 Ting Yang
Ting Yang Hongqi Xiao2†
Hongqi Xiao2†- 1Department of General Surgery, The Fourth Affiliated Hospital of Harbin Medical University, Harbin, China
- 2Department of General Surgery, The Second Affiliated Hospital of Harbin Medical University, Harbin, China
Preclinical and clinical antiangiogenic approaches, with multiple side effects such as resistance, have not been proved to be very successful in treating tumor blood vessels which are important targets for tumor therapy. Meanwhile, restoring aberrant tumor blood vessels, known as tumor vascular normalization, has been shown not only capable of reducing tumor invasion and metastasis but also of enhancing the effectiveness of chemotherapy, radiation therapy, and immunotherapy. In addition to the introduction of such methods of promoting tumor vascular normalization such as maintaining the balance between proangiogenic and antiangiogenic factors and targeting endothelial cell metabolism, microRNAs, and the extracellular matrix, the latest molecular mechanisms and the potential connections between them were primarily explored. In particular, the immunotherapy-induced normalization of blood vessels further promotes infiltration of immune effector cells, which in turn improves immunotherapy, thus forming an enhanced loop. Thus, immunotherapy in combination with antiangiogenic agents is recommended. Finally, we introduce the imaging technologies and serum markers, which can be used to determine the window for tumor vascular normalization.
Introduction
Since cancer ranks the top killer of human all over the world and approximately accounts for one out of every six deaths, cancer treatment is certain to be at the epicenter of medical research (1). Angiogenesis, which provides nutrients and oxygen for tumor proliferation, is a fundamental requirement for tumor growth, showing targeting tumor angiogenesis would be a promising method to inhibit tumor growth (2).
When exposed to excessive proangiogenic signaling and hypoxia, vascular endothelial cells (ECs), called “phalanx” cells in established vessels, can quickly adapt and switch their phenotype to the so-called “tip” and “stalk,” with tip cells acting as a guide to new blood vessels and extending filopodia and stalk cells being responsible for elongating new blood vessels (3, 4). Besides, cancers can initiate some underlying angiogenesis mechanisms. For example, cancer cells can induce the formation of new vessels or use preexisting vessels (vascular co-option) to maintain growth. The former includes two new vessels splitting from one when interstitial tissue pillars insert into the lumen (intussusception) and new vessels forming from bone marrow-derived endothelial progenitor cells (postnatal vasculogenesis). Vasculogenic mimicry and mosaic vessel formation refer to the transdifferentiation of tumor cells into ECs and the incorporation of tumor cells into the blood vessel wall, respectively (5–7). Although tumor vessels are abundant, the structure and function of these vessels are abnormal, thus creating a hypoxic tumor microenvironment (TME), impeding the infiltration and function of immune cells, and promoting tumor development and metastasis (2, 7). Conventional approaches have focused primarily on inhibiting angiogenesis in tumors by cutting off their blood supply (2, 8). Several antiangiogenic drugs have been approved for the treatment of cancer. However, these antiangiogenic drugs have limited efficacy and drug resistance. Moreover, some antiangiogenic drugs reduce oxygen supply, increase the production of proangiogenic factors, and promote pathological angiogenesis, thus leading to the increased tumor metastasis and relapse in some cancers (9, 10).
A good alternative therapy for antiangiogenic explored in this study is remodeling tumor blood vessels to restore their structure and function, known as vascular normalization, which can, by restoring tumor perfusion and reducing hypoxia, not only prevent cancer cells from acquiring the aggressive phenotypes under the hypoxic microenvironment but also be conducive to other cancer therapies, such as chemotherapy, radiotherapy, and immunotherapy (11) (Figure 1). In this review, the methods and mechanisms of promoting tumor vascular normalization in recent years are introduced, the relationship between tumor vascular normalization and immunotherapy is discussed, and the imaging technologies and serum markers used to determine the window for tumor vascular normalization are summarized.
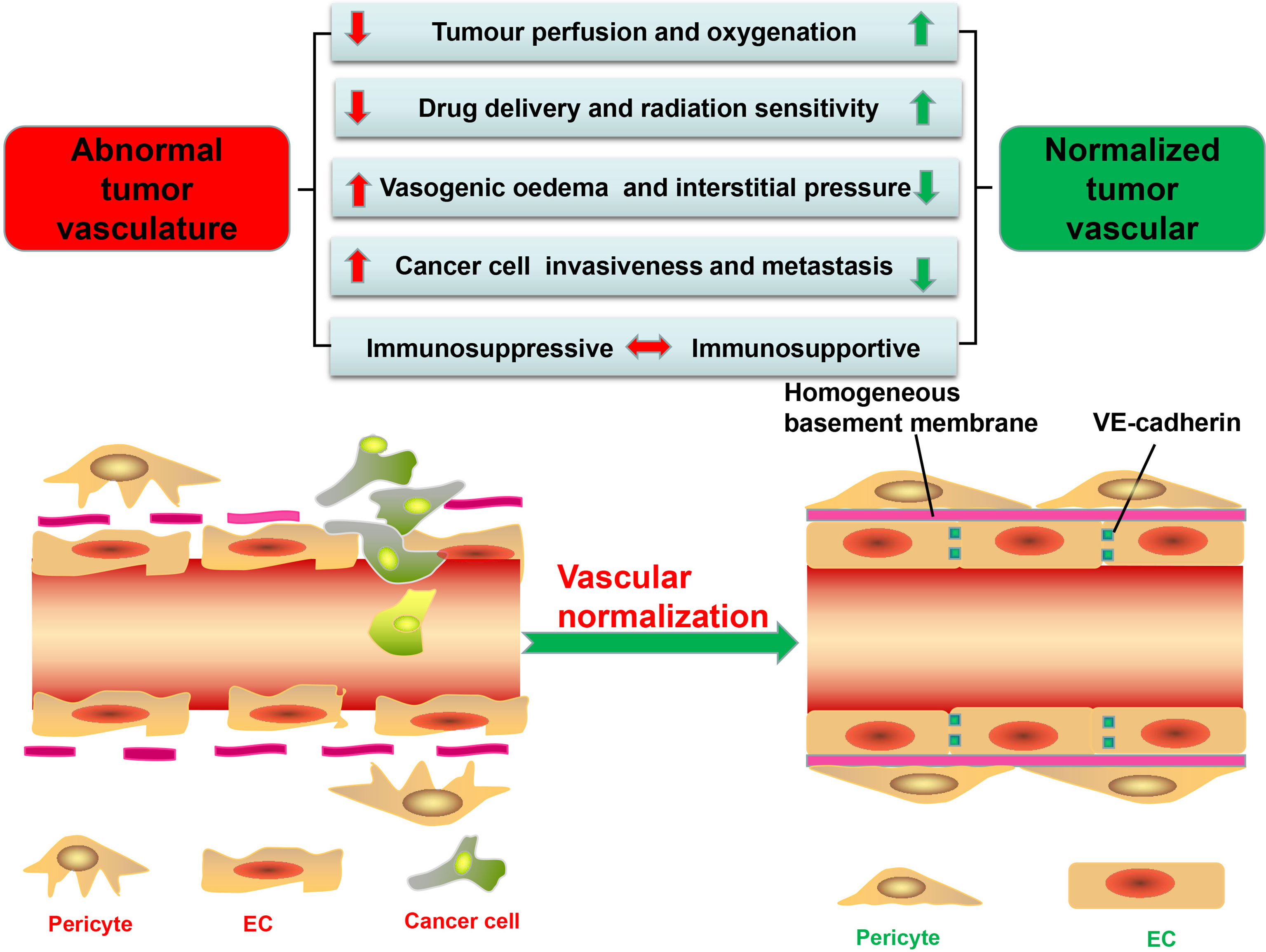
Figure 1 Significant differences in structure and function between tumor vessels and normal vessels. tECs have an irregular shape and lack VE-cadherin, leading to high permeability and disordered arrangement. Besides, tumor vessels have low pericyte coverage and are often separated from endothelial cells (ECs). The basement membrane of tumor vessels is also incomplete and discontinuous, further increasing vascular permeability. These structural abnormalities stimulate the formation of a hypoxic, acidic tumor microenvironment (TME); promote the proliferation and metastasis of tumor cells; and reduce the efficacy of tumor therapy. Furthermore, the abnormal TME promotes the immunosuppression. The methods of tumor vascular normalization introduced in this paper can restore the normal structure and function of blood vessels and enhance the efficacy of antitumor therapy.
Highly Abnormal Tumor Vessels
The Abnormal Vascular Structure of the Tumor
Tumor vessels, aberrant in structure and function and heterogeneous in lumen size and wall thickness, form a chaotic network characterized by permeability and tortuousness (12–14), with perivascular cells, comprised of pericytes and vascular smooth muscle cells, not only displaying abnormal morphology but also having low coverage rate on tumor vessels compared with normal vessels, thus making the wall of the vessel thin and disrupting cellular connections and signal transduction between perivascular cells with ECs and basement membrane (15, 16) (Figure 1). In normal tissues, pericytes, close to ECs, communicate by intimating cell-to-cell contact and mutually secreted factors, leading to an orderly arrangement of ECs and forming an effective and organized mature blood vessel network (17). While in the tumor vessels, they usually detach from the vascular walls, resulting in decreased vascular structural stability and increased vascular permeability (7, 18–20) (Figure 1).
In addition, tumor endothelial cells (tECs) lining vessels have weak junctions and are disorganized (7). Responsible for the connection between vascular ECs thus maintaining the barrier integrity is VE-cadherin, which can be broken down by proteolytic enzymes (such as matrix metalloproteinases, trypsin, or elastase) and inflammatory cytokines released by tumor cells (21, 22) (Figure 1). For example, histamine not only directly impairs human umbilical vein ECs but also indirectly damages VE-cadherin, thereby increasing the permeability of umbilical vein (23). Besides, excessive vascular endothelial growth factor (VEGF), produced by TME, can also increase vascular permeability by regulating several GTPases such as RhoA (24), with ECs immigrating from their resident site and leaving gaps in the absence of VE-cadherin (6, 12, 13). Finally, Ang overexpressed in ECs can decrease endothelial integrity and disrupt pericyte attachment to the endothelial wall (11, 25).
Although tumor blood vessels, compared with normal blood vessels, are almost covered by incomplete and discontinuous basement membrane, confocal and electron microscopy can help discover the vascular basement membrane in tumors with such distinctive abnormalities such as unusual thickness, multiple layers, and weak junction with tECs and pericytes (6, 26), leading to the conclusion that the structure of tumor blood vessels is loose and irregular with high permeability (Figure 1).
Abnormal Function of Tumor Blood Vessels
Despite a rich blood supply, the network of tumor blood vessels is chaotic and lacks the arteriole–capillary–venule hierarchical organization, leading to poor tissue perfusion (12, 15, 27) (Figure 1). Permeable tumor blood vessels facilitate plasma and proteins into the surrounding interstitial space, which not only enhances red blood concentration and blood viscosity but also elevates the interstitial fluid pressure in the TME, further hindering blood flow (27, 28). Moreover, the TME with high quantity of fibers and cells restricts tumor progress and induces the mechanical force which can compress or even collapse intratumoral vessels and severely retard the blood flow into and from tumors (7, 27, 29). The mechanical force also damages lymphatic networks and blocks lymphatic drainage of excessive interstitial fluid (15, 27). All these factors result in increased interstitial fluid pressure, which, in collaboration with poor perfusion, decreases oxygen and nutrient supply, leading to a hypoxic and acidic microenvironment conducive to screening out more aggressive tumor cells for survival, with their ability to metastasize and invade increased by undergoing an “epithelial to mesenchymal transition” (20, 30, 31) (Figure 1). However, abnormal blood vessels also disrupt the blood supply of the tumor, leaving drugs and cytotoxic T lymphocytes (CTLs) from the circulating blood unable to get into the tumors to play their full antitumor role (11, 32).
Methods of Tumor Vascular Normalization
Blocking Angiogenic Factors
The imbalance of signaling mediated by proangiogenic versus antiangiogenic molecules is one of the main mechanisms for abnormal tumor vascular (11, 33), due to the fact that, when in need of the nutrient and oxygen, tumor and stromal cells start secreting proangiogenic factors, such as the VEGF, angiopoietins, platelet-derived growth factors, and fibroblast growth factor, promoting the formation of excessive abnormal blood vessels (34, 35), indicating that targeting tumor blood vessels has become an attractive anticancer therapy approach. Antiangiogenic treatments are thought to have a good anticancer effect by inhibiting blood vessel production, thus leaving cancer cells in starvation with the supply of oxygen and nutrients blocked (8, 20, 36). However, there is a fact that antiangiogenic monotherapy is less effective in colorectal cancer, breast cancer, and nonsmall cell lung cancer, while when combined with chemotherapy drugs, anticancer effect increased significantly (37, 38), which seems to be contradictory to the traditional claim of antiangiogenesis reducing the blood perfusion and further restricting the intratumoral delivery of coadministered drugs. Besides, the hypoxic microenvironment caused by antiangiogenic therapy also renders tumor cells relatively resistant to chemoradiotherapy and even more aggressive (20, 39). By means of transiently normalizing tumor vessels with antiangiogenic therapy, the hypothesis called “vascular normalization” could explain this paradox (33).
Among all vascular angiogenic factors, VEGF, which was first identified as vascular permeability factor by Napoleone Ferrara, promotes EC proliferation and migration, sprouting via binding VEGF receptor–tyrosine kinase-dependent receptor on ECs (38, 40–42). The evidence, from preclinical and clinical studies, shows that blocking VEGF signaling can remodel abnormal tumor vasculature (13, 20, 43). For example, the Willett CG study showed that, after receiving a low dose of bevacizumab (5 mg/kg), patients with rectal cancer showed an increase in pericyte coverage of blood vessels and drug delivery and a decrease in permeability surface product after 12 days, while after receiving a higher dose of bevacizumab (10 mg/kg), patients showed no sign of such phenomenon, indicating that a low dose of bevacizumab not only reduced the formation of pathological blood vessels but also repaired defective blood vessels and normalize them (38, 44). This idea is also supported by bevacizumab combined with chemotherapy, with the overall and progress-free survival improved by adding a low dose, rather than a higher dose, of bevacizumab to chemotherapeutic drug in patients with metastatic colorectal cancer (37, 45, 46), indicating that an appropriate dosage of VEGF inhibitors can rebalance angiogenic signals in tumors, resulting in the formation of more mature blood vessels by actively recruiting pericytes and strengthening cell–cell connections (47, 48). Furthermore, anti-VEGF has been shown to increase platelet-derived growth factor receptor-β signaling and promote pericyte recruitment and decrease permeability (49). Regarding the phenomenon that anti-VEGF therapy increased pericyte coverage while causing vessel regression, Hanahan and Bergers postulated that this is an unconventional mode of resistance that the high coverage of pericytes can help tECs to survive and function, so that tumors can still grow slowly in antiangiogenesis treatments (50).
Despite that anti-VEGF therapy has the potential to restore abnormal tumor vascular normality, there are still some challenges, of which the major one is the time range and dose of anti-VEGF administration to achieve vascular normalization (15, 20), known as the “normalization window,” which was observed to last for only several days after treatment began, providing a narrow time window for the delivery of drugs (13, 20, 48, 51). However, tumors begin to grow again after adaptive resistance, preexisting nonresponsiveness, and antiangiogenic therapy drug withdrawal, leaving many patients failing to produce enduring clinical responses (48, 50, 52, 53). The transient effect of tumor vascular normalization might be associated either with excessively high and continuous administration of antiangiogenic drugs or the occurrence of drug resistance by activating other proangiogenic factors, such as angiopoietins, basic fibroblast growth factor, and transforming growth factor-b (TGF-β) (2, 13, 50, 54). High doses of antiangiogenic drugs degenerate blood vessels, increase tumor hypoxia, and stimulate invasion, infiltration, and metastasis of cancer cells (20, 55, 56), and the heterogeneity of the response of different tumors to VEGF inhibitors further increases the difficulty of achieving the normalization window. Moreover, the discovery of VEGF receptors on nonendothelial cells, specifically VEGFR2, further increases the complexity of VEGF research. MET, the receptor of hepatocyte growth factor, is correlated with increased tumor invasion and poor survival in glioblastoma multiforme. The study found that VEGF blockade restored and increased MET activity in a hypoxia-independent manner while enhancing mesenchymal characteristics in mouse models of glioblastoma multiforme (57, 58). It has been revealed in clinical practices that antiangiogenic drugs have caused some serious side effects, such as hypertension, venous thromboembolism, spontaneous internal bleeding, gastrointestinal perforation, cardiac toxicity, and endocrine dysfunction (54, 59, 60).
Receptor tyrosine kinase inhibitor, another class of antiangiogenic drugs such as sorafenib, was approved by the U.S. Food and Drug Administration to inhibit VEGFR and platelet-derived growth factor receptor-β, leading to the enhanced VE-cadherin junctions between ECs and pericyte recruitment (61–63). The mechanism of VEGFR2 blockade leads to vascular normalization via angiopoietin-1 (Ang1)/Tie2 signaling (64); thus, recent studies aimed at its role involved in the regulation of vascular normalization (13, 55, 65–67). Ang1, expressed primarily on perivascular cells, acts as a stabilizer for the tumor vasculature through Tie2 activation (68–70). However, Ang2, predominantly produced by ECs in hypoxic TME, not only competes with Ang1 for the binding of Tie2 and deactivates it, but also promotes tumor vessels sprouting in the presence of angiogenic growth factors including VEGF (71–74). Ang2 blockade promotes vessel normalization via reducing pericyte dropout and strengthening endothelial junctions of the tumor vasculature (71, 75, 76). Vascular endothelial protein tyrosine phosphatase (VE-PTP) inactivates Tie2, so blocking VE-PTP can promote tumor vascular maturation and normalization (77). Since single blockade of Ang2 may be insufficient to induce vessel normalization, dual blockade of Ang2 and VEGF can reprogram tumor-associated macrophages and promote the effector function of T cells, which can enhance the efficacy of tumor vascular normalization and has been shown to increase the efficacy of immune checkpoint blockade (ICB) (78–80). Dual blockade of Ang2 and VEGF has been shown to prolong survival in mouse models of glioblastoma, melanoma, pancreatic neuroendocrine tumors (PNETs), and metastatic or early stage (resected) breast cancer, except for rectal cancer (11, 81–83). These findings prove that the effect of dual blockade of Ang2 and VEGF is closely related to tumor type, stage, and TME. For the dosing and scheduling of VEGF and ANG2 blockers, further studies are needed (11). Besides, compared with Ang2 blockade alone, the vascular stabilization and perfusion were simultaneously increased by the combined targeting of Tie2 and Ang2 (84). However, research studies have shown that blocking both Ang1 and Ang2 might not be of benefit to patients (85–87). The possible reason is that blocking Ang1 might compromise the benefits of vascular normalization conferred by the blockade of the antagonistic TIE2 ligand ANG2 (11). Notably, both single and double blocking strategies are still posing the risks of increasing tumor hypoxia and promoting TME degradation (20).
