 Guangjie Zhao
Guangjie Zhao Qian Wang
Qian Wang- Department of Hematology, Huashan Hospital, Fudan University, Shanghai, China
The nucleoside analogs decitabine (5-AZA-dC) and azacitidine (5-AZA) have been developed as targeted therapies to reverse DNA methylation in different cancer types, and they significantly improve the survival of patients who are not suitable for traditional intensive chemotherapies or other treatment regimens. However, approximately 50% of patients have a response to hypomethylating agents (HMAs), and many patients have no response originally or in the process of treatment. Even though new combination regimens have been tested to overcome the resistance to 5-AZA-dC or 5-AZA, only a small proportion of patients benefited from these strategies, and the outcome was very poor. However, the mechanisms of the resistance remain unknown. Some studies only partially described management after failure and the mechanisms of resistance. Herein, we will review the clinical and molecular signatures of the HMA response, alternative treatment after failure, and the causes of resistance in hematological malignancies.
Introduction
DNA methylation, which adds a methyl group to the cytosine base in the context of a CpG dinucleotide by DNA methyltransferases (DNMTs), is a crucial epigenetic modification that regulates gene expression in normal tissue development, aging, and disease (1, 2). In many cancers, DNA methylation in CpG islands by DNMTs, which is associated with gene repression, is closely related to disease pathogenesis and progression (3). The Ten eleven translocation (TET) family mediates the formation of unmethylated cytosines through passive demethylation during cell division or active demethylation via the base excision repair pathway (4). Given that DNA methylation is a reversible process, expressing silenced genes and reprograming cells to a normal-like state by inhibiting DNMTs may have treatment potentials, leading to the research and development of DNMT inhibitors for cancer treatment. Two DNMT inhibitors, 5-AZA and 5-AZA-dC have been successfully applied in the clinic (5).
Hypomethylating agents (HMAs) are the first-line regimen for treating patients with intermediate and high-risk myelodysplastic syndrome (MDS) and significantly improve the overall survival (OS) (6). Some studies reported that HMA treatment in MDS cannot prolong the OS, but it was associated with improvement in patient-reported quality of life (QOL) and reduced leukemia transformation (7, 8). The application of HMAs to elderly patients with acute myeloid leukemia (AML) who were ineligible for intensive chemotherapy conferred an overall or disease-free survival advantage (9). However, there were clinically significant differences in the achievement of red blood cell transfusion independence or survival between azacitidine- and decitabine-treated older AML patients (10). In other tumors, such as ovarian cancer, melanoma, prostate cancer, and relapsed or refractory diffuse large B-cell lymphoma, HMAs are mainly used in combination with standard chemotherapy to improve the activities of cytotoxic drugs (11–15).
The active tri-phosphorylated metabolite of 5-AZA-dCTP, which is catalyzed and formed by intracellular kinases, is directly incorporated into DNA. For 5-AZA, the majority of 5-AZA-CTP is incorporated into RNA, whereas the remaining part is converted to 5-AZA-dCTP by ribonucleotide reductase (RNR) and is incorporated into DNA during replication (16). Since the cytotoxic action of 5-AZA-dC and 5-AZA is a result of its incorporation into DNA, it is an S-phase-specific agent and produces much greater cell kill of long phase cells than of plateau-phase cells (17, 18). In the process of DNA methylation, DNMTs establish covalent bond with the cytosine ring and are subsequently released after methylation, while the reaction is blocked with azacytosine. The covalent protein adducts impair the function of DNA and trigger DNA damage signaling, resulting in the degradation of “trapped” DNMTs by the proteasomal pathway (19). DNMT1 (preferentially), DNMT3A, and DNMT3B mutation at the catalytic site are still sensitive to 5-AZA-dCTP-mediated degradation. This indicates that covalent bond formation between DNMTs and 5-AZA-dCTP-incorporated DNA may not be necessary for its degradation (20), and the alternative process is not known. 5-AZA-dC treatment also increases the level of E3 ligase, TNF receptor-associated factor 6 (TRAF6), leading to the ubiquitination of DNMTs and lysosome-dependent degradation (21).
Anticancer activities of HMAs are executed by inducing the expression of tumor-repressor genes, stimulating immune responses, and reducing oncogene expression and angiogenesis, resulting in cell differentiation and death as well as inhibition of cell proliferation and the stem cell niche (22). However, the overall response rate (ORR) in intermediate- and high- risk MDS patients was approximately 50% or less (23), and some of them lost response during treatment. HMA treatment had no benefit in patients with chronic lymphocytic leukemia and non-Hodgkin’s lymphoma (24). The causes of the inefficiency and responsiveness remain unknow. In this review, we will shed light on the signatures predicting response to HMAs, combination strategies to overcome resistance, and the mechanism of resistance.
Predicting the Response to HMAs
Clinical Parameters
To evaluate the clinical response of patients with myelodysplasia or AML receiving treatment, the International Working Group (IWG) published and revised standard response criteria, including alteration of the natural history of the disease, cytogenetic response, hematologic improvement (HI), and quality of life (QOL) (25, 26). Complete remission (CR), partial remission (PR), and HI are standard clinical parameters for predicting response. Bone marrow blasts > 15%, abnormal karyotypes, and previous treatment with low-dose cytosine arabinoside have been reported to be independent indicators for lower response rates in MDS patients treated with 5-AZA (27). However, adverse cytogenetics (intermediate and poor), including chromosome 7 deletion, were confirmed to show higher response rates (28, 29). The WHO classification, and IPSS risk category were close to patient survival, but the response rate could not be predicted. In contrast, platelets ≥ 100 × 10^9/L and WBC<3.0 × 10^9/L before treatment, and platelet count recovery by the second cycle of 5-AZA-dC treatment can be used as an early predictive marker of response (30–32).
Mutation
A series of gene mutations drive clone expansion and malignant transformation (33) and are significantly prevalent in hematological disorders. Studies have shown that TET2 and DNMT3A mutations (29, 32, 34) are linked to improved response to HMAs in MDS and related disorders; P53 mutation also predicted 5-AZA-dC -induced complete remission in patients with MDS (35). In contrast, mutations in ASXL1, CBL, RAS and SF3B1 genes are not associated with the prediction of response to treatment (32). According to a study of 15 gene mutation analyses in CMML patients, no somatic mutations (SRSF2, TET2, ASXL1, NRAS, DNMT3A, RUNX1, U2AF1, TP53, JAK2, KIT, KRAS, SF3B1, EZH2, IDH1 and IDH2) were significantly correlated with response to 5-AZA-dC (36). FLT3-ITD mutation did not affect the overall response rate (ORR) in patients with AML (37). Notably, an unbiased framework on investigating the role of several mutations in predicting HMA resistance in MDS showed that EZH2 mutation predicted a lower response, while IDH1 mutation was linked to a higher response rate; seven different mutation combinations including ASXL1, NF1, EZH2, TET2, RUNX1, SRSF2 and BCOR predicted the resistance to HMAs (38, 39). Wu et al. reported the co-occurrence of RUNX1 and ASXL1 mutations that were associated with a poor response to HMAs (40), it was summarized in Table 1. This indicates that the HMA treatment response might be affected by two or more mutations.
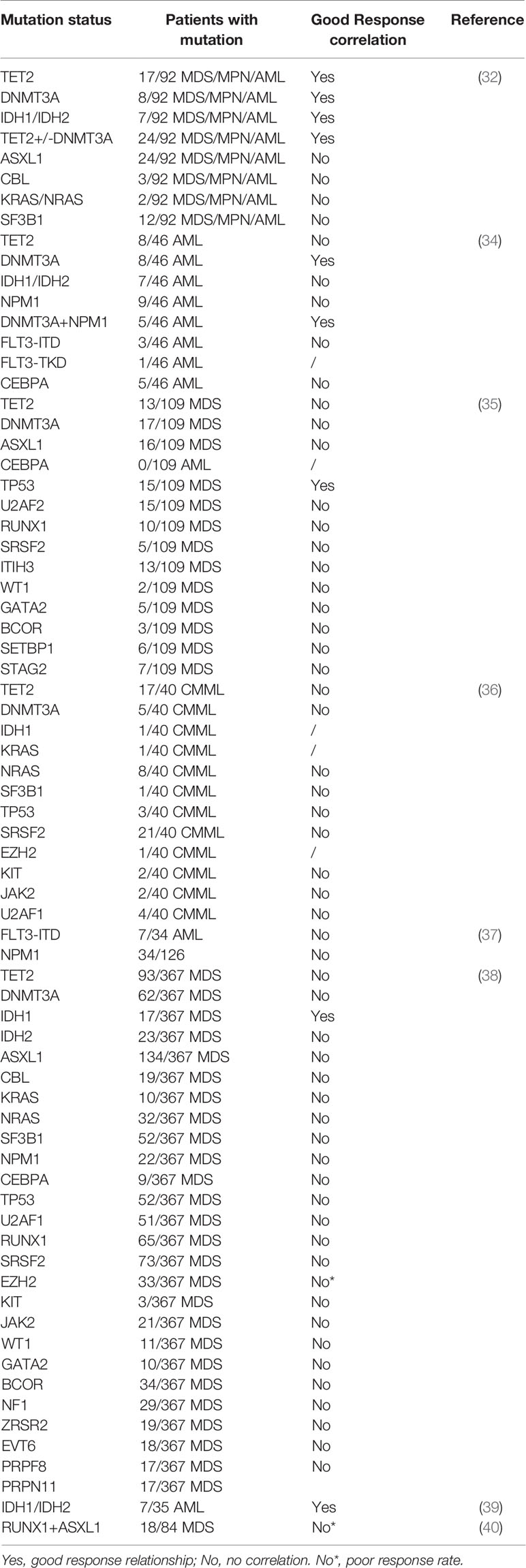
Table 1 Gene mutation and HMA response.
DNA Methylation
A critical mechanism of HMAs in anticancer treatment is demethylation. More researchers have explored the role of DNA methylation or demethylation in predicting the HMA response. Global hypomethylation (predominantly in CpG islands and CpG island-associated regions) was correlated with the response in AML patients treated with low-dose 5-AZA. However, for some genes such as LINE1, HOXA5, P15, and H19, the alteration of DNA methylation after treatment cannot predict response (41, 42). K Raj reported that the baseline level of P15 methylation was much lower in responders than non-responders (43). However, one study suggested that gene methylation (ERα, NOR, CDH1, NPM2, OLIG2, CDH13, CDNK2B, PGRA, PDZ and RIL) at baseline did not correlate with clinical response to 5-AZA-dC, in which a significant correlation between reduced methylation after more than four cycles of treatment and clinical responses was observed (44). Owing to the identification of some epigenetic enzyme mutations, and given that concurrent hypermethylation and hypomethylation after treatment complicated the predictive factors of the HMA response, DNA methylation cannot be used as a marker to predict the response to HMAs (45). While some methylation regions were still able to potentially predict the response, 167 differentially methylated regions (DMRs) of DNA at baseline, which were preferentially located at distal regulatory regions, distinguished responders from non-responders in CMML patients (36).
