 Fang Feng
Fang Feng Huiqin Han
Huiqin Han Shuqi Wu
Shuqi Wu Hui Wang
Hui Wang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Oncol. , 28 September 2021
Sec. Head and Neck Cancer
Volume 11 - 2021 | https://doi.org/10.3389/fonc.2021.718578
Iodide uptake and the metabolism of thyroid cells are regulated by thyrotropin (TSH)-TSH receptor (TSHR) signaling. Thus, it is necessary to elevate serum TSH levels by T4 withdraw or rTSH administration to facilitate radioiodide (131I) therapy for differentiated thyroid cancer (DTC). However, non-iodide-avid metastases of DTC which is dedifferentiated do not respond to stimulation by high levels of TSH, suggesting abnormal TSH-TSHR signal transduction in cancer cells. In addition, PI3K/AKT/mTOR signaling activation has been shown to be associated with the dedifferentiated phenotype of thyroid cancer, but the mechanism remains elusive. Therefore, in this study, we aimed to explore the role of abnormal TSH-TSHR signaling activation in regulating iodide uptake and cell mobility in thyroid cancer and its relationship with PI3K/AKT/mTOR signaling. We found that in thyroid cancer cells, TSH binds TSHR coupled to the Gα12/13 protein and then activates RhoA through interacting with leukemia associated RhoA guanine exchange factor (LARG). This results in a promigration tumorigenic phenotype independent of canonical TSHR-GαS signaling that regulates the expression of molecules involved in iodine uptake and metabolism. We observed that signaling pathways downstream of Gα12/13 signaling were increased, while that of Gαs signaling was decreased in thyroid cancer cells undergoing dedifferentiation compared to control cells following stimulation with different levels of TSH. PI3K/AKT/mTOR signaling activation enhanced Gα12/13 signaling through increasing LARG levels but also inhibited the expression of molecules downstream of Gαs signaling, including thyroid-specific molecules, and iodide uptake. In summary, our results demonstrate the noncanonical activation of TSH-TSHR signaling and its role in increasing the cell mobility and dedifferentiation of thyroid cancer through crosstalk with PI3K/AKT/mTOR signaling.
Thyroid cancer, over 90% of which is differentiated thyroid cancer (DTC), is one of the most rapidly increasing cancers worldwide. Although most patients with DTC show good prognosis and are cured by thyroidectomy followed by radioiodine (131I) ablation, 1-4% of patients at initial diagnosis and 7-23% of patients during follow-up develop distant metastasis (1, 2). To date, 131I therapy remains the first-line treatment for metastatic DTC, and a high level of 131I uptake is a marker of good prognosis (3, 4). However, in one-third of metastatic DTC patients, the cancer cells undergo dedifferentiation and lose their ability to accumulate 131I, with increased cell proliferation and invasion; therefore, these patients exhibit a poorer prognosis (3–6). In addition, 131I treatment is limited in thyroid cancer that is poorly differentiated or anaplastic thyroid cancer (ATC), which do not take up or take up very low levels of 131I (7), but the molecular mechanism remains elusive.
Iodide uptake and thyroid hormone (TH) biosynthesis, vital functions of thyroid cells, are mainly controlled by thyrotropin (TSH) and its receptor (TSHR). As a G protein coupled receptor (GPCR), TSHR can couple with all four G protein families to activate downstream signaling (8), though the Gαs-adenylyl cyclase-cyclic AMP (cAMP) and Gαq-calcium pathways have been reported to be the most physiologically relevant (9, 10). Classically, through coupling to Gαs and activating the cAMP-PKA pathway, TSH-TSHR stimulates the expression of various thyroid-specific genes involved in iodide metabolism, such as thyroglobulin (Tg), thyroperoxidase (TPO) and sodium-iodide symporter (NIS), in turn increasing the iodide uptake of normal or tumorous thyroid cells. Therefore, T4 withdrawal or rTSH administration is routinely utilized to increase serum TSH levels to facilitate 131I therapy for DTC patients. However, in patients with non-iodide-avid metastases, high levels of TSH cannot stimulate iodide uptake by the lesions. Our previous work has shown that TSHR expression levels play important roles in maintaining the differentiation of thyroid cancer cells (11–13). Nevertheless, in some cases, TSHR expression has been reported to remain normal (14). Moreover, few loss-of-function TSHR mutations in DTC tissue have been reported. Therefore, we hypothesized that the abnormal transduction of downstream TSHR signaling contributes to decreased iodide uptake and is related to the invasion phenotype.
Among G proteins, Gα12/13 is closely associated with increased cell mobility in some cancer tissues (15), but its role in TSHR signaling in thyroid cancer remains elusive. In addition, crosstalk between PI3K/AKT/mTOR signaling activation and multiple protumorigenic pathways has been well established in multiple cancer models, including thyroid cancers, and shown to play important roles in cancer initiation and progression (16). Importantly, PI3K/AKT/mTOR signaling activation is associated with reduced NIS expression and iodide uptake in thyroid cancer cells (17). These findings suggest that PI3K signaling is involved in crosstalk with TSHR signaling, which affects the differentiation of thyroid cancer cells. We therefore hypothesized that in addition to its canonical function in regulating iodide uptake and TH synthesis, the abnormal activation of TSHR signaling, such as that through Gα12/13 downstream signaling, promotes tumorigenesis and is associated with the dedifferentiation of DTC cells.
We utilized human thyroid cancer cell lines TPC1, BCPAP and FTC-133, grown in DMEM, RPMI-1640 and DMEM/F12 medium respectively, supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin (P/S). We also used a rat thyroid cell line PCCL3, cultured in Coon’s Modified F-12 medium (Sigma T8931), supplemented with 1% P/S, 5% FBS, 10mM NaHCO3, 2mM L-Glutamine, 1mU/ml TSH (Sigma I1882), 10μg/ml bovine insulin (Sigma), 10nM Hydrocortisone (Sigma H4001), 10ng/ml Somatostatin (Sigma S1763), 5μg/ml Transferrin (Sigma T8158) and 2 ng/ml L-Glycyl-Histidyl-Lysine (Sigma G1887). For wound healing assay, HBSS was used instead of FBS in the medium. All cell lines were maintained at 37°C and 5% CO2 culture conditions. All experiments were conducted with cells at passage numbers between 8 and 15. All cell lines were authenticated through the American Type Culture Collection (ATCC) human cell authentication service (ATCC® 135-XV™) and were 100% matched to the reported STR profiles in the DSMZ database (test date 19/08/2018).
Rapamycin, Y27632 were purchased from Sigma. LY294002 and MK-2206 were obtained from Selleckchem (Houston, TX). Dimethyl sulfoxide (DMSO, Sigma) served as the vehicle control for the experiments involving PI3K/AKT/mTOR inhibitor treatment.
RNA was extracted from the cell lines using the NucleoSpin® RNA Plus kit (MACHEREY-NAGEL GmbH & Co. KG, D̈ren, Germany) following the user manual, and reverse transcribed using Superscript III reverse transcriptase (Life Technologies). Primers were designed for gene transcripts of interest and cDNA quantified using SYBR Green (Life Technologies). We utilized the Applied Biosystems 7500 Real-Time PCR System. Results were analyzed using the standard ΔΔCT method.
Protein was extracted from whole cell lysates using the Mammalian Protein Extraction Reagent M-PER (Thermo Scientific Pierce, Rockford, IL) supplemented with a cocktail of protease and phosphatase inhibitors (Sigma) and quantified through the BCA protein assay (Thermo Scientific Pierce). Lysates were separated by SDS-PAGE and transferred onto nitrocellulose membranes. We probed for anti-TSHR (4C1) mouse monoclonal (Thermo Fisher Scientific) at 1:1000, anti-Gα12 (N3C3) rabbit polyclonal (GeneTex, Irvine, CA) at 1:1000, anti-Gα13 rabbit polyclonal (NewEast Bioscience, Malvern, PA) at 1:1000, anti-PTEN (6H2.1) mouse monoclonal (Cascade Bioscience, Winchester, MA) at 1:1000, anti-LARG mouse monoclonal (EMD Millipore, Temecula, CA) at 1:10000, anti Phospho-AKT (Ser473) rabbit polyclonal (Cell Signaling #9271L) at 1:1000, anti-Phospho-p70 S6 Kinase (Thr389) (1A5) mouse monoclonal (Cell Signaling #9206S) at 1:1000, anti Phospho-S6 ribosomal protein (S235/236) rabbit monoclonal (Cell Signaling #4858S) at 1:1000, anti-Phospho-cofilin (Abcam #ab12866) at 1:1000 and anti-GAPDH rabbit monoclonal (Cell Signaling #2118) at 1:20000 dilution. Blots were scanned digitally using the ChemiDoc™ touch imaging system (Bio-Rad, Hercules, CA). Densitometry was performed using ImageJ software.
Thyroglobulin levels in thyroid cancer cells were measured using radioimmunoassay according to the manufacturer’s instructions as described previously (11). Total proteins were extracted from thyroid cancer cells as described above and quantified by using the BCA method. Relative Tg content was calculated as the ratio of Tg concentration to total protein concentration.
cDNAs encoding the constitutively active Gα12 (Gα12Q229L) and Gα13 (Gα13Q226L) were subcloned into pCMV5 vector. Cells at 70% confluence were transiently transfected with plasmid using Lipofectamine 3000 reagent (Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions. The efficiency of transfection was determined using Western blotting and RT-PCR to evaluate protein and mRNA expression following cell collection. For PTEN transfection, we used GFP-tagged wild type PTEN plasmid (pcDNA3.1 vector). The empty vector pcDNA3.1 was set as the control. For transient overexpression, cell lines were transfected with Lipofectamine 3000 reagent (Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions. Cells were grown for at least 48 hours before harvesting. To generate stable cell lines, retrovirally-transduced cells were kept under 1 µg/ml Puromycin selection, and the different Puromycin-resistant cell colonies were pooled together for downstream interrogation.
