 Xanthene Miles
Xanthene Miles Charlot Vandevoorde
Charlot Vandevoorde Alistair Hunter
Alistair Hunter Julie Bolcaen
Julie Bolcaen
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 08 July 2021
Sec. Radiation Oncology
Volume 11 - 2021 | https://doi.org/10.3389/fonc.2021.703442
This article is part of the Research Topic Ionizing Radiation and Human Health: A Multifaceted Relationship View all 28 articles
Inhibition of the MDM2/X-p53 interaction is recognized as a potential anti-cancer strategy, including the treatment of glioblastoma (GB). In response to cellular stressors, such as DNA damage, the tumor suppression protein p53 is activated and responds by mediating cellular damage through DNA repair, cell cycle arrest and apoptosis. Hence, p53 activation plays a central role in cell survival and the effectiveness of cancer therapies. Alterations and reduced activity of p53 occur in 25-30% of primary GB tumors, but this number increases drastically to 60-70% in secondary GB. As a result, reactivating p53 is suggested as a treatment strategy, either by using targeted molecules to convert the mutant p53 back to its wild type form or by using MDM2 and MDMX (also known as MDM4) inhibitors. MDM2 down regulates p53 activity via ubiquitin-dependent degradation and is amplified or overexpressed in 14% of GB cases. Thus, suppression of MDM2 offers an opportunity for urgently needed new therapeutic interventions for GB. Numerous small molecule MDM2 inhibitors are currently undergoing clinical evaluation, either as monotherapy or in combination with chemotherapy and/or other targeted agents. In addition, considering the major role of both p53 and MDM2 in the downstream signaling response to radiation-induced DNA damage, the combination of MDM2 inhibitors with radiation may offer a valuable therapeutic radiosensitizing approach for GB therapy. This review covers the role of MDM2/X in cancer and more specifically in GB, followed by the rationale for the potential radiosensitizing effect of MDM2 inhibition. Finally, the current status of MDM2/X inhibition and p53 activation for the treatment of GB is given.
The classification of gliomas is traditionally based on histologic type and malignancy grade. It varies from low grade glioma, classified as benign with a high curative chance, to high grade glioma which is typically associated with rapid proliferation linked to disease evolution (grade I - IV). Since 2016, the World Health Organization (WHO) classification no longer relies solely on histological criteria but incorporated additional molecular biomarkers to improve diagnosis and prognosis of glioma patients. Especially the use of molecular techniques, such as arrays and next generation sequencing, play an integral role in the identification of mutations in gliomas (1, 2). Glioblastoma multiforme (GB) is classified as a grade IV, the highest grade in the WHO classification of brain tumors, and is the most common malignant central nervous system (CNS) tumor with a global incidence of 0.59–3.69 per 100 000 (3–5).
Despite numerous attempts over the past decade to find more effective treatments, the standard care for GB has remained essentially unchanged. This involves maximal safe surgical resection, external beam radiation therapy (EBRT) plus concomitant and adjuvant chemotherapy using the alkylating agent temozolomide (TMZ) - this is known as the Stupp protocol (6). Various avenues have been explored to improve GB therapy, such as targeting the immune system through gene therapy, viral vectors and targeted drug therapy to name a few (7, 8). Sadly, despite multiple clinical trials, median survival from diagnosis is still only 15-17 months (1, 6, 9–12). Treatment challenges often derive from the molecular and cellular heterogeneity inherent to these tumors. They include innate and acquired resistance with subpopulations of tumor cells harboring stem-like properties rendering them more resistant to therapy (13–15). Another major challenge in GB patients is tumor recurrence, which is unfortunately inevitable and results in a more aggressive and radioresistant secondary tumor. The standard of care for patients with recurrent GB is not well defined (1).
There has been an increased interest in the molecular pathogenesis of malignant tumors and this led to the development of monoclonal antibodies (mAbs) and small molecule (SM) inhibitors blocking critical pathways involved in tumor resistance and progression. These include the targeting of DNA repair pathways, cell cycle control enzymes/genes and their downstream pathways, as well as growth factor receptors (16, 17). Secondly, these targeted drugs can often function as radiosensitizers to enhance the cytotoxicity of subsequently administered radiation therapy (RT) while minimizing deleterious side effects towards surrounding normal tissues (18, 19).
In this review, the rationale for influencing the p53 and mouse double minute 2 (MDM2) pathway as a radiosensitizing and therapeutic strategy for GB will be covered. 84% of GB patients show a deregulation of the p53-MDM2 pathway (4, 20). MDM2 plays an imperative role in down regulating p53 activity via ubiquitin-dependent degradation and is amplified or overexpressed in 14% of GB cases. Hence, suppression of MDM2 through different approaches, offers an opportunity for urgently needed new therapeutic interventions for GB. In addition, the combination of MDM2 inhibitors with ionizing radiation (IR) may offer a valuable therapeutic radiosensitizing strategy by influencing the DNA damage response (21). Since the release of the structure of the MDM2–p53 interaction 25 years ago (22), numerous SM MDM2 inhibitors have been discovered and investigated, including SAR405838, HDM-201, NVP-CGM097, MK-8242, RG7112, RG7388, ALRN-6924 and AMG232 (23–31). Many of these inhibitors are currently being investigated in clinical trials as novel cancer treatments. The growing interest is reflected by the amount of reviews published in the last years (30–41). However, to date, only a limited number of MDM2 inhibitors have been tested for the treatment of GB or in combination with RT.
GB tumors have been identified as therapy resistant due to multiple molecular mechanisms including inadequate drug blood-brain barrier (BBB) passage, intra- and intertumoral heterogeneity, redundant signaling pathways resulting in rescue mechanisms, adaptive radioresistance and an immunosuppressive tumor micro-environment (TME) promoted by a chronic state of hypoxia (15, 42–44). Hypoxic niches limiting the formation of reactive oxygen species (ROS) and a hyperactivation of the DNA damage response machinery induced by glioma stem cells (GSC) contribute to glioma radioresistance (44, 45). In addition, a cross-talk between TME populations via shared pathways, such as STAT3, Wnt and Notch play a role (15, 46).
Alternative RT technologies to improve therapy effectiveness in GB, including dose escalation, a stereotactic radiosurgery boost, brachytherapy and boron neutron capture therapy, have failed to become incorporated in the routine management of newly diagnosed malignant glioma (47, 48). However, several technological advances can contribute to a reduction of RT induced acute and late normal tissue toxicity. Three major examples are intensity-modulated radiotherapy (IMRT), proton therapy (PT) and ultra-high dose rate (FLASH) RT, which are promising to reduce cognitive impairments that could negatively impact the quality of life of GB survivors (49, 50). Compared to photon-based therapies, dosimetric PT studies in gliomas have shown a dose reduction to nearby organs at risk (OARs) and a lower risk of developing RT-induced tumors, which could even further improve with advanced intensity modulated proton therapy (IMPT) (14, 51–53). However, this is of less importance in GB compared to low-grade gliomas, due to the low median survival of GB patients. A phase II trial which compares PT with IMRT in their ability to preserve brain function in patients with IDH mutant grade II/III glioma is currently running (NCT03180502) (54). In conjunction with that, the outcome of PT dose-escalation and randomized clinical trials of PT versus IMRT are also currently under investigation (NCT01854554, NCT04752280, NCT02179086, NCT03180502) (54, 55).
Compared to PT, the unique physical and biological properties of high linear energy transfer (LET) radiation, such as carbon ion radiotherapy (CIRT), are expected to overcome microenvironmental limitations present in GB, such as hypoxia, and confer an improved glioma and GSC killing ability (56–58). In GSC models, CIRT showed to overcome glioma radioresistance by eradicating stem cells, inducing anti-angiogenic effects and influencing the immune system (42, 59, 60). For the treatment of brain tumors, multiple clinical studies have suggested that CIRT is effective with a favorable toxicity profile, mainly through the delivery of a carbon ion boost following conventional RT or PT (61, 62). This led to the prospective CLEOPATRA Trial at Heidelberg Ion Therapy Center (HIT) and a Phase I/III clinical by the Shanghai Proton and Heavy Ion Center (NCT04536649) (42, 48, 61, 63). First results applying particle RT at a dose ≥60 gray-equivalents showed to be safe and potentially effective with an 18-month overall survival (OS) rate of 72.8% and progression free survival (PFS) rate of 59.8% (48). CIRT is also being investigated in recurrent GB, with results of the randomized phase I/II CINDERELLA trial pending (64). For recurrent high-grade glioma, the recent study of Eberle et al. deemed carbon ion reirradiation as safe and feasible (65).
In FLASH RT, the dose is delivered at ≥ 40 Gy/sec compared to dose rates of approximately 1-4 Gy/min in conventional EBRT (66). This technique provided encouraging results in an in vivo study using a murine GB model, but is currently still limited to superficial tumors using electron beams (67). New developments in FLASH proton and heavy ion beam therapy look promising and could pave the way to treat deeper seated tumors in a clinical context, such as GB (68, 69). The combination of FLASH with mini-beams, could even further increase the protection of healthy tissue and preserve anti-tumoral immunological reactions (70).
TP53 is markedly the most studied tumor-suppressor gene. It encodes the tumor suppressor protein p53 which, in light of its nature and action, has been defined as the “guardian of the genome”. It is a multifunctional transcription factor that can be activated through cellular stresses, such as hypoxia, DNA damage, or oncogene activation. Upon activation, p53 acts as a tumor suppressor and responds to cellular damage by mediating cell proliferation, arrest, DNA repair, metabolism, angiogenesis, senescence and apoptosis, as depicted in Figure 1 (20). The most critical downstream targets of p53 are the apoptotic proteins, as they are responsible for the activation of various cell death pathways (35). The activation of the latter plays a role in prohibiting the replication of damage-causing genetic lesions, as these could result in unconstrained cell growth and oncogenesis (71).
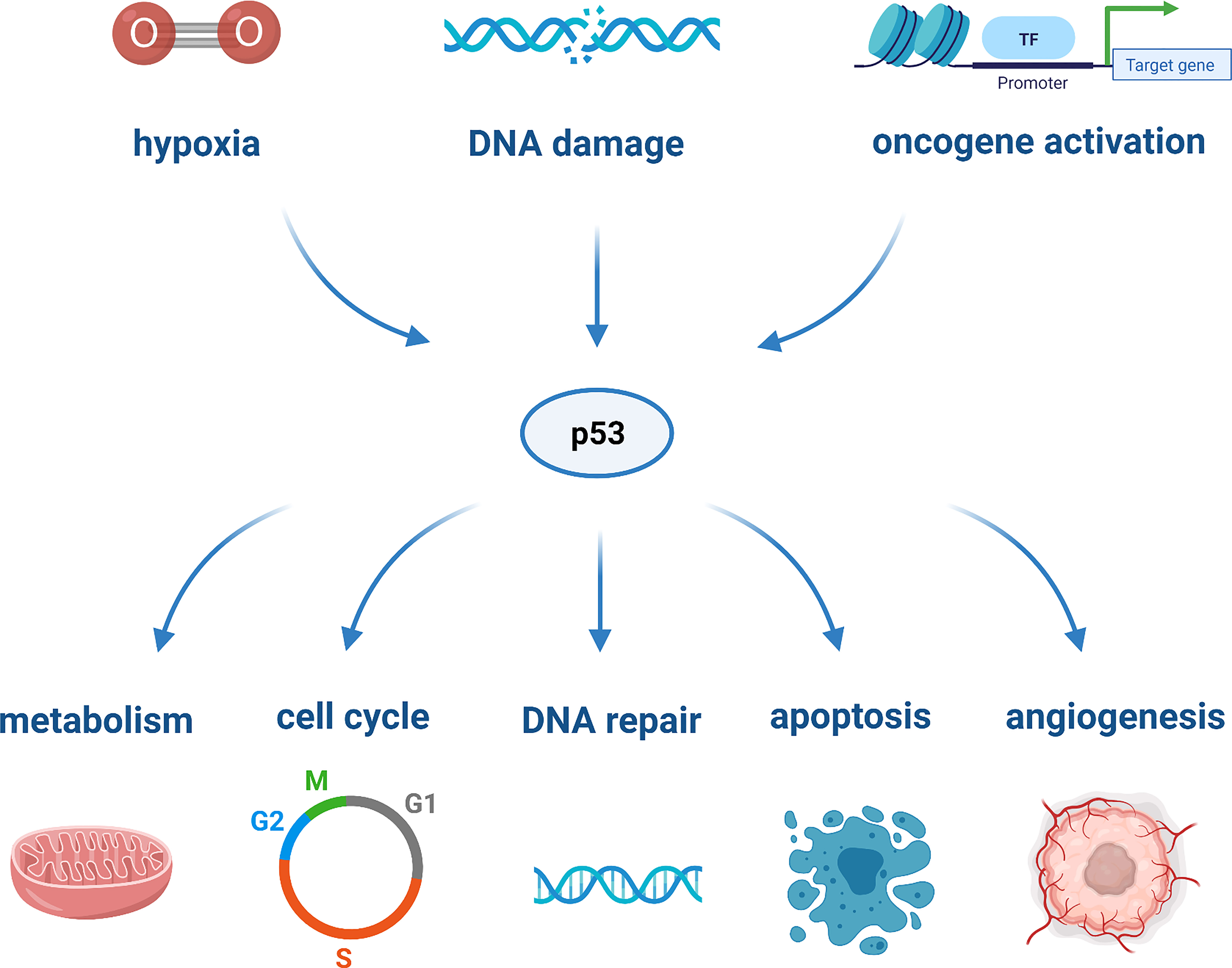
Figure 1 The various cellular processes regulated by p53 in response to cellular stressors.
In normal conditions and in the absence of cellular stress, cellular homeostasis is set to preserve low p53 levels. This level is regulated by MDM2, a E3 protein ligase which is responsible for p53 degradation through a ubiquitin-dependent pathway. When the amino-terminal domain of MDM2 binds to p53, the transcriptional activity of p53 is inhibited and the p53 protein complex is exported from the nucleus to the cytoplasm for degradation by cytoplasmic proteasomes. In this way, both the p53-mediated cell cycle arrest and the apoptosis functions of p53 are affected (Figure 2A) (72, 73). Hence, targeting the interaction between p53 and the E3 ligase MDM2 represents an attractive anti-cancer approach with the condition that the tumor is wild-type (wt) TP53 or functional TP53 is present (40). The p53-MDM2 pathway is also referred to as the p53-ARF-MDM2 pathway, since ARF (alternative reading frame), is a tumor suppressor that interacts with MDM2. This interaction prevents MDM2 shuttling between the nucleus and cytoplasm and thereby circumvents p53 degradation (76).