Inducing Angiostatic Factors
As mentioned above, inhibition of proangiogenic signals can promote the normalization of tumor vessels, which can also be achieved by increasing angiostatic factors such as tumor necrosis factor α (TNFα), thrombospondin-1 (TSP-1), and endostatin. Direct injection of low-dose TNF-α into tumors stabilizes tumor vascular network, improves vascular perfusion, and substantially enhances antitumor vaccination or adoptive T-cell therapy (88, 89). The tumor vasculature could be normalized and the effector tumor infiltrating lymphocyte (TIL) infiltration could be improved by a member of the tumor necrosis factor superfamily, LIGHT, also known as herpesvirus entry mediator ligand (90–92), which has been shown, when bound to a tumor vascular targeting peptide (VTP), to be able to repair abnormal tumor vasculature by increasing the expression of intercellular adhesion molecule-1 (ICAM-1), vascular cell adhesion protein-1 (VCAM-1), smooth muscle actin, caldesmon, and calponin (90, 93, 94). All of these lymphotoxin β receptor (LTβR)-dependent pericyte contractile markers can strengthen cell-to-cell connections and reduce leakage of blood vessels. LIGHT also activates the intratumoral macrophages to secrete TGF-β, which increases the synthesis of adhesion molecules in a Rho-kinase-dependent manner and modulates pericyte differentiation. LIGHT-driven tumor vascular normalization has been shown to be achieved by improving the pericyte coverage rate of blood vessels and enhancing the effect of cancer therapy (3, 90, 95, 96). Furthermore, LIGHT–LTβR signaling induces the formation of high endothelial venules, which are the primary sites where leukocyte, especially TIL, extravasates from blood vessels into target tissues (94, 97). Although some research studies have shown that LIGHT could be used as a monotherapy, the most effective way is to combine it with vaccinations or checkpoint inhibitors to promote vascular normalization and enhance cancer treatment (94, 98, 99). However, in clinical practices, a LIGHT-based therapy should take into consideration the interfering effect of the tumor necrosis factor receptor superfamily member 6b, known as decoy receptor 3 (DcR3), because LIGHT signaling could be attenuated by DcR3. Owing to the fact that DcR3 expression was detected in neither mice nor rats, further explorations need to be conducted for the purpose of confirming whether LIGHT plays a role in promoting vascular normalization in the presence of DcR3 (100–102).
TSP-1, produced by stromal cells and cancer cells, can induce tumor vascular normalization. ABT-510, one of the mimics of TSP-1, is able to promote the normalization of vascular structure and function without reducing vascular density (103). In addition to normalize tumor blood vessels by directly increasing TSP-1 levels, metronomic chemotherapy, defined as frequent administration at lower doses, also reprograms the tumor immunosuppressive microenvironment into immune stimulation, thus kills cancer cells and reduces the pressure on tumor blood vessels, resulting in the promotion of the tumor blood vessel normalization (20, 104–107).
Besides, studies have shown that TSP-1 related to the normalization of tumor vessels is induced by moderate aerobic exercise, which alone promoted melanoma growth but had no effect on pancreatic ductal adenocarcinoma growth, possibly owing to the specific effect of exercise on tumor growth. However, on the basis of further research, antitumor efficacy in both types of tumors was increased by combining exercise with chemotherapy, as a result of exercise-induced shear stress activating calcineurin–NFAT–TSP-1 signaling in ECs and promoting tumor vascular normalization in mouse models (108–111). Moreover, the level of TSP-1 had been found in recent studies to be usually elevated around normal vessels compared with abnormal vessels, suggesting that TSP-1 may be a marker of vascular normalization (112).
EC proliferation and migration, as well as the expression of VEGF, could be inhibited by an angiogenesis inhibitor, recombinant human endostatin (endostar), which was shown, with recent evidence, to possibly restore vascular homeostasis and induce vascular normalization in some cancer (112, 113). However, still unclear is the molecular mechanism, which, in studies, was shown that the Src signaling pathway and matrix metalloproteinase (MMP) might be the mechanism of its action (114, 115). As mentioned above, inhibiting proangiogenic signaling or enhancing antiangiogenic signaling can prune some abnormal vessels and fortify the remaining, resulting in a normalized vasculature (20).
Targeting EC Metabolism
Endothelial cell metabolism has become a new way to promote tumor vessel normalization. Although located next to the bloodstream and has the capacity to obtain oxygen easily, ECs surprisingly produce up to 85% of their total cellular adenosine-triphosphate (ATP) content via aerobic glycolysis—a phenomenon known as the Warburg effect (116–118). Angiogenesis requires abundant ATP and biomass to proliferate and migrate ECs, especially the active tip and stalk cells (119). However, ECs select glycolysis to produce ATP, which is less efficient than oxidative phosphorylation for the following reasons (116, 120): first, regulation of glycolytic flux occurs faster than oxidative phosphorylation, which ensures an immediate supply of energy when the ECs proliferate and migrate (121); second, ATP, only produced by glycolysis rather than oxidative phosphorylation, provides energy for EC migration, and the intermediates of glycolysis provide the precursors of biomass synthesis for EC proliferation (122–124); third, via aerobic glycolysis, metabolism reduces the release of reactive oxygen species and protects ECs from oxidative stress (116); and fourth, high glycolysis reduces oxygen consumption, thus ensuring adequate oxygen supply to surrounding cells (125, 126).
During the process of angiogenesis, stimulation by VEGF leads to increased glycolysis in ECs and displays increased expression of 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3 (PFKFB3) (116, 127). Hyperglycolysis accumulates excess lactic acid, which is utilized by ECs and activates hypoxia-inducible factor 1α (HIF-1α), thus leading to pathological angiogenesis (116). PFKFB3, an indirect component of the glycolytic pathway, can synthesize fructose-2,6-bisphosphate (F2,6BP), thus allosterically activating phosphofructokinase 1 (PFK1), a rate-limiting enzyme of glycolysis (116, 128, 129) (Figure 2). Compared with normal proliferating ECs, tECs show an even higher reliance on PFKFB3, of which the additional expression leads to a more immature and dysfunctional vasculature, via activating tip cell behavior and enhancing stalk cell proliferation (119, 130).
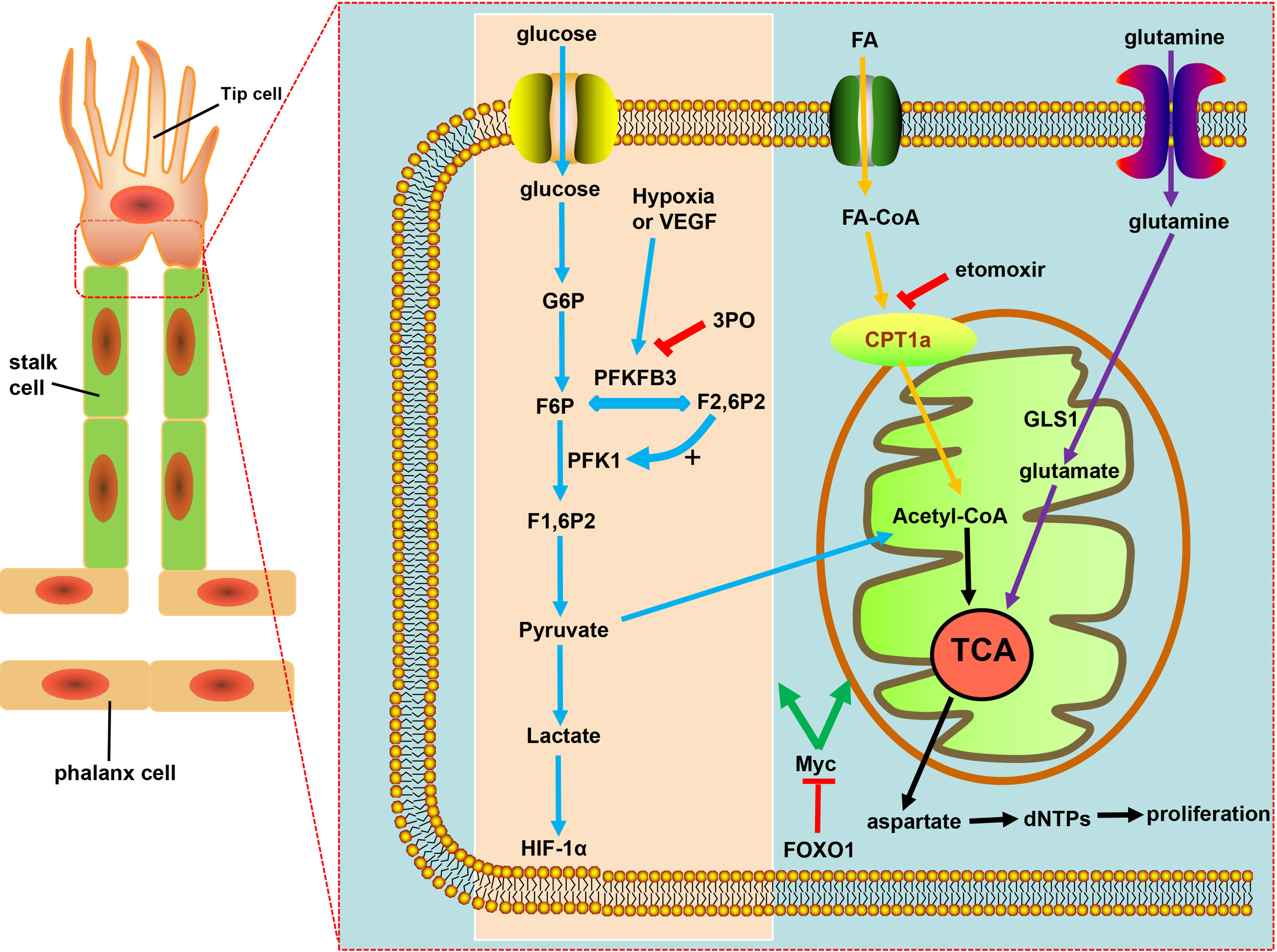
Figure 2 Targeting EC metabolism promotes tumor vascular normalization. Glycolysis fuels the proliferating ECs. PFKFB3, overexpressed under hypoxia and vascular endothelial growth factor (VEGF) stimulation, is a key regulator of the glycolytic activity in ECs. F2,6P2, produced from F6P by PFKFB3, is the main allosteric activator of PFK1, which further increases glycolysis to produce ATP necessary for angiogenesis. PFKB3 inhibitor 3PO reduces pathological angiogenesis. CPT1a-driven FAO and glutamine metabolism both regulate angiogenesis by replenishing the TCA cycle and producing aspartate for nucleotides and DNA synthesis. CPT1a inhibition with etomoxir can promote tumor vessel normalization. FOXO1 keeps EC quiescence and vascular homeostasis via c-Myc regulation, ultimately resulting in decreased glycolysis and impaired mitochondrial function.
The glycolysis of ECs was partially reduced by the inhibition of glycolytic activator, PFKFB3, either with low-dose pharmacological PFKFB3 blockade with the small molecule 3-(3-pyridinyl)-1-(4-pyridinyl)-2-propen-1-one (3PO) or by endothelial PFKFB3 haplodeficiency, with the pathological angiogenesis blocked without causing excessive vascular pruning (7, 131) (Figure 2). Moreover, PFKFB3 inhibition also downregulates the expression of adhesion molecules such as VCAM-1 and ICAM-1 by inhibiting NF-kB activity, thereby weakening the adhesive interaction between tumor cell and ECs, leading to the reduced cancer cell invasion and metastasis (119, 129, 132). Mechanistically, PFKFB3 blockade reduces the endocytosis of VE-cadherin and increases the expression of N-cadherin expression in pericytes, leaving the adhesion to ECs enhanced to keep more quiescent, thus promoting the barrier function of tumor blood vessels (132, 133). However, deletion of both PFKFB3 alleles in ECs or the excess of PFKFB3 inhibitor 3PO inhibited tumor growth due to reduced tumor perfusion and induced systemic toxicity, which proved that the inhibition degree of PFKFB3 determines its effect on the tumor vascular system (119, 134, 135).
Crucial to angiogenesis is, apart from the glycolytic pathway, fatty acid oxidation, on which stalk cells depend to promote vessel sprout elongation through deoxynucleotide triphosphate (dNTP) synthesis. Fatty acids (FAs) were imported into the mitochondria by carnitine palmitoyltransferase 1a (CPT1a), a rate-controlling enzyme of FA breakdown, then underwent β-oxidation to produce acetyl-CoA, which, in conjunction with anaplerotic substrate, sustains the tricarboxylic acid cycle (TCA) for dNTPs and DNA synthesis in proliferating ECs (119), resulting in the decrease in dNTP synthesis and blockade of pathological angiogenesis caused by pharmacological blockade of CPT1a with etomoxir as well as endothelial cell-selective genetic deletion of CPT1a (116, 136, 137) (Figure 2). ECs can harness glutamine metabolism to provide energy, and drugs that inhibit glutaminase (GLS1) can normalize tumor blood vessels (7, 138, 139) (Figure 2). Forkhead box O 1 (FOXO1), a member of the FOX transcription factors family, was found in recent investigations to keep EC quiescence by impairing mitochondrial function and reducing glycolysis, playing its role by inhibiting Myc protooncogene protein (c-Myc)—a key transcriptional factor in growth and anabolic metabolism (140–142), thus, due to the importance of FOXO1 in angiogenesis and homeostasis of the vessel, raising the question: whether FOXO1 is a good target of restoring tumor vessel normalization (Figure 2).
MicroRNAs
Encoded by endogenous genes with approximately 22 nucleotides, microRNAs are single-stranded noncoding RNAs that can regulate mRNA expression in the development and progression of tumors, of which some can restore the integrity of tumor vascular structure by regulating EC functions during angiogenesis (143–146). Considered to play a major role in controlling angiogenesis and vascular structural integrity, miR-126 is widely expressed in venous and arterial ECs and promotes tumor angiogenesis and maintains tumor vascular normalization by regulating downstream growth factors such as VEGF, basic fibroblast growth factor, and epidermal growth factor (147). For example, a single dose of miR-126 injected in a urine ischemic hindlimb model renders a significant reduction in capillary vessel density (148–150). MiR-15b and miR-16 in cells lead to decreased VEGFA expression affecting the blood vessel formation during tumor growth (151, 152). MiR-20b acts as a negative regulator of VEGF in breast cancer cells and nasopharyngeal carcinoma epithelioid cells under hypoxic conditions. The mechanism came into play as miR-20b inhibited the nuclear aggregation of HIF-1α and signal transducer and activation of transcription 3 (STAT3) (153, 154). The increase of HIF-1α and activated STAT3 has been shown to be effective in promoting tumor pathologic angiogenesis, while downregulation of HIF-1α and STAT3 did the opposite (155–157). MiR-100 decreases EC proliferation, migration, and invasion through the mammalian target of rapamycin (mTOR)/HIF‐1α/VEGF pathway in mesenchymal stem cells (158). MiR-107 regulates tumor suppressor gene by p53 in human colon cancer specimens. Yamakuchi et al. found that the overexpression of miR-107 in tumor cells reduced VEGF expression and inhibited tumor angiogenesis in HCT116 cells, and miR-107 could downregulate HIF-1β expression in human colon cancer specimens, leading to the results that miR-107 can mediate p53 regulation of hypoxic signaling and tumor angiogenesis (159). MiR-192 inhibits tumor angiogenesis, resulting in tumor regression and growth inhibition (160). MiR-221/222 can reduce EC migration and proliferation via targeting endothelial nitric oxide synthase (eNOS) and c-kit (161, 162), and miR-874 has been shown to be able to inhibit the tumor angiogenesis of gastric cancer cells in vitro and in vivo by targeting STAT3, and downregulation of miR-874 contributes to tumor angiogenesis (163). On the basis of the above analysis, it has a good prospect to promote vascular normalization through regulation of microRNAs (miRNAs). In Table 1, some miRNAs listed are shown to have great potential in promoting tumor vascular normalization.

Table 1 Some miRNAs associated with vessel normalization.
Targeting the Extracellular Matrix
Vascular normalization was also promoted by the normalization of the extracellular matrix (ECM) which contains a large number of fibers, including collagens, elastin, proteoglycans, glycoprotein, and fibronectin in the TME (164–166), owing to the fact that the dense ECM and hyperplasia of cancer cells create mechanical forces that squeeze the blood vessels, thus destroying the integrity of blood vessels and impairing perfusion (29, 164). The mechanical therapy, which reduced perivascular pressure by targeting ECM fibers or stromal cells, was proposed for the purpose of restoring vascular normalization, since degradation or depletion of ECM such as cancer-associated fibroblasts (CAFs) and hyaluronan had been shown to increase tumor perfusion and chemotherapy efficacy in preclinical studies (167–169). Desmoplasia was promoted by angiotensin II (Ang II) bond to Ang II type 1 receptor (AT1R) via activating TGF-β and upregulating the expression level of connective tissue growth factor in CAFs. These mechanisms regulate the generation of ECM. The angiotensin system inhibitor and direct TGF-β inhibition not only inhibit CAF activation and ECM production but also inhibit angiogenesis and enhance the effect of immunotherapy on some cancers (167, 170). As shown in studies, inhibiting CXCL12/CXCR4 could improve the distribution of chemotherapy agents by effectively relieving stress in tumors, while inducing stromal cell depletion does not relieve tumor solid stress (171, 172). In conclusion, matrix normalization is a good strategy to restore tumor vascular structure and potentiate chemotherapy (173–175).
Other Methods
The vascular normalization can also be promoted by, in addition to the above methods for tumor vascular normalization, targeting other molecules in the TME. For example, transient receptor potential vanilloid 4 (TRPV4), a mechanosensitive ion channel, is a mechanosensor of shear stress and cyclic strain commonly expressed in vascular ECs, of which, compared with normal ECs, the expression and function were decreased in tECs (176–182). Regarding TRPV4 knockout mice, the vascular density increased, the pericyte coverage decreased, and the tumor growth increased. On the contrary, overexpression or pharmacological activation of TRPV4 can normalize vasculature and increase drug delivery (178, 183–186). Importantly, TRPV4 activator inhibited tumor growth in mice when used in combination with the chemotherapeutic drug cisplatin (183, 184). The mechanism of TRPV4 inducing tumor vascular normalization might be through regulation of Rho/Rho kinase via modulating integrin activation, with TRPV4 overexpression significantly inhibiting Rho/Rho kinase activity, which possibly accounts for the recovery of mechanical sensitivity and the normalization of angiogenesis in tECs (183, 184, 187–190). It was found in further experiments that the deletion of TRPV4 significantly increased the basal Rho and promoted tEC proliferation and migration, indicating that TRPV4 can be used as a novel therapeutic target to promote tumor vascular normalization by regulating the Rho/Rho kinase pathway (185, 191).
With G-protein signaling 5 (Rgs5) mediating abnormal tumor angiogenesis and targeting Rgs5 promoting tumor vessel normalization, RGS5-deficient mice blood vessels showed increased pericyte coverage and reduced vessel permeability, which is conducive to reducing tumor hypoxia and promoting blood perfusion to the tumor (31, 192). Able to upregulate the expression of VEGFR2 and promote endothelial sprouting is a member of the sex-determining region Y-box (SOX) transcription factor family, transcription factor Sox17, of which deletions in tECs inhibited pathological angiogenesis and promoted tumor vascular normalization. Research found that Sox17 mutant mice improved drug delivery and inhibited tumor metastasis, suggesting that long-term tumor vascular normalization is conducive to inhibiting the malignant progression (193, 194).