Rho activity was measured using the RhoA pull-down activation assay kit (Cytoskeleton, Denver, CO). Cells were homogenized in the cell lysis buffer containing protease inhibitor cocktail. Lysates were cleared by centrifugation and the soluble fraction was incubated with 50μg of GST-Rhotekin beads, which can specifically pull down activated RhoA, for 1 hour at 4°C. Beads were collected by centrifugation for 1minute at 3,000×g and 4°C, washed, resuspended in Laemmli buffer, separated by SDS-PAGE, and then analyzed by western blotting using a mouse monoclonal anti-RhoA antibody (1:500 dilution). Relative variations in normalized RhoA-GTP level is defined as the ratio of the level of activated GTP-bound forms of RhoA and the level of total RhoA in NIS stable cells, divided by the same ratio in control cells.
Cells were pelleted and lysed with M-PER (Thermo Scientific Pierce) supplemented with a cocktail of protease and phosphatase inhibitors (Sigma-Aldrich). Protein lysates were collected by centrifugation at 13,000 RPM for 10 min at 4°C and pre-cleared by incubation with Thermo Protein A/G Dynabeads (Thermo Scientific Pierce) for 3 hours at 4°C on a rotator. Pre-cleared protein lysates were quantified with the BCA Protein Assay Kit (Thermo Scientific Pierce), and 2 mg/ml lysates were prepared. We used anti-Gα12/13, anti-LARG and anti-RhoA antibodies for pull-down and immunoblotting at the recommended dilutions. Cell lysates were separated by SDS-PAGE and transferred onto nitrocellulose membranes. Blots were scanned digitally using the ChemiDoc™ touch imaging system (Bio-Rad).
Thyroid cancer cells were seeded in 6 well plates and allowed to grow overnight. For TSHR, Gα12/13, LARG or PTEN knockdown, cells were transfected with TSHR, Gα12/13, LARG or PTEN siRNA smartpool (Dharmacon, Lafayette, CO) using Lipofectamine RNAiMAX (Thermo Fisher Scientific) according to the manufacturer’s instructions. We used ON-TARGET non-targeting siRNA pool (Dharmacon) as a control (siNT). Cells were collected for downstream analysis from 48 hours to 72 hours after knockdown transfection. We used Western blot and qRT-PCR analysis to confirm knockdown.
Cells were seeded on coverslips and were fixed with 4% paraformaldehyde for 2 minutes at room temperature, washed with PBS, and then incubated with 1% Triton X-100 for 2 minutes at room temperature. Cells were blocked with 10% goat serum (Vector Laboratories Inc, Burlingame, CA) for 1 hour at room temperature, and incubated with rabbit anti-LARG antibodies (at 1:500 dilution) overnight at 4°C. Secondary antibodies (Alexa 568) were incubated at 1:2000 dilution for 1 hour in the dark at room temperature. Coverslips were mounted using ProLong Gold Antifade mountant with DAPI (Invitrogen). Slides were visualized and images obtained using the Leica TCS SP5 II confocal microscope (Leica Microsystems, Heidelberg, Germany).
Intracellular cAMP levels were measured follow the method reported previously (18). Briefly, culture medium was removed 48 h after transfection or treatment with drugs and replaced by HEPES buffer for 30 min. Then, the medium was discarded and replaced with 0.1 M HCl. The cell extracts were dried in a vacuum concentrator, resuspended in water, and diluted appropriately for cAMP measurements by RIA. Duplicate samples were assayed in all experiments. Results are expressed as picomoles cAMP per milliliter. Experiments were performed in triplicates.
For wound-healing assays, confluent-transfected cells monolayers were scraped with a fine sterile pipette tip, placed in the cell incubator (5% CO2, 37°C). Image were acquired at 0 and 20 hours after scraping using a Leica DMI3000B microscope (Leica Microsystems). HBSS was used instead of FBS in the medium for suppressing cell proliferation. For migration inhibitor tests, Rho-associated coiled-coil kinase (ROCK) inhibitor Y-27632 at 30μM was added at 1 hour before scraping. The wound margin area was determined by image processing using ImageJ software. The average wound-healing speeds were calculated. Each condition was run in triplicate. Data represent means ± SEM from 3 independent experiments.
RAIU assay was performed follow the methods reported previously (19). Briefly, cells were pretreated with siRNA for 24h before the assay. At 24 h, cells were incubated with 2 μCi Na125I in 5 μM nonradioactive NaI for 30 min at 37°C with 5% CO2. Cells were then washed with cold Hank’s Balanced Salt Solution two times and then lysed in cold 95% ethanol for 20 min at room temperature. Cell lysate was collected and counted for radioactivity by the gamma-counter. RAIU not mediated by NIS was excluded by performing the assay in the presence of 3 mM perchlorate , a competitive inhibitor of iodide uptake by NIS. Experiments were performed in triplicates.
Experimental data between control and experimental cells are given as means ± SEM with n corresponding to the number of experiments performed. The Student’s t test was used for significance testing as indicated in figure legends. All statistical tests were two-sided, and p-values <0.05 were deemed significant.
TSH canonically regulates the functions of thyroid cells through binding TSHR, which then activates downstream signaling. To test our hypothesis that TSH-TSHR signaling is also associated with the cell migration phenotype in thyroid cancer cells, we performed a wound healing assay in the TPC1, BCPAP and FTC-133 thyroid cancer cell lines grown in medium containing 30 μU/ml TSH, which was observed to induce sufficient TSHR expression (Supplementary Figure S1). HBSS was used instead of FBS in the medium for suppressing cell proliferation. The cell migration rate was increased in all of the three cell lines after their incubation with TSH. The migration rate was increased 1.3-, 1.4- and 1.8-fold in TPC1, BCPAP and FTC-133 cells, respectively, after treatment with TSH compared to the migration rate of corresponding control cells (P=0.016, 0.004 and 0.003, respectively; Figure 1A). To further validate that this promigration effect occurs through TSHR signaling, we transiently knocked down TSHR in thyroid cancer cells through siRNA. These cells were cultured with 30 μU/ml TSH, after which we compared their migration rates. The migration rates of TPC1, BCPAP and FTC-133 cells at 72 h following TSHR siRNA knockdown were reduced by approximately 20%, 23% and 25%, respectively, compared to those in control cells transfected with siNT (P=0.007, 0.005 and 0.004; Figure 1B and Supplementary Figure S2). siRNA-mediated TSHR knockdown was confirmed by Western blotting and qRT-PCR (Figure 1C and Supplementary Figure S2). Taken together, these data provide evidence that TSH-TSHR signaling activation increases thyroid cancer cell migration.