Gene Expression
Gene expression may be a predictive marker for HMA response. In AML patients treated with 5-AZA-dC, different gene expression patterns can be used to identify the 5-AZA-dC response. Genes such as SLC24A3, MUM1, TNFSF9, DBN1, ABAT, and DDX52 were highly expressed, manifesting the response to treatment; in contrast, overexpression of IFI44L, IFI27, PDK4, MX1, FAS, and ITGB2 were uncorrelated to HMAs (46). CXCL4, CXCL7, CJUN, and CMYB were highly expressed in non-responder patients with CMML (36, 47). Although these genes are related to the inflammatory pathway, the mechanism by which the altered expression of these genes affects HMA responsiveness is not clear.
As described previously, DNMTs are implicated in DNA methylation, the expression levels of DNMTs might be related to the 5-AZA-dC response. In breast cancer, patients with high DNMT3A and DNMT3B protein expression levels, and to a less extent, DNMT1, were more sensitive to 5-AZA-dC (21). The target of DNMTs, micro-29b, was highly expressed in responsive AML patients and could predict the response (48). Gene expression mutually affects DNA methylation. Loss of MLL5, a novel histone lysine methyltransferase, was associated with resistance to low- dose 5-AZA-dC, reduced global DNA methylation in promoter regions, and reduced DNA demethylation (49). Reduced methylation of phosphoinositide-phospholipase C beta1 in the promoter region and subsequent high mRNA expression after 5-AZA treatment predicted the responsiveness (50).
Options After HMA Failure
Even though HMAs treatment showed prolonged survival in patients, the response was almost transient, many patients lost sensitivity within two years (51). After HMA failure, patients with MDS or AML receive traditional AML-like chemotherapies, which include low-dose cytarabine, the combination of cytarabine and daunorubicin, purine nucleoside analogs, or investigational treatment (inhibitors targeting PD-1, CTLA-4, Ras, BCL-2, IDH1 and IDH2 mutations, TLR-2, AXL, TGF-beta, spliceosome, NED88 activating enzyme), some of which also benefit from switching to another nucleoside analog (52–72). We have summarized this in Table 2. Novel forms of HMAs, which could be taken orally, could potentially change the routine of administration (73). Compared to intravenous administration, the oral form of cedazuridine/decitabine (ASTX727) produced similar decitabine exposure and efficacy and was recently approved by the FDA (74). However, there is still a lack of randomized controlled trials that compare the investigational drugs with and without HMA to a single use of HMA, and whether target therapies combined with HMA could improve the OS or ORR remains unknown. In addition, CD47, CD33/CD3 antibody, FLT3 inhibitors and autophagy inhibitors (ROC-325) are also being investigated for the treatment of naïve high-risk MDS/AML and refractory/relapsed AML (75–78), and autophagy inhibitors may be very promising treatment options for hematological malignancies in the future. The efficiency of these novel treatments in relapsed or refractory AML/MDS after HMA failure is unknown. We believe that the administration of these novel drugs should be based on the molecular characteristics of patients who relapse after HMA treatment.
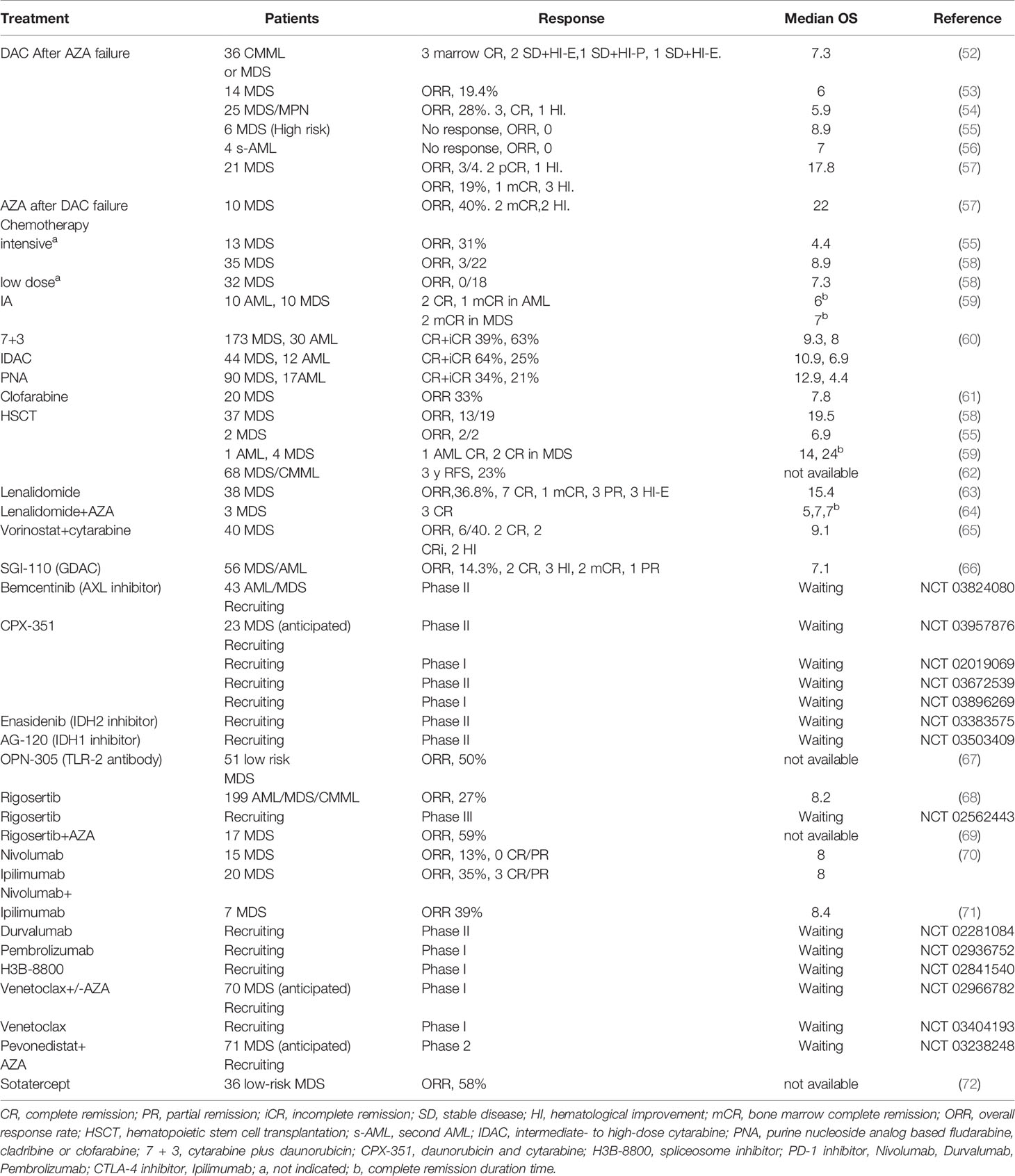
Table 2 Different strategies for patients with HMA failure.
Resistance To HMA
Membrane Transporters
Hydrophilic drugs, such as 5-AZA and 5-AZA-dC, are taken up into the cells by human membrane nucleoside transporters (hNTs), which include equilibrate nucleoside transporters (hENT1, hENT2, hENT3, hENT4) and concentrative nucleoside transporters (hCNT1, hCNT2, hCNT3) (79). However, 5-AZA and 5-AZA-dC exhibited different nucleoside transportability profiles. All seven hNTs showed the ability to transport 5-AZA, hCNT3 showed the highest transportation efficiency, whereas hENT1 and hENT2 showed modest and hCNT1 and hCNT3 showed poor transportation of 5-AZA-dC (80). In fact, hENT1 mainly takes up 5-AZA-dC in a Na+ independent manner, which determines the activity of 5-AZA-dC in human HCT116 cancer cells (81). The expression level of these genes may determine the concentration and activity of 5-AZA and 5-AZA-dC in the cells. A study of 12 patients showed that in primary AML blast cells, hENT1 mRNA expression was highly abundant compared to hCNT1, hCNT3, hENT2 and hCNT2, and the transportation of 5-AZA also predominantly depended on the activity of hENT1 (82). To determine how transporters mediate resistance to 5-AZA-dC, Wu et al. measured the expression levels of hENT1 and hENT2 in 98 patients with MDS and found that patients responding to 5-AZA-dC displayed significantly higher hENT1 expression levels than non-responders, whereas hENT2 did not (83). However, Qin et al. reported that the gene expression of hENT1, hENT2, hCNT3 was not different between responders and non-responders in 14 MDS patients, and these genes expression were also comparable at diagnosis and relapse (84). We still do not know whether these patients who achieved a response initially and finally relapsed showed reduced expression of hENTs in a large population, and whether MDS/AML patients with AZA resistance had low expression levels of hNTs compared to responders. Notably, the development of lipid nanocapsules (LNCs) to encapsulate 5-AZA-dC showed high activity in 5-AZA-dC resistant and sensitive leukemia cells and could potentially bypass these transporters (85).
Metabolism of HMA
After being transported into the cells, 5-AZA and 5-AZA-dC were catalyzed by a series of enzymes including deoxycytidine kinase (DCK), uridine cytidine kinase 2 (UCK2), cytidine deaminase (CDA), and carbamoyl-phosphate synthetase (CAD). 5-AZA-dC was converted to 5-AZA-dCTP and incorporated into DNA; however, most 5-AZA was incorporated into RNA as 5-AZA-CTP, and only 10%–20% is translated into 5-aza-dCTP after multistep catalyzation (21). Therefore, researchers believe that insufficient metabolites of 5-AZA and 5-AZA-dC might result in HMA resistance because of aberrant expression of metabolic genes. By measuring the expression of several genes encoding metabolic enzymes, Gu et al. found that mRNA expression of UCK2 and CDA increased in 5-AZA-dC treated MDS patients at relapse, while DCK expression was decreased compared to pre-treatment levels. In contrast, at relapse, DCK expression was upregulated while UCK2 and CDA expression was reduced. The expression of the de novo pyrimidine synthesis enzyme CAD increased in patients who resisted to both 5-AZA-dC and 5-AZA (86). In another study of 32 MDS patients who were either resistant or sensitized to 5-AZA-dC, DCK and CDA gene expression levels were comparable, but the ratio of CDA to DCK was significantly higher in non-responders than in responders, suggesting that this could be a mechanism of primary resistance (84). Mutations in genes that encode metabolic enzymes may affect HMA metabolism, and switching mutation of DCK from heterozygosity to homozygosity impaired the 5-AZA-dC sensitivity in the HL-60 cell line. However, no such mutation has been found in MDS patients (84, 87). In addition, the sterile alpha motif and histidine-aspartate domain-containing protein 1 (SAMHD1) is a 2’-deoxynucleoside-5’-triphosphate (dNTP) triphosphohydrolase that interacts with DAC-TP (not AZA-TP) and influences DAC efficacy in leukemia cells. SAMHD1 expression is inversely correlated with the clinical response to 5-AZA-dC in AML patients (88). We summarized them in the Figure 1A. This indicates that gene expression related to dNTP metabolism potentially interferes with 5-AZA or 5-AZA-dC activity and needs further investigation.