Oxygen levels can be sensed by an oxygen sensor, endothelial prolyl-hydroxylase 2 (PHD2), which regulates angiogenesis by targeting HIF (195). Via tightening the endothelial layer, the tumor vascular normalization was promoted by mice with haploid-deficient PHD2 in the ECs that were able to restore tumor vascular abnormalities by reducing CAF activation, matrix production, and contraction by CAFs (196, 197). As CAF-induced matrix deposition was a migration scaffold for cancer cell dissemination, PHD inhibition reduced lung metastasis. Moreover, PHD protein expression in lung metastases inhibited T-cell effector function and increased the recruitment of Treg cells, so its inhibition decreased Treg cell recruitment and enhanced the immune response (198, 199). C-Src, a member of the Src family kinases (SFKs), is a nonreceptor tyrosine kinase located in cells and is well known for its role in tumorigenesis. Recent findings indicated that the Src inhibitor dasatinib could maintain the integrity of ECs, thus showing the potential to restore tumor vascular normalization (200, 201). Lysophosphatidic acid receptor 4 (LPA4) could tighten endothelial cell–cell contact via promoting cortical actin fiber formation of ECs and stabilizing VE-cadherin, which strengthened the connections between ECs and reduced leakage of blood vessels, and when used in combination with chemotherapy drugs, LPA4 could enhance the antitumor effect (202, 203). A neural adhesion protein with the effect of promoting angiogenesis is, expressed in tumor ECs, endothelial glycoprotein L1 (L1CAM), of which the blockade with anti-L1CAM antibodies or lacking L1CAM resulted in reasonable distribution, complete structure, and normal function of tumor vessels (204). R-Ras is a GTPase that maintains EC survival and promotes vascular maturation through activation or overexpression of R-Ras in vascular smooth muscle cells or EC cells (205). Mediated by proangiogenic factors in tumors is the upregulation of MMPs, which damages vascular basement membrane making the vessel dysfunctional, indicating that inhibiting MMPs or targeting integrinα6 can block the action of MMPs and promote vascular basement membrane integrity (183, 206, 207). On the basis of studies, the antimalarial drug chloroquine had also shown promising results in normalizing tumor blood vessels in that chloroquine increased intratumoral oxygen, improved the efficacy of chemotherapy, and inhibited cancer cell metastasis, which relied mainly on an autophagy-independent, NOTCH1-reliant mechanism to promote tumor vessel normalization (208–210). Another chemotherapy drug with antiangiogenic effects, eribulin, could increase EC–pericyte interactions and form fortified vessels by modulating the expression of angiogenesis molecules (211, 212).
Taken together, new targets for promoting tumor vascular normalization might be provided by targeting the stroma, cancer cells, and other cells in the tumor environment.
The Relationship Between Tumor Blood Vessels and Immunity
A Vicious Circle Between Abnormal Tumor Blood Vessels and Immunosuppressive Microenvironment
Normal structure and function of blood vessels allow immune cells to enter tissues and destroy cancer cells, while the disorganized tumor vessels seem to form a selective immune cell barrier to limit the extravasation of leukocytes—especially cytotoxic T lymphocytes (CTLs)—into blood vessels (12, 20, 51, 213). Particularly, the process of immune cells adhering to the ECs and then transmigrating across the vessel wall is also blocked (214). For example, integrin ligands ICAM-1 and VCAM-1 were both expressed on ECs and immune cells, thus reducing the transport of immune effector cells and infiltration into the tumor core (215, 216). tECs, expressing the immune checkpoint protein-programmed cell death 1 ligand 1 (PD-L1), can bind to PD-1 expressed on T cells and thus inhibit its anticancer activity. The hypoxic TME leads to excessive lactic acid accumulation, thus impairing cytotoxic T-cell function via interfering with the production of interferon-γ (IFNγ) triggered by the T-cell receptor (TCR) (217–219) (Figure 3).
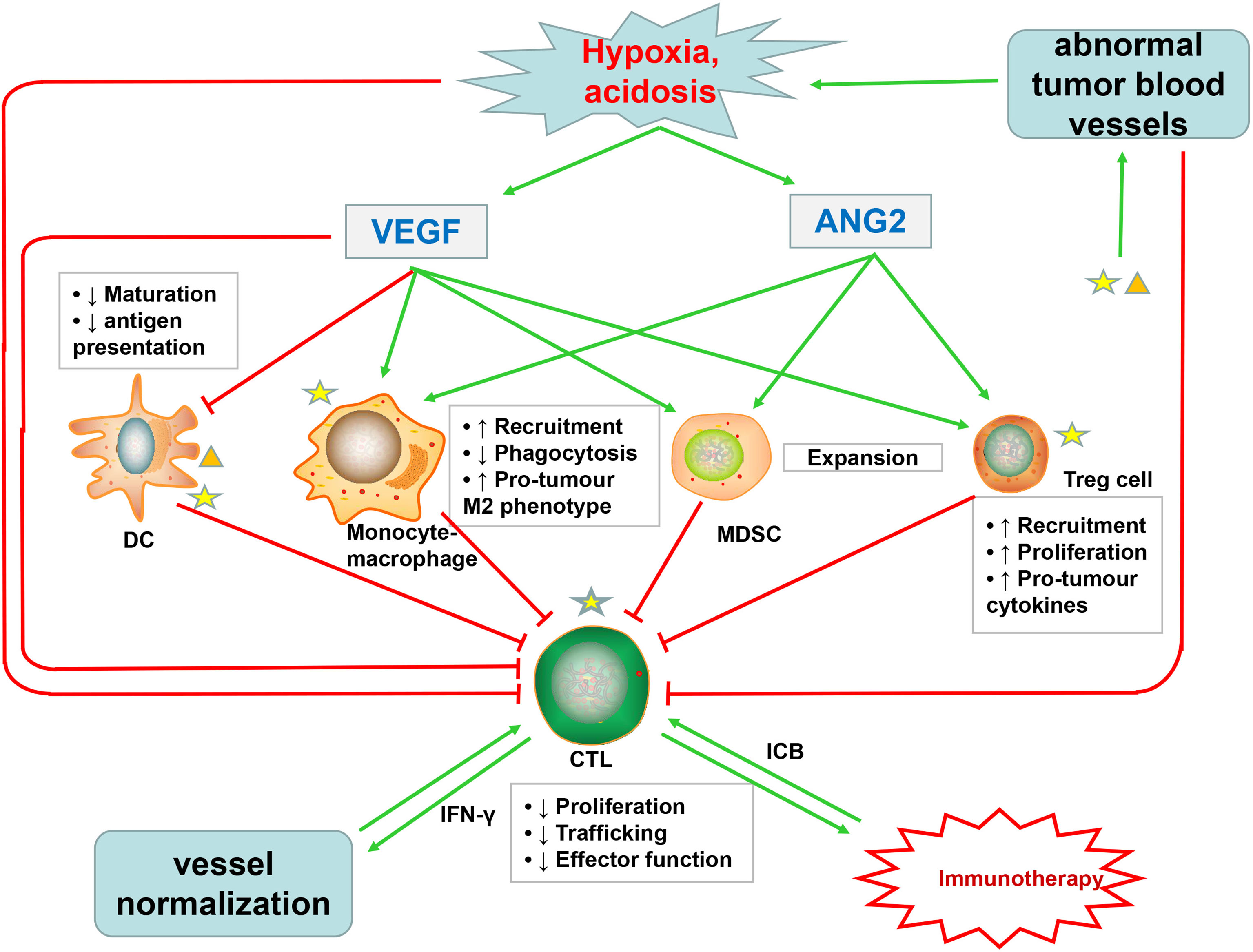
Figure 3 Abnormal tumor blood vessels not only inhibit the infiltration of cytotoxic T lymphocytes (CTLs) but also promote the formation of hypoxic, acidic TME, which affects the function of CTLs and increases the accumulation of VEGF and Ang2. VEGF, in addition to directly impairing the function of CTLs, also inhibits the maturation and antigen presentation of DC, which are necessary for priming of CTLs. Ang2 restricts anticancer activity by modulating the function of monocytes. Both VEGF and Ang2 promote the recruitment of immunosuppressive cells such as Treg cells, MDSCs, and TAMs. These immunosuppressive cells suppress the activity of CTLs, which promote tumor vascular normalization through secreting IFNγ. Immune cells, especially immunosuppressive cells, can produce excessive VEGF and Ang2, further promoting pathological angiogenesis. Immunotherapy with ICB can stimulate CTLs to produce IFNγ, further inducing tumor vascular normalization. The normal tumor blood vessels can promote the infiltration of CTLs and enhance tumor immunotherapy, thus forming a virtuous cycle.
The abnormal tumor vasculature not only directly affects the infiltration of immune effector cells but also indirectly promotes TME-mediated immune suppression through synthesis of proangiogenic factors, such as VEGF and Ang2 (20, 220). Excessive VEGF in TME promotes immunosuppression through at least four mechanisms: first, VEGF inhibits CTL trafficking and effector function by regulating the inhibitory checkpoints of T cells (221); second, VEGF inhibits dendritic cell (DC) maturation and antigen presentation, thus further hampering T-cell activation (222); third, VEGF promotes the recruitment and proliferation of immunosuppressive cells, such as Treg cells, myeloid-derived suppressor cells, and protumor M2-like tumor-associated macrophages (TAMs) (19, 223, 224); and fourth, as described above, VEGF promotes aberrant vasculature and causes hypoxia to locally and systemically foster immunosuppression (19, 33, 225) (Figure 3). In addition to VEGF, activated Ang2 signaling also plays a significant role in promoting tumor immunosuppression: for one thing, Ang2 facilitates the recruitment of MDSCs (myeloid-derived suppressor cells) (226), Treg cells (227), and TIE2-expressing monocytes via upregulating adhesion molecules between leukocytes and ECs (228); for another, Ang2 limits the anticancer activity of monocytes by inhibiting the secretion of TNF. These proangiogenic factors might cooperate to induce tumor immunosuppression (Figure 3). Moreover, an optimum level of VEGF and Ang2 blockade not only promotes tumor vascular normalization but also enhances the anticancer effect of immune cells (82, 83).
Furthermore, increased activation and recruitment of immunosuppressive cells can in turn induce more abnormal angiogenesis, which form a vicious circle of disrupted immune activation (229). MDSCs can not only secrete VEGF but also induce MMP9 to act on the ECM, of which both can enhance the proliferation and migration of ECs, thus promoting angiogenesis (230). As shown in studies, angiogenesis will be promoted when VEGFR1 and VEGFR2 expressed on DC cells bind to the VEGF-A, and TAMs and monocytes can also synthesize and secrete VEGF (231, 232) (Figure 3). Interestingly, recent studies have shown that immunosuppression, in turn, leads to resistance to antiangiogenic treatments (233–235).
A Virtuous Circle Between Immunotherapy and Vascular Normalization
Immunotherapy has proven effective in treating cancers and has become the standard for many cancers (236, 237), while recent findings indicated that stimulation of immune cells, especially T cells, plays an important role in the process of vascular normalization in transplant mammary tumors (238–240). Studies have shown that whether in CD4+ T-cell-deficient mice or CD8+ T-cell-deficient mice, the blood vessels were structurally and functionally abnormal, with the CD4+ T-cell-deficient mice particularly decreasing pericyte coverage and increasing permeability of the blood vessels, whereas manual injection of CD4+ T cells into tumor-bearing mice increased pericyte coverage of the blood vessels and reduced hypoxia, indicating the occurrence of tumor vascular normalization (238, 240). Through activating of immune checkpoints, cancer cells are able to block the initiation and activation of T cells. T cells, which were thought to be primarily affected by ICB with anti-PD1 and anti-cytotoxic T-lymphocyte-associated antigen 4 (CTLA4) antibodies, can promote abnormal tumor vessel normalization (241–243). The main molecular mechanism of this process is mediated by IFNγ (244), which not only inhibited tumor growth, but also acted on ECs to downregulate the expression of delta-like protein 4, thus inhibiting Notch signaling pathway, which is a key pathway of angiogenesis (245). Besides, IFNγ stimulation significantly reduced VEGF secretion in tumor-associated fibroblasts, leaving the angiogenesis further inhibited (246). Moreover, Tian et al. found a positive correlation between IFNγ secreted by Th1 cells and vascular normalization in various mouse models with vessel normalization or T-lymphocyte deficiencies (238) (Figure 3). ICB has been proved to be effective at improving overall survival in many cancers. However, in phase III clinical trials, ICB failed to improve overall survival in glioblastoma multiforme due in part to the immunosuppressive TME. Immune suppression was mediated in part by microglia, bone-marrow-derived myeloid cells, and granulocytes. These cells also enhanced tumor growth and resistance toward antiangiogenic therapy by expression of alternative proangiogenic factors such as CXCL2, IL8, and CD13 (247–251). In addition to T cells, the activation of eosinophils can also normalize tumor blood vessels, but the exact mechanism is unclear, which may be that the TAMs were polarized into an M1-like phenotype through eosinophil-derived IFNγ and TNF signaling, leading to a reduced low VEGF production (252). However, it was shown in a study on promoting IFNγ expression in murine fibrosarcoma and adenocarcinoma cells that that IFNγ directly inhibited angiogenesis and cut off blood flow to bring about intratumoral ischemia, by binding to IFNγ receptors on tECs. It is markedly different from the evidence that IFNγ promotes vascular normalization, which might be because the IFNγ expressed in tumor cells produced a higher and more persistent systemic concentration than the transient IFNγ elevation that can be stimulated by ICB (11, 89, 238).
Immunotherapy has the potential to promote vascular normalization, which in turn further promotes the improvement in immunotherapy effectiveness, thus forming a positive feedback loop. Restoring blood vessel normalization reduces interstitial fluid pressure and improves tumor perfusion, a process that not only increases the infiltration of immune cells within the tumor but also increases the supply of oxygen and nutrients. Adequate oxygen and nutrients can improve the overall anticancer immunotherapeutic response, as high oxygen can enhance the function of cytotoxic T cells (31, 225, 253, 254).
The Combination of Immunotherapy and Conventional Antiangiotherapy
In order to expand the advantages of immunotherapy, the combination of immunotherapy and antiangiotherapy was investigated to improve the effect of promoting vascular normalization. Since the appropriate dose of antiangiogenic drugs can induce vascular normalization and improve the delivery of therapeutic agents to tumors, the dose of ICB can be reduced, which was sometimes known to cause severe immune-related adverse events. Recently, statistics showed that anti-PD1/PDL1 combination trials has continued to increase over the past decade, especially the combination of anti-PD1/PDL1 with VEGF/VEGFR-targeted therapies has become the top combination treatment modality (11, 255, 256). Some preclinical studies demonstrated that the combination treatment of ICB and antiangiotherapy was significantly more effective than monotherapy (257), which is supported by the instance of the findings of Lieu et al. that the combination of bevacizumab and atezolizumab could normalize the TME and inhibit tumor growth in a Cloudman melanoma mouse transplant tumor model. Zhao et al. have proven that low-dose apatinib combined with anti-PD-L1 antibody could inhibit tumor growth and prolong the survival time of a syngeneic lung cancer mouse model (258). The findings of Allen et al. suggested that the combination treatment of anti-PD-L1 therapy and antiangiogenic therapy could improve clinical anticancer efficacy by creating positive feedback loops as well (254, 259). The results of the phase I clinical trials showed that bevacizumab combined with ipilimumab (anti-CTLA4) can improve vessel morphology and increase infiltration of DCs and cytotoxic T cells in melanoma tumors (260–262). Moreover, dual VEGF–Ang2 blockade has been shown to upregulate the expression of adhesion molecules during the window of vascular normalization, thereby facilitating the accumulation of anticancer T cells within multiple types of tumors in mice. Further studies revealed that dual VEGF–Ang2 blockade leads to the upregulation of PD-L1 on ECs and tumor cells, which may be a possible mechanism of resistance to dual VEGF–Ang2 blockade (11, 81). It further justifies the combination of antiangiotherapy and ICB. However, anti-VEGFR2 combined with anti-PD-L1 antibodies failed to improve survival in a glioblastoma model (259), and this lack of efficacy was attributed to a low incidence of high endothelial venules (263). Moreover, neither ICB nor anti-VEGF therapy was proved to be effective in highly desmoplastic tumors, such as cholangiocarcinoma and pancreatic ductal adenocarcinoma (20, 264). These different results suggest that vascular effects depend on tumor location and type, and more accurate measures of vascular normalization are required to determine the synergistic effect of immunotherapy and antiangiogenic therapy on tumor vascular normalization.
Markers of Vascular Normalization
Given that there is no consensus on the types and the dosages of drugs used to normalize blood vessels, and because dose and duration of treatment may rely on the types and sizes of the tumors, vascular densities, expression levels of proangiogenic growth factors, and the conditions of the patients, it is critical to find and identify quantifiable methods to “normalization” (265, 266). With regard to antiangiogenic therapy, higher than the optimal dose of antiangiogenic drugs leads to excessive tumor vessel regression, further aggravating tumor hypoxia, while lower than the optimal dose cannot promote the normalization of tumor vessels (64, 267). Thus, a better understanding of the time window for vascular normalization will help determine appropriate drugs and dosages. Presented here are several imaging technologies or markers that can assess the window for vascular normalization. The morphological changes of blood vessels in tumor tissues can be observed by means of traditional histochemistry and the use of intravital microscopy to track the changes of vascular network over time, of which both are invasive and yield no information on vascular function, making it very difficult to apply them in clinical practice (265, 266). With the window of tumor vascular normalization dynamically monitored, the changes of tumor vascular perfusion can be detected by the noninvasive imaging technology that is also feasible in clinical practice. The perfusion and the window for blood vessel normalization were detected by using MRI (DCE-MRI and BOLD-MRI), dynamic contrast-enhanced ultrasonography (DCE-US), endomicroscopy, computed tomography, and positron emission tomography (PET), etc. (268–273), with the functionality of tumor blood vessels and the ideal regimen required to achieve normalization determined with the dynamic contrast-enhanced magnetic resonance imaging (DCE-MRI) and the specific radiotracer 18F-MISO developed for PET (13, 274, 275).
