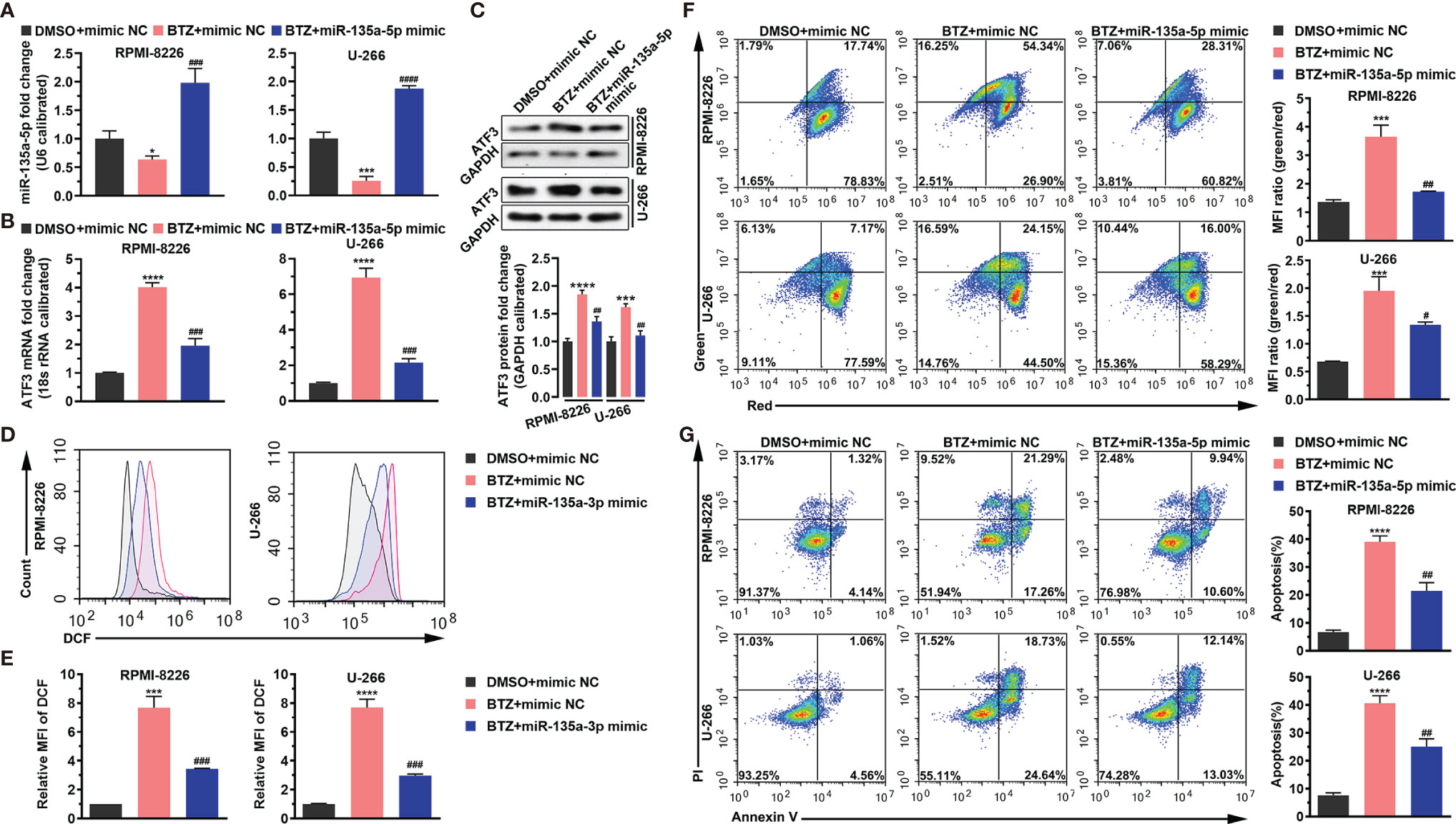
Figure 4 BTZ modulates apoptosis dependently on miR-135a-5p. Apoptosis was analyzed in three groups: (A). DMSO + mimics NC; (B) BTZ + mimics NC; (C) BTZ + miR-135a-5p mimics. (A) The miR-135a-5p level was analyzed by RT-qPCR in RPMI-8226 and U-266 cells of the three groups. (B) The ATF3 mRNA level was analyzed by RT-qPCR in RPMI-8226 and U-266 cells of the three groups. (C) The ATF3 protein level was analyzed in the three groups. Quantification of ATF3 band intensity, with values normalized to GAPDH. (D) The ROS level was measured by the Cellular ROS Assay Kit in the three groups. DCF was detected by FACS. (E) Quantitation of the ROS level indicated by DCF. (F) Left panel: The MMP was analyzed by the JC-1 assay. Right panel: Quantitation of the MMP level indicated by MFI. (G) Left panel: Apoptosis was analyzed by annexin-V PI staining. Right panel: Quantitation of the percentage of apoptotic cells. One-way ANOVA, followed by Tukey’s post-hoc test was used for statistical analysis. ***P < 0.001 and ****P < 0.0001 compared with the BTZ + mimic NC group; #P < 0.05, ##P < 0.01, ###P < 0.001, and ####P < 0.0001 compared with the BTZ + miR-135a-5p mimic group. *P < 0.05 compared with the BTZ+mimic NC group.