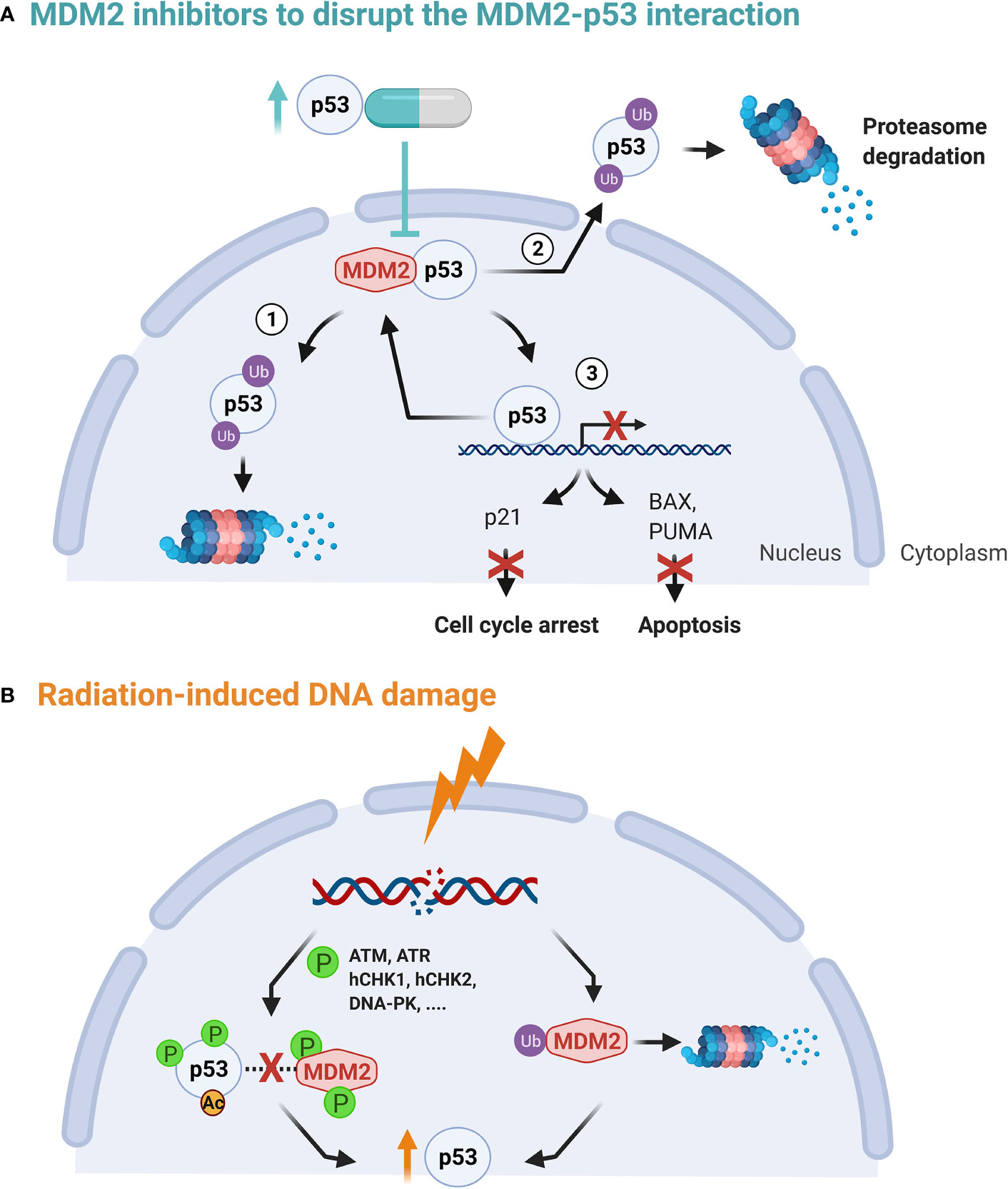
Figure 2 (A) The p53-MDM2 autoregulatory feedback loop. p53 stimulates MDM2 expression while MDM2, in turn, inhibits p53 activity by stimulating its degradation in the nucleus and the cytoplasm (1, 2), promoting its nuclear export (2) and blocking its transcriptional activity (3) (72, 73). (B) Upon DNA damage, both MDM2 auto-degradation and phosphorylation of p53 is activated. This in turn disrupts the MDM2 binding, increasing transcription activation and stability of the p53 protein. In addition, ATM phosphorylation of MDM2 is critical for MDM2 destabilization, leading to less p53 ubiquitination (74, 75).
Secondly, upon sensing DNA damage, ataxia telangiectasia mutated (ATM) becomes activated and induces phosphorylation of p53 and MDM2 directly or indirectly via checkpoint kinases, such as hCHK1 and hCHK2. The latter prevents their interaction and guarantees the stabilization of p53, see Figure 2B (72, 77). DNA damage has also shown to induce MDM2 auto-degradation (78). However, high levels of p53 in their turn activate transcription of downstream targets, including MDM2. Hence, the above mechanisms form a autoregulatory loop to control the amount of p53 and MDM2 proteins (74, 79, 80).
The MDM2 homologue protein MDMX (also known as MDM4) shares some similarity with MDM2 in the p53 binding domains, but they are not identical and MDMX exhibits no E3 ligase activity. MDMX is able to inactivate p53 in two ways: by binding to the N-terminus of p53 directly or by heterodimerization with MDM2 stimulating its ubiquitination function. This is called the p53-MDM2/MDMX loop, in which both MDM2 and MDMX act as inhibitors of p53’s tumor suppressor function (81, 82). At variance to MDM2, MDMX appears not to be transcriptionally regulated by p53, as explained by Marine et al. (83).
Alterations of the p53 pathway are common in multiple cancer types, including GB. It is clear that the most common cause for TP53 deregulation is due to MDM2 and MDMX amplification as well as missense mutations in the TP53 gene, which results in the demise of its role as a tumor suppressor. This area has been extensively reviewed by Zhang et al. (20). The complicated genetic profile of GB was confirmed by genomic profiling and the Cancer Genome Atlas project, which revealed a set of three core signaling pathways that are commonly altered in GB: the p53 pathway, the receptor tyrosine kinase/Ras/phosphoinositide 3-kinase (PI3K) signaling pathway, and the retinoblastoma (Rb) pathway (74, 77, 84, 85). Alterations of the p53 pathway play a key role in GB development, cell invasion, migration, proliferation, apoptosis, cancer cell stemness and resistance to TMZ treatment (86–88). Interestingly, genomic characterization of human GB genes and its core pathways showed that p53 signaling was altered in 87% of GB cases (84). More specifically, 84% of GB patients and 94% of GB cell lines showed a deregulation of the p53-ARF-MDM2 pathway (4, 20). In primary GBs, TP53 is relatively infrequently mutated (25-30%), while in secondary GB, alterations of p53 are observed in 60-70% of cases (89). The prevalence of TP53 mutations also depends on the GB molecular subtype: proneural (54%), mesenchymal (32%), neural (21%) and classical (0%) respectively (20, 90). However, even in p53 wt GB, p53 availability is frequently reduced because of interactions with overexpressed MDM2 proteins (86–88). An amplification and overexpression of MDM2 gene is observed in 14% of GB cases (84). Concerning MDMX, a 5- to 25-fold amplification in 2.4% of 208 glioma cases was assessed by Riemenschneider et al. and interestingly, all had a retained p53 wt status. Of these, none showed MDM2 amplification (91). Another study performed qPCR on 86 GB samples and found an amplification of the MDMX gene in 27% of these samples. They also observed a 28.6% MDMX amplification of low-grade astrocytic tumors and deduced that this could signify an early event in carcinogenesis (92, 93). Hence, reactivating p53 activity through inhibition of MDM2/X offers a tenable opportunity for therapeutic intervention in GB.
As previously mentioned, TP53 function can also be suppressed in p53 wt tumors via MDM2 overexpression, limiting the p53 protein to perform its tumor suppressor role and thereby promoting cancer progression (94, 95). As such, the re-activation of the p53 pathway is regarded as a plausible anti-cancer strategy and has the potential to increase the radiosensitivity of cancer cells. The main p53-based targeted therapies involve either the use of targeted molecules to convert the mutant (mut) p53 back to its wt form or MDM2 inhibitors which allow tumors with a p53 wt form but with MDM2 amplification to consequently restore p53 functioning (35, 71, 96).
One of the first attempts at understanding the mechanisms behind p53 reactivation entailed the phosphorylation and acetylation of its complex. Studies revealed that although the latter plays a role in weakening the p53-MDM2 interaction, it is not critical for p53 stabilization upon DNA damage (72). Consequently, the MDM2 protein itself became the principal target. Since the structure of the MDM2-p53 interaction has been revealed, multiple SM MDM2 inhibitors have been developed against the p53-binding pockets of MDM2 (95). These include nutlins, spiro-oxindole derivatives and piperidinone-containing compounds, such as MI-77301/SAR405838, APG-115, MK-8242, RG7112, RG7388, DS-3032b, and AMG232. An overview of different categories of MDM2 inhibitors, their design and the current status in the clinic has been reviewed elsewhere (31, 32, 34, 41, 74). Peptides have also been studied as potent inhibitors of the p53-MDM2 interaction and a number of these induced p53 mediated cell cycle arrest and apoptosis in solid cancers and hematological malignancies (88, 97–100). However, it is important to note that tumors harboring p53 mutations are not responsive in contrast to p53 wt tumors. Furthermore, sensitivity to MDM2 targeted therapy increases when p53 wt tumors also show MDM2 amplification (29, 98, 99). Clinical trials on MDM2 inhibitors are ongoing in acute myeloid leukemia (AML) (NCT02319369, NCT03634228), soft tissue sarcoma (NCT03217266), malignant salivary gland carcinoma (NCT03781986), pediatric cancers (NCT03654716) and small cell lung cancer (NCT04022876) (51, 52).
Additionally, MDMX antagonists have shown to inhibit the MDMX-p53 interaction. As an example, Pellegrino et al. identified a peptide that mimics the MDMX C-terminus, and binds MDM2, thereby blocking the MDMX/MDM2 complex (101). Importantly, the amount of MDMX influences the sensitivity to MDM2 inhibitors and the susceptibility to MDMX targeting appears to be dependent on the levels of p53 and especially of MDM2 (102, 103). Hence, studies have shown that combination therapy using MDM2/MDMX inhibitors result in a more effective anti-tumor reaction by more actively inducing apoptosis and cancer cell cycle arrest (81, 101, 104). For more extensive reviews on targeting MDM2 and MDMX in cancer therapy, see (30, 37, 74, 102, 105–107).
Two tumor characteristics enable a selection of patients who could benefit from MDM2- and MDMX-based therapies aimed at reactivating p53 function: a p53 wt status and overexpression of MDM2, MDMX or both. In addition, through the understanding of the dysregulation and functioning of MDM2 and MDMX in GB cancers, diagnostic and prognostic methods could be improved for a more personalized approach (29).
The concept behind radiosensitizers is based on their ability to enhance the radiosensitivity of cancer cells, resulting in increased radiation-induced cell killing. This can be achieved by targeting specific radiation response mechanisms, such as DNA repair mechanisms, and in the case of MDM2/X inhibitors, the p53 transcription factor pathway (18, 82). The actions of p53 are critical in determining the effectiveness of IR and/or chemotherapeutic agents (79). The cellular effects induced by IR are mediated by the DNA damage response (DDR) pathway, which facilitates MDM2-p53 signaling via activated kinases, such as ATM (see Figure 2). In cancer cells with p53 wt genes, the level of both MDM2 and p53 expression is directly correlated to the amount of IR induced DNA damage. Radiosensitive tissues have shown prolonged p53 signaling after IR, while more resistant tissues show transient p53 activation (108). Within the two major pathways in DNA double-strand break (DSB) repair, p53 interacts with both non-homologous end-joining (NHEJ) proteins as well as with protein RAD51 which plays a major role in homologous recombination (HR), influencing their expression (82, 109).
The effectiveness of IR to treat cancer is hampered by MDM2 mediated p53 inhibition, causing a decrease in DNA damage cell cycle arrest and apoptosis (110). As a result, MDM2 overexpression has been correlated with a decreased therapeutic response and failure of p53 to induce p21BAX expression has been linked to radioresistance in GB cells (79, 111). Blocking of the negative regulators MDM2 and MDMX could be a promising strategy to improve RT outcomes of wt TP53 GB - see Figures 2A, B. Sustaining p53 using MDM2/X inhibitors has shown radiosensitizing effects pre-clinically in lung cancer, prostate cancer, adenocarcinoma and colon cancer (21, 82, 108, 110, 112, 113). Remarkably, glioma cells lacking p53 wt function seem to be susceptible to IR-induced apoptosis due to an increased caspase-8 activity, which may be triggered by ceramide (114, 115).
Some critical factors will have to be considered when MDM2/X inhibition is combined with IR. Firstly, the effects on non-cancerous (brain) tissue have been poorly researched. Different cells/tissues can show different levels of apoptotic response to IR and the restoration of p53 in non-cancerous tissues levels after non-lethal DNA damage should take place rapidly to avoid unnecessary cell death. MDM2 inhibitors could however promote cell cycle arrest in non-cancerous cells and tissues that surround the tumor, without affecting tumor cells in case the tumor is p53 mut. However, the toxicity to healthy tissues might be limited since MDM2 inhibitors, such as nutlins (MI-219), have shown to activate p53 in normal tissues with limited p53 accumulation in contrast to a robust accumulation of p53 in normal tissues induced by chemo/radiotherapy (94, 116). An optimal approach would be to influence the dynamics of p53 differently between tumor and normal tissues following genotoxic therapies (108). Secondly, MDM2 has been reported to have p53-independent functions, also influencing the cell cycle, and DNA repair, amongst others (117). Particularly the interaction between MDM2 and the DNA repair complex (Mre11/Rad50/NBS1 or MRN) at DNA damaged sites is important concerning the response to IR. Nbs1 has been identified as a p53-independent MDM2 binding protein. This interaction in turn reduces DNA damage signaling levels and causes significant delays in DNA break repair, which might be an important side effect to take into consideration in the normal tissue response (118, 119).
For the aforementioned reasons, the synergy between MDM2 inhibitors combined with IR exposure may offer a more effective cancer treatment strategy, but more research is needed to reveal the exact mechanism of action and possible normal tissue toxicities (72). Two aspects must be considered: 1) MDM2 inhibitors may not be effective in GB tumors with inactivation of p53, 2) MDM2 inhibition combined with IR may lead to the radiosensitization of normal tissues (74, 116). Therefore, the targeted delivery of MDM2 inhibitors is crucial to induce targeted apoptosis of cancer cells and limit toxicity in normal tissues.
Different apoptotic signaling mechanisms and p53 dependency have been suggested between different radiation qualities (120–124). For increasing LET a tendency towards an increased apoptotic response has been observed (121, 125, 126). In normal human fibroblasts, the induction of TP53 and CDKN1A was dependent on the dose and LET (123). Also, p53 was slightly induced by both proton and X-ray irradiation, while a significant increase in protein expression of a downstream regulator of p53, CDKN1A, was seen after low-energy proton irradiation (127). A greater TP53 protein accumulation was observed after carbon ion exposure, compared to that of iso-doses of X-rays (123). In GB cell lines, X-rays, CIRT or alpha-particle IR all induced p53-dependent p21 accumulation (128).
Compared with photon radiation, PT has shown to induce more robust DNA damage and reduced cell cycle recovery from G2 arrest, leading to apoptosis and cytotoxicity (127, 129, 130). In addition, the mechanism of cell death induced by high LET CIRT is significantly different when compared to low LET radiation. This includes a greater ability of inducing the ceramide pathway and more complex DNA DSB damage resulting in increased levels of autophagy and apoptosis (131–133). High LET radiation phosphorylated p53 at serine 37, which is involved in cell death, more extensively compared to low LET irradiation (134). Different amounts of ROS induced by different radiation qualities will also impact the activation of p53, which can in turn activate cell survival and/or cell death processes (135).
Importantly, the presence of p53 seems to be crucial for the induction of apoptosis by PT, while the induction of apoptosis by high LET (in the order of 70 keV/µm) radiation, such as iron ions, was seen regardless of TP53 gene status in cancer cells (122, 124). Instead, in case of high LET radiation, caspase-9 activation plays a role in apoptosis enhancement in mutated p53 cancer cells and suppression of AKT (serine/threonine protein kinase B)-related signaling inhibits cell growth (122, 136). The response of GB cells to photon and CIRT irradiation also included an p53 independent G2/M phase arrest and subsequent appearance of mitotic catastrophe, while a ceramide-dependent-apoptotic cell death was observed (131). However, studies on p53 targeted drugs, such as MDM2 inhibitors, and the potential differences in radiosensitizing effects for different radiation qualities remain limited.
In non-GB cancer types, preclinical evidence has been provided of a RT sensitization effect induced by MDM2 inhibitors, including nutlins, serdemetan/JNJ-26854165, APG-115, PM2 and MI-219 (21, 112, 113, 116, 137–141). Interestingly, data showed that Nutlin-3 acted as a radiosensitizer under hypoxic conditions and as a radiosensitizer of tumor vasculature (140, 141). One of the main conclusions of this literature review is the fact that the combined strategy of MDM2/X inhibitors with RT is underexplored for GB. In p53 wt glioma cell lines, an enhanced radiosensitivity was observed when Nutlin-3 was combined with X-rays (142). RG7388 and RT also showed synergism, however, long-term treatment induces resistance (29). The RG7388/RT combination is also included in a phase I/IIa trial in patients with newly diagnosed GB without O(6)-methylguanine-DNA methyltransferase (MGMT) promotor methylation (N²M² (NOA-20), NCT03158389). Nutraceutical resveratrol, which has been reported to induce p53 and its downstream targets, acted as a radiosensitizing anticancer agent for highly radioresistant human SU-2 GSC both in vitro and in vivo (143, 144).
Despite the limited studies performed on the combination of IR and MDM2/X inhibitors for the treatment of GB, this section will give an extensive overview of all GB studies investigating MDM2/X inhibitors and other approaches to activate p53 (Table 1).