In addition to imaging techniques, serum-based biomarkers also provide a possibility for monitoring tumor vascular normalization windows. The serum level of soluble VEGFR (sFlt1), which is produced by ECs to finalize angiogenesis for maturation of neovasculature, was reported as a potential predictive marker to detect vascular normalization (199, 276). Ang1/Ang2 ratio is correlated with the degree of vascular normalization and may predict the degree of vascular maturation (66, 277). Apelin, an easily measured secreted protein whose expression is regulated by hypoxia, is overexpressed in many human cancers, including colon adenocarcinoma, nonsmall cell lung cancer, prostate cancer, and hepatocellular carcinoma. Apelin mRNA expression and plasma apelin levels were found in a preclinical study to reduce during the vessel normalization window induced by bevacizumab, indicating that apelin can be used as a potential indicator to identify the window of vascular normalization (210, 278–280). Despite the fact that the patients were reported to have higher levels of circulating type IV collagen in their blood when antiangiogenic therapy was efficient, and the intratumorale expression of P1GF was augmented during the gradual restoration of normalization of blood vessels (276, 281), all of the biomarkers proposed in recent years to measure the normalization of blood vessels have certain disadvantages, showing that, currently, there is no universally accepted method for identifying tumor window for vascular normalization. In Table 2, we list some imaging methods and serum markers that may monitor the normalization of tumor vessels and highlight their advantages and disadvantages.
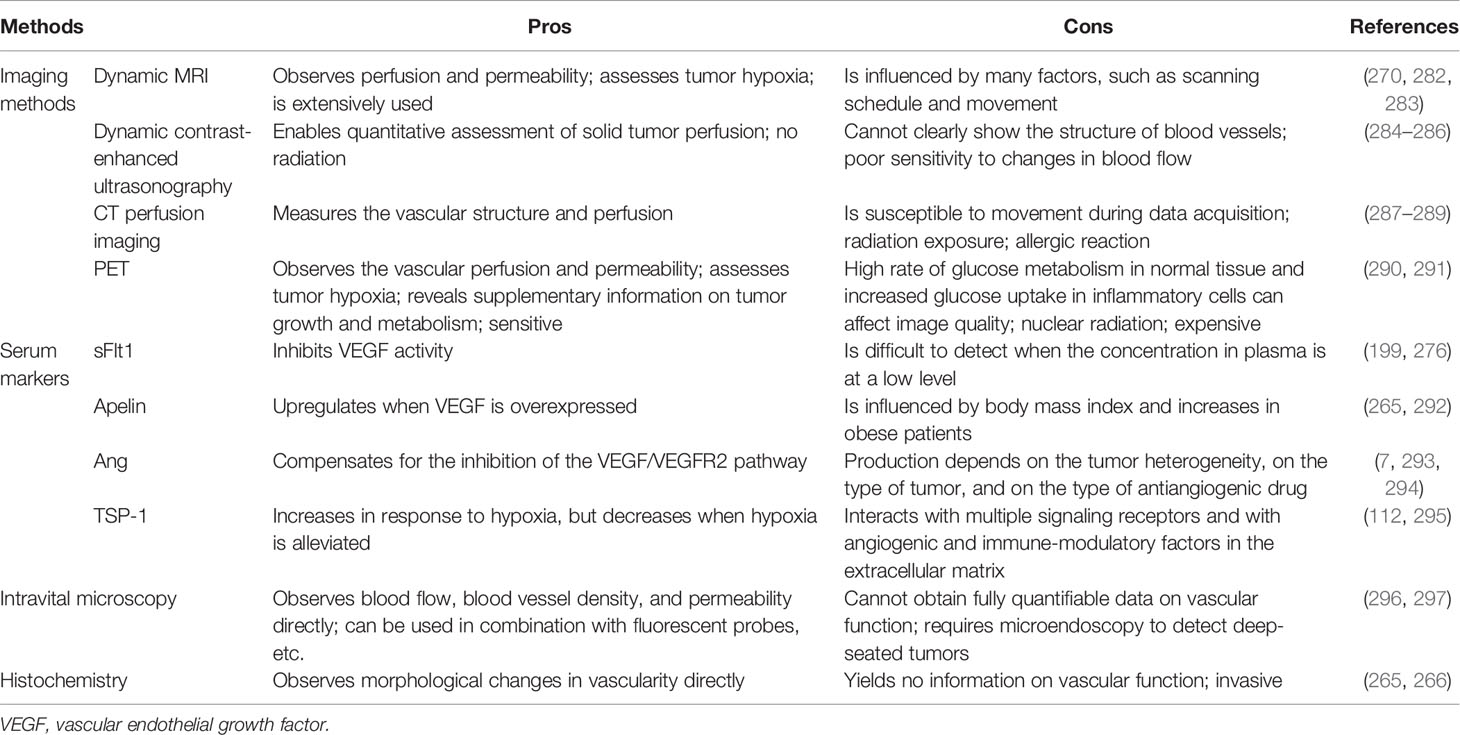
Table 2 The pros and cons of some vascular normalization testing methods.
Conclusion
The U.S. Food and Drug Administration approved bevacizumab, the first angiogenesis inhibitor, to treat metastatic colorectal cancer. Targeting tumor vessels has aroused the interest of a growing number of researchers because of its great potential in tumor therapy. Although antiangiogenic therapy has increased progression-free survival of patients in many types of cancers, the overall results indicated that overall survival improvement was very limited and can be considered worse as long-term antiangiogenic therapy in cancer patients could lead to toxicity and drug resistance, and discontinuation of antiangiogenic drugs might lead to rebound effects that would further aggravate tumor invasion and metastasis. Targeting tumor vascular normalization can not only overcome the shortcomings of antiangiogenic therapy but also enhance the anticancer effect when combined with radiotherapy, chemotherapy, and immunotherapy.
One of the keys to maintaining tumor vascular normalization is to keep the balance between proangiogenic factors and antiangiogenic factors: for one thing, although inhibiting proangiogenic factors such as VEGF and Ang2 have been extensively studied to induce vascular normalization, there is, owing to their dose dependence, the lack of advanced technologies or biomarkers to determine the window for vascular normalization posing the major challenge; for another, angiostatic factors such as TNF, TSP-1, and endostar have shown the potential to promote vascular normalization. A particular example is TSP-1, whose expression can be regulated by nonpharmacological means of aerobic exercise, which is a good adjuvant therapy in combination with other methods in the process of promoting vascular normalization. Targeting miRNAs and endothelial cell metabolism provides a novel and exciting but challenging way to achieve vascular normalization, which has become a research hot spot in recent years. Aside from the methods of vascular normalization, the degrees of normalization are also associated with the types of tumors, the duration of treatments, and the types and dosages of the drugs. The current possible methods are summarized in detail for judging vascular normalization. For the purpose of enhancing the persistence of vascular normalization and overcoming the toxicity and drug resistance that may result from monotherapy, future research on vascular normalization might be focused more on the combination of different approaches, indicating that the key to normalizing tumor blood vessels is selecting an appropriate combination of these methods or with anticancer strategies (including surgery, chemotherapy, radiation therapy), as well as selecting reasonable order and time and appropriate drug dosage in the combination therapy. However, these therapies have complex biological effects, and their combinations may pose a risk of increasing toxic side effects to patients. Thus, it is necessary to find more evidence to confirm whether the same effect can be achieved after the transition to clinical application, despite the therapeutic benefits of tumor vascular normalization demonstrated in many preclinical studies.
In light of the importance of blood vessels to tumors, targeting tumor blood vessels is certain to play a crucial role in future tumor therapy. Hence, in this review, the methods of vascular normalization and the main challenges are described in detail, and the possible imaging techniques and biomarkers for evaluating vascular normalization are also introduced, which will provide references for further research directions of researchers.
Author Contributions
TY developed the idea and drew the figures. All authors wrote the manuscript. HX and XG reviewed the manuscript and approved the final manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by grants to XG from The Special Supported Project of the Fourth Affiliated Hospital of Harbin Medical University [HYDSYTB201908].
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank Dr. Xinshu Dong for critical reading of the article and helpful suggestions. We apologized to those colleagues whose work is not specifically referenced, and gratefully acknowledge their contributions to our article.
References
1. Collaborators GBDCoD. Global, Regional, and National Age-Sex Specific Mortality for 264 Causes of Death, 1980-2016: A Systematic Analysis for the Global Burden of Disease Study 2016. Lancet (2017) 390(10100):1151–210. doi: 10.1016/S0140-6736(17)32152-9
2. Lugano R, Ramachandran M, Dimberg A. Tumor Angiogenesis: Causes, Consequences, Challenges and Opportunities. Cell Mol Life Sci (2020) 77(9):1745–70. doi: 10.1007/s00018-019-03351-7
3. Potente M, Gerhardt H, Carmeliet P. Basic and Therapeutic Aspects of Angiogenesis. Cell (2011) 146(6):873–87. doi: 10.1016/j.cell.2011.08.039
4. Yetkin-Arik B, Vogels IMC, Nowak-Sliwinska P, Weiss A, Houtkooper RH, Van Noorden CJF, et al. The Role of Glycolysis and Mitochondrial Respiration in the Formation and Functioning of Endothelial Tip Cells During Angiogenesis. Sci Rep (2019) 9(1):12608. doi: 10.1038/s41598-019-48676-2
5. Lupo G, Caporarello N, Olivieri M, Cristaldi M, Motta C, Bramanti V, et al. Anti-Angiogenic Therapy in Cancer: Downsides and New Pivots for Precision Medicine. Front Pharmacol (2016) 7:519. doi: 10.3389/fphar.2016.00519
6. Viallard C, Larrivee B. Tumor Angiogenesis and Vascular Normalization: Alternative Therapeutic Targets. Angiogenesis (2017) 20(4):409–26. doi: 10.1007/s10456-017-9562-9
7. Martin JD, Seano G, Jain RK. Normalizing Function of Tumor Vessels: Progress, Opportunities, and Challenges. Annu Rev Physiol (2019) 81:505–34. doi: 10.1146/annurev-physiol-020518-114700
8. Ferrara N, Kerbel RS. Angiogenesis as a Therapeutic Target. Nature (2005) 438(7070):967–74. doi: 10.1038/nature04483
9. Kuczynski EA, Vermeulen PB, Pezzella F, Kerbel RS, Reynolds AR. Vessel Co-Option in Cancer. Nat Rev Clin Oncol (2019) 16(8):469–93. doi: 10.1038/s41571-019-0181-9
10. Wang Q, Zorn JA, Kuriyan J. A Structural Atlas of Kinases Inhibited by Clinically Approved Drugs. Methods Enzymol (2014) 548:23–67. doi: 10.1016/B978-0-12-397918-6.00002-1
11. Fukumura D, Kloepper J, Amoozgar Z, Duda DG, Jain RK. Enhancing Cancer Immunotherapy Using Antiangiogenics: Opportunities and Challenges. Nat Rev Clin Oncol (2018) 15(5):325–40. doi: 10.1038/nrclinonc.2018.29
12. De Bock K, Cauwenberghs S, Carmeliet P. Vessel Abnormalization: Another Hallmark of Cancer? Molecular Mechanisms and Therapeutic Implications. Curr Opin Genet Dev (2011) 21(1):73–9. doi: 10.1016/j.gde.2010.10.008
13. Goel S, Duda DG, Xu L, Munn LL, Boucher Y, Fukumura D, et al. Normalization of the Vasculature for Treatment of Cancer and Other Diseases. Physiol Rev (2011) 91(3):1071–121. doi: 10.1152/physrev.00038.2010
14. Kim C, Yang H, Fukushima Y, Saw PE, Lee J, Park JS, et al. Vascular RhoJ is an Effective and Selective Target for Tumor Angiogenesis and Vascular Disruption. Cancer Cell (2014) 25(1):102–17. doi: 10.1016/j.ccr.2013.12.010
15. Jain RK. Normalizing Tumor Microenvironment to Treat Cancer: Bench to Bedside to Biomarkers. J Clin Oncol (2013) 31(17):2205–18. doi: 10.1200/JCO.2012.46.3653
16. Chauhan VP, Stylianopoulos T, Martin JD, Popovic Z, Chen O, Kamoun WS, et al. Normalization of Tumour Blood Vessels Improves the Delivery of Nanomedicines in a Size-Dependent Manner. Nat Nanotechnol (2012) 7(6):383–8. doi: 10.1038/nnano.2012.45
17. Bergers G, Song S. The Role of Pericytes in Blood-Vessel Formation and Maintenance. Neuro Oncol (2005) 7(4):452–64. doi: 10.1215/S1152851705000232
18. Morikawa S, Baluk P, Kaidoh T, Haskell A, Jain RK, McDonald DM. Abnormalities in Pericytes on Blood Vessels and Endothelial Sprouts in Tumors. Am J Pathol (2002) 160(3):985–1000. doi: 10.1016/s0002-9440(10)64920-6
19. Huang Y, Goel S, Duda DG, Fukumura D, Jain RK. Vascular Normalization as an Emerging Strategy to Enhance Cancer Immunotherapy. Cancer Res (2013) 73(10):2943–8. doi: 10.1158/0008-5472.CAN-12-4354
20. Jain RK. Antiangiogenesis Strategies Revisited: From Starving Tumors to Alleviating Hypoxia. Cancer Cell (2014) 26(5):605–22. doi: 10.1016/j.ccell.2014.10.006
21. Dejana E, Tournier-Lasserve E, Weinstein BM. The Control of Vascular Integrity by Endothelial Cell Junctions: Molecular Basis and Pathological Implications. Dev Cell (2009) 16(2):209–21. doi: 10.1016/j.devcel.2009.01.004
22. Ribatti D, Crivellato E. Mast Cells, Angiogenesis, and Tumour Growth. Biochim Biophys Acta (2012) 1822(1):2–8. doi: 10.1016/j.bbadis.2010.11.010
23. Guo M, Breslin JW, Wu MH, Gottardi CJ, Yuan SY. VE-Cadherin and Beta-Catenin Binding Dynamics During Histamine-Induced Endothelial Hyperpermeability. Am J Physiol Cell Physiol (2008) 294(4):C977–84. doi: 10.1152/ajpcell.90607.2007
24. Beckers CM, van Hinsbergh VW, van Nieuw Amerongen GP. Driving Rho GTPase Activity in Endothelial Cells Regulates Barrier Integrity. Thromb Haemost (2010) 103(1):40–55. doi: 10.1160/TH09-06-0403
25. Engblom C, Pfirschke C, Pittet MJ. The Role of Myeloid Cells in Cancer Therapies. Nat Rev Cancer (2016) 16(7):447–62. doi: 10.1038/nrc.2016.54
26. Baluk P, Hashizume H, McDonald DM. Cellular Abnormalities of Blood Vessels as Targets in Cancer. Curr Opin Genet Dev (2005) 15(1):102–11. doi: 10.1016/j.gde.2004.12.005
27. Stylianopoulos T, Munn LL, Jain RK. Reengineering the Physical Microenvironment of Tumors to Improve Drug Delivery and Efficacy: From Mathematical Modeling to Bench to Bedside. Trends Cancer (2018) 4(4):292–319. doi: 10.1016/j.trecan.2018.02.005
28. Jain RK, Martin JD, Stylianopoulos T. The Role of Mechanical Forces in Tumor Growth and Therapy. Annu Rev BioMed Eng (2014) 16:321–46. doi: 10.1146/annurev-bioeng-071813-105259
29. Stylianopoulos T, Jain RK. Combining Two Strategies to Improve Perfusion and Drug Delivery in Solid Tumors. Proc Natl Acad Sci USA (2013) 110(46):18632–7. doi: 10.1073/pnas.1318415110
30. Jain RK, Stylianopoulos T. Delivering Nanomedicine to Solid Tumors. Nat Rev Clin Oncol (2010) 7(11):653–64. doi: 10.1038/nrclinonc.2010.139
31. Hamzah J, Jugold M, Kiessling F, Rigby P, Manzur M, Marti HH, et al. Vascular Normalization in Rgs5-Deficient Tumours Promotes Immune Destruction. Nature (2008) 453(7193):410–4. doi: 10.1038/nature06868
32. Padera TP, Stoll BR, Tooredman JB, Capen D, di Tomaso E, Jain RK. Pathology: Cancer Cells Compress Intratumour Vessels. Nature (2004) 427(6976):695. doi: 10.1038/427695a
33. Jain RK. Normalization of Tumor Vasculature: An Emerging Concept in Antiangiogenic Therapy. Science (2005) 307(5706):58–62. doi: 10.1126/science.1104819
34. Carmeliet P, Jain RK. Molecular Mechanisms and Clinical Applications of Angiogenesis. Nature (2011) 473(7347):298–307. doi: 10.1038/nature10144
35. Rust R, Gantner C, Schwab ME. Pro- and Antiangiogenic Therapies: Current Status and Clinical Implications. FASEB J (2019) 33(1):34–48. doi: 10.1096/fj.201800640RR
36. Cao Y, Arbiser J, D’Amato RJ, D’Amore PA, Ingber DE, Kerbel R, et al. Forty-Year Journey of Angiogenesis Translational Research. Sci Transl Med (2011) 3(114):114rv3. doi: 10.1126/scitranslmed.3003149
37. Giantonio BJ, Catalano PJ, Meropol NJ, O’Dwyer PJ, Mitchell EP, Alberts SR, et al. Bevacizumab in Combination With Oxaliplatin, Fluorouracil, and Leucovorin (FOLFOX4) for Previously Treated Metastatic Colorectal Cancer: Results From the Eastern Cooperative Oncology Group Study E3200. J Clin Oncol (2007) 25(12):1539–44. doi: 10.1200/JCO.2006.09.6305
38. Jain RK, Duda DG, Clark JW, Loeffler JS. Lessons From Phase III Clinical Trials on Anti-VEGF Therapy for Cancer. Nat Clin Pract Oncol (2006) 3(1):24–40. doi: 10.1038/ncponc0403
39. Pennacchietti S, Michieli P, Galluzzo M, Mazzone M, Giordano S, Comoglio PM. Hypoxia Promotes Invasive Growth by Transcriptional Activation of the Met Protooncogene. Cancer Cell (2003) 3(4):347–61. doi: 10.1016/s1535-6108(03)00085-0
40. Ferrara N. Vascular Endothelial Growth Factor. Arterioscler Thromb Vasc Biol (2009) 29(6):789–91. doi: 10.1161/ATVBAHA.108.179663
41. Goel HL, Mercurio AM. VEGF Targets the Tumour Cell. Nat Rev Cancer (2013) 13(12):871–82. doi: 10.1038/nrc3627
42. Tugues S, Koch S, Gualandi L, Li X, Claesson-Welsh L. Vascular Endothelial Growth Factors and Receptors: Anti-Angiogenic Therapy in the Treatment of Cancer. Mol Aspects Med (2011) 32(2):88–111. doi: 10.1016/j.mam.2011.04.004
43. Melincovici CS, Bosca AB, Susman S, Marginean M, Mihu C, Istrate M, et al. Vascular Endothelial Growth Factor (VEGF) - Key Factor in Normal and Pathological Angiogenesis. Rom J Morphol Embryol (2018) 59(2):455–67.