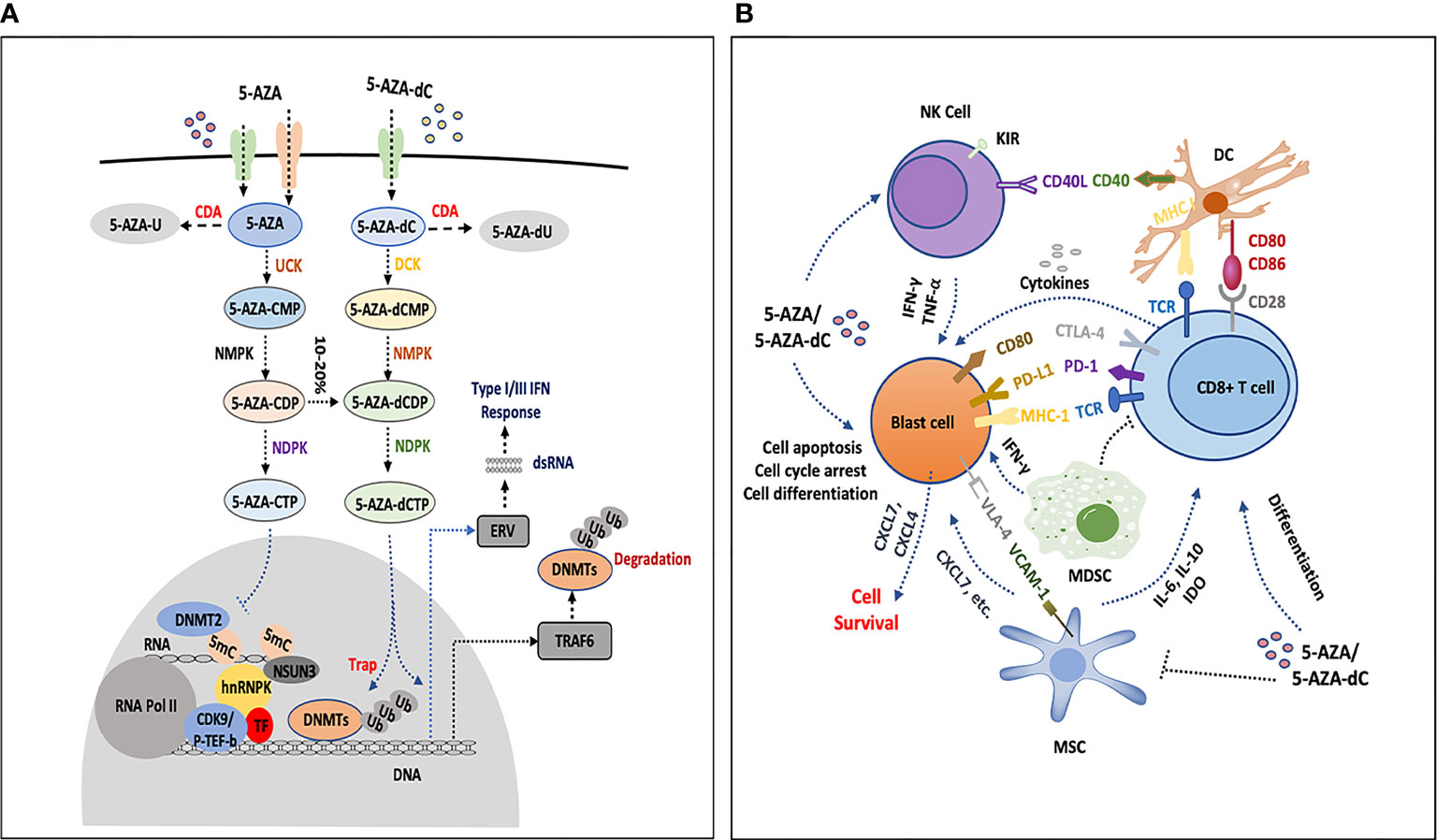
Figure 1 The influence of HMAs on bone marrow cells. (A) 5-AZA and 5-AZA-dC are transported into blast cells or immune cells (T cells, NK cells and DCs) by hENT1, hENT2, and hENT1-2, hCNT1 and hCNT3, respectively, part of which are catalyzed by CDA and result in degradation. The remaining AZA and 5-AZA-dC are catalyzed into 5-AZA-CMP and 5-AZA-dCMP by UCK and DCK, separately. In the following process, NMPK and NDPK play a role in the formation of 5-AZA-CTP and 5-AZA-dCTP. 5-AZA-CTP is mainly incorporated into RNA and possibly promotes the formation of 5-AZA sensitive chromatin structure, which consists of hnRNPK, DNMT2, NSUN3, CDK9/p-TEF-b and TF. 5-AZA-dCTP depletes DNMTs and increases gene expression, degradation of DNMTs is processed by proteosome pathway or by upregulating TRAF6 that leads to DNMTs ubiquitination, 5-AZA-dCTP is also activated and becomes hydrolyzed by SAMHD1 in leukemia. Simultaneously, 5-AZA-dCTP activates the transcription of ERV, induces dsRNA formation and facilitates type III interferon response. (B) 5-AZA-dC and 5-AZA inhibit NK cells by upregulating the inhibitory receptor KIR, or activate the antitumor activities by secreting some cytokines (IFN-Gamma or TNF-Alpha). HMAs induce the expression of CD40 and CD86 on DCs, which interact with CD8+ T cells by CD80/86-CD28 and TCR-MHC I CD8+ T cells communicate with blast cells by TCR-MHC I, PD-1-PDL1, and CTLA-4-CD80. HMAs increase the secretion of IFN-Gamma and antitumor activity of CD8+ T cells, and the expression of PD-1 and CTLA4 is upregulated as well, which participate in immune suppression. HMAs decrease the percentage of MDSCs in the bone marrow, which may promote the function of HMA. MSCs secrete IL-6, IL-10, IDO-1, etc., to inhibit CD8 T cells or directly interact with blast cells by VCAM1-VLA4. Leukemia cells express high level of CXCL4 and CXCL7 to promote the survival of leukemia cells under the treatment of HMAs. MSC can also secret CXCL7. These interactions and secretion of chemokines or cytokines in the bone marrow, which potentially mediate the resistance to HMAs, are still not completely understood.
AZA incorporation may also be associated with 5-AZA sensitivity. By applying AZA mass spectrometry (AZA-MS) to primary bone marrow samples of MDS/CMML patients undergoing 5-AZA therapy, responders showed greater incorporation of 5-AZA-CdR into DNA than non-responders; however, much higher free AZA and AZA-RNA were observed in non-responders, which may have resulted from a shift in azacitidine/cytidine nucleotide ratios in the cytoplasm of non-responders. RNA methylation was not changed in either group of patients (89). In this study, it showed that the incorporation of 5-AZA-dCTP into DNA affected anticancer activity. Given the difference in the cell cycle, cell viability and gene expression between 5-AZA and 5-AZA-dC (90), the 5-AZA-CTP-RNA should have its unneglectable function and worth investigating.
T Cells and Immune Response
Several groups have proposed that the clinical benefits for patients treated with HMA may be the result of direct cytotoxic and differential effects or immune responses for malignant cells. The role of HMA in the frequency and function of natural killer (NK) cells, T cells, and dendritic cells (DCs) has been reviewed previously (91). Overall, there is no consensus on how HMA affects the functionality of immune cells, and the role of DCs and NK cells in HMA responsiveness remains unknown. In this study, we mainly present the association of T cells with the HMA response, as briefly described in Figure 1B.
In a T-cell lymphoma mouse model, 5-AZA-dC stimulated CD80 expression in malignant cells and upgraded the cytolytic activity of IFN- γ- producing CD8+ T-cells (92). Low-dose 5-AZA-dC enhanced the activation and proliferation of human IFN-gamma+ T cells, as well as Th1 polarization and activity of cytotoxic T cells in solid tumor patients, increased IFN- γ+ T cells, and increased T-cell cytotoxicity predicted improved ORR and survival (93). In contrast to another study, Zhao et al. reported that 5-AZA-dC treatment was associated with increased expression of inhibitory receptors on T cells and reduced T cell population in elderly patients with AML. When comparing the differences in T cell differentiation and phenotypes between responders and non-responders, they found more naïve and central memory T cells, and inducible T cell costimulatory (ICOS)-expressing CD8+ T cells in responders, and found a specific immune signature predicted the response to 5-AZA-dC (94). These observations suggest IFN-γ+ T cells are implicated in the 5-AZA-dC response. Of note, 5-AZA-dC inhibited Gamma Delta T cell proliferation and cytotoxicity, and induced the expression of KIR2DL2/3 on Gamma Delta T cells, which were less toxic than negative cells (95), Suggesting that T cell activation was a double-edged sword and needed further analysis in HMA responders and non-responders.
Several studies have shown that 5-AZA upregulates the expression of tumor-specific antigens and cytotoxic T-lymphocytes. HMA induces the expression of inhibitory receptors on the surface, such as T cell programmed death-1 (PD-1), PD-L1, PDL-2 and CTLA-4 (96). Persistent expression and engagement of PD-1 and CTLA-4 results in T cell exhaustion and tumor immune evasion (97). The expression of these receptors was associated with resistance to 5-AZA-dC and 5-AZA treatment, and may be exploited by target therapies (96). Combination treatment with PD-1 antibody showed higher ORR than 5-AZA-dC or 5-AZA alone in older AML patients who were ineligible for intensive chemotherapy (98). HMA triggered the expression of endogenous retroviral (ERV) elements, and increased the transcription of double-stranded RNA, innate type I or III interferon response by the MDA5/MAVS/IRF7 pathway (99). However, the difference in ERV activation between responders and non-responders has not been explored in MDS/AML. In another study, when comparing the 5-AZA-dC responders and non-responders, there was no differential expression of PD-1 before and after treatment (94). In contrast to a report from Nahas et al. they showed guadecitabine (GDAC) negatively regulated inhibitory accessory cells by decreasing PD-1 expressing T cells and AML-mediated expansion of myeloid-derived suppressor cells (MDSCs) in mouse model, therapy with guadecitabine resulted in enhanced leukemia-specific immunity as well, as manifested by increased CD4+ and CD8+ cells expressing IFN-γ (100). These inconsistencies are likely due to different models in vitro or in vivo, functional assays, concentration administration, etc.