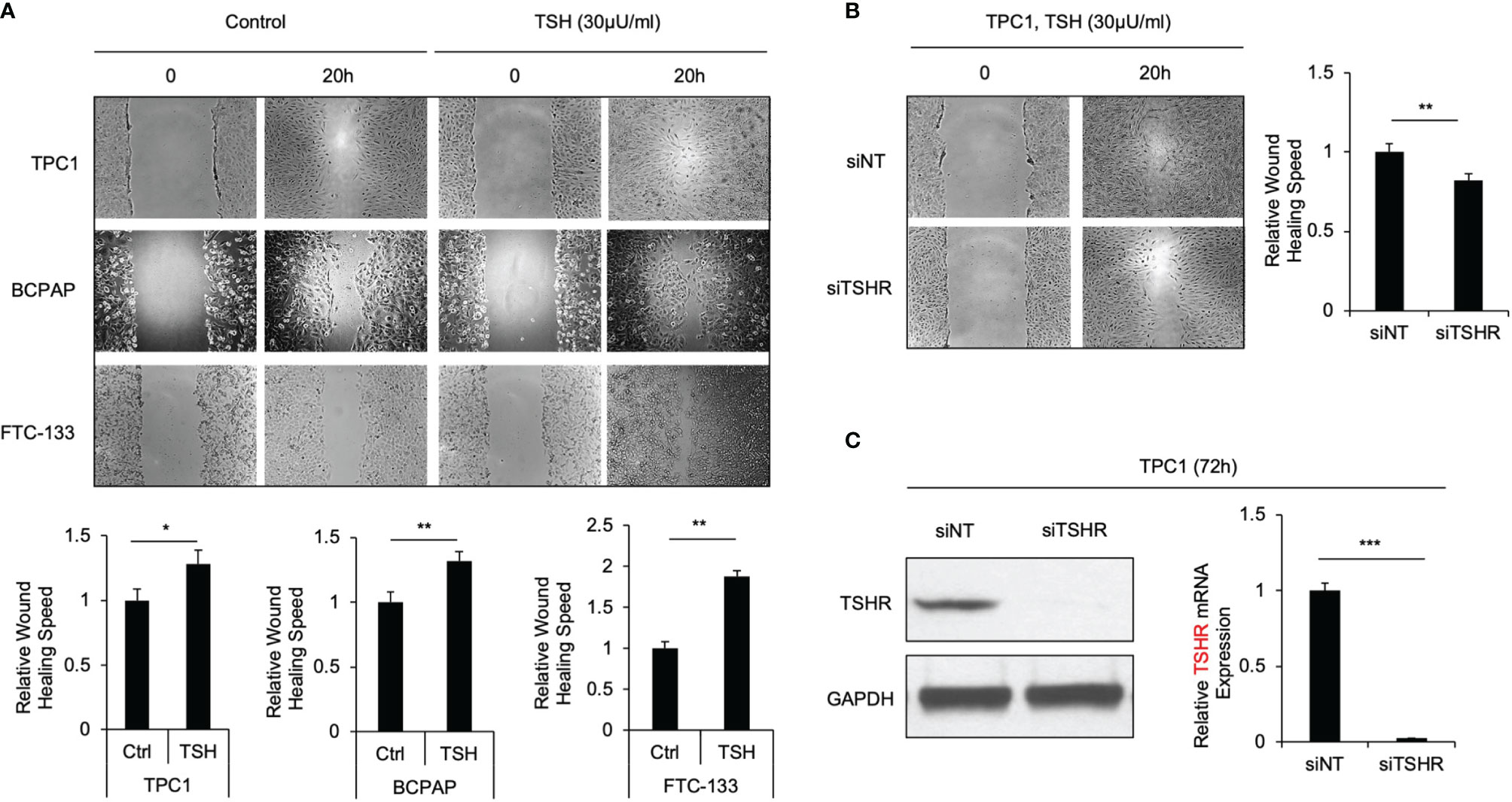
Figure 1 TSH-TSHR increase cell migration in thyroid cancer cells. (A) Wound-healing assay for TPC1, BCPAP and FTC-133 cells treated with 30μU/ml TSH or control with HBSS. Pictures were taken at 0 and 20 hours after wound scratch. The bar graphs represent relative wound healing rate of TPC1, BCPAP and FTC-133 cells with TSH treatment normalized with controls. Data represent means ± SEM of 3 independent experiments. (B) Wound-healing assay for TPC1 cells incubated with 30 μU/mL TSH transiently transfected with non-targeting siRNA control (siNT) or TSHR siRNA (siTSHR). Wound scratch was performed at 72 hours after siRNA transfection, and then picture was taken at that point and 20 hours later. Bar graph represents relative wound healing rate of TPC1 after siTSHR knockdown normalized with control. Data represent means ± SEM of 3 independent experiments. (C) Immunoblots of TSHR and GAPDH in TPC1 cells transfected with siNT and siTSHR for 72 hours. Bar charts represent relative TSHR mRNA levels in TPC1 cells transfected with siNT and siTSHR for 72 hours. Data represent means ± SEM of 3 independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001.
RhoA activation plays an important role in thyroid cancer cell motility, according to our most recent study (20). Hence, as the downstream activation of RhoA was observed to be relevant to the migration phenotype, we observed its effects in thyroid cancer cells treated with TSH with a GST-Rhotekin pulldown assay. The relative levels of the activated GTP-bound form of RhoA in TPC1, BCPAP and FTC-133 cells grown with 30 μU/ml TSH for 72 hours were significantly higher than those in control cells (2.2-, 2.5- and 2.6-fold; P=0.018, 0.009 and 0.001, respectively; Figure 2A). Furthermore, the Rho-associated kinase (ROCK) inhibitor Y-27632, which was conformed to decrease phospho-cofilin by immunoblot, suppressed the migration rate of thyroid cancer cells treated with TSH in the absent of FBS. In TPC1 cells, the migration rate was decreased 0.77-fold after treatment with Y-27632 compared to that in control cells (P<0.001, Figure 2B). These data further support the involvement of the RhoA-ROCK pathway in enhanced cell migration downstream of TSH-TSHR signaling.
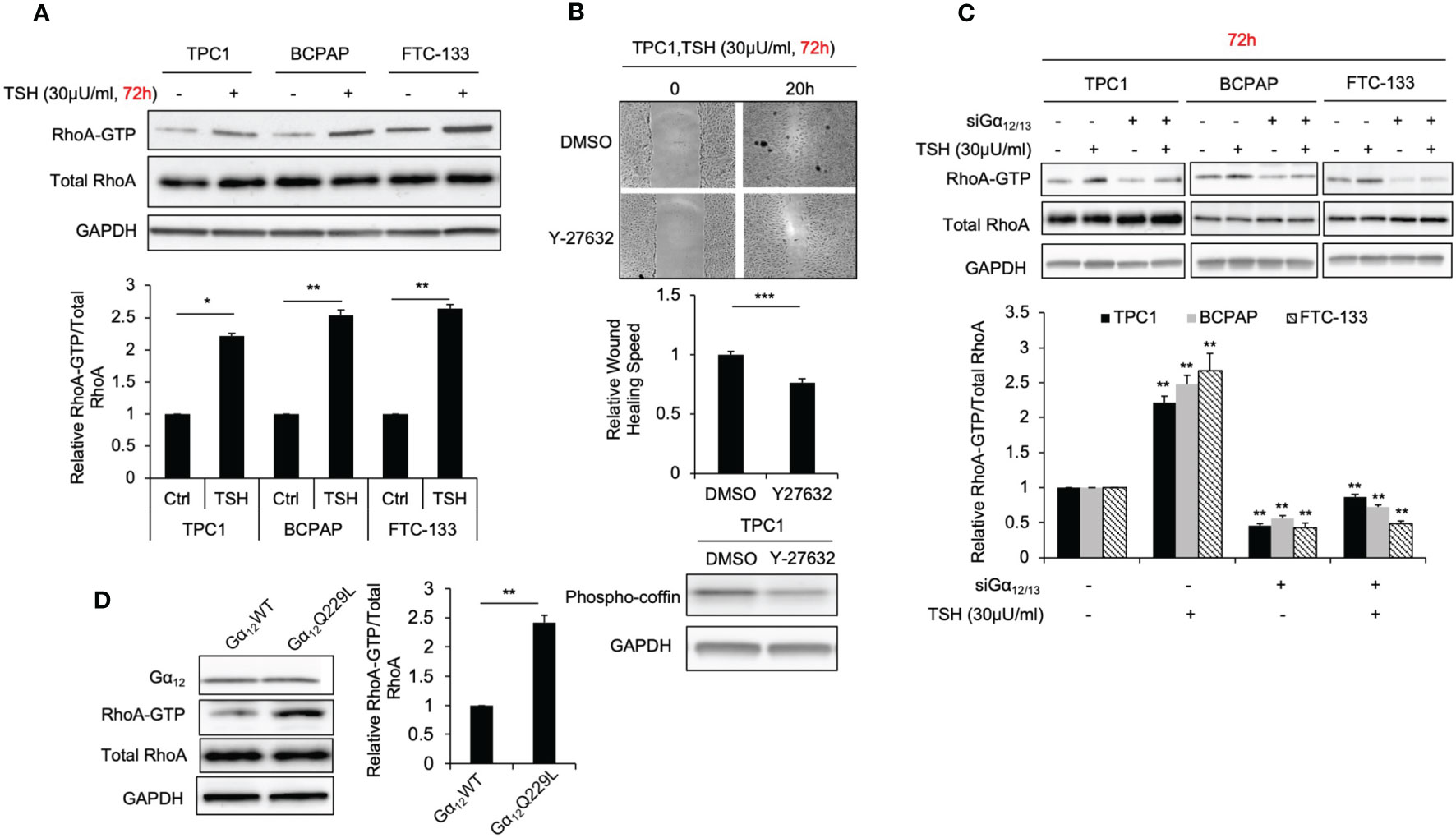
Figure 2 TSH increasing RhoA activation and cell migration is Gα12/13 dependent. (A) Immunoblots of RhoA and GAPDH from total cell lysates and RhoA after GST-Rhotekin pull-down (RhoA-GTP form) in TPC1, BCPAP and FTC-133 cells. Bar graphs, normalized ratios of active RhoA relative to total RhoA levels. Data represent means ± SEM of 3 independent experiments. (B) Wound-healing assay for TPC1 cells with treatment of 30 μU/mL TSH and 30μM Y27632 or control. Bar graph Data represents relative wound healing rate of TPC1 cells with treatment of 30 μU/mL TSH and 30μM Y27632 normalized with control. Immunoblots of phosphor-coffin and GAPDH in TPC1 cells incubated with 30 μU/mL TSH and 30μM Y27632 for 72 hours. and Data represent means ± SEM of 3 independent experiments. (C) Immunoblots of RhoA, Gα12 and GAPDH from total cell lysates and RhoA after GST-Rhotekin pull-down (RhoA-GTP form) in TPC1, BCPAP and FTC-133 transiently transfected with non-targeting siRNA (siNT) or Gα12 and Gα13 siRNA (si Gα12/13) with treatment of 30μU/mL TSH. GAPDH is used as a loading control for normalization. The bar graph represents normalized ratios of active RhoA relative to total RhoA levels. (D) Immunoblots of RhoA, Gα12 and GAPDH from total cell lysates and RhoA after GST-Rhotekin pull-down in TPC1 stably transfected with Gα12WT or Gα12Q229L. Bar graph, normalized ratios of active RhoA relative to total RhoA levels. *P < 0.05, **P < 0.01, ***P < 0.001.