44. Willett CG, Boucher Y, di Tomaso E, Duda DG, Munn LL, Tong RT, et al. Direct Evidence That the VEGF-Specific Antibody Bevacizumab has Antivascular Effects in Human Rectal Cancer. Nat Med (2004) 10(2):145–7. doi: 10.1038/nm988
45. Levin VA, Chan J, Datta M, Yee JL, Jain RK. Effect of Angiotensin System Inhibitors on Survival in Newly Diagnosed Glioma Patients and Recurrent Glioblastoma Patients Receiving Chemotherapy and/or Bevacizumab. J Neurooncol (2017) 134(2):325–30. doi: 10.1007/s11060-017-2528-3
46. Lu-Emerson C, Duda DG, Emblem KE, Taylor JW, Gerstner ER, Loeffler JS, et al. Lessons From Anti-Vascular Endothelial Growth Factor and Anti-Vascular Endothelial Growth Factor Receptor Trials in Patients With Glioblastoma. J Clin Oncol (2015) 33(10):1197–213. doi: 10.1200/JCO.2014.55.9575
47. Tolaney SM, Boucher Y, Duda DG, Martin JD, Seano G, Ancukiewicz M, et al. Role of Vascular Density and Normalization in Response to Neoadjuvant Bevacizumab and Chemotherapy in Breast Cancer Patients. Proc Natl Acad Sci USA (2015) 112(46):14325–30. doi: 10.1073/pnas.1518808112
48. Huang Y, Yuan J, Righi E, Kamoun WS, Ancukiewicz M, Nezivar J, et al. Vascular Normalizing Doses of Antiangiogenic Treatment Reprogram the Immunosuppressive Tumor Microenvironment and Enhance Immunotherapy. Proc Natl Acad Sci USA (2012) 109(43):17561–6. doi: 10.1073/pnas.1215397109
49. Greenberg JI, Shields DJ, Barillas SG, Acevedo LM, Murphy E, Huang J, et al. A Role for VEGF as a Negative Regulator of Pericyte Function and Vessel Maturation. Nature (2008) 456(7223):809–13. doi: 10.1038/nature07424
50. Bergers G, Hanahan D. Modes of Resistance to Anti-Angiogenic Therapy. Nat Rev Cancer (2008) 8(8):592–603. doi: 10.1038/nrc2442
51. Carmeliet P, Jain RK. Principles and Mechanisms of Vessel Normalization for Cancer and Other Angiogenic Diseases. Nat Rev Drug Discovery (2011) 10(6):417–27. doi: 10.1038/nrd3455
52. Rylova SN, Barnucz E, Fani M, Braun F, Werner M, Lassmann S, et al. Does Imaging Alphavbeta3 Integrin Expression With PET Detect Changes in Angiogenesis During Bevacizumab Therapy? J Nucl Med (2014) 55(11):1878–84. doi: 10.2967/jnumed.114.137570
53. Saltz LB, Lenz HJ, Kindler HL, Hochster HS, Wadler S, Hoff PM, et al. Randomized Phase II Trial of Cetuximab, Bevacizumab, and Irinotecan Compared With Cetuximab and Bevacizumab Alone in Irinotecan-Refractory Colorectal Cancer: The BOND-2 Study. J Clin Oncol (2007) 25(29):4557–61. doi: 10.1200/JCO.2007.12.0949
54. Teleanu RI, Chircov C, Grumezescu AM, Teleanu DM. Tumor Angiogenesis and Anti-Angiogenic Strategies for Cancer Treatment. J Clin Med (2019) 9(1). doi: 10.3390/jcm9010084
55. De Bock K, Mazzone M, Carmeliet P. Antiangiogenic Therapy, Hypoxia, and Metastasis: Risky Liaisons, or Not? Nat Rev Clin Oncol (2011) 8(7):393–404. doi: 10.1038/nrclinonc.2011.83
56. Ebos JM, Kerbel RS. Antiangiogenic Therapy: Impact on Invasion, Disease Progression, and Metastasis. Nat Rev Clin Oncol (2011) 8(4):210–21. doi: 10.1038/nrclinonc.2011.21
57. Lu KV, Chang JP, Parachoniak CA, Pandika MM, Aghi MK, Meyronet D, et al. VEGF Inhibits Tumor Cell Invasion and Mesenchymal Transition Through a MET/VEGFR2 Complex. Cancer Cell (2012) 22(1):21–35. doi: 10.1016/j.ccr.2012.05.037
58. Abounader R, Laterra J. Scatter Factor/Hepatocyte Growth Factor in Brain Tumor Growth and Angiogenesis. Neuro Oncol (2005) 7(4):436–51. doi: 10.1215/S1152851705000050
59. Chen HX, Cleck JN. Adverse Effects of Anticancer Agents That Target the VEGF Pathway. Nat Rev Clin Oncol (2009) 6(8):465–77. doi: 10.1038/nrclinonc.2009.94
60. Hapani S, Sher A, Chu D, Wu S. Increased Risk of Serious Hemorrhage With Bevacizumab in Cancer Patients: A Meta-Analysis. Oncology (2010) 79(1-2):27–38. doi: 10.1159/000314980
61. Wilhelm S, Carter C, Lynch M, Lowinger T, Dumas J, Smith RA, et al. Discovery and Development of Sorafenib: A Multikinase Inhibitor for Treating Cancer. Nat Rev Drug Discov (2006) 5(10):835–44. doi: 10.1038/nrd2130
62. Staton CA, Brown NJ, Reed MW. Current Status and Future Prospects for Anti-Angiogenic Therapies in Cancer. Expert Opin Drug Discov (2009) 4(9):961–79. doi: 10.1517/17460440903196737
63. Azzi S, Hebda JK, Gavard J. Vascular Permeability and Drug Delivery in Cancers. Front Oncol (2013) 3:211. doi: 10.3389/fonc.2013.00211
64. Winkler F, Kozin SV, Tong RT, Chae S-S, Booth MF, Garkavtsev I, et al. Kinetics of Vascular Normalization by VEGFR2 Blockade Governs Brain Tumor Response to Radiation. Cancer Cell (2004) 6(6):553–63. doi: 10.1016/j.ccr.2004.10.011
65. Mazzieri R, Pucci F, Moi D, Zonari E, Ranghetti A, Berti A, et al. Targeting the ANG2/TIE2 Axis Inhibits Tumor Growth and Metastasis by Impairing Angiogenesis and Disabling Rebounds of Proangiogenic Myeloid Cells. Cancer Cell (2011) 19(4):512–26. doi: 10.1016/j.ccr.2011.02.005
66. Augustin HG, Koh GY, Thurston G, Alitalo K. Control of Vascular Morphogenesis and Homeostasis Through the Angiopoietin-Tie System. Nat Rev Mol Cell Biol (2009) 10(3):165–77. doi: 10.1038/nrm2639
67. Huang H, Bhat A, Woodnutt G, Lappe R. Targeting the ANGPT-TIE2 Pathway in Malignancy. Nat Rev Cancer (2010) 10(8):575–85. doi: 10.1038/nrc2894
68. De Palma M, Naldini L. Angiopoietin-2 TIEs Up Macrophages in Tumor Angiogenesis. Clin Cancer Res (2011) 17(16):5226–32. doi: 10.1158/1078-0432.CCR-10-0171
69. Gerald D, Chintharlapalli S, Augustin HG, Benjamin LE. Angiopoietin-2: An Attractive Target for Improved Antiangiogenic Tumor Therapy. Cancer Res (2013) 73(6):1649–57. doi: 10.1158/0008-5472.CAN-12-4697
70. Gnudi L. Angiopoietins and Diabetic Nephropathy. Diabetologia (2016) 59(8):1616–20. doi: 10.1007/s00125-016-3995-3
71. Holopainen T, Saharinen P, D’Amico G, Lampinen A, Eklund L, Sormunen R, et al. Effects of Angiopoietin-2-Blocking Antibody on Endothelial Cell-Cell Junctions and Lung Metastasis. J Natl Cancer Inst (2012) 104(6):461–75. doi: 10.1093/jnci/djs009
72. Koh GY. Orchestral Actions of Angiopoietin-1 in Vascular Regeneration. Trends Mol Med (2013) 19(1):31–9. doi: 10.1016/j.molmed.2012.10.010
73. Srivastava K, Hu J, Korn C, Savant S, Teichert M, Kapel SS, et al. Postsurgical Adjuvant Tumor Therapy by Combining Anti-Angiopoietin-2 and Metronomic Chemotherapy Limits Metastatic Growth. Cancer Cell (2014) 26(6):880–95. doi: 10.1016/j.ccell.2014.11.005
74. Hansen TM, Singh H, Tahir TA, Brindle NP. Effects of Angiopoietins-1 and -2 on the Receptor Tyrosine Kinase Tie2 Are Differentially Regulated at the Endothelial Cell Surface. Cell Signal (2010) 22(3):527–32. doi: 10.1016/j.cellsig.2009.11.007
75. Daly C, Eichten A, Castanaro C, Pasnikowski E, Adler A, Lalani AS, et al. Angiopoietin-2 Functions as a Tie2 Agonist in Tumor Models, Where It Limits the Effects of VEGF Inhibition. Cancer Res (2013) 73(1):108–18. doi: 10.1158/0008-5472.Can-12-2064
76. Isidori AM, Venneri MA, Fiore D. Angiopoietin-1 and Angiopoietin-2 in Metabolic Disorders: Therapeutic Strategies to Restore the Highs and Lows of Angiogenesis in Diabetes. J Endocrinol Invest (2016) 39(11):1235–46. doi: 10.1007/s40618-016-0502-0
77. Goel S, Gupta N, Walcott BP, Snuderl M, Kesler CT, Kirkpatrick ND, et al. Effects of Vascular-Endothelial Protein Tyrosine Phosphatase Inhibition on Breast Cancer Vasculature and Metastatic Progression. J Natl Cancer Inst (2013) 105(16):1188–201. doi: 10.1093/jnci/djt164
78. Koh YJ, Kim HZ, Hwang SI, Lee JE, Oh N, Jung K, et al. Double Antiangiogenic Protein, DAAP, Targeting VEGF-A and Angiopoietins in Tumor Angiogenesis, Metastasis, and Vascular Leakage. Cancer Cell (2010) 18(2):171–84. doi: 10.1016/j.ccr.2010.07.001
79. Brown JL, Cao ZA, Pinzon-Ortiz M, Kendrew J, Reimer C, Wen S, et al. A Human Monoclonal Anti-ANG2 Antibody Leads to Broad Antitumor Activity in Combination With VEGF Inhibitors and Chemotherapy Agents in Preclinical Models. Mol Cancer Ther (2010) 9(1):145–56. doi: 10.1158/1535-7163.MCT-09-0554
80. Kienast Y, Klein C, Scheuer W, Raemsch R, Lorenzon E, Bernicke D, et al. Ang-2-VEGF-A CrossMab, a Novel Bispecific Human IgG1 Antibody Blocking VEGF-A and Ang-2 Functions Simultaneously, Mediates Potent Antitumor, Antiangiogenic, and Antimetastatic Efficacy. Clin Cancer Res (2013) 19(24):6730–40. doi: 10.1158/1078-0432.CCR-13-0081
81. Schmittnaegel M, Rigamonti N, Kadioglu E, Cassara A, Wyser Rmili C, Kiialainen A, et al. Dual Angiopoietin-2 and VEGFA Inhibition Elicits Antitumor Immunity That is Enhanced by PD-1 Checkpoint Blockade. Sci Transl Med (2017) 9(385). doi: 10.1126/scitranslmed.aak9670
82. Kloepper J, Riedemann L, Amoozgar Z, Seano G, Susek K, Yu V, et al. Ang-2/VEGF Bispecific Antibody Reprograms Macrophages and Resident Microglia to Anti-Tumor Phenotype and Prolongs Glioblastoma Survival. Proc Natl Acad Sci USA (2016) 113(16):4476–81. doi: 10.1073/pnas.1525360113
83. Peterson TE, Kirkpatrick ND, Huang Y, Farrar CT, Marijt KA, Kloepper J, et al. Dual Inhibition of Ang-2 and VEGF Receptors Normalizes Tumor Vasculature and Prolongs Survival in Glioblastoma by Altering Macrophages. Proc Natl Acad Sci USA (2016) 113(16):4470–5. doi: 10.1073/pnas.1525349113
84. Park JS, Kim IK, Han S, Park I, Kim C, Bae J, et al. Normalization of Tumor Vessels by Tie2 Activation and Ang2 Inhibition Enhances Drug Delivery and Produces a Favorable Tumor Microenvironment. Cancer Cell (2016) 30(6):953–67. doi: 10.1016/j.ccell.2016.10.018
85. Rini B, Szczylik C, Tannir NM, Koralewski P, Tomczak P, Deptala A, et al. AMG 386 in Combination With Sorafenib in Patients With Metastatic Clear Cell Carcinoma of the Kidney: A Randomized, Double-Blind, Placebo-Controlled, Phase 2 Study. Cancer (2012) 118(24):6152–61. doi: 10.1002/cncr.27632
86. Peeters M, Strickland AH, Lichinitser M, Suresh AV, Manikhas G, Shapiro J, et al. A Randomised, Double-Blind, Placebo-Controlled Phase 2 Study of Trebananib (AMG 386) in Combination With FOLFIRI in Patients With Previously Treated Metastatic Colorectal Carcinoma. Br J Cancer (2013) 108(3):503–11. doi: 10.1038/bjc.2012.594
87. Eatock MM, Tebbutt NC, Bampton CL, Strickland AH, Valladares-Ayerbes M, Swieboda-Sadlej A, et al. Phase II Randomized, Double-Blind, Placebo-Controlled Study of AMG 386 (Trebananib) in Combination With Cisplatin and Capecitabine in Patients With Metastatic Gastro-Oesophageal Cancer. Ann Oncol (2013) 24(3):710–8. doi: 10.1093/annonc/mds502
88. Johansson A, Hamzah J, Payne CJ, Ganss R. Tumor-Targeted TNFalpha Stabilizes Tumor Vessels and Enhances Active Immunotherapy. Proc Natl Acad Sci USA (2012) 109(20):7841–6. doi: 10.1073/pnas.1118296109
89. Kammertoens T, Friese C, Arina A, Idel C, Briesemeister D, Rothe M, et al. Tumour Ischaemia by Interferon-Gamma Resembles Physiological Blood Vessel Regression. Nature (2017) 545(7652):98–102. doi: 10.1038/nature22311
90. He B, Jabouille A, Steri V, Johansson-Percival A, Michael IP, Kotamraju VR, et al. Vascular Targeting of LIGHT Normalizes Blood Vessels in Primary Brain Cancer and Induces Intratumoural High Endothelial Venules. J Pathol (2018) 245(2):209–21. doi: 10.1002/path.5080
91. Mauri DN, Ebner R, Montgomery RI, Kochel KD, Cheung TC, Yu GL, et al. LIGHT, a New Member of the TNF Superfamily, and Lymphotoxin Alpha Are Ligands for Herpesvirus Entry Mediator. Immunity (1998) 8(1):21–30. doi: 10.1016/s1074-7613(00)80455-0
92. Wang J, Lo JC, Foster A, Yu P, Chen HM, Wang Y, et al. The Regulation of T Cell Homeostasis and Autoimmunity by T Cell–Derived LIGHT. J Clin Invest (2001) 108(12):1771–80. doi: 10.1172/jci200113827
93. Johansson-Percival A, Li ZJ, Lakhiani DD, He B, Wang X, Hamzah J, et al. Intratumoral LIGHT Restores Pericyte Contractile Properties and Vessel Integrity. Cell Rep (2015) 13(12):2687–98. doi: 10.1016/j.celrep.2015.12.004
94. Johansson-Percival A, He B, Li ZJ, Kjellen A, Russell K, Li J, et al. De Novo Induction of Intratumoral Lymphoid Structures and Vessel Normalization Enhances Immunotherapy in Resistant Tumors. Nat Immunol (2017) 18(11):1207–17. doi: 10.1038/ni.3836
95. Joshi NS, Akama-Garren EH, Lu Y, Lee DY, Chang GP, Li A, et al. Regulatory T Cells in Tumor-Associated Tertiary Lymphoid Structures Suppress Anti-Tumor T Cell Responses. Immunity (2015) 43(3):579–90. doi: 10.1016/j.immuni.2015.08.006
96. He B, Johansson-Percival A, Backhouse J, Li J, Lee GYF, Hamzah J, et al. Remodeling of Metastatic Vasculature Reduces Lung Colonization and Sensitizes Overt Metastases to Immunotherapy. Cell Rep (2020) 30(3):714–24.e5. doi: 10.1016/j.celrep.2019.12.013
97. Lu TT, Browning JL. Role of the Lymphotoxin/LIGHT System in the Development and Maintenance of Reticular Networks and Vasculature in Lymphoid Tissues. Front Immunol (2014) 5:47. doi: 10.3389/fimmu.2014.00047
98. Skeate JG, Otsmaa ME, Prins R, Fernandez DJ, Da Silva DM, Kast WM. TNFSF14: LIGHTing the Way for Effective Cancer Immunotherapy. Front Immunol (2020) 11:922. doi: 10.3389/fimmu.2020.00922
99. Abastado JP. The Next Challenge in Cancer Immunotherapy: Controlling T-Cell Traffic to the Tumor. Cancer Res (2012) 72(9):2159–61. doi: 10.1158/0008-5472.CAN-11-3538
100. Jiang M, Lin X, He R, Lin X, Liang L, Tang R, et al. Decoy Receptor 3 (DcR3) as a Biomarker of Tumor Deterioration in Female Reproductive Cancers: A Meta-Analysis. Med Sci Monit (2016) 22:1850–7. doi: 10.12659/msm.896226
101. Lin CK, Ting CC, Tsai WC, Chen YW, Hueng DY. A Tissue Microarray Study of Toll-Like Receptor 4, Decoy Receptor 3, and External Signal Regulated Kinase 1/2 Expressions in Astrocytoma. Indian J Pathol Microbiol (2016) 59(3):294–300. doi: 10.4103/0377-4929.188122
102. Huang S, Chen G, Dang Y, Chen LH. Overexpression of DcR3 and its Significance on Tumor Cell Differentiation and Proliferation in Glioma. ScientificWorldJournal (2014) 2014:605236. doi: 10.1155/2014/605236
103. Campbell NE, Greenaway J, Henkin J, Moorehead RA, Petrik J. The Thrombospondin-1 Mimetic ABT-510 Increases the Uptake and Effectiveness of Cisplatin and Paclitaxel in a Mouse Model of Epithelial Ovarian Cancer. Neoplasia (2010) 12(3):275–83. doi: 10.1593/neo.91880
104. Pasquier E, Kavallaris M, Andre N. Metronomic Chemotherapy: New Rationale for New Directions. Nat Rev Clin Oncol (2010) 7(8):455–65. doi: 10.1038/nrclinonc.2010.82
105. Andre N, Carre M, Pasquier E. Metronomics: Towards Personalized Chemotherapy? Nat Rev Clin Oncol (2014) 11(7):413–31. doi: 10.1038/nrclinonc.2014.89
106. Kerbel RS, Kamen BA. The Anti-Angiogenic Basis of Metronomic Chemotherapy. Nat Rev Cancer (2004) 4(6):423–36. doi: 10.1038/nrc1369