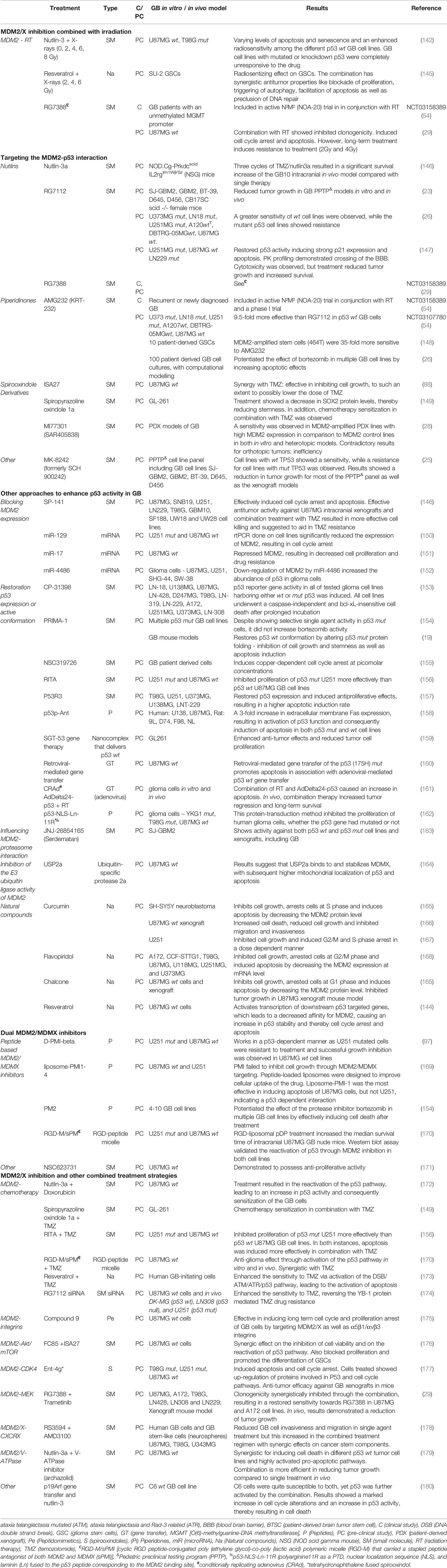
Table 1 Overview of single or combined GB treatment strategies with MDM2/X inhibitors.
Nutlin-3 is the first potent MDM2 SM inhibitor that was identified (181). Its analogue Nutlin-3a was effective at inhibiting GB cell growth, inducing varying levels of apoptosis and senescence, decreasing TMZ resistance and acting as a radiosensitizer (88, 142). The first modified MDM2 inhibitor that reached clinical trials was a more potent Nutlin analogue RG7112 (182). RG7112 showed a potential cell killing effect in GB both in vitro and in vivo, with up to a 44 times higher efficacy in MDM2-amplified and p53 wt GB cell lines (147, 183). In several Phase I trials in solid and hematological malignancies, RG7112 was successful in activating p53 and subsequently increasing the expression of downstream pro-apoptotic proteins. However, the higher dose that was required to attain satisfactory p53 activation caused significant toxicities (184–187). A second-generation nutlin analogue, RG7388 (idasanutlin), showed an increased potency, selectivity, and had a better pharmacokinetic profile. This SM inhibitor has been studied in both solid and hematological malignancies (188–190). RG7388 is included in the N²M² (NOA-20) trial in conjunction with RT with the aim to increase OS of patients with GB with an unmethylated MGMT promoter status (NCT03158389) (54).
After nutlins, piperidinone-based compounds were identified as potent MDM2-p53 interaction inhibitors. Their discovery and development for targeted cancer therapy has been reviewed elsewhere (34). AMG232 consists of a piperidinone scaffold which is similar to that of nutlins. AMG232, as a single therapy or in a combined treatment strategy, is under clinical evaluation for the treatment of advanced solid tumors, metastatic melanoma, multiple myeloma, soft tissue sarcoma and AML. At the moment, one clinical phase I trial is running in primary and recurrent GB (NCT03107780) (30, 54). In a phase I trial in p53 wt solid tumors which included GB, AML and multiple myeloma patients, AMG232 showed an acceptable patient tolerability and safety and favorable dose-proportional pharmacokinetics (191). AMG232 has also shown to increase the radiation response in several in vitro and in vivo experiments across a variety of p53 wt tumor types, but this was not studied in GB (21). However, it has been observed that AMG232 inhibition is more specific and highly regulated compared to RG7112 and its effect on GSCs was more potent (26, 148).
ISA27 has a spirooxoindolepyrrolidine core structure that has the ability to reactivate the antitumor capacities of p53 in GB cells by dissociating the MDM2-p53 complex. It has been shown to be non-toxic and it inhibited the growth of GB U87MG cells, with the implication that a lowering of the dose of TMZ as part of a combination therapy was suggested (88). The modified compound spiropyrazoline oxindole 1a was tested on the glioma cell line GL-261, alone and in combination with TMZ. These studies revealed an effective reduction in stemness through the reduction of the SOX2 protein levels, thereby promoting chemotherapy sensitization (149). Other spirooxindoles entered clinical trials and have been or are being studied in patients with advanced solid tumors and AML (MI77301(SAR405838), DS-3032b/milademetan, APG-115) (27, 54). In patient-derived xenograft (PDX) models of GB, the effectiveness of MI77301(SAR405838) was dependent on MDM2 expression but limited by poor distribution across the BBB (28). In a phase I study in patients with advanced solid tumors, MI77301 had an acceptable safety profile but had limited single agent activity (54, 192). Pre-clinically, other spirooxindoles are currently being evaluated, such as MI-219, MI-63, MI-319, MI-43, MI-88, MI-137, but none of them include GB results (32).
Novartis (Basel, Switzerland) designed a new category of MDM2 antagonists based on the dihydroisoquinolinone core which are being tested in clinical trials. These include CGM097 and HDM-201 (siremadlin) (38, 193, 194). A phase I study of CGM097 and HDM-201 in adult patients with selected advanced solid tumors was recently completed (NCT01760525, NCT02143635) (54, 195). Another SM inhibitor of the MDM2-p53 interaction, MK-8242 (SCH-900242), has been investigated in a phase I trial in patients with advanced p53 wt solid tumors and AML. An acceptable safety and tolerability was shown after MK-8242 treatment, with a successful activation of the p53 pathway (196). In GB, data is limited, but the compound was included in the in vitro pediatric preclinical testing program (PPTP) that included GB and proved to be effective in reducing tumor growth by inhibiting MDM2 expression (25).
Next to blocking the interaction between MDM2 and p53, other strategies have been studied in GB to enhance p53 activity: blocking MDM2 expression, restoring p53 expression or its active conformation, influencing the MDM2-proteasome interaction and inhibiting MDM2 ubiquitin ligase activity (197).
In vitro effects of the novel brain-penetrating SM MDM2 degrader SP-141 was assessed on numerous GB cell lines. Binding of SP-141 to MDM2, induces MDM2 auto-ubiquitination and proteasomal degradation and inhibits its expression (146). Because SP-141 crosses the BBB adequately and due to its ability to eliminate MDM2 irrespective of the p53 gene status, this compound gained interest as a GB therapy agent (146, 198). Treatment in vitro resulted in a marked decrease of MDM2 and increase in p53 as well as G2/M cell cycle arrest and apoptosis. The inhibition of brain tumor growth by SP-141 therapy was confirmed in vivo and the combination with TMZ showed a synergistic cell death ratio (146).
Small interfering RNA (siRNA) and microRNA (miRNA) are other possibilities to influence MDM2 expression (150, 174). The miRNA precursor miR-129 significantly reduced MDM2 expression in glioma cell lines, resulting in cell cycle arrest (150). miR-126 expression is abnormally low in glioma cells and miR-126 inhibits the course of glioma through targeted regulation of phosphatase and tensin homolog (PTEN)/PI3K/AKT and MDM2-p53 pathways, which, therefore, can be used as a new potential biomarker (199). miR-4486 has also shown to target MDM2 expression and increased the abundance of p53 in glioma cells (152). miR-17 transfected GB cells also showed a down-regulation of MDM2 expression, which resulted in an effective decrease in drug resistance and cell proliferation (151).
The current approaches for (re)activating p53’s tumor suppressor role using SMs were recently reviewed by Silva et al. (200). In GB, stabilizing the active conformation of p53 by altering mutant p53 protein folding, has been explored with the SMs CP-31398, PRIMA-1, P53R3, NSC319726 and RITA (Reactivation of p53 and Induction of Tumor cell Apoptosis). CP-31398 induced p53 reporter gene activity in all of the tested p53 wt and mutated glioma cell lines. High concentrations of CP-31398 resulted in the reduction of MDM2 mRNA expression (153). In GB cells, PRIMA-1 induces an inhibition of cell growth and stemness as well as apoptosis induction (20, 154). Its methylated analog PRIMA-1Met (APR-246) is currently being studied in a phase I/II study in combination with pembrolizumab in subjects with solid malignancies (NCT04383938) (54). However, compound P53R3 blocks glioma proliferation in a p53-dependent manner with a higher specificity and over a broader concentration range than PRIMA-1 (157). In vivo in GB, RITA showed synergistic effects when combined with TMZ and an inhibition of cell growth and stemness, as well as apoptosis induction. Interestingly, RITA acted independently of the p53 status (156). Protein expression studies showed that RITA suppressed cell proliferation by targeting the p53 associated protein ASK1 (156). Johansson et al. tested its efficacy in combination with the proteasome inhibitor bortezomib and despite showing specific single-agent activity in p53 mut cells, it did not strengthen bortezomib activity (154).
Since the p53 protein binds to DNA through a zinc-stabilized structurally complex domain, zinc plays a critical role in function of p53. It was shown that zinc aids in the transition of p53 mut into a functional conformation. In GB cells expressing the R273H mutation, this recovered their chemosensitivity (201). Also, NSC319726 was able to restore the p53(R175) mutant to a functional p53 wt structure by acting as a zinc ionophore. This compound arrests GB-patient-derived cells, mediated by its binding to copper (155). Restoration of p53 function was also shown in glioma cells in vitro and in vivo upon exposure to a peptide called p53p-Ant (COOH-terminal peptide of p53 linked to the truncated homeobox domain of Antennapedia). The Fas extrinsic apoptotic pathway seemed to play a role in cell death induced by this protein (158).
Another possible approach to induce p53 reactivation is targeted gene therapy. This strategy enhanced radiosensitivity of p53 wt human glioma cells (202). The introduction of p53 mut into p53 wt human glioma cells promotes adenoviral-mediated p53 wt (175H) gene transfer induced apoptosis (160). SGT-53 is a liposomal nanocomplex that delivers the p53 wt gene to tumor cells and has shown chemo-sensitization effects of GB in vitro and in vivo (28). However, the phase II trial of SGT-53 combined with TMZ in recurrent GB was terminated (NCT02340156) (54). The intratumoral administration of the adenovirus p53 gene was further explored in a phase I trial in patients with malignant primary glioma. However, a beneficial anti-tumor effect but widespread distribution of this agent remains a significant goal (159, 203). Nutlin-3 drug treatment combined with p19Arf gene transduction further activated p53 compared to single therapy in C6 GB cells. This vector is able to introduce p19Arf into p53 wt glioma cells, inducing viral expression of p19Arf with a subsequent activation of p53 (180). The adenovirus AdDelta24-p53, which encodes the p53 protein and only replicates in Rb mutant cells, achieved potent anti-glioma effects in vitro when combined with RT (161). As an alternative for gene therapy, trans-membrane peptide therapy showed promising results in glioma cells. This technique uses a peptide derived from the MDM2 binding site of p53 (162).
Next to a direct MDM2-p53 interaction regulating the stability and ubiquitylation of p53, MDM2 also links with multiple subunits of the 26S proteasome increasing proteasomal turnover of p53. This lead to an increased interest in targeting the MDM2-26S proteasomal subunit interactions (106). This is achieved by SM JNJ-26854165 (Serdemetan), which binds the RING domain of MDM2. Results showed activity against both p53 wt and p53 mut GB cell lines and xenografts. However, a phase I clinical trial in advanced or refractory tumors did not proceed to phase II (204).
Multiple inhibitors of ubiquitin E3 ligases and deubiquitinating enzymes (DUBs) have been found to have potential anti-cancer properties. As reviewed by Antao et al., ‘thus far, USP2a, USP4, USP5, USP7, USP9X, USP10, USP11, USP15, USP24, USP29, and USP49 have been linked with p53 regulation’ (205). In vitro in glioma, the binding of USP2a to MDMX increased the mitochondrial location of p53 and stimulated apoptosis (164).
A handful of natural compounds/nutraceuticals have been studied for their MDM2 inhibitory or p53 activating effects in GB, as reviewed by Qin et al. (165). The BBB permeable nutraceutical curcumin has shown to exert anti-proliferative effects on glioma cells by modulating TP53/MDM2/MDMX/p14ARF signaling. In particular, curcumin upregulates p53 expression in GB in vitro and induces cell cycle arrest in a p53-dependent manner (167, 206). Pre-clinically in GB, chalcone derivatives and flavopiridol have shown to decrease MDM2 protein level or inhibit MDM2 expression at mRNA level, respectively (168, 207). Resveratrol showed inhibitory effects on the growth and metastatic capacity of both GB and GSCs, by partially acting through AKT inhibition and p53 activation, and suppressed GB growth in vivo (144).
For optimal efficacy, concomitant targeting of both MDM2 and MDMX may be necessary, since overexpression of MDMX can act as a MDM2 substitute, causing drug resistance (41, 208, 209). MDM2/X dual inhibitors have been reviewed elsewhere (37, 103). In a study by Chen et al., NSC623731 was identified as the most potent dual specificity inhibitor via virtual screening and computational models and demonstrated anti-proliferative activity on the U87MG p53 wt GB cell line (171). In combined treatment strategies dual MDM2/X inhibitor RS3594 and CXCRX inhibition presented synergic effects against GB pre-clinically (178). Other MDMX/2 inhibitors which have, to the best of our knowledge, not been studied for GB include SJ-172550, XI-006, XI-011, RO-2443, RO-5963, WK23 and WK298 (36, 37, 210, 211). RO-2443 and its chemically optimized analog RO-5963 are indolyl hydantoins which appeared to be MDM2/X antagonists with promising preclinical results (36, 210). In adult patients with advanced or metastatic solid tumors and in pediatric cancer, a phase I trial evaluating the MDM2/X inhibitor ALRN-6924 is currently active (NCT03725436, NCT03654716) (54).
Peptides and peptidomimetics in the p53/MDM2/MDMX circuitry are also emerging as interesting anti-cancer compounds given their increased selectivity linked to less toxicity and a lower propensity in developing cancer resistance, when compared to SMs (103). Liu et al. tested the D-peptide inhibitors of the p53-MDM2 interaction DPMI-α and DPMI-#xD835;#xDEFD; on U87MG and U251 GB cell lines and results confirmed p53 targeting. Interestingly, this group showed that D-peptide antagonists of MDM2 exert anti-GB effects in vivo, when encapsulated in liposomes linked to an integrin-targeting cyclic-RGD (Arg-Gly-Asp) peptide (212). Subsequently, a series of d-amino acid mutational PMI analogues, PMI-1-4, were reported to have a higher proteolytic resistance and showed increased anti-tumor effects in vitro. Liposome-PMI-1 showed a stronger inhibitory activity against the U87MG p53 wt cell lines than Nutlin-3, without an effect on the U251 p53 mut GB cell line (169). PM2 potentiated the effect of protease inhibitor bortezomib in multiple GB cell lines by effectively inducing cell death after treatment. Interestingly, PM2 also radiosensitized p53 wt tumors but this needs to be confirmed in GB (137). ATSP-704, a progenitor of the first stapled α-helical peptide entering clinical trials, binds both MDM2 and MDMX with high affinities and effectively activates the p53 pathway in tumors in vitro and in vivo but was not studied in GB. However, in vivo, [3H]-ATSP-7041 did not distribute to the brain and CNS tissues (213). Chen et al. tried to circumvent the BBB penetration issue by developing a cyclic RGD peptide-conjugated poly (ethylene glycol)-co-poly(lactic acid) polymeric micelle (RGD-M) that carried a stapled peptide antagonist of both MDM2 and MDMX (sPMI). RGD-M/sPMI inhibited GB growth both in vitro and in vivo (214).
Although MDM2 inhibition has shown promising anti-cancer effects, not all p53 wt cell lines are sensitive to this treatment strategy and induction of apoptosis in p53 wt cell lines is sometimes limited (27, 189). In addition, therapeutic effects have been documented to be short-term due to acquired resistance or acquisition of p53 mutations (38, 107, 215). Hence, a combined treatment strategy might be necessary to reach optimal therapeutic effectiveness. Kocik et al., recently reviewed the current status of drug combinations to support MDM2 antagonists. These include targeted therapy, DNA damaging agents (chemical or IR) and apoptosis inducers. Targeted therapy strategies included tyrosine kinase inhibitors, Ras/Raf/MEK/MAPK inhibitors, cyclin-dependent kinase (CDK) inhibitors and PI3K/AKT/mTOR inhibitors. Dual inhibitors that have been reported to co-inhibit MDM2 include proteasome, histone deacetylases (HDAC), ATPase, XIAP, zinc, antibiotics, NF-κB pathway, translocator protein (TSPO), heat shock protein (HSP) inhibitors, integrin and mitotic inhibitors. Apoptotic inducers included BCL-2 inhibitors and tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) agonists (38, 175, 176, 216–218).