To explore the signaling pathways through which BTZ modulated MM cell apoptosis, we examined the CHOP transcription factor related to endoplasmic reticulum (ER) stress-induced apoptosis, CHOP-dependent death receptor 5 (DR5), and Noxa, a proapoptotic BH3-only protein. The results showed that miR-135a-5p reversed the increases in promoter activities of CHOP, DR5, and Noxa mediated by BTZ and their mutants decreased the activities (Figure 5A) Furthermore, miR-135a-5p attenuated the BTZ-induced increases in CHOP, DR5 and Noxa mRNA levels (Figure 5B). Similarly, miR-135a-5p reversed the enhancing effects of BTZ on CHOP, DR5 and Noxa protein levels (Figures 5C, D).
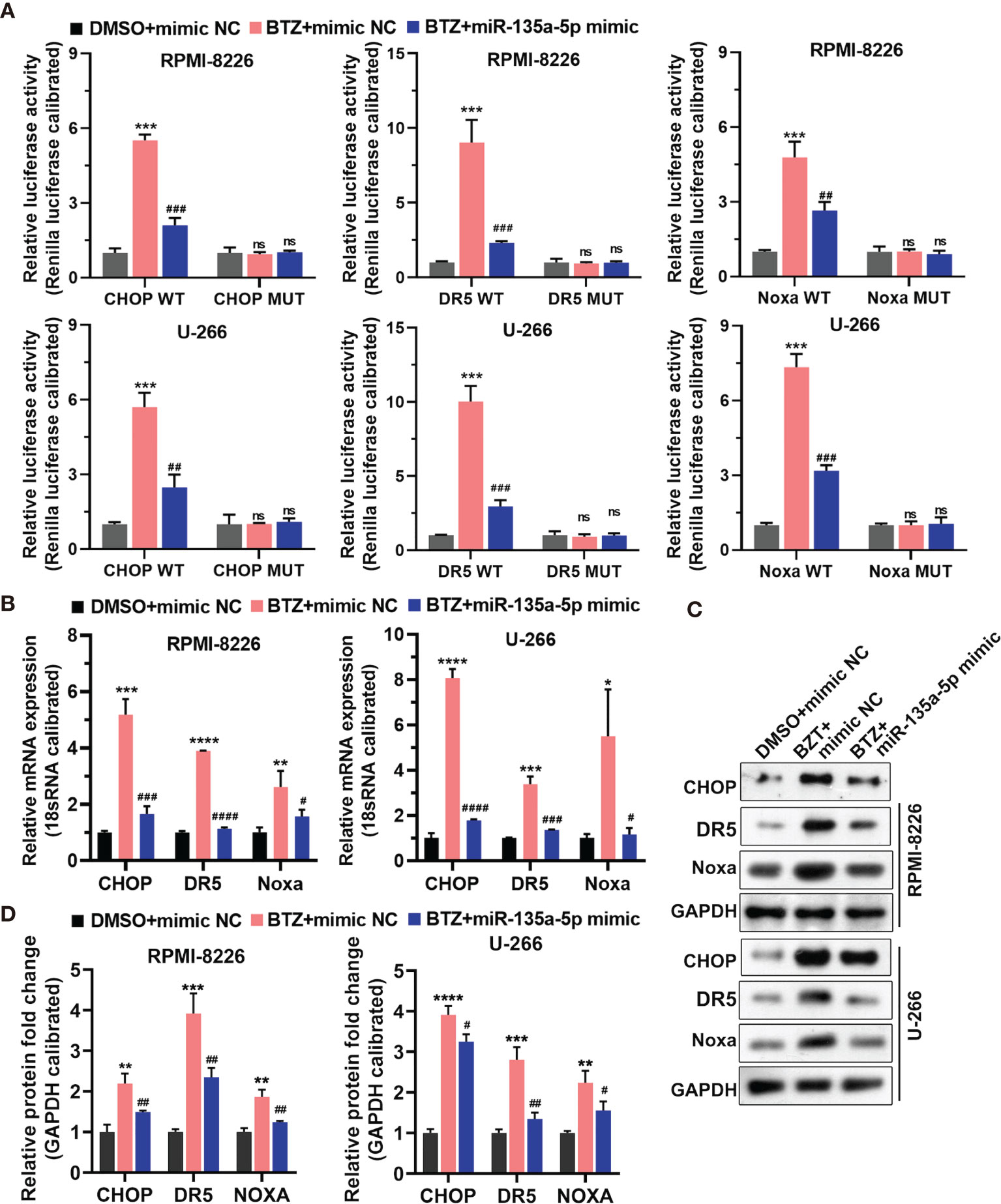
Figure 5 Expression of CHOP, DR5, and Noxa mRNA was analyzed in three groups: (A) DMSO + mimics NC; (B) BTZ + mimics NC; (C) BTZ + miR-135a-5p mimics. (A) The relative luciferase activity of CHOP, DR5, and Noxa was detected in RPMI-8226 and U-266 cells of the three groups. (B) CHOP, DR5, and Noxa mRNA levels were analyzed by RT-qPCR in the three groups. (C) CHOP, DR5 and Noxa protein levels were measured by western blotting in the three groups. (D) Quantification of CHOP, DR5, and Noxa band intensity, with values normalized to GAPDH. One-way ANOVA followed by Tukey’s post-hoc test was used for statistical analysis. Ns, no significance; *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001 compared with the BTZ + mimic NC group; #P < 0.05, ##P < 0.01, ###P < 0.001, and ####P < 0.0001 compared with BTZ + miR-135a-5p mimic group.
