 Miriam Gaggianesi1†
Miriam Gaggianesi1† Simone Di Franco1†
Simone Di Franco1† Vincenzo Davide Pantina1
Vincenzo Davide Pantina1 Gaetana Porcelli2
Gaetana Porcelli2 Caterina D'Accardo2
Caterina D'Accardo2 Francesco Verona2
Francesco Verona2 Veronica Veschi1
Veronica Veschi1 Lorenzo Colarossi3Naida Faldetta4Giuseppe Pistone2Maria Rita Bongiorno2
Lorenzo Colarossi3Naida Faldetta4Giuseppe Pistone2Maria Rita Bongiorno2 Matilde Todaro2
Matilde Todaro2 Giorgio Stassi1*
Giorgio Stassi1*- 1Department of Surgical Oncological and Stomatological Sciences (DICHIRONS), University of Palermo, Palermo, Italy
- 2Department of Health Promotion Sciences, Internal Medicine and Medical Specialties (PROMISE), University of Palermo, Palermo, Italy
- 3Pathology Unit, Mediterranean Institute of Oncology, Catania, Italy
- 4Department of Surgery, Villa Sofia-Cervello Hospital, Palermo, Italy
Despite the recent advances in cancer patient management and in the development of targeted therapies, systemic chemotherapy is currently used as a first-line treatment for many cancer types. After an initial partial response, patients become refractory to standard therapy fostering rapid tumor progression. Compelling evidence highlights that the resistance to chemotherapeutic regimens is a peculiarity of a subpopulation of cancer cells within tumor mass, known as cancer stem cells (CSCs). This cellular compartment is endowed with tumor-initiating and metastasis formation capabilities. CSC chemoresistance is sustained by a plethora of grow factors and cytokines released by neighboring tumor microenvironment (TME), which is mainly composed by adipocytes, cancer-associated fibroblasts (CAFs), immune and endothelial cells. TME strengthens CSC refractoriness to standard and targeted therapies by enhancing survival signaling pathways, DNA repair machinery, expression of drug efflux transporters and anti-apoptotic proteins. In the last years many efforts have been made to understand CSC-TME crosstalk and develop therapeutic strategy halting this interplay. Here, we report the combinatorial approaches, which perturb the interaction network between CSCs and the different component of TME.
Introduction
Despite huge progress has been made in the development and optimization of anti-tumor therapies, cancer remains the second leading cause of death worldwide. Intra- and inter-tumor heterogeneity represents the main hurdle for cancer treatment. For this reason, the comprehension of the molecular and phenotypic differences among different cancer types may help to improve the prognosis of cancer patients upon therapy. Two models have been proposed to explain the origin of tumor heterogeneity (1). According to the stochastic model, each cell within the tumor mass can become tumorigenic by acquiring specific (epi)genetic alterations. Conversely, in the hierarchical model tumor heterogeneity arises from a subpopulation of cancer cells, termed cancer stem cells (CSCs), able to self-renew and differentiate into phenotypically and functionally distinct cells. CSCs share most of the features with normal stem cells, but their self-renewal capacity is typically deregulated (2, 3). Therefore, CSCs represent the roots which feed tumor initiation and sustain metastatic spread, therapeutic resistance and recurrence (4, 5). Chemotherapy is a pivotal treatment for solid tumors and aims to counteract all the active proliferative cells, including both healthy and malignant cells (6). Compelling evidence have demonstrated that CSCs are endowed with i) high expression of ATP-binding cassette (ABC) transporter and anti-apoptotic molecules, ii) aberrant activation of proliferative and survival signaling pathway and iii) a proficient DNA repair machinery are the main mechanisms inducing multidrug resistance (MDR) (1). Interestingly, recent studies have shown that tumor microenvironment (TME) could generate a protective niche for tumor cells from drugs, leading to chemoresistance. In addition to the intrinsic characteristics of CSCs, the interaction with the TME must be taken into account because it is involved in the regulation of signaling pathway and resistance to therapy, representing a potential target for novel therapeutic approaches (7). In this review, we will illustrate TME protective effects against chemotherapic drugs and the most updated strategies for targeted therapies alone or in combination to disrupt the CSCs/TME interaction.
Cancer Stem Cells as a Shield to Elude Chemotherapeutic Agents
Different hypotheses have been made about the origin of CSCs, as a direct consequence of (epi)genetic alterations in the healthy stem cell compartment, or from progenitor/differentiated cells through the dysregulation of stemness-related pathways (8).
The pioneering studies conducted by Tilland McCulloch in early ‘60s demonstrated the existence of hematopoietic stem cells, opening the era of stem cell research (9). Later in 1994, Lapidot et al., provided the first evidence of CSC presence in acute myeloid leukemia (AML). AML cells were fractioned according to the expression of cell surface markers CD38 and CD34 and the obtained different subpopulations were injected into immunocompromised mice. They noticed that only the CD34+/CD38- subpopulation was able to engraft in mice reflecting many features of human AML (10, 11). The first demonstration of CSC existence in solid tumors was provided in breast cancer (BC) (12) and later in brain, colon, thyroid and other tumors (13, 14), pointing out that cancer cell transplantation into immunocompromised mice is the gold standard assay to identify and characterize CSCs (15). Compelling evidence point out that CSCs are responsible for the failure of the conventional therapies, due to aberrant activation of signaling pathways, high expression of efflux transporters/anti-apoptotic molecules, and enhanced DNA-damage repair machinery (4, 16, 17).
Stemness-Related Pathways Involved in CSC Chemoresistance
Deregulation of developmental and proliferative pathways, such as Hedgehog (HH), Wnt/β-catenin and Hippo, sustains CSC growth and chemoresistance (18). The HH pathway has been shown to regulate the properties of CSCs in various neoplasms through the up-regulation of stemness-related genes (Nanog, Oct4, Sox2 and Bmi1) (19, 20). In colorectal cancer (CRC) HH-GLI pathway activation fostered CSC survival and sustained in vivo growth and metastatic ability (21). In BC, the CD44+/CD24- subpopulation isolated from tumor xenografts displayed high expression levels of HH signaling molecules compared to more differentiated cell subsets (22). In glioma, the activation of Notch and HH pathway mediated the resistance to temozolomide treatment in CD133+ CSCs (23). Aberrant activation of the Wnt/β-catenin signaling pathway has been mainly linked to development of CRC (24, 25) and detected also in other tumor types, as hepatocellular and BC (26). Recently, it has been demonstrated that knockdown of Wnt1 decreases the expression of CD44, Aldehyde dehydrogenase 1 (ALDH1) and Sca-1 stemness genes, thus leading to the reduction of CSC subpopulation and tumor sphere formation in BC cells (27). Several studies linked Wnt/β-catenin signaling and chemoresistance (28). The overexpression of Frizzled1 (FZD1), a receptor of Wnt ligands, increased ABCB1 transporter and mediates MDR in neuroblastoma and BC (29, 30). Moreover, LGR5, a Wnt target gene, promoted resistance to 5-fluoruracil (5-FU) treatment in CSCs (31, 32). Recent studies have revealed a complex crosstalk between Wnt and Hippo-YAP/TAZ pathways. Hippo pathway via YAP/TAZ activation led to the induction of CSC properties in BC cells (33). In a very elegant study, Cheung et al demonstrated that the Hippo kinases LATS1/2 and MST1/2 maintain Lgr5+ CSCs phenotype and sustain the activation of Wnt/β-catenin signaling pathway in CRC (34).
Alterations of Apoptotic Pathways and DNA Damage Repair Machinery in Chemoresistant CSCs
Alterations of apoptotic pathways and DNA damage repair machinery are among the principal mechanisms underlying CSC-mediated chemoresistance. Apoptosis regulates tissue development and homeostasis and is finely regulated by a network of signals that are crucial for cell fate. The ratio between apoptotic and anti-apoptotic protein levels defines the sensitivity of malignant cells to apoptotic stimuli and contributes to CSCs resistance to anticancer treatments (35). A weakened expression of death receptors (DRs) was observed in CSCs from different tumors compared to differentiated counterparts. In AML, the CD34+ CD38- stem-like subpopulation display a lower expression of FAS and FAS ligand (FAS-L) than CD38+ differentiated cells, triggering chemoresistance (36, 37). FAS and FAS-L reduced expression was also observed in glioma stem cells (GSCs) and the use of a synthetic FAS-L, Apo010, in combination with temozolomide induced apoptosis in glioblastoma (GBM) stem-like cells (38, 39). Moreover, the use of recombinant soluble TRAIL (TNF related apoptosis inducing ligand), in combination with bortezomib reduced the colony formation capacity of GSCs and impaired tumor growth in a mouse model of GBM (40). Unfortunately, the short half-life of soluble TRAIL in plasma reduces its efficacy. An interesting approach to overcome this effect is the use of TRAIL-engineered mesenchymal stromal cells, which induce apoptosis and curtail the colony forming ability of lung and breast cancer stem-like cells (41, 42). However, CSCs usually exhibited TRAIL resistance, due to c-FLIP over-expression. In BC and GBM, c-FLIP up-regulation sustained resistance to TRAIL therapy and the use of siRNA specific for c-Flip lessened self-renewal and tumorigenic potential of breast CSCs (43–45). The inhibitor of apoptosis (IAP) proteins were found to be over-expressed in CD133+ GBM stem cells compared to the CD133- compartment and their inhibition, by using small molecules, enhanced apoptosis in γ-irradiated cells (46, 47). In GBM patients, the IAP protein, survivin, was demonstrated to be mainly expressed in patient-derived GBM stem cells compared to differentiated cells, with a predominant localization in the core of tumor mass and associated with the expression of CD133, SOX2 and MELK (48). In addition, our group demonstrated that highly chemoresistant colorectal CSCs are characterized by the autocrine production of IL-4 that boosts survivin expression (49, 50). On the other hand, the dysregulation of Bcl2 family, composed by anti-apoptotic (Bcl2, Bcl-xL and Mcl-1) and pro-apoptotic (Bak, Bax, Bid, Bim, Bic, Noxa and PUMA) factors, has been found in CSCs (51). In particular, the stem-like compartment expressed higher expression level of Bcl2 and Bcl-xL compared to differentiated cancer cells (46, 52). Moreover, in breast CSCs the activation of αvβ3/Src/Slug signaling pathway leads to inactivation of PUMA through SLUG, a PUMA repressor. The pharmacological inhibition of Src with dasatinib enhanced PUMA expression levels, reducing self-renewal and colony formation capacity and increasing sensitivity to apoptosis (53, 54). On the contrary, the interaction of PUMA with Bcl2 and Bcl-xL limited its anti-apoptotic activity and a combined treatment of Src and Bcl2 inhibitors increased apoptosis, thus reducing chemoresistance (55).
Chemotherapeutic drugs mainly target differentiated tumor cells, while sparing CSCs, characterized by a highly efficient DNA damage response (DDR) system able to repair DNA damage induced by radio- and chemotherapies (56, 57). In accordance, cisplatin (CIS) treatment led to an enrichment of CSC subpopulation in ovarian and lung cancers, confirming that chemotherapy efficiently eliminates rapidly dividing differentiated/progenitor cells (58, 59). DNA damage promoted the activation of ataxia-telangiectasia-mutated (ATM), Rad17, Chk1 and Chk2 checkpoint proteins. Experimental evidence showed that CD133+ GSCs are radio-resistant compared to CD133- tumor cells, due to a more efficient checkpoint protein activation in response of DNA damage (60). Another study reported that GBM stem cells after irradiation increase the expression levels of L1CAM (CD171), which in turn up-regulates NBS1, an important component of MRN complex implicating in the early activation of ATM in response to DNA damage (61). Knockdown of L1CAM reduced the activating phosphorylation of ATM and Chk2 in response to IR-induced DNA damage, sensitizing GBM stem cells to radiation and reducing in vitro tumor sphere formation (62). In addition to GSCs, alteration of DDR pathway has been described in CSCs from different tumor types, including CRC (63). CD133+ lung cancer cells are resistant to ionizing radiation treatment due to an up-regulation of genes involved in double strand break repair, such as Rad51, BRCA1 and Exo1 (64). Moreover, invasive CD133+ stem-like cells isolated from pancreatic cancer cell lines displayed higher expression levels of gene involved in the BRCA1-mediated DNA repair pathway and resistance to gemcitabine (GEM) treatment compared to CD133- subpopulation (65). In a syngeneic p53null mice mammary gland tumor model, the Lin-/CD29High/CD24High subpopulation was characterized by increased expression levels of DDR and DNA repair genes (66). Furthermore, Liu et al. demonstrated that CSCs, isolated from BRCA1-mutant BC cell lines, displayed resistance to PARP inhibitors and were characterized by the overexpression of RAD51. The use of a shRNA targeting RAD51 sensitized triple negative BC cells to olaparib treatment (67).
Another mechanism of CSC resistance to anticancer therapies is represented by the up-regulation of detoxifying enzymes and drug efflux pump expression levels. ALDH superfamily is responsible for oxidizing aldehydes to carboxylic acids and retinol to retinoic acid allowing the detoxification from drug and the reactive oxygen species (ROS). ALDH1 is the main isoform of the ALDH superfamily enzymes and is one of the markers used for the identification of the CSCs (68–71). In BC patients, ALDH1-positive CSCs were selected after neoadjuvant treatment and their presence within the tumor could predict resistance to chemotherapy (72). In breast CSCs, the resistance to doxorubicin and paclitaxel treatment is related to the over-expression of ABCB1 efflux pump (73). Moreover, ABCB1 confers resistance to carfilzomib in multiple myeloma stem cells (74). Indeed, high expression levels of ABCB5 were found in malignant melanoma initiating cell resistant to doxorubicin treatment (75) (Table 1).
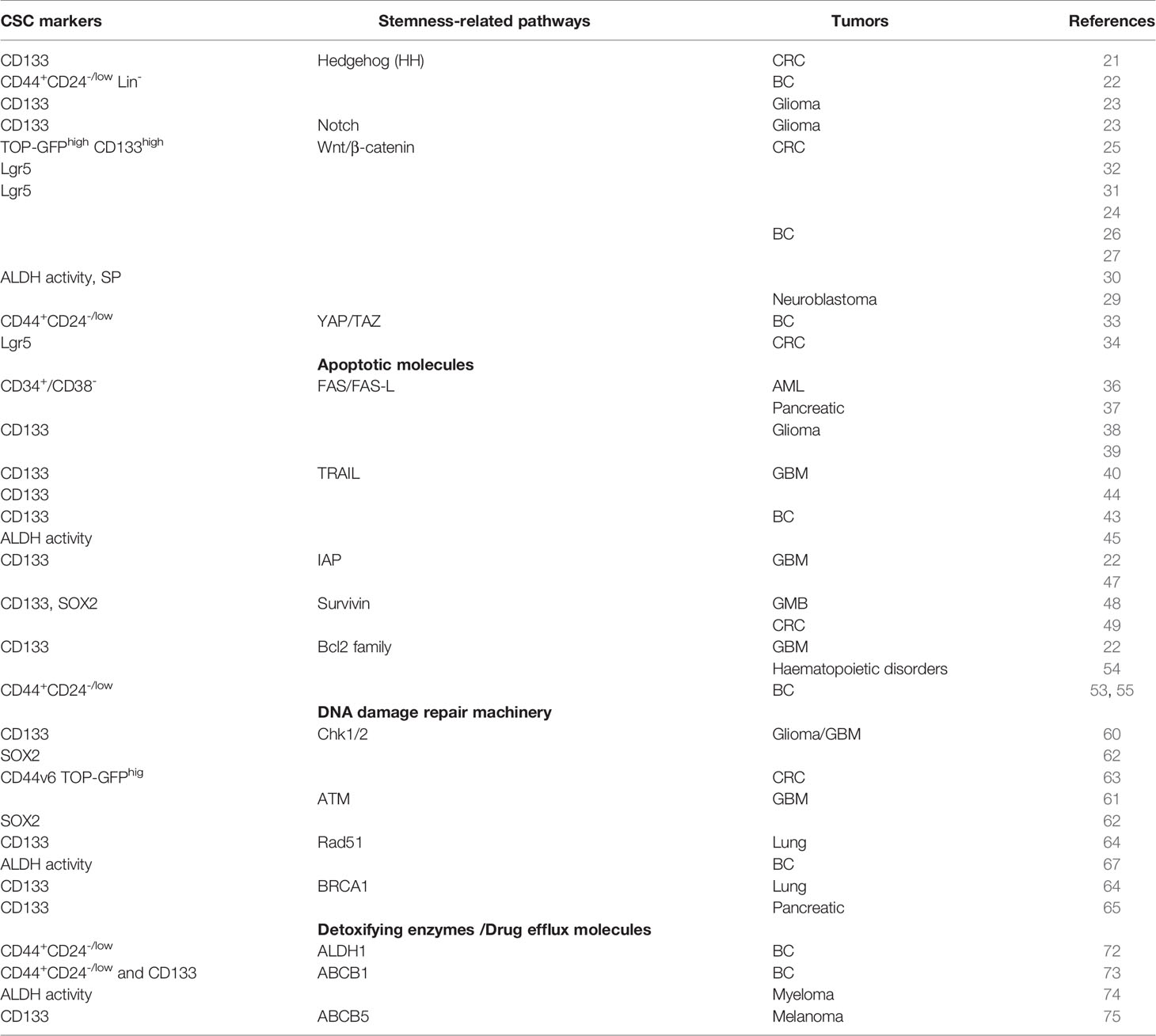
Table 1 Cancer stem cell biomarkers correlated to chemoresistance.
The Cancer Stem Cells-Tumor Microenvironment Interaction: a Hidden Hurdle in Chemotherapy Efficacy
CSCs require the cooperation of surrounding microenvironmental cells to promote tumor initiation, metastasis formation and drug resistance. Recent evidence highlighted the importance of TME cell education and recruitment as essential events for tumor dissemination. In fact, cancer cells prime stromal cells, which in turn maintain and boost CSC subpopulation (76). In particular, CSC features are regulated by autocrine and paracrine interactions between tumor cells and TME, mainly composed by extracellular matrix, cancer-associated fibroblasts (CAFs), cancer-associated adipocytes (CAAs), immune and endothelial cells. In addition to the intrinsic characteristic of CSCs, the understanding of tumor-TME cell interactions could provide actionable candidates for the development of novel therapeutic approaches (Figure 1).