Saiki et al. screened an 1169-compound library for potential compounds that synergize with MDM2 inhibition in inducing tumor cell death with the goal to circumvent resistance. They observed a robust synergy in inducing apoptosis with MEK or PI3K inhibitors, BH3 mimetics, BCR-ABL antagonists, and HDAC inhibitors (219). A phase II study combining MDM2 inhibitors in combination with immunotherapy, such as pembrolizumab (targeting programmed cell death protein 1) are currently undertaken in patients with advanced solid tumors, where p53 mutation status is an inclusion criteria (NCT03611868) (54, 220). Promising MDM2 inhibitor combination strategies for the treatment of GB will be briefly summarized in this section.
The synergism of combining MDM2 inhibition with chemotherapeutics has shown to be effective in AML and multiple trials are running in diverse tumor types (NCT04190550, NCT03725436, NCT03031730, NCT04113616, NCT04275518) (54). In GB, multiple pre-clinical studies have already proven that MDM2 inhibition induces chemosensitization, including Nutlin-3a, RG7112, spiropyrazoline oxindole 1a, RITA, SP-141 and SGT-53 therapy (146, 149, 159, 160, 174, 217, 221). Nutlin-3a enhanced antitumor activity of TMZ in a humanized intracranial patient-derived xenograft model of GB (222). Nutlin-3a-loaded targeted micelles in combination with doxorubicin or the RGD MDM2/X targeting peptide-conjugated micelle (RGD-M/sPMI) in combination with TMZ showed effective synergism against GB in vitro and in vivo (170, 172). Resveratrol also enhanced the sensitivity of TMZ resistant GB-initiating cells via the activation of the DSB/ATM/ataxia telangiectasia and Rad-3 related (ATR)/p53 pathway. However, blocking NF-κB-MGMT pathway thereby averting TMZ-resistance also plays a role (223). Genetic inhibition of MDM2 expression of glioma cells in vitro and in vivo by siRNA technologies or chemical inhibition by RG7112 also increased TMZ sensitivity of glioma cells, reversing the YB-1 protein mediated TMZ drug resistance (174).
Merlino et al. investigated the effectiveness of peptidomimetic compounds targeting MDM2/X as well as α5β1/αvβ3 integrins. Studies were conducted on p53 wt glioma cells and showed that compound 9 was the most effective in inducing long term cell cycle and proliferation arrest of cancer cells. Results also revealed a consequent reduction in cell invasion and migration, thereby confirming its potential as a novel class of integrin/MDM inhibitors (175).
The interplay between the p53-MDM2 pathway and the PI3K/AKT pathway plays an important role in the determination of cell death and/or survival since this network involves two tumor suppressor genes (TP53 and PTEN) and two oncogenes (MDM2 and AKT) (224–226). AKT has shown to enhance MDM2 mediated p53 degradation (227). Data obtained from The Cancer Genome Atlas revealed that ∼88% of GB have activated PI3K pathways, which is linked with a poor prognosis (169, 228).
Among the different GB subtypes, the mesenchymal type shows the highest drug resistance, most frequent PTEN mutations (37%) and hyperactivation of PI3K/AKT (90). Daniele et al. explored the outcome of targeting both pathways by treating U87MG cells with the AKT/mTOR inhibitor FC85 in combination with the established MDM2 inhibitor ISA27 in an attempt to effectively treat GB by targeting their stem cells. Results showed a synergic effect on the inhibition of cell viability and on the reactivation of the p53 pathway leading to increased cell killing. Co-therapy also resulted in promoting differentiation, blocking proliferation and consequently apoptosis of GSCs (176). Synergy between MDM2 and PI3K/AKT/mTOR antagonists was also shown in liposarcoma and AML (226, 229). Interestingly, Saiki et al. noted that PI3K pathway mutations are not a prerequisite for this synergistic effect (219).
Dual inhibitor ent-4g was developed to target both MDM2 and CDK4. Gene expression studies were performed on U251 GB cell lines and a noteworthy alteration in the cell cycle and p53 signaling pathways were observed. Flow cytometric results showed apoptotic induction and cell cycle arrest. This was confirmed in GB xenografts (230).
Pre-clinically, the MDM2 inhibitor RG7388 has shown promising results for the treatment of GB and synergism with irradiation but acquired resistance limits its potential. Combined treatment with the MEK inhibitor trametinib resulted in a restored sensitivity towards RG7388 therapy and a decrease in tumor growth in vivo (29).
Daniele et al. investigated the potential synergy between CXCR4 antagonists and MDM2/X inhibitors for GB therapy. The dual MDM2/X inhibitor RS3594 and the CXCR4 antagonist AMD3100 presented synergic effects on cancer stem components and appears to be a valuable strategy to inhibit GB proliferation and reduce invasiveness (178).
Inhibition of the proton pump V‐ATPase (vacuolar-type ATPase) by archazolid has shown to induce p53 protein levels in cancer cells. Subsequently, evidence was found that archazolid and nutlin‐3a combined therapy increased cell death in multiple p53 wt tumor cell lines and robustly activated IGFBP3 and Bax pro‐apoptotic pathways inducing caspase‐9 and PARP inactivation. Interestingly, the combination was more efficient in reducing U87MG GB growth in vivo compared to single dose treatment (179).
There is an urgency to develop novel agents directed at relevant pathways to increase effectiveness of GB therapy (231). Since 84% of GB patients show a deregulation of the p53-ARF-MDM2 pathway, the avenue of upregulating p53 and downregulating MDM2 has been explored extensively (4, 20). However, current data on single MDM2/X therapy in GB (see Table 1) is mostly preclinical and only a few clinical trials with MDM2 inhibitors are running in GB patients (NCT03107780, NCT03158389) (54, 154).
In addition, despite the acknowledged rationale, limited data is available on the use of MDM2/X inhibitors as radiosensitizers for the treatment of GB. p53 activation using MDM2/X inhibitors has shown radiosensitizing effects pre-clinically in lung cancer, prostate cancer, adenocarcinoma and colon cancer (21, 82, 108, 110, 112, 113). The first in vitro results on p53 wt GB cells show a potential synergy, but acquired resistance could be an issue (29, 142). This is further explored in GB patients under the active N²M² (NOA-20) trial, which investigates RT and molecularly matched targeted therapies, including RG7388 (idasanutlin) (NCT03158389) (NCT03158389) (54).
Importantly, dual inhibition of MDMX/MDM2 could help achieve full activation of p53, increasing therapeutic efficacy. In particular, inhibition of the p53-MDMX interaction presents an excellent opportunity for overcoming MDM2 inhibitor resistance when cancer cells overexpress MDMX (36). However, dual inhibitory drug development is proving to be challenging mainly due to the difference in the size of the Leu26 subpocket in MDM2 and MDMX (31). In addition, specific potent MDMX inhibitors are rare. There has been a recent trend in the emergence of peptides and peptidomimetics as attractive molecules due to their advantages compared to SMs, including their selectivity and tolerability, however, major drawbacks remain their intrinsic instability and their delivery to the target, including BBB crossing. Accordingly, only a few are currently in clinical trials compared to numerous SMs (103). The transfer of drug molecules to the tumor site could be improved using a wide range of carriers: liposomes, solid lipids nanoparticles, dendrimers, polymers, silicon or carbon materials and magnetic nanoparticles (232). As an example, DPMI-α16, a D-peptide inhibitor of the p53–MDM2 interaction, encapsulated in liposomes decorated via a poly(ethylene glycol) spacer with a cyclic RGD peptide was effective in GB models (97, 103). Convection-enhanced delivery is also an option to improve delivery of targeted drugs to GB, applying local drug delivery that bypasses the BBB, while limiting associated systemic toxicities (233).
In light of recent RT developments and the promising role of particle therapy in GB treatment, more research is also needed to discover variations between different radiation qualities in inducing apoptosis signaling mechanisms, dependency of the p53 and MDM2 status and ROS production (120, 121). It is still not clear what determinants render cells susceptible towards cell death in response to MDM2 inhibitors, aside from functional p53 (39). More research will also help to clarify the determination of cell fate by the MDM2-p53 axis after DNA damage and other pathways in which the MDM2 protein and its diverse isoforms are involved. In cancer drug design, the p53 independent function of MDM2 in NBS1 regulation should be considered (39, 74, 234). For more open questions on the function of MDM2, see the recent publication of Dobbelstein et al. (39). In this regard, p53 targeted drugs including MDM2 inhibitors could elucidate new information.
Challenges such as acquired resistance and toxicity upon MDM2/X inhibition are not overcome yet, including effects on healthy tissues (29). New ways to interfere with MDM2 function are currently being developed, including proteolysis targeting chimera (PROTAC) degraders. However, it remains unclear whether these will improve efficacy without substantially increasing toxicity in human cancer patients (31, 39). Acquired resistance could be overcome by targeting multiple pathways concomitantly due to pathway redundancy, known to be present in GB. The first multi-targeted therapy strategies are only starting for GB and the ideal combination of inhibitors is unknown. Others drugs that might be worth to explore include MDM2 inhibitors with potent DNA damage repair pathway inhibitors targeting e.g. PARP, ATM, ATR, Checkpoint kinases CHK1, CHK2, WEE1, DNA-dependent protein kinase (DNA-PK) or other cell cycle pathway inhibitors targeting e.g Aurora kinase A and B, Polo-like kinase 1, RAD51 (4, 235, 236).
The main factor to select patients that are likely to benefit from MDM2/X treatment is the p53 status of the tumor and the level of MDM2 expression, although a combination of gene signatures might be necessary (27). For example, the CDKN2A gene encoding for tumor suppressor ARF that blocks MDM2 (76). MDM2 overexpression with or without gene amplification(s) is observed mainly in GB without p53 gene mutations (237). Up until now, the prognostic significance of MDM2 expression in GB is not confirmed (238). However, recent phase 1 clinical trials with SM MDM2 antagonists have indicated significant association between pre-treatment MDM2 expression levels and therapeutic response in patients with AML (25). Hence, there is a need for non-invasive predictive biomarkers for MDM2 targeted therapies. Fluorescence in situ hybridization and immunohistochemistry, the most commonly used methods for assessing MDM2 gene amplification and MDM2 protein overexpression in tumors, respectively, are invasive and do not permit monitoring the treatment response in vivo (239).
To address these needs, positron emission tomography (PET) and single-photon emission computed tomography (SPECT) radiotracers are promising to foresee a non-invasive way of imaging not only MDM2 but also other DNA damage repair proteins. This would lead to a more personalized approach, including treatment follow-up after MDM2/X therapy. PET/SPECT imaging agents for the oncoprotein MDM2 and p53 are limited at the moment. MDM2 antisense oligonucleotides were radiolabeled with [99mTc], MDM2 inhibitor SP-141 was radiolabeled with [18F] and the peptide PM2 was radiolabeled with [125I], all in a pre-clinical stage (137, 240). Next to diagnostic information that radiolabeled MDM2/X inhibitors can reveal, they could also be useful for targeted radionuclide therapy when labelled with therapeutic radionuclides. In this way it would be possible to combine MDM2/X targeted treatment with targeted IR, taking advantage of the possible radiosensitizing effect of the combined treatment (241). However, this is a field that needs further investigation and more preclinical research.
JB: conception, literature search, write-up, revising, and approval. XM: conception, literature search, write-up, and approval. CV: conception, interpretation, revising, and approval. AH: interpretation, revising, and approval. All authors contributed to the article and approved the submitted version.
This research was funded by the International Atomic Energy Agency (IAEA CRP E35010 (22248)).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures were created with BioRender.com.
1. Weller M, van den Bent M, Tonn JC, Stupp R, Preusser M, Cohen-jonathan-moyal E, et al. Review European Association for Neuro-Oncology (EANO) Guideline on the Diagnosis and Treatment of Adult Astrocytic and Oligodendroglial Gliomas. Lancet Oncol (2017) 18:315–29. doi: 10.1016/S1470-2045(17)30194-8
2. Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, et al. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A Summary. Acta Neuropathol (2016) 131:803–20. doi: 10.1007/s00401-016-1545-1
3. Ostrom QT, Gittleman H, Stetson L, Virk SM, Barnholtz-Sloan J. Epidemiology of Gliomas. In: Raizer J, Parsa A, editors. Current Understanding and Treatment of Gliomas. Springer, Switzerland Springer International Publishing Switzerland (2015). p. 1–14.