The 5-AZA not only induces a cytotoxic CD8+ T-cell response, but also stimulates a shift from cytotoxic to regulatory T cells with a functional phenotype in proinflammatory Th1 cells, indicating a potent inhibition of tumor -specific T cell immunity by 5-AZA (101, 102). Given the inhibitory and activating effects of HMA on T cells, further research is needed to explore its role in mediating HMA responsiveness.
Bone Marrow-Derived Cells
Bone marrow-derived cells, mesenchymal stromal cells (MSCs) and myeloid-derived suppressor cells (MDSCs), are essential parts of the bone marrow microenvironment in regulating the immune response (103). In the myeloma microenvironment, 5-AZA-dC treatment inhibited tumor growth and enhanced T cell infiltration by depleting monocytic myeloid-derived suppressor cells (M-MDSCs) (104), which has also been shown in AML (100). Nevertheless, the percentage of M-MDSCs in non-responsive patients is uncertain. The phenotypes, transcriptome, and epigenomics of MSCs in MDS were significantly different from those of healthy donors, following 5-AZA treatment, and the gene expression pattern of MSCs from MDS patients with response was closely clustered with that of healthy donors. MSCs from patients who failed to respond to 5-AZA and could not be programmed by HMA were associated with rapid adverse disease transformation (105). MSCs play a role in immune suppression by secreting high levels of indoleamine 2, 3-dioxygenase (IDO-1), or other cytokines, which are generated by DCs and tumor cells as well. IDO-1 expression was associated with the failure of 5-AZA treatment through immunosuppression, which reduced the number of infiltrating CD8+ T cells and shortened the overall survival in high-risk MDS patients (106). IDO-1 inhibitors were investigated alone or combination with immune checkpoint inhibitors in the clinic (107). In addition, MSCs also secret CXCL7 that may promote the survival of cells (108), it was supported by the report that leukemia cells express high levels of CXCL4 and CXCL7 which is associated with the HMA non-responsiveness (36). The impact of MSCs on HMA responsiveness, the microenvironment in MDS/AML, and differential mediators secreted by MSCs that are implicated in HMA non-response remain uncertain.
Hematopoietic Stem Progenitor Cell
Accumulating evidence suggests that leukemia stem cells (LSCs) are responsible for chemoresistance and leukemia relapse, as they can self-renew and to differentiate into the heterogeneous lineages of leukemia cells (109). Even though HMA substantially reduced the LSC-containing population in patients with CR/iCR, it cannot eradicate LSC, which will finally re-expand when relapse occurs. In non-responders, there was no significant reduction in the size of the LSC-containing population (110). In MDS patients with monosomy 7, the clonal involvement in dominant CD45RA+ progenitor populations was not reduced following the 5-AZA response, which indicated the resistance of this compartment (111). Using RNA sequencing performed on HSPC cells, Unnikrishnan et al. found that cell cycle arrest predicts resistance to 5-AZA, and with 5-AZA response, the inflammatory pathway was activated. Although 5-AZA did not completely eliminate dysplastic clones upon response, it changed the clonal contribution, which enabled previously dormant clones with a lower mutational burden (112). Therefore, targeting LSCs may potentially improve HMA efficiency and prevent disease relapse. The LSC targets CD44, CD47, CD33, CD96, TIM-3, and CD123 antibodies are undergoing investigation (113, 114); in particular, TIM-3 antibody, MBG-453, which targets LSCs and leukemia blast cells, is used to combinate with HMA, and has shown encouraging response and durability (115). This will facilitate its clinical application and further research on LSC target therapies.
Conclusion
Clinical studies have shown that patients with intermediate-and high-risk MDS or elderly old AML receive HMA treatment preferentially. HMAs have also been widely used in low-risk MDS (116), in combination with other chemotherapies to enhance the activity of cytotoxic drugs in leukemia, lymphoma and other solid tumors (117–119), as maintenance therapy after allogeneic hematopoietic stem cell transplantation (allo-HSCT), and as part of conditioning regimens before allo-HSCT (120–122). Researchers have been exploring how to predict the response, cause, and outcome of resistance because of non-responsiveness in some patients. Clinical parameters, DNA methylation, gene expression signatures, and specific immune cell counterparts are promising markers for predicting response. MicroRNAs, such as microRNA-181 (123), microRNA-29c (124), microRNA-124 (125), and microRNA-29b (48) were also associated with HMA response. However, the role of long-noncoding RNAs (lncRNAs) in predicting HMA responsiveness and inducing the resistance is unknown.
New combination strategies have been developed to alleviate the resistance of HMAs and demonstrate the advantages of their safety and efficiency. It is important that more clinical trials are conducted to better understand of the mechanisms of resistance. Recently, Cheng et al. found 5-AZA-resistant MDS and AML patients showed a significant increase in RNA:m5C and NSUN1/BRD4-associated active chromatin. HnRNPK interacts with the lineage-determining transcription factors (TFs), GATA1, SPI1/PU.1, and CDK9/P-TEFb to recruit RNA-polymerase-II at nascent RNA, leading to the formation of an AZA-sensitive chromatin structure (Figure 1A) (126). Notably, 5-AZA inhibited cytosine 38 methylation of tRNA, a major substrate of DNMT2, resulting in tRNA hypomethylation (127). These studies suggested that 5-AZA was involved in RNA demethylation and that RNA demethylation affects the sensitivity to HMAs by modeling chromatin.
Importantly, 5-AZA-dC and 5-AZA induce cell cycle arrest at G1 phase via p21 and G2/M phase via p38 MAPK kinase pathway (128). However, some reports showed both drugs induced a G2/M-arrest in P39 and HL-60 leukemia cell lines, but not in KG-1 and MDS-1 cells (129). They both inhibit cell proliferation by increasing genes expression, such as Cylcin-D, p21, MyoD (130). In addition, the gene expression of HO-1 affects the efficacy of 5-AZA-dC by decreasing the expression of cell cycle related protein p15 (131). High expression of the melanocyte late-differentiation driver, SOX9, also upregulates the expression of cyclin-dependent kinase inhibitors (CDKN) p27/CDKN1B and p21/CDKN1A that mediate cell cycle exit with differentiation (132). When comparing the gene expression differences between MDS patients with HMA non-responsiveness and HMA responsiveness, Ashwin U, et al. found that cell cycle quiescence of hematopoietic progenitors marked AZA non-responders, targeting cell cycle quiescence might overcome AZA resistance (112). In CMML patients, HMA non-responders have high expression of CXCL4 and CXCL7, both of them are related to cell cycle activity (36). It indicates that cell cycle plays the essential role in HMA resistance and deserves further investigation.
The immune cells, T cells, NK cells, and DCs are essential components of the bone marrow microenvironment, which can be programmed by HMAs and participate in resistance, Figure 1B. The monocyte subset repartition after treatment is also a useful tool for predicting HMA response (133). These cells may antagonize the function of HMAs and promote leukemia cell survival by interacting with leukemia cells directly or indirectly secreting a variety of cytokines and chemokines, or in the opposite way. The functioning of these cells in the bone marrow to confer HMA resistance is not completely understood. Single-cell sequencing has the advantage of distinguishing novel cell populations and plotting gene expression patterns of different cell types, which will lay the foundation for exploring the mechanism of HMA resistance.
Author Contributions
GZ and QW conceived and wrote the manuscript. GZ and SL did the figure and table. XW reviewed this manuscript. All authors contributed to the article and approved the submitted version.