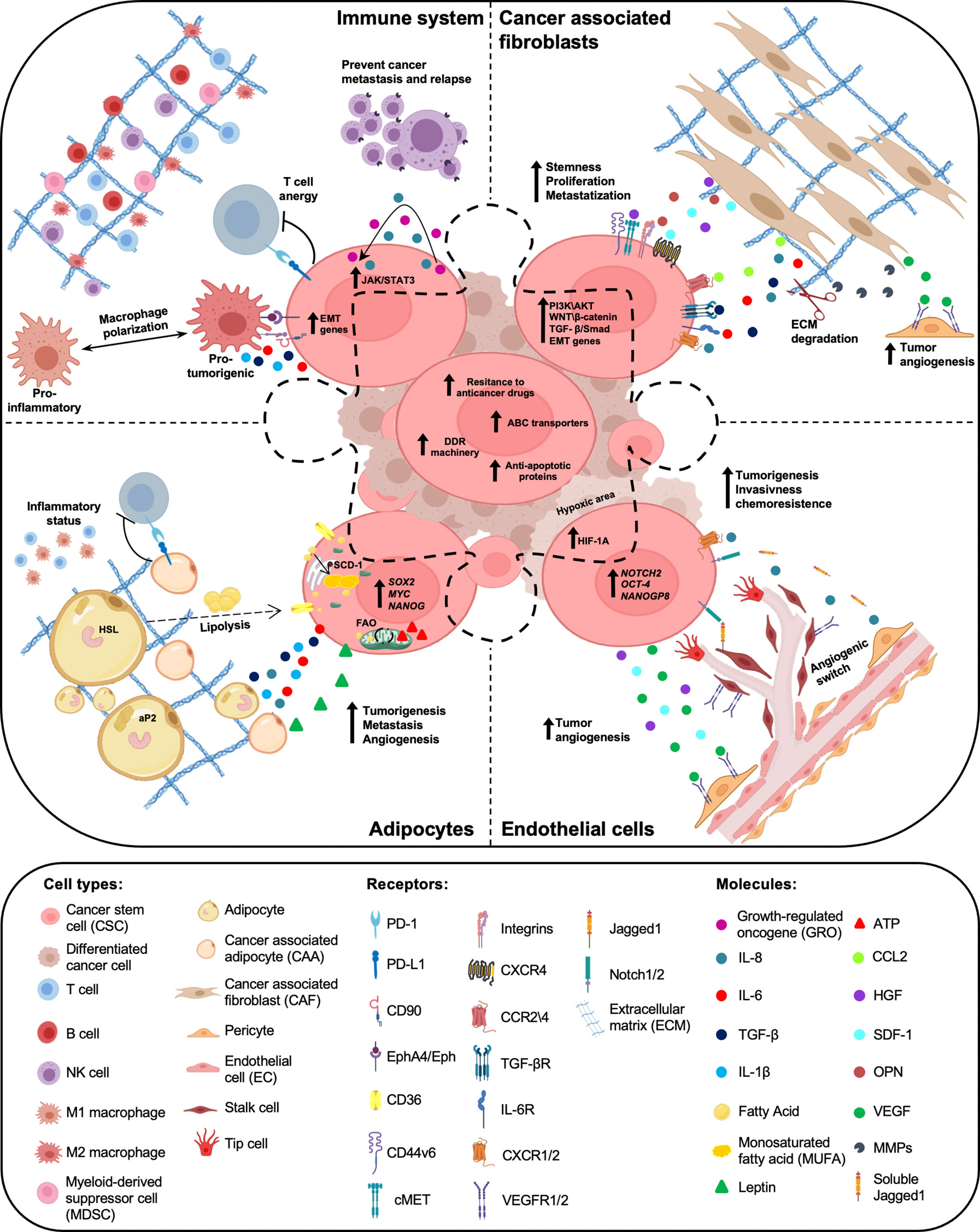
Figure 1 Crosstalk between cancer stem cells (CSCs) and tumor microenvironment (TME) components. Within the tumor mass, a subpopulation of cancer cells, called cancer stem cells (CSCs), are endowed with high resistance to anticancer therapies, due to elevated expression levels of ABC transporters, anti-apoptotic proteins and a proficient DNA damage repair (DDR) machinery. Tumor microenvironment (TME), mainly composed by cancer associated fibroblasts (CAFs), adipocytes, immune and endothelial cells, has a key role in the maintenance of CSC peculiarities. Cytokines and chemokines produced by both CSCs and TME cells boost cancer cell growth, prompt chemoresistance and promote tumor progression and relapse.
CAF Role in Inducing CSC-Mediated Resistance to Therapy
CAFs are the major component of TME involved in the complex network of tumor-stroma evolution and tumorigenesis (77, 78). Several studies demonstrated that CAFs can originate from the activation of resident fibroblast or derive from the conversion of adipocytes, endothelial cells, pericytes and bone marrow-derived mesenchymal stem cells (79). CAFs provide numerous molecules, soluble factors and proteases playing an important role in ECM synthesis or remodeling, but also pro-inflammatory cytokines, chemokines, and growth factors. Notably, CAF secreted factors are involved in a tight crosstalk with CSCs, governing their self-renewal capacity, plasticity and chemoresistance (80–85). Several in vitro experiments highlighted that CAFs guarantee a CSC reservoir in different tumors, such as breast, lung, colorectal, gastric and liver, enhancing stem markers expression (CD44, Sox2, Bmi-1), sphere formation, self-renewal and sustaining CSC pool expansion (86–90). Moreover, CAF conditioned medium influenced the tumorigenic behavior and the aggressiveness of CSCs (91). For these reasons, CAFs represent a cellular subtype on which particular attention is being paid to predict patients’ outcome and to design new target therapies.
CAF-secreted factors, which include chemokines, cytokines, growth factors, proteins and exosomes, influence and sustain CSCs aggressiveness by modulating their stemness features. CCL2 supported CSC self-renewal activating NOTCH signaling pathway and the co-injection of CAF and breast CSCs into the mammary fat pads of NOD/SCID/IL-2Rg-null mice enhanced CSC tumorigenic potential, unveiling the CCL2 driving role in BC (83). In addition, SDF-1 interacting with its receptor (CXCR4), highly expressed on CSC surface, regulated stem phenotype through the activation of Wnt/β-catenin and PI3K/AKT signaling pathways and boosted the proliferation of CD44+/CD24- BC cells (92). In agreement, CXCR4+ cells were more prone to reach the stem phenotype and properties, in comparison to CXCR4- cells (90).
Among the variety of cytokines and growth factors secreted by CAFs, a great number of studies highlighted the IL-6 and IL-8 essential role in the maintenance of stem-like features of cancer cells and in the promotion of tumor growth, metastasis formation, and chemoresistance (93, 94).
CAFs also supported the aggressive behavior of cancer cells through the secretion of TGF-β. In different tumor types, TGF-β induced the acquisition of a stem-like phenotype, promoted EMT and chemoresistance via the activation of TGF-β/Smad signaling pathway (88, 95).
Hepatocyte growth factor (HGF), another important molecules secreted by CAFs, promotes cancer cell invasiveness. In hepatocellular carcinoma, HGF sustained cancer cell stemness through the activation of MET/FRA1/HEY1 cascade (89). Our group recently demonstrated that HGF, SDF-1, and OPN released by CAFs were able to reprogram CD44v6- progenitor cells in metastatic CD44v6+ CSCs by activating Wnt/β-catenin and PI3K/AKT signaling pathways (90).
Synthesis and remodeling of extracellular matrix (ECM) represents an important function of CAFs in TME. Malanchi et al. demonstrated in a murine BC model that lung fibroblasts, activated by infiltrating CSCs, produce periostin which boosts Wnt pathway sustaining metastatic colonization (96). Moreover, in a syngeneic BC mouse model S100A4+ lung CAFs secreted VEGF-A and tenascin-C, which mediate angiogenesis and CSC survival, respectively (97). In addition to the production of ECM components, CAFs secreted metalloproteases, a family of enzymes able to degrade and remodel ECM, favoring cancer cell invasion (98).
Numerous studies highlighted that CAFs can support CSC chemoresistance in different solid tumors. Co-culture experiments performed with freshly isolated colorectal CSCs showed that CAFs secrete high levels of TGF-β2 and IL-6, which in turn prompt the transcription of GLI-2, promoting resistance to 5-fluorouracil/oxaliplatin (5-FU/oxa) treatment (99). In breast and lung tumors, the CD10+/GPR77+ CAF subpopulation secreted both IL-6 and IL-8, which induce CSC enrichment and chemoresistance to CIS treatment (100). Moreover, in triple negative BC mice models, cancer cells reprogrammed CAFs through the secretion of HH ligand. CAFs, in turn, triggered the acquisition of chemoresistance through FGF5 secretion and the production of fibrillar collagen (101). In head and neck small cellular cancer, CAF-secreted periostin bound PTK7, a receptor expressed on cancer cell surface, favoring CSC invasion and proliferation through the activation PTK7–Wnt/β-Catenin signaling pathway. Notably, PTK7/periostin interaction enhanced erlotinib chemoresistance and the formation of lung metastasis (102). Recently, our group demonstrated that in colorectal CSCs CAF-secreted cytokines confer resistance to PI3K/AKT inhibitors (103) (Table 2). Given the key role of CAFs in both CSC maintenance and drug refractoriness, the use of therapeutic strategies blocking CAFs-CSCs crosstalk could improve patients’ survival. In resistant BC cells, trastuzumab treatment activated an IL-6/STAT-3/NF-κB inflammatory loop, which correlates with the expansion of the CSC subpopulation. The administration of an anti-IL-6 receptor antibody reverted the stem-like phenotype of tumor cells (104). Zong et al. reported that the use of MEDI5117, an anti IL-6 antibody, in combination with chemotherapy or gefitinib impairs tumor growth in mice injected with NSCLC cells. MEDI5117 also displayed robust activity against trastuzumab-resistant HER2 tumor cells by targeting the CD44+/CD24- population (119).
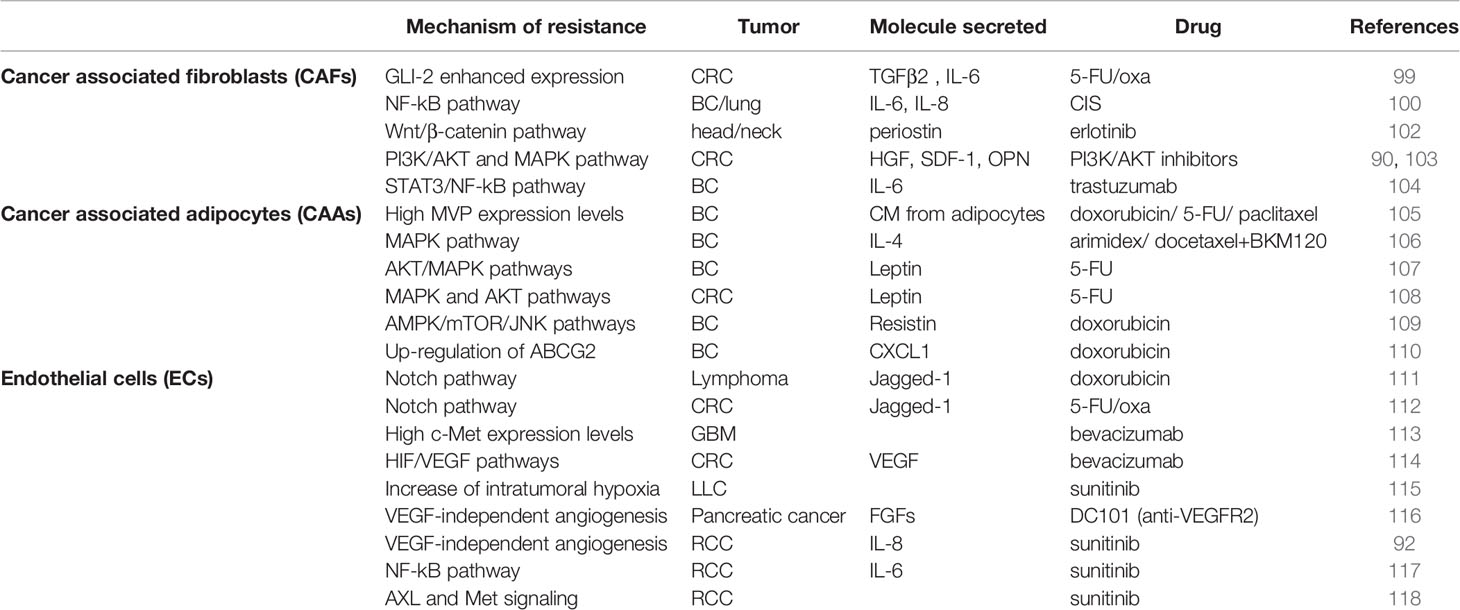
Table 2 Molecular mechanisms prompting cancer stem cell resistance to standard and targeted therapies.
A novel strategy to counteract IL-6 downstream pathway is represented by the use of specific oligonucleotide decoy specific for STAT3, which display encouraging anticancer effects. In EGFR inhibitors-resistant NSCLC cells, the treatment with a cyclic STAT3 decoy (CS3D) impaired in vitro proliferation and tumor formation (120). Likewise, AZD9150, a STAT3 antisense oligonucleotide, sustained antitumor activity in lymphoma and NSCLC preclinical models. Based on these promising results, AZD9150 was used as single agent in a Phase I clinical trial including patients with advanced lymphoma and NSCLC (121). Moreover, the double inhibition of IL-6 and IL-8 in combination with docetaxel in CD10+/GPR77+ CAFs impaired tumor growth in a patient-derived xenograft (PDX) model of BC (100). In human BC PDX, the use of a specific antibody against IL-8 receptor, CXCR-1, or an inhibitory molecule against to CXCR-1 and CXCR-2, repertaxin, favored the eradication of CSC pool, thus impeding tumor progression. In particular, ALDH+ and CD24-/CD44+ levels were reduced by ≥ 20% in 4/17 and 9/17 patients (122). Two independent research groups described that the use of CXCR-2 inhibitors, AZ13381758 and SB225002, suppresses tumor progression and hampers chemotherapy resistance in BC and pancreatic adenocarcinoma, respectively (123, 124).
The use of smoothened inhibitors (SMOi), in combination with docetaxel, in triple negative BC PDX and in a Phase I clinical trial (EDALINE) reduced metastasis formation and displayed clinical benefits, respectively (101). In addition, vismodegib, a HH inhibitor, triggered apoptosis and decreased both CAF and CSC proliferation in breast, colon and prostate cancer (125–127). In head and neck cancer, the combination of anti-PTK7 and erlotinib highly reduced tumor growth compared to single agent treatment (102).
In gastric cancer, TGFβ1 neutralizing antibody or TGFβR inhibitor (Ki26894) reduced the side population fraction, able to exclude fluorescence dye, even in presence of CAF conditioned medium (88). Alike, treatment with AMD3100 (plerixafor), a CXCR4 antagonist, blocked SDF1/CXCR4 interaction leading to a regression of CSC subpopulation in breast, colon and prostate cancer (125–127). A Phase I study on a cohort of cancer patients with worse prognosis showed that treatment with a cMET pan-inhibitor, capmatinib (INC280), displays anticancer activity in 8/44 patients (128). In preclinical studies, treatment with WNT/β-catenin inhibitors, iLGK974, Wnt-C59, and cyclosporin A, impaired CSC survival in different cancer types (129–131). In this context, we have recently demonstrated that the use of a variant of BMP7 with enhanced stability (BMP7v) induced the differentiation of CD44v6+ cells, suppressed Wnt pathway activity and sensitized CSCs to standard and target therapies (132). Recently, we demonstrated that cytokines secreted by CAFs boosted resistance to PI3K/AKT inhibitors in colorectal CSCs and this protective effect was overcome by the triple targeting of Her2, PI3K and MEK (103).
Interestingly, new therapeutic approaches focus on the direct depletion of CAFs. The targeting of FAP+ CAFs could represents a new promising target therapy (133). In agreement, FAP+ CAF inhibition and depletion with the use of the dipeptidyl peptidase inhibitor PT100 led to a reduction of the crosstalk between CAF and pro-tumorigenic immune or endothelial cells by enhancing oxa treatment efficacy in colon cancer mice models (134). Interestingly, FAP could be used as an antigen for CAR-T anticancer treatment strategy. For instance, treatment with CAR-T against FAP+ CAFs promoted growth arrest in in vivo models of lung cancer xenografts and syngeneic murine pancreatic cancers (135).
Adipocyte-Released Factors Strengthening CSC Chemotherapy Refractoriness
Adipose tissue (AT) is a specialized soft connective tissue consisting of about 90% of adipocytes cells and for the remaining part by adipose derived stem cells (ADSCs), endothelial cells, pericytes, fibroblasts and immune cells (macrophages, dendritic cells, lymphocytes). AT can be divided according to anatomic localization in three different subtypes: subcutaneous, visceral and intramuscular. In addition, adipose depots may be sub-classified in white (WAT) and brown AT (BAT), which is characterized by a dark color due to the presence of vessels and a high number of mitochondria (136). For these reasons, BAT is mainly implicated in thermogenic regulation, maintaining the appropriate balance between energy storage and consumption. AT originates from the mesoderm, whose cells give rise to adipocyte and the myogenic lineages. Specifically, white adipocytes derived from the adipogenic MYF5 negative cells, whereas brown adipocytes from myogenic MYF5 positive cells (137). White and brown cells could be discriminated in accordance with the expression of specific markers, with white adipose cells expressing leptin and S100B, lacking UCP-1 expression, and brown adipose cells characterized by PPAR gamma and UCP-1 (138). In the last years, WAT, which was traditionally considered as an energy storage tissue, due to the triglycerides and cholesterol contained in intracellular droplets, has been demonstrated to represent the biggest human endocrine organ, with the production and release of hormones, growth factors, cytokines and adipokines. Accordingly, a conspicuous secretion of these factors is observed in obesity conditions (139, 140). Nowadays, obesity represents a global health problem and constantly increases in all countries of the world (141, 142). It has been demonstrated that overweight and obesity correlate with the onset of several solid tumors, including esophagus, pancreatic, colon, breast, endometrium, ovarian and kidney, suggesting an association between these conditions and tumor initiation (143, 144). In obese subjects, adipocytes increase their dimension (hypertrophy) both in subcutaneous and visceral ATs, whereas only visceral is characterized by an increase of adipocyte number (hyperplasia) (145).