4. Pearson JRD, Regad T. Targeting Cellular Pathways in Glioblastoma Multiforme. Nat Publ Gr (2017) 2:1–11. doi: 10.1038/sigtrans.2017.40
5. Wirsching HG, Galanis E, Weller M. Glioblastoma. Handb Clin Neurol (Elsevier) (2016) 134:381–97. doi: 10.1016/B978-0-12-802997-8.00023-2
6. Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Cairncross G, et al. Radiotherapy Plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N Engl J Med (2015) 352:987–96. doi: 10.1056/NEJMoa043330
7. Tan AC, Ashley DM, López GY, Malinzak M, Friedman HS, Khasraw M. Management of Glioblastoma: State of the Art and Future Directions. CA Cancer J Clin (2020) 70:299–312. doi: 10.3322/caac.21613
8. Touat M, Idbaih A, Sanson M, Ligon KL. Glioblastoma Targeted Therapy: Updated Approaches From Recent Biological Insights. Ann Oncol (2017) 28:1457–72. doi: 10.1093/annonc/mdx106
9. Stupp R, Hegi M, Gorlia T, Kim C, Nabors L, Reardon D, et al. Cilengitide Combined With Standard Treatment for Patients With Newly Diagnosed Glioblastoma With Methylated MGMT Promoter (CENTRIC EORTC 26071-22072 Study): A Multicentre, Randomised, Open-Label, Phase 3 Trial. Lancet Oncol (2014) 15:1100–8. doi: 10.1016/S1470-2045(14)70379-1
10. Chinot OL, Wick W, Mason W, Henriksson R, Saran F, Nishikawa R, et al. Bevacizumab Plus Radiotherapy–Temozolomide for Newly Diagnosed Glioblastoma. N Eng J Med (2014) 370:709–22. doi: 10.1016/S1470-2045(14)70379-1
11. Gilbert MR, Wang M, Aldape KD, Stupp R, Hegi ME, Jaeckle KA, et al. Dose-Dense Temozolomide for Newly Diagnosed Glioblastoma: A Randomized Phase III Clinical Trial. J Clin Oncol (2013) 31:4085–92. doi: 10.1200/JCO.2013.49.6968
12. Westphal M, Heese O, Steinbach JP, Schnell O, Schackert G, Mehdorn M, et al. Clinical Trial A Randomised, Open Label Phase III Trial With Nimotuzumab, an Anti-Epidermal Growth Factor Receptor Monoclonal Antibody in the Treatment of Newly Diagnosed Adult Glioblastoma. Eur J Cancer (2015) 51:522–32. doi: 10.1016/j.ejca.2014.12.019
13. Huse JT, Holland EC. Targeting Brain Cancer: Advances in the Molecular Pathology of Malignant Glioma and Medulloblastoma. Nat Rev Cancer (2010) 10:319–31. doi: 10.1038/nrc2818
14. Caragher SP, Sachdev S, Ahmed AU. Radiotherapy and Glioma Stem Cells: Searching for Chinks in Cellular Armor. Curr Stem Cell Rep (2017) 3:348–57. doi: 10.1007/s40778-017-0102-8
15. Ou A, Yung WKA, Majd N. Molecular Mechanisms of Treatment Resistance in Glioblastoma. Int J Mol Sci (2020) 22:351. doi: 10.3390/ijms22010351
16. Tan SK, Jermakowicz A, Mookhtiar AK, Nemeroff CB, Schürer SC, Ayad NG. Drug Repositioning in Glioblastoma : A Pathway Perspective. Front Pharmacol (2018) 9:218. doi: 10.3389/fphar.2018.00218
17. Rees JH. Diagnosis and Treatment in Neuro-Oncology: An Oncological Perspective. Br J Radiol (2011) 84:S082–89. doi: 10.1259/bjr/18061999
18. Wang H, Mu X, He H, Zhang X. Cancer Radiosensitizers. Trends Pharmacol Sci (2018) 39:24–48. doi: 10.1016/j.tips.2017.11.003
19. Kareliotis G, Tremi I, Kaitatzi M, Drakaki E, Serafetinides AA, Makropoulou M, et al. Combined Radiation Strategies for Novel and Enhanced Cancer Treatment. Int J Radiat Biol (2020) 96:1087–103. doi: 10.1080/09553002.2020.1787544
20. Zhang Y, Dube C, Gibert M, Cruickshanks N, Wang B, Coughlan M, et al. The P53 Pathway in Glioblastoma. Cancers (2018) 10:297. doi: 10.3390/cancers10090297
21. Werner LR, Huang S, Francis DM, Armstrong EA, Ma F, Li C, et al. Small Molecule Inhibition of MDM2-P53 Interaction Augments Radiation Response in Human Tumors. Mol Cancer Ther (2015) 14:1994–2003. doi: 10.1158/1535-7163.MCT-14-1056-T
22. Kussie PH, Gorina S, Marechal V, Elenbaas B, Moreau J, Levine AJ, et al. Structure of the MDM2 Oncoprotein Bound to the P53 Tumor Suppressor Transactivation Domain. Science (1996) 274:948–53. doi: 10.1126/science.274.5289.948
23. Carol H, Reynolds CP, Kang MH, Keir ST, Maris JM, Gorlick R, et al Initial Testing of the MDM2 Inhibitor RG7112 by the Pediatric Preclinical Testing Program. Pediatr Blood Cancer (2013) 60:633–41. doi: 10.1002/pbc.24235
24. Ferretti S, Berger M, Rebmann R, Sterker D, Masuya K, Jeay S. NVP-CGM097: A Novel P53-Mdm2 Inhibitor Exhibiting Potent Antitumor Activity in Mouse Models of Human Cancer. Cancer Res (2014) 74:4638. doi: 10.1158/1538-7445.AM2014-4638
25. Kang MH, Reynolds CP, Kolb EA, Gorlick R, Carol H, Lock R, et al. Initial Testing (Stage 1) of MK-8242-A Novel MDM2 Inhibitor-By the Pediatric Preclinical Testing Program. Pediatr Blood Cancer (2016) 63:1744–52. doi: 10.1002/pbc.26064
26. Her NG, Oh JW, Oh YJ, Han S, Cho HJ, Lee Y, et al. Potent Effect of the MDM2 Inhibitor AMG232 on Suppression of Glioblastoma Stem Cells. Cell Death Dis (2018) 9:1–12. doi: 10.1038/s41419-018-0825-1
27. Ishizawa J, Nakamaru K, Seki T, Tazaki K, Kojima K, Chachad D, et al. Predictive Gene Signatures Determine Tumor Sensitivity to MDM2 Inhibition. Cancer Res (2018) 78:2721–31. doi: 10.1158/0008-5472.CAN-17-0949
28. Kim M, Ma DJ, Calligaris D, Zhang S, Feathers RW, Vaubel RA, et al. Efficacy of the MDM2 Inhibitor SAR405838 in Glioblastoma Is Limited by Poor Distribution Across the Blood–Brain Barrier. Mol Cancer Ther (2018) 17:1893–901. doi: 10.1158/1535-7163.MCT-17-0600
29. Berberich A, Kessler T, Thome CM, Pusch S, Hielscher T, Sahm F, et al. Targeting Resistance Against the MDM2 Inhibitor RG7388 in Glioblastoma Cells by the MEK Inhibitor Trametinib. Clin Cancer Res (2019) 25:253–65. doi: 10.1158/1078-0432.CCR-18-1580
30. Tisato V, Voltan R, Gonelli A, Secchiero P, Zauli G. MDM2/X Inhibitors Under Clinical Evaluation: Perspectives for the Management of Hematological Malignancies and Pediatric Cancer. J Hematol Oncol (2017) 10:1–17. doi: 10.1186/s13045-017-0500-5
31. Fang Y, Liao G, Yu B. Small-Molecule MDM2/X Inhibitors and PROTAC Degraders for Cancer Therapy: Advances and Perspectives. Acta Pharm Sin B (2020) 10:1253–78. doi: 10.1016/j.apsb.2020.01.003
32. Liu Y, Wang X, Wang G, Yang Y, Yuan Y, Ouyang L. The Past, Present and Future of Potential Small-Molecule Drugs Targeting P53-MDM2/MDMX for Cancer Therapy. Eur J Med Chem (2019) 176:92–104. doi: 10.1016/j.ejmech.2019.05.018
33. Ribeiro CJA, Rodrigues CMP, Moreira R, Santos MMM. Chemical Variations on the P53 Reactivation Theme. Pharmaceuticals (2016) 9:25. doi: 10.3390/ph9020025
34. Liao G, Yang D, Ma L, Li W, Hu L, Zeng L, et al. The Development of Piperidinones as Potent MDM2-P53 Protein-Protein Interaction Inhibitors for Cancer Therapy. Eur J Med Chem (2018) 159:1–9. doi: 10.1016/j.ejmech.2018.09.044
35. Beloglazkina A, Zyk N, Majouga A, Beloglazkina E. Recent Small-Molecule Inhibitors of the P53-MDM2 Protein-Protein Interaction. Molecules (2020) 25:1211. doi: 10.3390/molecules25051211
36. Anifowose A, Agbowuro AA, Yang X, Wang B. Anticancer Strategies by Upregulating P53 Through Inhibition of its Ubiquitination by MDM2. Med Chem Res (2020) 29:1105–21. doi: 10.1007/s00044-020-02574-9
37. Espadinha M, Barcherini V, Lopes EA, Santos MMM. An Update on MDMX and Dual Mdm2/X Inhibitors. Curr Top Med Chem (2018) 18:647–60. doi: 10.2174/1568026618666180604080119
38. Kocik J, Machula M, Wisniewska A, Surmiak E, Holak TA, Skalniak L. Helping the Released Guardian: Drug Combinations for Supporting the Anticancer Activity of HDM2 (MDM2) Antagonists. Cancers (2019) 11:1–39. doi: 10.3390/cancers11071014
39. Dobbelstein M, Levine AJ. Mdm2: Open Questions. Cancer Sci (2020) 111:2203–11. doi: 10.1111/cas.14433
40. Konopleva M, Martinelli G, Daver N, Papayannidis C, Wei A, Higgins B, et al. MDM2 Inhibition: An Important Step Forward in Cancer Therapy. Leukemia (2020) 34:2858–74. doi: 10.1038/s41375-020-0949-z
41. Burgess A, Chia KM, Haupt S, Thomas D, Haupt Y, Lim E. Clinical Overview of MDM2/X-Targeted Therapies. Front Oncol (2016) 6:1–7. doi: 10.3389/fonc.2016.00007
42. Kong L, Gao J, Hu J, Lu R, Yang J, Qiu X, et al. Carbon Ion Radiotherapy Boost in the Treatment of Glioblastoma: A Randomized Phase I/III Clinical Trial ChiCTR-OID-17013702 ChiCTR-OID. Cancer Commun (2019) 39:1–12. doi: 10.1186/s40880-019-0351-2
43. Ali MY, Oliva CR, Noman ASM, Allen BG, Goswami PC, Zakharia Y, et al. Radioresistance in Glioblastoma and the Development of Radiosensitizers. Cancers (2020) 12:1–29. doi: 10.3390/cancers12092511
45. Piper K, DePledge L, Karsy M, Cobbs C. Glioma Stem Cells as Immunotherapeutic Targets: Advancements and Challenges. Front Oncol (2021) 11:1–13. doi: 10.3389/fonc.2021.615704
46. Chédeville AL, Madureira PA. The Role of Hypoxia in Glioblastoma Radiotherapy Resistance. Cancers (2021) 13:1–16. doi: 10.3390/cancers13030542
47. Ziu M, Kim BYS, Jiang W, Ryken T, Olson JJ. The Role of Radiation Therapy in Treatment of Adults With Newly Diagnosed Glioblastoma Multiforme: A Systematic Review and Evidence-Based Clinical Practice Guideline Update. J Neuro-Oncol (2020) 150:215–67. doi: 10.1007/s11060-020-03612-7
48. Kong L, Wu J, Gao J, Qiu X, Yang J, Hu J, et al. Particle Radiation Therapy in the Management of Malignant Glioma: Early Experience at the Shanghai Proton and Heavy Ion Center. Cancer (2020) 126:2802–10. doi: 10.1002/cncr.32828
49. Marnitz S, Wlodarczyk W, Neumann O, Koehler C, Weihrauch M, Budach V, et al. Which Technique for Radiation Is Most Beneficial for Patients With Locally Advanced Cervical Cancer? Intensity modulated proton therapy versus intensity modulated photon treatment, helical tomotherapy and volumetric arc therapy for primary radiation - an intraindividual comparison. Radiat Oncol (2015) 10:1–9. doi: 10.1186/s13014-015-0402-z
50. v van de Schoot AJAJ, de Boer P, Crama KF, Visser J, Stalpers LJA, Rasch CRN, et al. Dosimetric Advantages of Proton Therapy Compared With Photon Therapy Using an Adaptive Strategy in Cervical Cancer. Acta Oncol (2016) 55:892–9. doi: 10.3109/0284186X.2016.1139179
51. Grosshans DR, Mohan R, Gondi V, Shih HA, Mahajan A, Brown PD. The Role of Image-Guided Intensity Modulated Proton Therapy in Glioma. Neuro Oncol (2017) 19:ii30–7. doi: 10.1093/neuonc/nox002
52. Adeberg S, Harrabi SB, Verma V, Bernhardt D, Grau N, Debus J, et al. Treatment of Meningioma and Glioma With Protons and Carbon Ions. Radiat Oncol (2017) 12:1–7. doi: 10.1186/s13014-017-0924-7
53. Dennis ER, Bussiere MR, Niemierko A, Lu MW, Fullerton BC, Loeffler JS, et al. A Comparison of Critical Structure Dose and Toxicity Risks in Patients With Low Grade Gliomas Treated With IMRT Versus Proton Radiation Therapy. Technol Cancer Res Treat (2013) 12:1–9. doi: 10.7785/tcrt.2012.500276
54. ClinicalTrials.gov [Internet]. Available at: https://clinicaltrials.gov/.
55. Mizumoto M, Yamamoto T, Takano S, Ishikawa E, Matsumura A, Ishikawa H, et al. Long-Term Survival After Treatment of Glioblastoma Multiforme With Hyperfractionated Concomitant Boost Proton Beam Therapy. Pract Radiat Oncol (2015) 5:e9–16. doi: 10.1016/j.prro.2014.03.012
56. Tinganelli W, Durante M. Carbon Ion Radiobiology. Cancers (2020) 12:3022. doi: 10.3390/cancers12103022
57. Antonovic L, Lindblom E, Dasu A, Bassler N, Furusawa Y, Toma-Dasu I. Clinical Oxygen Enhancement Ratio of Tumors in Carbon Ion Radiotherapy: The Influence of Local Oxygenation Changes. J Radiat Res (2014) 55:902–11. doi: 10.1093/jrr/rru020
58. Malouff TD, Mahajan A, Krishnan S, Beltran C, Seneviratne DS, Trifiletti DM. Carbon Ion Therapy: A Modern Review of an Emerging Technology. Front Oncol (2020) 10:82. doi: 10.3389/fonc.2020.00082
59. Chiblak S, Tang Z, Lemke D, Knoll M, Dokic I, Warta R, et al. Carbon Irradiation Overcomes Glioma Radioresistance by Eradicating Stem Cells and Forming an Antiangiogenic and Immunopermissive Niche. J Clin Invest (2019) 4:1–14. doi: 10.1172/jci.insight.123837
60. Wenzl T, Wilkens JJ. Modelling of the Oxygen Enhancement Ratio for Ion Beam Radiation Therapy. Phys Med Biol (2011) 56:3251–68. doi: 10.1088/0031-9155/56/11/006
61. Combs SE, Bruckner T, Mizoe J, Kamada T, Tsujii H, Kieser M, et al. Comparison of Carbon Ion Radiotherapy to Photon Radiation Alone or in Combination With Temozolomide in Patients With High-Grade Gliomas : Explorative Hypothesis-Generating Retrospective Analysis. Radiother Oncol (2013) 108:132–5. doi: 10.1016/j.radonc.2013.06.026
62. Mizoe J-E, Tsujii H, Hasegawa A, Yanagi T, Takagi R, Kamada T, et al. Phase I/II Clinical Trial of Carbon Ion Radiotherapy for Malignant Gliomas: Combined X-Ray Radiotherapy, Chemotherapy, and Carbon Ion Radiotherapy. Int J Radiat Oncol (2007) 69:390–6. doi: 10.1016/j.ijrobp.2007.03.003
63. Combs SE, Kieser M, Rieken S, Habermehl D, Jäkel O, Haberer T, et al. Randomized Phase II Study Evaluating a Carbon Ion Boost Applied After Combined Radiochemotherapy With Temozolomide Versus a Proton Boost After Radiochemotherapy With Temozolomide in Patients With Primary Glioblastoma : The CLEOPATRA Trial. BMC Cancer (2010) 10:1–9. doi: 10.1186/1471-2407-10-478
64. Combs SE, Burkholder I, Edler L, Rieken S, Habermehl D, Jäkel O, et al. Randomised Phase I/II Study to Evaluate Carbon Ion Radiotherapy Versus Fractionated Stereotactic Radiotherapy in Patients With Recurrent or Progressive Gliomas: The CINDERELLA Trial. BMC Cancer (2010) 10:533. doi: 10.1186/1471-2407-10-533
65. Eberle F, Engenhart-cabillic R, Jensen AD, Carl B, Stein M. Carbon Ion Beam Reirradiation in Recurrent High-Grade Glioma. Cancer Manag Res (2020) 12:633–9. doi: 10.2147/CMAR.S217824
66. Wilson JD, Hammond EM, Higgins GS, Petersson K. Ultra-High Dose Rate (FLASH) Radiotherapy: Silver Bullet or Fool’s Gold? Front Oncol (2020) 9:1563. doi: 10.3389/fonc.2019.01563
67. Bourhis J, Montay-gruel P, Gonçalves P, Bailat C, Petit B, Ollivier J, et al. Clinical Translation of FLASH Radiotherapy : Why and How ? Radiother Oncol (2019) 139:11–7. doi: 10.1016/j.radonc.2019.04.008
68. Colangelo NW, Azzam EI. The Importance and Clinical Implications of FLASH Ultra-High Dose-Rate Studies for Proton and Heavy Ion Radiotherapy. Radiat Res (2020) 193:1–4. doi: 10.1667/RR15537.1
69. Hughes JR, Parsons JL. FLASH Radiotherapy : Current Knowledge and Future Insights Using Proton-Beam Therapy. Int J Mol Sci (2020) 21:6492. doi: 10.3390/ijms21186492
70. Mazal A, Prezado Y, Ares C, De Marzi L, Patriarca A, Miralbell R, et al. Proton Therapy Special Feature : Review Article FLASH and Minibeams in Radiation Therapy : The Effect of Microstructures on Time and Space and Their Potential Application to Protontherapy. Br J Radiol (2020) 93:1–17. doi: 10.1259/bjr.20190807
71. Canon J, Osgood T, Olson SH, Saiki AY, Robertson R, Yu D, et al. The MDM2 Inhibitor AMG 232 Demonstrates Robust Antitumor Efficacy and Potentiates the Activity of P53-Inducing Cytotoxic Agents. Mol Cancer Ther (2015) 14:649–58. doi: 10.1158/1535-7163.MCT-14-0710
73. Nag S, Qin J, Srivenugopal KS, Wang M, Zhang R. The MDM2-P53 Pathway Revisited. J BioMed Res (2013) 27:254–71. doi: 10.7555/JBR.27.20130030
74. Li W, Peng X, Lang J, Xu C. Targeting Mouse Double Minute 2: Current Concepts in DNA Damage Repair and Therapeutic Approaches in Cancer. Front Pharmacol (2020) 11:1–18. doi: 10.3389/fphar.2020.00631
75. Carr MI, Roderick JE, Gannon HS, Kelliher MA, Jones SN Mdm2 Phosphorylation Regulates Its Stability and Has Contrasting Effects on Oncogene and Radiation-Induced Tumorigenesis. Cell Rep (2016) 16:2618–29. doi: 10.1016/j.celrep.2016.08.014
76. Maggi LB, Winkeler CL, Miceli AP, Apicelli AJ, Brady SN, Kuchenreuther MJ, et al. ARF Tumor Suppression in the Nucleolus. Biochim Biophys Acta (2014) 1842:831–9. doi: 10.1016/j.bbadis.2014.01.016
77. Carr MI, Roderick JE, Gannon HS, Kelliher MA, Jones SN. Mdm2 Phosphorylation Regulates Its Stability and Has Contrasting Effects on Oncogene and Radiation-Induced Tumorigenesis. Cell Rep (2016) 16:2618–29. doi: 10.1016/j.celrep.2016.08.014
78. Stommel JM, Wahl GM. Accelerated MDM2 Auto-Degradation Induced by DNA-Damage Kinases Is Required for P53 Activation. EMBO J (2004) 23:1547–56. doi: 10.1038/sj.emboj.7600145
79. Tollini LA, Jin A, Park J, Zhang Y. Regulation of P53 by Mdm2 E3 Ligase Function Is Dispensable in Embryogenesis and Development But Essential in Response to DNA Damage. Cancer Cell (2015) 26:235–47. doi: 10.1016/j.ccr.2014.06.006
80. Kruse JP, Gu W. Modes of P53 Regulation. Cell (2009) 137:609–22. doi: 10.1016/j.cell.2009.04.050
81. Yu D-H, Xu Z-Y, Mo S, Yuan L, Cheng X-D, Qin J-J. Targeting MDMX for Cancer Therapy: Rationale, Strategies, and Challenges. Front Oncol (2020) 10:1389. doi: 10.3389/fonc.2020.01389
82. Spiegelberg D, Mortensen AC, Lundsten S, Brown CJ, Lane DP, Nestor M The MDM2/MDMX-P53 Antagonist PM2 Radiosensitizes Wild-Type P53 Tumors. Cancer Res (2018) 78:5084–93. doi: 10.1158/0008-5472.CAN-18-0440
83. Marine J CW, Dyer MA, Jochemsen AG. MDMX: From Bench to Bedside. J Cell Sci (2007) 120:371–8. doi: 10.1242/jcs.03362
84. McLendon R, Friedman A, Bigner D, Van Meir EG, Brat DJ, Mastrogianakis GM, et al. Comprehensive Genomic Characterization Defines Human Glioblastoma Genes and Core Pathways. Nature (2008) 455:1061–8. doi: 10.1038/nature07385
85. Parsons DW, Jones S, Zhang X, Lin JC-H, Leary RJ, Angenendt P, et al. An Integrated Genomic Analysis of Human Glioblastoma Multiforme. Science (2008) 321:1807–12. doi: 10.1126/science.1164382
86. Ohgaki H, Kleihues P. Genetic Alterations and Signaling Pathways in the Evolution of Gliomas. Cancer Sci (2009) 100:2235–41. doi: 10.1111/j.1349-7006.2009.01308.x
87. Cerami E, Demir E, Schultz N, Taylor BS, Sander C. Automated Network Analysis Identifies Core Pathways in Glioblastoma. PloS One (2010) 5:e8918. doi: 10.1371/journal.pone.0008918
88. Costa B, Bendinelli S, Gabelloni P, Da Pozzo E, Daniele S, Scatena F, et al. Human Glioblastoma Multiforme: P53 Reactivation by a Novel MDM2 Inhibitor. PloS One (2013) 8:e72281. doi: 10.1371/journal.pone.0072281
89. England B, Huang T, Karsy M. Current Understanding of the Role and Targeting of Tumor Suppressor P53 in Glioblastoma Multiforme. Tumor Biol (2013) 34:2063–74. doi: 10.1007/s13277-013-0871-3
90. Verhaak RGW, Hoadley KA, Purdom E, Wang V, Wilkerson MD, Miller CR, et al. Integrated Genomic Analysis Identifies Clinically Relevant Subtypes of Glioblastoma Characterized by Abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell (2011) 17:1–25. doi: 10.1016/j.ccr.2009.12.020
91. Riemenschneider MJ, Büschges R, Wolter M, Reifenberger J, Boström J, Kraus JA, et al. Amplification and Overexpression of the MDM4 (MDMX) Gene From 1q32 in a Subset of Malignant Gliomas Without TP53 Mutation or MDM2 Amplification. Cancer Res (1999) 59:6091–6.