MM is a life-threatening cancer that can be effectively treated by BTZ. However, BTZ resistance may develop over the course of therapy. Full exploration of the detailed mechanism of BTZ effectiveness and tolerance in MM would be beneficial to patients. In the present study, we found that BTZ inhibited cell proliferation and expression of ATF3 was significantly increased under BTZ treatment in a dose-dependent manner. This indicated that ATF3 might function as an important regulator in BTZ effectiveness against MM. This result was consistent with a previous report in which BTZ resistance was increased after silencing ATF3 expression (25).Additionally, expression of ATF3 is closely related to the BTZ treatment response and progression-free survival (25). We also found that knockdown of ATF3 reduced the effects of BTZ on apoptosis, which further supports the hypothesis that ATF3 plays an important role in BTZ treatment of MM.
It was very interesting to observe the co-occurrence of increased apoptotic and necrotic cells induced by BTZ, which was partially blocked by shATF3. We believe that BTZ-induced apoptosis can be considered a hallmark of MM treatment. In the BTZ treatment process, MM cells defensively trigger non-apoptotic cell death pathways, such as necrosis, autophagy, and mitotic catastrophe, to resistant apoptosis induced by BTZ. Such tumorigenesis may be associated with mitotic kinases, the ATP level, and mitochondria. While co-occurrence of apoptosis and necrosis is poorly understood, we consider that such a condition is mediated by the network of internal/external stress factors and ATP/plasma membrane permeabilization and the MMP. In the current study, it was surprising that ATF3 knockdown caused a significant decrease in necrosis induced by BTZ in U266 cells, but not RPMI-8226 cells, which suggested a distinct role of ATF3 in the different MM cell lines. Therefore, we should further investigate the role of ATF3 in death signal stimulation to reveal shared pathways.