In obese conditions, WAT secrete high amount of hormones, adipokines and pro-inflammatory cytokines, such as leptin, IGF-1, HGF, TNF-α, IL1β, IL-4, IL-6, IL-8, plasminogen activator inhibitor 1 (PAI-1) and CCL-2. This promotes both a chronic inflammatory state and a tumor-permissive microenvironment, which in turn induce tumorigenesis, neo-angiogenesis via VEGF release and metastatic progression (106, 134, 146–150). Furthermore, adipokines locally recruit monocytes, macrophages, lymphocytes and other immune cells, which increase the inflammatory status in the AT particularly in obese subjects (151). The adipocytes’ role in TME has been broadly studied in the context of BC. The established crosstalk between BC cells and the close AT cells increases the production of cytokines with proinflammatory activity. Picon-Ruiz et al. demonstrated that tumor cells, after exposure to proinflammatory cytokines, are characterized by the activation of ALDH1 and an increment of mammosphere formation capacity, which are correlated with the increase of CSC number and metastasis formation in in vivo settings. These processes are driven by Src oncogene, which activates the transcription of SOX2, MYC and NANOG, well-known stem cell markers (152). In agreement with these observations, we have previously demonstrated that the release of IL-4 sustains breast CSCs invasion, tumorigenic potential, and drug resistance (106). Moreover, in obese conditions, adipocytes released elevated levels of leptin, which trigger the activation of many stemness-related molecular pathways, as Notch, Wnt/B-catenin, OCT4,SOX2, Nanog and ALDH1 up-regulation (153–156). In intestinal epithelial cells, the activation of Wnt pathway determines the expansion of crypt stem cells and favors progenitor proliferation (153–156). Breast CSCs harness higher lipid metabolism than differentiated cancer cells and used long chain fatty acids as an energy source (157). This population is characterized by an increased β-oxidation activity, which produces numerous metabolic intermediates used in ATP production (154). Recent studies have shown that ovarian and colorectal CSCs retain a high amount of fatty acids within lipid droplets to maintain their stem-like features (158). This population is rich of monounsaturated fatty acids (MUFAs), generated by stearoyl-CoA desaturase-1 (SCD1), which are metabolic markers of CSCs (159, 160). The inhibition of SCD1 decreased ovarian CSC phenotype, impairing the expression of SOX2, Nanog and Oct4, sphere forming capacity and tumorigenic potential (161–163). In melanoma, lipids released by adipocytes induced metabolic reprogramming, enhancing cell proliferation (164).
In addition, tumor cells prime peritumoral adipocytes, boosting intense lipolysis. In fact, these adipocytes, called cancer associated adipocytes (CAAs), show both in vitro and in vivo smaller cell sizes and irregular shapes with an expanded ECM and over-expression of collagen IV. They are also characterized by an increased secretion of proinflammatory factors and numerous high-energy metabolites, free fatty acids, ketone bodies, pyruvate, and lactate (165, 166). Several studies show that CAAs activate Wnt/β-catenin pathway, leading to the loss of terminal adipocyte differentiation markers such as adiponectin (APN), resistin, hormone-sensitive lipase (HSL) and adipocyte protein 2 (aP2) (166). In particular, CAAs have some characteristics of the senescence-associated secretory phenotype (SASP), such as the release of proinflammatory factors (167). CAA-released leptin determined the activation of STAT3-CPT1-fatty acid β-oxidation (FAO) in CSCs, with an increased use of fatty acids as an energy source. The in vivo blocking of this signaling pathway led to a reduction of stem-like features and a re-sensitization of breast tumor cells to chemotherapy (107).
Recent studies showed that obesity could be associated with treatment-related toxicity (168), thus, lower doses of chemotherapeutic drugs are administered to obese patients, compromising therapy efficacy and leading to resistance development (169, 170). These observations highlighted that the body max index (BMI) is not the appropriate parameter to determine the dose of chemotherapy, because it does not take into account the altered pharmacokinetics and pharmacodynamics in obese patients. A meta-analysis revealed that obese patients treated with full chemotherapy doses, estimated using actual body weight, showed lesser toxicity compared to normal weight subject (171). Lehuédé et al. observed that adipocytes promote in vitro resistance to doxorubicin, paclitaxel and 5-FU in BC cells and this phenomenon is amplified by adipocytes isolated from obese women (105). It has been demonstrated that the adipocytes, to accomplish their protective effect on BC cells treated with doxorubicin, increase the production and secretion of resistin mediating AMPK/mTOR and JNK signaling pathway activation (109). In addition, doxorubicin may influence adipocyte functions, deregulating adipokine secretion and thus altering lipogenesis and lipolysis (170). Yeh et al. observed that pre-adipocytes promote doxorubicin resistance in triple negative BC by secreting CXCL1, which determines over-expression of ABCG2 (110). Moreover, high concentrations of leptin increased colorectal CSCs survival and the resistance to 5-FU treatment (108) (Table 2).
In addition to cytotoxic drugs, adipocytes are implicated in the resistance to multiple therapies, including radiotherapy, hormonal therapy, immunotherapy, and chemotherapy (172–174). CAAs expressed high levels of PD-L1 and in turn protected cancer cell from the anti-tumor activity of CD8+ T lymphocytes (151). Of note, the use of immune checkpoint inhibitors in BC displays limited efficacy, probably due to the presence of surrounding AT. The inhibition of adipogenic processes increased anti PD-L1 or anti PD-1 activity (175). Moreover, it was demonstrated that IL-6 secreted by mammary adipose tissue up-regulated Chk1 signaling pathway in BC cells, promoting resistance to radiotherapy (138). Therefore, the targeting of tumor-released factors which induce the activation of adipocytes in CAAs could improve patient outcomes. In BC cachectic patients, the secretion of miR-155 by tumor cells restored adipocyte metabolism, reducing PPARγ expression levels, and was associated with tumor progression. The administration of propranolol impaired the release of exosomes containing miR-155, thus restoring PPARγ in adipocytes (176, 177).
Moreover, targeting the metabolic dependence of cancer cells on adipocytes could be a therapeutic strategy to lessen tumor progression (178). In melanoma cells, the treatment with fatty acid transport protein 1 inhibitor impaired the invasive capacity of tumor cells promoted by adipocyte conditioned medium (179). In addition, CD36 inhibition in ovarian cancer cells reduced their in vitro and in vivo invasive capabilities sustained by CAAs (180). Moreover, Masko et al. pointed out that the combination of standard treatments with drugs interfering with adipocyte metabolism, like statins, has promising therapeutic relevance in prostate cancer treatment (181, 182). The administration of a high fat diet, instead of a normal one, in mice treated with diethylnitrosamine promoted hepatocellular carcinoma development, increasing STAT3 activation and IL-6 production. This phenotype was counteracted using acyclic retinoids (183, 184). Metformin is an anti-hyperglycemic agent, indicated for obesity-related type 2 diabetes, which determines the inhibition of the hepatic gluconeogenesis pathway, through the activation of AMPK. Moreover, metformin reduced the circulating levels of androgen, estrogen, insulin and sensitized BC cells to chemo and radiotherapy through a selective killing of stemness compartment (NCT02874430) (185–187).
Immune Cells Rewiring Therapies
Immune system is an interesting network composed of specialized immune cells (ICs), cytokines, chemokines, and lymphoid organs, which, all together, contribute to immune response. The principal function of immune system is to discriminate “self” from “non-self” components. In TME, ICs affect both cancer development and immunological surveillance, influencing patients’ clinical outcome (188). ICs are classified in effector and non-effector cells, with the first category including natural killer (NK) cells, B and T lymphocytes, involved in the adoptive immune response along with the killing of cancer cells. The presence of T cytotoxic CD8+, T helper 1 (Th1) CD4+, B and NK cells within the TME is associated with a positive patients’ outcome in many cancers (189, 190). It is well known that NK cells act directly on the tumor cells, hampering their proliferation and dissemination. Compelling evidence demonstrated that NK cells eradicate CSCs at mestatatic sites, preventing tumor progression and relapse (191–194). Differently, the reduction of NK cells was associated with a worse outcome (195).
Non effector cells include antigen-presenting cells (APCs), regulatory T cells (Treg), tumor-associated macrophages (TAMs) and myeloid-derived suppressor cells (MDSCs), which support tumor growth, progression, and dissemination, hampering immune response.
The most abundant IC subset within the TME is represented by TAMs, which modulate the innate immune response in the context of tumor. TAMs own a phenotypic plasticity, thus transit from M1 to M2 phenotypes, and viceversa. M1 TAMs are involved in activated proinflammatory pathway and counteract tumor growth, while the M2 are engaged in anti-inflammatory response, largely promoting angiogenesis and tissue remodeling and sustaining tumor progression (196, 197).
Several studies highlighted that M2 TAMs are characterized by the expression of specific markers, such as CD163 and CD206 (198). In BC the release of TNF-α, IL-6 and IL-1β in the TME sustained M2 macrophages, which boost tumor initiation, dissemination and metastasis formation (199). In addition, Rodriguez-Garcia et al. highlighted the role of folate receptor β (FRβ) in TAMs cells. They demonstrated that immunosuppressive M2 TAMs expressing FRβ promote tumor progression in a mouse model of ovarian cancer, pointing out FRβ as a potential therapeutic target in combination with chemo- or immunotherapy (200). Furthermore, the expression of FRβ in M2 TAMs correlated with a poor prognosis also in pancreatic cancer (201). A recent study showed that BC cells, through TNF-α and IL-1β releasing in TME, induce the production of CCL8 by pro-tumorigenic TAMs and this crosstalk correlates with worse outcomes (202). These data confirm that M2 phenotype of TAMs plays a pivotal role in sustaining tumor growth and therefore could be a potential target. Overall, many studies highlighted that TAMs are responsible, in addition with other factors and cells available in the TME, for the increase of CSC subpopulation, leading to chemotherapy resistance. The induction of EMT and the over-expression of stem cell markers, such as CD90/Thy1 and EphA4, mediated the crosstalk between CSCs and TAMs. In addition, in different types of cancers and in particular in BC, the maintenance of a stem-like phenotype is also correlated to the presence of M2 macrophages in the TME (203, 204). In osteosarcoma, Xue-jing Shao et al. described the contribution of CD209+ M2 macrophages in tumor initiation and CSC maintenance, corroborating the possibility that the blockage of M2 macrophages depletes CSC subpopulation in the tumor bulk and, at the same time, inhibits tumor progression (205).
In liver cancer, the activation of oncoprotein Yes-associated protein (YAP) in CSCs correlated with both tumorigenesis and TAM recruitment, indicating that the blocking of M2 macrophage or YAP could be an efficacious therapeutic strategy (206).
In BC the inflammatory process predisposes to the malignant transformation, inducing the release chemokines, such as IL-8 and growth-regulated oncogene (GRO), which activate JAK/STAT3 pathway and in turn maintain CSC-like cell phenotype (207). Larionova et al. dissected the contributions of M2 macrophages in chemoresistance, showing that the depletion of M2 macrophages or M2-to-M1 re-polarization improves therapy efficacy of conventional cytotoxic drugs and/or immunotherapy, enhancing immune response (197).
It is widely demonstrated that cytotoxic chemotherapeutic drugs weaken immune system homeostasis (208). Simultaneously, over the last ten years, several studies highlighted the effects of chemotherapeutic agents regarding increased immunogenicity of human cancer cells and the role of chemotherapies in activating antitumor immune responses (209, 210). Different studies shed light on the role of NK cells within tumors and the influence of TME and chemotherapy on innate lymphoid cells. The high presence of NK cells in the TME correlate with an increased patients’ survival in different types of cancer, such as HER2-positive and triple negative BCs (211). It has been demonstrated that NK function could be regulated by chemotherapy (212). Recent studies have reported that different chemotherapeutic compounds, such as GEM, positively regulate NK cell functions. In lung cancers, the use of low-dose GEM enhanced the release of INF-γ and at the same time activated NK cells (213). In in vivo models of pancreatic cancer, the use of GEM as adjuvant chemotherapy improved mice overall survival with a reduction of tumor burden bulk. Thereafter, GEM induced a decrease of MDSCs and, on the other hand, increased the anti-tumor capability of NK cells (214, 215). In the last decades, to eradicate cancer cells many therapeutic strategies were focused on the re-activation of ICs, in particular of T cells. Cancer immunotherapies comprehend different approaches including the immune checkpoint blockade, with anti programmed death 1 (PD-1)/PD-ligand 1 (PD-L1)/cytotoxic T lymphocyte antigen 4 (CTLA4) antibodies, and adoptive cellular therapies (216). The following data illustrate that the use of cytotoxic chemotherapy combined with immunotherapy could block signaling factors or targets essential for CSC-mediated tumor progression and dissemination (190, 217).
In order to counteract fast cancer cell proliferation and enhance immune response, Orecchioni et al. tested the synergic effect of 5-FU, cyclophosphamide (CPX) or vinorelbine in combination with checkpoint inhibitors. In immunocompetent mice, the treatment with all the three chemotherapic drugs influenced the number of circulating ICs, in particular reducing MDSCs, APC cells, Treg, whereas increasing NK cells. The combination of chemotherapy and anti PD-L1 in mice injected with triple negative BC and B cell lymphoma cells reduced tumor growth and metastasis formation compared to the control group (218).
Many studies have been carried out to characterize the immunomodulatory properties of GEM. In pancreatic cancer, GEM induced a decrease in MDSCs and Treg, albeit did not counteract effector lymphocytes. Although GEM influences infiltrating ICs, generating an unfavorable condition for tumor growth, it was not sufficient as single agent and needed to be combined with immunotherapy to enhance immune response (219). In fact, in in vivo models GEM in combination with immunotherapy reduced the number of immunosuppressive cells, enhancing CD8+ T cells and promoting tumor cell elimination (220). These results pointed out that GEM is an immune checkpoint inhibitor-compatible drug, and this combination treatment reactivates the immune response with the goal of killing active proliferating cells (221).
Chemotherapy resistance is nowadays a sensitive issue that led several scientists to look for the causes of this phenomenon and the possibility to counteract the failure of chemotherapy drugs linked with the CSC subgroup. In NSCLC adenocarcinoma, the use of pemetrexed firstly stimulated the host antitumor immunity and simultaneously induced in vitro immunogenic cancer cell death (ICD), leading to improved antitumor immune response (222). In the KEYNOTE–021G trial, the use of pemetrexed and carboplatin (CARB) plus anti PD-1 antibody promoted immune response through the recruitment of infiltrating T cells, the reduction of APC cells, as well as elicited ICD in patients affected by NSCLC, improving their clinical outcome (NCT 02039674).
It has been demonstrated that CIS, oxa and CARB are able to stimulate antitumor immunity by promoting the enhancement of CD8+ T and APC cells with concomitant down-regulation of Treg and MDSC subpopulations. This effect, prompted by platinum derivatives drugs, improved the sensitivity of tumor cells to immunotherapy (223).
In bladder cancer cell lines, the use of CIS increased the expression level of PD-L1 through the activation of c-Jun, one of the activator protein-1 (AP-1) subunits, via ERK1/2. These data showed that chemotherapy in combination with immunotherapy (anti PD-L1) preempts cancer relapse blocking AP-1 oncogene factor (224).
The Keynote-407 multicentric study investigated the use of immune checkpoint inhibitors alone or in combination with CARB and paclitaxel in squamous NSCLC patients. The response rate and median progression-free survival increased in patients treated with checkpoint inhibitors plus chemotherapy instead of placebo groups (NCT02775435). The use of these combinatorial regimes recruited T, NK, and APC cells with a concomitant reduction of MDSCs and Treg in the TME (225).
The results of a Phase III clinical trial, which includes recurrent inoperable or metastatic triple-negative BC patients, reported that pembrolizumab plus chemotherapy (paclitaxel, GEM) improved the median progression-free survival (NCT02220894) (226). Moreover, in an ongoing Phase III randomized trial in patients affected by metastatic CRC the use of chemotherapy (FOLFOX) in combination with immunotherapy (atezolizumab, anti PD-L1) was tested in order to hamper cancer progression and improve immune system response, in particular cytotoxic CD8+ T cells (NCT02912559) (227). All these data highlighted that immunotherapy, in association with standard chemotherapy, has erupted as a novel therapeutic strategy to counteract tumor growth and chemoresistance.
In addition to the use of immune checkpoint inhibitors, another promising therapeutic approach is the chimeric antigen receptor T-cell (CAR-T) therapy. This methodology is based on the use of patients T cells engineered with vectors carrying a CAR specifically expressed on cancer cells. This genetic modification allows T cells, after re-infusion in patients, to efficiently recognize and kill cancer cells (228). To date, five different generations of CARs, characterized by differences in their intracellular domain, have been developed. Specifically, the first generation of CARs presented only the CD3ζ domain, which in the second generation was conjugated with a costimulatory domain, such as CD28 or 4-1BB, to improve their proliferation and cytotoxic potential. The third and fourth generation differed from the second generation for the addition of CD137/CD134 or IL-2 inducer domain, respectively. To improve CAR-T proliferation and survival, the last generation display a STAT3 inducer domain in combination with CD3ζ-CD28 and IL-2 inducer (229, 230). Promising data showed that CAR-T cells, engineered for the most abundant surface antigen expressed on CSCs, efficiently target cancer cells mainly in liquid tumors. CAR-T cell-based clinical trials displayed huge remission rates in patients with B cell hematologic malignancies (231). In the ELIANA trial, children and young patients with refractory B-cell acute lymphoblastic leukemia (ALL) were infused with autologous T cells engineered with a CD19 CAR (CTL019, tisagenlecleucel), achieving durable remission with transient toxic effects (NCT02435849) (232). The anti CD19 CAR-T cell therapy also displayed remarkable results in adult patients with diffuse large B-cell lymphoma (DLBCL) (NCT02445248) (233). Despite the encouraging results obtained in the treatment of hematological tumors, limited successes have been reached with solid ones. This is probably due to the immunosuppressive role of TME and the heterogeneous expression of targetable antigens (234). Nevertheless, several CAR-T clinical trials have been approved for the treatment of solid cancers. The high expression levels of EpCAM have been associated with local growth and dissemination in different cancers, including breast and colorectal tumors (230). Zhang et al. described that use of CAR-T cells, targeting EpCAM+ cancer cells, induces tumor strinkage in in vivo CRC models (235). Moreover, in a Phase I clinical trial the use of a CD133 CAR-T cells induced, after the first infusion, the reduction of tumor growth and the partial remission or stable disease for the treatment in hepatocellular, pancreatic and CRC patients (NCT02541370) (236). Besides hitting cancer cells, CAR-T could also be engineered to target components of TME. In murine ovarian carcinoma cell lines, the use of CAR-T targeting FRβ induced a selective depletion of M2 TAMs and, at the same time, led to the recruitment of inflammatory cytokine and precursor myeloid cells. Despite the clinical benefits obtained in term of durable remission, the majority of CAR-T cell therapies displayed high grade toxic effects, such as cytokine-release syndrome and neurotoxicity (237, 238). Therefore, the next milestone on CAR-T cell therapies is the optimization of clinical approaches and engineering strategies to improve safety and efficacy.
Role of Tumor Angiogenesis in Chemotherapy Failure
The aberrant and rapid growth of cancer cells requires a continuous demand of nutrient and oxygen, which generate hypoxic area in the TME. To restore an adequate oxygen supply, CSCs boosted HIF-1A expression levels which mediate the secretion of VEGF-A, SDF-1 and HGF, recruiting VEGF receptors (VEGFRs)-expressing endothelial cells (ECs) and promoting tumor angiogenesis (239–241). Through this process, VEGF signaling activates the proliferation and survival of ECs, determining the increase of vessel permeability and supporting the metabolic needs of cancer cells (242). Moreover, the VEGF secreted by CSCs recruited mesenchymal stem cells inducing their differentiation into ECs (243).