107. Mpekris F, Baish JW, Stylianopoulos T, Jain RK. Role of Vascular Normalization in Benefit From Metronomic Chemotherapy. Proc Natl Acad Sci USA (2017) 114(8):1994–9. doi: 10.1073/pnas.1700340114
108. Schadler KL, Thomas NJ, Galie PA, Bhang DH, Roby KC, Addai P, et al. Tumor Vessel Normalization After Aerobic Exercise Enhances Chemotherapeutic Efficacy. Oncotarget (2016) 7(40):65429–40. doi: 10.18632/oncotarget.11748
109. Elice F, Rodeghiero F. Side Effects of Anti-Angiogenic Drugs. Thromb Res (2012) 129:S50–S3. doi: 10.1016/s0049-3848(12)70016-6
110. Paez-Ribes M, Allen E, Hudock J, Takeda T, Okuyama H, Vinals F, et al. Antiangiogenic Therapy Elicits Malignant Progression of Tumors to Increased Local Invasion and Distant Metastasis. Cancer Cell (2009) 15(3):220–31. doi: 10.1016/j.ccr.2009.01.027
111. Betof AS, Lascola CD, Weitzel D, Landon C, Scarbrough PM, Devi GR, et al. Modulation of Murine Breast Tumor Vascularity, Hypoxia and Chemotherapeutic Response by Exercise. J Natl Cancer Inst (2015) 107(5). doi: 10.1093/jnci/djv040
112. Yang W, Li W, Pan F, Yang XY, Zhao X, Qin L, et al. TSP-1 as a Novel Biological Marker of Tumor Vasculature Normalization in Colon Carcinoma Induced by Endostar. Oncol Lett (2020) 19(3):2107–14. doi: 10.3892/ol.2020.11320
113. Guan L. Endostar Rebuilding Vascular Homeostasis and Enhancing Chemotherapy Efficacy in Cervical Cancer Treatment. Onco Targets Ther (2020) 13:12811–27. doi: 10.2147/OTT.S277644
114. Yu M, Han Y, Zhuo H, Zhang S. Endostar, a Modified Endostatin Induces Vascular Normalization to Improve Chemotherapy Efficacy Through Suppression of Src Signaling Pathway. Cancer Biother Radiopharm (2018) 33(4):131–8. doi: 10.1089/cbr.2017.2399
115. Pan JH, Zhu S, Huang J, Liang J, Zhang D, Zhao X, et al. Monitoring the Process of Endostar-Induced Tumor Vascular Normalization by Non-Contrast Intravoxel Incoherent Motion Diffusion-Weighted MRI. Front Oncol (2018) 8:524. doi: 10.3389/fonc.2018.00524
116. De Bock K, Georgiadou M, Schoors S, Kuchnio A, Wong BW, Cantelmo AR, et al. Role of PFKFB3-Driven Glycolysis in Vessel Sprouting. Cell (2013) 154(3):651–63. doi: 10.1016/j.cell.2013.06.037
117. Krutzfeldt A, Spahr R, Mertens S, Siegmund B, Piper HM. Metabolism of Exogenous Substrates by Coronary Endothelial Cells in Culture. J Mol Cell Cardiol (1990) 22(12):1393–404. doi: 10.1016/0022-2828(90)90984-a
118. Culic O, Gruwel ML, Schrader J. Energy Turnover of Vascular Endothelial Cells. Am J Physiol (1997) 273(1 Pt 1):C205–13. doi: 10.1152/ajpcell.1997.273.1.C205
119. Cantelmo AR, Conradi LC, Brajic A, Goveia J, Kalucka J, Pircher A, et al. Inhibition of the Glycolytic Activator PFKFB3 in Endothelium Induces Tumor Vessel Normalization, Impairs Metastasis, and Improves Chemotherapy. Cancer Cell (2016) 30(6):968–85. doi: 10.1016/j.ccell.2016.10.006
120. Groschner LN, Waldeck-Weiermair M, Malli R, Graier WF. Endothelial Mitochondria–Less Respiration, More Integration. Pflugers Arch (2012) 464(1):63–76. doi: 10.1007/s00424-012-1085-z
121. Pfeiffer T, Schuster S, Bonhoeffer S. Cooperation and Competition in the Evolution of ATP-Producing Pathways. Science (2001) 292(5516):504–7. doi: 10.1126/science.1058079
122. Yizhak K, Le Devedec SE, Rogkoti VM, Baenke F, de Boer VC, Frezza C, et al. A Computational Study of the Warburg Effect Identifies Metabolic Targets Inhibiting Cancer Migration. Mol Syst Biol (2014) 10:744. doi: 10.15252/msb.20134993
123. Vander Heiden MG. Targeting Cancer Metabolism: A Therapeutic Window Opens. Nat Rev Drug Discovery (2011) 10(9):671–84. doi: 10.1038/nrd3504
124. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science (2009) 324(5930):1029–33. doi: 10.1126/science.1160809
125. Eelen G, de Zeeuw P, Simons M, Carmeliet P. Endothelial Cell Metabolism in Normal and Diseased Vasculature. Circ Res (2015) 116(7):1231–44. doi: 10.1161/CIRCRESAHA.116.302855
126. Ghesquiere B, Wong BW, Kuchnio A, Carmeliet P. Metabolism of Stromal and Immune Cells in Health and Disease. Nature (2014) 511(7508):167–76. doi: 10.1038/nature13312
127. Bartrons R, Simon-Molas H, Rodriguez-Garcia A, Castano E, Navarro-Sabate A, Manzano A, et al. Fructose 2,6-Bisphosphate in Cancer Cell Metabolism. Front Oncol (2018) 8:331. doi: 10.3389/fonc.2018.00331
128. Schoors S, Cantelmo AR, Georgiadou M, Stapor P, Wang X, Quaegebeur A, et al. Incomplete and Transitory Decrease of Glycolysis: A New Paradigm for Anti-Angiogenic Therapy? Cell Cycle (2014) 13(1):16–22. doi: 10.4161/cc.27519
129. Schoors S, De Bock K, Cantelmo AR, Georgiadou M, Ghesquiere B, Cauwenberghs S, et al. Partial and Transient Reduction of Glycolysis by PFKFB3 Blockade Reduces Pathological Angiogenesis. Cell Metab (2014) 19(1):37–48. doi: 10.1016/j.cmet.2013.11.008
130. Jayaraman A, Kumar P, Marin S, de Atauri P, Mateo F, MT T, et al. Untargeted Metabolomics Reveals Distinct Metabolic Reprogramming in Endothelial Cells Co-Cultured With CSC and Non-CSC Prostate Cancer Cell Subpopulations. PloS One (2018) 13(2):e0192175. doi: 10.1371/journal.pone.0192175
131. Fitzgerald G, Soro-Arnaiz I, De Bock K. The Warburg Effect in Endothelial Cells and its Potential as an Anti-Angiogenic Target in Cancer. Front Cell Dev Biol (2018) 6:100. doi: 10.3389/fcell.2018.00100
132. Matsumoto K, Noda T, Kobayashi S, Sakano Y, Yokota Y, Iwagami Y, et al. Inhibition of Glycolytic Activator PFKFB3 Suppresses Tumor Growth and Induces Tumor Vessel Normalization in Hepatocellular Carcinoma. Cancer Lett (2021) 500:29–40. doi: 10.1016/j.canlet.2020.12.011
133. Xu Y, An X, Guo X, Habtetsion TG, Wang Y, Xu X, et al. Endothelial PFKFB3 Plays a Critical Role in Angiogenesis. Arterioscler Thromb Vasc Biol (2014) 34(6):1231–9. doi: 10.1161/ATVBAHA.113.303041
134. Conradi LC, Brajic A, Cantelmo AR, Bouche A, Kalucka J, Pircher A, et al. Tumor Vessel Disintegration by Maximum Tolerable PFKFB3 Blockade. Angiogenesis (2017) 20(4):599–613. doi: 10.1007/s10456-017-9573-6
135. Zhang J, Xue W, Xu K, Yi L, Guo Y, Xie T, et al. Dual Inhibition of PFKFB3 and VEGF Normalizes Tumor Vasculature, Reduces Lactate Production, and Improves Chemotherapy in Glioblastoma: Insights From Protein Expression Profiling and MRI. Theranostics (2020) 10(16):7245–59. doi: 10.7150/thno.44427
136. Hulsmann WC, Dubelaar ML. Aspects of Fatty Acid Metabolism in Vascular Endothelial Cells. Biochimie (1988) 70(5):681–6. doi: 10.1016/0300-9084(88)90253-2
137. Schoors S, Bruning U, Missiaen R, Queiroz KC, Borgers G, Elia I, et al. Fatty Acid Carbon is Essential for dNTP Synthesis in Endothelial Cells. Nature (2015) 520(7546):192–7. doi: 10.1038/nature14362
138. Huang H, Vandekeere S, Kalucka J, Bierhansl L, Zecchin A, Bruning U, et al. Role of Glutamine and Interlinked Asparagine Metabolism in Vessel Formation. EMBO J (2017) 36(16):2334–52. doi: 10.15252/embj.201695518
139. Vander Heiden MG. Exploiting Tumor Metabolism: Challenges for Clinical Translation. J Clin Invest (2013) 123(9):3648–51. doi: 10.1172/JCI72391
140. Coomans de Brachene A, Demoulin JB. FOXO Transcription Factors in Cancer Development and Therapy. Cell Mol Life Sci (2016) 73(6):1159–72. doi: 10.1007/s00018-015-2112-y
141. Potente M, Urbich C, Sasaki K, Hofmann WK, Heeschen C, Aicher A, et al. Involvement of Foxo Transcription Factors in Angiogenesis and Postnatal Neovascularization. J Clin Invest (2005) 115(9):2382–92. doi: 10.1172/JCI23126
142. Wilhelm K, Happel K, Eelen G, Schoors S, Oellerich MF, Lim R, et al. FOXO1 Couples Metabolic Activity and Growth State in the Vascular Endothelium. Nature (2016) 529(7585):216–20. doi: 10.1038/nature16498
143. Choudhry H, Harris AL, McIntyre A. The Tumour Hypoxia Induced non-Coding Transcriptome. Mol Aspects Med (2016) 47-48:35–53. doi: 10.1016/j.mam.2016.01.003
144. Camps C, Saini HK, Mole DR, Choudhry H, Reczko M, Guerra-Assuncao JA, et al. Integrated Analysis of microRNA and mRNA Expression and Association With HIF Binding Reveals the Complexity of microRNA Expression Regulation Under Hypoxia. Mol Cancer (2014) 13:28. doi: 10.1186/1476-4598-13-28
145. Orso F, Quirico L, Dettori D, Coppo R, Virga F, Ferreira LC, et al. Role of miRNAs in Tumor and Endothelial Cell Interactions During Tumor Progression. Semin Cancer Biol (2020) 60:214–24. doi: 10.1016/j.semcancer.2019.07.024
146. Salinas-Vera YM, Marchat LA, Gallardo-Rincon D, Ruiz-Garcia E, Astudillo-De La Vega H, Echavarria-Zepeda R, et al. AngiomiRs: MicroRNAs Driving Angiogenesis in Cancer (Review). Int J Mol Med (2019) 43(2):657–70. doi: 10.3892/ijmm.2018.4003
147. Annese T, Tamma R, De Giorgis M, Ribatti D. microRNAs Biogenesis, Functions and Role in Tumor Angiogenesis. Front Oncol (2020) 10:581007. doi: 10.3389/fonc.2020.581007
148. Wang S, Aurora AB, Johnson BA, Qi X, McAnally J, Hill JA, et al. The Endothelial-Specific microRNA miR-126 Governs Vascular Integrity and Angiogenesis. Dev Cell (2008) 15(2):261–71. doi: 10.1016/j.devcel.2008.07.002
149. Fish JE, Santoro MM, Morton SU, Yu S, Yeh R-F, Wythe JD, et al. miR-126 Regulates Angiogenic Signaling and Vascular Integrity. Dev Cell (2008) 15(2):272–84. doi: 10.1016/j.devcel.2008.07.008
150. van Solingen C, Seghers L, Bijkerk R, Duijs JM, Roeten MK, van Oeveren-Rietdijk AM, et al. Antagomir-Mediated Silencing of Endothelial Cell Specific microRNA-126 Impairs Ischemia-Induced Angiogenesis. J Cell Mol Med (2009) 13(8A):1577–85. doi: 10.1111/j.1582-4934.2008.00613.x
151. Sun CY, She XM, Qin Y, Chu ZB, Chen L, Ai LS, et al. miR-15a and miR-16 Affect the Angiogenesis of Multiple Myeloma by. Carcinogenesis (2013) 34(2):426–35. doi: 10.1093/carcin/bgs333
152. Tiwari A, Mukherjee B, Dixit M. MicroRNA Key to Angiogenesis Regulation: MiRNA Biology and Therapy. Curr Cancer Drug Targets (2018) 18(3):266–77. doi: 10.2174/1568009617666170630142725
153. Hua Z, Lv Q, Ye W, Wong CK, Cai G, Gu D, et al. MiRNA-Directed Regulation of VEGF and Other Angiogenic Factors Under Hypoxia. PloS One (2006) 1:e116. doi: 10.1371/journal.pone.0000116
154. Cascio S, D’Andrea A, Ferla R, Surmacz E, Gulotta E, Amodeo V, et al. miR-20b Modulates VEGF Expression by Targeting HIF-1 Alpha and STAT3 in MCF-7 Breast Cancer Cells. J Cell Physiol (2010) 224(1):242–9. doi: 10.1002/jcp.22126
155. Azoitei N, Becher A, Steinestel K, Rouhi A, Diepold K, Genze F, et al. PKM2 Promotes Tumor Angiogenesis by Regulating HIF-1alpha Through NF-kappaB Activation. Mol Cancer (2016) 15:3. doi: 10.1186/s12943-015-0490-2
156. Yang Y, Sun M, Wang L, Jiao B. HIFs, Angiogenesis, and Cancer. J Cell Biochem (2013) 114(5):967–74. doi: 10.1002/jcb.24438
157. Niu G, Wright KL, Huang M, Song L, Haura E, Turkson J, et al. Constitutive Stat3 Activity Up-Regulates VEGF Expression and Tumor Angiogenesis. Oncogene (2002) 21(13):2000–8. doi: 10.1038/sj.onc.1205260
158. Grundmann S, Hans FP, Kinniry S, Heinke J, Helbing T, Bluhm F, et al. MicroRNA-100 Regulates Neovascularization by Suppression of Mammalian Target of Rapamycin in Endothelial and Vascular Smooth Muscle Cells. Circulation (2011) 123(9):999–1009. doi: 10.1161/CIRCULATIONAHA.110.000323
159. Yamakuchi M, Lotterman CD, Bao C, Hruban RH, Karim B, Mendell JT, et al. P53-Induced microRNA-107 Inhibits HIF-1 and Tumor Angiogenesis. Proc Natl Acad Sci USA (2010) 107(14):6334–9. doi: 10.1073/pnas.0911082107
160. Wu SY, Rupaimoole R, Shen F, Pradeep S, Pecot CV, Ivan C, et al. A miR-192-EGR1-HOXB9 Regulatory Network Controls the Angiogenic Switch in Cancer. Nat Commun (2016) 7:11169. doi: 10.1038/ncomms11169
161. Poliseno L, Tuccoli A, Mariani L, Evangelista M, Citti L, Woods K, et al. MicroRNAs Modulate the Angiogenic Properties of HUVECs. Blood (2006) 108(9):3068–71. doi: 10.1182/blood-2006-01-012369
162. Pan Z, Tian Y, Niu G, Cao C. Role of microRNAs in Remodeling the Tumor Microenvironment (Review). Int J Oncol (2020) 56(2):407–16. doi: 10.3892/ijo.2019.4952
163. Zhang X, Tang J, Zhi X, Xie K, Wang W, Li Z, et al. miR-874 Functions as a Tumor Suppressor by Inhibiting Angiogenesis Through STAT3/VEGF-A Pathway in Gastric Cancer. Oncotarget (2015) 6(3):1605–17. doi: 10.18632/oncotarget.2748
164. Stylianopoulos T, Martin JD, Chauhan VP, Jain SR, Diop-Frimpong B, Bardeesy N, et al. Causes, Consequences, and Remedies for Growth-Induced Solid Stress in Murine and Human Tumors. Proc Natl Acad Sci USA (2012) 109(38):15101–8. doi: 10.1073/pnas.1213353109
165. Mpekris F, Papageorgis P, Polydorou C, Voutouri C, Kalli M, Pirentis AP, et al. Sonic-Hedgehog Pathway Inhibition Normalizes Desmoplastic Tumor Microenvironment to Improve Chemo- and Nanotherapy. J Control Release (2017) 261:105–12. doi: 10.1016/j.jconrel.2017.06.022
166. Mortezaee K. Normalization in Tumor Ecosystem: Opportunities and Challenges. Cell Biol Int (2021). doi: 10.1002/cbin.11655
167. Pinter M, Jain RK. Targeting the Renin-Angiotensin System to Improve Cancer Treatment: Implications for Immunotherapy. Sci Transl Med (2017) 9(410). doi: 10.1126/scitranslmed.aan5616
168. Chauhan VP, Chen IX, Tong R, Ng MR, Martin JD, Naxerova K, et al. Reprogramming the Microenvironment With Tumor-Selective Angiotensin Blockers Enhances Cancer Immunotherapy. Proc Natl Acad Sci USA (2019) 116(22):10674–80. doi: 10.1073/pnas.1819889116
169. Ilie MD, Vasiljevic A, Raverot G, Bertolino P. The Microenvironment of Pituitary Tumors-Biological and Therapeutic Implications. Cancers (Basel) (2019) 11(10). doi: 10.3390/cancers11101605
170. Chauhan VP, Martin JD, Liu H, Lacorre DA, Jain SR, Kozin SV, et al. Angiotensin Inhibition Enhances Drug Delivery and Potentiates Chemotherapy by Decompressing Tumour Blood Vessels. Nat Commun (2013) 4:2516. doi: 10.1038/ncomms3516
171. Chen IX, Chauhan VP, Posada J, Ng MR, Wu MW, Adstamongkonkul P, et al. Blocking CXCR4 Alleviates Desmoplasia, Increases T-Lymphocyte Infiltration, and Improves Immunotherapy in Metastatic Breast Cancer. Proc Natl Acad Sci USA (2019) 116(10):4558–66. doi: 10.1073/pnas.1815515116
172. Martin JD, Fukumura D, Duda DG, Boucher Y, Jain RK. Reengineering the Tumor Microenvironment to Alleviate Hypoxia and Overcome Cancer Heterogeneity. Cold Spring Harb Perspect Med (2016) 6(12). doi: 10.1101/cshperspect.a027094
173. Zhao Y, Cao J, Melamed A, Worley M, Gockley A, Jones D, et al. Losartan Treatment Enhances Chemotherapy Efficacy and Reduces Ascites in Ovarian Cancer Models by Normalizing the Tumor Stroma. Proc Natl Acad Sci USA (2019) 116(6):2210–9. doi: 10.1073/pnas.1818357116
174. Polydorou C, Mpekris F, Papageorgis P, Voutouri C, Stylianopoulos T. Pirfenidone Normalizes the Tumor Microenvironment to Improve Chemotherapy. Oncotarget (2017) 8(15):24506–17. doi: 10.18632/oncotarget.15534