Furthermore, we identified miR-135a-5p as an miRNA that directly targeted ATF3 mRNA and downregulated its expression under BTZ treatment, which indicated that miR-135a-5p may act as a regulatory microRNA. Finally, we investigated the effects of miR-135a-5p and ATF3 mRNA on BTZ treatment of MM cells. The results demonstrated that miR-135a-5p reduced the effects of BTZ on the apoptotic process, which might influence expression of CHOP, DR5, and Noxa proteins. It is well recognized that transcriptional activation of CHOP is a major event in ER stress-mediated apoptosis. Moreover, ER stress-induced CHOP activation is associated with DR5 in human cancer cells. CHOP-mediated DR5 transcription is dependent on several transcription factors such as ATF3 and ATF4, Therefore, we determined whether BTZ-induced apoptosis was mediated through the ATF3-CHOP/DR5 axis. Additionally, Noxa gene expression is mediated by ATF3 in Noxa-dependent intrinsic apoptosis, i.e., the ATF3-CHOP/Noxa axis. For example, the CHOP/NOXA/Mcl-1 axis contributes to LiCl-induced apoptosis (26), and ATF4-triggered Noxa- and DR5-dependent extrinsic apoptosis have been seen in fangchinoline-treated esophageal squamous cells (27). Because ER stress in BTZ-treated MM cell lines is beyond their self-defense capacity, the MM cell activates apoptotic signals to remove damage organelles (28). We hypothesize that CHOP/Noxa and CHOP/DR5 axes may be triggered by an ATF3-dependent pattern.
ATF3 is highly prominent in cancer that includes breast, laryngeal, and lung cancers (29). Conversely, other studies have demonstrated that ATF3 functions as a tumor suppressor in other cancers that include esophageal, colon, and bladder cancers (30). In addition, ATF3 and Smad are synergistically involved in JNK-induced apoptosis of MM cells (31). In the brain, BTZ induces oxidative stress, mitochondrial dysfunction, and apoptosis by the activation of ATF3 (32). Moreover, and enhanced interaction between c-Jun and ATF3 promotes BTZ-induced mechanical allodynia (4).
An miRNA can regulate numerous genes and act as either a tumor suppressor or oncogenic miRNA. In MM, combined overexpression of miR-520g and miR-520h inhibits bortezomib-resistant tumor growth in vivo (19). Expression of miR-135a-5p has been found in patients with colorectal cancer (33) and acts as a mechanistic regulator in patients with HCV (34). Additionally, miR-135a-5p inhibits the proliferation of head and neck cancer cells and promotes apoptosis (35). Moreover, miR-135a-3p inhibits ovarian tumorigenesis by regulating CCR2 (36). The proliferation, migration, and invasion of glioma cells are regulated by the miR-135a-5p axis, which provides viable therapeutic avenues for the treatment of glioma (37). MiR-135a-5p may also act as a tumor suppressor in the development of colorectal adenomas and promote apoptosis of colon cancer cells (38). Here, we demonstrated that miR-135a-5p also targeted ATF3 (39). Our results also revealed the importance of miR-135a-5p in BTZ drug sensitivity in MM.
The present study has some limitations such as a lack of rescue experiments and interference experiments of ATF3 mRNA and protein in the cell lines. The MM cell lines for functional assessment were also limited and there was a shortcoming of evidence from BTZ-resistant MM cells in terms of the regulation of ATF3 and miR-135a-5p. Overall, our results suggested crosstalk between miR-135a-5p and ATF3 in BTZ-induced apoptosis and necrotic cell death. It is unknown whether the regulation was in a dual direction or only one direction. Although the detailed association of BTZ and crosstalk of miR-135a-5p and ATF3 remain to be explored further, to the best of our knowledge, this study is the first to demonstrate an interaction of ATF3 and miR-135a-5p in BTZ treatment of MM. In conclusion, these results revealed miR-135a-5p/ATF3 crosstalk in BTZ treatment of MM and suggests a new regulatory protein interactome to potentially provide a therapeutic option for MM.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
XL, Rc, and ZC made substantial contributions to the concept and design of the study as well as the manuscript. XL, CH, XN, QC, YT, XF, YL, CN, XX, and XW conducted the experiments and data analysis. All authors contributed to the article and approved the submitted version.