These observations indicate that CSCs play a fundamental role in determining the TME through an important crosstalk with mesenchymal cells and ECs associated with the tumor. In normal conditions, angiogenesis, which has key role during embryonic development and tissue repair, is finely regulated by a poise between pro- and anti-angiogenic factors (244). This process is characterized by a dynamic and complex sequence of events which involve two main cells type: proliferating stalk cells and the highly invasive and motile endothelial tip cells. At the end of vessel formation, pericytes and vascular smooth muscle cells are recruited to stabilize newly formed blood vessels (245). The alteration of normal angiogenesis is a hallmark of cancer which leads to important changes and transformation inside TME and is connected with tumor progression (246). In 1971, Folkman used for the first time the expression “tumor angiogenesis” to describe blood vessel sprouting mediated by activated ECs nearly tumor mass (247). The new tumoral vessels are characterized by chaotic organization and weak interactions between pericytes and ECs favoring vascular leakiness, which is one of the most important barriers for efficient drug delivery in solid tumors (245, 248, 249). Several studies described that CSCs could trans-differentiate and promote the formation of new vessels without the recruitment of ECs. In the 1999, Maniotis et al. described for the first time this phenomenon, called vascular mimicry, in melanoma (250). Thereafter, other groups described the trans-differentiation of CSCs in ECs and pericytes in other tumors, like GBM, colon, and BC (4, 243, 251–253). Calabrese et al. described a close ECs-brain CSCs interaction in the perivascular niche, which maintains the self-renewal capacity of CD133+ stem-like cells and supports xenograft tumor growth. The treatment with anti-angiogenic drugs impaired CSC features (254). In a 3D system, brain ECs secreted IL-8 which promotes the expression of stem cell markers and boosts the invasive potential of patient-derived GBM cells (255). Moreover, ECs isolated from different organs increased the stem-like phenotype of CRC cells and the expression levels of OCT4 and NANOGP8 mediated by AKT activation (256). Many studies highlighted the key role of NOTCH signaling pathway in ECs/CSCs cross-talk, which prompts stem-like phenotype and tumor progression in cancer cells (257).
In GBM, the juxtacrine signaling between NOTCH ligand-expressing ECs and tumor cells exposing Notch1 receptor boosted in vitro and in vivo growth of cancer stem-like cells (258). Cao et al. pointed out that FGF4, produced by lymphoma cells, induces the expression of Jagged-1 on ECs, which in turn promotes Notch2 activation in cancer cells, increasing their tumorigenic and invasive capacity (111). A similar mechanism has been described in breast CSCs (259, 260). In a complementary manner, ECs released a soluble Jagged-1 which activates Notch signaling in colorectal CSCs, enhancing their tumorigenic and metastatic potential (112).
The below reported studies pointed out that ECs not only promote stem-like phenotype in cancer cells, but also play a key role in the resistance to chemotherapy. The activation of Notch pathway triggered by ECs confers resistance to doxorubicin treatment in aggressive lymphoma cells (111). Moreover, EC conditioned medium drove refractoriness to 5-FU and oxa treatment in colorectal CSCs (112). Therefore, the possibility to counteract chemoresistance mediated by ECs/CSCs interaction could ameliorate the management of cancer patients. Compelling evidence showed that anti-angiogenic treatments not only contrast the formation of new blood vessels, but also improve the quality of existing blood vessels, enhancing blood perfusion and consequently the exposure of cancer cells to chemotherapeutic treatments (261). The majority of anti-angiogenic therapies is represented by monoclonal antibodies directed against EC membrane molecules, while other compounds targeted intracellular components, inhibiting their activation (261).
In the last decades, numerous anti-angiogenic drugs have been tested in clinical trials and approved by the FDA. Bevacizumab is a humanized monoclonal antibody directed against VEGF-A, approved in 2004, and used in clinic for treatment of different tumors, such as GBM, colorectal, ovarian and BC (262, 263). Bevacizumab prevents the VEGF-A/VEGFR interaction and thus impairs the activation of VEGF signaling pathways in ECs. In vivo studies have shown that bevacizumab inhibited the spouting of blood vessel, induced the regression of newly formed vessels, and normalized the morphology of preexisting ones to improve the administration of cytotoxic chemotherapy (242). However, the treatment with bevacizumab did not display significant improvement of patients’ overall survival in advanced BC (264). The inefficacy of bevacizumab treatment was also observed in colorectal and brain tumors due to the increased expression levels or activation of alternative angiogenic factors and signaling pathways, respectively (113, 114). In particular, Lu et al. demonstrated that in GBM CSCs, VEGF inhibits cell invasiveness by blocking HGF receptor (Met)/VEGFR2 interaction and recruiting PTP1B phosphatase, which promotes Met dephosphorylation. The treatment with bevacizumab led to Met signaling pathway activation and to the acquisition of a mesenchymal-like phenotype in GBM CSCs (265). Although angiogenesis inhibition initially reduce tumor growth and prevent metastasis formation, these effects are transitory and associated with tumor relapse and recurrence (266).
There are several explanations for the failure of anti-angiogenic therapies. One possible cause is the induction of intra-tumoral hypoxia related to decreased number of blood vessels and the over-expression of HIF-1A, which promotes and sustains CSC features and paradoxically reactivate neo-angiogenesis (267, 268).
These observations suggest that anti-angiogenic therapy used as single agent not only favors tumor growth and progression, but also induces therapy resistance. In fact, angiogenic inhibitors induced deep changes in vascular morphology involving the down-regulation of junction proteins and a reduction of pericyte number and functionality (115).
Another important limiting factor of anti-angiogenic drug efficacy is the activation of VEGF-independent pro-angiogenic signaling pathways in pancreatic tumors (116).
Other possible strategies able to interfere with tumor angiogenesis consist in the use of tyrosine receptor kinase inhibitors molecules (269). Sorafenib is an inhibitor of numerous tyrosine kinases, including Ras and VEGFR family and platelet-derived growth factor receptor β (PDGFR-β). In a Phase III study, the treatment with sorafenib increased the overall median survival of hepatocellular carcinoma patients (270). Moreover, in the DECISION trial patients with radioactive iodine-refractory thyroid cancer treated with sorafenib display an improved progression free survival compared to the placebo group (NCT00984282) (271). Unfortunately, mechanisms of resistance to sorafenib treatment similar to those described above for bevacizumab have been reported (272, 273). In addition, the intratumoral hypoxia generated by sorafenib treatment enhanced the expression of PD-L1 in cancer cells and the recruitment of TAMs (274).
Another tyrosine kinase inhibitor used as anti-angiogenic molecule is sunitinib, which targets PDGFR, VEGFRs and c-kit. Suninitib has been approved for the treatment of imatinib-resistant gastrointestinal stromal tumor (GIST) and metastatic renal cell carcinoma (RCC), displaying an increased response rate compared to placebo patient group (275). Nevertheless, patients rapidly acquire resistance to treatment (276). Huang et al. generated a sunitinib-resistant RCC xenograft model and observed high microvessel density together with increased serum levels of IL-8, suggesting that patients with elevated IL-8 levels display intrinsic resistance to sunitinib (277). Moreover, sunitinib treatment induced a stem-like phenotype and refractoriness in RCC cells through the activation of PAK1/NF-κB/IL-6 signaling axis (117). In addition, the chronic administration of sunitinib in RCC cells promoted EMT, invasion and angiogenesis via the activation of MET and AXL. This sunitinib-induced phenotype was suppressed by cabozantinib treatment (118) (Table 2).
Based on the poor clinical efficacy of VEGF pathway inhibitors, in the last years alternative strategies have been tested to impair tumor angiogenesis. Small molecules (rebastinib) and monoclonal antibodies (MEDI3617, demcizumab, enoticumab and MEDI0639) targeting ANGPT2/TIE2 and Notch ligand–receptor interactions have been tested and approved for the treatment of advanced solid tumors (278, 279).
Given that CSCs can activate ECs through different stimuli, the simultaneous targeting or the subsequent multiple targeting of several angiogenic factors could represent an important perspective in the innovation of anti-angiogenic therapies avoiding the above described resistance mechanisms (261). In particular, in many clinical trials the treatment with anti-angiogenic compounds displays clinical effects only in early stage, due to a ‘selection’ of functional vessels among the newly tumor vessels. Therefore, this treatment-induced ‘therapeutic window’ could be an advantage for the administration of standard and targeted therapies (278).
Another important therapeutic strategy used to counteract the resistance to the combination of anti-angiogenic and cytotoxic drugs is the metronomic chemotherapy, based on the continuous administration of low chemotherapy doses. This approach hinders CSC/TME interactions, targeting both cancer cells and tumor-associated ECs (261, 280).
Concluding Remarks and Future Perspectives
Despite the great advances made in early diagnosis and the development of targeted therapies, which increase patients’ survival rates, the metastatic disease remains incurable. This is mainly due to primary or acquired resistance to chemotherapeutic drugs and the presence of TME. Compelling evidence highlights that the inefficacy of anti-cancer therapy results from the refractoriness of a subpopulation of tumor cells, called CSCs, which are endowed with stem-like features including tumor-initiating and metastasis formation capabilities. In addition to the intrinsic characteristics of CSCs, interactions with TME are crucially involved in the resistance to chemo and targeted therapies. The mechanisms sustaining CSC/TME crosstalk and the limitations of targeting this complex signaling network have been comprehensively described in this review. Specifically, the reasons of treatment failure using the most recently available compounds targeting both CSCs or TME components have been reported. Of note, CSC plasticity and ability to adapt to the metabolic demand are the major hurdles in targeting CSC/TME interplay. Therefore, additional studies are needed to develop potential promising strategies to overcome cancer progression and drug refractory.
Author Contributions
MG, SDF, and GS conceived and wrote the manuscript. VDP, GPo, CDA, FV, VV, LC, NF, GPi, MRB, and MT wrote the manuscript.All authors contributed to the article and approved the submitted version.
Funding
This work was supported by grants from AIRC (21492) to MT and AIRC IG (21445) and PRIN (2017WNKSLR) to GS.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Francesco Calò for graphic images editing. GPo, CDA, and FV are students of the Molecular and Clinical Medicine PhD Program. VV is a research fellow funded by European Union-FESR FSE, PON Ricerca e Innovazione 2014–2020 (AIM line 1).
References
1. Batlle E, Clevers H. Cancer Stem Cells Revisited. Nat Med (2017) 23(10):1124–34. doi: 10.1038/nm.4409
2. Rich JN. Cancer Stem Cells: Understanding Tumor Hierarchy and Heterogeneity. Med (Baltimore) (2016) 95(1 Suppl 1):S2–7. doi: 10.1097/MD.0000000000004764
3. Valent P, Bonnet D, De Maria R, Lapidot T, Copland M, Melo JV, et al. Cancer Stem Cell Definitions and Terminology: The Devil Is in the Details. Nat Rev Cancer (2012) 12(11):767–75. doi: 10.1038/nrc3368
4. Turdo A, Veschi V, Gaggianesi M, Chinnici A, Bianca P, Todaro M, et al. Meeting the Challenge of Targeting Cancer Stem Cells. Front Cell Dev Biol (2019) 7:16. doi: 10.3389/fcell.2019.00016
5. Kreso A, Dick JE. Evolution of the Cancer Stem Cell Model. Cell Stem Cell (2014) 14(3):275–91. doi: 10.1016/j.stem.2014.02.006
6. Rebe C, Ghiringhelli F. Cytotoxic Effects of Chemotherapy on Cancer and Immune Cells: How can it be Modulated to Generate Novel Therapeutic Strategies? Future Oncol (2015) 11(19):2645–54. doi: 10.2217/fon.15.198
7. Catalano V, Turdo A, Di Franco S, Dieli F, Todaro M, Stassi G. Tumor and Its Microenvironment: A Synergistic Interplay. Semin Cancer Biol (2013) 23(6 Pt B):522–32. doi: 10.1016/j.semcancer.2013.08.007
8. Walcher L, Kistenmacher AK, Suo H, Kitte R, Dluczek S, Strauss A, et al. Cancer Stem Cells-Origins and Biomarkers: Perspectives for Targeted Personalized Therapies. Front Immunol (2020) 11:1280. doi: 10.3389/fimmu.2020.01280
9. Till JE, Mc CE. A Direct Measurement of the Radiation Sensitivity of Normal Mouse Bone Marrow Cells. Radiat Res (1961) 14:213–22. doi: 10.2307/3570892
10. Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, Caceres-Cortes J, et al. A Cell Initiating Human Acute Myeloid Leukaemia After Transplantation Into SCID Mice. Nature (1994) 367(6464):645–8. doi: 10.1038/367645a0
11. Bonnet D, Dick JE. Human Acute Myeloid Leukemia Is Organized as a Hierarchy That Originates From a Primitive Hematopoietic Cell. Nat Med (1997) 3(7):730–7. doi: 10.1038/nm0797-730
12. Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective Identification of Tumorigenic Breast Cancer Cells. Proc Natl Acad Sci USA (2003) 100(7):3983–8. doi: 10.1073/pnas.0530291100
13. Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, et al. Identification of Human Brain Tumour Initiating Cells. Nature (2004) 432(7015):396–401. doi: 10.1038/nature03128
14. Ricci-Vitiani L, Lombardi DG, Pilozzi E, Biffoni M, Todaro M, Peschle C, et al. Identification and Expansion of Human Colon-Cancer-Initiating Cells. Nature (2007) 445(7123):111–5. doi: 10.1038/nature05384
15. Clarke MF, Dick JE, Dirks PB, Eaves CJ, Jamieson CH, Jones DL, et al. Cancer Stem Cells–Perspectives on Current Status and Future Directions: AACR Workshop on Cancer Stem Cells. Cancer Res (2006) 66(19):9339–44. doi: 10.1158/0008-5472.CAN-06-3126
16. Veschi V, Verona F, Lo Iacono M, D’Accardo C, Porcelli G, Turdo A, et al. Cancer Stem Cells in Thyroid Tumors: From the Origin to Metastasis. Front Endocrinol (Lausanne) (2020) 11:566. doi: 10.3389/fendo.2020.00566
17. Di Franco S, Todaro M, Dieli F, Stassi G. Colorectal Cancer Defeating? Challenge Accepted! Mol Aspects Med (2014) 39:61–81. doi: 10.1016/j.mam.2013.07.001
18. Clara JA, Monge C, Yang Y, Takebe N. Targeting Signalling Pathways and the Immune Microenvironment of Cancer Stem Cells - a Clinical Update. Nat Rev Clin Oncol (2020) 17(4):204–32. doi: 10.1038/s41571-019-0293-2
19. Merchant AA, Matsui W. Targeting Hedgehog–a Cancer Stem Cell Pathway. Clin Cancer Res (2010) 16(12):3130–40. doi: 10.1158/1078-0432.CCR-09-2846
20. Cochrane CR, Szczepny A, Watkins DN, Cain JE. Hedgehog Signaling in the Maintenance of Cancer Stem Cells. Cancers (Basel) (2015) 7(3):1554–85. doi: 10.3390/cancers7030851
21. Varnat F, Duquet A, Malerba M, Zbinden M, Mas C, Gervaz P, et al. Human Colon Cancer Epithelial Cells Harbour Active HEDGEHOG-GLI Signalling That Is Essential for Tumour Growth, Recurrence, Metastasis and Stem Cell Survival and Expansion. EMBO Mol Med (2009) 1(6-7):338–51. doi: 10.1002/emmm.200900039
22. Liu S, Dontu G, Mantle ID, Patel S, Ahn NS, Jackson KW, et al. Hedgehog Signaling and Bmi-1 Regulate Self-Renewal of Normal and Malignant Human Mammary Stem Cells. Cancer Res (2006) 66(12):6063–71. doi: 10.1158/0008-5472.CAN-06-0054
23. Ulasov IV, Nandi S, Dey M, Sonabend AM, Lesniak MS. Inhibition of Sonic Hedgehog and Notch Pathways Enhances Sensitivity of CD133(+) Glioma Stem Cells to Temozolomide Therapy. Mol Med (2011) 17(1-2):103–12. doi: 10.2119/molmed.2010.00062
24. Bienz M, Clevers H. Linking Colorectal Cancer to Wnt Signaling. Cell (2000) 103(2):311–20. doi: 10.1016/S0092-8674(00)00122-7
25. Vermeulen L, De Sousa EMF, van der Heijden M, Cameron K, de Jong JH, Borovski T, et al. Wnt Activity Defines Colon Cancer Stem Cells and Is Regulated by the Microenvironment. Nat Cell Biol (2010) 12(5):468–76. doi: 10.1038/ncb2048
26. De Sousa e Melo F, Vermeulen L. Wnt Signaling in Cancer Stem Cell Biology. Cancers (Basel) (2016) 8(7):60. doi: 10.3390/cancers8070060
27. Jang GB, Kim JY, Cho SD, Park KS, Jung JY, Lee HY, et al. Blockade of Wnt/beta-Catenin Signaling Suppresses Breast Cancer Metastasis by Inhibiting CSC-Like Phenotype. Sci Rep (2015) 5:12465. doi: 10.1038/srep12465
28. Martin-Orozco E, Sanchez-Fernandez A, Ortiz-Parra I, Ayala-San Nicolas M. WNT Signaling in Tumors: The Way to Evade Drugs and Immunity. Front Immunol (2019) 10:2854. doi: 10.3389/fimmu.2019.02854
29. Flahaut M, Meier R, Coulon A, Nardou KA, Niggli FK, Martinet D, et al. The Wnt Receptor FZD1 Mediates Chemoresistance in Neuroblastoma Through Activation of the Wnt/beta-Catenin Pathway. Oncogene (2009) 28(23):2245–56. doi: 10.1038/onc.2009.80
30. Zhang H, Zhang X, Wu X, Li W, Su P, Cheng H, et al. Interference of Frizzled 1 (FZD1) Reverses Multidrug Resistance in Breast Cancer Cells Through the Wnt/beta-Catenin Pathway. Cancer Lett (2012) 323(1):106–13. doi: 10.1016/j.canlet.2012.03.039
31. Cao HZ, Liu XF, Yang WT, Chen Q, Zheng PS. LGR5 Promotes Cancer Stem Cell Traits and Chemoresistance in Cervical Cancer. Cell Death Dis (2017) 8(9):e3039. doi: 10.1038/cddis.2017.393
32. Liu YS, Hsu HC, Tseng KC, Chen HC, Chen SJ. Lgr5 Promotes Cancer Stemness and Confers Chemoresistance Through ABCB1 in Colorectal Cancer. BioMed Pharmacother (2013) 67(8):791–9. doi: 10.1016/j.biopha.2013.08.001
33. Cordenonsi M, Zanconato F, Azzolin L, Forcato M, Rosato A, Frasson C, et al. The Hippo Transducer TAZ Confers Cancer Stem Cell-Related Traits on Breast Cancer Cells. Cell (2011) 147(4):759–72. doi: 10.1016/j.cell.2011.09.048
34. Cheung P, Xiol J, Dill MT, Yuan WC, Panero R, Roper J, et al. Regenerative Reprogramming of the Intestinal Stem Cell State via Hippo Signaling Suppresses Metastatic Colorectal Cancer. Cell Stem Cell (2020) 27(4):590–604 e9. doi: 10.1016/j.stem.2020.07.003
35. Catalano V, Gaggianesi M, Spina V, Iovino F, Dieli F, Stassi G, et al. Colorectal Cancer Stem Cells and Cell Death. Cancers (Basel) (2011) 3(2):1929–46. doi: 10.3390/cancers3021929
36. Costello RT, Mallet F, Gaugler B, Sainty D, Arnoulet C, Gastaut JA, et al. Human Acute Myeloid Leukemia CD34+/CD38- Progenitor Cells Have Decreased Sensitivity to Chemotherapy and Fas-Induced Apoptosis, Reduced Immunogenicity, and Impaired Dendritic Cell Transformation Capacities. Cancer Res (2000) 60(16):4403–11.