As a GPCR, TSHR can couple the members of four G protein families to activate downstream signaling. Among these G protein family members, Gα12/13 has been reported to be associated with the migration phenotype (21). To investigate whether Gα12/13 is involved in the mechanism by which TSHR signaling regulates RhoA activation and enhances cell migration, we measured the relative RhoA-GTP levels in TPC1, BCPAP and FTC-133 cells transiently transfected with Gα12 and/or Gα13 siRNA or siNT treated with TSH or control. Gα12 and Gα13 knockdown was confirmed by Western blotting (Supplementary Figure S3). Again, we observed that cells treated with TSH showed high levels of RhoA-GTP. After Gα12/13 knockdown, this increase in RhoA-GTP levels was abrogated (Figure 2C). To further confirm that Gα12/13 is involved in RhoA activation, we transiently and stably transfected TPC1 cells with plasmid encoding active Gα12Q229L or Gα13Q226L mutant and then compared the RhoA-GTP levels (22). The RhoA-GTP level was increased 2.3-fold (P=0.005) in TPC1 cells stably transfected with Gα12Q229L plasmid compared to that in the control cells transfected with Gα12WT plasmid (Figure 2D). Similar results were observed in cells transfected with Gα13Q226L plasmid. Taken together, these data show that TSH-TSHR enhances RhoA activation and downstream cellular migration through Gα12/13.
RhoA activation is controlled by Rho guanine exchange factors (RhoGEFs) (23), and some GPCRs coupled to G proteins were found to interact with RhoGEFs, activating their downstream signaling. Our previous study revealed that the RhoGEF leukemia associated RhoA guanine exchange factor (LARG) is involved in thyroid tumorigenesis (20). To determine whether Gα12/13 can interact with LARG in thyroid cancer cells, we performed immunoprecipitation experiments. LARG was detected in Gα12 or Gα13 immunoprecipitates from whole-cell lysates of the thyroid cancer cell lines (Figure 3A). Then, to confirm that the Gα12/13-mediated increase in RhoA-GTP was dependent on LARG, we performed a RhoA activation assay in TPC1 cells stably transfected with Gα12Q229L or Gα12WT plasmid and then transiently transfected with siLARG or siNT. Again, Gα12Q229L-transfected cells showed a higher level of RhoA-GTP than control Gα12WT-transfected cells. After LARG knockdown, the RhoA-GTP level in all the cells decreased, and this decrease was more substantial in cells expressing Gα12Q229L. The RhoA-GTP level decreased by 0.67- and 0.46-fold in cells expressing Gα12WT and Gα12Q229L, respectively (P=0.004, Figure 3B). Taken together, our data indicate that TSH-TSHR couples with Gα12/13 and then interacts with LARG to activate RhoA, enhancing the migration of thyroid cancer cells.
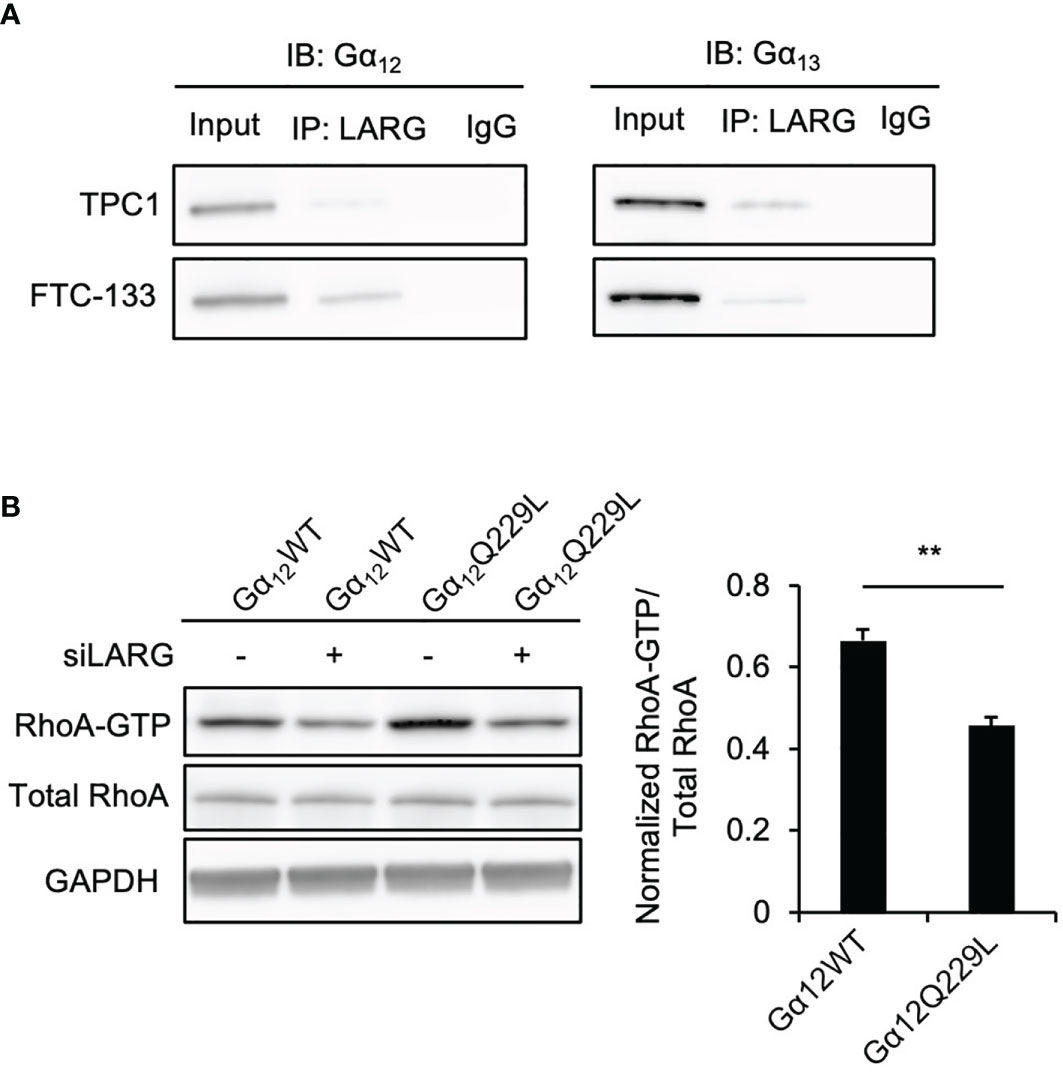
Figure 3 Gα12/13 interacts with LARG to activate RhoA in thyroid cancer cells. (A) Co-immunoprecipitation (IP) of Gα12/13 -LARG using lysates prepared from thyroid cancer cells. Input, 20 µg of total proteins from each cell line. The exposure time was 2 minutes. Left panel, Gα12 immunoblots from lysates immunoprecipitated with anti-LARG or control IgG in TPC1 and FTC-133 cells. Right panel, Gα13 immunoblots from lysates immunoprecipitated with anti-LARG or control IgG in TPC1 and FTC-133 cells. Data are representative of 3 sets of independent experiments. (B) Immunoblots of RhoA, and GAPDH from total cell lysates and RhoA after GST-Rhotekin pull-down (RhoA-GTP form) in TPC1 stably transfected with Gα12WT or Gα12Q229L then transiently transfected with siNT or siLARG. The bar graph represents relative active RhoA levels in TPC1 cells stably transfected with Gα12WT or Gα12Q229L and then transiently transfected with siLARG normalized to control. Data represent means ± SEM of 3 independent experiments. **P < 0.01.
TSH-TSHR signaling is known to increase iodide uptake in thyroid cells through the activation of Gαs-cAMP and downstream signaling; however, the role of Gα12/13 in iodide uptake remains elusive. Interestingly, we observed that transfection of the rat thyroid cell line PCCL3 with endogenous NIS expression with active Gα12Q229L significantly reduced iodide uptake (Figure 4A). Compared to control cells transfected with Gα12WT plasmid, cells transfected with Gα12Q229L plasmid showed decreased iodide uptake by approximately 49% (P=0.004). By qRT-PCR, Western blotting and Tg radioimmunoassay (RIA), we observed that both the mRNA and protein expression levels of the thyroid-specific genes TTF-1, PAX-8, Tg, TPO and NIS (Figures 4B, C) were significantly decreased in cells transfected with Gα12Q229L plasmid compared to cells transfected with Gα12WT plasmid. The protein levels were decreased 0.65-, 0.70-, 0.62-, 0.31- and 0.53-fold, respectively (all P<0.05). Similar results were observed in cells transfected with Gα13Q226L plasmid compared to control cells. Furthermore, TSH increased iodide uptake more significantly in cells in which Gα12/13 was knocked down compared to control cells; iodide uptake increased by 3.4- and 1.5-fold in cells transfected with siGα12/13 and siNT, respectively (P<0.001) (Figure 4D). These data indicate that the activation of Gα12/13 signaling might inhibit the expression of thyroid-specific molecules and reduce iodide uptake of thyroid cells. Then, we investigated cAMP levels in cells transfected with Gα12Q229L plasmid. Compared to control cells transfected with Gα12WT plasmid, cells transfected with Gα12Q229L plasmid did not show significantly changed cAMP levels, suggesting that Gα12/13 activation after thyroid-specific molecular expression and iodide uptake is independent of cAMP levels (Figure 4E).