92. Arjona D, Bello MJ, Alonso ME, Isla A, De Campos JM, Vaquero J, et al. Real-Time Quantitative PCR Analysis of Regions Involved in Gene Amplification Reveals Gene Overdose in Low-Grade Astrocytic Gliomas. Diagn Mol Pathol (2005) 14:224–9. doi: 10.1097/01.pas.0000177799.58336.1a
94. Shangary S, Wang S. Targeting the MDM2-P53 Interaction for Cancer Therapy. Clin Cancer Res (2008) 14:5318–24. doi: 10.1158/1078-0432.CCR-07-5136
95. Karni-Schmidt O, Lokshin M, Prives C. The Roles of MDM2 and MDMX in Cancer. Annu Rev Pathol Mech Dis (2016) 11:617–44. doi: 10.1146/annurev-pathol-012414-040349
96. Sullivan KD, Gallant-Behm CL, Henry RE, Fraikin J-L, Espinosa JM. The P53 Circuit Board. Biochem Biophys Res Commun (2012) 1825:229–44. doi: 10.1016/j.bbcan.2012.01.004
97. Liu M, Li C, Pazgier M, Li C, Mao Y, Lv Y, et al D-Peptide Inhibitors of the P53-MDM2 Interaction for Targeted Molecular Therapy of Malignant Neoplasms. Proc Natl Acad Sci USA (2010) 107:14321–6. doi: 10.1073/pnas.1008930107
98. Shangary S, Wang S. Small-Molecule Inhibitors of the MDM2-P53 Protein-Protein Interaction to Reactivate P53 Function: A Novel Approach for Cancer Therapy. Annu Rev Pharmacol Toxicol (2009) 49:223–41. doi: 10.1146/annurev.pharmtox.48.113006.094723
99. Henze J, Mühlenberg T, Simon S, Grabellus F, Rubin B, Taeger G, et al. P53 Modulation as a Therapeutic Strategy in Gastrointestinal Stromal Tumors. PloS One (2012) 7:e37776. doi: 10.1371/journal.pone.0037776
100. Li J, Kurokawa M. Regulation of MDM2 Stability After DNA Damage. J Cell Physiol (2015) 230:2318–27. doi: 10.1002/jcp.24994
101. Pellegrino M, Mancini F, Lucà R, Coletti A, Giacchè N, Manni I, et al. Targeting the MDM2/MDM4 Interaction Interface as a Promising Approach for P53 Reactivation Therapy. Cancer Res (2015) 75:4560–72. doi: 10.1158/0008-5472.CAN-15-0439
102. Mancini F, Di Conza G, Monti O, Macchiarulo A, Pellicciari R, Pontecorvi A, et al. Puzzling Over MDM4 – P53 Network. Int J Biochem Cell Biol (2010) 42:1080–3. doi: 10.1016/j.biocel.2010.04.010
103. Teveroni E, Lucà R, Pellegrino M, Cioli G, Pontecorvi A, Moretti F. Peptides and Peptidomimetics in the P53/MDM2/MDM4 Circuitry - a Patent Review. Expert Opin Ther Pat (2016) 26:1417–29. doi: 10.1080/13543776.2017.1233179
104. Marine JC, Francoz S, Maetens M, Wahl G, Toledo F, Lozano G. Keeping P53 in Check: Essential and Synergistic Functions of Mdm2 and Mdm4. Cell Death Differ (2006) 13:927–34. doi: 10.1038/sj.cdd.4401912
105. Toledo F, Wahl GM. Regulating the P53 Pathway: In Vitro Hypotheses, In Vivo Veritas. Cancer (2006) 6:909–23. doi: 10.1038/nrc2012
106. Zhang Q, Zeng S, Lu H. Targeting P53-MDM2-MDMX Loop for Cancer Therapy. Subcell Biochem (2014) 85:281–319. doi: 10.1007/978-94-017-9211-0_16
107. Skalniak L, Surmiak E, Holak TA. Expert Opinion on Therapeutic Patents A Therapeutic Patent Overview of MDM2/X- Targeted Therapies (2014-2018). Expert Opin Ther Pat (2019) 29:151–70. doi: 10.1080/13543776.2019.1582645
108. Stewart-ornstein J, Iwamoto Y, Miller MA, Prytyskach MA, Ferretti S, Holzer P, et al. P53 Dynamics Vary Between Tissues and Are Linked With Radiation Sensitivity. Nat Commun (2021) 12:1–11. doi: 10.1038/s41467-021-21145-z
109. Williams AB. P53 in the DNA-Damage-Repair Process. Cold Spring Harb Perspect Med (2016) 6:a026070. doi: 10.1101/cshperspect.a026070
111. Shu HKG, Kim MM, Chen P, Furman F, Julin CM, Israel MA. The Intrinsic Radioresistance of Glioblastoma-Derived Cell Lines Is Associated With a Failure of P53 to Induce p21BAX Expression. Proc Natl Acad Sci USA (1998) 95:14453–8. doi: 10.1073/pnas.95.24.14453
112. Luo H, Yount C, Lang H, Yang A, Riemer EC, Lyons K, et al. Lung Cancer Activation of P53 With Nutlin-3a Radiosensitizes Lung Cancer Cells via Enhancing Radiation-Induced Premature Senescence. Lung Cancer (2013) 81:167–73. doi: 10.1016/j.lungcan.2013.04.017
113. Feng F, Zhang Z, Kothari V, Evans JR, Jackson WC, Chen W, et al. MDM2 Inhibition Sensitizes Prostate Cancer Cells to Androgen Ablation and Radiotherapy in a P53-Dependent Manner. Neoplasia (2016) 18:213–22. doi: 10.1016/j.neo.2016.01.006
114. Afshar G, Jelluma N, Yang X, Basila D, Arvold ND, Karlsson A, et al. Radiation-Induced Caspase-8 Mediates P53-Independent Apoptosis in Glioma Cells. Cancer Res (2006) 66:4223–33. doi: 10.1158/0008-5472.CAN-05-1283
115. Hara S, Nakashima S, Kiyono T, Sawada M, Yoshimura S-I, Iwama T, et al. Ceramide Triggers Caspase Activation During Gamma-Radiation-Induced Apoptosis of Human Glioma Cells Lacking Functional P53. Oncol Rep (2004) 12:119–23. doi: 10.3892/or.12.1.119
116. Impicciatore G, Sancilio S, Miscia S, Di Pietro R. Nutlins and Ionizing Radiation in Cancer Therapy. Curr Pharm Des (2010) 2:1427–42. doi: 10.2174/138161210791033932
118. Eischen CM. Role of Mdm 2 and Mdmx in DNA Repair. J Mol Cell Biol (2017) 9:69–73. doi: 10.1093/jmcb/mjw052
119. Alt JR, Bouska A, Fernandez MR, Cerny RL, Xiao H, Eischen CM. Mdm2 Binds to Nbs1 at Sites of DNA Damage and Regulates Double Strand Break Repair. J Biol Chem (2005) 280:18771–81. doi: 10.1074/jbc.M413387200
120. Tomao F, Papa A, Rossi L, Zaccarelli E, Caruso D, Zoratto F, et al. Angiogenesis and Antiangiogenic Agents in Cervical Cancer. Onco Targets Ther (2014) 7:2237–48. doi: 10.2147/OTT.S68286
121. Tommasino F, Durante M. Proton Radiobiology. Cancers (2015) 7:353–81. doi: 10.3390/cancers7010353
122. Yamakawa N, Takahashi A, Mori E, Imai Y, Furusawa Y, Ohnishi K, et al. High LET Radiation Enhances Apoptosis in Mutated P53 Cancer Cells Through Caspase-9 Activation. Cancer Sci (2008) 99:1455–60. doi: 10.1111/j.1349-7006.2008.00818.x
123. Fournier AC, Wiese C, Fournier C, Wiese C. Accumulation of the Cell Cycle Regulators TP53 and CDKN1A (P21) in Human Fibroblasts After Exposure to Low- and High-LET Radiation Accumulation of the Cell Cycle Regulators TP53 and CDKN1A (P21) in Human Fibroblasts After Exposure to Low- and High-LET. Radiat Res (2004) 161:675–84. doi: 10.1667/RR3182
124. Lee KB, Kim KR, Huh TL, Lee YM. Proton Induces Apoptosis of Hypoxic Tumor Cells by the P53-Dependent and P38/JNK MAPK Signaling Pathways. Int J Oncol (2008) 33:1247–56. doi: 10.3892/ijo_00000115
125. Di Pietro C, Piro S, Tabb G, Ragusa M, Di Pietro V, Zimmitti V, et al. Cellular and Molecular Effects of Protons : Apoptosis Induction and Potential Implications for Cancer Therapy. Apoptosis (2006) 11:57–66. doi: 10.1007/s10495-005-3346-1
126. Ristic-Fira AM, Todorovic DV, Koricanac LB, Petrovic IM, Valastro LM, Cirrone PGA, et al. Response of a Human Melanoma Cell Line to Low and High Ionizing Radiation. Ann N Y Acad Sci (2007) 1095:165–74. doi: 10.1196/annals.1397.020
127. Antoccia A, Sgura A, Berardinelli F, Cavinato M, Cherubini R, Gerardi S, et al. Cell Cycle Perturbations and Genotoxic Effects in Human Primary Fibroblasts Induced by Low-Energy Protons and X/γ-Rays. J Radiat Res (2009) 50:457–68. doi: 10.1269/jrr.09008
128. Takahashi A, Ohnishi K, Tsuji K, Matsumoto H, Aoki H. WAF1 Accumulation by Carbon-Ion Beam and a -Particle Irradiation in Human Glioblastoma Cultured Cells. Int J Radiat Biol (2000) 76:335–41. doi: 10.1080/095530000138673
129. Chaudhary P, Marshall TI, Currell FJ, Kacperek A, Schettino G, Prise KM. Variations in the Processing of DNA Double-Strand Breaks Along 60-MeV Therapeutic Proton Beams. Int J Radiat Oncol Biol Phys (2016) 95:86–94. doi: 10.1016/j.ijrobp.2015.07.2279
130. Mitteer AR, Wang Y, Shah J, Gordon S, Fager M, Butter PP, et al. Proton Beam Radiation Induces DNA Damage and Cell Apoptosis in Glioma Stem Cells Through Reactive Oxygen Species. Sci Rep (2015) 5:1–12. doi: 10.1038/srep13961
131. Ferrandon S, Magné N, Battiston-Montagne P, Hau-Desbat NH, Diaz O, Beuve M, et al. Cellular and Molecular Portrait of Eleven Human Glioblastoma Cell Lines Under Photon and Carbon Ion Irradiation. Cancer Lett (2015) 360:10–6. doi: 10.1016/j.canlet.2015.01.025
132. Maalouf M, Alphonse G, Colliaux A, Beuve M, Trajkovic-Bodennec S, Battiston-Montagne P, et al. Different Mechanisms of Cell Death in Radiosensitive and Radioresistant P53 Mutated Head and Neck Squamous Cell Carcinoma Cell Lines Exposed to Carbon Ions and X-Rays. Int J Radiat Oncol Biol Phys (2009) 74:200–9. doi: 10.1016/j.ijrobp.2009.01.012
133. Alphonse G, Maalouf M, Battiston-Montagne P, Ardail D, Beuve M, Rousson R, et al. P53-Independent Early and Late Apoptosis Is Mediated by Ceramide After Exposure of Tumor Cells to Photon or Carbon Ion Irradiation. BMC Cancer (2013) 13:151. doi: 10.1186/1471-2407-13-151
134. Niemantsverdriet M, van Goethem M-J, Bron R, Hogewerf W, Brandenburg S, Langendijk JA, et al. High and Low LET Radiation Differentially Induce Normal Tissue Damage Signals. Int J Radiat Oncol Biol Phys (2012) 83:1291–7. doi: 10.1016/j.ijrobp.2011.09.057
135. Redza-Dutordoir M, Averill-Bates DA. Activation of Apoptosis Signalling Pathways by Reactive Oxygen Species. Biochim Biophys Acta - Mol Cell Res (2016) 1863:2977–92. doi: 10.1016/j.bbamcr.2016.09.012
136. Nakagawa Y, Takahashi A, Kajihara A, Yamakawa N, Imai Y, Ota I, et al. Depression of P53-Independent Akt Survival Signals in Human Oral Cancer Cells Bearing Mutated P53 Gene After Exposure to High-LET Radiation. Biochem Biophys Res Commun (2012) 423:654–60. doi: 10.1016/j.bbrc.2012.06.004
137. Spiegelberg D, Mortensen AC, Lundsten S, Brown CJ, Lane DP, Nestor M. The MDM2/MDMX-P53 Antagonist PM2 Radiosensitizes Wild-Type P53 Tumors. Cancer Res (2018) 78:5084–94. doi: 10.1158/0008-5472.CAN-18-0440
138. Mendrysa SM, McElwee MK, Michalowski J, O’Leary KA, Young KM, Perry ME. Mdm2 Is Critical for Inhibition of P53 During Lymphopoiesis and the Response to Ionizing Irradiation. Mol Cell Biol (2003) 23:462–72. doi: 10.1128/MCB.23.2.462-473.2003
139. Chargari C, Leteur C, Angevin E, Bashir T, Schoentjes B, Arts J, et al. Preclinical Assessment of JNJ-26854165 (Serdemetan), a Novel Tryptamine Compound With Radiosensitizing Activity In Vitro and in Tumor Xenografts. Cancer Lett (2011) 312:209–18. doi: 10.1016/j.canlet.2011.08.011
140. Supiot S, Hill RP, Bristow RG. Nutlin-3 Radiosensitizes Hypoxic Prostate Cancer Cells Independent of P53. Mol Cancer Ther (2008) 7:993–9. doi: 10.1158/1535-7163.MCT-07-0442
141. Cao C, Shinohara ET, Subhawong TK, Geng L, Kim KW, Albert JM, et al. Radiosensitization of Lung Cancer by Nutlin, an Inhibitor of Murine Double Minute 2. Mol Cancer Ther (2006) 5:411–7. doi: 10.1158/1535-7163.MCT-05-0356
142. Villalonga-Planells R, Coll-Mulet L, Martínez-Soler F, Castaño E, Acebes JJ, Giménez-Bonafé P, et al. Activation of P53 by Nutlin-3a Induces Apoptosis and Cellular Senescence in Human Glioblastoma Multiforme. PloS One (2011) 6:e18588. doi: 10.1371/journal.pone.0018588
143. Richard SA. The Therapeutic Potential of Resveratrol in Gliomas. Adv Biosci Clin Med (2019) 7:44. doi: 10.7575/aiac.abcmed.v.7n.2p.44
144. Clark PA, Bhattacharya S, Elmayan A, Darjatmoko SR, Thuro BA, Yan MB, et al. Resveratrol Targeting of AKT and P53 in Glioblastoma and Glioblastoma Stem-Like Cells to Suppress Growth and Infiltration. J Neurosurg (2017) 126:1448–60. doi: 10.3171/2016.1.JNS152077
145. Wang L, Long L, Wang W, Liang Z. Resveratrol, a Potential Radiation Sensitizer for Glioma Stem Cells Both In Vitro and In Vivo. J Pharmacol Sci (2015) 129:216–25. doi: 10.1016/j.jphs.2015.11.001
146. Punganuru S, Arutla V, Zhao W, Rajaei M, Deokar H, Zhang R, et al. Targeted Brain Tumor Therapy by Inhibiting the MDM2 Oncogene: In Vitro and In Vivo Antitumor Activity and Mechanism of Action. Cells (2020) 9:1–14. doi: 10.3390/cells9071592