37. Santofimia-Castano P, Iovanna J. Combating Pancreatic Cancer Chemoresistance by Triggering Multiple Cell Death Pathways. Pancreatology (2021) 21(3):522–9. doi: 10.1016/j.pan.2021.01.010
38. Tao J, Qiu B, Zhang D, Wang Y. Expression Levels of Fas/Fas-L mRNA in Human Brain Glioma Stem Cells. Mol Med Rep (2012) 5(5):1202–6. doi: 10.3892/mmr.2012.791
39. Eisele G, Wolpert F, Decrey G, Weller M. APO010, a Synthetic Hexameric CD95 Ligand, Induces Death of Human Glioblastoma Stem-Like Cells. Anticancer Res (2013) 33(9):3563–71.
40. Unterkircher T, Cristofanon S, Vellanki SH, Nonnenmacher L, Karpel-Massler G, Wirtz CR, et al. Bortezomib Primes Glioblastoma, Including Glioblastoma Stem Cells, for TRAIL by Increasing Tbid Stability and Mitochondrial Apoptosis. Clin Cancer Res (2011) 17(12):4019–30. doi: 10.1158/1078-0432.CCR-11-0075
41. Loebinger MR, Eddaoudi A, Davies D, Janes SM. Mesenchymal Stem Cell Delivery of TRAIL can Eliminate Metastatic Cancer. Cancer Res (2009) 69(10):4134–42. doi: 10.1158/0008-5472.CAN-08-4698
42. Loebinger MR, Sage EK, Davies D, Janes SM. TRAIL-Expressing Mesenchymal Stem Cells Kill the Putative Cancer Stem Cell Population. Br J Cancer (2010) 103(11):1692–7. doi: 10.1038/sj.bjc.6605952
43. Zobalova R, McDermott L, Stantic M, Prokopova K, Dong LF, Neuzil J. CD133-Positive Cells Are Resistant to TRAIL Due to Up-Regulation of FLIP. Biochem Biophys Res Commun (2008) 373(4):567–71. doi: 10.1016/j.bbrc.2008.06.073
44. Ding L, Yuan C, Wei F, Wang G, Zhang J, Bellail AC, et al. Cisplatin Restores TRAIL Apoptotic Pathway in Glioblastoma-Derived Stem Cells Through Up-Regulation of DR5 and Down-Regulation of C-FLIP. Cancer Invest (2011) 29(8):511–20. doi: 10.3109/07357907.2011.605412
45. Piggott L, Omidvar N, Marti Perez S, French R, Eberl M, Clarkson RW. Suppression of Apoptosis Inhibitor C-FLIP Selectively Eliminates Breast Cancer Stem Cell Activity in Response to the Anti-Cancer Agent, TRAIL. Breast Cancer Res (2011) 13(5):R88. doi: 10.1186/bcr2945
46. Liu G, Yuan X, Zeng Z, Tunici P, Ng H, Abdulkadir IR, et al. Analysis of Gene Expression and Chemoresistance of CD133+ Cancer Stem Cells in Glioblastoma. Mol Cancer (2006) 5:67. doi: 10.1186/1476-4598-5-67
47. Vellanki SH, Grabrucker A, Liebau S, Proepper C, Eramo A, Braun V, et al. Small-Molecule XIAP Inhibitors Enhance Gamma-Irradiation-Induced Apoptosis in Glioblastoma. Neoplasia (2009) 11(8):743–52. doi: 10.1593/neo.09436
48. Guvenc H, Pavlyukov MS, Joshi K, Kurt H, Banasavadi-Siddegowda YK, Mao P, et al. Impairment of Glioma Stem Cell Survival and Growth by a Novel Inhibitor for Survivin-Ran Protein Complex. Clin Cancer Res (2013) 19(3):631–42. doi: 10.1158/1078-0432.CCR-12-0647
49. Di Stefano AB, Iovino F, Lombardo Y, Eterno V, Hoger T, Dieli F, et al. Survivin Is Regulated by Interleukin-4 in Colon Cancer Stem Cells. J Cell Physiol (2010) 225(2):555–61. doi: 10.1002/jcp.22238
50. Todaro M, Alea MP, Di Stefano AB, Cammareri P, Vermeulen L, Iovino F, et al. Colon Cancer Stem Cells Dictate Tumor Growth and Resist Cell Death by Production of Interleukin-4. Cell Stem Cell (2007) 1(4):389–402. doi: 10.1016/j.stem.2007.08.001
51. Wang YH, Scadden DT. Harnessing the Apoptotic Programs in Cancer Stem-Like Cells. EMBO Rep (2015) 16(9):1084–98. doi: 10.15252/embr.201439675
52. Cross AH, Naismith RT. Established and Novel Disease-Modifying Treatments in Multiple Sclerosis. J Intern Med (2014) 275(4):350–63. doi: 10.1111/joim.12203
53. Sun Q, Lesperance J, Wettersten H, Luterstein E, DeRose YS, Welm A, et al. Proapoptotic PUMA Targets Stem-Like Breast Cancer Cells to Suppress Metastasis. J Clin Invest (2018) 128(1):531–44. doi: 10.1172/JCI93707
54. Wu WS, Heinrichs S, Xu D, Garrison SP, Zambetti GP, Adams JM, et al. Slug Antagonizes P53-Mediated Apoptosis of Hematopoietic Progenitors by Repressing Puma. Cell (2005) 123(4):641–53. doi: 10.1016/j.cell.2005.09.029
55. Sun Q, Wang Y, Desgrosellier JS. Combined Bcl-2/Src Inhibition Synergize to Deplete Stem-Like Breast Cancer Cells. Cancer Lett (2019) 457:40–6. doi: 10.1016/j.canlet.2019.05.004
56. Wang QE. DNA Damage Responses in Cancer Stem Cells: Implications for Cancer Therapeutic Strategies. World J Biol Chem (2015) 6(3):57–64. doi: 10.4331/wjbc.v6.i3.57
57. Schulz A, Meyer F, Dubrovska A, Borgmann K. Cancer Stem Cells and Radioresistance: DNA Repair and Beyond. Cancers (Basel) (2019) 11(6). doi: 10.3390/cancers11060862
58. Rizzo S, Hersey JM, Mellor P, Dai W, Santos-Silva A, Liber D, et al. Ovarian Cancer Stem Cell-Like Side Populations Are Enriched Following Chemotherapy and Overexpress EZH2. Mol Cancer Ther (2011) 10(2):325–35. doi: 10.1158/1535-7163.MCT-10-0788
59. Levina V, Marrangoni AM, DeMarco R, Gorelik E, Lokshin AE. Drug-Selected Human Lung Cancer Stem Cells: Cytokine Network, Tumorigenic and Metastatic Properties. PloS One (2008) 3(8):e3077. doi: 10.1371/journal.pone.0003077
60. Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, et al. Glioma Stem Cells Promote Radioresistance by Preferential Activation of the DNA Damage Response. Nature (2006) 444(7120):756–60. doi: 10.1038/nature05236
61. Helleday T, Petermann E, Lundin C, Hodgson B, Sharma RA. DNA Repair Pathways as Targets for Cancer Therapy. Nat Rev Cancer (2008) 8(3):193–204. doi: 10.1038/nrc2342
62. Cheng L, Wu Q, Huang Z, Guryanova OA, Huang Q, Shou W, et al. L1CAM Regulates DNA Damage Checkpoint Response of Glioblastoma Stem Cells Through NBS1. EMBO J (2011) 30(5):800–13. doi: 10.1038/emboj.2011.10
63. Di Franco S, Parrino B, Gaggianesi M, Pantina VD, Bianca P, Nicotra A, et al. CHK1 Inhibitor Sensitizes Resistant Colorectal Cancer Stem Cells to Nortopsentin. iScience (2021) 24(6):1–18. doi: 10.1016/j.isci.2021.102664
64. Desai A, Webb B, Gerson SL. CD133+ Cells Contribute to Radioresistance via Altered Regulation of DNA Repair Genes in Human Lung Cancer Cells. Radiother Oncol (2014) 110(3):538–45. doi: 10.1016/j.radonc.2013.10.040
65. Mathews LA, Cabarcas SM, Hurt EM, Zhang X, Jaffee EM, Farrar WL. Increased Expression of DNA Repair Genes in Invasive Human Pancreatic Cancer Cells. Pancreas (2011) 40(5):730–9. doi: 10.1097/MPA.0b013e31821ae25b
66. Zhang M, Behbod F, Atkinson RL, Landis MD, Kittrell F, Edwards D, et al. Identification of Tumor-Initiating Cells in a P53-Null Mouse Model of Breast Cancer. Cancer Res (2008) 68(12):4674–82. doi: 10.1158/0008-5472.CAN-07-6353
67. Liu Y, Burness ML, Martin-Trevino R, Guy J, Bai S, Harouaka R, et al. RAD51 Mediates Resistance of Cancer Stem Cells to PARP Inhibition in Triple-Negative Breast Cancer. Clin Cancer Res (2017) 23(2):514–22. doi: 10.1158/1078-0432.CCR-15-1348
68. Ginestier C, Hur MH, Charafe-Jauffret E, Monville F, Dutcher J, Brown M, et al. ALDH1 Is a Marker of Normal and Malignant Human Mammary Stem Cells and a Predictor of Poor Clinical Outcome. Cell Stem Cell (2007) 1(5):555–67. doi: 10.1016/j.stem.2007.08.014
69. Ma S, Chan KW, Lee TK, Tang KH, Wo JY, Zheng BJ, et al. Aldehyde Dehydrogenase Discriminates the CD133 Liver Cancer Stem Cell Populations. Mol Cancer Res (2008) 6(7):1146–53. doi: 10.1158/1541-7786.MCR-08-0035
70. Todaro M, Iovino F, Eterno V, Cammareri P, Gambara G, Espina V, et al. Tumorigenic and Metastatic Activity of Human Thyroid Cancer Stem Cells. Cancer Res (2010) 70(21):8874–85. doi: 10.1158/0008-5472.CAN-10-1994
71. Vishnubalaji R, Manikandan M, Fahad M, Hamam R, Alfayez M, Kassem M, et al. Molecular Profiling of ALDH1(+) Colorectal Cancer Stem Cells Reveals Preferential Activation of MAPK, FAK, and Oxidative Stress Pro-Survival Signalling Pathways. Oncotarget (2018) 9(17):13551–64. doi: 10.18632/oncotarget.24420
72. Tanei T, Morimoto K, Shimazu K, Kim SJ, Tanji Y, Taguchi T, et al. Association of Breast Cancer Stem Cells Identified by Aldehyde Dehydrogenase 1 Expression With Resistance to Sequential Paclitaxel and Epirubicin-Based Chemotherapy for Breast Cancers. Clin Cancer Res (2009) 15(12):4234–41. doi: 10.1158/1078-0432.CCR-08-1479
73. Wright MH, Calcagno AM, Salcido CD, Carlson MD, Ambudkar SV, Varticovski L. Brca1 Breast Tumors Contain Distinct CD44+/CD24- and CD133+ Cells With Cancer Stem Cell Characteristics. Breast Cancer Res (2008) 10(1):R10. doi: 10.1186/bcr1855
74. Hawley TS, Riz I, Yang W, Wakabayashi Y, Depalma L, Chang YT, et al. Identification of an ABCB1 (P-Glycoprotein)-Positive Carfilzomib-Resistant Myeloma Subpopulation by the Pluripotent Stem Cell Fluorescent Dye Cdy1. Am J Hematol (2013) 88(4):265–72. doi: 10.1002/ajh.23387
75. Frank NY, Margaryan A, Huang Y, Schatton T, Waaga-Gasser AM, Gasser M, et al. ABCB5-Mediated Doxorubicin Transport and Chemoresistance in Human Malignant Melanoma. Cancer Res (2005) 65(10):4320–33. doi: 10.1158/0008-5472.CAN-04-3327
76. Anderson NM, Simon MC. The Tumor Microenvironment. Curr Biol (2020) 30(16):R921–R5. doi: 10.1016/j.cub.2020.06.081
77. Fiori ME, Di Franco S, Villanova L, Bianca P, Stassi G, De Maria R. Cancer-Associated Fibroblasts as Abettors of Tumor Progression at the Crossroads of EMT and Therapy Resistance. Mol Cancer (2019) 18(1):70. doi: 10.1186/s12943-019-0994-2
78. Kalluri R, Zeisberg M. Fibroblasts in Cancer. Nat Rev Cancer (2006) 6(5):392–401. doi: 10.1038/nrc1877
79. Park D, Sahai E, Rullan A. SnapShot: Cancer-Associated Fibroblasts. Cell (2020) 181(2):486–e1. doi: 10.1016/j.cell.2020.03.013
80. LeBleu VS, Kalluri R. A Peek Into Cancer-Associated Fibroblasts: Origins, Functions and Translational Impact. Dis Model Mech (2018) 11(4). doi: 10.1242/dmm.029447
81. Nurmik M, Ullmann P, Rodriguez F, Haan S, Letellier E. In Search of Definitions: Cancer-Associated Fibroblasts and Their Markers. Int J Cancer (2020) 146(4):895–905. doi: 10.1002/ijc.32193
82. Iliopoulos D, Hirsch HA, Wang G, Struhl K. Inducible Formation of Breast Cancer Stem Cells and Their Dynamic Equilibrium With non-Stem Cancer Cells via IL6 Secretion. Proc Natl Acad Sci USA (2011) 108(4):1397–402. doi: 10.1073/pnas.1018898108
83. Tsuyada A, Chow A, Wu J, Somlo G, Chu P, Loera S, et al. CCL2 Mediates Cross-Talk Between Cancer Cells and Stromal Fibroblasts That Regulates Breast Cancer Stem Cells. Cancer Res (2012) 72(11):2768–79. doi: 10.1158/0008-5472.CAN-11-3567
84. Shi Y, Gao W, Lytle NK, Huang P, Yuan X, Dann AM, et al. Targeting LIF-Mediated Paracrine Interaction for Pancreatic Cancer Therapy and Monitoring. Nature (2019) 569(7754):131–5. doi: 10.1038/s41586-019-1130-6
85. Chen WJ, Ho CC, Chang YL, Chen HY, Lin CA, Ling TY, et al. Cancer-Associated Fibroblasts Regulate the Plasticity of Lung Cancer Stemness via Paracrine Signalling. Nat Commun (2014) 5:3472. doi: 10.1038/ncomms4472
86. Liao CP, Chen LY, Luethy A, Kim Y, Kani K, MacLeod AR, et al. Androgen Receptor in Cancer-Associated Fibroblasts Influences Stemness in Cancer Cells. Endocr Relat Cancer (2017) 24(4):157–70. doi: 10.1530/ERC-16-0138
87. Burke CA. A Statistical View of Clinical Trials in Chronic Hepatitis B. J Hepatol (1986) 3 Suppl 2:S261–7. doi: 10.1016/S0168-8278(86)80130-1
88. Hasegawa T, Yashiro M, Nishii T, Matsuoka J, Fuyuhiro Y, Morisaki T, et al. Cancer-Associated Fibroblasts Might Sustain the Stemness of Scirrhous Gastric Cancer Cells via Transforming Growth Factor-Beta Signaling. Int J Cancer (2014) 134(8):1785–95. doi: 10.1002/ijc.28520
89. Lau EY, Lo J, Cheng BY, Ma MK, Lee JM, Ng JK, et al. Cancer-Associated Fibroblasts Regulate Tumor-Initiating Cell Plasticity in Hepatocellular Carcinoma Through C-Met/FRA1/HEY1 Signaling. Cell Rep (2016) 15(6):1175–89. doi: 10.1016/j.celrep.2016.04.019
90. Todaro M, Gaggianesi M, Catalano V, Benfante A, Iovino F, Biffoni M, et al. CD44v6 Is a Marker of Constitutive and Reprogrammed Cancer Stem Cells Driving Colon Cancer Metastasis. Cell Stem Cell (2014) 14(3):342–56. doi: 10.1016/j.stem.2014.01.009
91. Chen X, Song E. Turning Foes to Friends: Targeting Cancer-Associated Fibroblasts. Nat Rev Drug Discovery (2019) 18(2):99–115. doi: 10.1038/s41573-018-0004-1
92. Huang M, Li Y, Zhang H, Nan F. Breast Cancer Stromal Fibroblasts Promote the Generation of CD44+CD24- Cells Through SDF-1/CXCR4 Interaction. J Exp Clin Cancer Res (2010) 29:80. doi: 10.1186/1756-9966-29-80
93. Li Y, Wang R, Xiong S, Wang X, Zhao Z, Bai S, et al. Cancer-Associated Fibroblasts Promote the Stemness of CD24(+) Liver Cells via Paracrine Signaling. J Mol Med (Berl) (2019) 97(2):243–55. doi: 10.1007/s00109-018-1731-9
94. Singh JK, Farnie G, Bundred NJ, Simoes BM, Shergill A, Landberg G, et al. Targeting CXCR1/2 Significantly Reduces Breast Cancer Stem Cell Activity and Increases the Efficacy of Inhibiting HER2 via HER2-Dependent and -Independent Mechanisms. Clin Cancer Res (2013) 19(3):643–56. doi: 10.1158/1078-0432.CCR-12-1063
95. Yu Y, Xiao CH, Tan LD, Wang QS, Li XQ, Feng YM. Cancer-Associated Fibroblasts Induce Epithelial-Mesenchymal Transition of Breast Cancer Cells Through Paracrine TGF-Beta Signalling. Br J Cancer (2014) 110(3):724–32. doi: 10.1038/bjc.2013.768
96. Malanchi I, Santamaria-Martinez A, Susanto E, Peng H, Lehr HA, Delaloye JF, et al. Interactions Between Cancer Stem Cells and Their Niche Govern Metastatic Colonization. Nature (2011) 481(7379):85–9. doi: 10.1038/nature10694
97. O’Connell JT, Sugimoto H, Cooke VG, MacDonald BA, Mehta AI, LeBleu VS, et al. VEGF-A and Tenascin-C Produced by S100A4+ Stromal Cells Are Important for Metastatic Colonization. Proc Natl Acad Sci USA (2011) 108(38):16002–7. doi: 10.1073/pnas.1109493108
98. Sahai E, Astsaturov I, Cukierman E, DeNardo DG, Egeblad M, Evans RM, et al. A Framework for Advancing Our Understanding of Cancer-Associated Fibroblasts. Nat Rev Cancer (2020) 20(3):174–86. doi: 10.1038/s41568-019-0238-1
99. Tang YA, Chen YF, Bao Y, Mahara S, Yatim S, Oguz G, et al. Hypoxic Tumor Microenvironment Activates GLI2 via HIF-1alpha and TGF-Beta2 to Promote Chemoresistance in Colorectal Cancer. Proc Natl Acad Sci USA (2018) 115(26):E5990–9. doi: 10.1073/pnas.1801348115
100. Su S, Chen J, Yao H, Liu J, Yu S, Lao L, et al. CD10(+)GPR77(+) Cancer-Associated Fibroblasts Promote Cancer Formation and Chemoresistance by Sustaining Cancer Stemness. Cell (2018) 172(4):841–56 e16. doi: 10.1016/j.cell.2018.01.009
101. Cazet AS, Hui MN, Elsworth BL, Wu SZ, Roden D, Chan CL, et al. Targeting Stromal Remodeling and Cancer Stem Cell Plasticity Overcomes Chemoresistance in Triple Negative Breast Cancer. Nat Commun (2018) 9(1):2897. doi: 10.1530/oncolabs.1.P009
102. Yu B, Wu K, Wang X, Zhang J, Wang L, Jiang Y, et al. Periostin Secreted by Cancer-Associated Fibroblasts Promotes Cancer Stemness in Head and Neck Cancer by Activating Protein Tyrosine Kinase 7. Cell Death Dis (2018) 9(11):1082. doi: 10.1038/s41419-018-1116-6
103. Mangiapane LR, Nicotra A, Turdo A, Gaggianesi M, Bianca P, Di Franco S, et al. PI3K-Driven HER2 Expression Is a Potential Therapeutic Target in Colorectal Cancer Stem Cells. Gut (2021) p. 1–10. doi: 10.1136/gutjnl-2020-323553
104. Korkaya H, Kim GI, Davis A, Malik F, Henry NL, Ithimakin S, et al. Activation of an IL6 Inflammatory Loop Mediates Trastuzumab Resistance in HER2+ Breast Cancer by Expanding the Cancer Stem Cell Population. Mol Cell (2012) 47(4):570–84. doi: 10.1016/j.molcel.2012.06.014
105. Lehuede C, Li X, Dauvillier S, Vaysse C, Franchet C, Clement E, et al. Adipocytes Promote Breast Cancer Resistance to Chemotherapy, a Process Amplified by Obesity: Role of the Major Vault Protein (MVP). Breast Cancer Res (2019) 21(1):7. doi: 10.1186/s13058-018-1088-6
106. Gaggianesi M, Turdo A, Chinnici A, Lipari E, Apuzzo T, Benfante A, et al. IL4 Primes the Dynamics of Breast Cancer Progression via DUSP4 Inhibition. Cancer Res (2017) 77(12):3268–79. doi: 10.1158/0008-5472.CAN-16-3126
107. Wang T, Fahrmann JF, Lee H, Li YJ, Tripathi SC, Yue C, et al. JAK/STAT3-Regulated Fatty Acid Beta-Oxidation Is Critical for Breast Cancer Stem Cell Self-Renewal and Chemoresistance. Cell Metab (2018) 27(1):136–50.e5. doi: 10.1016/j.cmet.2017.11.001
108. Bartucci M, Svensson S, Ricci-Vitiani L, Dattilo R, Biffoni M, Signore M, et al. Obesity Hormone Leptin Induces Growth and Interferes With the Cytotoxic Effects of 5-Fluorouracil in Colorectal Tumor Stem Cells. Endocr Relat Cancer (2010) 17(3):823–33. doi: 10.1677/ERC-10-0083
109. Liu Z, Shi A, Song D, Han B, Zhang Z, Ma L, et al. Resistin Confers Resistance to Doxorubicin-Induced Apoptosis in Human Breast Cancer Cells Through Autophagy Induction. Am J Cancer Res (2017) 7(3):574–83.