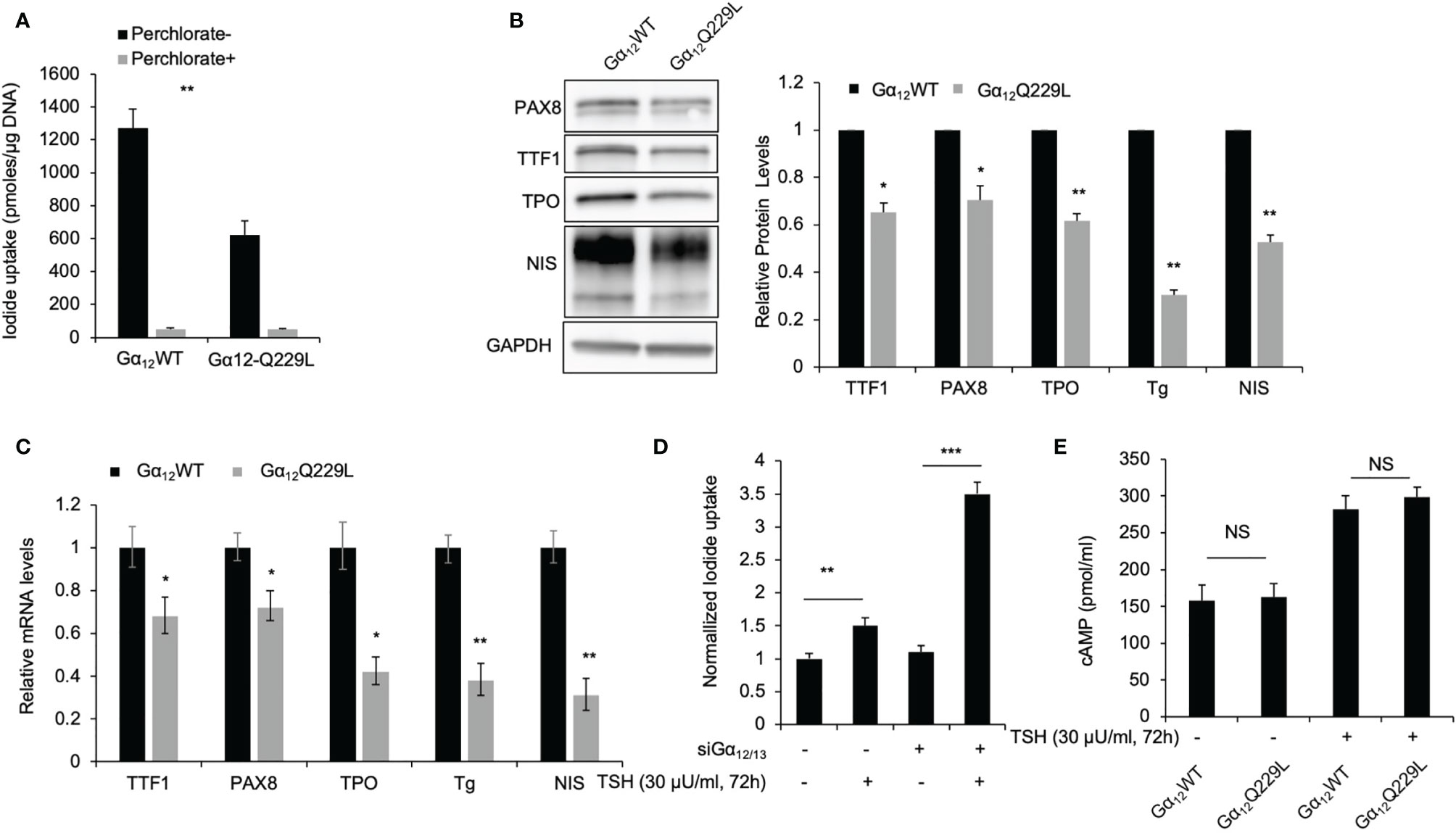
Figure 4 Constitutively active Gα12/13 is associated with thyroid specific maker loss and decreased iodide uptake. (A) Radioiodide uptake of PCCL3 cells stably transfected with Gα12Q229L or Gα12WT. Data represent means ± SEM of 3 independent experiments. (B) Immunoblots of TTF1, PAX8, TPO and NIS in PCCL3 cells stably transfected with Gα12Q229L or Gα12WT. GAPDH is used as a loading control for normalization. Bar charts, Relative TTF1, PAX8, TPO, Tg and NIS protein levels in PCCL3 cells stably transfected with Gα12Q229L or Gα12WT. Tg levels were detect by RIA. Data represent means ± SEM of 3 independent experiments. (C) Bar charts, Relative mRNA levels of TTF1, PAX8, TPO, Tg and NIS in PCCL3 cells stably transfected with Gα12Q229L or Gα12WT. Data represent means ± SEM of 3 independent experiments. (D) Normalized radioiodide uptake of PCCL3 cells transiently transfected with siGα12/13 or siNT then treated with 30μU/ml TSH or control. Data represent means ± SEM of 3 independent experiments. (E) Bar chart, cAMP levels in PCCL3 cells stably transfected with Gα12Q229L or Gα12WT incubated with 30 μU/ml TSH. Data are representative of 3 sets of independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001, NS, not significant.
Our previous studies revealed that thyroid cancer cells cultured in vitro underwent dedifferentiation exhibited decreased iodide uptake and expression of thyroid-specific molecules (11, 12). Since Gα12/13 and Gαs signaling had the opposite effect in regulating iodide uptake, we then investigated the levels of RhoA activation associated with Gα12/13 and cAMP in our cell models of dedifferentiation compared to control cells. Again, our data confirmed that in TPC1 and FTC-133 cells with stable NIS expression, NIS and TSHR protein levels were lower in cells at passage 10 than in those at passage 5 (Figure 5A). Consistent with the change in TSHR level, in TPC1-FL hNIS and FTC-133-FL hNIS cells, the cAMP level was decreased by 0.78- and 0.65-fold, respectively (P=0.016 and 0.018, respectively; Figure 5B), in dedifferentiated cells compared to control cells. However, relative RhoA-GTP levels were higher in dedifferentiated cells than in control cells, with an increase of 1.48- and 1.62-fold, respectively (P=0.021 and 0.019, respectively; Figure 5C).
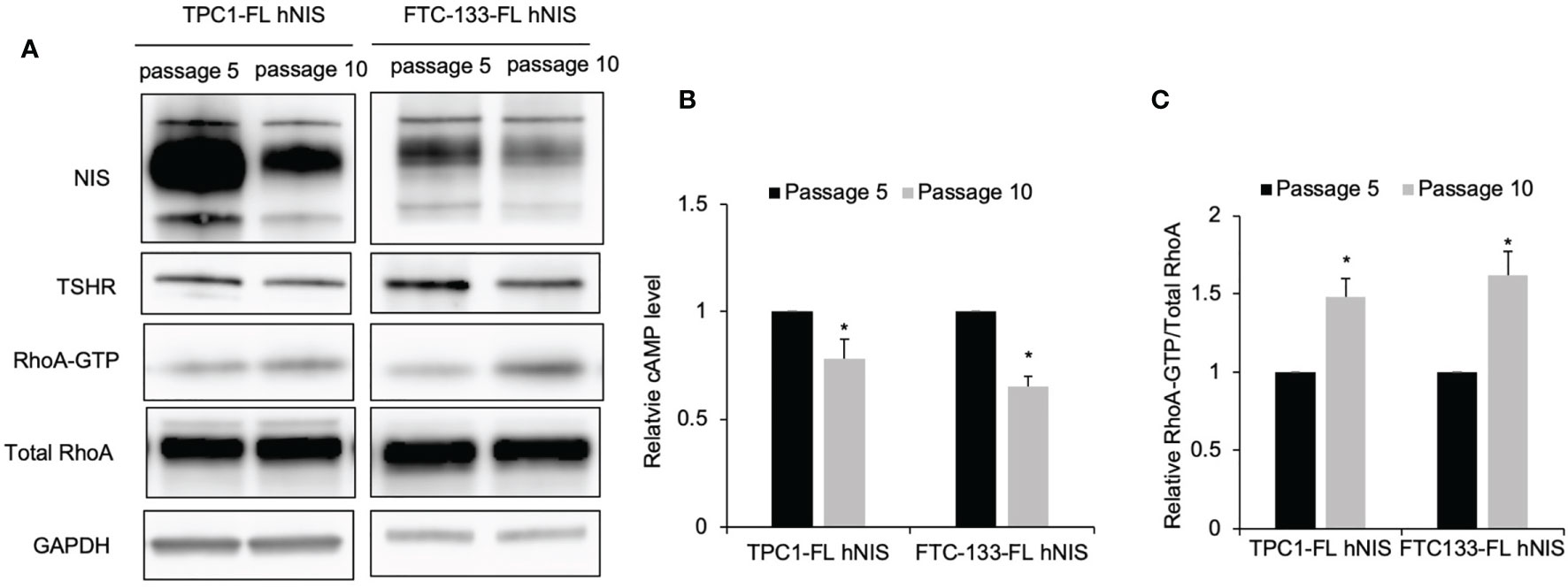
Figure 5 Gαs signaling is inhibited while Gα12/13 activation in thyroid cancer cells undergoing dedifferentiation. (A) Immunoblot of NIS, TSHR, RhoA and GAPDH from total cell lysates and RhoA after GST-Rhotekin pull-down (RhoA-GTP form) in FTC-133-FL hNIS and TPC1-FL hNIS cells at passage 5 or passage 10. GAPDH is used as a loading control for normalization. Data are representative of 3 sets of independent experiments. (B) Bar chart, cAMP levels in FTC-133-FL hNIS and TPC1-FL hNIS cells at passage 5 or passage 10. Data are representative of 3 sets of independent experiments. (C) Bar graphs, normalized ratios of active RhoA relative to total RhoA levels. Data represent means ± SEM of 3 independent experiments. *P < 0.05.