Funding
This project is founded by National Nature Science Foundation of China NSFC 81100389 and 82170136.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Greenberg MVC, Bourc'his D. The Diverse Roles of DNA Methylation in Mammalian Development and Disease. Nat Rev Mol Cell Biol (2019) 20(10):590–607. doi: 10.1038/s41580-019-0159-6
2. Field AE, Robertson NA, Wang T, Havas A, Ideker T, Adams PD. DNA Methylation Clocks in Aging: Categories, Causes, and Consequences. Mol Cell (2018) 71(6):882–95. doi: 10.1016/j.molcel.2018.08.008
3. McMahon KW, Karunasena E, Ahuja N. The Roles of DNA Methylation in the Stages of Cancer. Cancer J (2017) 23(5):257–61. doi: 10.1097/PPO.0000000000000279
4. Feng Y, Li X, Cassady K, Zou Z, Zhang X. TET2 Function in Hematopoietic Malignancies, Immune Regulation, and DNA Repair. Front Oncol (2019) 9:210. doi: 10.3389/fonc.2019.00210
5. Navada SC, Steinmann J, Lübbert M, Silverman LR. Clinical Development of Demethylating Agents in Hematology. J Clin Invest (2014) 124(1):40–6. doi: 10.1172/JCI69739
6. Fenaux P, Mufti GJ, Hellstrom-Lindberg E, Santini V, Finelli C, Giagounidis A, et al. Efficacy of Azacitidine Compared With That of Conventional Care Regimens in the Treatment of Higher-Risk Myelodysplastic Syndromes: A Randomized, Open-Label, Phase III Study. Lancet Oncol (2009) 10(3):223–32. doi: 10.1016/S1470-2045(09)70003-8
7. Silverman LR, Demakos EP, Peterson BL, Kornblith AB, Holland JC, Odchimar-Reissig R, et al. Randomized Controlled Trial of Azacitidine in Patients With the Myelodysplastic Syndrome: A Study of the Cancer and Leukemia Group B. J Clin Oncol (2002) 20(10):2429–40. doi: 10.1200/JCO.2002.04.117
8. Lübbert M, Suciu S, Baila L, Rüter BH, Platzbecker U, Giagounidis A, et al. Low-Dose Decitabine Versus Best Supportive Care in Elderly Patients With Intermediate- or High-Risk Myelodysplastic Syndrome (MDS) Ineligible for Intensive Chemotherapy: Final Results of the Randomized Phase III Study of the European Organization for Research and Treatment of Cancer Leukemia Group and the German MDS Study Group. J Clin Oncol (2011) 29(15):1987–96. doi: 10.1200/JCO.2010.30.9245
9. Al-Ali HK, Jaekel N, Niederwieser D. The Role of Hypomethylating Agents in the Treatment of Elderly Patients With AML. J Geriatr Oncol (2014) 5(1):89–105. doi: 10.1016/j.jgo.2013.08.004
10. Zeidan AM, Wang R, Wang X, Shallis RM, Podoltsev NA, Bewersdorf JP, et al. Clinical Outcomes of Older Patients With AML Receiving Hypomethylating Agents: A Large Population-Based Study in the United States. Blood Adv (2020) 4(10):2192–201. doi: 10.1182/bloodadvances.2020001779
11. Zhang Y, Mei Q, Liu Y, Li X, Brock MV, Chen M, et al. The Safety, Efficacy, and Treatment Outcomes of a Combination of Low Dose Decitabine Treatment in Patients With Recurrent Ovarian Cancer. Oncoimmunology (2017) 6(9):e1323619. doi: 10.1080/2162402X.2017.1323619
12. Glasspool RM, Brown R, Gore ME, Rustin GJ, McNeish IA, Wilson RH, et al. A Randomised, Phase II Trial of the DNA-Hypomethylating Agent 5-Aza-2’-Deoxycytidine (Decitabine) in Combination With Carboplatin vs Carboplatin Alone in Patients With Recurrent, Partially Platinum-Sensitive Ovarian Cancer. Br J Cancer (2014) 110(8):1923–9. doi: 10.1038/bjc.2014.116
13. Tawbi HA, Beumer JH, Tarhini AA, Moschos S, Buch SC, Egorin MJ, et al. Safety and Efficacy of Decitabine in Combination With Temozolomide in Metastatic Melanoma: A Phase I/II Study and Pharmacokinetic Analysis. Ann Oncol (2013) 24(4):1112–9. doi: 10.1093/annonc/mds591
14. Nervi C, De Marinis E, Codacci-Pisanelli G. Epigenetic Treatment of Solid Tumors: A Review of Clinical Trials. Clin Epigenet (2015) 7:127–47. doi: 10.1186/s13148-015-0157-2
15. Zhang XD, Hu JX, Chen QJ, Zhang MZ, Young KH. Decitabine Can Improve the Efficacy of Second-Line Chemotherapy in Relapsed or Refractory Diffuse Large B Cell Lymphoma. Blood (2019) 134:5221. doi: 10.1182/blood-2019-128424
16. Agrawal K, Das V, Vyas P, Hajdúch M. Nucleosidic DNA Demethylating Epigenetic Drugs: A Comprehensive Review From Discovery to Clinic. Pharmacol Ther (2018) 188:45–79. doi: 10.1016/j.pharmthera.2018.02.006
17. Momparler RL. Pharmacology of 5-Aza-2'-Deoxycytidine (Decitabine). Semin Hematol (2005) 42(3 Suppl 2):S9–16. doi: 10.1053/j.seminhematol
18. Momparler RL, Samson J, Momparler LF, Rivard GE. Cell Cycle Effects and Cellular Pharmacology of 5-Aza-2'-Deoxycytidine. Cancer Chemother Pharmacol (1984) 13(3):191–4. doi: 10.1007/BF00269027
19. Stresemann C, Lyko F. Modes of Action of the DNA Methyltransferase Inhibitors Azacytidine and Decitabine. Int J Cancer (2008) 123(3):8–13. doi: 10.1002/ijc.23607
20. Ghoshal K, Datta J, Majumder S, Bai S, Kutay H, Motiwala T, et al. 5-Aza-Deoxycytidine Induces Selective Degradation of DNA Methyltransferase 1 by a Proteasomal Pathway That Requires the KEN Box, Bromo-Adjacent Homology Domain, and Nuclear Localization Signal. Mol Cell Biol (2005) 25(11):4727–41. doi: 10.1128/MCB.25.11.4727-4741.2005
21. Yu J, Qin B, Moyer AM, Nowsheen S, Liu T, Qin S, et al. DNA Methyltransferase Expression in Triple-Negative Breast Cancer Predicts Sensitivity to Decitabine. J Clin Invest (2018) 128(6):2376–88. doi: 10.1172/JCI97924
22. Sato T, Issa JJ, Kropf P. DNA Hypomethylating Drugs in Cancer Therapy. Cold Spring Harb Perspect Med (2017) 7(5):a026948. doi: 10.1101/cshperspect.a026948
23. Garcia JS, Jain N, Godley LA. An Update on the Safety and Efficacy of Decitabine in the Treatment of Myelodysplastic Syndromes. Onco Targets Ther (2010) 3:1–13. doi: 10.2147/ott.s7222
24. Blum KA, Liu Z, Lucas DM, Chen P, Xie Z, Baiocchi R, et al. Phase I Trial of Low Dose Decitabine Targeting DNA Hypermethylation in Patients With Chronic Lymphocytic Leukemia and Non-Hodgkin’s Lymphoma: Dose-Limiting Myelosuppression Without Evidence of DNA Hypomethylation. Br J Haematol (2010) 150(2):189–95. doi: 10.1111/j.1365-2141.2010.08213.x
25. Cheson BD, Greenberg PL, Bennett JM, Lowenberg B, Wijermans PW, Nimer SD, et al. Clinical Application and Proposal for Modification of the International Working Group (IWG) Response Criteria in Myelodysplasia. Blood (2006) 108(2):419–25. doi: 10.1182/blood-2005-10-4149
26. Cheson BD, Bennett JM, Kopecky KJ, Büchner T, Willman CL, Estey EH, et al. Revised Recommendations of the International Working Group for Diagnosis, Standardization of Response Criteria, Treatment Outcomes, and Reporting Standards for Therapeutic Trials in Acute Myeloid Leukemia. Clin Oncol (2003) 21(24):4642–9. doi: 10.1200/JCO.2003.04.036
27. Itzykson R, Thépot S, Quesnel B, Dreyfus F, Beyne-Rauzy O, Turlure P, et al. Prognostic Factors for Response and Overall Survival in 282 Patients With Higher-Risk Myelodysplastic Syndromes Treated With Azacitidine. Blood (2011) 117(2):403–11. doi: 10.1182/blood-2010-06-289280
28. Kantarjian H, Issa JP, Rosenfeld CS, Bennett JM, Albitar M, DiPersio J, et al. Decitabine Improves Patient Outcomes in Myelodysplastic Syndromes. Cancer (2006) 106(8):1794–803. doi: 10.1002/cncr.21792
29. Itzykson R, Kosmider O, Cluzeau T, Mansat-De Mas V, Dreyfus F, Beyne-Rauzy O, et al. Impact of TET2 Mutations on Response Rate to Azacitidine in Myelodysplastic Syndromes and Low Blast Count Acute Myeloid Leukemias. Leukemia (2011) 25(7):1147–52. doi: 10.1038/leu.2011.71
30. Jung HA, Maeng CH, Kim M, Kim S, Jung CW, Jang JH. Platelet Response During the Second Cycle of Decitabine Treatment Predicts Response and Survival for Myelodysplastic Syndrome Patients. Oncotarget (2015) 6(18):16653–62. doi: 10.18632/oncotarget.3914
31. van der Helm LH, Alhan C, Wijermans PW, van Marwijk Kooy M, Schaafsma R, Biemond BJ, et al. Platelet Doubling After the First Azacitidine Cycle Is a Promising Predictor for Response in Myelodysplastic Syndromes (MDS), Chronic Myelomonocytic Leukaemia (CMML) and Acute Myeloid Leukaemia (AML) Patients in the Dutch Azacitidine Compassionate Named Patient Programme. Br J Haematol (2011) 155(5):599–606. doi: 10.1111/j.1365-2141.2011.08893.x
32. Traina F, Visconte V, Elson P, Tabarroki A, Jankowska AM, Hasrouni E, et al. Impact of Molecular Mutations on Treatment Response to DNMT Inhibitors in Myelodysplasia and Related Neoplasms. Leukemia (2014) 28(1):78–87. doi: 10.1038/leu.2013.269
33. Bowman RL, Busque L, Levine RL. Clonal Hematopoiesis and Evolution to Hematopoietic Malignancies. Cell Stem Cell (2018) 22(2):157–70. doi: 10.1016/j.stem.2018.01.011
34. Metzeler KH, Walker A, Geyer S, Garzon R, Klisovic RB, Bloomfield CD, et al. DNMT3A Mutations and Response to the Hypomethylating Agent Decitabine in Acute Myeloid Leukemia. Leukemia (2012) 26(5):1106–7. doi: 10.1038/leu.2011.342
35. Chang CK, Zhao YS, Xu F, Guo J, Zhang Z, He Q, et al. TP53 Mutations Predict Decitabine-Induced Complete Responses in Patients With Myelodysplastic Syndromes. Br J Haematol (2017) 176(4):600–8. doi: 10.1111/bjh.14455
36. Meldi K, Qin T, Buchi F, Droin N, Sotzen J, Micol JB, et al. Specific Molecular Signatures Predict Decitabine Response in Chronic Myelomonocytic Leukemia. J Clin Invest (2015) 125(5):1857–72. doi: 10.1172/JCI78752
37. Prata PH, Bally C, Prebet T, Recher C, Venton G, Thomas X, et al. NPM1 Mutation Is Not Associated With Prolonged Complete Remission in Acute Myeloid Leukemia Patients Treated With Hypomethylating Agents. Haematologica (2018) 103(10):e455–7. doi: 10.3324/haematol.2018.189886
38. Nazha A, Sekeres MA, Bejar R, Rauh MJ, Othus M, Komrokji RS, et al. Genomic Biomarkers to Predict Resistance to Hypomethylating Agents in Patients With Myelodysplastic Syndromes Using Artificial Intelligence. JCO Precis Oncol (2019) 3:PO.19.00119. doi: 10.1200/PO.19.00119
39. Emadi A, Faramand R, Carter-Cooper B, Tolu S, Ford LA, Lapidus RG, et al. Presence of Isocitrate Dehydrogenase Mutations may Predict Clinical Response to Hypomethylating Agents in Patients With Acute Myeloid Leukemia. Am J Hematol (2015) 90(5):77–9. doi: 10.1002/ajh.23965
40. Wu P, Weng J, Li M, Lu Z, Deng C, Sun Q, et al. Co-Occurrence of RUNX1 and ASXL1 Mutations Underlie Poor Response and Outcome for MDS Patients Treated With HMAs. Am J Transl Res (2019) 11(6):3651–3658.eCollection 2019.