110. Yeh WL, Tsai CF, Chen DR. Peri-Foci Adipose-Derived Stem Cells Promote Chemoresistance in Breast Cancer. Stem Cell Res Ther (2017) 8(1):177. doi: 10.1186/s13287-017-0630-2
111. Cao Z, Ding BS, Guo P, Lee SB, Butler JM, Casey SC, et al. Angiocrine Factors Deployed by Tumor Vascular Niche Induce B Cell Lymphoma Invasiveness and Chemoresistance. Cancer Cell (2014) 25(3):350–65. doi: 10.1016/j.ccr.2014.02.005
112. Lu J, Ye X, Fan F, Xia L, Bhattacharya R, Bellister S, et al. Endothelial Cells Promote the Colorectal Cancer Stem Cell Phenotype Through a Soluble Form of Jagged-1. Cancer Cell (2013) 23(2):171–85. doi: 10.1016/j.ccr.2012.12.021
113. Jahangiri A, De Lay M, Miller LM, Carbonell WS, Hu YL, Lu K, et al. Gene Expression Profile Identifies Tyrosine Kinase C-Met as a Targetable Mediator of Antiangiogenic Therapy Resistance. Clin Cancer Res (2013) 19(7):1773–83. doi: 10.1158/1078-0432.CCR-12-1281
114. Mesange P, Poindessous V, Sabbah M, Escargueil AE, de Gramont A, Larsen AK. Intrinsic Bevacizumab Resistance Is Associated With Prolonged Activation of Autocrine VEGF Signaling and Hypoxia Tolerance in Colorectal Cancer Cells and Can be Overcome by Nintedanib, a Small Molecule Angiokinase Inhibitor. Oncotarget (2014) 5(13):4709–21. doi: 10.18632/oncotarget.1671
115. Rovida A, Castiglioni V, Decio A, Scarlato V, Scanziani E, Giavazzi R, et al. Chemotherapy Counteracts Metastatic Dissemination Induced by Antiangiogenic Treatment in Mice. Mol Cancer Ther (2013) 12(10):2237–47. doi: 10.1158/1535-7163.MCT-13-0244
116. Casanovas O, Hicklin DJ, Bergers G, Hanahan D. Drug Resistance by Evasion of Antiangiogenic Targeting of VEGF Signaling in Late-Stage Pancreatic Islet Tumors. Cancer Cell (2005) 8(4):299–309. doi: 10.1016/j.ccr.2005.09.005
117. Zhu Y, Liu H, Xu L, An H, Liu W, Liu Y, et al. P21-Activated Kinase 1 Determines Stem-Like Phenotype and Sunitinib Resistance via NF-Kappab/IL-6 Activation in Renal Cell Carcinoma. Cell Death Dis (2015) 6:e1637. doi: 10.1038/cddis.2015.2
118. Zhou L, Liu XD, Sun M, Zhang X, German P, Bai S, et al. Targeting MET and AXL Overcomes Resistance to Sunitinib Therapy in Renal Cell Carcinoma. Oncogene (2016) 35(21):2687–97. doi: 10.1038/onc.2015.343
119. Zhong H, Davis A, Ouzounova M, Carrasco RA, Chen C, Breen S, et al. A Novel IL6 Antibody Sensitizes Multiple Tumor Types to Chemotherapy Including Trastuzumab-Resistant Tumors. Cancer Res (2016) 76(2):480–90. doi: 10.1158/0008-5472.CAN-15-0883
120. Njatcha C, Farooqui M, Kornberg A, Johnson DE, Grandis JR, Siegfried JM. STAT3 Cyclic Decoy Demonstrates Robust Antitumor Effects in Non-Small Cell Lung Cancer. Mol Cancer Ther (2018) 17(9):1917–26. doi: 10.1158/1535-7163.MCT-17-1194
121. Hong D, Kurzrock R, Kim Y, Woessner R, Younes A, Nemunaitis J, et al. AZD9150, a Next-Generation Antisense Oligonucleotide Inhibitor of STAT3 With Early Evidence of Clinical Activity in Lymphoma and Lung Cancer. Sci Transl Med (2015) 7(314):314ra185. doi: 10.1126/scitranslmed.aac5272
122. Goldstein LJ, Perez RP, Yardley D, Han LK, Reuben JM, Gao H, et al. A Window-of-Opportunity Trial of the CXCR1/2 Inhibitor Reparixin in Operable HER-2-Negative Breast Cancer. Breast Cancer Res (2020) 22(1):4. doi: 10.1186/s13058-019-1243-8
123. Chan TS, Hsu CC, Pai VC, Liao WY, Huang SS, Tan KT, et al. Metronomic Chemotherapy Prevents Therapy-Induced Stromal Activation and Induction of Tumor-Initiating Cells. J Exp Med (2016) 213(13):2967–88. doi: 10.1084/jem.20151665
124. Steele CW, Karim SA, Leach JDG, Bailey P, Upstill-Goddard R, Rishi L, et al. CXCR2 Inhibition Profoundly Suppresses Metastases and Augments Immunotherapy in Pancreatic Ductal Adenocarcinoma. Cancer Cell (2016) 29(6):832–45. doi: 10.1016/j.ccell.2016.04.014
125. Valenti G, Quinn HM, Heynen G, Lan L, Holland JD, Vogel R, et al. Cancer Stem Cells Regulate Cancer-Associated Fibroblasts via Activation of Hedgehog Signaling in Mammary Gland Tumors. Cancer Res (2017) 77(8):2134–47. doi: 10.1158/0008-5472.CAN-15-3490
126. Wu C, Hu S, Cheng J, Wang G, Tao K. Smoothened Antagonist GDC-0449 (Vismodegib) Inhibits Proliferation and Triggers Apoptosis in Colon Cancer Cell Lines. Exp Ther Med (2017) 13(5):2529–36. doi: 10.3892/etm.2017.4282
127. Tong W, Qiu L, Qi M, Liu J, Hu K, Lin W, et al. GANT-61 and GDC-0449 Induce Apoptosis of Prostate Cancer Stem Cells Through a GLI-Dependent Mechanism. J Cell Biochem (2018) 119(4):3641–52. doi: 10.1002/jcb.26572
128. Esaki T, Hirai F, Makiyama A, Seto T, Bando H, Naito Y, et al. Phase I Dose-Escalation Study of Capmatinib (INC280) in Japanese Patients With Advanced Solid Tumors. Cancer Sci (2019) 110(4):1340–51. doi: 10.1111/cas.13956
129. Kahlert UD, Suwala AK, Koch K, Natsumeda M, Orr BA, Hayashi M, et al. Pharmacologic Wnt Inhibition Reduces Proliferation, Survival, and Clonogenicity of Glioblastoma Cells. J Neuropathol Exp Neurol (2015) 74(9):889–900. doi: 10.1097/NEN.0000000000000227
130. Cheng Y, Phoon YP, Jin X, Chong SY, Ip JC, Wong BW, et al. Wnt-C59 Arrests Stemness and Suppresses Growth of Nasopharyngeal Carcinoma in Mice by Inhibiting the Wnt Pathway in the Tumor Microenvironment. Oncotarget (2015) 6(16):14428–39. doi: 10.18632/oncotarget.3982
131. Wang G, Shen J, Sun J, Jiang Z, Fan J, Wang H, et al. Cyclophilin A Maintains Glioma-Initiating Cell Stemness by Regulating Wnt/beta-Catenin Signaling. Clin Cancer Res (2017) 23(21):6640–9. doi: 10.1158/1078-0432.CCR-17-0774
132. Veschi V, Mangiapane LR, Nicotra A, Di Franco S, Scavo E, Apuzzo T, et al. Targeting Chemoresistant Colorectal Cancer via Systemic Administration of a BMP7 Variant. Oncogene (2020) 39(5):987–1003. doi: 10.1038/s41388-019-1047-4
133. Santos AM, Jung J, Aziz N, Kissil JL, Pure E. Targeting Fibroblast Activation Protein Inhibits Tumor Stromagenesis and Growth in Mice. J Clin Invest (2009) 119(12):3613–25. doi: 10.1172/JCI38988
134. Deng T, Lyon CJ, Bergin S, Caligiuri MA, Hsueh WA. Obesity, Inflammation, and Cancer. Annu Rev Pathol (2016) 11:421–49. doi: 10.1146/annurev-pathol-012615-044359
135. Lo A, Wang LS, Scholler J, Monslow J, Avery D, Newick K, et al. Tumor-Promoting Desmoplasia Is Disrupted by Depleting FAP-Expressing Stromal Cells. Cancer Res (2015) 75(14):2800–10. doi: 10.1158/0008-5472.CAN-14-3041
136. Lee MJ, Wu Y, Fried SK. Adipose Tissue Heterogeneity: Implication of Depot Differences in Adipose Tissue for Obesity Complications. Mol Aspects Med (2013) 34(1):1–11. doi: 10.1016/j.mam.2012.10.001
137. Petrovic N, Walden TB, Shabalina IG, Timmons JA, Cannon B, Nedergaard J. Chronic Peroxisome Proliferator-Activated Receptor Gamma (PPARgamma) Activation of Epididymally Derived White Adipocyte Cultures Reveals a Population of Thermogenically Competent, UCP1-Containing Adipocytes Molecularly Distinct From Classic Brown Adipocytes. J Biol Chem (2010) 285(10):7153–64. doi: 10.1074/jbc.M109.053942
138. Kothari C, Diorio C, Durocher F. The Importance of Breast Adipose Tissue in Breast Cancer. Int J Mol Sci (2020) 21(16). doi: 10.3390/ijms21165760
139. Rajala MW, Scherer PE. Minireview: The Adipocyte–at the Crossroads of Energy Homeostasis, Inflammation, and Atherosclerosis. Endocrinology (2003) 144(9):3765–73. doi: 10.1210/en.2003-0580
140. MacDougald OA, Burant CF. The Rapidly Expanding Family of Adipokines. Cell Metab (2007) 6(3):159–61. doi: 10.1016/j.cmet.2007.08.010
141. Wang Y, Tang B, Long L, Luo P, Xiang W, Li X, et al. Improvement of Obesity-Associated Disorders by a Small-Molecule Drug Targeting Mitochondria of Adipose Tissue Macrophages. Nat Commun (2021) 12(1):102. doi: 10.1038/s41467-020-20315-9
142. Collaboration NCDRF. Trends in Adult Body-Mass Index in 200 Countries From 1975 to 2014: A Pooled Analysis of 1698 Population-Based Measurement Studies With 19.2 Million Participants. Lancet (2016) 387(10026):1377–96. doi: 10.1016/S0140-6736(16)30054-X
143. Kyrgiou M, Kalliala I, Markozannes G, Gunter MJ, Paraskevaidis E, Gabra H, et al. Adiposity and Cancer at Major Anatomical Sites: Umbrella Review of the Literature. BMJ (2017) 356:j477. doi: 10.1136/bmj.j477
144. Lauby-Secretan B, Scoccianti C, Loomis D, Grosse Y, Bianchini F, Straif K, et al. Body Fatness and Cancer–Viewpoint of the IARC Working Group. N Engl J Med (2016) 375(8):794–8. doi: 10.1056/NEJMsr1606602
146. Bochet L, Meulle A, Imbert S, Salles B, Valet P, Muller C. Cancer-Associated Adipocytes Promotes Breast Tumor Radioresistance. Biochem Biophys Res Commun (2011) 411(1):102–6. doi: 10.1016/j.bbrc.2011.06.101
147. Angelo LS, Kurzrock R. Vascular Endothelial Growth Factor and Its Relationship to Inflammatory Mediators. Clin Cancer Res (2007) 13(10):2825–30. doi: 10.1158/1078-0432.CCR-06-2416
148. Incio J, Ligibel JA, McManus DT, Suboj P, Jung K, Kawaguchi K, et al. Obesity Promotes Resistance to Anti-VEGF Therapy in Breast Cancer by Up-Regulating IL-6 and Potentially FGF-2. Sci Transl Med (2018) 10(432). doi: 10.1126/scitranslmed.aag0945
149. Picon-Ruiz M, Morata-Tarifa C, Valle-Goffin JJ, Friedman ER, Slingerland JM. Obesity and Adverse Breast Cancer Risk and Outcome: Mechanistic Insights and Strategies for Intervention. CA Cancer J Clin (2017) 67(5):378–97. doi: 10.3322/caac.21405
150. Quail DF, Dannenberg AJ. The Obese Adipose Tissue Microenvironment in Cancer Development and Progression. Nat Rev Endocrinol (2019) 15(3):139–54. doi: 10.1038/s41574-018-0126-x
151. Wu Q, Li B, Li J, Sun S, Yuan J, Sun S. Cancer-Associated Adipocytes as Immunomodulators in Cancer. Biomark Res (2021) 9(1):2. doi: 10.1186/s40364-020-00257-6
152. Picon-Ruiz M, Pan C, Drews-Elger K, Jang K, Besser AH, Zhao D, et al. Interactions Between Adipocytes and Breast Cancer Cells Stimulate Cytokine Production and Drive Src/Sox2/miR-302b-Mediated Malignant Progression. Cancer Res (2016) 76(2):491–504. doi: 10.1158/0008-5472.CAN-15-0927
153. Wolfson B, Eades G, Zhou Q. Adipocyte Activation of Cancer Stem Cell Signaling in Breast Cancer. World J Biol Chem (2015) 6(2):39–47. doi: 10.4331/wjbc.v6.i2.39
154. Li H, Feng Z, He ML. Lipid Metabolism Alteration Contributes to and Maintains the Properties of Cancer Stem Cells. Theranostics (2020) 10(16):7053–69. doi: 10.7150/thno.41388
155. Mao J, Hu X, Xiao Y, Yang C, Ding Y, Hou N, et al. Overnutrition Stimulates Intestinal Epithelium Proliferation Through Beta-Catenin Signaling in Obese Mice. Diabetes (2013) 62(11):3736–46. doi: 10.2337/db13-0035
156. Chen D, Liu S, Ma H, Liang X, Ma H, Yan X, et al. Paracrine Factors From Adipose-Mesenchymal Stem Cells Enhance Metastatic Capacity Through Wnt Signaling Pathway in a Colon Cancer Cell Co-Culture Model. Cancer Cell Int (2015) 15:42. doi: 10.1186/s12935-015-0198-9
157. Wang YY, Attane C, Milhas D, Dirat B, Dauvillier S, Guerard A, et al. Mammary Adipocytes Stimulate Breast Cancer Invasion Through Metabolic Remodeling of Tumor Cells. JCI Insight (2017) 2(4):e87489. doi: 10.1172/jci.insight.87489
158. Tirinato L, Liberale C, Di Franco S, Candeloro P, Benfante A, La Rocca R, et al. Lipid Droplets: A New Player in Colorectal Cancer Stem Cells Unveiled by Spectroscopic Imaging. Stem Cells (2015) 33(1):35–44. doi: 10.1002/stem.1837
159. Mukherjee A, Kenny HA, Lengyel E. Unsaturated Fatty Acids Maintain Cancer Cell Stemness. Cell Stem Cell (2017) 20(3):291–2. doi: 10.1016/j.stem.2017.02.008
160. Li J, Condello S, Thomes-Pepin J, Ma X, Xia Y, Hurley TD, et al. Lipid Desaturation Is a Metabolic Marker and Therapeutic Target of Ovarian Cancer Stem Cells. Cell Stem Cell (2017) 20(3):303–14 e5. doi: 10.1016/j.stem.2016.11.004
161. Bezuidenhout N, Shoshan M. A Shifty Target: Tumor-Initiating Cells and Their Metabolism. Int J Mol Sci (2019) 20(21). doi: 10.3390/ijms20215370
162. Potze L, Di Franco S, Grandela C, Pras-Raves ML, Picavet DI, van Veen HA, et al. Betulinic Acid Induces a Novel Cell Death Pathway That Depends on Cardiolipin Modification. Oncogene (2016) 35(4):427–37. doi: 10.1038/onc.2015.102
163. Potze L, di Franco S, Kessler JH, Stassi G, Medema JP. Betulinic Acid Kills Colon Cancer Stem Cells. Curr Stem Cell Res Ther (2016) 11(5):427–33. doi: 10.2174/1574888X11666151203223512
164. Cao Y. Adipocyte and Lipid Metabolism in Cancer Drug Resistance. J Clin Invest (2019) 129(8):3006–17. doi: 10.1172/JCI127201
165. Wu Q, Li B, Li Z, Li J, Sun S, Sun S. Cancer-Associated Adipocytes: Key Players in Breast Cancer Progression. J Hematol Oncol (2019) 12(1):95. doi: 10.1186/s13045-019-0778-6
166. Dirat B, Bochet L, Dabek M, Daviaud D, Dauvillier S, Majed B, et al. Cancer-Associated Adipocytes Exhibit an Activated Phenotype and Contribute to Breast Cancer Invasion. Cancer Res (2011) 71(7):2455–65. doi: 10.1158/0008-5472.CAN-10-3323
167. Wu Q, Li B, Sun S, Sun S. Unraveling Adipocytes and Cancer Links: Is There a Role for Senescence? Front Cell Dev Biol (2020) 8:282. doi: 10.3389/fcell.2020.00282
168. Renehan AG, Harvie M, Cutress RI, Leitzmann M, Pischon T, Howell S, et al. How to Manage the Obese Patient With Cancer. J Clin Oncol (2016) 34(35):4284–94. doi: 10.1200/JCO.2016.69.1899
169. Colleoni M, Li S, Gelber RD, Price KN, Coates AS, Castiglione-Gertsch M, et al. Relation Between Chemotherapy Dose, Oestrogen Receptor Expression, and Body-Mass Index. Lancet (2005) 366(9491):1108–10. doi: 10.1016/S0140-6736(05)67110-3