175. Papageorgis P, Polydorou C, Mpekris F, Voutouri C, Agathokleous E, Kapnissi-Christodoulou CP, et al. Tranilast-Induced Stress Alleviation in Solid Tumors Improves the Efficacy of Chemo- and Nanotherapeutics in a Size-Independent Manner. Sci Rep (2017) 7:46140. doi: 10.1038/srep46140
176. Kohler R, Heyken WT, Heinau P, Schubert R, Si H, Kacik M, et al. Evidence for a Functional Role of Endothelial Transient Receptor Potential V4 in Shear Stress-Induced Vasodilatation. Arterioscler Thromb Vasc Biol (2006) 26(7):1495–502. doi: 10.1161/01.ATV.0000225698.36212.6a
177. Mendoza SA, Fang J, Gutterman DD, Wilcox DA, Bubolz AH, Li R, et al. TRPV4-Mediated Endothelial Ca2+ Influx and Vasodilation in Response to Shear Stress. Am J Physiol Heart Circ Physiol (2010) 298(2):H466–76. doi: 10.1152/ajpheart.00854.2009
178. Thodeti CK, Matthews B, Ravi A, Mammoto A, Ghosh K, Bracha AL, et al. TRPV4 Channels Mediate Cyclic Strain-Induced Endothelial Cell Reorientation Through Integrin-to-Integrin Signaling. Circ Res (2009) 104(9):1123–30. doi: 10.1161/CIRCRESAHA.108.192930
179. Troidl C, Troidl K, Schierling W, Cai WJ, Nef H, Mollmann H, et al. Trpv4 Induces Collateral Vessel Growth During Regeneration of the Arterial Circulation. J Cell Mol Med (2009) 13(8B):2613–21. doi: 10.1111/j.1582-4934.2008.00579.x
180. Matthews BD, Thodeti CK, Tytell JD, Mammoto A, Overby DR, Ingber DE. Ultra-Rapid Activation of TRPV4 Ion Channels by Mechanical Forces Applied to Cell Surface Beta1 Integrins. Integr Biol (Camb) (2010) 2(9):435–42. doi: 10.1039/c0ib00034e
181. Hartmannsgruber V, Heyken WT, Kacik M, Kaistha A, Grgic I, Harteneck C, et al. Arterial Response to Shear Stress Critically Depends on Endothelial TRPV4 Expression. PloS One (2007) 2(9):e827. doi: 10.1371/journal.pone.0000827
182. Baylie RL, Brayden JE. TRPV Channels and Vascular Function. Acta Physiol (Oxf) (2011) 203(1):99–116. doi: 10.1111/j.1748-1716.2010.02217.x
183. Adapala RK, Thoppil RJ, Ghosh K, Cappelli HC, Dudley AC, Paruchuri S, et al. Activation of Mechanosensitive Ion Channel TRPV4 Normalizes Tumor Vasculature and Improves Cancer Therapy. Oncogene (2016) 35(3):314–22. doi: 10.1038/onc.2015.83
184. Thoppil RJ, Cappelli HC, Adapala RK, Kanugula AK, Paruchuri S, Thodeti CK. TRPV4 Channels Regulate Tumor Angiogenesis via Modulation of Rho/Rho Kinase Pathway. Oncotarget (2016) 7(18):25849–61. doi: 10.18632/oncotarget.8405
185. Shapovalov G, Lehen’kyi V, Skryma R, Prevarskaya N. TRP Channels in Cell Survival and Cell Death in Normal and Transformed Cells. Cell Calcium (2011) 50(3):295–302. doi: 10.1016/j.ceca.2011.05.006
186. Adapala RK, Thoppil RJ, Luther DJ, Paruchuri S, Meszaros JG, Chilian WM, et al. TRPV4 Channels Mediate Cardiac Fibroblast Differentiation by Integrating Mechanical and Soluble Signals. J Mol Cell Cardiol (2013) 54:45–52. doi: 10.1016/j.yjmcc.2012.10.016
187. Kusuyama J, Bandow K, Shamoto M, Kakimoto K, Ohnishi T, Matsuguchi T. Low Intensity Pulsed Ultrasound (LIPUS) Influences the Multilineage Differentiation of Mesenchymal Stem and Progenitor Cell Lines Through ROCK-Cot/Tpl2-MEK-ERK Signaling Pathway. J Biol Chem (2014) 289(15):10330–44. doi: 10.1074/jbc.M113.546382
188. Samarakoon R, Chitnis SS, Higgins SP, Higgins CE, Krepinsky JC, Higgins PJ. Redox-Induced Src Kinase and Caveolin-1 Signaling in TGF-Beta1-Initiated SMAD2/3 Activation and PAI-1 Expression. PloS One (2011) 6(7):e22896. doi: 10.1371/journal.pone.0022896
189. Sanz-Moreno V, Gaggioli C, Yeo M, Albrengues J, Wallberg F, Viros A, et al. ROCK and JAK1 Signaling Cooperate to Control Actomyosin Contractility in Tumor Cells and Stroma. Cancer Cell (2011) 20(2):229–45. doi: 10.1016/j.ccr.2011.06.018
190. Li X, Cheng Y, Wang Z, Zhou J, Jia Y, He X, et al. Calcium and TRPV4 Promote Metastasis by Regulating Cytoskeleton Through the RhoA/ROCK1 Pathway in Endometrial Cancer. Cell Death Dis (2020) 11(11):1009. doi: 10.1038/s41419-020-03181-7
191. Thoppil RJ, Adapala RK, Cappelli HC, Kondeti V, Dudley AC, Gary Meszaros J, et al. TRPV4 Channel Activation Selectively Inhibits Tumor Endothelial Cell Proliferation. Sci Rep (2015) 5:14257. doi: 10.1038/srep14257
192. Sladojevic N, Yu B, Liao JK. Regulator of G-Protein Signaling 5 Maintains Brain Endothelial Cell Function in Focal Cerebral Ischemia. J Am Heart Assoc (2020) 9(18):e017533. doi: 10.1161/JAHA.120.017533
193. Yang H, Lee S, Lee S, Kim K, Yang Y, Kim JH, et al. Sox17 Promotes Tumor Angiogenesis and Destabilizes Tumor Vessels in Mice. J Clin Invest (2013) 123(1):418–31. doi: 10.1172/JCI64547
194. Tan DS, Holzner M, Weng M, Srivastava Y, Jauch R. SOX17 in Cellular Reprogramming and Cancer. Semin Cancer Biol (2020) 67(Pt 1):65–73. doi: 10.1016/j.semcancer.2019.08.008
195. Kaelin WG Jr., Ratcliffe PJ. Oxygen Sensing by Metazoans: The Central Role of the HIF Hydroxylase Pathway. Mol Cell (2008) 30(4):393–402. doi: 10.1016/j.molcel.2008.04.009
196. De Bock K, De Smet F, Leite De Oliveira R, Anthonis K, Carmeliet P. Endothelial Oxygen Sensors Regulate Tumor Vessel Abnormalization by Instructing Phalanx Endothelial Cells. J Mol Med (Berl) (2009) 87(6):561–9. doi: 10.1007/s00109-009-0482-z
197. Kuchnio A, Moens S, Bruning U, Kuchnio K, Cruys B, Thienpont B, et al. The Cancer Cell Oxygen Sensor PHD2 Promotes Metastasis via Activation of Cancer-Associated Fibroblasts. Cell Rep (2015) 12(6):992–1005. doi: 10.1016/j.celrep.2015.07.010
198. Clever D, Roychoudhuri R, Constantinides MG, Askenase MH, Sukumar M, Klebanoff CA, et al. Oxygen Sensing by T Cells Establishes an Immunologically Tolerant Metastatic Niche. Cell (2016) 166(5):1117–31.e14. doi: 10.1016/j.cell.2016.07.032
199. Mazzone M, Dettori D, de Oliveira RL, Loges S, Schmidt T, Jonckx B, et al. Heterozygous Deficiency of PHD2 Restores Tumor Oxygenation and Inhibits Metastasis via Endothelial Normalization. Cell (2009) 136(5):839–51. doi: 10.1016/j.cell.2009.01.020
200. Patel A, Sabbineni H, Clarke A, Somanath PR. Novel Roles of Src in Cancer Cell Epithelial-to-Mesenchymal Transition, Vascular Permeability, Microinvasion and Metastasis. Life Sci (2016) 157:52–61. doi: 10.1016/j.lfs.2016.05.036
201. Caner A, Asik E, Ozpolat B. SRC Signaling in Cancer and Tumor Microenvironment. Adv Exp Med Biol (2021) 1270:57–71. doi: 10.1007/978-3-030-47189-7_4
202. Takara K, Eino D, Ando K, Yasuda D, Naito H, Tsukada Y, et al. Lysophosphatidic Acid Receptor 4 Activation Augments Drug Delivery in Tumors by Tightening Endothelial Cell-Cell Contact. Cell Rep (2017) 20(9):2072–86. doi: 10.1016/j.celrep.2017.07.080
203. Yasuda D, Kobayashi D, Akahoshi N, Ohto-Nakanishi T, Yoshioka K, Takuwa Y, et al. Lysophosphatidic Acid-Induced YAP/TAZ Activation Promotes Developmental Angiogenesis by Repressing Notch Ligand Dll4. J Clin Invest (2019) 129(10):4332–49. doi: 10.1172/JCI121955
204. Magrini E, Villa A, Angiolini F, Doni A, Mazzarol G, Rudini N, et al. Endothelial Deficiency of L1 Reduces Tumor Angiogenesis and Promotes Vessel Normalization. J Clin Invest (2014) 124(10):4335–50. doi: 10.1172/JCI70683
205. Sawada J, Urakami T, Li F, Urakami A, Zhu W, Fukuda M, et al. Small GTPase R-Ras Regulates Integrity and Functionality of Tumor Blood Vessels. Cancer Cell (2012) 22(2):235–49. doi: 10.1016/j.ccr.2012.06.013
206. Ager EI, Kozin SV, Kirkpatrick ND, Seano G, Kodack DP, Askoxylakis V, et al. Blockade of MMP14 Activity in Murine Breast Carcinomas: Implications for Macrophages, Vessels, and Radiotherapy. J Natl Cancer Inst (2015) 107(4). doi: 10.1093/jnci/djv017
207. Primo L, Seano G, Roca C, Maione F, Gagliardi PA, Sessa R, et al. Increased Expression of Alpha6 Integrin in Endothelial Cells Unveils a Proangiogenic Role for Basement Membrane. Cancer Res (2010) 70(14):5759–69. doi: 10.1158/0008-5472.CAN-10-0507
208. Maes H, Rubio N, Garg AD, Agostinis P. Autophagy: Shaping the Tumor Microenvironment and Therapeutic Response. Trends Mol Med (2013) 19(7):428–46. doi: 10.1016/j.molmed.2013.04.005
209. Maes H, Kuchnio A, Carmeliet P, Agostinis P. Chloroquine Anticancer Activity is Mediated by Autophagy-Independent Effects on the Tumor Vasculature. Mol Cell Oncol (2016) 3(1):e970097. doi: 10.4161/23723548.2014.970097
210. Maes H, Kuchnio A, Peric A, Moens S, Nys K, De Bock K, et al. Tumor Vessel Normalization by Chloroquine Independent of Autophagy. Cancer Cell (2014) 26(2):190–206. doi: 10.1016/j.ccr.2014.06.025
211. Ueda S, Saeki T, Takeuchi H, Shigekawa T, Yamane T, Kuji I, et al. In Vivo Imaging of Eribulin-Induced Reoxygenation in Advanced Breast Cancer Patients: A Comparison to Bevacizumab. Br J Cancer (2016) 114(11):1212–8. doi: 10.1038/bjc.2016.122
212. Funahashi Y, Okamoto K, Adachi Y, Semba T, Uesugi M, Ozawa Y, et al. Eribulin Mesylate Reduces Tumor Microenvironment Abnormality by Vascular Remodeling in Preclinical Human Breast Cancer Models. Cancer Sci (2014) 105(10):1334–42. doi: 10.1111/cas.12488
213. Rahma OE, Hodi FS. The Intersection Between Tumor Angiogenesis and Immune Suppression. Clin Cancer Res (2019) 25(18):5449–57. doi: 10.1158/1078-0432.CCR-18-1543
214. Chen DS, Mellman I. Oncology Meets Immunology: The Cancer-Immunity Cycle. Immunity (2013) 39(1):1–10. doi: 10.1016/j.immuni.2013.07.012
215. Jain RK, Koenig GC, Dellian M, Fukumura D, Munn LL, Melder RJ. Leukocyte-Endothelial Adhesion and Angiogenesis in Tumors. Cancer Metastasis Rev (1996) 15(2):195–204. doi: 10.1007/BF00437472
216. Hendry SA, Farnsworth RH, Solomon B, Achen MG, Stacker SA, Fox SB. The Role of the Tumor Vasculature in the Host Immune Response: Implications for Therapeutic Strategies Targeting the Tumor Microenvironment. Front Immunol (2016) 7:621. doi: 10.3389/fimmu.2016.00621
217. Mendler AN, Hu B, Prinz PU, Kreutz M, Gottfried E, Noessner E. Tumor Lactic Acidosis Suppresses CTL Function by Inhibition of P38 and JNK/c-Jun Activation. Int J Cancer (2012) 131(3):633–40. doi: 10.1002/ijc.26410
218. Gottfried E, Kunz-Schughart LA, Ebner S, Mueller-Klieser W, Hoves S, Andreesen R, et al. Tumor-Derived Lactic Acid Modulates Dendritic Cell Activation and Antigen Expression. Blood (2006) 107(5):2013–21. doi: 10.1182/blood-2005-05-1795
219. Fischer K, Hoffmann P, Voelkl S, Meidenbauer N, Ammer J, Edinger M, et al. Inhibitory Effect of Tumor Cell-Derived Lactic Acid on Human T Cells. Blood (2007) 109(9):3812–9. doi: 10.1182/blood-2006-07-035972
220. Guo F, Cui J. Anti-Angiogenesis: Opening a New Window for Immunotherapy. Life Sci (2020) 258:118163. doi: 10.1016/j.lfs.2020.118163
221. Voron T, Colussi O, Marcheteau E, Pernot S, Nizard M, Pointet AL, et al. VEGF-A Modulates Expression of Inhibitory Checkpoints on CD8+ T Cells in Tumors. J Exp Med (2015) 212(2):139–48. doi: 10.1084/jem.20140559
222. Gabrilovich D, Ishida T, Oyama T, Ran S, Kravtsov V, Nadaf S, et al. Vascular Endothelial Growth Factor Inhibits the Development of Dendritic Cells and Dramatically Affects the Differentiation of Multiple Hematopoietic Lineages In Vivo. Blood (1998) 92(11):4150–66. doi: 10.1182/blood.V92.11.4150
223. Chaudhary B, Khaled YS, Ammori BJ, Elkord E. Neuropilin 1: Function and Therapeutic Potential in Cancer. Cancer Immunol Immunother (2014) 63(2):81–99. doi: 10.1007/s00262-013-1500-0
224. Maenhout SK, Thielemans K, Aerts JL. Location, Location, Location: Functional and Phenotypic Heterogeneity Between Tumor-Infiltrating and non-Infiltrating Myeloid-Derived Suppressor Cells. Oncoimmunology (2014) 3(10):e956579. doi: 10.4161/21624011.2014.956579
225. Huang Y, Yuan J, Righi E, Kamoun WS, Ancukiewicz M, Nezivar J, et al. Vascular Normalizing Doses of Antiangiogenic Treatment Reprogram the Immunosuppressive Tumor Microenvironment and Enhance Immunotherapy. Proc Natl Acad Sci (2012) 109(43):17561–6. doi: 10.1073/pnas.1215397109
226. Scholz A, Lang V, Henschler R, Czabanka M, Vajkoczy P, Chavakis E, et al. Angiopoietin-2 Promotes Myeloid Cell Infiltration in a Beta(2)-Integrin-Dependent Manner. Blood (2011) 118(18):5050–9. doi: 10.1182/blood-2011-03-343293
227. Coffelt SB, Chen YY, Muthana M, Welford AF, Tal AO, Scholz A, et al. Angiopoietin 2 Stimulates TIE2-Expressing Monocytes to Suppress T Cell Activation and to Promote Regulatory T Cell Expansion. J Immunol (2011) 186(7):4183–90. doi: 10.4049/jimmunol.1002802
228. De Palma M, Venneri MA, Galli R, Sergi Sergi L, Politi LS, Sampaolesi M, et al. Tie2 Identifies a Hematopoietic Lineage of Proangiogenic Monocytes Required for Tumor Vessel Formation and a Mesenchymal Population of Pericyte Progenitors. Cancer Cell (2005) 8(3):211–26. doi: 10.1016/j.ccr.2005.08.002
229. Albini A, Bruno A, Noonan DM, Mortara L. Contribution to Tumor Angiogenesis From Innate Immune Cells Within the Tumor Microenvironment: Implications for Immunotherapy. Front Immunol (2018) 9:527. doi: 10.3389/fimmu.2018.00527
230. Yang L, DeBusk LM, Fukuda K, Fingleton B, Green-Jarvis B, Shyr Y, et al. Expansion of Myeloid Immune Suppressor Gr+CD11b+ Cells in Tumor-Bearing Host Directly Promotes Tumor Angiogenesis. Cancer Cell (2004) 6(4):409–21. doi: 10.1016/j.ccr.2004.08.031
231. Owen JL, Mohamadzadeh M. Macrophages and Chemokines as Mediators of Angiogenesis. Front Physiol (2013) 4:159. doi: 10.3389/fphys.2013.00159
232. Huang Y, Kim BYS, Chan CK, Hahn SM, Weissman IL, Jiang W. Improving Immune-Vascular Crosstalk for Cancer Immunotherapy. Nat Rev Immunol (2018) 18(3):195–203. doi: 10.1038/nri.2017.145
233. Jung K, Heishi T, Khan OF, Kowalski PS, Incio J, Rahbari NN, et al. Ly6Clo Monocytes Drive Immunosuppression and Confer Resistance to Anti-VEGFR2 Cancer Therapy. J Clin Invest (2017) 127(8):3039–51. doi: 10.1172/JCI93182
234. Jung K, Heishi T, Incio J, Huang Y, Beech EY, Pinter M, et al. Targeting CXCR4-Dependent Immunosuppressive Ly6C(low) Monocytes Improves Antiangiogenic Therapy in Colorectal Cancer. Proc Natl Acad Sci USA (2017) 114(39):10455–60. doi: 10.1073/pnas.1710754114
235. Rahbari NN, Kedrin D, Incio J, Liu H, Ho WW, Nia HT, et al. Anti-VEGF Therapy Induces ECM Remodeling and Mechanical Barriers to Therapy in Colorectal Cancer Liver Metastases. Sci Transl Med (2016) 8(360):360ra135. doi: 10.1126/scitranslmed.aaf5219
236. Munn LL, Jain RK. Vascular Regulation of Antitumor Immunity. Science (2019) 365(6453):544–5. doi: 10.1126/science.aaw7875
237. Xin Yu J, Hubbard-Lucey VM, Tang J. Immuno-Oncology Drug Development Goes Global. Nat Rev Drug Discovery (2019) 18(12):899–900. doi: 10.1038/d41573-019-00167-9