147. Verreault M, Schmitt C, Goldwirt L, Pelton K, Haidar S, Levasseur C, et al. Preclinical Efficacy of the MDM2 Inhibitor RG7112 in MDM2-Amplified and TP53 Wild-Type Glioblastomas. Clin Cancer Res (2016) 22:1185–96. doi: 10.1158/1078-0432.CCR-15-1015
148. Gluck WL, Gounder MM, Frank R, Eskens F, Blay JY, Cassier PA, et al. Phase 1 Study of the MDM2 Inhibitor AMG 232 in Patients With Advanced P53 Wild-Type Solid Tumors or Multiple Myeloma. Invest New Drugs (2019) 38:831–43. doi: 10.1007/s10637-019-00840-1
149. Amaral JD, Silva D, Rodrigues CMP, Solá S, Santos MMM. A Novel Small Molecule P53 Stabilizer for Brain Cell Differentiation. Front Chem (2019) 7:1–14. doi: 10.3389/fchem.2019.00015
150. Moradimotlagh A, Arefian E, Rezazadeh Valojerdi R, Ghaemi S, Jamshidi Adegani F, Soleimani M. MicroRNA-129 Inhibits Glioma Cell Growth by Targeting CDK4, CDK6, and MDM2. Mol Ther Nucleic Acids (2020) 19:759–64. doi: 10.1016/j.omtn.2019.11.033
151. Li H, Yang BB. Stress Response of Glioblastoma Cells Mediated by miR-17-5p Targeting PTEN and the Passenger Strand miR-17-3p Targeting MDM2. Oncotarget (2012) 3:1653–68. doi: 10.18632/oncotarget.810
152. Liu J, Cheng LG, Li HG. LncRNA SNHG20 Promoted the Proliferation of Glioma Cells via Sponging miR-4486 to Regulate the MDM2-P53 Pathway. Eur Rev Med Pharmacol Sci (2019) 23:5323–31. doi: 10.26355/eurrev_201906_18199
153. Wischhusen J, Naumann U, Ohgaki H, Rastinejad F, Weller M. CP-31398, a Novel P53-Stabilizing Agent, Induces P53-Dependent and P53-Independent Glioma Cell Death. Oncogene (2003) 22:8233–45. doi: 10.1038/sj.onc.1207198
154. Johansson P, Krona C, Kundu S, Doroszko M, Baskaran S, Schmidt L, et al. A Patient-Derived Cell Atlas Informs Precision Targeting of Glioblastoma. Cell Rep (2020) 32:107897. doi: 10.1016/j.celrep.2020.107897
155. Shimada K, Reznik E, Stokes ME, Krishnamoorthy L, Bos H, Song Y, et al. Copper-Binding Small Molecule Induces Oxidative Stress and Cell Cycle Arrest in Glioblastoma-Patient-Derived Cells. Cell Chem Biol (2019) 25:585–94. doi: 10.1016/j.chembiol.2018.02.010
156. Wu Q, Cao Z, Xiao W, Zhu L, Xie Q, Li L, et al. Study on Therapeutic Action and Mechanism of TMZ Combined With RITA Against Glioblastoma. Cell Physiol Biochem (2019) 51:2536–46. doi: 10.1159/000495923
157. Weinmann L, Wischhusen J, Demma MJ, Naumann U, Roth P, DasMahapatra B, et al. A Novel P53 Rescue Compound Induces P53-Dependent Growth Arrest and Sensitises Glioma Cells to Apo2L/TRAIL-Induced Apoptosis. Cell Death Differ (2008) 15:718–29. doi: 10.1038/sj.cdd.4402301
158. Senatus PB, Li Y, Mandigo C, Nichols G, Moise G, Mao Y, et al. Restoration of P53 Function for Selective Fas-Mediated Apotosis in Human and Rat Glioma Cells In Vitro and In Vivo by a P53 COOH-Terminal Peptide. Mol Cancer Ther (2006) 5:20–8. doi: 10.1158/1535-7163.MCT-05-0181
159. Kim SS, Harford JB, Moghe M, Rait A, Chang EH. Combination With SGT-53 Overcomes Tumor Resistance to a Checkpoint Inhibitor. Oncoimmunology (2018) 7:1–14. doi: 10.1080/2162402X.2018.1484982
160. Cerrato JA, Yung WKA, Liu TJ. Introduction of Mutant P53 Into a Wild-Type P53-Expressing Glioma Cell Line Confers Sensitivity to Ad-P53-Induced Apoptosis. Neuro Oncol (2001) 3:113–22. doi: 10.1215/15228517-3-2-113
161. Idema S, Lamfers MLM, van Beusechem VW, Noske DP, Heukelom S, Moeniralm S, et al. AdDelta24 and the P53-Expressing Variant AdDelta24-P53 Achieve Potent Anti-Tumor Activity in Glioma When Combined With Radiotherapy. J Gene Med (2007) 9:1046–56. doi: 10.1002/jgm.1113
162. Yamada S, Kanno H, Kawahara N. Trans-Membrane Peptide Therapy for Malignant Glioma by Use of a Peptide Derived From the MDM2 Binding Site of P53. J Neurooncol (2012) 109:7–14. doi: 10.1007/s11060-012-0860-1
163. Smith MA, Gorlick R, Kolb EA, Lock R, Carol H, Maris JM, et al. Initial Testing of JNJ-26854165 (Serdemetan) by the Pediatric Preclinical Testing Program. Pediatr Blood Cancer (2012) 59:329–32. doi: 10.1002/pbc.23319
164. Wang CL, Wang JY, Liu ZY, Ma XM, Wang XW, Jin H, et al. Ubiquitin-Specific Protease 2a Stabilizes MDM4 and Facilitates the P53-Mediated Intrinsic Apoptotic Pathway in Glioblastoma. Carcinogenesis (2014) 35:1500–9. doi: 10.1093/carcin/bgu015
165. Qin JJ, Li X, Hunt C, Wang W, Wang H, Zhang R. Natural Products Targeting the P53-MDM2 Pathway and Mutant P53: Recent Advances and Implications in Cancer Medicine. Genes Dis (2018) 5:204–19. doi: 10.1016/j.gendis.2018.07.002
166. Sordillo LA, Sordillo PP, Helson L. Curcumin for the Treatment of Glioblastoma. Anticancer Res (2015) 35:6373–8.
167. Liu E, Wu J, Cao W, Zhang J, Liu W, Jiang X, et al. Curcumin Induces G2/M Cell Cycle Arrest in a P53-Dependent Manner and Upregulates ING4 Expression in Human Glioma. J Neurooncol (2007) 85:263–70. doi: 10.1007/s11060-007-9421-4
168. Alonso M, Tamasdan C, Miller DC, Newcomb EW. Flavopiridol Induces Apoptosis in Glioma Cell Lines Independent of Retinoblastoma and P53 Tumor Suppressor Pathway Alterations by a Caspase-Independent Pathway. Mol Cancer Ther (2003) 2:139–50.
169. Li X, Liu C, Chen S, Hu H, Su J, Zou Y. D-Amino acid mutation of PMI as potent dual peptide inhibitors of p53-MDM2/MDMX interactions. Bioorg Med Chem Lett (2017) 27:4678–81. doi: 10.1016/j.bmcl.2017.09.014
170. Chen X, Tai L, Gao J, Qian J, Zhang M, Li B, et alA Stapled Peptide Antagonist of MDM2 Carried by Polymeric Micelles Sensitizes Glioblastoma to Temozolomide Treatment Through P53 Activation. J Control Release (2015) 218:29–35. doi: 10.1016/j.jconrel.2015.09.061
171. Chen S, Li X, Yuan W, Zou Y, Guo Z, Chai Y, et al. Rapid Identification of Dual P53-MDM2/MDMX Interaction Inhibitors Through Virtual Screening and Hit-Based Substructure Search. RSC Adv (2017) 7:9989–97. doi: 10.1039/C7RA00473G
172. Sarisozen C, Tan Y, Liu J, Bilir C, Shen L, Filipzcak N, et al. MDM2 Antagonist-Loaded Targeted Micelles in Combination With Doxorubicin: Effective Synergism Against Human Glioblastoma via P53 Re-Activation. J Drug Target (2019) 27:624–33. doi: 10.1080/1061186X.2019.1570518
173. Li H, Liu Y, Jiao Y, Guo A, Xu X, Qu X, et al. Resveratrol Sensitizes Glioblastoma-Initiating Cells to Temozolomide by Inducing Cell Apoptosis and Promoting Differentiation. Oncol Rep (2016) 35:343–51. doi: 10.3892/or.2015.4346
174. Tong H, Zhao K, Zhang J, Zhu J, Xiao J. YB-1 Modulates the Drug Resistance of Glioma Cells by Activation of MDM2/p53 Pathway. Drug Des Devel Ther (2019) 13:317–26. doi: 10.2147/DDDT.S185514
175. Merlino F, Daniele S, La Pietra V, Di Maro S, Di Leva FS, Brancaccio D, et al. Simultaneous Targeting of RGD-Integrins and Dual Murine Double Minute Proteins in Glioblastoma Multiforme. J Med Chem (2018) 61:4791–809. doi: 10.1021/acs.jmedchem.8b00004
176. Daniele S, Costa B, Zappelli E, Da Pozzo E, Sestito S, Nesi G, et al. Combined Inhibition of AKT/mTOR and MDM2 Enhances Glioblastoma Multiforme Cell Apoptosis and Differentiation of Cancer Stem Cells. Sci Rep (2015) 5:1–14. doi: 10.1038/srep09956
177. Wang B, Peng F, Huang W, Zhou J, Zhang NRational Drug Design, Synthesis, and Biological Evaluation of Novel Chiral Tetrahydronaphthalene-Fused Spirooxindole as MDM2-CDK4 Dual Inhibitor Against Glioblastoma.Acta Pharm Sin B (2020) 10:1492–510. doi: 10.1016/j.apsb.2019.12.013
178. Daniele S, La Pietra V, Piccarducci R, Pietrobono D, Cavallini C, D’Amore VM, et al. CXCR4 Antagonism Sensitizes Cancer Cells to Novel Indole-Based MDM2/4 Inhibitors in Glioblastoma Multiforme. Eur J Pharmacol (2021) 897:173936. doi: 10.1016/j.ejphar.2021.173936
179. Schneider LS, Ulrich M, Lehr T, Menche D. MDM2 Antagonist Nutlin-3a Sensitizes Tumors to V-ATPase Inhibition. Mol Oncol (2016) 10:1054–62. doi: 10.1016/j.molonc.2016.04.005
180. Merkel CA, da Silva Soares RB, de Carvalho ACV, Zanatta DB, Bajgelman MC, Fratini P, et al. Activation of Endogenous P53 by Combined p19Arf Gene Transfer and Nutlin-3 Drug Treatment Modalities in the Murine Cell Lines B16 and C6. BMC Cancer (2010) 10:361. doi: 10.1186/1471-2407-10-316
181. Vassilev L, Vu B, Graves B, Klein C, Fotouhi N, Liu E. In Vivo Activation of the P53 Pathway by Small-Molecule Antagonists of MDM2. Science (2004) 303:844–8. doi: 10.1126/science.1092472
182. Vu B, Wovkulich P, Pizzolato G, Lovey A, Ding Q, Jiang N, et al. Discovery of RG7112: A Small-Molecule MDM2 Inhibitor in Clinical Development. ACS Med Chem Lett (2013) 4:466–9. doi: 10.1021/ml4000657
183. Carol H, Reynolds P, Kang M, Keir ST, Maris JM, Houghton PJ, et al. Initial Testing of the MDM2 Inhibitor RG7112 by the Pediatric Preclinical Testing Program. Pediatr Blood Cancer (2012) 60:633–41. doi: 10.1002/pbc.24235
184. Andreeff M, Kelly KR, Yee K, Assouline S, Strair R, Popplewell L, et al. Results of the Phase I Trial of RG7112, a Small-Molecule MDM2 Antagonist in Leukemia. Clin Cancer Res (2016) 22:868–76. doi: 10.1158/1078-0432.CCR-15-0481
185. Kurzrock R, Blay J-Y, Bui Nguyen B, Wagner AJ, Maki RG, Schwartz GK, et al. A phase I Study of MDM2 Antagonist RG7112 in Patients (Pts) With Relapsed/Refractory Solid Tumors. J Clin Oncol (2012) 30:e13600. doi: 10.1200/jco.2012.30.15_suppl.e13600
186. Ray-Coquard I, Blay J-Y, Italiano A, Le Cesne A, Penel N, Zhi J, et al. Effect of the MDM2 Antagonist RG7112 on the P53 Pathway in Patients With MDM2-Amplified, Well-Differentiated or Dedifferentiated Liposarcoma: An Exploratory Proof-of-Mechanism Study. Lancet Oncol (2012) 13:1133–40. doi: 10.1016/S1470-2045(12)70474-6
187. Khurana A, Shafer DA. MDM2 Antagonists as a Novel Treatment Option for Acute Myeloid Leukemia: Perspectives on the Therapeutic Potential of Idasanutlin (RG7388). Onco Targets Ther (2019) 12:2903–10. doi: 10.2147/OTT.S172315
188. Ding Q, Zhang Z, Liu JJ, Jiang N, Zhang J, Ross TM, et al. Discovery of RG7388, a Potent and Selective P53-MDM2 Inhibitor in Clinical Development. J Med Chem (2013) 56:5979–83. doi: 10.1021/jm400487c
189. Tovar C, Rosinski J, Filipovic Z, Higgins B, Kolinsky K, Hilton H, et al. Small-Molecule MDM2 Antagonists Reveal Aberrant P53 Signaling in Cancer: Implications for Therapy. Proc Natl Acad Sci USA (2006) 103:1888–93. doi: 10.1073/pnas.0507493103
190. Montesinos P, Beckermann BM, Catalani O, Esteve J, Gamel K, Konopleva MY, et al. MIRROS: A Randomized, Placebo-Controlled, Phase III Trial of Cytarabine ± Idasanutlin in Relapsed or Refractory Acute Myeloid Leukemia. Future Oncol (2020) 16:807–15. doi: 10.2217/fon-2020-0044
191. Erba HP, Becker PS, Shami PJ, Grunwald MR, Flesher DL, Zhu M, et al. Phase 1b Study of the MDM2 Inhibitor AMG 232 With or Without Trametinib in Relapsed/Refractory Acute Myeloid Leukemia. Blood Adv (2019) 3:1939–49. doi: 10.1182/bloodadvances.2019030916
192. de Jonge M, de Weger VA, Dickson MA, Langenberg M, Le Cesne A, Wagner AJ, et al. A Phase I Study of SAR405838, a Novel Human Double Minute 2 (HDM2) Antagonist, in Patients With Solid Tumours. Eur J Cancer (2017) 76:144–51. doi: 10.1016/j.ejca.2017.02.005
193. Holzer P, Masuya K, Furet P, Kallen J, Valat-Stachyra T, Ferretti S, et al. Discovery of a Dihydroisoquinolinone Derivative (NVP-CGM097): A Highly Potent and Selective MDM2 Inhibitor Undergoing Phase 1 Clinical Trials in P53wt Tumors. J Med Chem (2015) 58:6348–58. doi: 10.1021/acs.jmedchem.5b00810
194. Furet P, Masuya K, Kallen J, Stachyra-Valat T, Ruetz S, Guagnano V, et al. Discovery of a Novel Class of Highly Potent Inhibitors of the P53–MDM2 Interaction by Structure-Based Design Starting From a Conformational Argument. Bioorg Med Chem Lett (2016) 26:4837–41. doi: 10.1016/j.bmcl.2016.08.010