170. Mentoor I, Engelbrecht AM, van Jaarsveld PJ, Nell T. Chemoresistance: Intricate Interplay Between Breast Tumor Cells and Adipocytes in the Tumor Microenvironment. Front Endocrinol (Lausanne) (2018) 9:758. doi: 10.3389/fendo.2018.00758
171. Hourdequin KC, Schpero WL, McKenna DR, Piazik BL, Larson RJ. Toxic Effect of Chemotherapy Dosing Using Actual Body Weight in Obese Versus Normal-Weight Patients: A Systematic Review and Meta-Analysis. Ann Oncol (2013) 24(12):2952–62. doi: 10.1093/annonc/mdt294
172. Osman MA, Hennessy BT. Obesity Correlation With Metastases Development and Response to First-Line Metastatic Chemotherapy in Breast Cancer. Clin Med Insights Oncol (2015) 9:105–12. doi: 10.4137/CMO.S32812
173. Krisnawan VE, Stanley JA, Schwarz JK, DeNardo DG. Tumor Microenvironment as a Regulator of Radiation Therapy: New Insights Into Stromal-Mediated Radioresistance. Cancers (Basel) (2020) 12(10). doi: 10.3390/cancers12102916
174. Zhao C, Wu M, Zeng N, Xiong M, Hu W, Lv W, et al. Cancer-Associated Adipocytes: Emerging Supporters in Breast Cancer. J Exp Clin Cancer Res (2020) 39(1):156. doi: 10.1186/s13046-020-01666-z
175. Wu B, Sun X, Gupta HB, Yuan B, Li J, Ge F, et al. Adipose PD-L1 Modulates PD-1/PD-L1 Checkpoint Blockade Immunotherapy Efficacy in Breast Cancer. Oncoimmunology (2018) 7(11):e1500107. doi: 10.1080/2162402X.2018.1500107
176. Wu Q, Sun S, Li Z, Yang Q, Li B, Zhu S, et al. Tumour-Originated Exosomal miR-155 Triggers Cancer-Associated Cachexia to Promote Tumour Progression. Mol Cancer (2018) 17(1):155. doi: 10.1186/s12943-018-0899-5
177. Wu Q, Sun S, Li Z, Yang Q, Li B, Zhu S, et al. Breast Cancer-Released Exosomes Trigger Cancer-Associated Cachexia to Promote Tumor Progression. Adipocyte (2019) 8(1):31–45. doi: 10.1186/s12943-018-0899-5
178. Turdo A, Porcelli G, D’Accardo C, Franco SD, Verona F, Forte S, et al. Metabolic Escape Routes of Cancer Stem Cells and Therapeutic Opportunities. Cancers (Basel) (2020) 12(6). doi: 10.3390/cancers12061436
179. Zhang M, Di Martino JS, Bowman RL, Campbell NR, Baksh SC, Simon-Vermot T, et al. Adipocyte-Derived Lipids Mediate Melanoma Progression via FATP Proteins. Cancer Discovery (2018) 8(8):1006–25. doi: 10.1158/2159-8290.CD-17-1371
180. Ladanyi A, Mukherjee A, Kenny HA, Johnson A, Mitra AK, Sundaresan S, et al. Adipocyte-Induced CD36 Expression Drives Ovarian Cancer Progression and Metastasis. Oncogene (2018) 37(17):2285–301. doi: 10.1038/s41388-017-0093-z
181. Yang J, Li C, Shen Y, Zhou H, Shao Y, Zhu W, et al. Impact of Statin Use on Cancer-Specific Mortality and Recurrence: A Meta-Analysis of 60 Observational Studies. Med (Baltimore) (2020) 99(14):e19596. doi: 10.1097/MD.0000000000019596
182. Masko EM, Alfaqih MA, Solomon KR, Barry WT, Newgard CB, Muehlbauer MJ, et al. Evidence for Feedback Regulation Following Cholesterol Lowering Therapy in a Prostate Cancer Xenograft Model. Prostate (2017) 77(5):446–57. doi: 10.1002/pros.23282
183. Shirakami Y, Sakai H, Shimizu M. Retinoid Roles in Blocking Hepatocellular Carcinoma. Hepatobil Surg Nutr (2015) 4(4):222–8. doi: 10.3978/j.issn.2304-3881.2015.05.01
184. Park EJ, Lee JH, Yu GY, He G, Ali SR, Holzer RG, et al. Dietary and Genetic Obesity Promote Liver Inflammation and Tumorigenesis by Enhancing IL-6 and TNF Expression. Cell (2010) 140(2):197–208. doi: 10.1016/j.cell.2009.12.052
185. Klil-Drori AJ, Azoulay L, Pollak MN. Cancer, Obesity, Diabetes, and Antidiabetic Drugs: Is the Fog Clearing? Nat Rev Clin Oncol (2017) 14(2):85–99. doi: 10.1038/nrclinonc.2016.120
186. Saraei P, Asadi I, Kakar MA, Moradi-Kor N. The Beneficial Effects of Metformin on Cancer Prevention and Therapy: A Comprehensive Review of Recent Advances. Cancer Manag Res (2019) 11:3295–313. doi: 10.2147/CMAR.S200059
187. Hirsch HA, Iliopoulos D, Tsichlis PN, Struhl K. Metformin Selectively Targets Cancer Stem Cells, and Acts Together With Chemotherapy to Block Tumor Growth and Prolong Remission. Cancer Res (2009) 69(19):7507–11. doi: 10.1158/0008-5472.CAN-09-2994
188. Codony-Servat J, Rosell R. Cancer Stem Cells and Immunoresistance: Clinical Implications and Solutions. Transl Lung Cancer Res (2015) 4(6):689–703. doi: 10.3978/j.issn.2218-6751.2015.12.11
189. Gajewski TF, Schreiber H, Fu YX. Innate and Adaptive Immune Cells in the Tumor Microenvironment. Nat Immunol (2013) 14(10):1014–22. doi: 10.1038/ni.2703
190. Pages F, Galon J, Dieu-Nosjean MC, Tartour E, Sautes-Fridman C, Fridman WH. Immune Infiltration in Human Tumors: A Prognostic Factor That Should Not be Ignored. Oncogene (2010) 29(8):1093–102. doi: 10.1038/onc.2009.416
191. Grossenbacher SK, Canter RJ, Murphy WJ. Natural Killer Cell Immunotherapy to Target Stem-Like Tumor Cells. J Immunother Cancer (2016) 4:19. doi: 10.1186/s40425-016-0124-2
192. Cristiani CM, Turdo A, Ventura V, Apuzzo T, Capone M, Madonna G, et al. Accumulation of Circulating CCR7(+) Natural Killer Cells Marks Melanoma Evolution and Reveals a CCL19-Dependent Metastatic Pathway. Cancer Immunol Res (2019) 7(5):841–52. doi: 10.1158/2326-6066.CIR-18-0651
193. Melaiu O, Lucarini V, Cifaldi L, Fruci D. Influence of the Tumor Microenvironment on NK Cell Function in Solid Tumors. Front Immunol (2019) 10:3038. doi: 10.3389/fimmu.2019.03038
194. Tallerico R, Todaro M, Di Franco S, Maccalli C, Garofalo C, Sottile R, et al. Human NK Cells Selective Targeting of Colon Cancer-Initiating Cells: A Role for Natural Cytotoxicity Receptors and MHC Class I Molecules. J Immunol (2013) 190(5):2381–90. doi: 10.4049/jimmunol.1201542
195. Spiegel A, Brooks MW, Houshyar S, Reinhardt F, Ardolino M, Fessler E, et al. Neutrophils Suppress Intraluminal NK Cell-Mediated Tumor Cell Clearance and Enhance Extravasation of Disseminated Carcinoma Cells. Cancer Discovery (2016) 6(6):630–49. doi: 10.1158/2159-8290.CD-15-1157
196. Madden EC, Gorman AM, Logue SE, Samali A. Tumour Cell Secretome in Chemoresistance and Tumour Recurrence. Trends Cancer (2020) 6(6):489–505. doi: 10.1016/j.trecan.2020.02.020
197. Larionova I, Cherdyntseva N, Liu T, Patysheva M, Rakina M, Kzhyshkowska J. Interaction of Tumor-Associated Macrophages and Cancer Chemotherapy. Oncoimmunology (2019) 8(7):1596004. doi: 10.1080/2162402X.2019.1596004
198. Nawaz A, Aminuddin A, Kado T, Takikawa A, Yamamoto S, Tsuneyama K, et al. CD206(+) M2-Like Macrophages Regulate Systemic Glucose Metabolism by Inhibiting Proliferation of Adipocyte Progenitors. Nat Commun (2017) 8(1):286. doi: 10.1038/s41467-017-00231-1
199. Williams CB, Yeh ES, Soloff AC. Tumor-Associated Macrophages: Unwitting Accomplices in Breast Cancer Malignancy. NPJ Breast Cancer (2016) 2. doi: 10.1038/npjbcancer.2015.25
200. Rodriguez-Garcia A, Lynn RC, Poussin M, Eiva MA, Shaw LC, O’Connor RS, et al. CAR-T Cell-Mediated Depletion of Immunosuppressive Tumor-Associated Macrophages Promotes Endogenous Antitumor Immunity and Augments Adoptive Immunotherapy. Nat Commun (2021) 12(1):877. doi: 10.1038/s41467-021-20893-2
201. Kurahara H, Takao S, Kuwahata T, Nagai T, Ding Q, Maeda K, et al. Clinical Significance of Folate Receptor Beta-Expressing Tumor-Associated Macrophages in Pancreatic Cancer. Ann Surg Oncol (2012) 19(7):2264–71. doi: 10.1245/s10434-012-2263-0
202. Cassetta L, Fragkogianni S, Sims AH, Swierczak A, Forrester LM, Zhang H, et al. Human Tumor-Associated Macrophage and Monocyte Transcriptional Landscapes Reveal Cancer-Specific Reprogramming, Biomarkers, and Therapeutic Targets. Cancer Cell (2019) 35(4):588–602 e10. doi: 10.1016/j.ccell.2019.02.009
203. Fico F, Santamaria-Martinez A. The Tumor Microenvironment as a Driving Force of Breast Cancer Stem Cell Plasticity. Cancers (Basel) (2020) 12(12). doi: 10.3390/cancers12123863
204. Lu H, Clauser KR, Tam WL, Frose J, Ye X, Eaton EN, et al. A Breast Cancer Stem Cell Niche Supported by Juxtacrine Signalling From Monocytes and Macrophages. Nat Cell Biol (2014) 16(11):1105–17. doi: 10.1038/ncb3041
205. Shao XJ, Xiang SF, Chen YQ, Zhang N, Cao J, Zhu H, et al. Inhibition of M2-Like Macrophages by All-Trans Retinoic Acid Prevents Cancer Initiation and Stemness in Osteosarcoma Cells. Acta Pharmacol Sin (2019) 40(10):1343–50. doi: 10.1038/s41401-019-0262-4
206. Guo X, Zhao Y, Yan H, Yang Y, Shen S, Dai X, et al. Single Tumor-Initiating Cells Evade Immune Clearance by Recruiting Type II Macrophages. Genes Dev (2017) 31(3):247–59. doi: 10.1101/gad.294348.116
207. Valeta-Magara A, Gadi A, Volta V, Walters B, Arju R, Giashuddin S, et al. Inflammatory Breast Cancer Promotes Development of M2 Tumor-Associated Macrophages and Cancer Mesenchymal Cells Through a Complex Chemokine Network. Cancer Res (2019) 79(13):3360–71. doi: 10.1158/0008-5472.CAN-17-2158
208. Liu WM, Fowler DW, Smith P, Dalgleish AG. Pre-Treatment With Chemotherapy Can Enhance the Antigenicity and Immunogenicity of Tumours by Promoting Adaptive Immune Responses. Br J Cancer (2010) 102(1):115–23. doi: 10.1038/sj.bjc.6605465
209. Galluzzi L, Buque A, Kepp O, Zitvogel L, Kroemer G. Immunological Effects of Conventional Chemotherapy and Targeted Anticancer Agents. Cancer Cell (2015) 28(6):690–714. doi: 10.1016/j.ccell.2015.10.012
210. Kaneno R, Shurin GV, Kaneno FM, Naiditch H, Luo J, Shurin MR. Chemotherapeutic Agents in Low Noncytotoxic Concentrations Increase Immunogenicity of Human Colon Cancer Cells. Cell Oncol (Dordr) (2011) 34(2):97–106. doi: 10.1007/s13402-010-0005-5
211. Denkert C, von Minckwitz G, Darb-Esfahani S, Lederer B, Heppner BI, Weber KE, et al. Tumour-Infiltrating Lymphocytes and Prognosis in Different Subtypes of Breast Cancer: A Pooled Analysis of 3771 Patients Treated With Neoadjuvant Therapy. Lancet Oncol (2018) 19(1):40–50. doi: 10.1016/S1470-2045(17)30904-X
212. Zingoni A, Fionda C, Borrelli C, Cippitelli M, Santoni A, Soriani A. Natural Killer Cell Response to Chemotherapy-Stressed Cancer Cells: Role in Tumor Immunosurveillance. Front Immunol (2017) 8:1194. doi: 10.3389/fimmu.2017.01194
213. Zhang X, Wang D, Li Z, Jiao D, Jin L, Cong J, et al. Low-Dose Gemcitabine Treatment Enhances Immunogenicity and Natural Killer Cell-Driven Tumor Immunity in Lung Cancer. Front Immunol (2020) 11:331. doi: 10.3389/fimmu.2020.00331
214. Gurlevik E, Fleischmann-Mundt B, Brooks J, Demir IE, Steiger K, Ribback S, et al. Administration of Gemcitabine After Pancreatic Tumor Resection in Mice Induces an Antitumor Immune Response Mediated by Natural Killer Cells. Gastroenterology (2016) 151(2):338–50.e7. doi: 10.1053/j.gastro.2016.05.004
215. Dawson DW, Fernandez-Zapico ME. Gemcitabine Activates Natural Killer Cells to Attenuate Pancreatic Cancer Recurrence. Gastroenterology (2016) 151(2):234–7. doi: 10.1053/j.gastro.2016.06.032
216. Waldman AD, Fritz JM, Lenardo MJ. A Guide to Cancer Immunotherapy: From T Cell Basic Science to Clinical Practice. Nat Rev Immunol (2020) 20(11):651–68. doi: 10.1038/s41577-020-0306-5
217. Galluzzi L, Humeau J, Buque A, Zitvogel L, Kroemer G. Immunostimulation With Chemotherapy in the Era of Immune Checkpoint Inhibitors. Nat Rev Clin Oncol (2020) 17(12):725–41. doi: 10.1038/s41571-020-0413-z
218. Orecchioni S, Talarico G, Labanca V, Calleri A, Mancuso P, Bertolini F. Vinorelbine, Cyclophosphamide and 5-FU Effects on the Circulating and Intratumoural Landscape of Immune Cells Improve Anti-PD-L1 Efficacy in Preclinical Models of Breast Cancer and Lymphoma. Br J Cancer (2018) 118(10):1329–36. doi: 10.1038/s41416-018-0076-z
219. Eriksson E, Wenthe J, Irenaeus S, Loskog A, Ullenhag G. Gemcitabine Reduces MDSCs, Tregs and TGFbeta-1 While Restoring the Teff/Treg Ratio in Patients With Pancreatic Cancer. J Transl Med (2016) 14(1):282. doi: 10.1186/s12967-016-1037-z
220. Chang LS, Yan WL, Chang YW, Yeh YC, Chen HW, Leng CH, et al. Gemcitabine Enhances Antitumor Efficacy of Recombinant Lipoimmunogen-Based Immunotherapy. Oncoimmunology (2016) 5(3):e1095433. doi: 10.1080/2162402X.2015.1095433
221. Riley RS, June CH, Langer R, Mitchell MJ. Delivery Technologies for Cancer Immunotherapy. Nat Rev Drug Discov (2019) 18(3):175–96. doi: 10.1038/s41573-018-0006-z
222. Fumet JD, Limagne E, Thibaudin M, Ghiringhelli F. Immunogenic Cell Death and Elimination of Immunosuppressive Cells: A Double-Edged Sword of Chemotherapy. Cancers (Basel) (2020) 12(9). doi: 10.3390/cancers12092637
223. Schaer DA, Geeganage S, Amaladas N, Lu ZH, Rasmussen ER, Sonyi A, et al. The Folate Pathway Inhibitor Pemetrexed Pleiotropically Enhances Effects of Cancer Immunotherapy. Clin Cancer Res (2019) 25(23):7175–88. doi: 10.1158/1078-0432.CCR-19-0433
224. Tsai TF, Lin JF, Lin YC, Chou KY, Chen HE, Ho CY, et al. Cisplatin Contributes to Programmed Death-Ligand 1 Expression in Bladder Cancer Through ERK1/2-AP-1 Signaling Pathway. Biosci Rep (2019) 39(9). doi: 10.1042/BSR20190362
225. Viteri S, Cabrera-Galvez C, Rosell R. Keynote 407: The Combination of Pembrolizumab and Chemotherapy Cracks the Shell of Squamous Cell Lung Cancer. Transl Lung Cancer Res (2020) 9(3):828–32. doi: 10.21037/tlcr-20-400