The present study was supported by grants from the Natural Science Foundation of Fujian Province (grant no. 2018J05151 and 2019J01626).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank Mitchell Arico from Liwen Bianji (Edanz) (https://www.liwenbianji.cn) for editing the language of a draft of this manuscript.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2021.720261/full#supplementary-material
2. Kazandjian D. Multiple Myeloma Epidemiology and Survival: A Unique Malignancy. Semin Oncol (2016) 43(6):676–81. doi: 10.1053/j.seminoncol.2016.11.004
3. Pawlyn C, Davies FE. Toward Personalized Treatment in Multiple Myeloma Based on Molecular Characteristics. Blood (2019) 133(7):660–75. doi: 10.1182/blood-2018-09-825331
4. Liu C, Luan S, OuYang H, Huang Z, Wu S, Ma C, et al. Upregulation of CCL2 via ATF3/c-Jun Interaction Mediated the Bortezomib-Induced Peripheral Neuropathy. Brain Behav Immun (2016) 53:96–104. doi: 10.1016/j.bbi.2015.11.004
5. Kawabata S, Gills JJ, Mercado-Matos JR, Lopiccolo J, Wilson W 3rd, Hollander MC, et al. Synergistic Effects of Nelfinavir and Bortezomib on Proteotoxic Death of NSCLC and Multiple Myeloma Cells. Cell Death Dis (2012) 3:e353. doi: 10.1038/cddis.2012.87
6. Shen Y, Lu L, Xu J, Meng W, Qing Y, Liu Y, et al. Bortezomib Induces Apoptosis of Endometrial Cancer Cells Through microRNA-17-5p by Targeting P21. Cell Biol Int (2013) 37(10):1114–21. doi: 10.1002/cbin.10139
7. Campanella A, Santambrogio P, Fontana F, Frenquelli M, Cenci S, Marcatti M, et al. Iron Increases the Susceptibility of Multiple Myeloma Cells to Bortezomib. Haematologica (2013) 98(6):971–9. doi: 10.3324/haematol.2012.074872
8. Bordini J, Galvan S, Ponzoni M, Bertilaccio MT, Chesi M, Bergsagel PL, et al. Induction of Iron Excess Restricts Malignant Plasma Cells Expansion and Potentiates Bortezomib Effect in Models of Multiple Myeloma. Leukemia (2017) 31(4):967–70. doi: 10.1038/leu.2016.346
9. Hao M, Zhang L, An G, Meng H, Han Y, Xie Z, et al. Bone Marrow Stromal Cells Protect Myeloma Cells From Bortezomib Induced Apoptosis by Suppressing microRNA-15a Expression. Leuk Lymphoma (2011) 52(9):1787–94. doi: 10.3109/10428194.2011.576791
10. Jagannathan S, Vad N, Vallabhapurapu S, Vallabhapurapu S, Anderson KC, Driscoll JJ. MiR-29b Replacement Inhibits Proteasomes and Disrupts Aggresome+Autophagosome Formation to Enhance the Antimyeloma Benefit of Bortezomib. Leukemia (2015) 29(3):727–38. doi: 10.1038/leu.2014.279
11. Narita T, Ri M, Masaki A, Mori F, Ito A, Kusumoto S, et al. Lower Expression of Activating Transcription Factors 3 and 4 Correlates With Shorter Progression-Free Survival in Multiple Myeloma Patients Receiving Bortezomib Plus Dexamethasone Therapy. Blood Cancer J (2015) 5:e373. doi: 10.1038/bcj.2015.98
12. Zhang K, Xu Z, Sun Z. Identification of the Key Genes Connected With Plasma Cells of Multiple Myeloma Using Expression Profiles. Onco Targets Ther (2015) 8:1795–803. doi: 10.2147/OTT.S80075
13. Shringarpure R, Catley L, Bhole D, Burger R, Podar K, Tai YT, et al. Gene Expression Analysis of B-Lymphoma Cells Resistant and Sensitive to Bortezomib. Br J Haematol (2006) 134(2):145–56. doi: 10.1111/j.1365-2141.2006.06132.x