Our previous studies also revealed that PTEN/PI3K signaling affect the iodide uptake function of thyroid cancer cells. To test our hypothesis that PTEN/PI3K signaling is involved in crosstalk with the TSHR pathway in thyroid cancer cells, we first knocked down PTEN, the key regulator of PI3K signaling, by siRNA-mediated transfection in TPC1 and BCPAP cells. The loss of PTEN resulted in increased LARG levels in these thyroid cancer cell lines. The relative expression of LARG in TPC1 and BCPAP cells after PTEN knockdown was increased approximately 1.2- and 1.5-fold compared to that in control cells (P=0.015 and 0.009, respectively; Figure 6A and Supplementary Figure S4). In addition, the LARG level in FTC-133 cells (PTEN null) transiently transfected with PTEN plasmid was decreased 0.82-fold compared to that in cells transfected with the vector control (P=0.013; Figure 6A). Consistently, in TPC1 cells, treatment with the PI3K inhibitor LY294002 decreased LARG protein levels by 20% (P=0.025; Figure 6B). However, following treatment with the AKT inhibitor MK-2206 and the mTOR inhibitor rapamycin, LARG levels were not significantly difference compared to those in control cells, suggesting that LARG was regulated by the PI3K level. Moreover, by immunofluorescence, in TPC1 cells in which PTEN had been knocked down, LARG was predominately localized in the submembrane region (Figure 6C), where it can interact with Gα12/13. Consistently, in FTC-133 cells, PTEN transfection resulted in increased intracellular LARG localization (Figure 6D). Together, these data indicate that in thyroid cancer cells, PTEN/PI3K signaling activation facilitates Gα12/13-LARG-RhoA activation by increasing LARG levels and promoting the localization of LARG in the submembrane region.

Figure 6 PTEN/PI3K is associated with LARG levels and submembrane localization in thyroid cancer cells. (A) Immunoblot of LARG using total lysate from TPC1 cells transiently transfected with siNT or siPTEN, and FTC-133 cells transiently transfected with empty vector or PTEN plasmid for 72 hours. GAPDH was used as a loading control. Bar chart shows the relative LARG protein levels in TPC1 after siPTEN knockdown compared to control, and in FTC-133 cells after PTEN transfection compared to control. Data represent means ± SEM of 3 independent experiments. (B) Immunoblot of LARG in TPC1 cells treated with LY294002, MK-2206, rapamycin or DMSO. Bar chart, LARG protein levels normalized to GAPDH. Data represent means ± SEM of 3 independent experiments. (C) Confocal microscopy of LARG immunofluorescence staining in TPC1 cells transiently transfected with siNT or siPTEN for 72 hours. Scale bar, 25 µm. Data are representative of 3 sets of independent experiments. (D) Confocal microscopy of LARG immunofluorescence staining in FTC-133 cells transiently transfected with PTEN-GFP or empty vector for 48 hours. Scale bar, 10 µm. Data are representative of 3 sets of independent experiments. (E) Immunoblot of phospho-Akt, Akt, phospho-mTOR, mTOR, phospho-p70 S6K, p70 S6K, phosphor-ribosomal S6, ribosomal S6 and GAPDH in TPC1 cells stably transfected with Gα12Q229L or Gα12WT. Data are representative of 3 sets of independent experiments. (F) Bar chart, cAMP levels in TPC1 cells transiently transfected with siNT or siPTEN, with or without TSH for 72 hours. Data represent means ± SEM of 3 independent experiments. (G) Bar chart, cAMP levels in TPC1 cells treated with PI3K inhibitor LY294002 or DMSO, with or without TSH for 72 hours. Data represent means ± SEM of 3 independent experiments. *P < 0.05, NS, not significant.
Interestingly, in TPC1 cells, we observed that after active Gα12Q229L plasmid transfection, the expression levels of molecules downstream of PI3K (phospho-AKT, phospho-mTOR, phospho-P70 S6K and phosphor-ribosomal S6) were increased compared to those in control cells transfected with Gα12WT plasmid, suggesting that Gα12/13 activation is associated with the activation of molecules downstream of PI3K (Figure 6E).
Following the observation of crosstalk between PI3K and molecules downstream of TSHR-Gα12/13 signaling, we then investigated the effect of PI3K signaling on TSHR-Gαs-cAMP signaling. We did not observe a significant difference in measured intracellular cAMP levels between PTEN-knockdown TPC1 cells and control cells after siNT transfection treated with or without TSH (Figure 6F). Consistently, there was no significant difference in intracellular cAMP levels between cells treated with the PI3K inhibitor LY294002 and control cells (Figure 6G). Since our and others’ previous studies have shown that PI3K/AKT/mTOR signaling activation negatively regulates iodide uptake through regulating NIS expression and localization (20), we speculate that PI3K/AKT/mTOR signaling inhibits molecules downstream of Gαs signaling.
Our observations reveal that TSH-TSHR increases thyroid cancer cell mobility through activating the Gα12/13 signaling pathway. The best known functions of TSH-TSHR signaling are its regulation of the proliferation and differentiation of thyroid follicular cells and iodide uptake and TH secretion by activation of the Gαs-cAMP-PKA pathway (24). In fact, as a GPCR, TSHR can couple to the members of all four G protein families (8). For example, TSHR couples to Gαq and then activates the phospholipase C (PLC) pathway to stimulate iodination by stimulating H2O2 generation. However, the role of Gα12/13 in TSHR signaling has rarely been reported. Our data show that a high level of TSH increased the migration rate of thyroid cancer cells, which was abrogated after Gα12/13 knockdown. Furthermore, overexpression of the active mutant Gα12Q229L or Gα13Q226L also resulted in altered migration and dedifferentiation phenotypes. These data provide sufficient evidence that Gα12/13 is involved in the role of TSH-TSHR signaling in increasing the motility of thyroid cancer cells. Our data also suggest that TSHR signaling plays a protumorigenic role. TSHR expression has been reported to be associated with unfavorable prognosis in other cancers, such as liver cancer (25), but whether this is also the case in thyroid cancer remains unknown.
Our data demonstrated that TSH-TSHR-Gα12/13 signaling increases the migration of thyroid cancer cells through activating RhoA. RhoA activation, similar to the activation of other Rho family GTPases, plays an important role in enhancing cancer cell motility. Evidence supports the notion that in a number of cell lines, Gα12/13 can mediate signals through coupling GPCRs to Rho GTPase activation (26, 27). RhoA activation is controlled by RhoGEFs (23, 28). Our previous study revealed that a novel RhoGEF, LARG, is involved in tumorigenesis in thyroid cancer (24). Here, we observed that LARG interacts with Gα12/13 and activates RhoA in thyroid cancer cells and that this process is dependent on stimulation by high levels of TSH. This suggests that LARG mediates signal transduction from TSH-TSHR-Gα12/13 to RhoA activation, in turn increasing the migration of thyroid cancer cells.
Interestingly, while we observed that overexpression of the active mutant Gα12Q229L activated RhoA signaling to increase cell migration, it also resulted in decreased expression of the thyroid-specific molecules TTF1, PAX8, TPO, Tg and NIS and consistent iodide uptake in PCCL3 cells, independent of cAMP levels. Our data suggest that the activation of noncanonical TSHR-Gα12/13 signaling inhibits the expression of thyroid differentiation markers, thus promoting the dedifferentiation of thyroid cancer cells, although the mechanism warrants further study. One possible mechanism is the inhibited expression of thyroid-specific markers due to the downstream activation of RhoA. In fact, Medinaet et al. found that FRTL-5 cells expressing a constitutively active form of RhoA (RhoA QL) showed decreased levels of Tg and TTF-1 compared to those in cells expressing WT RhoA (29).
An investigation of how noncanonical Gα12/13 signaling is preferentially activated in thyroid cancer cells would be intriguing. Gain- or loss-of-function mutations in TSHR have been reported to be associated with the abnormal activation of downstream signaling (30, 31). For example, in transfected COS-7 cells, L653A TSHR expression was shown to reduce the downstream production of different G proteins to a different extent, as stimulated cAMP was decreased by 62%, while stimulated IP production was decreased 12% compared to that in cells expressing WT TSHR (32). This suggests that some mutations affect specific G protein signal transduction pathways. However, few TSHR mutations in thyroid cancer tissue have been reported. We speculate that crosstalk between pathways downstream of TSHR and other tumorigenesis signaling pathways plays an important role in aberrant TSHR signal transduction.
PI3K/AKT/mTOR signaling activation, which is negatively regulated by the tumor suppressor gene PTEN, enhances cell proliferation and migration in multiple cancer cells, including thyroid cancer cells (16). Importantly, PI3K/AKT/mTOR signaling is also associated with decreased iodide uptake in thyroid cancer cells (17). We investigated the relationship between PI3K/AKT/mTOR signaling and TSHR signaling and found that PI3K/AKT/mTOR signaling activation does not affect pathways upstream of Gαs and Gα12/13. However, PI3K/AKT/mTOR signaling exhibited crosstalk with pathways downstream of Gα12/13 by increasing LARG levels. Interestingly, the loss of PTEN was also found to promote LARG localization in the submembrane region, particularly under the migration leading edge, where it can interact with and activate RhoA to mediate cell migration. We also observed that active mutant Gα12Q229 overexpression resulted in the activation of molecules downstream of PI3K signaling, further proving that these two signaling pathways form a network to regulate the migration and differentiation of thyroid cancer cells.