41. Yang AS, Doshi KD, Choi SW, Mason JB, Mannari RK, Gharybian V, et al. DNA Methylation Changes After 5-Aza-2-Deoxycytidine Therapy in Patients With Leukemia. Cancer Res (2006) 66(10):5495–503. doi: 10.1158/0008-5472.CAN-05-2385
42. Yan P, Frankhouser D, Murphy M, Tam HH, Rodriguez B, Curfman J, et al. Genome-Wide Methylation Profiling in Decitabine-Treated Patients With Acute Myeloid Leukemia. Blood (2012) 120(12):2466–74. doi: 10.1182/blood-2012-05-429175
43. Raj K, John A, Ho A, Chronis C, Khan S, Samuel J, et al. CDKN2B Methylation Status and Isolated Chromosome 7 Abnormalities Predict Responses to Treatment With 5-Azacytidine. Leukemia (2007) 21(9):1937–44. doi: 10.1038/sj.leu.2404796
44. Shen L, Kantarjian H, Guo Y, Lin E, Shan J, Huang X, et al. DNA Methylation Predicts Survival and Response to Therapy in Patients With Myelodysplastic Syndromes. J Clin Oncol (2010) 28(4):605–13. doi: 10.1200/JCO.2009.23.4781
45. Voso MT, Santini V, Fabiani E, Fianchi L, Criscuolo M, Falconi G, et al. Why Methylation Is Not a Marker Predictive of Response to Hypomethylating Agents. Haematologica (2014) 99(4):613–9. doi: 10.3324/haematol.2013.099549
46. Bohl SR, Claus R, Dolnik A, Schlenk RF, Dohner K, Hackanson B, et al. Decitabine Response Associated Gene Expression Patterns in Acute Myeloid Leukemia (AML). Blood (2013) 122(21):3756. doi: 10.1182/blood.V122.21.3756.3756
47. Braun T, Itzykson R, Renneville A, de Renzis B, Dreyfus F, Laribi K, et al. Molecular Predictors of Response to Decitabine in Advanced Chronic Myelomonocytic Leukemia: A Phase 2 Trial. Blood (2011) 118(14):3824–31. doi: 10.1182/blood-2011-05-352039
48. Blum W, Garzon R, Klisovic RB, Schwind S, Walker A, Geyer S, et al. Clinical Response and miR-29b Predictive Significance in Older AML Patients Treated With a 10-Day Schedule of Decitabine. Proc Natl Acad Sci (2010) 107(16):7473–8. doi: 10.1073/pnas.1002650107
49. Yun H, Damm F, Yap D, Schwarzer A, Chaturvedi A, Jyotsana N, et al. Impact of MLL5 Expression on Decitabine Efficacy and DNA Methylation in Acute Myeloid Leukemia. Haematologica (2014) 99(9):1456–64. doi: 10.3324/haematol.2013.101386
50. Follo MY, Finelli C, Mongiorgi S, Clissa C, Bosi C, Testoni N, et al. Reduction of Phosphoinositide-Phospholipase C Beta1 Methylation Predicts the Responsiveness to Azacitidine in High-Risk MDS. Proc Natl Acad Sci (2009) 106(39):16811–6. doi: 10.1073/pnas.0907109106
51. Zeidan AM, Kharfan-Dabaja MA, Komrokji RS. Beyond Hypomethylating Agents Failure in Patients With Myelodysplastic Syndromes. Curr Opin Hematol (2014) 21(2):123–30. doi: 10.1097/MOH.0000000000000016
52. Braun T, Cherait A, Berthon C, Wilekens C, Park S, Rigal M, et al. Treatment With Decitabine (DAC) After Azacitidine (AZA) Failure in High-Risk Myelodysplastic Syndrome (MDS) and Advanced Chronic Myelomonocytic Leukemia (CMML). Blood (2013) 122(21):2796. doi: 10.1182/blood.V122.21.2796.2796
53. Borthakur G, Ahdab SE, Ravandi F, Faderl S, Ferrajoli A, Newman B, et al. Activity of Decitabine in Patients With Myelodysplastic Syndrome Previously Treated With Azacitidine. Leuk Lymphoma (2008) 49(4):690–5. doi: 10.1080/10428190701882146
54. Duong VH, Bhatnagar B, Zandberg DP, Vannorsdall EJ, Tidwell ML, Chen Q, et al. Lack of Objective Response of Myelodysplastic Syndromes and Acute Myeloid Leukemia to Decitabine After Failure of Azacitidine. Leuk Lymphoma (2015) 56(6):1718–22. doi: 10.3109/10428194.2014.966708
55. Duong VH, Lin K, Reljic T, Kumar A, Al Ali NH, Lancet JE, et al. Poor Outcome of Patients With Myelodysplastic Syndrome After Azacitidine Treatment Failure. Clin Lymphoma Myeloma Leukemia (2013) 13(6):711–5. doi: 10.1016/j.clml.2013.07.007
56. Guaragna G, Rinaldi E, Spina A, Girasoli M, Coppi MR, Brocca MC, et al. Decitabine as S-AML Treatment Following Azacytidine Failure: A Single Center Experience. HemaSphere (2019) 3(1):803–4. doi: 10.1097/01.HS9.0000565500.88230.d0
57. Komrokji RS, Apuri S, Al Ali N, Padron E, Pinilla-Ibarz J, List HF, et al. Evidence for Selective Benefit of Sequential Treatment With Azanucleosides in Patients With Myelodysplastic Syndromes (MDS). Clin Lymphoma Myeloma Leukemia (2017) 17(4):211–4. doi: 10.1016/j.clml.2016.10.003
58. Prebet T, Gore SD, Esterni B, Gardin C, Itzykson R, Thepot S, et al. Outcome of High-Risk Myelodysplastic Syndrome After Azacitidine Treatment Failure. JCO (2011) 29(24):3322–7. doi: 10.1200/JCO.2011.35.8135
59. Jabbour E, Garcia-Manero G, Batty N, Shan J, O'Brien S, Cortes J, et al. Outcome of Patients With Myelodysplastic Syndrome After Failure of Decitabine Therapy. Cancer (2010) 116(16):3830–4. doi: 10.1002/cncr.25247
60. Ball B, Komrokji RS, Adès L, Sekeres MA, DeZern AE, Pleyer L, et al. Evaluation of Induction Chemotherapies After Hypomethylating Agent Failure in Myelodysplastic Syndromes and Acute Myeloid Leukemia. Blood Adv (2018) 2(16):2063–71. doi: 10.1182/bloodadvances.2018015529
61. Faderl S, Garcia-Manero G, Estrov Z, Ravandi F, Borthakur G, Cortes JE, et al. Oral Clofarabine in the Treatment of Patients With Higher-Risk Myelodysplastic Syndrome. J Clin Oncol (2010) 28(16):2755–60. doi: 10.1200/JCO.2009.26.3509
62. Festuccia M, Baker K, Gooley TA, Sandmaier BM, Deeg HJ, Scott BL, et al. Hematopoietic Cell Transplantation in Myelodysplastic Syndromes After Treatment With Hypomethylating Agents. Biol Blood Marrow Transplant (2017) 23(9):1509–14. doi: 10.1016/j.bbmt.2017.05.034
63. Kim H, Lee J-H, Lee WS, Kim I, Moon JH, Choi CW, et al. Lenalidomide as a Second-Line Therapy After Failure of Hypomethylating Agents in Patients With Myelodysplastic Syndrome. Br J Hematol (2019) 186:e151–5. doi: 10.1111/bjh.15991
64. Sekeres MA, O'Keefe C, List AF, Paulic K, Afable M 2nd, Englehaupt R, et al. Demonstration of Additional Benefit in Adding Lenalidomide to Azacitidine in Patients With Higher-Risk Myelodysplastic Syndromes. Am J Hematol (2011) 86(1):102–3. doi: 10.1002/ajh.21891
65. Prebet T, Braun T, Beyne-Rauzy O, Dreyfus F, Stammatoullas A, Wattel E, et al. Combination of Vorinostat and Low Dose Cytarabine for Patients With Azacitidine-Refractory Relapsed High-Risk Myelodysplastic Syndromes. Leuk Res (2014) 38(1):29–33. doi: 10.1016/j.leukres.2013.07.023
66. Sébert M, Renneville A, Bally C, Peterlin P, Beyne-Rauzy O, Legros L, et al. A Phase II Study of Guadecitabine in Higher-Risk Myelodysplastic Syndrome and Low Blast Count Acute Myeloid Leukemia After Azacitidine Failure. Haematologica (2019) 104(8):1565–71. doi: 10.3324/haematol.2018.207118
67. Garcia-Manero G, Jabbour EJ, Konopleva MY, Daver NG, Borthakur G, DiNardo CD, et al. A Clinical Study of Tomaralimab (OPN-305), A Toll-Like Receptor 2 (TLR-2) Antibody, in Heavily Pre-Treated Transfusion Dependent Patients With Lower Risk Myelodysplastic Syndromes (MDS) That Have Received and Failed on Prior Hypomethylating Agent (HMA) Therapy. Blood (2018) 132(Suppl 1):798. doi: 10.1182/blood-2018-99-119805
68. Garcia-Manero G, Fenaux P, Al-Kali A, Baer MR, Sekeres MA, Roboz GJ, et al. Rigosertib Versus Best Supportive Care for Patients With High-Risk Myelodysplastic Syndromes After Failure of Hypomethylating Drugs (ONTIME): A Randomised, Controlled, Phase 3 Trial. Lancet Oncol (2016) 17(4):496–508. doi: 10.1016/S1470-2045(16)00009-7
69. Navada SC, Garcia-Manero G, Atallah EL, Rajeh MN, Shammo JM, Griffiths EA, et al. Phase 2 Expansion Study of Oral Rigosertib Combined With Azacitidine (AZA) in Patients With Higher-Risk (HR) Myelodysplastic Syndromes (MDS): Efficacy and Safety Results in HMA Treatment Naïve & Relapsed/Refractory (Ref) Patients. Blood (2018) 132(Suppl 1):230. doi: 10.1182/blood-2019-131676
70. Garcia-Manero G, Sasaki K, Montalban-Bravo G, Daver NG, Jabbour EJ, Alvarado Y, et al. A Phase II Study of Nivolumab or Ipilimumab With or Without Azacitidine for Patients With Myelodysplastic Syndrome (MDS). Blood (2018) 132(Supplement 1):465. doi: 10.1182/blood-2018-99-119424