14. Ri M. Endoplasmic-Reticulum Stress Pathway-Associated Mechanisms of Action of Proteasome Inhibitors in Multiple Myeloma. Int J Hematol (2016) 104(3):273–80. doi: 10.1007/s12185-016-2016-0
15. Amodio N, Gallo Cantafio ME, Botta C, Agosti V, Federico C, Caracciolo D, et al. Replacement of miR-155 Elicits Tumor Suppressive Activity and Antagonizes Bortezomib Resistance in Multiple Myeloma. Cancers (Basel) (2019) 11(2):236. doi: 10.3390/cancers11020236
16. Li F, Hao M, Feng X, Zang M, Qin Y, Yi S, et al. Downregulated miR-33b Is a Novel Predictor Associated With Disease Progression and Poor Prognosis in Multiple Myeloma. Leuk Res (2015) 39(7):793–9. doi: 10.1016/j.leukres.2015.04.010
17. Incoronato M, Garofalo M, Urso L, Romano G, Quintavalle C, Zanca C, et al. miR-212 Increases Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand Sensitivity in Non-Small Cell Lung Cancer by Targeting the Antiapoptotic Protein PED. Cancer Res (2010) 70(9):3638–46. doi: 10.1158/0008-5472.CAN-09-3341
18. Ballabio E, Armesto M, Breeze CE, Manterola L, Arestin M, Tramonti D, et al. Bortezomib Action in Multiple Myeloma: microRNA-Mediated Synergy (and miR-27a/CDK5 Driven Sensitivity)? Blood Cancer J (2012) 2:e83. doi: 10.1038/bcj.2012.31
19. Yuan X, Ma R, Yang S, Jiang L, Wang Z, Zhu Z, et al. miR-520g and miR-520h Overcome Bortezomib Resistance in Multiple Myeloma via Suppressing APE1. Cell Cycle (2019) 18(14):1660–9. doi: 10.1080/15384101.2019.1632138
20. Lerner M, Haneklaus M, Harada M, Grander D. MiR-200c Regulates Noxa Expression and Sensitivity to Proteasomal Inhibitors. PloS One (2012) 7(5):e36490. doi: 10.1371/journal.pone.0036490
21. Shi B, Yan W, Liu G, Guo Y. MicroRNA-488 Inhibits Tongue Squamous Carcinoma Cell Invasion and EMT by Directly Targeting ATF3. Cell Mol Biol Lett (2018) 23:28. doi: 10.1186/s11658-018-0094-0
22. Han Y, Zhang K, Hong Y, Wang J, Liu Q, Zhang Z, et al. miR-342-3p Promotes Osteogenic Differentiation via Targeting ATF3. FEBS Lett (2018) 592(24):4051–65. doi: 10.1002/1873-3468.13282
23. Lan YF, Chen HH, Lai PF, Cheng CF, Huang YT, Lee YC, et al. MicroRNA-494 Reduces ATF3 Expression and Promotes AKI. J Am Soc Nephrol (2012) 23(12):2012–23. doi: 10.1681/ASN.2012050438
24. Livak KJ, Schmittgen TD. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods (2001) 25(4):402–8. doi: 10.1006/meth.2001.1262
25. Narita T, Ri M, Masaki A, Mori F, Ito A, Kusumoto S, et al. Lower Expression of Activating Transcription Factors 3 and 4 Correlates With Shorter Progression-Free Survival in Multiple Myeloma Patients Receiving Bortezomib Plus Dexamethasone Therapy. Blood Cancer J (2015) 5(12):e373. doi: 10.1038/bcj.2015.98
26. Zhang Q, Zhang Q, Li H, Zhao X, Zhang H. LiCl Induces Apoptosis via CHOP/NOXA/Mcl-1 Axis in Human Choroidal Melanoma Cells. Cancer Cell Int (2021) 21(1):96. doi: 10.1186/s12935-021-01778-2
27. Zhang Y, Wang S, Chen Y, Zhang J, Yang J, Xian J, et al. Fangchinoline Inhibits Human Esophageal Cancer by Transactivating ATF4 to Trigger Both Noxa-Dependent Intrinsic and DR5-Dependent Extrinsic Apoptosis. Front Oncol (2021) 11:666549. doi: 10.3389/fonc.2021.666549
28. Łuczkowska K, Rogińska D, Ulańczyk Z, Paczkowska E, Schmidt CA, Machaliński B. Molecular Mechanisms of Bortezomib Action: Novel Evidence for the miRNA-mRNA Interaction Involvement. Int J Mol Sci (2020) 21(1):350. doi: 10.3390/ijms21010350