We also carried out experiments to explore the mechanism by which Gα12/13 activation collaborates with PI3K signaling to decrease thyroid-specific gene expression and iodide uptake. We observed that PI3K signaling inhibition by the pharmacologic drug LY294002, PTEN knockdown by siRNA and constitutively active Gα12/13 overexpression did not change cAMP levels, which normally accumulate due to Gαs and activate PKA to regulate thyroid-specific gene transcription. However, some groups have reported that treatment with PI3K inhibitors, as well as inhibitors of molecules downstream of PI3K, such AKT and mTOR, increases thyroid-specific gene expression and iodide uptake in thyroid cells (19, 33). Based on our observation that constitutively active Gα12/13 decreased thyroid-specific protein expression and activated PI3K signaling, we speculate that Gα12/13 collaborates with PI3K/AKT/mTOR signaling to regulate pathways downstream of the cAMP pathway. However, the exact target of this regulation warrants further study.
It has been reported that BCPAP cells harbor BRAFV600E mutation (34). However, the effect of TSH on migration phenotype is similar as TPC1 cells which have wild type BRAF. Interestingly, we observed the effect of TSH is more significant in FTC-133 cells which PTEN is null. Thus, whether BRAF-MAPK signaling is associated with Gα12/13 signaling warrant further study. Some research has reported BRAF mutation associated with MAPK activation affect GαS signaling to inhibit iodine uptake of thyroid cancer cells (35). We speculate that PI3K signaling play more import roles in the transform from well differentiated in to poor- or un-differentiated thyroid cancer especially the migration phenotype.
In summary, we found that in thyroid cancer cells, TSHR couples to Gα12/13, interacts with LARG and then activates RhoA to increase migration. In addition, constitutively active Gα12/13 expression decreased thyroid-specific gene expression and iodide uptake in thyroid cells. PI3K signaling activation increased LARG levels in cancer cells and altered its localization to facilitate the interaction of LARG with Gα12/13 and enhance TSHR-Gα12/13 signaling. Gα12/13 signaling participates with PI3K to inhibit the pathways downstream Gαs signaling (Figure 7). Together, our data suggest that in thyroid cancer, noncanonical activation of TSH-TSHR signaling through Gα12/13 plays an important role in cell motility and the dedifferentiation of thyroid cancer. These findings have broader implications as they are relevant to the function of TSHR in thyroid cancer and understanding of the dedifferentiation of thyroid cancer in parallel with invasion.
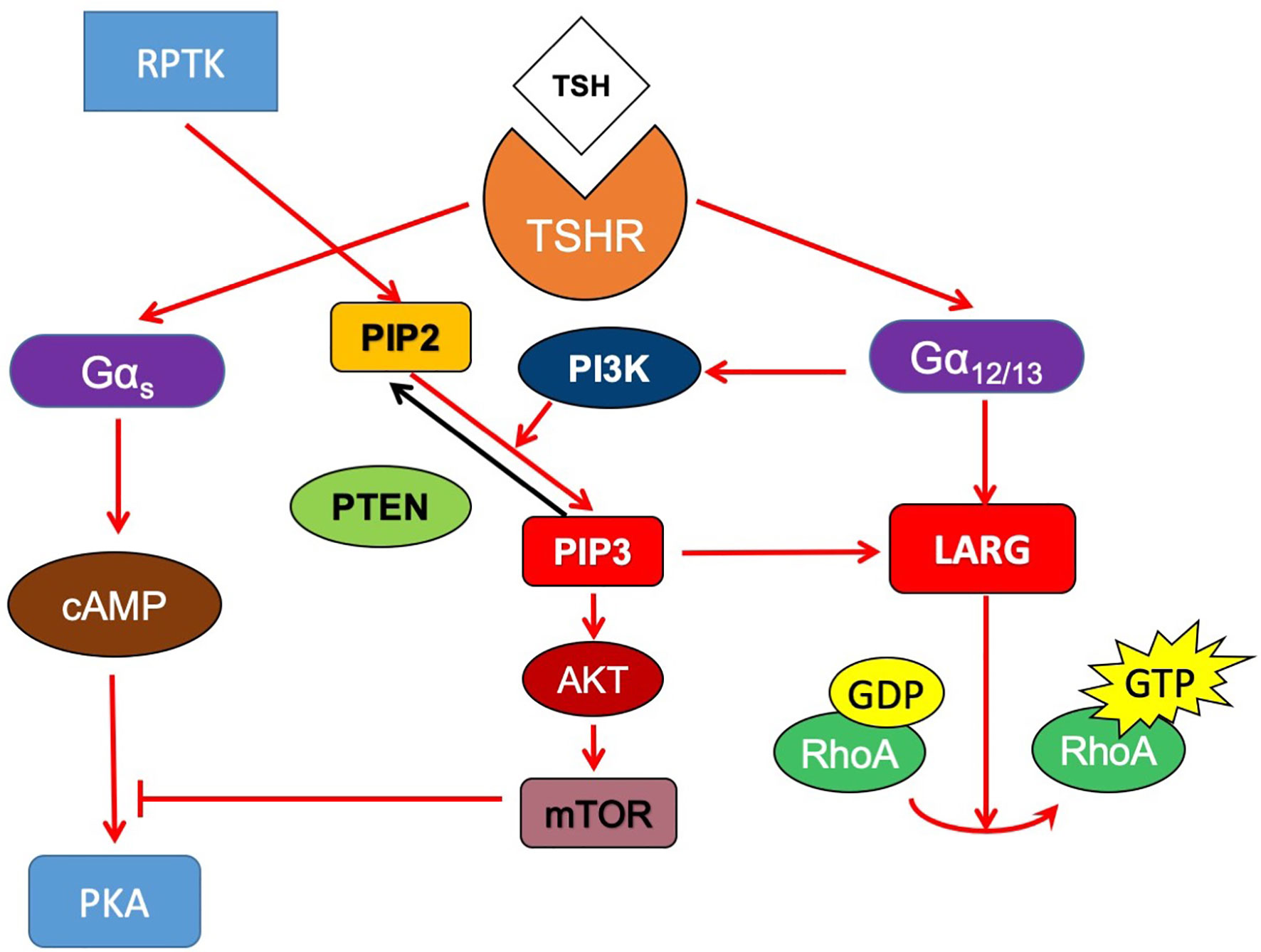
Figure 7 Proposed signaling model of crosstalk between TSHR and PI3K signaling pathway. In thyroid cancer cells, TSHR couples to Gα12/13, interacts with LARG and then activates RhoA to increase migration. PI3K signaling activation increased LARG levels in cancer cells and altered its localization to facilitate the interaction of LARG with Gα12/13 and enhance TSHR-Gα12/13 signaling. Gα12/13 signaling also might participate with PI3K to inhibit the pathways downstream Gαs signaling.
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Conception and design: FF. Development of methodology: HH and FF. Acquisition of data: HH. Analysis and interpretation of data: HH and SW. Writing, review and/or revision of the manuscript: FF, HH, SW, and HW. Administrative, technical, or material support: FF and HW. Study supervision: FF and HW. All authors contributed to the article and approved the submitted version.