71. Manero GG, Bravo GM, Sasaki K, Daver NG, Jabbour EJ, Alvarado Y, et al. Double Immune Checkpoint Inhibitor Blockade With Nivolumab and Ipilimumab with or Without Azacitidine in Patients With Myelodysplastic Syndrome (MDS). Blood (2018) 132(suppl 1):1831. doi: 10.1182/blood-2018-99-118948
72. Komrokji R, Garcia-Manero G, Ades L, Prebet T, Steensma DP, Jurcic JG, et al. Sotatercept With Long-Term Extension for the Treatment of Anemia in Patients With Lower-Risk Myelodysplastic Syndromes: A Phase 2, Dose-Ranging Trial. Lancet Haematol (2018) 5(2):e63–72. doi: 10.1016/S2352-3026(18)30002-4
73. Ziemba A, Hayes E, Freeman BB 3rd, Ye T, Pizzorno G. Development of an Oral Form of Azacytidine: 2’3’5’triacetyl-5-Azacytidine. Chemother Res Pract (2011) 2011:965826. doi: 10.1155/2011/965826
74. Garcia-Manero G, Griffiths EA, Steensma DP, Roboz GJ, Wells R, McCloskey J, et al. Oral Cedazuridine/Decitabine for MDS and CMML: A Phase 2 Pharmacokinetic/Pharmacodynamic Randomized Crossover Study. Blood (2020) 136(6):674–83. doi: 10.1182/blood.2019004143
75. Antar AI, Otrock ZK, Jabbour E, Mohty M, Bazarbachi A. FLT3 Inhibitors in Acute Myeloid Leukemia: Ten Frequently Asked Questions. Leukemia (2020) 34:682–96. doi: 10.1038/s41375-019-0694-3
76. Sallman DA, Donnellan WB, Asch AS, Lee DJ, Malki MA, Marcucci G, et al. The First-in-Class Anti-CD47 Antibody Hu5F9-G4 Is Active and Well Tolerated Alone or With Azacitidine in AML and MDS Patients: Initial Phase 1b Results. JCO (2020) 37(15):7009. doi: 10.1200/JCO.2019.37.15_suppl.7009
77. Jitschin R, Saul D, Braun M, Tohumeken S, Völkl S, Kischel R, et al. CD33/CD3-Bispecific T-Cell Engaging (BiTE®) Antibody Construct Targets Monocytic AML Myeloid-Derived Suppressor Cells. J Immunother Cancer (2018) 6:116. doi: 10.1186/s40425-018-0432-9
78. Nawrocki ST, Han Y, Visconte V, Przychodzen B, Espitia CM, Phillips J, et al. The Novel Autophagy Inhibitor ROC-325 Augments the Antileukemic Activity of Azacitidine. Leukemia (2019) 33:2971–4. doi: 10.1038/s41375-019-0529-2
79. Pastor-Anglada M, Pérez-Torras S. Nucleoside Transporter Proteins as Biomarkers of Drug Responsiveness and Drug Targets. Front Pharmacol (2015) 6:13. doi: 10.3389/fphar.2015.00013
80. Damaraju VL, Mowles D, Yao S, Ng A, Young JD, Cass CE, et al. Role of Human Nucleoside Transporters in the Uptake and Cytotoxicity of Azacitidine and Decitabine. Nucleosides Nucleotides Nucleic Acids (2012) 31(3):236–55. doi: 10.1080/15257770.2011.652330
81. Ueda K, Hosokawa M, Iwakawa S. Cellular Uptake of Decitabine by Equilibrative Nucleoside Transporters in HCT116 Cells. Biol Pharm Bull (2015) 38(8):1113–9. doi: 10.1248/bpb.b14-00622
82. Hummel-Eisenbeiss J, Hascher A, Hals PA, Sandvold ML, Müller-Tidow C, Lyko F, et al. The Role of Human Equilibrative Nucleoside Transporter 1 on the Cellular Transport of the DNA Methyltransferase Inhibitors 5-Azacytidine and CP-4200 in Human Leukemia Cells. Mol Pharmacol (2013) 84(3):438–50. doi: 10.1124/mol.113.086801
83. Wu L, Shi W, Li X, Chang C, Xu F, He Q, et al. High Expression of the Human Equilibrative Nucleoside Transporter 1 Gene Predicts a Good Response to Decitabine in Patients With Myelodysplastic Syndrome. Transl Med (2016) 14:66. doi: 10.1186/s12967-016-0817-9
84. Qin T, Castoro R, El Ahdab S, Jelinek J, Wang X, Si J, et al. Mechanisms of Resistance to Decitabine in the Myelodysplastic Syndrome. PloS One (2011) 6(8):e23372. doi: 10.1371/journal.pone.0023372
85. Briot T, Roger E, Lautram N, Verger A, Clavreul A, Lagarce F. Development and In Vitro Evaluations of New Decitabine Nanocarriers for the Treatment of Acute Myeloid Leukemia. Int J Nanomedicine (2017) 12:8427–42. doi: 10.2147/IJN.S147659
86. Gu X, Tohme R, Tomlinson B, Sakre N, Hasipek M, Durkin L, et al. Decitabine- and 5-Azacytidine Resistance Emerges From Adaptive Responses of the Pyrimidine Metabolism Network. Leukemia (2021) 35(4):1023–36. doi: 10.1038/s41375-020-1003-x
87. Qin T, Jelinek J, Si J, Shu J, Issa JP. Mechanisms of Resistance to 5-Aza-2'-Deoxycytidine in Human Cancer Cell Lines. Blood (2009) 113(3):659–67. doi: 10.1182/blood-2008-02-140038
88. Oellerich T, Schneider C, Thomas D, Knecht KM, Buzovetsky O, Kaderali L, et al. Selective Inactivation of Hypomethylating Agents by SAMHD1 Provides a Rationale for Therapeutic Stratification in AML. Nat Commun (2019) 10(1):3475. doi: 10.1038/s41467-019-11413-4
89. Unnikrishnan A, Vo ANQ, Pickford R, Raftery MJ, Nunez AC, Verma A, et al. AZA-MS: A Novel Multiparameter Mass Spectrometry Method to Determine the Intracellular Dynamics of Azacitidine Therapy In Vivo. Leukemia (2018) 32:900–10. doi: 10.1038/leu.2017.340
90. Hollenbach PW, Nguyen AN, Brady H, Williams M, Ning Y, Richard N, et al. A Comparison of Azacitidine and Decitabine Activities in Acute Myeloid Leukemia Cell Lines. PloS One (2010) 5(2):e9001. doi: 10.1371/journal.pone.0009001
91. Lindblad KE, Goswami M, Hourigan CS, Oetjen KA. Immunological Effects of Hypomethylating Agents. Expert Rev Hematol (2017) 10(8):745–52. doi: 10.1080/17474086.2017.1346470
92. Wang LX, Mei ZY, Zhou JH, Yao YS, Li YH, Xu YH, et al. Low Dose Decitabine Treatment Induces CD80 Expression in Cancer Cells and Stimulates Tumor Specific Cytotoxic T Lymphocyte Responses. PloS One (2013) 8(5):e62924. doi: 10.1371/journal.pone.0062924
93. Li X, Zhang Y, Chen M, Mei Q, Liu Y, Feng K, et al. Increased IFN-Gamma Positive T Cells Are Responsible for the Clinical Responses of Low-Dose DNA Demethylating Agent Decitabine Antitumor Therapy. Clin Cancer Res (2017) 23(20):6031–43. doi: 10.1158/1078-0432.CCR-17-1201
94. Zhao C, Jia B, Wang M, Schell TD, Claxton DF, Ehmann WC, et al. Multi-Dimensional Analysis Identifies an Immune Signature Predicting Response to Decitabine Treatment in Elderly Patients With AML. Br J Haematol (2020) 188(5):674–84. doi: 10.1111/bjh.16228
95. Niu C, Li M, Zhu S, Chen Y, Zhou L, Xu D, et al. Decitabine Inhibits Gamma Delta T Cell Cytotoxicity by Promoting Kir2Dl2/3 Expression. Front Immunol (2018) 9:617. doi: 10.3389/fimmu.2018.00617
96. Yang H, Bueso-Ramos C, DiNardo C, Estecio MR, Davanlou M, Geng QR, et al. Expression of PD-L1, PD-L2, PD-1 and CTLA4 in Myelodysplastic Syndromes Is Enhanced by Treatment With Hypomethylating Agents. Leukemia (2014) 28(6):1280–8. doi: 10.1038/leu.2013.355
97. Wherry EJ, Kurachi M. Molecular and Cellular Insights Into T Cell Exhaustion. Nat Rev Immunol (2015) 15(8):486–99. doi: 10.1038/nri3862
98. Daver N, Basu S, Garcia-Manero G, Cortes JE, Ravandi F, Jabbour EJ, et al. Phase IB/II Study of Nivolumab in Combination With Azacytidine in Patients With Relapsed Acute Myeloid Leukemia. Blood (2016) 128(22):763. doi: 10.1182/blood.V128.22.763.763
99. Roulois D, Loo Yau H, Singhania R, Wang Y, Danesh A, Shen SY, et al. DNA-Demethylating Agents Target Colorectal Cancer Cells by Inducing Viral Mimicry by Endogenous Transcripts. Cell (2015) 162(5):961–73. doi: 10.1016/j.cell.2015.07.056
100. Nahas MR, Stroopinsky D, Rosenblatt J, Cole L, Pyzer AR, Anastasiadou E, et al. Hypomethylating Agent Alters the Immune Microenvironment in Acute Myeloid Leukaemia (AML) and Enhances the Immunogenicity of a Dendritic Cell/AML Vaccine. Br J Haematol (2019) 185(4):679–90. doi: 10.1111/bjh.15818
101. Sẗbig T, Badbaran A, Luetkens T, Hildebrandt Y, Atanackovic D, Binder TMC, et al. 5-Azacytidine Promotes an Inhibitory T-Cell Phenotype and Impairs Immune Mediated Antileukemic Activity. Mediator Inflamm (2014) 2014:1–12. doi: 10.1155/2014/418292
102. Goodyear OC, Dennis M, Jilani NY, Loke J, Siddique S, Ryan G, et al. Azacitidine Augments Expansion of Regulatory T Cells After Allogeneic Stem Cell Transplantation in Patients With Acute Myeloid Leukemia (AML). Blood (2012) 119(14):3361–9. doi: 10.1182/blood-2011-09-377044
103. Vladimirovna IL, Sosunova E, Nikolaev A, Nenasheva T. Mesenchymal Stem Cells and Myeloid Derived Suppressor Cells: Common Traits in Immune Regulation. J Immunol Res (2016) 2016:7121580. doi: 10.1155/2016/7121580
104. Zhou J, Shen Q, Lin H, Hu L, Li G, Zhang X. Decitabine Shows Potent Anti-Myeloma Activity by Depleting Monocytic Myeloid-Derived Suppressor Cells in the Myeloma Microenvironment. J Cancer Res Clin Oncol (2019) 145(2):329–36. doi: 10.1007/s00432-018-2790-6