29. Song Q, Chen Q, Wang Q, Yang L, Lv D, Jin G, et al. ATF-3/miR-590/GOLPH3 Signaling Pathway Regulates Proliferation of Breast Cancer. BMC Cancer (2018) 18(1):255. doi: 10.1186/s12885-018-4031-4
30. Xie JJ, Xie YM, Chen B, Pan F, Guo JC, Zhao Q, et al. ATF3 Functions as a Novel Tumor Suppressor With Prognostic Significance in Esophageal Squamous Cell Carcinoma. Oncotarget (2014) 5(18):8569–82. doi: 10.18632/oncotarget.2322
31. Kashiwakura Y, Ochiai K, Watanabe M, Abarzua F, Sakaguchi M, Takaoka M, et al. Down-Regulation of Inhibition of Differentiation-1 via Activation of Activating Transcription Factor 3 and Smad Regulates REIC/Dickkopf-3-Induced Apoptosis. Cancer Res (2008) 68(20):8333–41. doi: 10.1158/0008-5472.CAN-08-0080
32. Yin Y, Qi X, Qiao Y, Liu H, Yan Z, Li H, et al. The Association of Neuronal Stress With Activating Transcription Factor 3 in Dorsal Root Ganglion of In Vivo and In Vitro Models of Bortezomib- Induced Neuropathy. Curr Cancer Drug Targets (2019) 19(1):50–64. doi: 10.2174/1568009618666181003170027
33. Wang Q, Zhang H, Shen X, Ju S. Serum microRNA-135a-5p as an Auxiliary Diagnostic Biomarker for Colorectal Cancer. Ann Clin Biochem (2017) 54(1):76–85. doi: 10.1177/0004563216638108
34. Van Renne N, Roca Suarez AA, Duong FHT, Gondeau C, Calabrese D, Fontaine N, et al. miR-135a-5p-Mediated Downregulation of Protein Tyrosine Phosphatase Receptor Delta Is a Candidate Driver of HCV-Associated Hepatocarcinogenesis. Gut (2018) 67(5):953–62. doi: 10.1136/gutjnl-2016-312270
35. Guo LM, Ding GF, Xu W, Ge H, Jiang Y, Chen XJ, et al. MiR-135a-5p Represses Proliferation of HNSCC by Targeting HOXA10. Cancer Biol Ther (2018) 19(11):973–83. doi: 10.1080/15384047.2018.1450112
36. Duan S, Dong X, Hai J, Jiang J, Wang W, Yang J, et al. MicroRNA-135a-3p Is Downregulated and Serves as a Tumour Suppressor in Ovarian Cancer by Targeting CCR2. BioMed Pharmacother (2018) 107:712–20. doi: 10.1016/j.biopha.2018.08.044
37. Feng L, Lin T, Che H, Wang X. Long Noncoding RNA DANCR Knockdown Inhibits Proliferation, Migration and Invasion of Glioma by Regulating miR-135a-5p/BMI1. Cancer Cell Int (2020) 20:53. doi: 10.1186/s12935-020-1123-4
38. Wang J, Yang J, Zhang H, Liao Y, Xu D, Ma S. Effects of miR-135a-5p and miR-141 on Proliferation, Invasion and Apoptosis of Colorectal Cancer SW620 Cells. Oncol Lett (2020) 20(1):914–20. doi: 10.3892/ol.2020.11598
Keywords: multiple myeloma, bortezomib, miR-135a-5p, ATF3, apoptosis
Citation: Lai X, Huang C, Nie X, Chen Q, Tang Y, Fu X, Lin Y, Nie C, Xu X, Wang X, Chen R and Chen Z (2021) Bortezomib Inhibits Multiple Myeloma Cells by Transactivating ATF3 to Trigger miR-135a-5p- Dependent Apoptosis. Front. Oncol. 11:720261. doi: 10.3389/fonc.2021.720261
Received: 04 June 2021; Accepted: 06 September 2021;
Published: 22 September 2021.
Edited by:
Guru Prasad Maiti, Oklahoma Medical Research Foundation, United StatesReviewed by:
Garima Pandey, Moffitt Cancer Center, United StatesCopyright © 2021 Lai, Huang, Nie, Chen, Tang, Fu, Lin, Nie, Xu, Wang, Chen and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zichun Chen, Y2hlbnppY2h1bjAwM0AxNjMuY29t; Renli Chen, Y2hlbnJlbmxpMDA2QHNpbmEuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.