195. Bauer S, Demetri G, Jeay S, Dummer R, Guerreiro N, Tan DS, et al. Open-Label, Multi-Center, Dose Escalation Study of Oral NVP-CGM097, a P53/HDM2-Protein-Protein Interaction Inhibitor, in adult patients with selected advanced solid tumors. Ann Oncol (2016) 27:vi116. doi: 10.1093/annonc/mdw368.09
196. Ravandi F, Gojo I, Patnaik M, Minden M, Kantarjian H, Knox C, et al. A Phase I Trial of the Human Double Minute 2 Inhibitor (MK-8242) in Patients With Refractory/Recurrent Acute Myelogenous Leukemia (AML). Leuk Res (2016) 48:92–100. doi: 10.1016/j.leukres.2016.07.004
197. Wang W, Hu Y. Small Molecule Agents Targeting the P53-MDM2 Pathway for Cancer Therapy. Med Res Rev (2012) 32:1159–96. doi: 10.1002/med.20236
198. Nag S, Qin JJ, Voruganti S, Wang MH, Sharma H, Patil S, et al. Development and Validation of a Rapid HPLC Method for Quantitation of SP-141, a Novel Pyrido[B]Indole Anticancer Agent, and an Initial Pharmacokinetic Study in Mice. BioMed Chromatogr (2015) 29:654–63. doi: 10.1002/bmc.3327
199. Chen SR, Cai WP, Dai XJ, Guo AS, Chen HP, Lin GS, et al. Research on miR-126 in Glioma Targeted Regulation of PTEN/PI3K/Akt and MDM2-P53 Pathways. Eur Rev Med Pharmacol Sci (2019) 23:3461–70. doi: 10.26355/eurrev_201904_17711
200. Silva JL, Lima CGS, Rangel LP, Ferretti GDS, Pauli FP, Ribeiro RCB, et al. Recent Synthetic Approaches Towards Small Molecule Reactivators of P53. Biomolecules (2020) 10:635. doi: 10.3390/biom10040635
201. Puca R, Nardinocchi L, Porru M, Simon AJ, Rechavi G, Leonetti C, et al. Restoring P53 Active Conformation by Zinc Increases the Response of Mutant P53 Tumor Cells to Anticancer Drugs. Cell Cycle (2011) 10:1679–89. doi: 10.4161/cc.10.10.15642
202. Lang FF, Yung WK, Raju U, Libunao F, Terry NH, Tofilon PJ. Enhancement of Radiosensitivity of Wild-Type P53 Human Glioma Cells by Adenovirus-Mediated Delivery of the P53 Gene. J Neurosurg (1998) 89:125–32. doi: 10.3171/jns.1998.89.1.0125
203. Lang FF, Bruner JM, Fuller GN, Aldape K, Prados MD, Chang S, et al. Phase I Trial of Adenovirus-Mediated P53 Gene Therapy for Recurrent Glioma: Biological and Clinical Results. J Clin Oncol (2003) 21:2508–18. doi: 10.1200/JCO.2003.21.13.2508
204. Tabernero J, Dirix L, Schöfski P, Cervantes A, Lopez-Martin JA, Capdevila J, et al. A Phase I First-in-Human Pharmacokinetic and Pharmacodynamic Study of Serdemetan in Patients With Advanced Solid Tumors. Clin Cancer Res (2011) 17:6313–21. doi: 10.1158/1078-0432.CCR-11-1101
205. Antao AM, Tyagi A, Kim K, Ramakrishna S. Advances in Deubiquitinating Enzyme Inhibition and Applications in Cancer Therapeutics. Cancers (2020) 12:1–34. doi: 10.3390/cancers12061579
206. Ryskalin L, Biagioni F, Busceti CL, Lazzeri G, Frati A, Fornai F. The Multi-Faceted Effect of Curcumin in Glioblastoma From Rescuing Cell Clearance to Autophagy-Independent Effects. Molecules (2020) 25:1–17. doi: 10.3390/molecules25204839
207. Loch-Neckel G, Bicca MA, Leal PC, Mascarello A, Siqueira JM, Calixto JB. In Vitro and In Vivo Anti-Glioma Activity of a Chalcone-Quinoxaline Hybrid. Eur J Med Chem (2015) 90:93–100. doi: 10.1016/j.ejmech.2014.11.014
208. Bernal F, Wade M, Godes M, Davis TN, Whitehead DG, Kung AL, et al. A Stapled P53 Helix Overcomes HDMX-Mediated Suppression of P53. Cancer Cell (2010) 18:411–22. doi: 10.1016/j.ccr.2010.10.024
209. Brown CJ, Lain S, Verma CS, Fersht AR, Lane DP. Awakening Guardian Angels: Drugging the P53 Pathway. Nat Rev Cancer (2009) 9:862–73. doi: 10.1038/nrc2763
210. Graves B, Thompson T, Xia M, Janson C, Lukacs C, Deo D, et al. Activation of the P53 Pathway by Small-Molecule- Induced MDM2 and MDMX Dimerization. Proc Natl Acad Sci USA (2012) 109:11788–93. doi: 10.1073/pnas.1203789109
211. Popowicz GM, Czarna A, Wolf S, Wang K, Wang W, Dömling A, et al. Structures of Low Molecular Weight Inhibitors Bound to MDMX and MDM2 Reveal New Approaches for P53-MDMX/MDM2 Antagonist Drug Discovery Structures of Low Molecular Weight Inhibitors Bound to MDMX and MDM2 Reveal New Approaches for P53-MDMX/MDM2 Antagon. Cell Cycle (2010) 9:1104–11. doi: 10.4161/cc.9.6.10956
212. Liu M, Li C, Pazgier M, Li C, Mao Y, Lv Y, et alD-Peptide Inhibitors of the P53-MDM2 Interaction for Targeted Molecular Therapy of Malignant Neoplasms.Proc Natl Acad Sci USA (2010) 107:14321–6. doi: 10.1073/pnas.1008930107
213. Chang YS, Graves B, Guerlavais V, Tovar C, Packman K, To K-H, et al. Stapled α–Helical Peptide Drug Development: A Potent Dual Inhibitor of MDM2 and MDMX for P53-Dependent Cancer Therapy. Proc Natl Acad Sci USA (2013) 110:E3445. doi: 10.1073/pnas.1303002110
214. Chen X, Tai L, Gao J, Qian J, Zhang M, Li B, et al. A Stapled Peptide Antagonist of MDM2 Carried by Polymeric Micelles Sensitizes Glioblastoma to Temozolomide Treatment Through P53 Activation. J Control Release (2015) 218:29–35. doi: 10.1016/j.jconrel.2015.09.061
215. Michaelis M, Rothweiler F, Schneider C, Rothenburger T, Mernberger M, Nist A, et al. Long-Term Cultivation Using Ineffective MDM2 Inhibitor Concentrations Alters the Drug Sensitivity Profiles of PL21 Leukaemia Cells. BioMed Sci (2020) 1:1–10. doi: 10.1017/exp.2019.1
216. Zhuang C, Miao Z, Wu Y, Guo Z, Li J, Yao J, et al. Double-Edged Swords as Cancer Therapeutics: Novel, Orally Active, Small Molecules Simultaneously Inhibit P53-MDM2 Interaction and the NF-κb Pathway. J Med Chem (2014) 57:567–77. doi: 10.1021/jm401800k
217. Gu L, Zhang H, Liu T, Zhou S, Du Y, Xiong J, et al. Discovery of Dual Inhibitors of MDM2 and XIAP for Cancer Treatment. Cancer Cell (2016) 30:623–36. doi: 10.1016/j.ccell.2016.08.015
218. He S, Dong G, Wu S, Fang K, Miao Z, Wang W, et al. Small Molecules Simultaneously Inhibiting P53-Murine Double Minute 2 (MDM2) Interaction and Histone Deacetylases (HDACs): Discovery of Novel Multitargeting Antitumor Agents. J Med Chem (2018) 61:7245–60. doi: 10.1021/acs.jmedchem.8b00664
219. Saiki AY, Caenepeel S, Yu D, Lofgren JA, Osgood T, Robertson R, et al. MDM2 Antagonists Synergize Broadly and Robustly With Compounds Targeting Fundamental Oncogenic Signaling Pathways. Oncotarget (2014) 5:2030–43. doi: 10.18632/oncotarget.1918
220. Fang DD, Tang Q, Kong Y, Wang Q, Gu J, Fang X, et al. MDM2 Inhibitor APG-115 Synergizes With PD-1 Blockade Through Enhancing Antitumor Immunity in the Tumor Microenvironment. J Immunother Cancer (2019) 7:327. doi: 10.1186/s40425-019-0750-6
221. Wang H, Mu X, He H, Zhang XD. Cancer Radiosensitizers. Trends Pharmacol Sci (2018) 39:24–48. doi: 10.1016/j.tips.2017.11.003
222. Wang H, Cai S, Bailey BJ, Saadatzadeh MR, Ding J, Tonsing-Carter E, et al. Combination Therapy in a Xenograft Model of Glioblastoma: Enhancement of the Antitumor Activity of Temozolomide by an MDM2 Antagonist. J Neurosurg (2017) 126:446–59. doi: 10.3171/2016.1.JNS152513
223. Huang H, Lin H, Zhang X, Li J. Resveratrol Reverses Temozolomide Resistance by Downregulation of MGMT in T98G Glioblastoma Cells by the NF- κ B-Dependent Pathway. Oncol Rep (2012) 27:2050–6. doi: 10.3892/or.2012.1715
224. Mayo LD, Donner DB. A Phosphatidylinositol 3-KinaseAkt Pathway Promotes Translocation of Mdm2 From the Cytoplasm to the Nucleus. Proc Natl Acad Sci USA (2001) 98:11598–603. doi: 10.1073/pnas.181181198
225. Trotman L, Pandolfi P. PTEN and P53 : Who Will Get the Upper Hand ? Cancer Cell (2003) 3:97–9. doi: 10.1016/S1535-6108(03)00022-9
226. Laroche A, Chaire V, Algeo M, Karanian M, Fourneaux B, Italiano A. MDM2 Antagonists Synergize With PI3K/mTOR Inhibition in Well- Differentiated/Dedifferentiated Liposarcomas. Oncotarget (2017) 8:53968–77. doi: 10.18632/oncotarget.16345
227. Abraham AG, O’Neill E. PI3K/Akt-Mediated Regulation of P53 in Cancer. Biochem Soc Trans (2014) 42:798–803. doi: 10.1042/BST20140070
228. The Cancer Genome Atlas Program [Internet]. Available at: https://www.cancer.gov/about-nci/organization/ccg/research/structural-genomics/tcga.
229. Kojima K, Shimanuki M, Shikami M, Samudio I, Andreeff M, Nakakuma H. The Dual PI3 Kinase/mTOR Inhibitor PI-103 Prevents P53 Induction by Mdm2 Inhibition But Enhances P53-Mediated Mitochondrial Apoptosis in P53 Wild-Type AML. Leukemia (2008) 22:1728–36. doi: 10.1038/leu.2008.158
230. Wang B, Peng F, Huang W, Zhou J, Zhang N. Rational Drug Design, synthesis, and biological evaluation of novel chiral tetrahydronaphthalene-fused spirooxindole as MDM2-CDK4 dual inhibitor against glioblastoma Acta Pharmaceutica Sinica B (2020) 10(8):1492–510. doi: 10.1016/j.apsb.2019.12.013
231. Wang H, Xu T, Jiang Y, Yan Y, Fu D, Chen J. The Challenges and the Promise of Molecular Targeted Therapy in Malignant Glioma. Neoplasia (2015) 17:239–55. doi: 10.1016/j.neo.2015.02.002
232. Wilczewska AZ, Niemirowicz K, Markiewicz KH, Car H. Nanoparticles as Drug Delivery Systems. Pharmacol Rep (2012) 64:1020–37. doi: 10.1016/S1734-1140(12)70901-5
233. D’Amico RS, Aghi MK, Vogelbaum MA, Bruce JN. Convection-Enhanced Drug Delivery for Glioblastoma: A Review. J Neurooncol (2021) 151:415–27. doi: 10.1007/s11060-020-03408-9
234. Bouska A, Eischen CM. Mdm2 Affects Genome Stability Independent of P53. Cancer Res (2009) 69:1697–701. doi: 10.1158/0008-5472.CAN-08-3732
235. Frosina G, Marubbi D, Marcello D, Vecchio D, Daga A. Critical Reviews in Oncology Hematology The Efficacy and Toxicity of ATM Inhibition in Glioblastoma Initiating Cells- Driven Tumor Models. Crit Rev Oncol Hematol (2019) 138:214–22. doi: 10.1016/j.critrevonc.2019.04.015
236. Timme CR, Rath BH, O’Neill JW, Camphausen K, Tofilon PJ. The DNA-PK Inhibitor VX-984 Enhances the Radiosensitivity of Glioblastoma Cells Grown In Vitro and as Orthotopic Xenografts. Mol Cancer Ther (2018) 17:1207–16. doi: 10.1158/1535-7163.MCT-17-1267
237. Schiebe M, Ohneseit P, Hoffmann W, Meyermann R, Rodemann H-P, Bamberg M. Analysis of Mdm2 and P53 Gene Alterations in Glioblastomas and its Correlation With Clinical Factors. J Neurooncol (2000) 49:197–203. doi: 10.1023/A:1006410702284
238. Limam S, Missaoui N, Abdessayed N, Mestiri S, Selmi B, Mokni M, et al. Prognostic Significance of MGMT Methylation and Expression of MGMT, P53, EGFR, MDM2 and PTEN in Glioblastoma Multiforme. Ann Biol Clin (2019) 77:307–17. doi: 10.1684/abc.2019.1448
239. Liu Z, Miao Z, Li J. A Fluorescent Probe for Imaging p53 – MDM2 Protein – Protein Interaction. Chem Biol Drug Des (2014) 85:411–7. doi: 10.1111/cbdd.12434
240. Chitneni SK, Zhou Z, Watts BE, Zalutsky MR. Feasibility of Developing Radiotracers for MDM2: Synthesis and Preliminary Evaluation of an 18F-Labeled Analogue of the MDM2 Inhibitor SP-141. Pharmaceuticals (2021) 14:358. doi: 10.3390/ph14040358
Keywords: p53, MDM2 & MDMX, radiation, glioblastoma, radiosensitizer, radiotherapy, targeted therapy
Citation: Miles X, Vandevoorde C, Hunter A and Bolcaen J (2021) MDM2/X Inhibitors as Radiosensitizers for Glioblastoma Targeted Therapy. Front. Oncol. 11:703442. doi: 10.3389/fonc.2021.703442
Received: 30 April 2021; Accepted: 24 June 2021;
Published: 08 July 2021.
Edited by:
Lorenzo Manti, University of Naples Federico II, ItalyReviewed by:
Géza Sáfrány, Frédéric Joliot-Curie National Research Institute for Radiobiology and Radiohygiene, HungaryCopyright © 2021 Miles, Vandevoorde, Hunter and Bolcaen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Julie Bolcaen, amJvbGNhZW5AdGxhYnMuYWMuemE=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.