105. Poon Z, Dighe N, Venkatesan SS, Cheung AMS, Fan X, Bari S, et al. Bone Marrow MSCs in MDS: Contribution Towards Dysfunctional Hematopoiesis and Potential Targets for Disease Response to Hypomethylating Therapy. Leukemia (2019) 33(6):1487–500. doi: 10.1038/s41375-018-0310-y
106. Müller-Thomas C, Heider M, Piontek G, Schlensog M, Bassermann F, Kirchner T, et al. Prognostic Value of Indoleamine 2,3 Dioxygenase in Patients With Higher Risk Myelodysplastic Syndromes Treated With Azacytidine. Br J Haematol (2020) 190(3):361–70. doi: 10.1111/bjh.16652
107. Leisch M, Greil R, Pleyer L. IDO in MDS/AML Disease Progression and its Role in Resistance to Azacitidine: A Potential New Drug Target. Br J Haematol (2020) 190(3):314–7. doi: 10.1111/bjh.16710
108. Liu S, Ginestier C, Ou SJ, Clouthier SG, Patel SH, Monville F, et al. Breast Cancer Stem Cells Are Regulated by Mesenchymal Stem Cells Through Cytokine Networks. Cancer Res (2011) 71(2):614–24. doi: 10.1158/0008-5472.CAN-10-0538
109. Wang X, Huang S, Chen JL. Understanding of Leukemic Stem Cells and Their Clinical Implications. Mol Cancer (2017) 16(1):2. doi: 10.1186/s12943-016-0574-7
110. Craddock C, Quek L, Goardon N, Freeman S, Siddique S, Raghavan M, et al. Azacitidine Fails to Eradicate Leukemic Stem/Progenitor Cell Populations in Patients With Acute Myeloid Leukemia and Myelodysplasia. Leukemia (2013) 27(5):1028–36. doi: 10.1038/leu.2012.312
111. Dimitriou M, Woll PS, Mortera-Blanco T, Karimi M, Wedge DC, Doolittle H, et al. Perturbed Hematopoietic Stem and Progenitor Cell Hierarchy in Myelodysplastic Syndromes Patients With Monosomy 7 as the Sole Cytogenetic Abnormality. Oncotarget (2016) 7(45):72685–98. doi: 10.18632/oncotarget.12234
112. Unnikrishnan A, Papaemmanuil E, Beck D, Deshpande NP, Verma A, Kumari A, et al. Integrative Genomics Identifies the Molecular Basis of Resistance to Azacitidine Therapy in Myelodysplastic Syndromes. Cell Rep (2017) 20(3):572–85. doi: 10.1016/j.celrep.2017.06.067
113. Pollyea DA, Gutman JA, Gore L, Smith CA, Jordan CT. Targeting Acute Myeloid Leukemia Stem Cells: A Review and Principles for the Development of Clinical Trials. Haematologica (2014) 99(8):1277–84. doi: 10.3324/haematol.2013.085209
114. Yanagisawa B, Ghiaur G, Smith BD, Jones RJ. Translating Leukemia Stem Cells Into the Clinic: Harmonizing the Heterogeneity. Exp Hematol (2016) 44(12):1130–7. doi: 10.1016/j.exphem.2016.08.010
115. Brunner A, Borate U, Esteve J, Porkka K, Knapper S, Vey N, et al. Anti-TIM3 Antibody MBG-453 in Combination With Hypomethylating Agents in Patients With High-Risk Myelodysplastic Syndrome and Acute Myeloid Leukemia: A Phase I Study. EHA Library (2020) 295005:S185. doi: 10.1016/S2152-2650(20)30728-X
116. Jabbour E, Short NJ, Montalban-Bravo G, Huang X, Bueso-Ramos C, Qiao W, et al. Randomized Phase 2 Study of Low-Dose Decitabine vs Low-Dose Azacitidine in Lower-Risk MDS and MDS/MPN. Blood (2017) 130(13):1514–22. doi: 10.1182/blood-2017-06-788497
117. Roboz GJ, Ritchie EK, Dault Y, Lam L, Marshall DC, Cruz NM, et al. Phase I Trial of Plerixafor Combined With Decitabine in Newly Diagnosed Older Patients With Acute Myeloid Leukemia. Haematologica (2018) 103(8):1308–16. doi: 10.3324/haematol.2017.183418
118. Pera B, Tang T, Marullo R, Yang SN, Ahn H, Patel J, et al. Combinatorial Epigenetic Therapy in Diffuse Large B Cell Lymphoma Pre-Clinical Models and Patients. Clin Epigenet (2016) 8:79. doi: 10.1186/s13148-016-0245-y
119. Linnekamp JF, Butter R, Spijker R, Medema JP, van Laarhoven HWM. Clinical and Biological Effects of Demethylating Agents on Solid Tumours– A Systematic Review. Cancer Treat Rev (2017) 54:10–23. doi: 10.1016/j.ctrv.2017.01.004
120. Pusic I, Choi J, Fiala MA, Gao F, Holt M, Cashen AF, et al. Maintenance Therapy With Decitabine After Allogeneic Stem Cell Transplantation for Acute Myelogenous Leukemia and Myelodysplastic Syndrome. Biol Blood Marrow Transplant (2015) 21(10):1761–9. doi: 10.1016/j.bbmt.2015.05.026
121. Wang QY, Li Y, Liang ZY, Yin Y, Liu W, Wang Q, et al. Decitabine-Containing Conditioning Regimen for Allogeneic Hematopoietic Stem Cell Transplantation in Patients With Intermediate and High-Risk Myelodysplastic Syndrome/Acute Myeloid Leukemia: Potential Decrease in the Incidence of Acute Graft Versus Host Disease. Cancer Manag Res (2019) 11:10195–203. doi: 10.2147/CMAR.S229768
122. Murakami K, Ueno H, Okabe T, Kagoo T, Boku S, Yano T, et al. Bridging-To-Transplant With Azacitidine for Myelodysplastic Syndrome and Acute Myeloid Leukemia, Reduces the Incidence of Acute Graft-Versus-Host Disease. Hematol Rep (2017) 9(2):7114. doi: 10.4081/hr.2017.7114
123. Butrym A, Rybka J, Baczyńska D, Poręba R, Mazur G, Kuliczkowski K. Expression of microRNA−181 Determines Response to Treatment With Azacitidine and Predicts Survival in Elderly Patients With Acute Myeloid Leukaemia. Oncol Lett (2016) 12(4):2296–300. doi: 10.3892/ol.2016.4970
124. Butrym A, Rybka J, Baczyńska D, Poręba R, Kuliczkowski K, Mazur G. Clinical Response to Azacitidine Therapy Depends on microRNA-29c (miR-29c) Expression in Older Acute Myeloid Leukemia (AML) Patients. Oncotarget (2016) 7(21):30250–7. doi: 10.18632/oncotarget.7172
125. Castoro RJ, Dekmezian M, Saraf AJ, Watanabe Y, Chung W, Adhab SE, et al. MicroRNA 124 and Its Role in Response to Epigenetic Therapy in Patients With Acute Mylogenous Leukemia and Myelodysplastic Syndrome. Blood (2008) 112(11):598. doi: 10.1182/blood.V112.11.598.598
126. Cheng JX, Chen L, Li Y, Cloe A, Yue M, Wei J, et al. RNA Cytosine Methylation and Methyltransferases Mediate Chromatin Organization and 5-Azacytidine Response and Resistance in Leukaemia. Nat Commun (2018) 9(1):1163. doi: 10.1038/s41467-018-03513-4
127. Schaefer M, Hagemann S, Hanna K, Lyko F. Azacytidine Inhibits RNA Methylation at DNMT2 Target Sites in Human Cancer Cell Lines. Cancer Res (2009) 69(20):8127–32. doi: 10.1158/0008-5472.CAN-09-0458
128. Lavelle D, DeSimone J, Hankewych M, Kousnetzova T, Chen YH. Decitabine Induces Cell Cycle Arrest at the G1 Phase via P21(WAF1) and the G2/M Phase via the P38 MAP Kinase Pathway. Leuk Res (2003) 27(11):999–1007. doi: 10.1016/s0145-2126(03)00068-7
129. Simone B, Lionel A, Nicolas T, Lorenzo G, Stephane B, Pierre F, et al. The Capacity of the Hypomethylating Agents Azacytidine and Decitabine to Induce Apoptosis and Cell Cycle Arrest Depends on the Activation of the DNA Damage Response Pathway. Blood (2008) 112(11):1655. doi: 10.1182/blood.V112.11.1655.1655
130. Montesano A, Luzi L, Senesi P, Terruzzi I. Modulation of Cell Cycle Progression by 5-Azacytidine Is Associated With Early Myogenesis Induction in Murine Myoblasts. Int J Biol Sci (2013) 9(4):391–402. doi: 10.7150/ijbs.4729
131. Ma D, Fang Q, Wang P, Gao R, Sun J, Li Y, et al. Downregulation of HO-1 Promoted Apoptosis Induced by Decitabine via Increasing P15ink4b Promoter Demethylation in Myelodysplastic Syndrome. Gene Ther (2015) 22(4):287–96. doi: 10.1038/gt.2015.1
132. Alcazar O, Achberger S, Aldrich W, Hu Z, Negrotto S, Saunthararajah Y, et al. Epigenetic Regulation by Decitabine of Melanoma Differentiation In Vitro and In Vivo. Int J Cancer (2012) 131(1):18–29. doi: 10.1002/ijc.26320
Keywords: decitabine, azacitidine, hypomethylating agents, resistance, acute myeloid leukemia, myelodysplastic syndrome
Citation: Zhao G, Wang Q, Li S and Wang X (2021) Resistance to Hypomethylating Agents in Myelodysplastic Syndrome and Acute Myeloid Leukemia From Clinical Data and Molecular Mechanism. Front. Oncol. 11:706030. doi: 10.3389/fonc.2021.706030
Received: 06 May 2021; Accepted: 08 September 2021;
Published: 28 September 2021.
Edited by:
Jean El Cheikh, American University of Beirut Medical Center, LebanonReviewed by:
Ciprian Tomuleasa, Iuliu Hațieganu University of Medicine and Pharmacy, RomaniaAmit Verma, Albert Einstein College of Medicine, United States
Copyright © 2021 Zhao, Wang, Li and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaoqin Wang, d2FuZ3hpYW9xaW5Ac2htdS5lZHUuY24=