This study was funded by the National Natural Science Found of China (grant 81974269) and Biomedical-engineering Cross Fund of Shanghai Jiao Tong University (grant YG2019QNA39).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank Dr. Hongliang Fu for helpful discussions, Xiaoran Mei and Miaomiao Xu for assistance in cell culture and Western blot analysis.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2021.718578/full#supplementary-material
1. Sampson E, Brierley JD, Le LW, Rotstein L, Tsang RW. Clinical Management and Outcome of Papillary and Follicular (Differentiated) Thyroid Cancer Presenting With Distant Metastasis at Diagnosis. Cancer (2007) 110:1451–6. doi: 10.1002/cncr.22956
2. Hong CM, Lee WK, Jeong SY, Lee SW, Ahn BC, Lee J. Superiority of Delayed Risk Stratification in Differentiated Thyroid Cancer After Total Thyroidectomy and Radioactive Iodine Ablation. Nucl Med Commun (2014) 35:1119–26. doi: 10.1097/MNM.0000000000000183
3. Kim DH, Jung JH, Son SH, Kim CY, Hong CM, Jeong SY, et al. Difference of Clinical and Radiological Characteristics According to Radioiodine Avidity in Pulmonary Metastases of Differentiated Thyroid Cancer. Nucl Med Mol Imaging (2014) 48:55–62. doi: 10.1007/s13139-013-0239-z
4. Ahn BC. Personalized Medicine Based on Theranostic Radioiodine Molecular Imaging for Differentiated Thyroid Cancer. BioMed Res Int (2016) 2016:1680464. doi: 10.1155/2016/1680464
5. Durante C, Haddy N, Baudin E, Leboulleux S, Hartl D, Travagli JP, et al. Long-Term Outcome of 444 Patients With Distant Metastases From Papillary and Follicular Thyroid Carcinoma: Benefits and Limits of Radioiodine Therapy. J Clin Endocrinol Metab (2006) 91:2892–9. doi: 10.1210/jc.2005-2838
6. Hong CM, Ahn BC, Jeong SY, Lee SW, Lee J. Distant Metastatic Lesions in Patients With Differentiated Thyroid Carcinoma. Clinical Implications of Radioiodine and FDG Uptake. Nuklearmedizin. Nucl Med (2013) 52:121–9. doi: 10.3413/Nukmed-0541-12-11
7. Chung JK, Youn HW, Kang JH, Lee HY, Kang KW. Sodium Iodide Symporter and the Radioiodine Treatment of Thyroid Carcinoma. Nucl Med Mol Imaging (2010) 44:4–14. doi: 10.1007/s13139-009-0016-1
8. Laugwitz KL, Allgeier A, Offermanns S, Spicher K, Van Sande J, Dumont JE, et al. The Human Thyrotropin Receptor: A Heptahelical Receptor Capable of Stimulating Members of All Four G Protein Families. Proc Natl Acad Sci U S A (1996) 93:116–20. doi: 10.1073/pnas.93.1.116
9. Laurent E, Mockel J, Van Sande J, Graff I, Dumont JE. Dual Activation by Thyrotropin of the Phospholipase C and Cyclic AMP Cascades in Human Thyroid. Mol Cell Endocrinol (1987) 52:273–8. doi: 10.1016/0303-7207(87)90055-4
10. Raspe E, Laurent E, Andry G, Dumont JE. ATP, Bradykinin, TRH and TSH Activate the Ca(2+)-Phosphatidylinositol Cascade of Human Thyrocytes in Primary Culture. Mol Cell Endocrinol (1991) 81:175–83. doi: 10.1016/0303-7207(91)90216-F
11. Feng F, Wang H, Fu H, Wu S, Ye Z, Chen S, et al. Dedifferentiation of Differentiated Thyroid Carcinoma Cell Line FTC-133 Is Enhanced by 131I Pretreatment. Nucl Med Biol (2011) 38:1053–8. doi: 10.1016/j.nucmedbio.2011.03.001
12. Feng F, Wang H, Hou S, Fu H. Re-Induction of Cell Differentiation and (131)I Uptake in Dedifferentiated FTC-133 Cell Line by TSHR Gene Transfection. Nucl Med Biol (2012) 39:1261–5. doi: 10.1016/j.nucmedbio.2012.07.004
13. Cheng W, Feng F, Ma C, Wang H. The Effect of Antagonizing RGD-Binding Integrin Activity in Papillary Thyroid Cancer Cell Lines. Onco Targets Ther (2016) 9:1415–23. doi: 10.2147/OTT.S99166
14. Bevan S, Pal T, Greenberg CR, Green H, Wixey J, Bignell G, et al. A Comprehensive Analysis of MNG1, TCO1, fPTC, PTEN, TSHR, and TRKA in Familial Nonmedullary Thyroid Cancer: Confirmation of Linkage to TCO1. J Clin Endocrinol Metab (2001) 86:3701–4. doi: 10.1210/jcem.86.8.7725
15. Yang YM, Lee S, Nam CW, Ha JH, Jayaraman M, Dhanasekaran DN, et al. G(alpha)12/13 Inhibition Enhances the Anticancer Effect of Bortezomib Through PSMB5 Downregulation. Carcinogenesis (2010) 31:1230–7. doi: 10.1093/carcin/bgq097
16. Song MS, Salmena L, Pandolfi PP. The Functions and Regulation of the PTEN Tumour Suppressor. Nat Rev Mol Cell Biol (2012) 13:283–96. doi: 10.1038/nrm3330
17. Jin S, Yang YT, Bao W. Signaling Pathways in Thyroid Cancer. Vitam Horm (2018) 106:501–15. doi: 10.1016/bs.vh.2017.05.002
18. Vlaeminck-Guillem V, Ho SC, Rodien P, Vassart G, Costagliola S. Activation of the cAMP Pathway by the TSH Receptor Involves Switching of the Ectodomain From a Tethered Inverse Agonist to an Agonist. Mol Endocrinol (2002) 16:736–46. doi: 10.1210/mend.16.4.0816
19. Liu YY, Zhang X, Ringel MD, Jhiang SM. Modulation of Sodium Iodide Symporter Expression and Function by LY294002, Akti-1/2 and Rapamycin in Thyroid Cells. Endocr Relat Cancer (2012) 19:291–304. doi: 10.1530/ERC-11-0288
20. Feng F, Yehia L, Ni Y, Chang YS, Jhiang SM, Eng C. A Nonpump Function of Sodium Iodide Symporter in Thyroid Cancer via Cross-Talk With PTEN Signaling. Cancer Res (2018) 78:6121–33. doi: 10.1158/0008-5472.CAN-18-1954
21. Hart MJ, Jiang X, Kozasa T, Roscoe W, Singer WD, Gilman AG, et al. Direct Stimulation of the Guanine Nucleotide Exchange Activity of P115 RhoGEF by Galpha13. Science (1998) 280:2112–4. doi: 10.1126/science.280.5372.2112
22. Suzuki N, Nakamura S, Mano H, Kozasa T. Galpha 12 Activates Rho GTPase Through Tyrosine-Phosphorylated Leukemia-Associated RhoGEF. Proc Natl Acad Sci USA (2003) 100:733–8. doi: 10.1073/pnas.0234057100
23. Cook DR, Rossman KL, Der CJ. Rho Guanine Nucleotide Exchange Factors: Regulators of Rho GTPase Activity in Development and Disease. Oncogene (2014) 33:4021–35. doi: 10.1038/onc.2013.362
24. Dumont JE, Lamy F, Roger P, Maenhaut C. Physiological and Pathological Regulation of Thyroid Cell Proliferation and Differentiation by Thyrotropin and Other Factors. Physiol Rev (1992) 72:667–97. doi: 10.1152/physrev.1992.72.3.667
25. Shih YL, Huang YH, Lin KH, Chu YD, Yeh CT. Identification of Functional Thyroid Stimulating Hormone Receptor and TSHR Gene Mutations in Hepatocellular Carcinoma. Anticancer Res (2018) 38:2793–802. doi: 10.21873/anticanres.12523
26. Gohla A, Harhammer R, Schultz G. The G-Protein G13 But Not G12 Mediates Signaling From Lysophosphatidic Acid Receptor via Epidermal Growth Factor Receptor to Rho. J Biol Chem (1998) 273:4653–9. doi: 10.1074/jbc.273.8.4653
27. Kranenburg O, Poland M, van Horck FP, Drechsel D, Hall A, Moolenaar WH. Activation of RhoA by Lysophosphatidic Acid and Galpha12/13 Subunits in Neuronal Cells: Induction of Neurite Retraction. Mol Biol Cell (1999) 10:1851–7. doi: 10.1091/mbc.10.6.1851
28. Amin E, Dubey BN, Zhang SC, Gremer L, Dvorsky R, Moll JM, et al. Rho-Kinase: Regulation, (Dys)Function, and Inhibition. Biol Chem (2013) 394:1399–410. doi: 10.1515/hsz-2013-0181
29. Medina DL, Rivas M, Cruz P, Barroso I, Regadera J, Santisteban P. RhoA Activation Promotes Transformation and Loss of Thyroid Cell Differentiation Interfering With Thyroid Transcription Factor-1 Activity. Mol Endocrinol (2002) 16:33–44. doi: 10.1210/mend.16.1.0749
30. Jaeschke H, Undeutsch H, Patyra K, Lof C, Eszlinger M, Khalil M, et al. Hyperthyroidism and Papillary Thyroid Carcinoma in Thyrotropin Receptor D633H Mutant Mice. Thyroid (2018) 28:1372–86. doi: 10.1089/thy.2018.0041
31. Roberts SA, Moon JE, Dauber A, Smith JR. Novel Germline Mutation (Leu512Met) in the Thyrotropin Receptor Gene (TSHR) Leading to Sporadic Non-Autoimmune Hyperthyroidism. J Pediatr Endocrinol Metab (2017) 30:343–7. doi: 10.1515/jpem-2016-0185
32. Sugisawa C, Abe K, Sunaga Y, Taniyama M, Hasegawa T, Narumi S. Identification of Compound Heterozygous TSHR Mutations (R109Q and R450H) in a Patient With Nonclassic TSH Resistance and Functional Characterization of the Mutant Receptors. Clin Pediatr Endocrinol (2018) 27:123–30. doi: 10.1297/cpe.27.123
33. Kogai T, Sajid-Crockett S, Newmarch LS, Liu YY, Brent GA. Phosphoinositide-3-Kinase Inhibition Induces Sodium/Iodide Symporter Expression in Rat Thyroid Cells and Human Papillary Thyroid Cancer Cells. J Endocrinol (2008) 199:243–52. doi: 10.1677/JOE-08-0333
34. Manuel Saiselet SF, Tarabichi M, Dom G, Hébrant A, van Staveren WCG, Maenhaut C. Thyroid Cancer Cell Lines: An Overview. Front Endocrinol (Lausanne) (2012) 3:133. doi: 10.3389/fendo.2012.00133
Keywords: differentiated thyroid carcinoma, thyrotropin receptor, radioiodine, dedifferentiation, migration
Citation: Feng F, Han H, Wu S and Wang H (2021) Crosstalk Between Abnormal TSHR Signaling Activation and PTEN/PI3K in the Dedifferentiation of Thyroid Cancer Cells. Front. Oncol. 11:718578. doi: 10.3389/fonc.2021.718578
Received: 01 June 2021; Accepted: 08 September 2021;
Published: 28 September 2021.
Edited by:
Laura Sterian Ward, State University of Campinas, BrazilReviewed by:
Janete Maria Cerutti, Federal University of São Paulo, BrazilCopyright © 2021 Feng, Han, Wu and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fang Feng, ZmVuZ2ZhbmdAeGluaHVhbWVkLmNvbS5jbg==; Hui Wang, d2FuZ2h1aUB4aW5odWFtZWQuY29tLmNu
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.