238. Tian L, Goldstein A, Wang H, Ching Lo H, Sun Kim I, Welte T, et al. Mutual Regulation of Tumour Vessel Normalization and Immunostimulatory Reprogramming. Nature (2017) 544(7649):250–4. doi: 10.1038/nature21724
239. Zheng X, Fang Z, Liu X, Deng S, Zhou P, Wang X, et al. Increased Vessel Perfusion Predicts the Efficacy of Immune Checkpoint Blockade. J Clin Invest (2018) 128(5):2104–15. doi: 10.1172/JCI96582
240. De Palma M, Jain RK. CD4(+) T Cell Activation and Vascular Normalization: Two Sides of the Same Coin? Immunity (2017) 46(5):773–5. doi: 10.1016/j.immuni.2017.04.015
241. Curran MA, Montalvo W, Yagita H, Allison JP. PD-1 and CTLA-4 Combination Blockade Expands Infiltrating T Cells and Reduces Regulatory T and Myeloid Cells Within B16 Melanoma Tumors. Proc Natl Acad Sci USA (2010) 107(9):4275–80. doi: 10.1073/pnas.0915174107
242. Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, et al. Tumor-Associated B7-H1 Promotes T-Cell Apoptosis: A Potential Mechanism of Immune Evasion. Nat Med (2002) 8(8):793–800. doi: 10.1038/nm730
243. Huang AC, Postow MA, Orlowski RJ, Mick R, Bengsch B, Manne S, et al. T-Cell Invigoration to Tumour Burden Ratio Associated With Anti-PD-1 Response. Nature (2017) 545(7652):60–5. doi: 10.1038/nature22079
244. Chen DS, Mellman I. Elements of Cancer Immunity and the Cancer-Immune Set Point. Nature (2017) 541(7637):321–30. doi: 10.1038/nature21349
245. Deng J, Liu X, Rong L, Ni C, Li X, Yang W, et al. IFNgamma-Responsiveness of Endothelial Cells Leads to Efficient Angiostasis in Tumours Involving Down-Regulation of Dll4. J Pathol (2014) 233(2):170–82. doi: 10.1002/path.4340
246. Lu Y, Yang W, Qin C, Zhang L, Deng J, Liu S, et al. Responsiveness of Stromal Fibroblasts to IFN-Gamma Blocks Tumor Growth via Angiostasis. J Immunol (2009) 183(10):6413–21. doi: 10.4049/jimmunol.0901073
247. Krishnan S, Amoozgar Z, Fukumura D, Jain RK. Implications of a Granulocyte-High Glioblastoma Microenvironment in Immune Suppression and Therapy Resistance(Dagger). J Pathol (2021) 254(2):105–8. doi: 10.1002/path.5637
248. Blank A, Kremenetskaia I, Urbantat RM, Acker G, Turkowski K, Radke J, et al. Microglia/macrophages Express Alternative Proangiogenic Factors Depending on Granulocyte Content in Human Glioblastoma. J Pathol (2021) 253(2):160–73. doi: 10.1002/path.5569
249. Duckworth C, Zhang L, Carroll SL, Ethier SP, Cheung HW. Overexpression of GAB2 in Ovarian Cancer Cells Promotes Tumor Growth and Angiogenesis by Upregulating Chemokine Expression. Oncogene (2016) 35(31):4036–47. doi: 10.1038/onc.2015.472
250. Liang J, Piao Y, Holmes L, Fuller GN, Henry V, Tiao N, et al. Neutrophils Promote the Malignant Glioma Phenotype Through S100A4. Clin Cancer Res (2014) 20(1):187–98. doi: 10.1158/1078-0432.CCR-13-1279
251. Zhao J, Chen AX, Gartrell RD, Silverman AM, Aparicio L, Chu T, et al. Immune and Genomic Correlates of Response to Anti-PD-1 Immunotherapy in Glioblastoma. Nat Med (2019) 25(3):462–9. doi: 10.1038/s41591-019-0349-y
252. Carretero R, Sektioglu IM, Garbi N, Salgado OC, Beckhove P, Hammerling GJ. Eosinophils Orchestrate Cancer Rejection by Normalizing Tumor Vessels and Enhancing Infiltration of CD8(+) T Cells. Nat Immunol (2015) 16(6):609–17. doi: 10.1038/ni.3159
253. Hatfield SM, Kjaergaard J, Lukashev D, Schreiber TH, Belikoff B, Abbott R, et al. Immunological Mechanisms of the Antitumor Effects of Supplemental Oxygenation. Sci Transl Med (2015) 7(277):277ra30. doi: 10.1126/scitranslmed.aaa1260
254. Liu Z, Wang Y, Huang Y, Kim BYS, Shan H, Wu D, et al. Tumor Vasculatures: A New Target for Cancer Immunotherapy. Trends Pharmacol Sci (2019) 40(9):613–23. doi: 10.1016/j.tips.2019.07.001
255. Upadhaya S, Neftelino ST, Hodge JP, Oliva C, Campbell JR, Yu JX. Combinations Take Centre Stage in PD1/PDL1 Inhibitor Clinical Trials. Nat Rev Drug Discovery (2021) 20(3):168–9. doi: 10.1038/d41573-020-00204-y
256. Nishino M, Ramaiya NH, Hatabu H, Hodi FS. Monitoring Immune-Checkpoint Blockade: Response Evaluation and Biomarker Development. Nat Rev Clin Oncol (2017) 14(11):655–68. doi: 10.1038/nrclinonc.2017.88
257. Yi M, Jiao D, Qin S, Chu Q, Wu K, Li A. Synergistic Effect of Immune Checkpoint Blockade and Anti-Angiogenesis in Cancer Treatment. Mol Cancer (2019) 18(1):60. doi: 10.1186/s12943-019-0974-6
258. Zhao S, Ren S, Jiang T, Zhu B, Li X, Zhao C, et al. Low-Dose Apatinib Optimizes Tumor Microenvironment and Potentiates Antitumor Effect of PD-1/PD-L1 Blockade in Lung Cancer. Cancer Immunol Res (2019) 7(4):630–43. doi: 10.1158/2326-6066.CIR-17-0640
259. Allen E, Jabouille A, Rivera LB, Lodewijckx I, Missiaen R, Steri V. Combined Antiangiogenic and Anti-PD-L1 Therapy Stimulates Tumor Immunity Through HEV Formation. Sci Transl Med. (2017) 9(385). doi: 10.1126/scitranslmed.aak9679
260. Georganaki M, van Hooren L, Dimberg A. Vascular Targeting to Increase the Efficiency of Immune Checkpoint Blockade in Cancer. Front Immunol (2018) 9:3081. doi: 10.3389/fimmu.2018.03081
261. Ott PA, Hodi FS, Buchbinder EI. Inhibition of Immune Checkpoints and Vascular Endothelial Growth Factor as Combination Therapy for Metastatic Melanoma: An Overview of Rationale, Preclinical Evidence, and Initial Clinical Data. Front Oncol (2015) 5:202. doi: 10.3389/fonc.2015.00202
262. Hodi FS, Lawrence D, Lezcano C, Wu X, Zhou J, Sasada T, et al. Bevacizumab Plus Ipilimumab in Patients With Metastatic Melanoma. Cancer Immunol Res (2014) 2(7):632–42. doi: 10.1158/2326-6066.CIR-14-0053
263. Elizabeth AAJ, Rivera LB, Lodewijckx I, Missiaen R, Steri V, Feyen K, et al. Combined Antiangiogenic and Anti–PD-L1 Therapy Stimulates Tumor Immunity Through HEV Formation. Sci Trans Med (2017) doi: 10.1126/scitranslmed.aak9679.
264. Salmon H, Franciszkiewicz K, Damotte D, Dieu-Nosjean MC, Validire P, Trautmann A, et al. Matrix Architecture Defines the Preferential Localization and Migration of T Cells Into the Stroma of Human Lung Tumors. J Clin Invest (2012) 122(3):899–910. doi: 10.1172/JCI45817
265. Zhang L, Takara K, Yamakawa D, Kidoya H, Takakura N. Apelin as a Marker for Monitoring the Tumor Vessel Normalization Window During Antiangiogenic Therapy. Cancer Sci (2015) 107(1):36–44. doi: 10.1111/cas.12836
266. El Alaoui-Lasmaili K, Faivre B. Antiangiogenic Therapy: Markers of Response, “Normalization” and Resistance. Crit Rev Oncol Hematol (2018) 128:118–29. doi: 10.1016/j.critrevonc.2018.06.001
267. Franco M, Man S, Chen L, Emmenegger U, Shaked Y, Cheung AM, et al. Targeted Anti-Vascular Endothelial Growth Factor Receptor-2 Therapy Leads to Short-Term and Long-Term Impairment of Vascular Function and Increase in Tumor Hypoxia. Cancer Res (2006) 66(7):3639–48. doi: 10.1158/0008-5472.CAN-05-3295
268. Zahra MA, Hollingsworth KG, Sala E, Lomas DJ, Tan LT. Dynamic Contrast-Enhanced MRI as a Predictor of Tumour Response to Radiotherapy. Lancet Oncol (2007) 8(1):63–74. doi: 10.1016/s1470-2045(06)71012-9
269. Bowen SR, Yuh WTC, Hippe DS, Wu W, Partridge SC, Elias S, et al. Tumor Radiomic Heterogeneity: Multiparametric Functional Imaging to Characterize Variability and Predict Response Following Cervical Cancer Radiation Therapy. J Magn Reson Imaging (2018) 47(5):1388–96. doi: 10.1002/jmri.25874
270. Price JM, Robinson SP, Koh DM. Imaging Hypoxia in Tumours With Advanced MRI. Q J Nucl Med Mol Imaging (2013) 57(3):257–70.
271. Wang L, Lu YH, Wang TY. [Value of Ultrasound-Targeted Vascular Endothelial Growth Factor Receptor-2 in Non-Invasive Monitoring of Anti-Angiogenic Response in Nude Mice With Subcutaneous Xenograft Model]. Zhonghua Zhong Liu Za Zhi (2020) 42(10):856–60. doi: 10.3760/cma.j.cn112152-20191230-00854
272. Lassau N, Chebil M, Chami L, Bidault S, Girard E, Roche A. Dynamic Contrast-Enhanced Ultrasonography (DCE-US): A New Tool for the Early Evaluation of Antiangiogenic Treatment. Target Oncol (2010) 5(1):53–8. doi: 10.1007/s11523-010-0136-7
273. De Palma GD, Maione F, Esposito D, Luglio G, Giglio MC, Siciliano S, et al. In Vivo Assessment of Tumour Angiogenesis in Colorectal Cancer: The Role of Confocal Laser Endomicroscopy. Colorectal Dis (2016) 18(2):O66–73. doi: 10.1111/codi.13222
274. Chen BB, Lu YS, Lin CH, Chen WW, Wu PF, Hsu CY, et al. A Pilot Study to Determine the Timing and Effect of Bevacizumab on Vascular Normalization of Metastatic Brain Tumors in Breast Cancer. BMC Cancer (2016) 16:466. doi: 10.1186/s12885-016-2494-8
275. Lin A, Hahn SM. Hypoxia Imaging Markers and Applications for Radiation Treatment Planning. Semin Nucl Med (2012) 42(5):343–52. doi: 10.1053/j.semnuclmed.2012.04.002
276. Duda DG, Willett CG, Ancukiewicz M, di Tomaso E, Shah M, Czito BG, et al. Plasma Soluble VEGFR-1 is a Potential Dual Biomarker of Response and Toxicity for Bevacizumab With Chemoradiation in Locally Advanced Rectal Cancer. Oncologist (2010) 15(6):577–83. doi: 10.1634/theoncologist.2010-0029
277. Nixon AB, Pang H, Starr MD, Friedman PN, Bertagnolli MM, Kindler HL, et al. Prognostic and Predictive Blood-Based Biomarkers in Patients With Advanced Pancreatic Cancer: Results From CALGB80303 (Alliance). Clin Cancer Res (2013) 19(24):6957–66. doi: 10.1158/1078-0432.CCR-13-0926
278. Picault FX, Chaves-Almagro C, Projetti F, Prats H, Masri B, Audigier Y. Tumour Co-Expression of Apelin and its Receptor is the Basis of an Autocrine Loop Involved in the Growth of Colon Adenocarcinomas. Eur J Cancer (2014) 50(3):663–74. doi: 10.1016/j.ejca.2013.11.017
279. Berta J, Kenessey I, Dobos J, Tovari J, Klepetko W, Jan Ankersmit H, et al. Apelin Expression in Human non-Small Cell Lung Cancer: Role in Angiogenesis and Prognosis. J Thorac Oncol (2010) 5(8):1120–9. doi: 10.1097/JTO.0b013e3181e2c1ff
280. Wan Y, Zeng ZC, Xi M, Wan S, Hua W, Liu YL, et al. Dysregulated microRNA-224/Apelin Axis Associated With Aggressive Progression and Poor Prognosis in Patients With Prostate Cancer. Hum Pathol (2015) 46(2):295–303. doi: 10.1016/j.humpath.2014.10.027
281. Ma Q, Gu JT, Wang B, Feng J, Yang L, Kang XW, et al. PlGF Signaling and Macrophage Repolarization Contribute to the Anti-Neoplastic Effect of Metformin. Eur J Pharmacol (2019) 863:172696. doi: 10.1016/j.ejphar.2019.172696
282. Piludu F, Marzi S, Pace A, Villani V, Fabi A, Carapella CM, et al. Early Biomarkers From Dynamic Contrast-Enhanced Magnetic Resonance Imaging to Predict the Response to Antiangiogenic Therapy in High-Grade Gliomas. Neuroradiology (2015) 57(12):1269–80. doi: 10.1007/s00234-015-1582-9
283. Liang JY, Xiao ZY, Zhang D, Ma MJ, Huang JX, Shi CZ, et al. [Value of Dynamic MRI in Monitoring the Microenvironmental Changes of Anti-Vascular Therapy in a Xenograft Model]. Zhonghua Yi Xue Za Zhi (2020) 100(1):51–6. doi: 10.3760/cma.j.issn.0376-2491.2020.01.012
284. Lassau N, Coiffier B, Kind M, Vilgrain V, Lacroix J, Cuinet M, et al. Selection of an Early Biomarker for Vascular Normalization Using Dynamic Contrast-Enhanced Ultrasonography to Predict Outcomes of Metastatic Patients Treated With Bevacizumab. Ann Oncol (2016) 27(10):1922–8. doi: 10.1093/annonc/mdw280
285. Amadori M, Barone D, Scarpi E, Oboldi D, Amadori E, Bandi G, et al. Dynamic Contrast-Enhanced Ultrasonography (D-CEUS) for the Early Prediction of Bevacizumab Efficacy in Patients With Metastatic Colorectal Cancer. Eur Radiol (2018) 28(7):2969–78. doi: 10.1007/s00330-017-5254-5
286. Lassau N, Chami L, Chebil M, Benatsou B, Bidault S, Girard E, et al. Dynamic Contrast-Enhanced Ultrasonography (DCE-US) and Anti-Angiogenic Treatments. Discovery Med (2011) 11(56):18–24.
287. Kuleznewa J, Nikitin B, Orlova N, Lesko K. Vascular Involvement Assessment in Patients With Hilar Cholangiocarcinoma by 128-Row Multidetector Computed Tomography. Ann Hepatobil Pancreat Surg (2021) 25(Suppl 1):S408. doi: 10.14701/ahbps.EP-211
288. Kim JI, Lee HJ, Kim YJ, Kim KG, Lee KW, Lee JH, et al. Multiparametric Monitoring of Early Response to Antiangiogenic Therapy: A Sequential Perfusion CT and PET/CT Study in a Rabbit VX2 Tumor Model. ScientificWorldJournal (2014) 2014:701954. doi: 10.1155/2014/701954
289. Dighe S, Castellano E, Blake H, Jeyadevan N, Koh MU, Orten M, et al. Perfusion CT to Assess Angiogenesis in Colon Cancer: Technical Limitations and Practical Challenges. Br J Radiol (2012) 85(1018):e814–25. doi: 10.1259/bjr/19855447
290. Hernandez-Agudo E, Mondejar T, Soto-Montenegro ML, Megias D, Mouron S, Sanchez J, et al. Monitoring Vascular Normalization Induced by Antiangiogenic Treatment With (18)F-Fluoromisonidazole-PET. Mol Oncol (2016) 10(5):704–18. doi: 10.1016/j.molonc.2015.12.011
291. Nedergaard MK, Kristoffersen K, Michaelsen SR, Madsen J, Poulsen HS, Stockhausen MT, et al. The Use of Longitudinal 18F-FET MicroPET Imaging to Evaluate Response to Irinotecan in Orthotopic Human Glioblastoma Multiforme Xenografts. PloS One (2014) 9(2):e100009. doi: 10.1371/journal.pone.0100009
292. Gourgue F, Derouane F, van Marcke C, Villar E, Dano H, Desmet L, et al. Tumor Apelin and Obesity are Associated With Reduced Neoadjuvant Chemotherapy Response in a Cohort of Breast Cancer Patients. Sci Rep (2021) 11(1):9922. doi: 10.1038/s41598-021-89385-z
293. Duda DG. Molecular Biomarkers of Response to Antiangiogenic Therapy for Cancer. ISRN Cell Biol (2012) 2012. doi: 10.5402/2012/587259
294. van Beijnum JR, Nowak-Sliwinska P, Huijbers EJ, Thijssen VL, Griffioen AW. The Great Escape; the Hallmarks of Resistance to Antiangiogenic Therapy. Pharmacol Rev (2015) 67(2):441–61. doi: 10.1124/pr.114.010215
295. Kaur S, Bronson SM, Pal-Nath D, Miller TW, Soto-Pantoja DR, Roberts DD. Functions of Thrombospondin-1 in the Tumor Microenvironment. Int J Mol Sci (2021) 22(9). doi: 10.3390/ijms22094570
296. Lunt SJ, Gray C, Reyes-Aldasoro CC, Matcher SJ, Tozer GM. Application of Intravital Microscopy in Studies of Tumor Microcirculation. J BioMed Opt (2010) 15(1):011113. doi: 10.1117/1.3281674
Keywords: antiangiogenesis, tumor vessel normalization, immunotherapy, microRNA, endothelial cell metabolism, extracellular matrix
Citation: Yang T, Xiao H, Liu X, Wang Z, Zhang Q, Wei N and Guo X (2021) Vascular Normalization: A New Window Opened for Cancer Therapies. Front. Oncol. 11:719836. doi: 10.3389/fonc.2021.719836
Received: 03 June 2021; Accepted: 23 July 2021;
Published: 12 August 2021.
Edited by:
Giorgio Seano, Institut Curie, FranceReviewed by:
Shanmugarajan Krishnan, Massachusetts General Hospital, United StatesBarbara Muz, Washington University in St. Louis, United States
Copyright © 2021 Yang, Xiao, Liu, Wang, Zhang, Wei and Guo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xinggang Guo, Z3hnMjM2NTI1NUAxMjYuY29t
†These authors have contributed equally to this work