226. Cortes J, Cescon DW, Rugo HS, Nowecki Z, Im SA, Yusof MM, et al. Pembrolizumab Plus Chemotherapy Versus Placebo Plus Chemotherapy for Previously Untreated Locally Recurrent Inoperable or Metastatic Triple-Negative Breast Cancer (KEYNOTE-355): A Randomised, Placebo-Controlled, Double-Blind, Phase 3 Clinical Trial. Lancet (2020) 396(10265):1817–28. doi: 10.1016/S0140-6736(20)32531-9
227. Sinicrope FA, Ou F-S, Zemla T, Nixon AB, Mody K, Levasseur A, et al. Randomized Trial of Standard Chemotherapy Alone or Combined With Atezolizumab as Adjuvant Therapy for Patients With Stage III Colon Cancer and Deficient Mismatch Repair (ATOMIC, Alliance A021502). J Clin Oncol (2019) 37(15_suppl):e15169–e. doi: 10.1200/JCO.2019.37.15_suppl.e15169
228. Rafiq S, Hackett CS, Brentjens RJ. Engineering Strategies to Overcome the Current Roadblocks in CAR T Cell Therapy. Nat Rev Clin Oncol (2020) 17(3):147–67. doi: 10.1038/s41571-019-0297-y
229. Larson RC, Maus MV. Recent Advances and Discoveries in the Mechanisms and Functions of CAR T Cells. Nat Rev Cancer (2021) 21(3):145–61. doi: 10.1038/s41568-020-00323-z
230. Alhabbab RY. Targeting Cancer Stem Cells by Genetically Engineered Chimeric Antigen Receptor T Cells. Front Genet (2020) 11:312. doi: 10.3389/fgene.2020.00312
231. D’Aloia MM, Zizzari IG, Sacchetti B, Pierelli L, Alimandi M. CAR-T Cells: The Long and Winding Road to Solid Tumors. Cell Death Dis (2018) 9(3):282. doi: 10.1038/s41419-018-0278-6
232. Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in Children and Young Adults With B-Cell Lymphoblastic Leukemia. N Engl J Med (2018) 378(5):439–48. doi: 10.1056/NEJMoa1709866
233. Schuster SJ, Bishop MR, Tam CS, Waller EK, Borchmann P, McGuirk JP, et al. Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. N Engl J Med (2019) 380(1):45–56. doi: 10.1056/NEJMoa1804980
234. Wagner J, Wickman E, DeRenzo C, Gottschalk S. CAR T Cell Therapy for Solid Tumors: Bright Future or Dark Reality? Mol Ther (2020) 28(11):2320–39. doi: 10.1016/j.ymthe.2020.09.015
235. Zhang BL, Li D, Gong YL, Huang Y, Qin DY, Jiang L, et al. Preclinical Evaluation of Chimeric Antigen Receptor-Modified T Cells Specific to Epithelial Cell Adhesion Molecule for Treating Colorectal Cancer. Hum Gene Ther (2019) 30(4):402–12. doi: 10.1089/hum.2018.229
236. Wang Y, Chen M, Wu Z, Tong C, Dai H, Guo Y, et al. CD133-Directed CAR T Cells for Advanced Metastasis Malignancies: A Phase I Trial. Oncoimmunology (2018) 7(7):e1440169. doi: 10.1080/2162402X.2018.1440169
237. Santomasso BD, Park JH, Salloum D, Riviere I, Flynn J, Mead E, et al. Clinical and Biological Correlates of Neurotoxicity Associated With CAR T-Cell Therapy in Patients With B-Cell Acute Lymphoblastic Leukemia. Cancer Discovery (2018) 8(8):958–71. doi: 10.1158/2159-8290.CD-17-1319
238. Neelapu SS, Tummala S, Kebriaei P, Wierda W, Gutierrez C, Locke FL, et al. Chimeric Antigen Receptor T-Cell Therapy - Assessment and Management of Toxicities. Nat Rev Clin Oncol (2018) 15(1):47–62. doi: 10.1038/nrclinonc.2017.148
239. Folkins C, Shaked Y, Man S, Tang T, Lee CR, Zhu Z, et al. Glioma Tumor Stem-Like Cells Promote Tumor Angiogenesis and Vasculogenesis via Vascular Endothelial Growth Factor and Stromal-Derived Factor 1. Cancer Res (2009) 69(18):7243–51. doi: 10.1158/0008-5472.CAN-09-0167
240. Pugh CW, Ratcliffe PJ. Regulation of Angiogenesis by Hypoxia: Role of the HIF System. Nat Med (2003) 9(6):677–84. doi: 10.1038/nm0603-677
241. Zhang YW, Su Y, Volpert OV, Vande Woude GF. Hepatocyte Growth Factor/Scatter Factor Mediates Angiogenesis Through Positive VEGF and Negative Thrombospondin 1 Regulation. Proc Natl Acad Sci USA (2003) 100(22):12718–23. doi: 10.1073/pnas.2135113100
242. Garcia J, Hurwitz HI, Sandler AB, Miles D, Coleman RL, Deurloo R, et al. Bevacizumab (Avastin(R)) in Cancer Treatment: A Review of 15 Years of Clinical Experience and Future Outlook. Cancer Treat Rev (2020) 86:102017. doi: 10.1016/j.ctrv.2020.102017
243. Ricci-Vitiani L, Pallini R, Biffoni M, Todaro M, Invernici G, Cenci T, et al. Tumour Vascularization via Endothelial Differentiation of Glioblastoma Stem-Like Cells. Nature (2010) 468(7325):824–8. doi: 10.1038/nature09557
244. De Palma M, Biziato D, Petrova TV. Microenvironmental Regulation of Tumour Angiogenesis. Nat Rev Cancer (2017) 17(8):457–74. doi: 10.1038/nrc.2017.51
245. Schaaf MB, Garg AD, Agostinis P. Defining the Role of the Tumor Vasculature in Antitumor Immunity and Immunotherapy. Cell Death Dis (2018) 9(2):115. doi: 10.1038/s41419-017-0061-0
246. Hanahan D, Weinberg RA. Hallmarks of Cancer: The Next Generation. Cell (2011) 144(5):646–74. doi: 10.1016/j.cell.2011.02.013
247. Folkman J. Tumor Angiogenesis: Therapeutic Implications. N Engl J Med (1971) 285(21):1182–6. doi: 10.1056/NEJM197111182852108
248. Sawada J, Urakami T, Li F, Urakami A, Zhu W, Fukuda M, et al. Small GTPase R-Ras Regulates Integrity and Functionality of Tumor Blood Vessels. Cancer Cell (2012) 22(2):235–49. doi: 10.1016/j.ccr.2012.06.013
249. Morikawa S, Baluk P, Kaidoh T, Haskell A, Jain RK, McDonald DM. Abnormalities in Pericytes on Blood Vessels and Endothelial Sprouts in Tumors. Am J Pathol (2002) 160(3):985–1000. doi: 10.1016/S0002-9440(10)64920-6
250. Maniotis AJ, Folberg R, Hess A, Seftor EA, Gardner LM, Pe’er J, et al. Vascular Channel Formation by Human Melanoma Cells In Vivo and In Vitro: Vasculogenic Mimicry. Am J Pathol (1999) 155(3):739–52. doi: 10.1016/S0002-9440(10)65173-5
251. Bussolati B, Grange C, Sapino A, Camussi G. Endothelial Cell Differentiation of Human Breast Tumour Stem/Progenitor Cells. J Cell Mol Med (2009) 13(2):309–19. doi: 10.1111/j.1582-4934.2008.00338.x
252. Cheng L, Huang Z, Zhou W, Wu Q, Donnola S, Liu JK, et al. Glioblastoma Stem Cells Generate Vascular Pericytes to Support Vessel Function and Tumor Growth. Cell (2013) 153(1):139–52. doi: 10.1016/j.cell.2013.02.021
253. Shangguan W, Fan C, Chen X, Lu R, Liu Y, Li Y, et al. Endothelium Originated From Colorectal Cancer Stem Cells Constitute Cancer Blood Vessels. Cancer Sci (2017) 108(7):1357–67. doi: 10.1111/cas.13262
254. Calabrese C, Poppleton H, Kocak M, Hogg TL, Fuller C, Hamner B, et al. A Perivascular Niche for Brain Tumor Stem Cells. Cancer Cell (2007) 11(1):69–82. doi: 10.1016/j.ccr.2006.11.020
255. McCoy MG, Nyanyo D, Hung CK, Goerger JP, RZ W, Williams RM, et al. Endothelial Cells Promote 3D Invasion of GBM by IL-8-Dependent Induction of Cancer Stem Cell Properties. Sci Rep (2019) 9(1):9069. doi: 10.1038/s41598-019-45535-y
256. Wang R, Bhattacharya R, Ye X, Fan F, Boulbes DR, Xia L, et al. Endothelial Cells Activate the Cancer Stem Cell-Associated NANOGP8 Pathway in Colorectal Cancer Cells in a Paracrine Fashion. Mol Oncol (2017) 11(8):1023–34. doi: 10.1002/1878-0261.12071
257. Akil A, Gutierrez-Garcia AK, Guenter R, Rose JB, Beck AW, Chen H, et al. Notch Signaling in Vascular Endothelial Cells, Angiogenesis, and Tumor Progression: An Update and Prospective. Front Cell Dev Biol (2021) 9:642352. doi: 10.3389/fcell.2021.642352
258. Zhu TS, Costello MA, Talsma CE, Flack CG, Crowley JG, Hamm LL, et al. Endothelial Cells Create a Stem Cell Niche in Glioblastoma by Providing NOTCH Ligands That Nurture Self-Renewal of Cancer Stem-Like Cells. Cancer Res (2011) 71(18):6061–72. doi: 10.1158/0008-5472.CAN-10-4269
259. Ghiabi P, Jiang J, Pasquier J, Maleki M, Abu-Kaoud N, Rafii S, et al. Endothelial Cells Provide a Notch-Dependent Pro-Tumoral Niche for Enhancing Breast Cancer Survival, Stemness and Pro-Metastatic Properties. PloS One (2014) 9(11):e112424. doi: 10.1371/journal.pone.0112424
260. Jiang H, Zhou C, Zhang Z, Wang Q, Wei H, Shi W, et al. Jagged1-Notch1-Deployed Tumor Perivascular Niche Promotes Breast Cancer Stem Cell Phenotype Through Zeb1. Nat Commun (2020) 11(1):5129. doi: 10.1038/s41467-020-18860-4
261. Ma J, Waxman DJ. Combination of Antiangiogenesis With Chemotherapy for More Effective Cancer Treatment. Mol Cancer Ther (2008) 7(12):3670–84. doi: 10.1158/1535-7163.MCT-08-0715
262. Tewari KS, Sill MW, Long HJ 3rd, Penson RT, Huang H, Ramondetta LM, et al. Improved Survival With Bevacizumab in Advanced Cervical Cancer. N Engl J Med (2014) 370(8):734–43. doi: 10.1056/NEJMoa1309748
263. Ferrara N, Hillan KJ, Gerber HP, Novotny W. Discovery and Development of Bevacizumab, an Anti-VEGF Antibody for Treating Cancer. Nat Rev Drug Discovery (2004) 3(5):391–400. doi: 10.1038/nrd1381
264. Aalders KC, Tryfonidis K, Senkus E, Cardoso F. Anti-Angiogenic Treatment in Breast Cancer: Facts, Successes, Failures and Future Perspectives. Cancer Treat Rev (2017) 53:98–110. doi: 10.1016/j.ctrv.2016.12.009
265. Lu KV, Chang JP, Parachoniak CA, Pandika MM, Aghi MK, Meyronet D, et al. VEGF Inhibits Tumor Cell Invasion and Mesenchymal Transition Through a MET/VEGFR2 Complex. Cancer Cell (2012) 22(1):21–35. doi: 10.1016/j.ccr.2012.05.037
266. Lupo G, Caporarello N, Olivieri M, Cristaldi M, Motta C, Bramanti V, et al. Anti-Angiogenic Therapy in Cancer: Downsides and New Pivots for Precision Medicine. Front Pharmacol (2016) 7:519. doi: 10.3389/fphar.2016.00519
267. Hida K, Ohga N, Akiyama K, Maishi N, Hida Y. Heterogeneity of Tumor Endothelial Cells. Cancer Sci (2013) 104(11):1391–5. doi: 10.1111/cas.12251
268. Lizarraga-Verdugo E, Avendano-Felix M, Bermudez M, Ramos-Payan R, Perez-Plasencia C, Aguilar-Medina M. Cancer Stem Cells and Its Role in Angiogenesis and Vasculogenic Mimicry in Gastrointestinal Cancers. Front Oncol (2020) 10:413. doi: 10.3389/fonc.2020.00413
269. Al-Abd AM, Alamoudi AJ, Abdel-Naim AB, Neamatallah TA, Ashour OM. Anti-Angiogenic Agents for the Treatment of Solid Tumors: Potential Pathways, Therapy and Current Strategies - A Review. J Adv Res (2017) 8(6):591–605. doi: 10.1016/j.jare.2017.06.006
270. Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF, et al. Sorafenib in Advanced Hepatocellular Carcinoma. N Engl J Med (2008) 359(4):378–90. doi: 10.1056/NEJMoa0708857
271. Brose MS, Nutting CM, Jarzab B, Elisei R, Siena S, Bastholt L, et al. Sorafenib in Radioactive Iodine-Refractory, Locally Advanced or Metastatic Differentiated Thyroid Cancer: A Randomised, Double-Blind, Phase 3 Trial. Lancet (2014) 384(9940):319–28. doi: 10.1016/S0140-6736(14)60421-9
272. Dong XF, Liu TQ, Zhi XT, Zou J, Zhong JT, Li T, et al. COX-2/PGE2 Axis Regulates HIF2alpha Activity to Promote Hepatocellular Carcinoma Hypoxic Response and Reduce the Sensitivity of Sorafenib Treatment. Clin Cancer Res (2018) 24(13):3204–16. doi: 10.1158/1078-0432.CCR-17-2725
273. Chen J, Duda DG. Overcoming Sorafenib Treatment-Resistance in Hepatocellular Carcinoma: A Future Perspective at a Time of Rapidly Changing Treatment Paradigms. EBioMedicine (2020) 52:102644. doi: 10.1016/j.ebiom.2020.102644
274. Chen Y, Ramjiawan RR, Reiberger T, Ng MR, Hato T, Huang Y, et al. CXCR4 Inhibition in Tumor Microenvironment Facilitates Anti-Programmed Death Receptor-1 Immunotherapy in Sorafenib-Treated Hepatocellular Carcinoma in Mice. Hepatology (2015) 61(5):1591–602. doi: 10.1002/hep.27665
275. Goodman VL, Rock EP, Dagher R, Ramchandani RP, Abraham S, Gobburu JV, et al. Approval Summary: Sunitinib for the Treatment of Imatinib Refractory or Intolerant Gastrointestinal Stromal Tumors and Advanced Renal Cell Carcinoma. Clin Cancer Res (2007) 13(5):1367–73. doi: 10.1158/1078-0432.CCR-06-2328
276. Makhov P, Joshi S, Ghatalia P, Kutikov A, Uzzo RG, Kolenko VM. Resistance to Systemic Therapies in Clear Cell Renal Cell Carcinoma: Mechanisms and Management Strategies. Mol Cancer Ther (2018) 17(7):1355–64. doi: 10.1158/1535-7163.MCT-17-1299
277. Huang D, Ding Y, Zhou M, Rini BI, Petillo D, Qian CN, et al. Interleukin-8 Mediates Resistance to Antiangiogenic Agent Sunitinib in Renal Cell Carcinoma. Cancer Res (2010) 70(3):1063–71. doi: 10.1158/0008-5472.CAN-09-3965
278. Jayson GC, Kerbel R, Ellis LM, Harris AL. Antiangiogenic Therapy in Oncology: Current Status and Future Directions. Lancet (2016) 388(10043):518–29. doi: 10.1016/S0140-6736(15)01088-0
279. Gillen J, Richardson D, Moore K. Angiopoietin-1 and Angiopoietin-2 Inhibitors: Clinical Development. Curr Oncol Rep (2019) 21(3):22. doi: 10.1007/s11912-019-0771-9
Keywords: cancer stem cells, tumor microenvironment, anticancer drugs, chemoresistance, targeted therapy
Citation: Gaggianesi M, Di Franco S, Pantina VD, Porcelli G, D'Accardo C, Verona F, Veschi V, Colarossi L, Faldetta N, Pistone G, Bongiorno MR, Todaro M and Stassi G (2021) Messing Up the Cancer Stem Cell Chemoresistance Mechanisms Supported by Tumor Microenvironment. Front. Oncol. 11:702642. doi: 10.3389/fonc.2021.702642
Received: 29 April 2021; Accepted: 05 July 2021;
Published: 20 July 2021.
Edited by:
Nadiah Abu, National University of Malaysia, MalaysiaReviewed by:
Xiuping Yu, Louisiana State University Health Shreveport, United StatesHaojun Luo, Chongqing Medical University, China
Yibo Fan, The First Affiliated Hospital of China Medical University, China
Copyright © 2021 Gaggianesi, Di Franco, Pantina, Porcelli, D'Accardo, Verona, Veschi, Colarossi, Faldetta, Pistone, Bongiorno, Todaro and Stassi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Giorgio Stassi, Z2lvcmdpby5zdGFzc2lAdW5pcGEuaXQ=
†These authors have contributed equally to this work